

Abstract
Lewis acid catalysts have been shown to promote carbonyl–olefin metathesis through a critical four-membered-ring oxetane intermediate. Recently, Brønsted-acid catalysis of related substrates was similarly proposed to result in a transient oxetane, which fragments within a single elementary step via a postulated oxygen-atom transfer mechanism. Herein, careful quantum chemical investigations show that Brønsted acid (triflic acid, TfOH) instead invokes a mechanistic switch to a carbonyl-ene reaction, and oxygen-atom transfer is uncompetitive. TfOH’s conjugate base is also found to rearrange H atoms and allow isomerization of the carbocations that appear after the carbonyl-ene reaction. The mechanism explains available experimental information, including the skipped diene species that appear transiently before product formation. The present study clarifies the mechanism for activation of intramolecular carbonyl–olefin substrates by Brønsted acids and provides important insights that will help develop this exciting class of catalysts.
Graphical Abstract
INTRODUCTION
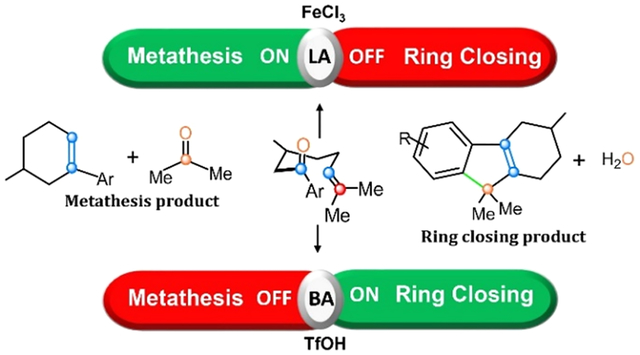
Carbonyl–olefin metathesis (COM),1–15 an analogue of the well-developed olefin metathesis reaction,16 is an evolving synthetic method for constructing carbon–carbon bonds from simple starting materials.17–25 The routine practice of COM was limited prior to reports of a bicyclic hydrazine catalyst26 that effects the transformation catalytically. More recently, Schindler and co-workers described a general catalytic approach involving earth-abundant Lewis acid (LA) catalysts (typically FeCl3), allowing catalytic intramolecular COM reactions involving aliphatic and aromatic substrates. These efficient catalysts have allowed COM to proceed at room temperature to yield a variety of cyclohexene or cyclopentene products.1,6,27–30 Similar COM reactions have also been observed via Brønsted-acid (HCl) catalysis within a supramolecular container.31 In closely related work, Schindler’s group also found that Brønsted-acid catalysts (triflic acid, TfOH) promote a related but qualitatively different mode of reactivity32 where carbonyl–olefin (CO) substrates are selectively transformed into tetrahydrofluorene (TDF) products33 in a single synthetic step. This transformation is a significant advance over strategies to form TDF that involves a series of transition-metal-catalyzed cycloaddition reactions,34,35 while also avoiding unwanted polymerization and isomerization of olefins.32,36 The TfOH-catalyzed transformation was shown to be applicable to a variety of electronically and sterically functionalized CO substrates, highlighting the potential of this promising synthetic approach.32
These recent works complement existing strategies for activation and transformation of carbonyl–olefin containing substrates (1). For instance, carbonyl-ene (CE)37–41 or Prins type41–45 reactions leading to cyclic unsaturated (2) or saturated alcohols (3), respectively, are the most commonly encountered reactions between carbonyl and olefin functional groups (Figure 1A,B).46,47 In contrast, Schindler and others27–30,32 have reported that LA activation can allow access to oxetane intermediates (4) which are otherwise only accessible under photochemical conditions (Figure 1C).46,48
Figure 1.
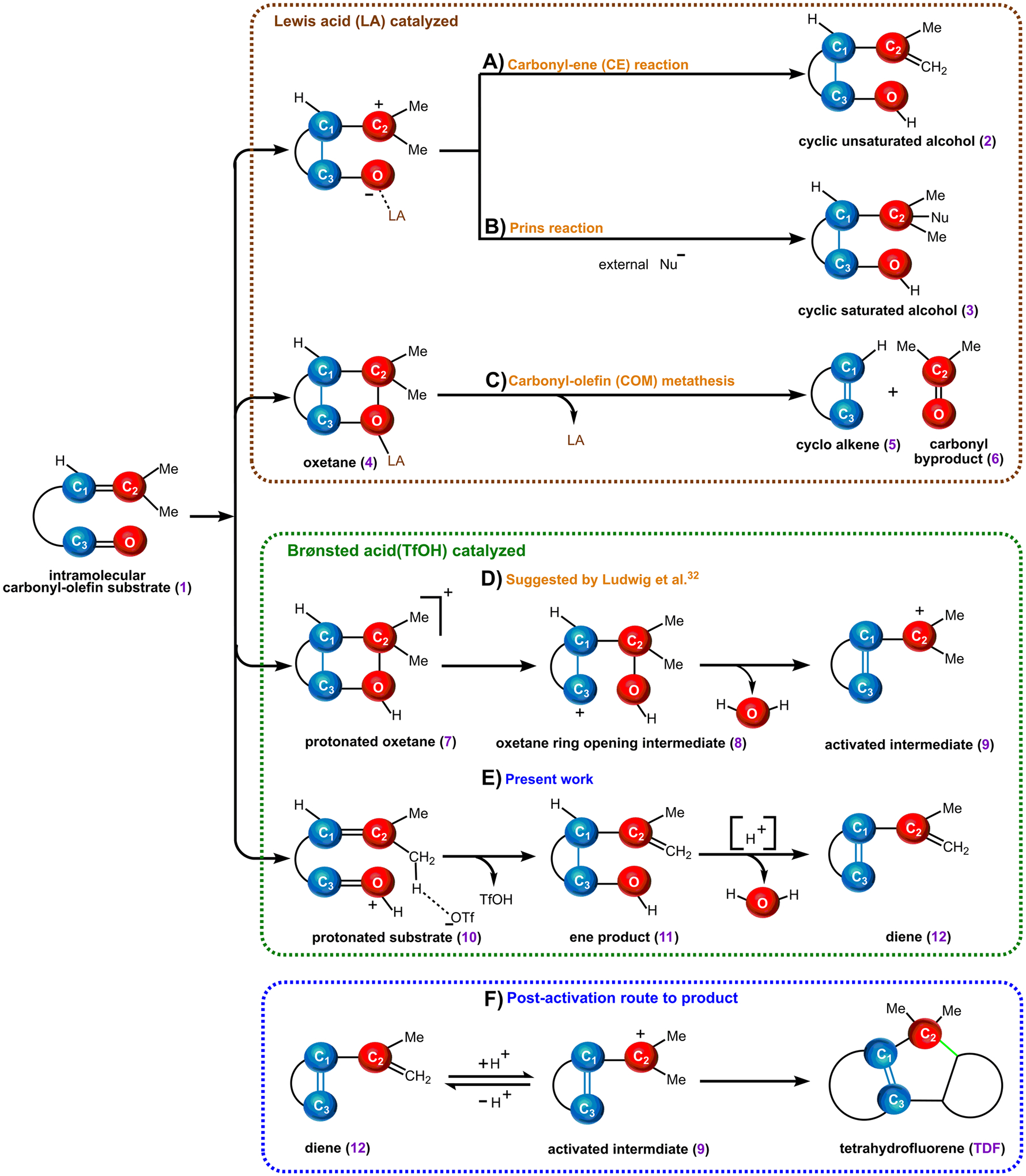
Reactivity of carbonyls and olefins: (A) carbonyl-ene (CE) reaction; (B) Prins reaction; (C) carbonyl–olefin metathesis (COM); (D) interrupted carbonyl–olefin metathesis suggested by Ludwig et al.;32 (E) our proposed mechanism; (F) postactivation route to the product.
The involvement of the oxetane intermediate has been implicated as crucial to the LA-catalyzed COM reaction27–30,32 and therefore serves as a centerpiece that distinguishes this class of catalysis from other synthetic strategies. Our mechanistic studies indicated that the oxetane intermediate is generated through a rate-limiting concerted, asynchronous [2 + 2]-cycloaddition pathway.28 A subsequent reverse [2 + 2]-cycloaddition channel breaks up the oxetane to yield the COM product, i.e., cyclic alkene (5) and carbonyl byproduct (6).27–29 The activation of CO substrates by the Brønsted acid TfOH was suggested to occur through a related pathway, where protonated oxetane (7) forms via turnover limiting [2 + 2] cycloaddition, which then immediately fragments.31 Through this single elementary step, it was proposed that protonation of the carbonyl (1) triggers an intramolecular oxygen-atom transfer (OAT) to yield an oxetane ring-opening intermediate (8) (Figure 1D). The postulated OAT is followed by the elimination of water, which allows the activated carbocationic intermediate (9) to transform into TDF (Figure 1F, right).32 Therefore, this pathway appears to exploit the oxetane for a new transformation when Brønsted acid is involved, in comparison to LA activation of the same functional group to achieve COM. OAT, however, is unusual and merits additional investigation to verify its mechanistic relevance compared to the other pathways of Figure 1.
Herein, we propose that a less surprising pathway involving the protonated substrate (10) resulting in the ene product (11) is more likely than the postulated OAT mechanism (Figure 1E). In order to arrive at this mechanism, an improved model of the reaction was required, based on the treatment of the TfOH catalyst in the simulation. The prior study modeled TfOH as a proton and did not consider the conjugate base.32 TfOH, however, is unlikely to be a strong enough acid in low dielectric solvent (such as benzene (ε = 2.27) used in the experiment32) to permit full counterion separation from the proton, despite its low pKa in water (pKa = −14.7 ± 2.0, ε = 78.35).49 The study that follows will show that the triflate anion (OTf) plays an active role in the reaction in low-polarity media and is necessary for the model to yield the correct mechanism for activation. The present study will then point to the involvement of an allylic carbocation (AC) as essential to afford TDF via Friedel–Crafts alkylation (FCA).31 This finding will provide an explanation for the role of the experimentally observed diene species (12, Figure 1E), which converts into the activated carbocationic intermediate (9) en route to TDF (Figure 1F, left).
RESULTS AND DISCUSSION
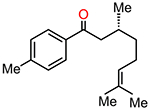
To begin investigating the mechanism for the reaction between CO and TfOH, the experimentally employed substrate 3,7-dimethyl-1-(p-tolyl)oct-6-en-1-one was complexed with TfOH, producing species 13 (Figure 2). Protonation of the substrate is essential for its activation,32 but this event was not accurately captured by standard gas-phase geometry optimization, with the proton remaining on TfOH rather than transferring to the substrate. This is consistent with TfOH being a much weaker acid in the gas phase53,54 compared to the solvent phase. Nevertheless, when solvent effects are included, protonation of the carbonyl substrate by TfOH results in the formation of ion-pair species 14. This species is slightly uphill by 0.8 kcal/mol, indicating that 14 is in thermodynamic equilibrium with complex 13. Separation of the protonated substrate from the OTf in 14 is unlikely in benzene, due to the associated cost of 39.3 kcal/mol for breaking up the ion pair. Therefore, mechanisms relying on ion-pair separation are unfeasible, at least in the low-dielectric-constant solvent. This observation, in turn, points toward the necessity of considering specific interactions of the reaction intermediates with the conjugate base throughout the simulation. The optimized geometry of 14 (displayed in Figure 3A) shows OTf can simultaneously interact with the protonated carbonyl group and the alkene methyl group. This suggests possible active participation of OTf in abstracting a proton from the proximal methyl group. Such a pathway was indeed found, where OTf permits a concerted CE reaction via TS14–15 (Figure 3B). TS14–15 has a barrier of 13.8 kcal/mol and results in the formation of intermediate 15 where the TfOH is hydrogen-bonded to the hydroxyl group (Figure 2). Therefore, transforming from 13 to 15 is a CE reaction, with a moderate barrier that can be overcome at room temperature. To the best of our knowledge, this active role of OTf in the CE reaction has never been demonstrated explicitly.55
Figure 2.
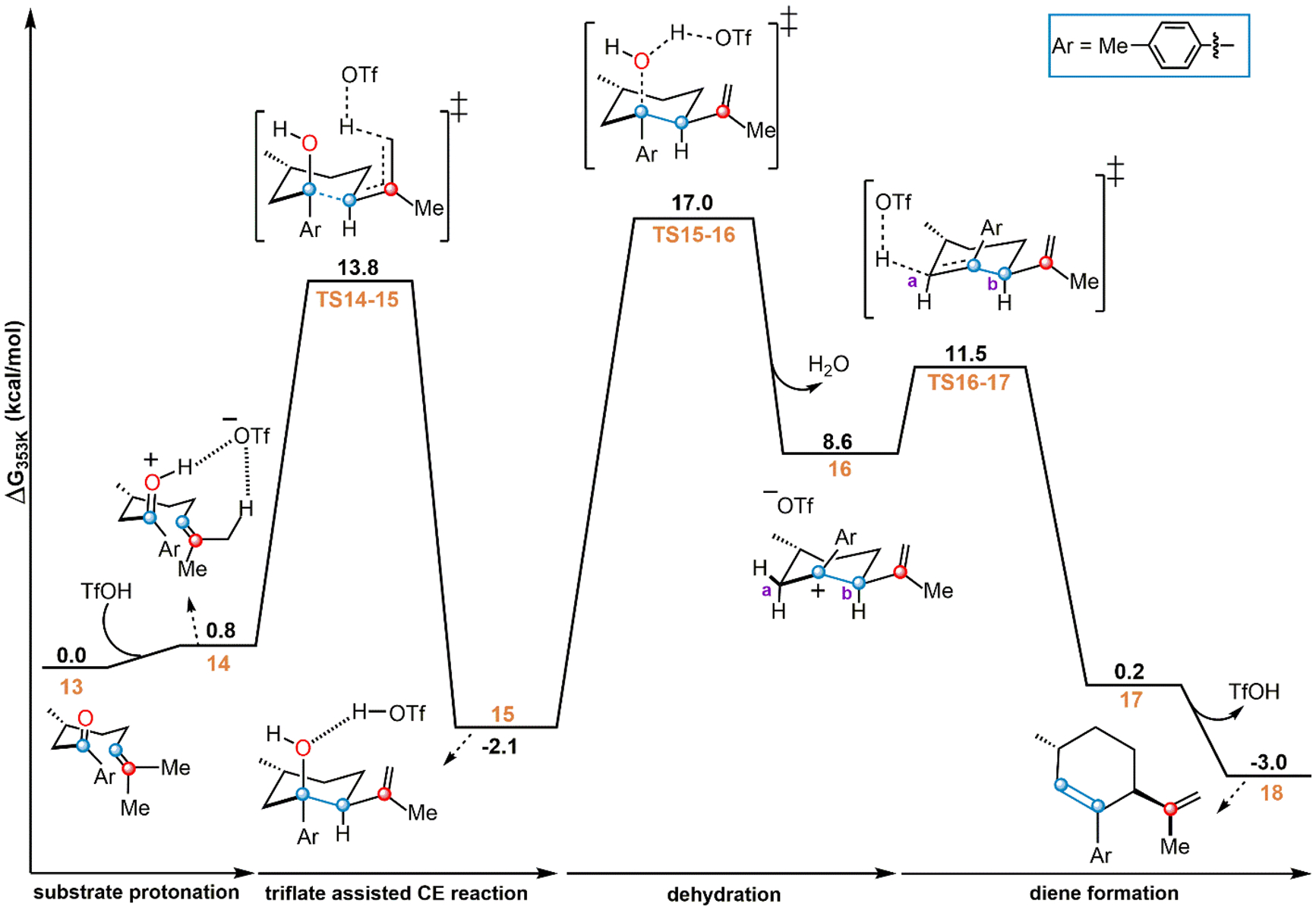
Reaction profile for activation via carbonyl-ene (CE) reaction. Free energies from ωB97X-D/6–311++G**/CPCM (benzene).
Figure 3.
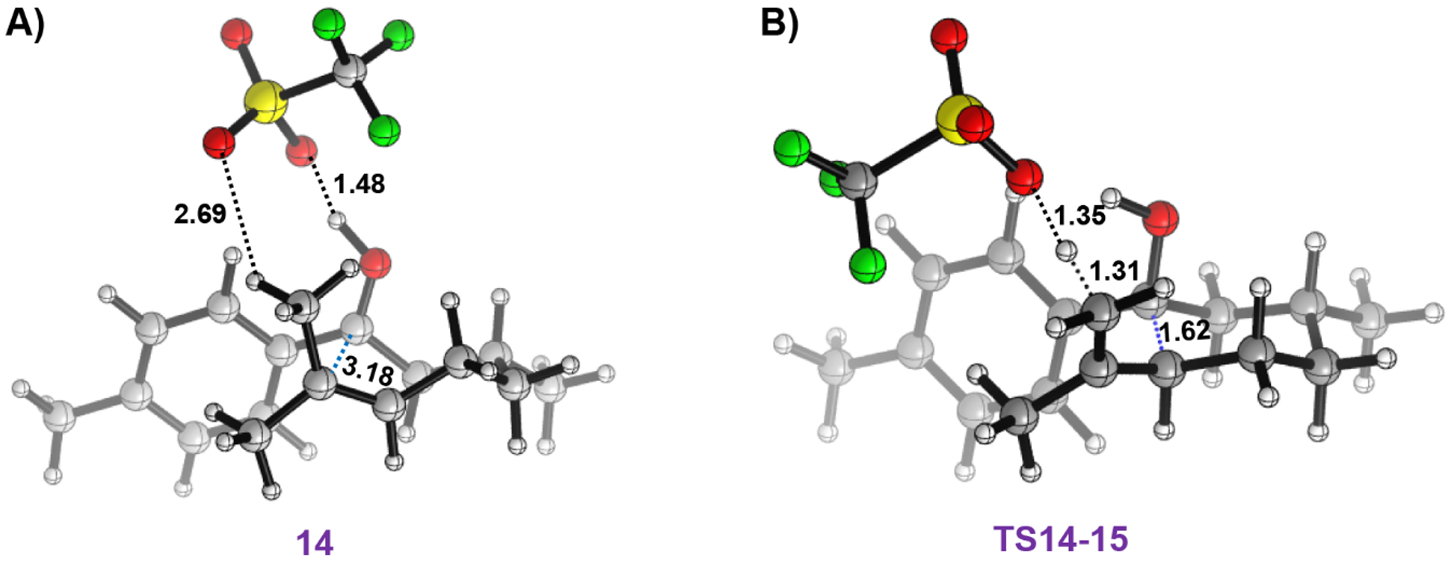
Optimized 3D structure of (A) species 14 and (B) TS14–15. Bond lengths are in Å. Color code: C gray, H white, O red, S yellow, F green.
Interestingly, intermediate 15 (Figure 2) is analogous to the mechanistic probe molecule, 19, whose reactivity was monitored experimentally to gather key mechanistic details (Scheme 1).32 The probe revealed that, under the reaction conditions (80 °C), species 19 can reversibly transform into the starting aryl ketone (20) and the product TDF (21, Scheme 1A, left). This supports 15 as being a plausible intermediate connecting 20 and product. In addition, experiments detected an unexpected diene product (22) at a lower temperature (25 °C) (Scheme 1A, right), which is also observed starting from 20, where 22 is found to be a transient species prior to TDF (21) formation (Scheme 1B).31 Our study indicates that intermediate 15 is slightly downhill (−2.1 kcal/mol) and hence it can revert back to its precursor 13 by overcoming a barrier of 15.9 kcal/mol (Figure 2). Thus, our prediction is consistent with experimental results that suggest the reversible transformation of the mechanistic probe molecule 19 into the starting aryl ketone (20) (Scheme 1A).
Scheme 1.
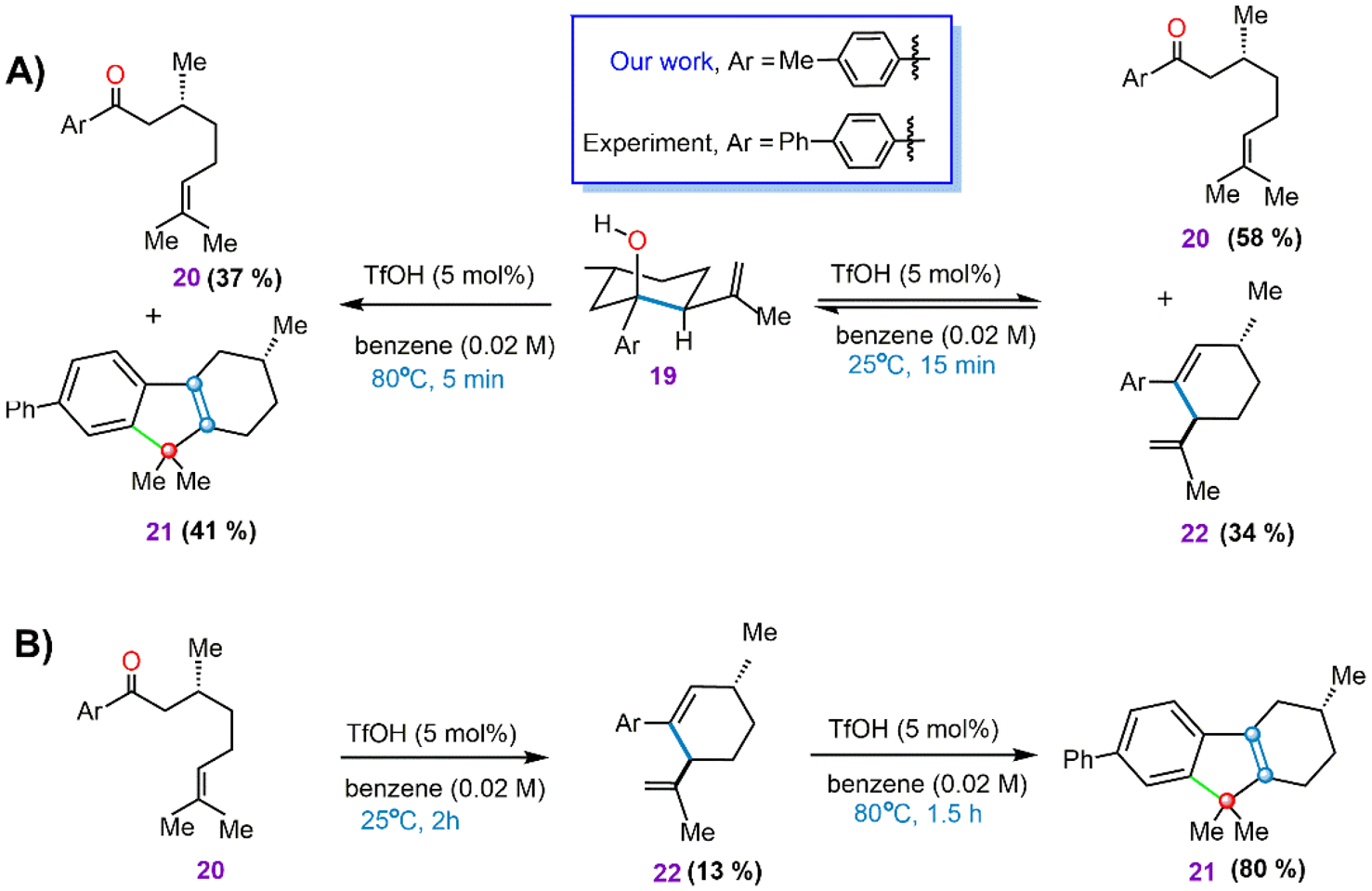
Important Experimental Results Obtained by Ludwig et al.,32 Showing (A) the Mechanistic Probe Intermediate (19) and (B) Differences in Reactivity at High and Low Temperatures
Figure 2 shows that species 18, which is analogous to the observed diene 22 (Scheme 1), can be formed from 15 via dehydration followed by deprotonation. A water molecule is released from 15 through TS15–16 at a barrier of 19.1 kcal/mol, resulting in the formation of the benzylic carbocation, 16, and OTf. The generation of the carbocationic species 16 is predicted to be 8.6 kcal/mol uphill in the free energy landscape.
Moving further along the reaction path of Figure 2, OTf can abstract a proton from one of the two α carbons attached to the positively charged carbon in 16. Deprotonation from the left α carbon (site a, Figure 2) is kinetically preferred due to the proximal location of the nascent OTf and proceeds via TS16–17 at a computed barrier of 13.6 kcal/mol. TS16–17 forms complex 17 which then separates to give TfOH and the diene 18. Because 18 is only −3.0 kcal/mol below the starting materials, it can reversibly transform back to its precursor at a predicted barrier of 14.5 kcal/mol. This indicates that the initial buildup of the experimentally observed diene species (22, Scheme 1B) is driven by kinetic preference. Furthermore, species 15 is the key intermediate that connects the substrate and transient diene product (18) through the CE reaction followed by dehydration. The CE reaction, therefore, appears as a plausible pathway for the activation of CO substrates by TfOH.
To evaluate the likelihood that oxetane (23) formation may compete with the CE mechanism, TS14–23 was located (Figure 4). Interestingly, the presence of the triflate allows a qualitatively different mechanism for oxetane formation compared to a bare proton: the oxetane is a stable intermediate, as TS14–23 involves deprotonation of the newly formed four-membered ring. Regardless, TS14–23 has a free energy barrier of 20.6 kcal/mol (Figure 4) and can be ruled out, since it is more than 6 kcal/mol above TS14–15. Additional computations of electronically and sterically differentiated CO substrates show the CE activation mechanism is favored over the oxetane channel (Table 1). This evidence suggests that OAT does not occur through the oxetane-forming transition state. Overall, the inactivity of the oxetane channel and predicted relevance of the CE activation mechanism agree with experimental observations that show a new reaction product (TDF) but not the usual metathesis product under Brønsted acid catalysis.32
Figure 4.
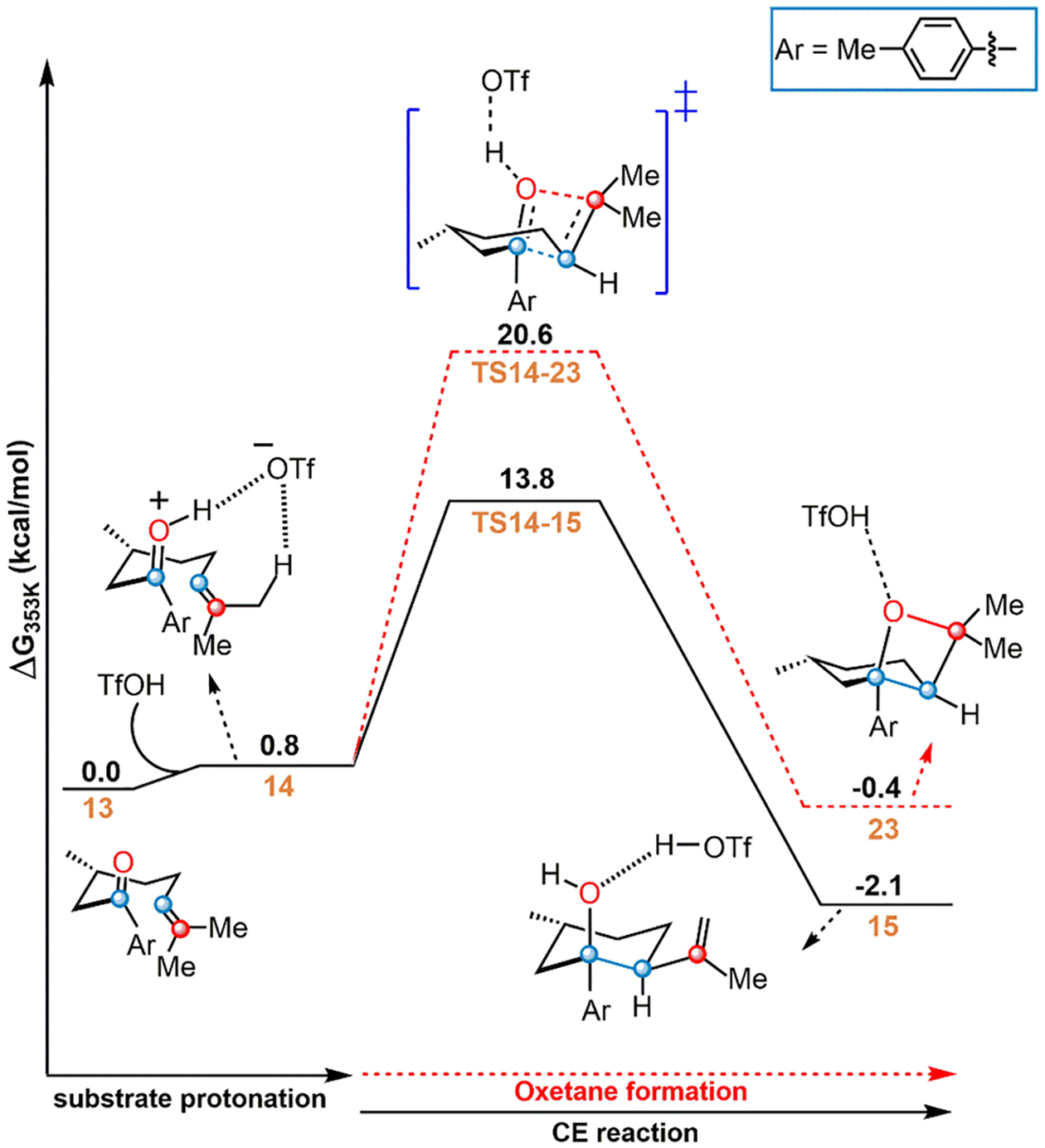
Reaction profile for oxetane formation. Free energies from ωB97X-D/6–311++G**/CPCM (benzene).
Table 1.
Free Energy Barriers for CE Reaction (TS14–15) Compared to Those of Oxetane Formation (TS14–23) for a Range of Carbonyl–Olefin Substratesa
| Substrate |
(kcal/mol) |
(kcal/mol) |
(kcal/mol) |
|
|---|---|---|---|---|
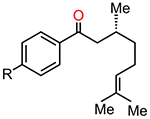 |
R = Me | 13.8 | 20.6 | 6.8 |
| R =OH | 14.3 | 20.8 | 6.5 | |
| R = OMe | 13.9 | 21.0 | 7.1 | |
| R = F | 13.4 | 19.9 | 6.5 | |
| R = tBu | 15.8 | 21.6 | 5.8 |
Free energies from ωB97X-D/6–311++G**/CPCM (benzene).
After revealing the preferred activation channel of the protonated reactant complex (14), the reactivity of its successor, diene intermediate 18, was investigated (Figure 5). In short, this pathway proceeds through ion-pair reorganization to access the crucial AC, which subsequently undergoes FCA to afford the final product, TDF. The reverse reaction from 18 to 16 initiates the postactivation channel for 18 (Figure 1F). From 16, OTf moves to the opposite face of the activated substrate, producing ion pair 24. The movement of the OTf brings the OTf close to the α hydrogen (site b) adjacent to the carbocation center (Figure 5), and thereby deprotonation from site b becomes feasible. From 24, deprotonation from the α-carbon center (site b, Figure 5) proceeds through TS24–26 with a free energy barrier of 15.8 kcal/mol above 18, leading to another diene intermediate (26). From 26, rotation around the C–C bond via TS26–27 leads to yet another diene intermediate (27) where the exocyclic double bond is close to TfOH. We note in passing that two of the steps along the pathway from 18 to 27 may interact with a second equivalent of TfOH; these details are provided in the Supporting Information (sections S2 and S3). Interestingly, the structure of the diene intermediate 18 is analogous to what was observed in the experiment (see the discussion in section S4 of the Supporting Information).32
Figure 5.
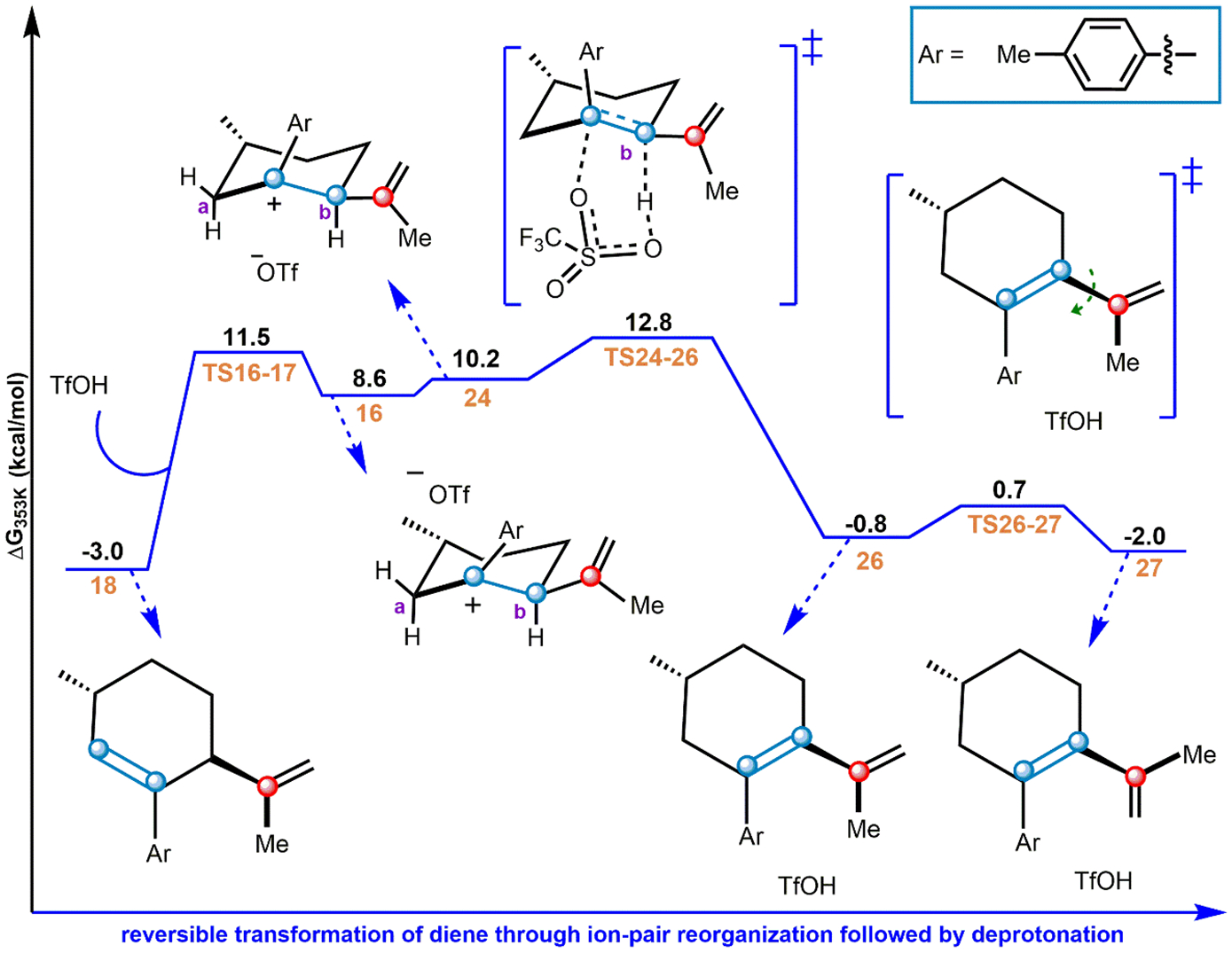
Reorganization of the diene intermediate. Free energies from ωB97X-D/6–311++G**/CPCM (benzene).
Following isomerization of the diene to reach 27, protonation of the exocyclic alkene generates the AC, 28, by overcoming a free energy barrier of 6.1 kcal/mol (Figure 6). 28 can transform into a charge-neutral species 29 through OTf binding to the carbocation center via TS28–29 with a barrier of 4.0 kcal/mol above 28. Rotation around the C–C bond followed by the C–O cleavage via TS29–30 (ΔG⧧ = 4.5 kcal/mol above 29) leads to 30, where the position of OTf is on the opposite face as that of 28. This positional change is required because FCA from 28 would have a misaligned phenyl group (see sections S5 and S6 of the Supporting Information), which must be first rotated for FCA to proceed with a low barrier. Species 30, however, bypasses this high barrier rotation and better aligns OTf for a feasible FCA step. After forming species 30, FCA via TS30–31 occurs with a barrier of 10.5 kcal/mol to produce the TDF product bound to TfOH. Finally, dissociation of the catalyst to form free TDF (32) is predicted to be a barrierless process. Formation of 32 is thermodynamically highly favorable compared to the starting materials (ΔG = −28.5 kcal/mol), and the reverse reaction is unfavorable from this point in the reaction sequence. The proposed mechanism for product formation thus agrees with ref 32’s invocation of FCA after substrate activation but arrives at the activated intermediates through CE rather than OAT. Moreover, the FCA step (TS30–31) has a higher activation barrier (10.5 kcal/mol, Figure 6) compared to the elimination from the AC, 28 (TS27–28, 5.8 kcal/mol above 28). Thus, this result is consistent with the experimental findings from the tertiary alcohol mechanistic probe where the elimination route from the AC is preferred over FCA at low temperature.32
Figure 6.
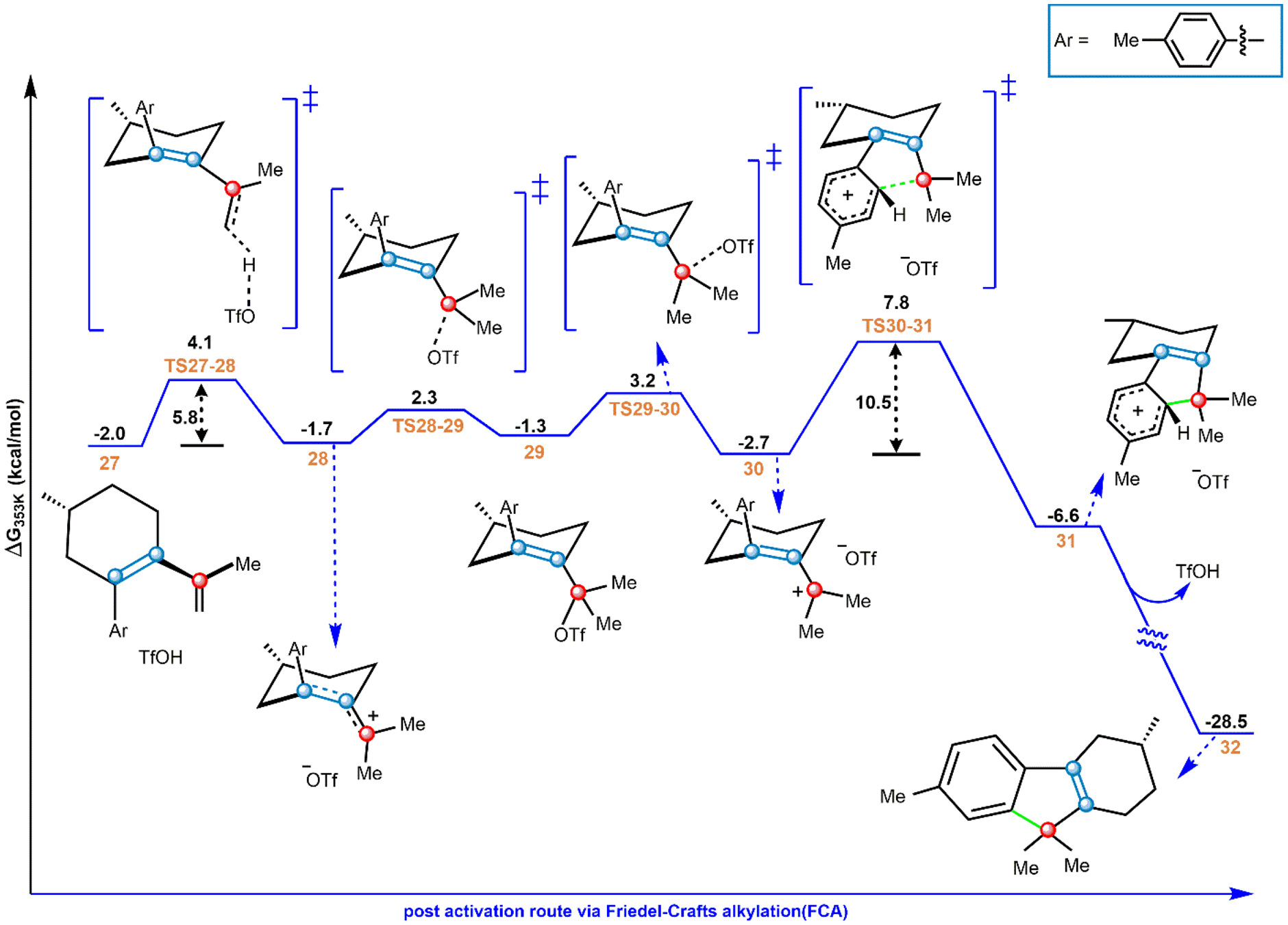
Reaction profile for postactivation of the diene. Free energies from ωB97X-D/6–311++G**/CPCM (benzene).
The proposed pathway for activation and FCA of intramolecular CO substrates by Brønsted acid catalysis requires nontrivial interactions with the conjugate base (OTf) to open up the crucial CE reaction channel. This mechanism implies that the nature of the Brønsted acid can have a significant influence on the barrier for methyl proton abstraction. While increasing the acidity (HClO4) had little impact on the activation barrier for TS14–15, a somewhat weaker acid (FSO3H) reduces the barrier by 1.4 kcal/mol (Table 2). The weaker acid (FSO3H) results in a slightly lower barrier because the conjugate base (FSO3 anion) is a stronger base, facilitating the proton abstraction from the methyl group in TS14–15. Reducing the acidity further, however, eventually hinders the protonation of the substrate and leads to an increase in the reaction barrier (e.g., CH3SO3H). This suggests a “Goldilocks” effect may be in play, where a specific range of pKa values are needed to effect the CE activation channel.58 Due to the non-oxidizing nature of their conjugate bases, FSO3H or TfOH might be advantageous in avoiding unwanted reaction channels.
Table 2.
Evaluation of Various Brønsted Acidsa
| Substrate | Bronstod Acid | pKa (in water) |
(kcal/mol) |
|---|---|---|---|
 |
TfOH(CF3SO3H) | −14.7 ± 2.048 | 13.8 |
| HCIO4 | −15.2 ± 2.048 | 12.8 | |
| FSO3H | −10.055 | 12.4 | |
| HSO3H | NA | 21.4 | |
| CH3SO3H | −2.056 | 22.3 |
Free energy barrier for the CE (TS14–15) reaction. Calculated at ωB97X-D/6–311++G**/CPCM (benzene).
CONCLUSION
The overall mechanism for Brønsted-acid-catalyzed transformation of CO substrates is shown in Figure 7. The ion pair 14 is critical to the CE reaction outcompeting oxetane formation, which allows TS14–15 to be predicted as the preferred pathway. While oxetane formation is favorable with other catalysts,28 it is not viable according to our simulations. Acid-catalyzed dehydration of 15 leads immediately to diene intermediates such as 18, which were observed in the experiment.32 This mechanism is also consistent with experimental studies of the probe molecule 19, shown in Scheme 1, which converts into starting material or diene under mild conditions or forms product under the higher-temperature reaction conditions. The simulations reported in this Article, therefore, provide a satisfactory description of the overall reaction mechanism, and this mechanism is consistent with experimental observations.32 This mechanism, therefore, will be especially valuable in guiding further experimental endeavors, as the invocation of a qualitatively different reaction mechanism will require new considerations in designing the next stage of experiments. Specific items that might be interesting for further in-depth analysis are the potential binding modes of OTf anions to the carbocationic intermediates (e.g., species 29) and kinetic isotope effects that may result from proton transfers during the activation event. The latter studies likely will differentiate the previously proposed OAT mechanism from the herein-proposed CE reaction for activation of CO substrates by Brønsted acids.
Figure 7.
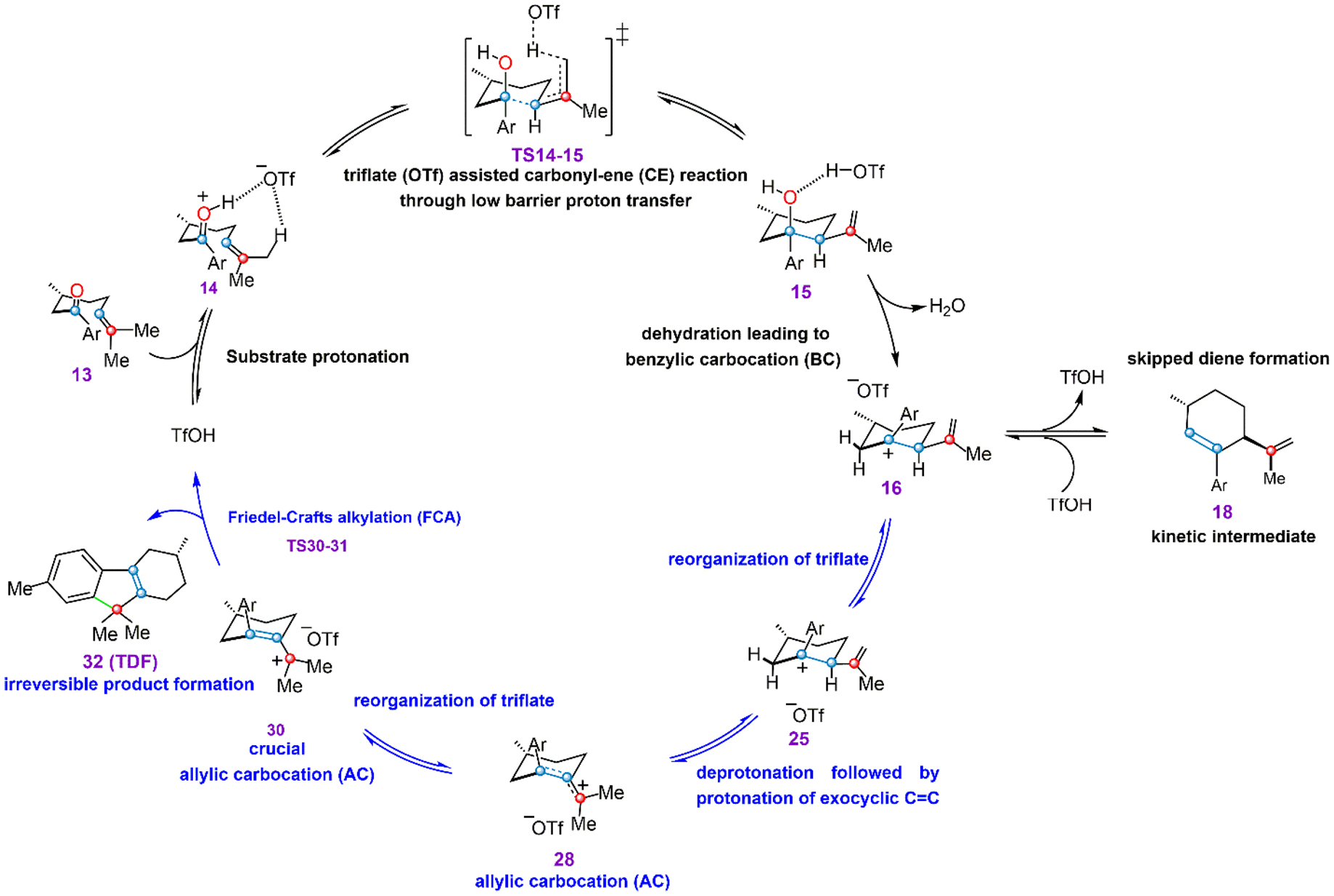
Proposed mechanism for TfOH-catalyzed reactivity of carbonyl–olefin substrates.
COMPUTATIONAL METHODS
Computational investigations were carried out using the reaction discovery tools of the Zimmerman group, in particular, the growing string method (GSM). The GSM searches for plausible elementary steps along a qualitatively defined reaction coordinate and then locates minimum energy reaction paths and the associated transition states,50–52 all without requiring detailed prior knowledge of the transition state structures. Reported energies for intermediates and activation barriers are solvent-phase free energies obtained using the ωB97X-D density functional (see section S1 in the Supporting Information for full computational details).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the NIH for support under R35-GM-128830.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.0c03021
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c03021.
Computational details, Cartesian coordinates, and energies of all intermediate and transition states investigated, ion-pair reorganization mechanism, and alternative Friedel–Crafts alkylation mechanism (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Becker MR; Watson RB; Schindler CS Beyond olefins: new metathesis directions for synthesis. Chem. Soc. Rev 2018, 47 (21), 7867–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Carless HAJ; Trivedi HS New ring expansion reaction of 2-t-butyloxetans. J. Chem. Soc., Chem. Commun 1979, No. 8, 382–383. [Google Scholar]
- (3).D’Auria M; Racioppi R; Viggiani L Paternò-Büchi reaction between furan and heterocyclic aldehydes: oxetane formation vs. metathesis. Photochem. Photobiol. Sci 2010, 9 (8), 1134–1138. [DOI] [PubMed] [Google Scholar]
- (4).Jones G; Acquadro MA; Carmody MA Long-chain enals via carbonyl-olefin metathesis. An application in pheromone synthesis. J. Chem. Soc., Chem. Commun 1975, 0 (6), 206–207. [Google Scholar]
- (5).Jones G; Schwartz SB; Marton MT Regiospecific thermal cleavage of some oxetan photoadducts: carbonyl-olefin metathesis in sequential photochemical and thermal steps. J. Chem. Soc., Chem. Commun 1973, No. 11, 374–375. [Google Scholar]
- (6).Ludwig JR; Schindler CS Lewis Acid Catalyzed Carbonyl-Olefin Metathesis. Synlett 2017, 28 (13), 1501–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pérez-Ruiz R; Gil S; Miranda MA Stereodifferentiation in the Photochemical Cycloreversion of Diastereomeric Methoxynaph-thalene-Oxetane Dyads. J. Org. Chem 2005, 70 (4), 1376–1381. [DOI] [PubMed] [Google Scholar]
- (8).Pérez-Ruiz R; Miranda MA; Alle R; Meerholz K; Griesbeck AG An efficient carbonyl-alkene metathesis of bicyclic oxetanes: photoinduced electron transfer reduction of the Paternò-Büchi adducts from 2,3-dihydrofuran and aromatic aldehydes. Photochem. Photobiol. Sci 2006, 5 (1), 51–55. [DOI] [PubMed] [Google Scholar]
- (9).Riehl PS; Schindler CS Lewis Acid-Catalyzed Carbonyl-Olefin Metathesis. Trends Chem. 2019, 1 (2), 272–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schopov I; Jossifov C A carbonyl-olefin exchange reaction — new route to polyconjugated polymers, 1. A new synthesis of polyphenylacetylene. Makromol. Chem., Rapid Commun 1983, 4 (10), 659–662. [Google Scholar]
- (11).Soicke A; Slavov N; Neudörfl J-M; Schmalz H-G Metal-Free Intramolecular Carbonyl-Olefin Metathesis of ortho-Prenylaryl Ketones. Synlett 2011, 2011 (17), 2487–2490. [Google Scholar]
- (12).Valiulin RA; Arisco TM; Kutateladze AG Double-Tandem [4π+2π]η[2π+2π]η[4π+2π]η[2π+2π] Synthetic Sequence with Photoprotolytic Oxametathesis and Photoepoxidation in the Chromone Series. J. Org. Chem 2011, 76 (5), 1319–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Valiulin RA; Arisco TM; Kutateladze AG Photoinduced Intramolecular Cyclopentanation vs Photoprotolytic Oxametathesis in Polycyclic Alkenes Outfitted with Conformationally Constrained Aroylmethyl Chromophores. J. Org. Chem 2013, 78 (5), 2012–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).van Schaik H-P; Vijn R-J; Bickelhaupt F Acid-Catalyzed Olefination of Benzaldehyde. Angew. Chem., Int. Ed. Engl 1994, 33 (15–16), 1611–1612. [Google Scholar]
- (15).Naidu VR; Bah J; Franzén J Direct Organocatalytic Oxo-Metathesis, a trans-Selective Carbocation-Catalyzed Olefination of Aldehydes. Eur. J. Org. Chem 2015, 2015 (8), 1834–1839. [Google Scholar]
- (16).Grubbs RH; Wenzel AG Handbook of metathesis; Wiley-VCH: Weinheim, Germany, 2015; Vol. 1. [Google Scholar]
- (17).Heller ST; Kiho T; Narayan ARH; Sarpong R Protic-Solvent-Mediated Cycloisomerization of Quinoline and Isoquinoline Propargylic Alcohols: Syntheses of (±)-3-Demethoxyerythratidinone and (±)-Cocculidine. Angew. Chem., Int. Ed 2013, 52 (42), 11129–11133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hong B; Li H; Wu J; Zhang J; Lei X Total Syntheses of (−)-Huperzine Q and (+)-Lycopladines B and C. Angew. Chem 2015, 127 (3), 1025–1029. [DOI] [PubMed] [Google Scholar]
- (19).Iyer K; Rainier JD Olefinic Ester and Diene Ring-Closing Metathesis Using a Reduced Titanium Alkylidene. J. Am. Chem. Soc 2007, 129 (42), 12604–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Majumder U; Rainier JD Olefinic-ester cyclizations using Takai-Utimoto reduced titanium alkylidenes. Tetrahedron Lett. 2005, 46 (42), 7209–7211. [Google Scholar]
- (21).Nicolaou KC; Postema MHD; Claiborne CF Olefin Metathesis in Cyclic Ether Formation. Direct Conversion of Olefinic Esters to Cyclic Enol Ethers with Tebbe-Type Reagents. J. Am. Chem. Soc 1996, 118 (6), 1565–1566. [Google Scholar]
- (22).Rainier JD; Allwein SP An Iterative Approach to Fused Ether Ring Systems. J. Org. Chem 1998, 63 (16), 5310–5311. [Google Scholar]
- (23).Rainier JD; Allwein SP; Cox JM C-Glycosides to Fused Polycyclic Ethers. A Formal Synthesis of (±)-Hemibrevetoxin B. J. Org. Chem 2001, 66 (4), 1380–1386. [DOI] [PubMed] [Google Scholar]
- (24).Stille JR; Grubbs RH Synthesis of (.+−.)-.DELTA.9,12-capnellene using titanium reagents. J. Am. Chem. Soc 1986, 108 (4), 855–856. [Google Scholar]
- (25).Stille JR; Santarsiero BD; Grubbs RH Rearrangement of bicyclo[2.2.1]heptane ring systems by titanocene alkylidene complexes to bicyclo[3.2.0]heptane enol ethers. Total synthesis of (.+−.)-.DELTA.9(12)-capnellene. J. Org. Chem 1990, 55 (3), 843–862. [Google Scholar]
- (26).Griffith AK; Vanos CM; Lambert TH Organocatalytic Carbonyl-Olefin Metathesis. J. Am. Chem. Soc 2012, 134 (45), 18581–18584. [DOI] [PubMed] [Google Scholar]
- (27).Albright H; Riehl PS; McAtee CC; Reid JP; Ludwig JR; Karp LA; Zimmerman PM; Sigman MS; Schindler CS Catalytic Carbonyl-Olefin Metathesis of Aliphatic Ketones: Iron(III) Homo-Dimers as Lewis Acidic Superelectrophiles. J. Am. Chem. Soc 2019, 141 (4), 1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ludwig JR; Phan S; McAtee CC; Zimmerman PM; Devery JJ; Schindler CS Mechanistic Investigations of the Iron(III)-Catalyzed Carbonyl-Olefin Metathesis Reaction. J. Am. Chem. Soc 2017, 139 (31), 10832–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ludwig JR; Zimmerman PM; Gianino JB; Schindler CS Iron(III)-catalysed carbonyl-olefin metathesis. Nature 2016, 533, 374. [DOI] [PubMed] [Google Scholar]
- (30).Ma L; Li W; Xi H; Bai X; Ma E; Yan X; Li Z FeCl3-Catalyzed Ring-Closing Carbonyl-Olefin Metathesis. Angew. Chem., Int. Ed 2016, 55 (35), 10410–10413. [DOI] [PubMed] [Google Scholar]
- (31).Catti L; Tiefenbacher K Brønsted Acid-Catalyzed Carbonyl-Olefin Metathesis inside a Self-Assembled Supramolecular Host. Angew. Chem., Int. Ed 2018, 57 (44), 14589–14592. [DOI] [PubMed] [Google Scholar]
- (32).Ludwig JR; Watson RB; Nasrallah DJ; Gianino JB; Zimmerman PM; Wiscons RA; Schindler CS Interrupted carbonyl-olefin metathesis via oxygen atom transfer. Science 2018, 361 (6409), 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lu Z; Yang M; Chen P; Xiong X; Li A Total Synthesis of Hapalindole-Type Natural Products. Angew. Chem., Int. Ed 2014, 53 (50), 13840–13844. [DOI] [PubMed] [Google Scholar]
- (34).Lemière G; Gandon V; Agenet N; Goddard J-P; de Kozak A; Aubert C; Fensterbank L; Malacria M Gold(I)- and Gold(III)-Catalyzed Cycloisomerization of Allenynes: A Remarkable Halide Effect. Angew. Chem., Int. Ed 2006, 45 (45), 7596–7599. [DOI] [PubMed] [Google Scholar]
- (35).Wilkerson-Hill SM; Lavados CM; Sarpong R The Diels-Alder reactivity of 2-vinylindenes: synthesis of functionalized tetrahydrofluorenes. Tetrahedron 2016, 72 (26), 3635–3640. [Google Scholar]
- (36).Snider BB Lewis-acid catalyzed ene reactions. Acc. Chem. Res 1980, 13 (11), 426–432. [Google Scholar]
- (37).Alder K; Pascher F; Schmitz A Über die Anlagerung von Maleinsäure-anhydrid und Azodicarbonsäure-ester an einfach ungesättigte Koh an einfach ungesattigte Kohlenwasserstoffe. Zur Kenntnis von Substitutionsvorgängen in der Allyl-Stellung. Ber. Dtsch. Chem. Ges. B 1943, 76, 27–53. [Google Scholar]
- (38).Clarke ML; France MB The carbonyl ene reaction. Tetrahedron 2008, 64 (38), 9003–9031. [Google Scholar]
- (39).Ho C-Y; Schleicher KD; Chan C-W; Jamison TF Catalytic Addition of Simple Alkenes to Carbonyl Compounds by Use of Group 10 Metals. Synlett 2009, 2009 (16), 2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mikami K; Shimizu M Asymmetric ene reactions in organic synthesis. Chem. Rev 1992, 92 (5), 1021–1050. [Google Scholar]
- (41).Trost BM Comprehensive organic synthesis; Pergamon: 1991. [Google Scholar]
- (42).Arundale E; Mikeska LA The Olefin-Aldehyde Condensation. The Prins Reaction. Chem. Rev 1952, 51 (3), 505–555. [Google Scholar]
- (43).Pastor IM; Yus M The Prins Reaction: Advances and Applications. Curr. Org. Chem 2007, 11 (10), 925–957. [Google Scholar]
- (44).Kriewitz O Ueber Addition von Formaldehyd an einige Terpene. Ber. Dtsch. Chem. Ges 1899, 32 (1), 57–60. [Google Scholar]
- (45).Olier C; Kaafarani M; Gastaldi S; Bertrand MP Synthesis of tetrahydropyrans and related heterocycles via prins cyclization; extension to aza-prins cyclization. Tetrahedron 2010, 66 (2), 413–445. [Google Scholar]
- (46).Comprehensive organic synthesis, Vol. 5; Pergamon Press: Oxford, U.K., 1991. [Google Scholar]
- (47).Wyatt P; Warren S Organic synthesis: strategy and control; Wiley: 2013. [Google Scholar]
- (48).Büchi G; Inman CG; Lipinsky ES Light-catalyzed Organic Reactions. I. The Reaction of Carbonyl Compounds with 2-Methyl-2-butene in the Presence of Ultraviolet Light. J. Am. Chem. Soc 1954, 76 (17), 4327–4331. [Google Scholar]
- (49).Trummal A; Lipping L; Kaljurand I; Koppel IA; Leito I Acidity of Strong Acids in Water and Dimethyl Sulfoxide. J. Phys. Chem. A 2016, 120 (20), 3663–3669. [DOI] [PubMed] [Google Scholar]
- (50).Zimmerman P Reliable Transition State Searches Integrated with the Growing String Method. J. Chem. Theory Comput 2013, 9 (7), 3043–3050. [DOI] [PubMed] [Google Scholar]
- (51).Zimmerman PM Growing string method with interpolation and optimization in internal coordinates: Method and examples. J. Chem. Phys 2013, 138 (18), 184102. [DOI] [PubMed] [Google Scholar]
- (52).Zimmerman PM Single-ended transition state finding with the growing string method. J. Comput. Chem 2015, 36 (9), 601–611. [DOI] [PubMed] [Google Scholar]
- (53).March J Advanced organic chemistry. Reactions, mechanisms and structure, 3rd ed.; Wiley: New York, 1985. [Google Scholar]
- (54).Raamat E; Kaupmees K; Ovsjannikov G; Trummal A; Kütt A; Saame J; Koppel I; Kaljurand I; Lipping L; Rodima T; Pihl V; Koppel IA; Leito I Acidities of strong neutral Brønsted acids in different media. J. Phys. Org. Chem 2013, 26 (2), 162–170. [Google Scholar]
- (55).Tremel P; Iacobucci C; Massi L; Olivero S; Gal JF; Duñach E Catalytic intramolecular carbonyl-ene reaction with ketones: evidence for a retro-ene process. New J. Chem 2015, 39 (9), 7453–7458. [Google Scholar]
- (56).Acid dissociation constant. https://www.chemeurope.com/en/encyclopedia/Acid_dissociation_constant.html (accessed Feb 5, 2020).
- (57).Ionization Constants of Heteroatom Organic Acids. https://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/acidity2.htm (accessed Feb 5, 2020).
- (58).Reid JP; Goodman JM Goldilocks Catalysts: Computational Insights into the Role of the 3,3′ Substituents on the Selectivity of BINOL-Derived Phosphoric Acid Catalysts. J. Am. Chem. Soc 2016, 138 (25), 7910–7917. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.