

Abstract
BACKGROUND
The gut-liver axis has attracted much interest in the context of chronic liver disease pathogenesis. Prebiotics such as dietary fibers were shown to attenuate non-alcoholic fatty liver disease (NAFLD) by modulating gut microbiota. Partially hydrolyzed guar gum (PHGG), a water-soluble dietary fiber, has been reported to alleviate the symptoms of various intestinal diseases and metabolic syndromes. However, its effects on NAFLD remain to be fully elucidated.
AIM
To determine whether treatment with PHGG attenuates NAFLD development in mice through the gut-liver axis.
METHODS
Seven-week-old male C57BL/6J mice with increased intestinal permeability were fed a control or atherogenic (Ath) diet (a mouse model of NAFLD) for 8 wk, with or without 5% PHGG. Increased intestinal permeability was induced through chronic intermittent administration of low-dose dextran sulfate sodium. Body weight, liver weight, macroscopic findings in the liver, blood biochemistry [aspartate aminotransferase (AST) and alanine aminotransferase (ALT), total cholesterol, triglyceride, free fatty acids, and glucose levels], liver histology, myeloperoxidase activity in liver tissue, mRNA expression in the liver and intestine, serum endotoxin levels in the portal vein, intestinal permeability, and microbiota and short-chain fatty acid (SCFA) profiles in the cecal samples were investigated.
RESULTS
Mice with increased intestinal permeability subjected to the Ath diet showed significantly increased serum AST and ALT levels, liver fat accumulation, liver inflammatory (tumor necrosis factor-α and monocyte chemotactic protein-1) and fibrogenic (collagen 1a1 and α smooth muscle actin) marker levels, and liver myeloperoxidase activity, which were significantly attenuated by PHGG treatment. Furthermore, the Ath diet combined with increased intestinal permeability resulted in elevated portal endotoxin levels and activated toll-like receptor (TLR) 4 and TLR9 expression, confirming that intestinal permeability was significantly elevated, as observed by evaluating the lumen-to-blood clearance of fluorescein isothiocyanate-conjugated dextran. PHGG treatment did not affect fatty acid metabolism in the liver. However, it decreased lipopolysaccharide signaling through the gut-liver axis. In addition, it significantly increased the abundance of cecal Bacteroides and Clostridium subcluster XIVa. Treatment with PHGG markedly increased the levels of SCFAs, particularly, butyric acid, acetic acid, propionic acid, and formic acid, in the cecal samples.
CONCLUSION
PHGG partially prevented NAFLD development in mice through the gut-liver axis by modulating microbiota and downstream SCFA profiles.
Keywords: Non-alcoholic fatty liver disease, Partially hydrolyzed guar gum, Gut-liver axis, Intestinal barrier integrity, Microbiota, Short-chain fatty acids
Core Tip: The gut-liver axis has attracted much interest in the context of chronic liver disease pathogenesis, including non-alcoholic fatty liver disease (NAFLD). Here, by using a mouse model of NAFLD induced by an atherogenic diet combined with increased intestinal permeability, we report that partially hydrolyzed guar gum (PHGG), a water-soluble dietary fiber, prevented NAFLD development in mice partially through the gut-liver axis by modulating the microbiota and downstream short-chain fatty acid profiles, highlighting that treatment with PHGG might show great promise as a therapeutic strategy for NAFLD.
INTRODUCTION
The incidence of obesity is currently increasing owing to changing dietary habits, consequently resulting in the development of a fatty liver irrespective of alcohol consumption, a condition known as non-alcoholic fatty liver disease (NAFLD)[1]. NAFLD is an acquired metabolic stress-related liver disease that is strongly associated with obesity, dyslipidemia, hypertension, type 2 diabetes mellitus, and metabolic syndrome[2]. It was originally considered a benign disease. However, 37% of patients with NAFLD were recently reported to progress to non-alcoholic steatohepatitis (NASH) and consequent cirrhosis. Multiple studies have shown the pathogenesis and pathophysiology of NAFLD[3,4]. Various treatments such as vitamin E[5], anti-diabetic agents[6], farnesoid X receptor ligand[7], bile acid sequestrant, and apical sodium-dependent bile acid transporter inhibitors are under investigation[4], but there is no established standard treatment at present.
The mechanisms underlying NAFLD/NASH are far from being clarified and the supposed mechanisms that are at the basis of numerous attempts to cure NAFLD until now have ended, at best, in modest results[8]. The “gut-liver axis”, which is potentially involved in the pathogenesis of NAFLD, has attracted much interest in the context of chronic liver disease[9]. Serum levels of lipopolysaccharide (LPS), derived from gram-negative bacteria in the gut, are elevated in patients with NASH and in animal models of NASH[10,11]. Additionally, activation of toll-like receptor (TLR) 4 in the liver plays an important role in NASH development in mice[12]. Furthermore, increased intestinal permeability is highlighted as the primary cause of liver injury in NASH[13]. Gäbele et al[14] reported that increased intestinal permeability via dextran sulfate sodium (DSS) treatment induces colitis, increases portal LPS levels, and enhances hepatic inflammation and fibrogenesis in an experimental NASH model[14]. In addition to these studies, recent evidence strongly supports a close link between gut microbiota and NAFLD. The severity of NAFLD is reportedly associated with gut dysbiosis and a shift in the metabolic function of the gut microbiota[3]. Therefore, many NAFLD studies have focused on gut dysbiosis, microbiota composition, and on treatments targeting the gut-liver axis that include antibiotics, probiotics, prebiotics, and synbiotics[15].
Probiotic and prebiotic supplements, which are inexpensive and safe for humans, may be considered as possible interventions for NAFLD. Probiotics are live microbial food supplements or bacterial components that are suggested to improve liver function and reduce liver fat in NASH patients[16,17]. In contrast, prebiotics refer to a group of non-digestible carbohydrates that alter the composition and activity of the gut microbiota[18]. The beneficial effects of prebiotics on NAFLD have been demonstrated in a few human studies[19]. Furthermore, a recent review showed that clinical trials using prebiotics such as oligofructose, oat bran, and inulin might not be applicable[4]. Therefore, further studies are required to demonstrate the effect of prebiotics on NAFLD.
Partially hydrolyzed guar gum (PHGG) is a water-soluble dietary fiber prepared from guar gum, a galactomannan obtained from the seeds of Cyamopsis tetragonoloba through microbial endo-β-mannanase activity to reduce its high viscosity[20]. PHGG treatment has been reported to improve the symptoms associated with both constipation- and diarrhea-predominant forms of irritable bowel syndrome[21]. The beneficial effects of dietary PHGG have also been demonstrated in the treatment of cholera[22], small intestinal bacterial overgrowth[23], pediatric functional gastrointestinal disorders[24], and metabolic syndrome-related functions such as aberrant lipid and glucose metabolism[25,26]. PHGG remains undigested in the upper gastrointestinal tract and is fermented by colonic bacteria, resulting in the production of short-chain fatty acids (SCFAs), particularly butyrate. It has recently been reported that PHGG increases butyrate-producing bacteria in the intestine[27]. More recently, PHGG treatment has been reported to alter the composition of the human microbiota, along with increasing the abundance of metabolites such as butyrate, acetate, and various amino acids[28]. To this end, PHGG should exert beneficial health effects on the host through alterations to the gut microbiota and SCFA production. However, the mechanisms by which PHGG prevents NAFLD development are yet to be elucidated. In this study, we used a murine model of NAFLD induced by an atherogenic (Ath) diet and an increase in intestinal permeability, and investigated the protective mechanisms by which PHGG modulates the gut microbiota.
MATERIALS AND METHODS
Experimental design
Seven-week-old male C57BL/6J mice were purchased from Shimizu Laboratory Supplies (Kyoto, Japan). The mice were caged individually in a room maintained at 18-24 °C with 40%-70% relative humidity and a 12-h light/dark cycle. The mice were allowed free access to drinking water and were fed with rodent diet CE-2 (Nihon Clea, Tokyo, Japan) during their 1-wk acclimatization period. Mouse maintenance and all subsequent experimental procedures were performed in accordance with the National Institutes of Health guidelines for the use of experimental animals. All the programs were approved by the Animal Care Committee of the Kyoto Prefectural University of Medicine (Kyoto, Japan, M2020-126). For NAFLD induction, mice were fed for 8 wk with an Ath diet as described by Matsuzawa et al[29], which resulted in hepatic steatosis and inflammation. The Ath diet was prepared by the addition of cocoa butter, cholesterol, and cholate to corticotropin-releasing factor-1 (CRF-1) (Oritental Yeast, Tokyo, Japan). Increased intestinal permeability was induced through the administration of 0.5% DSS (MW 36000-50000, MP Biomedicals, Illkirch, France) in drinking water as described previously[14]. DSS was administered in cycles, each consisting of 5-d DSS administration followed by a 9-d interval with normal drinking water. The cycles were repeated throughout the experimental period of 8 wk. Mice were randomly allocated to the following groups: (1) control mice fed standard chow (CRF-1; Control); (2) control mice fed a CRF-1 diet containing 5% PHGG (Control + PHGG); (3) NAFLD mice fed the Ath diet along with DSS induction (NAFLD); and (4) NAFLD mice supplemented with 5% PHGG (NAFLD + PHGG). Ingredient and fat contents of CRF-1 and the Ath diet are shown in Table 1. The commercial PHGG preparation (Sunfiber) used in this study was obtained from Taiyo Kagaku (Yokkaichi, Japan). The preparation was obtained by treating guar gum with β-endogalactomannase from a strain of Aspergillus niger. Mice were sacrificed after being anesthetized with ketamine and xylazine. Blood samples were collected from the portal vein for serum endotoxin measurements through opposite cannulation with a 23-gauge needle or from the inferior vena cava for serum chemistry measurements followed by exsanguination, and the organs and cecal contents were harvested.
Table 1.
Ingredients and fat content of the experimental diets
|
Ingredients (g/kg)
|
CRF-1
|
Ath
|
| Moisture (g/kg) | 82 | 74.5 |
| Protein (g/kg) | 219 | 199 |
| Fat (g/kg) | 54 | 141.5 |
| Crude ash (g/kg) | 63 | 57 |
| Crude fiber (g/kg) | 29 | 26 |
| Soluble nitrogen free extract (g/kg) | 553 | 502 |
| Total (g) | 1000 | 1000 |
| Fat content | CRF-1 | Ath |
| Fatty acid (g/kg) | 54 | 124 |
| Cholesterol (g/kg) | 12.5 | |
| Cholate (g/kg) | 5 |
Ath: Atherogenic; CRF-1: Corticotropin-releasing factor-1.
Serum biochemistry and liver histology
Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured using commercial assay kits (SRL, Tokyo, Japan) with an automated chemical analyzer (Olympus Automated Chemistry Analyzer AU5400, Tokyo, Japan). For hematoxylin and eosin (H&E) staining, liver samples were fixed in formalin, embedded in paraffin, sectioned at 5 μm, and stained with H&E. Histopathological changes were evaluated under a light microscope (Olympus BX50, Tokyo, Japan) in a blinded manner.
Measurement of myeloperoxidase activity
Tissue-associated myeloperoxidase (MPO) activity was measured in the liver tissue by a modified method reported by Grisham et al[30] as an index of neutrophil accumulation[30]. Liver tissue fragments were homogenized in 1 mL of 10 mmol/L potassium phosphate buffer (pH 7.8) containing 30 mmol/L KCl using a Teflon Potter-Elvehjem homogenizer. MPO activity was then determined. Briefly, liver homogenates were centrifuged at 20000´g for 15 min at 4 °C. The resulting pellet was then re-homogenized in 0.3 mL of 50 mmol/L potassium phosphate buffer (pH 5.4) containing 0.5% hexadecyltrimethylammonium bromide. The mixture was then centrifuged at 20000´g for 15 min at 4 °C, and the supernatants were collected. MPO activity was assessed by measuring the H2O2-dependent oxidation of 3,3',5,5'-tetramethylbenzidine. One unit of enzyme activity was defined as the amount of MPO that changed the absorbance by 1.0/min at 460 nm and 37 °C. Total protein in the tissue homogenates was measured using a Bio-Rad Protein Assay kit (Bio-Rad Laboratories, K. K., Tokyo, Japan) according to the manufacturer’s protocol.
Analysis of mRNA expression in the liver and intestine
Hepatic and intestinal mRNA expression was determined using real-time polymerase chain reaction (PCR). Total RNA was extracted using the acid-guanidinium-phenol chloroform method (Isogen kit; Nippon Gene, Tokyo, Japan). Extracted RNA was stored at −80 °C until use in real-time PCR. For the latter, 1 μg of RNA was reverse-transcribed to first-strand complementary DNA (cDNA) using a high capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, United States). Real-time PCR was carried out on a 7300 real-time PCR system (Applied Biosystems, Foster City, CA, United States) using the DNA-binding dye SYBR Green to detect the PCR products with the primers shown in Table 2. Gene expression was normalized to that of b-actin as an internal control.
Table 2.
Primer sequences for the targeted mouse genes
|
Gene
|
Forward primer
|
Reverse primer
|
| m_βactin | TATCCACCTTCCAGCAGATGT | AGCTCAGTAACAGTCCGCCTA |
| m_TNF-α | ATCCGCGACGTGGAACTG | ACCGCCTGGAGTTCTGGAA |
| m_MCP-1 | CTTCCTCCACCACCATGCA | CCAGCCGGCAACTGTGA |
| m_αSMA | GTCCCAGACATCAGGGAGTAA | TCGGATACTTCAGCGTCAGGA |
| m_collagen1a1 | GCTTCACCTACAGCACCCTTGT | GATGACTGTCTTGCCCCAAGTT |
| m_Acc | CTGGCTGCATCCATTATGTCA | TGGTAGACTGCCCGTGTGAA |
| m_Cpt2 | GGGCGAGCTTCAGCATATG | GGCCCATCGCTGCTTCTT |
| m_PPARα | CGATGCTGTCCTCCTTGATGA | CGCGTGTGATAAAGCCATTG |
| m_PPARγ | CTTGCTGTGGGGATGTCTCA | ATCTCCGCCAACAGCTTCTC |
| m_TLR4 | CCTCTGCCTTCACTACAGAGACTTT | CACAATAACCTTCCGGCTCTTG |
| m_TLR9 | CCTTCGTGGTGTTCGATAAGG | CACCCGCAGCTCGTTATACA |
TNF-α: Tumor necrosis factor-α; MCP-1: Monocyte chemotactic protein 1; αSMA: α smooth muscle actin; ACC: Acetyl-CoA carboxylase; CPT2: Carnitine O-palmitoyltransferase II; PPAR: Peroxisome proliferator-activated receptor; TLR: Toll-like receptor.
Serum endotoxin levels in the portal vein
After the experimental period of 8 wk, blood samples were collected from the portal vein. The chromogenic Limulus Amebocyte Lysate (LAL) Endotoxin Assay kit (GenScript, Piscataway, NJ, United States) was used to measure serum endotoxin levels. In brief, the samples, blanks, or standards were incubated with reconstituted LAL at 37 °C for 45 min followed by incubation with a reconstituted chromogenic substrate solution at 37 °C for 6 min. After adding the stop solution, the absorbance of each reaction was recorded at 545 nm with a microplate reader (Spectramax M2; Molecular Devices, Sunnyvale, CA, United States). Distilled water was used as the blank.
Intestinal permeability assay
Intestinal permeability was determined by measuring the lumen-to-blood clearance of fluorescein isothiocyanate (FITC)-conjugated dextran according to a previously published method, with some modifications[31,32]. In brief, after the experimental period of 8 wk, mice were anesthetized with ketamine and xylazine, and FITC-conjugated dextran (1 mg/mL; molecular mass, 4 kDa; Sigma Aldrich, St. Louis, MO, United States) in poly(butylene succinate) was administered from the proximal duodenum in a total volume of 0.5 mL. One hour after administration, blood samples were collected, centrifuged, and stored at -80 °C. The serum was then examined at the excitation/emission wavelengths of 490 nm/525 nm using a spectrofluorometer (Spectramax M2; Molecular Devices, Sunnyvale, CA, United States).
DNA extraction from mouse fecal samples
After the experimental period of 8 wk, mouse fecal samples were collected from the cecum, suspended in lysis buffer [4 mol/L guanidinium thiocyanate, 100 mmol/L tris-HCl (pH 9.0), 40 mmol/L ethylenediaminetetraacetic acid, and 0.001% bromothymol blue], and shaken in the presence of zirconia beads. DNA was extracted from the resultant suspension (Magtration System 12GC and GC series MagDEA DNA 200; Precision System Science, Chiba, Japan).
PCR amplification and terminal restriction fragment length polymorphism analysis
The 16S rRNA gene was amplified from fecal DNA using the 516F (5′-TGCCAGCAGCCGCGGTA-3′; Escherichia coli positions 516 to 532) and 1510R (5′-GGTTACCTTGTTACGACTT-3′; Escherichia coli positions 1510 to 1492) primers. The 5′-ends of the forward primers were labeled with 6'-carboxyfluorescein synthesized by Applied Biosystems (Life Technologies, Thermo Fisher Scientific, Waltham, MA). PCR amplification of the DNA samples (10 ng of each DNA sample) was performed according to a previously described protocol[33]. The amplified 16S rDNA genes were purified (MultiScreen PCR micro96 Plate; Merck Millipore, Tokyo, Japan) and dissolved in 40 μL of distilled water. The restriction enzymes for restriction fragment length polymorphism (RFLP) were selected according to a previously described protocol[34]. Purified PCR products were digested for 3 h with 10 U of the restriction enzyme BslI at 55 °C. The length of the terminal RF (T-RF) was determined using a genetic analyzer (ABI PRISM 3130xl; Applied Biosystems, Life Technologies, Thermo Fisher Scientific) and analysis software (GeneMapper; Applied Biosystems, Life Technologies, Thermo Fisher Scientific, Waltham, MA, United States). Standard-size markers were used (e.g., MapMarker X-Rhodamine Labeled 50-1000 bp; BioVentures, Murfreesboro, TN, United States). An operational taxonomic unit (OTU) was used to describe the clusters of clone sequences that differed from the known species by approximately 2%, which was at least 98% similar to the members of their cluster[35]. As the apparent size of identical T-RFs can vary over a range of 1-3 bp, major T-RFs that were similar in size to within 1-3 bp, were grouped as OTUs[34]. The major T-RFs were identified using a computer simulation with a T-RFLP analysis program and a phylogenetic assignment database for the T-RFLP analysis of human colonic microbiota[36].
Determination of organic acid concentration in cecal contents
A portion of the cecal contents (0.5 g) was suspended in 0.5 mL of 14% perchloric acid to remove proteins. After centrifugation at 10000 × g for 5 min at 4 °C, the supernatant was filtered through a cellulose acetate membrane filter with a pore size of 0.45 μm, and its organic acid content was then analyzed with ion-exclusion high performance liquid chromatography as described by Ushida and Sakata[37].
Statistical analyses
Data are expressed as the mean ± SD. Overall differences between groups were determined by one-way analysis of variance (ANOVA). Differences between individual groups were determined using Tukey’s multiple comparison tests when one-way ANOVA indicated a significant difference. Target gene copy numbers, estimated using real-time PCR, and the concentration of organic acids were analyzed using the Steel-Dwass method after the Friedman test. A P value < 0.05 was considered statistically significant. All analyses were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA, United States) for Macintosh.
RESULTS
Effects of PHGG on body weight, liver weight, and biochemical parameters
Mice with increased intestinal permeability exposed to the Ath diet showed significant increases in liver weight, liver/body ratios, AST, ALT, and total cholesterol levels, as well as a significant decrease in body weight and triglyceride levels, as shown in Table 3. Treatment with PHGG did not change the NAFLD model-induced change in body weight, liver weight, liver/body ratio, and total cholesterol levels but significantly improved the increase in AST and ALT levels.
Table 3.
Characteristics of the experimental groups
|
Characteristics
|
Control
|
Control + PHGG
|
NAFLD
|
NAFLD + PHGG
|
| Body weight (g) | 25.7 ± 0.5 | 25.2 ± 1.1 | 20.6 ± 0.8a | 20.8 ± 1.6a |
| Liver weight (g) | 1.17 ± 0.05 | 1.03 ± 0.08 | 1.47 ± 0.11a | 1.55 ± 0.13a |
| Liver weight (% body) | 4.45 ± 0.13 | 4.13 ± 0.32 | 7.13 ± 0.40a | 7.44 ± 0.61a |
| Aspartate aminotransferase (IU/L) | 72.0 ± 7.1 | 73.1 ± 11.9 | 133.1 ± 69.1a | 83.5 ± 24.1a,b |
| Alanine aminotransferase (IU/L) | 23.4 ± 2.9 | 20.0 ± 3.4 | 112.9 ± 77.0a | 46.3 ± 15.9a,b |
| Total cholesterol (mg/dL) | 97.7 ± 6.6 | 83.7 ± 10.8 | 359.7 ± 95.0a | 306.7 ± 26.5a |
| Triglyceride (mg/dL) | 88.9 ± 25.4 | 71.1 ± 25.1 | 16.6 ± 4.0a | 36.2 ± 25.7a,b |
| Free fatty acids (mEQ/L) | 1668 ± 257 | 1334 ± 254a | 1254 ± 201a | 1119 ± 310a |
| Glucose (mg/dL) | 70 ± 53 | 73 ± 16 | 73 ± 19 | 95 ± 32 |
Data are presented as the mean ± SD.
P < 0.05 significant differences compared to the control group.
P < 0.05 significant differences between the non-alcoholic fatty liver disease (NAFLD) group and the NAFLD + Partially hydrolyzed guar gum group. Non-alcoholic fatty liver disease group was induced by high fat diet with increased intestinal permeability. NAFLD: Non-alcoholic fatty liver disease; PHGG: Partially hydrolyzed guar gum.
Effects of PHGG on liver damage, inflammation, and fibrosis
Macroscopic observation showed that livers from mice subjected to the Ath diet with increased intestinal permeability were visibly lighter and fatty compared to those from mice subjected to a control diet (Figure 1A). Treatment with PHGG did not induce a dramatic change, but partly reverted some morphological alterations. Histological analysis of the liver tissues indicated that the fat content in mice fed a control diet with or without PHGG treatment were normal without inflammatory infiltration (Figure 1B). In contrast, mice with increased intestinal permeability exposed to the Ath diet showed both increased fat content and inflammatory infiltration (Figure 1B). These alterations were prevented by PHGG treatment (Figure 1B). The observed inflammatory infiltration was supported by the MPO activity results, which confirmed that treatment with PHGG attenuated liver damage in the NAFLD model (Figure 1C). Histological analysis of intestine and colon tissues indicated that mice with increased intestinal permeability exposed to the Ath diet did not show any morphological changes compared to the control mice (data not shown). Figure 2 shows hepatic inflammation and fibrosis in the NAFLD model. Tumor necrosis factor-α (TNF-α; (TNF-α; Figure 2A) and monocyte chemotactic protein-1 (MCP-1; (MCP-1; Figure 2B) mRNA expression was significantly higher in the liver tissue of mice with increased intestinal permeability exposed to the Ath diet than in that of control mice. This increase was significantly attenuated by treatment with PHGG. With respect to hepatic fibrosis, collagen 1a1 (Figure 2C) and α smooth muscle actin (αSMA; (αSMA; Figure 2D) mRNA expression was significantly elevated in the liver tissue of mice with NAFLD compared to that in control mice. Treatment with PHGG clearly decreased collagen 1a1 mRNA expression in the NAFLD model.
Figure 1.

Effects of partially hydrolyzed guar gum on the non-alcoholic fatty liver disease model. A: Representative macroscopic findings in the liver. Non-alcoholic fatty liver disease was induced in mice subjected to the atherogenic diet with increased intestinal permeability for 8 wk; B: Representative histological findings in the liver. Magnification, × 100. Hematoxylin and eosin staining; C: Myeloperoxidase (MPO) activity. The liver was homogenized and MPO activity was determined as an index of neutrophil accumulation in the liver. Data represent the mean ± SD of seven mice. aP < 0.05 significant differences compared to the control group; bP < 0.05 significant differences between the 2 selected groups. NAFLD: Non-alcoholic fatty liver disease; PHGG: Partially hydrolyzed guar gum; MPO: Myeloperoxidase.
Figure 2.

Effects of partially hydrolyzed guar gum on hepatic inflammation and fibrosis in the non-alcoholic fatty liver disease model. A: Tumor necrosis factor-α; B: Monocyte chemotactic protein-1; C: Collagen 1a1; D: α smooth muscle actin mRNA expression in the liver. mRNA expression was evaluated using real-time polymerase chain reaction. Data represent the mean ± SD of seven mice. aP < 0.05 significant differences compared to the control group; bP < 0.05 significant differences between the 2 selected groups. NAFLD: Non-alcoholic fatty liver disease; PHGG: Partially hydrolyzed guar gum; TNF-α: Tumor necrosis factor-α; MCP-1: Monocyte chemotactic protein-1; αSMA: α smooth muscle actin.
Effects of PHGG on lipid synthesis and degradation in the liver
Enzymes of the lipid synthesis and degradation pathways in the liver were further assessed as shown in Figure 3. Mice with increased intestinal permeability exposed to the Ath diet showed upregulation of acetyl-CoA carboxylase and peroxisome proliferator-activated receptor γ (PPARγ) and downregulation of carnitine O-palmitoyltransferase II and PPARα mRNA expression. Treatment with PHGG did not change these effects. Moreover, enzymes related to lipid absorption, such as acetyl-coenzyme A acetyltransferase 2, Niemann-Pick C1-like 1, and fatty acid binding protein, were evaluated in the intestine. Ath diet combined with increased intestinal permeability, and treatment with PHGG, did not affect the levels of these enzymes (data not shown).
Figure 3.

Effects of partially hydrolyzed guar gum on lipid synthesis and degradation in the non-alcoholic fatty liver disease model. A: Acetyl-CoA carboxylase; B: Carnitine O-palmitoyltransferase II; C: Peroxisome proliferator-activated receptor (PPAR) α; D: PPARγ mRNA expression in the liver. mRNA expression was evaluated using real-time polymerase chain reaction. Data represent the mean ± SD of seven mice. aP < 0.05 significant differences compared to the control group. NAFLD: Non-alcoholic fatty liver disease; PHGG: Partially hydrolyzed guar gum; ACC: Acetyl-CoA carboxylase; PPAR: Peroxisome proliferator-activated receptor; CPT2: Carnitine O-palmitoyltransferase II.
Effects of PHGG on regulation of the gut-liver axis
The concept of the gut-liver axis has received considerable attention in recent years. In this study, chronic administration of 0.5% DSS did not result in morphological changes such as mucosal disruption. However, it induced some effects on the liver, thereby leading to NAFLD development. We therefore examined the expression of TLRs in the liver. The results showed that the Ath diet combined with increased intestinal permeability significantly increased the mRNA expression of TLR4 and TLR9 in the liver, and that treatment with PHGG significantly attenuated this upregulation (Figure 4A and B). These results are consistent with the endotoxin levels determined in the portal vein (Figure 4C). To determine whether our NAFLD model or PHGG treatment resulted in altered intestinal permeability, we evaluated the lumen-to-blood clearance of FITC-conjugated dextran (FD4 flux). As shown in Figure 5, the FD4 flux was significantly elevated in the NAFLD model mice, and this increase was significantly reduced by treatment with PHGG.
Figure 4.

Effects of partially hydrolyzed guar gum on toll-like receptor signaling through the gut-liver axis. A: Toll-like receptor (TLR) 4 mRNA expression; B: TLR9 mRNA expression in the liver. mRNA expression was evaluated using real-time polymerase chain reaction; C: Endotoxin levels in the portal vein. Endotoxin levels were measured using the chromogenic Limulus Amebocyte Lysate Endotoxin Assay kit. Data represent the mean ± SD of seven mice. aP < 0.05 significant differences compared to the control group; bP < 0.05 significant differences between the 2 selected groups. NAFLD: Non-alcoholic fatty liver disease; PHGG: Partially hydrolyzed guar gum; TLR: Toll-like receptor.
Figure 5.
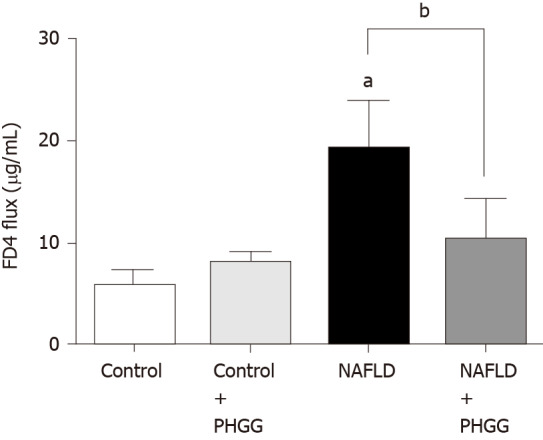
Effects of partially hydrolyzed guar gum on intestinal permeability in the non-alcoholic fatty liver disease model. Lumen-to-blood clearance of fluorescein isothiocyanate-conjugated dextran was evaluated by injection into the duodenum and collection of blood after 60 min. Fluorescence intensity in the serum was measured with a spectrofluorometer. Data represent the mean ± SD of seven mice. aP < 0.05 significant differences compared to the control group; bP < 0.05 significant differences between the 2 selected groups. NAFLD: Non-alcoholic fatty liver disease; PHGG: Partially hydrolyzed guar gum.
Effects of PHGG on SCFA profiles and cecal bacteria
The SCFA profiles of the cecal contents were determined using ion-exclusion high performance liquid chromatography, the results of which are summarized in Table 4. Under normal conditions, treatment with PHGG significantly increased the acetic acid, formic acid, propionic acid, and butyric acid contents. In contrast, isovaleric acid content was significantly decreased after PHGG treatment. The Ath diet together with increased intestinal permeability significantly elevated succinic acid, lactic acid, propionic acid, isobutyric acid, and isovaleric acid contents and significantly reduced formic acid, acetic acid, and butyric acid contents. In the NAFLD model, PHGG treatment significantly upregulated formic acid levels and significantly downregulated isobutyric acid, isovaleric acid, and valeric acid contents.
Table 4.
Partially hydrolyzed guar gum-mediated profiles of short-chain fatty acids in cecal contents
|
Short chain fatty acids (mmol/g)
|
Control
|
Control + PHGG
|
NAFLD
|
NAFLD + PHGG
|
| Succinic acid | 0.82 ± 0.34 | 0.85 ± 0.36 | 5.08 ± 2.27a | 5.28 ± 2.20a |
| Lactic acid | 0.05 ± 0.08 | 0.09 ± 0.13 | 0.40 ± 0.46a | 0.45 ± 0.39a |
| Formic acid | 0.89 ± 0.09 | 1.90 ± 0.06a | 0.72 ± 0.16a | 0.83 ± 0.28b |
| Acetic acid | 41.2 ± 6.0 | 47.9 ± 8.8a | 32.8 ± 5.3a | 33.1 ± 7.9a |
| Propionic acid | 8.18 ± 1.14 | 9.16 ± 1.04a | 9.76 ± 2.56a | 9.82 ± 3.39a |
| Isobutyric acid | 1.19 ± 0.11 | 1.04 ± 0.11 | 1.44 ± 0.20a | 1.06 ± 0.28b |
| Butyric acid | 8.86 ± 2.42 | 13.81 ± 5.14a | 3.91 ± 0.58a | 3.34 ± 1.09a |
| Isovaleric acid | 0.33 ± 0.14 | 0.12 ± 0.11a | 1.05 ± 0.40a | 0.38 ± 0.40b |
| Valeric acid | 0.49 ± 0.18 | 0.48 ± 0.14 | 0.50 ± 0.22 | 0.18 ± 0.17a,b |
Data are presented as the mean ± SD.
P < 0.05 significant differences compared to the control group.
P < 0.05 significant differences between the non-alcoholic fatty liver disease (NAFLD) group and the NAFLD + Partially hydrolyzed guar gum group. PHGG: Partially hydrolyzed guar gum; NAFLD: Non-alcoholic fatty liver disease.
To determine whether treatment with PHGG altered the cecal bacteria profiles, we examined the variation in eight representative bacterial groups using T-RFLP analysis. Chronic administration of 0.5% DSS caused a dramatic decrease in the number of cecal bacteria, and therefore, cecal bacterial profiles were only analyzed in the control and control + PHGG groups. As shown in Figure 6, the administration of PHGG altered the cecal bacteria profiles (Figure 6A) and significantly increased the cecal Bacteroides (Figure 6B) and Clostridium subcluster XIVa (Figure 6C) compared with that in the control mice. Conversely, PHGG treatment significantly reduced the cecal Prevotella, Bifidobacterium (Figure 6D), and Clostridium cluster XVIII (Figure 6E).
Figure 6.

Effects of partially hydrolyzed guar gum on cecal microbiota after administration. A: Percentage of the total identified sequences per group. Microbiota profiles in the cecal contents were analyzed using terminal restriction fragment-length polymorphism; B–E: Changes in the microbiota composition. Bars represent the mean ± SD of seven mice. aP < 0.05 significant differences compared to the control group. PHGG: Partially hydrolyzed guar gum.
DISCUSSION
Our results clearly indicate that increased intestinal permeability in combination with the administration of Ath diet resulted in NAFLD development through the gut-liver axis and that treatment with PHGG significantly attenuated NAFLD development in mice. The protective effects of PHGG might result in part from the modulation of microbiota composition and SCFA profiles, leading to attenuation of the increased intestinal permeability. A variety of animal models for NAFLD and NASH, including models induced by high-fat, high-fructose, high-cholesterol, high-sucrose, Ath, choline- and L-amino acid-deficient, and methionine- and choline-deficient diets, have been used to investigate the pathophysiology of NAFLD and NASH, indicating the important role of diet in these clinical states. However, because of the complex, multidirectional pathophysiology involved in NAFLD, the perfect animal model representing the complete NAFLD spectrum in a workable time frame does not appear to exist[38]. Moreover, due to the substantial physiological differences between rodents and humans, animal models should be used with caution.
Accumulating evidence indicates the significance of alterations in intestinal permeability in NAFLD and NASH pathogenesis. An increase in intestinal epithelial cell permeability is reported to allow the translocation of microbial products into the portal vein, subsequently inducing hepatic inflammation[14,39]. Luther et al[31] demonstrated that an early phase of hepatic injury and inflammation contributes to altered intestinal permeability[31]. Furthermore, Mouries et al[40] demonstrated that disruption of the gut vascular barrier is an early event in NASH pathogenesis and is a consequence of diet-induced dysbiosis[40]. These data suggest that communication between the gut and liver plays an important role in NAFLD development. In this context, the murine NAFLD model described in the present study appears to be valuable for investigating the pathophysiology of this disease. Our study also clearly shows that increased intestinal permeability combined with the Ath diet resulted in increased liver weight, AST/ALT, total cholesterol levels, and lipid accumulation in the liver, as well as the upregulation of pro-inflammatory cytokines/chemokines such as TNF-α and MCP-1, neutrophil accumulation, lipid synthesis, and hepatic fibrogenesis. These results are consistent with those of a previous study[14]. Several cytokines, such as interleukin (IL)-6, IL-1b, and TNF-α, have been recently suggested to be involved in the so-called chronic low-grade inflammation or meta inflammation of NAFLD/NASH[41,42]. Our study further demonstrates that increased intestinal permeability in combination with the Ath diet resulted in elevated portal endotoxin levels and activated the expression of TLR4 and TLR9, confirming that intestinal permeability was significantly elevated in this NAFLD model. Indeed, the FD4 flux, a marker of lumen-to-serum permeability, was significantly increased in our NAFLD model. These findings are consistent with those of a recent review[4,15,43]. Interestingly, our study also shows that treatment with PHGG significantly attenuated the development of NAFLD by decreasing TLR signaling and its downstream molecules, such as pro-inflammatory cytokines, despite continued lipid synthesis and degradation, suggesting that treatment with PHGG might have therapeutic effects on NAFLD through the gut-liver axis. Another study has also shown that a prebiotic, fermentable, dietary fructo-oligosaccharide alleviates hepatic steatosis[44]. However, in this study, the protection appeared to be mediated by reduced fatty acid oxidation and cholesterol accumulation. Thus, the protective mechanisms by which various prebiotics modulate NAFLD remain to be fully elucidated.
Accumulating evidence suggests a relationship between the intestinal microbiota and NAFLD, and modification of the composition of the former could play a role in the development and progression of the latter[45]. Le Roy et al[46] have shown that in mice fed the Ath diet, the gut microbiota composition alters lipid metabolism in the liver[46]. De Minicis et al[47] also demonstrated that the Ath diet model subjected to bile duct ligation shows an increase in the abundance of gram-negative bacteria, a reduced Bacteroidetes/Firmicutes ratio, the complete disappearance of Bifidobacteriaceae, and increased abundance of gram-negative Proteobacteria (especially Enterobacteriaceae)[47]. In our experiments, we quantified the Clostridium subcluster XIVa, Clostridium cluster IV, and Bacteroides as the SCFA-producing indigenous bacteria, Prevotella as resident microbiota, Bifidobacterium as a beneficial bacterium, Lactobacillales as the lactate-producing bacteria, Clostridium cluster XI as the harmful bacteria, including Clostridium difficile, and the Clostridium cluster XVIII as pathogenic bacteria such as Escherichia coli in regard to the cecal bacteria profiles. Additionally, the compositional alterations in intestinal bacteria might also change their metabolic capacity. Colonic fermentation of carbohydrates results in the generation of SCFAs. Recent evidence suggests that supplementation with sodium butyrate protects mice from NASH development[48]. Moreover, a recent review indicated the beneficial effects of commensal bacteria on the intestinal epithelial barrier and the importance of butyrate-producing bacteria for intestinal health[49]. A recent study also showed that SCFAs (acetate, propionate, and butyrate) stimulate and protect the intestinal barrier function[50].
In our study, treatment with PHGG significantly increased the abundance of cecal Bacteroides and Clostridium subcluster XIVa. Our results are partially in agreement with previous human experiments showing that PHGG increases the abundance of butyrate-producing bacteria, including Clostridium subcluster XIVa, in the intestine[27]. Moreover, treatment with PHGG resulted in a significant increase in SCFA levels, particularly butyric acid, acetic acid, propionic acid, and formic acid, in the cecal content. These results are consistent with those of a previous study[27]. PHGG-mediated increases in SCFA levels might exert protective effects on the intestinal barrier function. These observations are supported by a recent study[50], although it lacked direct data.
Our study has the following limitations: (1) the dramatic changes in microbiota and SCFA profiles in the current NAFLD model resulting from the adverse effects of DSS administration; (2) the method of analysis of microbiota, T-RFLP analysis; (3) the lack of determination of IL-6 and IL-1b (other markers of low-grade inflammation of NAFLD/NASH); and (4) the modest sample size of each group associated with mean differences and SD in some experiments. Further investigations are therefore required.
CONCLUSION
In summary, our data indicate that PHGG inhibits NAFLD development in mice, which is associated with decreased lipid accumulation, pro-inflammatory cytokine/chemokine production, and neutrophil infiltration in the liver. These findings might be related to the gut-liver axis, through the protection of intestinal barrier integrity, as well as cecal bacteria and SCFA profile alterations. Particularly, treatment with PHGG promotes an SCFA-producing bacterial group of Clostridium subcluster XIV and Bacteroides. Further studies are warranted to elucidate the detailed mechanisms involved in this phenomenon; however, treatment with PHGG in humans could show great promise as a therapeutic strategy for NAFLD.
ARTICLE HIGHLIGHTS
Research background
Non-alcoholic fatty liver disease (NAFLD) is caused by a variety of pathogenesis, and may involve the gut-liver axis, which has attracted much attention. Prebiotics such as dietary fibers were shown to attenuate NAFLD by modulating gut microbiota. Partially hydrolyzed guar gum (PHGG), a water-soluble dietary fiber, is associated with alteration of gut microbiota and the production of short-chain fatty acids (SCFAs), and has been reported to alleviate the symptoms of various intestinal diseases and metabolic syndrome.
Research motivation
PHGG should exert beneficial health effects on the host through alterations to the gut microbiota and SCFA production. However, its effects on NAFLD remain to be fully elucidated.
Research objectives
The present study aimed to determine whether treatment with PHGG attenuates NAFLD development in mice through the gut-liver axis.
Research methods
Male C57BL/6J mice with increased intestinal permeability by chronic intermittent administration of low-dose dextran sulfate sodium were fed a control or atherogenic (Ath) diet (a mouse model of NAFLD) for 8 wk, with or without 5% PHGG. Body weight, liver weight, macroscopic findings in the liver, blood biochemistry, liver histology, myeloperoxidase activity in liver tissue, mRNA expression in the liver and intestine, serum endotoxin levels in the portal vein, intestinal permeability, and microbiota and SCFA profiles in the cecal samples were investigated.
Research results
Mice subjected to a Ath diet with increased intestinal permeability showed significantly increased serum aspartate aminotransferase and alanine aminotransferase levels, liver fat accumulation, liver inflammatory (tumor necrosis factor-α and monocyte chemotactic protein-1) and fibrogenic (collagen 1a1 and α smooth muscle actin) marker levels, and liver myeloperoxidase activity, which were significantly attenuated by PHGG treatment. Moreover, increased intestinal permeability in combination with the Ath diet resulted in increased portal endotoxin levels and activated toll-like receptor (TLR) 4 and TLR9 expression. PHGG administration did not affect fatty acid metabolism in the liver, but decreased lipopolysaccharide signaling through the gut-liver axis. The administration of PHGG altered the cecal bacteria profiles and significantly increased the cecal Bacteroides and Clostridium subcluster XIVa. Treatment with PHGG markedly increased the levels of SCFAs, particularly, butyric acid, acetic acid, propionic acid, and formic acid, in the cecal samples.
Research conclusions
Treatment with PHGG attenuated NAFLD development in mice through the gut-liver axis by modulating microbiota and downstream SCFA profiles.
Research perspectives
By indicating that PHGG administration inhibits NAFLD development through the gut-liver axis, the present study showed a possible treatment strategy of NAFLD in humans.
Footnotes
Institutional review board statement: At our institution, the attached “Institutional Animal Care and Use Committee Approval Form” also serves as the Institutional Review Board Approval Form. The study was reviewed and approved by the Institutional Review Board at Kyoto Prefectural University of Medicine.
Institutional animal care and use committee statement: All procedures involving animal subject is approved by the Animal Care Committee of the Kyoto Prefectural University of Medicine, Kyoto, Japan, No. M2020-126.
Conflict-of-interest statement: All authors have nothing to disclose.
ARRIVE guidelines statement: The authors have read the ARRIVE guidelines, and the manuscript was prepared and revised according to the ARRIVE guidelines.
Manuscript source: Unsolicited manuscript
Peer-review started: February 6, 2021
First decision: February 27, 2021
Article in press: April 21, 2021
Specialty type: Gastroenterology and hepatology
Country/Territory of origin: Japan
Peer-review report’s scientific quality classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C, C
Grade D (Fair): 0
Grade E (Poor): 0
P-Reviewer: Tarantino G S-Editor: Fan JR L-Editor: A P-Editor: Liu JH
Contributor Information
Shun Takayama, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Kazuhiro Katada, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Tomohisa Takagi, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan; Department of Medical Innovation and Translational Medical Science, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Takaya Iida, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Tomohiro Ueda, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Katsura Mizushima, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Yasuki Higashimura, Department of Food Science, Ishikawa Prefectural University, Nonoichi 921-8836, Japan.
Mayuko Morita, Department of Health Care Nutrition, Showa Gakuin Junior College, Ichikawa 272-0823, Japan.
Tetsuya Okayama, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Kazuhiro Kamada, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan. katada@koto.kpu-m.ac.jp.
Kazuhiko Uchiyama, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Osamu Handa, Division of Gastroenterology, Department of Internal Medicine, Kawasaki Medical School, Kurashiki 701-0192, Japan.
Takeshi Ishikawa, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Zenta Yasukawa, Department of Nutrition, Taiyo Kagaku Co. Ltd, Yokkaichi 510-0844, Japan.
Tsutomu Okubo, Department of Nutrition, Taiyo Kagaku Co. Ltd, Yokkaichi 510-0844, Japan.
Yoshito Itoh, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Yuji Naito, Department of Molecular Gastroenterology and Hepatology, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Data sharing statement
No additional data are available.
References
- 1.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 2.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ American Gastroenterological Association; American Association for the Study of Liver Diseases; American College of Gastroenterologyh. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, Hunault G, Oberti F, Calès P, Diehl AM. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764–775. doi: 10.1002/hep.28356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu H, Lin A, Kong M, Yao X, Yin M, Xia H, Ma J, Liu H. Intestinal microbiome and NAFLD: molecular insights and therapeutic perspectives. J Gastroenterol. 2020;55:142–158. doi: 10.1007/s00535-019-01649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, Abrams SH, Scheimann AO, Sanyal AJ, Chalasani N, Tonascia J, Ünalp A, Clark JM, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR Nonalcoholic Steatohepatitis Clinical Research Network. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA. 2011;305:1659–1668. doi: 10.1001/jama.2011.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR NASH CRN. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarantino G, Citro V, Capone D. Nonalcoholic Fatty Liver Disease: A Challenge from Mechanisms to Therapy. J Clin Med. 2019;9 doi: 10.3390/jcm9010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeuzem S. Gut-liver axis. Int J Colorectal Dis. 2000;15:59–82. doi: 10.1007/s003840050236. [DOI] [PubMed] [Google Scholar]
- 10.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 11.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, Vecchio FM, Rapaccini G, Gasbarrini G, Day CP, Grieco A. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 12.Csak T, Velayudham A, Hritz I, Petrasek J, Levin I, Lippai D, Catalano D, Mandrekar P, Dolganiuc A, Kurt-Jones E, Szabo G. Deficiency in myeloid differentiation factor-2 and toll-like receptor 4 expression attenuates nonalcoholic steatohepatitis and fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2011;300:G433–G441. doi: 10.1152/ajpgi.00163.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590:447–458. doi: 10.1113/jphysiol.2011.219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gäbele E, Dostert K, Hofmann C, Wiest R, Schölmerich J, Hellerbrand C, Obermeier F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J Hepatol. 2011;55:1391–1399. doi: 10.1016/j.jhep.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 15.Safari Z, Gérard P. The links between the gut microbiome and non-alcoholic fatty liver disease (NAFLD) Cell Mol Life Sci. 2019;76:1541–1558. doi: 10.1007/s00018-019-03011-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aller R, De Luis DA, Izaola O, Conde R, Gonzalez Sagrado M, Primo D, De La Fuente B, Gonzalez J. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: a double blind randomized clinical trial. Eur Rev Med Pharmacol Sci. 2011;15:1090–1095. [PubMed] [Google Scholar]
- 17.Malaguarnera M, Vacante M, Antic T, Giordano M, Chisari G, Acquaviva R, Mastrojeni S, Malaguarnera G, Mistretta A, Li Volti G, Galvano F. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. 2012;57:545–553. doi: 10.1007/s10620-011-1887-4. [DOI] [PubMed] [Google Scholar]
- 18.Roberfroid M, Gibson GR, Hoyles L, McCartney AL, Rastall R, Rowland I, Wolvers D, Watzl B, Szajewska H, Stahl B, Guarner F, Respondek F, Whelan K, Coxam V, Davicco MJ, Léotoing L, Wittrant Y, Delzenne NM, Cani PD, Neyrinck AM, Meheust A. Prebiotic effects: metabolic and health benefits. Br J Nutr. 2010;104 Suppl 2:S1–63. doi: 10.1017/S0007114510003363. [DOI] [PubMed] [Google Scholar]
- 19.Parnell JA, Raman M, Rioux KP, Reimer RA. The potential role of prebiotic fibre for treatment and management of non-alcoholic fatty liver disease and associated obesity and insulin resistance. Liver Int. 2012;32:701–711. doi: 10.1111/j.1478-3231.2011.02730.x. [DOI] [PubMed] [Google Scholar]
- 20.Yoon SJ, Chu DC, Raj Juneja L. Chemical and physical properties, safety and application of partially hydrolized guar gum as dietary fiber. J Clin Biochem Nutr. 2008;42:1–7. doi: 10.3164/jcbn.2008001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giannini EG, Mansi C, Dulbecco P, Savarino V. Role of partially hydrolyzed guar gum in the treatment of irritable bowel syndrome. Nutrition. 2006;22:334–342. doi: 10.1016/j.nut.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Alam NH, Ashraf H, Sarker SA, Olesen M, Troup J, Salam MA, Gyr N, Meier R. Efficacy of partially hydrolyzed guar gum-added oral rehydration solution in the treatment of severe cholera in adults. Digestion. 2008;78:24–29. doi: 10.1159/000152844. [DOI] [PubMed] [Google Scholar]
- 23.Furnari M, Parodi A, Gemignani L, Giannini EG, Marenco S, Savarino E, Assandri L, Fazio V, Bonfanti D, Inferrera S, Savarino V. Clinical trial: the combination of rifaximin with partially hydrolysed guar gum is more effective than rifaximin alone in eradicating small intestinal bacterial overgrowth. Aliment Pharmacol Ther. 2010;32:1000–1006. doi: 10.1111/j.1365-2036.2010.04436.x. [DOI] [PubMed] [Google Scholar]
- 24.Romano C, Comito D, Famiani A, Calamarà S, Loddo I. Partially hydrolyzed guar gum in pediatric functional abdominal pain. World J Gastroenterol. 2013;19:235–240. doi: 10.3748/wjg.v19.i2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dall'Alba V, Silva FM, Antonio JP, Steemburgo T, Royer CP, Almeida JC, Gross JL, Azevedo MJ. Improvement of the metabolic syndrome profile by soluble fibre - guar gum - in patients with type 2 diabetes: a randomised clinical trial. Br J Nutr. 2013;110:1601–1610. doi: 10.1017/S0007114513001025. [DOI] [PubMed] [Google Scholar]
- 26.Yasukawa Z, Naito Y, Takagi T, Mizushima K, Tokunaga M, Ishihara N, R Juneja L, Yoshikawa T. Partially hydrolyzed guar gum affects the expression of genes involved in host defense functions and cholesterol absorption in colonic mucosa of db/db male mice. J Clin Biochem Nutr. 2012;51:33–38. doi: 10.3164/jcbn.11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohashi Y, Sumitani K, Tokunaga M, Ishihara N, Okubo T, Fujisawa T. Consumption of partially hydrolysed guar gum stimulates Bifidobacteria and butyrate-producing bacteria in the human large intestine. Benef Microbes. 2015;6:451–455. doi: 10.3920/BM2014.0118. [DOI] [PubMed] [Google Scholar]
- 28.Reider SJ, Moosmang S, Tragust J, Trgovec-Greif L, Tragust S, Perschy L, Przysiecki N, Sturm S, Tilg H, Stuppner H, Rattei T, Moschen AR. Prebiotic Effects of Partially Hydrolyzed Guar Gum on the Composition and Function of the Human Microbiota-Results from the PAGODA Trial. Nutrients. 2020;12 doi: 10.3390/nu12051257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y, Miyamoto K, Kaneko S. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 30.Grisham MB, Hernandez LA, Granger DN. Xanthine oxidase and neutrophil infiltration in intestinal ischemia. Am J Physiol. 1986;251:G567–G574. doi: 10.1152/ajpgi.1986.251.4.G567. [DOI] [PubMed] [Google Scholar]
- 31.Luther J, Garber JJ, Khalili H, Dave M, Bale SS, Jindal R, Motola DL, Luther S, Bohr S, Jeoung SW, Deshpande V, Singh G, Turner JR, Yarmush ML, Chung RT, Patel SJ. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol Gastroenterol Hepatol. 2015;1:222–232. doi: 10.1016/j.jcmgh.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagahama S, Korenaga D, Honda M, Inutsuka S, Sugimachi K. Assessment of the intestinal permeability after a gastrectomy and the oral administration of anticancer drugs in rats: nitric oxide release in response to gut injury. Surgery. 2002;131:S92–S97. doi: 10.1067/msy.2002.119310. [DOI] [PubMed] [Google Scholar]
- 33.Kish L, Hotte N, Kaplan GG, Vincent R, Tso R, Gänzle M, Rioux KP, Thiesen A, Barkema HW, Wine E, Madsen KL. Environmental particulate matter induces murine intestinal inflammatory responses and alters the gut microbiome. PLoS One. 2013;8:e62220. doi: 10.1371/journal.pone.0062220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagashima K, Hisada T, Sato M, Mochizuki J. Application of new primer-enzyme combinations to terminal restriction fragment length polymorphism profiling of bacterial populations in human feces. Appl Environ Microbiol. 2003;69:1251–1262. doi: 10.1128/AEM.69.2.1251-1262.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Doré J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagahama K, Mochizuki J, Suzuki S, Shimomura K. Phylogenetic Analysis of 16S ribosomal RNA gene sequences from human fecal microbiota and improved utility of terminal restriction fragment length polymorphism profiling. Biosci Microflora. 2006;25:99–107. [Google Scholar]
- 37.Ushida K, Sakata T. Effect of pH on oligosaccharide fermentation by porcine cecal digesta. Animal Sci Technol . 1998;69:100–107. [Google Scholar]
- 38.Van Herck MA, Vonghia L, Francque SM. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter's Guide. Nutrients. 2017;9:1072. doi: 10.3390/nu9101072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, Wiest R, Mileti E, Galbiati M, Invernizzi P, Adorini L, Penna G, Rescigno M. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol. 2019;71:1216–1228. doi: 10.1016/j.jhep.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pessentheiner AR, Ducasa GM, Gordts PLSM. Proteoglycans in Obesity-Associated Metabolic Dysfunction and Meta-Inflammation. Front Immunol. 2020;11:769. doi: 10.3389/fimmu.2020.00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Power Guerra N, Müller L, Pilz K, Glatzel A, Jenderny D, Janowitz D, Vollmar B, Kuhla A. Dietary-Induced Low-Grade Inflammation in the Liver. Biomedicines. 2020;8 doi: 10.3390/biomedicines8120587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carotti S, Guarino MP, Vespasiani-Gentilucci U, Morini S. Starring role of toll-like receptor-4 activation in the gut-liver axis. World J Gastrointest Pathophysiol. 2015;6:99–109. doi: 10.4291/wjgp.v6.i4.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pachikian BD, Essaghir A, Demoulin JB, Catry E, Neyrinck AM, Dewulf EM, Sohet FM, Portois L, Clerbaux LA, Carpentier YA, Possemiers S, Bommer GT, Cani PD, Delzenne NM. Prebiotic approach alleviates hepatic steatosis: implication of fatty acid oxidative and cholesterol synthesis pathways. Mol Nutr Food Res. 2013;57:347–359. doi: 10.1002/mnfr.201200364. [DOI] [PubMed] [Google Scholar]
- 45.Abdou RM, Zhu L, Baker RD, Baker SS. Gut Microbiota of Nonalcoholic Fatty Liver Disease. Dig Dis Sci. 2016;61:1268–1281. doi: 10.1007/s10620-016-4045-1. [DOI] [PubMed] [Google Scholar]
- 46.Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, Perlemuter G, Cassard-Doulcier AM, Gérard P. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. 2013;62:1787–1794. doi: 10.1136/gutjnl-2012-303816. [DOI] [PubMed] [Google Scholar]
- 47.De Minicis S, Rychlicki C, Agostinelli L, Saccomanno S, Candelaresi C, Trozzi L, Mingarelli E, Facinelli B, Magi G, Palmieri C, Marzioni M, Benedetti A, Svegliati-Baroni G. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology. 2014;59:1738–1749. doi: 10.1002/hep.26695. [DOI] [PubMed] [Google Scholar]
- 48.Jin CJ, Sellmann C, Engstler AJ, Ziegenhardt D, Bergheim I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH) Br J Nutr. 2015;114:1745–1755. doi: 10.1017/S0007114515003621. [DOI] [PubMed] [Google Scholar]
- 49.Hiippala K, Jouhten H, Ronkainen A, Hartikainen A, Kainulainen V, Jalanka J, Satokari R. The Potential of Gut Commensals in Reinforcing Intestinal Barrier Function and Alleviating Inflammation. Nutrients. 2018;10 doi: 10.3390/nu10080988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng Y, Wang Y, Wang P, Huang Y, Wang F. Short-Chain Fatty Acids Manifest Stimulative and Protective Effects on Intestinal Barrier Function Through the Inhibition of NLRP3 Inflammasome and Autophagy. Cell Physiol Biochem. 2018;49:190–205. doi: 10.1159/000492853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No additional data are available.