

Abstract
Exposures to genotoxic carcinogens and reactive species result in strand breaks and a spectrum of covalent modifications to DNA that can induce mutations and contribute to the initiation and progression of cancer. Measurements of DNA damage within tissue or tumor samples can serve as a biomarker for exposures or assess changes in DNA repair capacity relevant in cancer development and treatment. Numerous methods to characterize DNA damage exist. However, these methods are primarily applicable to isolated DNA or cultured cells, often require a substantial amount of material, and may be limited to the detection and quantification of only a handful of DNA adducts. Here, we used the Repair Assisted Damage Detection (RADD) assay to detect and excise DNA adducts using a cocktail of DNA repair enzymes, then the damage site within the genome are tagged for detection using a modified nucleotide. We previously demonstrated the RADD assay can detect lesions within isolated DNA and fixed cells, and now RADD can be used to detect DNA adducts and DNA strand breaks in formalin-fixed paraffin-embedded (FFPE) tissue samples. We verified the ability of the RADD assay to detect DNA damage in tissue by exogenously inducing DNA damage with X-rays and restriction enzymes. We also showed that RADD can be multiplexed with antibodies to detect cell cycle markers or other proteins of interest. Finally, we showed that RADD can detect DNA damage within clinically relevant ovarian tumor samples. RADD is a flexible and easy-to-use assay that allows relative damage levels to be determined within FFPE samples and allows the heterogeneity of DNA adducts and strand breaks within clinically relevant samples to be measured.
Graphical Abstract
1. Introduction
Measurements of DNA adducts and strand breaks have been used extensively for toxicological characterization of man-made chemicals and environmental agents. DNA damage measurements are also used to study cancer etiology. However, clear epidemiological links between DNA damage and cancer have been hard to validate due to methodological challenges in adduct and strand break measurements.
Current DNA adduct detection schemes are limited due to the complexity and diversity of adducts formed. Liquid chromatography and mass spectrometry (LC-MS) have been used extensively to identify and quantify specific DNA adducts. LC-MS methods have allowed precise quantitation of adduct levels and have significantly advanced our understanding of the structure and lifetime of DNA adducts. Yet, these techniques require expert users, expensive equipment, often employ isotopic labeling for precise quantitation, and require microgram quantities of isolated DNA [1–3]. Additionally, these methodologies do not detect strand breaks or allow multiple types of DNA damage to be assessed.
More general detection of DNA strand breaks and adducts is possible with antibody-based strategies, comet assays, and enzymatic detection by terminal deoxynucleotidyl transferase (TdT). Antibody strategies can be applied to isolated DNA, in cells, or in fixed tissues. While antibodies exist for strand break signals (γH2AX or 53BP-1) and some DNA lesions (thymine dimer, cyclobutane pyrimidine dimers, etc.), these techniques are limited by the small number of highly specific antibodies that have been validated and may be difficult to multiplex due to incompatibilities in fixation or staining procedures. Comet assay or Single Cell Gel Electrophoresis allows more specific strand break detection in cells, eliminating the requirements for specific antibodies and with modifications can detect alkali labile sites, oxidative base damage, and DNA cross-linking [4,5]. While CometChip is improving some of the standardization and reproducibility issues sometimes encountered, comet is not compatible with fixed samples or stored isolated DNA [6–8]. DNA strand breaks can also be detected with terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and in situ DNA end labeling (ISEL) [9–11]. However, just like the other methods, there are drawbacks to using TUNEL or ISEL because they are highly specific for 3’-OH ends, do not detect specific lesions, and are more commonly used for the detection of apoptosis rather than damage detection.
These well-accepted DNA strand break and adduct measurement techniques have clear limitations. One important limitation is that they are not often compatible with archival tissue samples, which can prevent organ-specific analysis and hamper the association of DNA adducts or strand breaks to disease. With the important role DNA strand breaks and DNA adducts play in mutagenesis and carcinogenesis, new methods are needed to bridge the gap between detecting single DNA adducts with extreme precision and detecting large numbers of adducts and strand breaks in a single method. New methods also need to be applicable across biological samples and be accessible without the requirement for highly specialized equipment or expertise.
Given the limitations of current DNA damage detection methods, we have developed and validated a new methodology for the broad-spectrum detection of DNA damage within biological samples, termed Repair Assisted Damage Detection (RADD) [12–14]. The RADD assay combines the natural efficiency and specificity of DNA repair enzymes to detect, excise, and tag DNA adducts and strand breaks. For RADD, damaged DNA can be isolated from cells or tissues using standard genomic DNA isolation techniques [12,14] or kept in situ in cells [13,15]. Once DNA is purified or cells are fixed and permeabilized, a cocktail of DNA repair enzymes are used to detect and excise DNA adducts. We have optimized the repair cocktail to detect a broad-spectrum of DNA adducts induced by alkylation, oxidation, UV, and deamination events (Table 1) [13].
Table 1.
Enzymes used for damage detection
| Enzyme | Substrate Specificity |
|---|---|
| Uracil DNA glycosylase (UDG) | Uracil |
| Formamidopyrimidine [Fapy]-DNA glycosylase (FPG) | Oxidative base lesions, aflatoxin adducts |
| T4 Pyrimidine dimer glycosylase (T4PDG) | Cyclobutane pyrimidine dimers, 6–4 photoproducts, abasic (AP) sites |
| Endonuclease IV (EndoIV) | AP sites and diesterease activity modifies 3’ phosphates |
| 3-alkyladenine DNA glycosylase (AAG) | Alkylated base lesions, hypoxanthine |
| Klenow DNA polymerase large fragment (exo-) | Incorporates modified dUTPs at gaps and strand breaks |
The RADD assay is accomplished in two steps. In the first step, the optimized repair cocktail of AAG, FPG, UDG, T4PDG, and EndoIV is added to the cells and allowed to react, removing DNA adducts and leaving gaps in the DNA sequence where these damage sites were located (Figure 1). Then gap-filling with a deoxyuridine triphosphate (dUTP) analog is completed with Klenow DNA polymerase I (3’->5’ exo- fragment) for in situ labeling of DNA strand breaks and DNA adducts. The nature of the analog inserted is dictated by the downstream analysis desired. We have incorporated ATTO-550-dUTP, digoxigenin-labeled dUTP, and DBCO-dUTP (for Click chemistry) into isolated DNA and cells [12–15]. Detection of the DNA damage occurs by combing and imaging the isolated repaired DNA or by imaging the fluorescent content of repaired cells with confocal microscopy or flow cytometry.
Figure 1. RADD detection of DNA damage within FFPE samples.
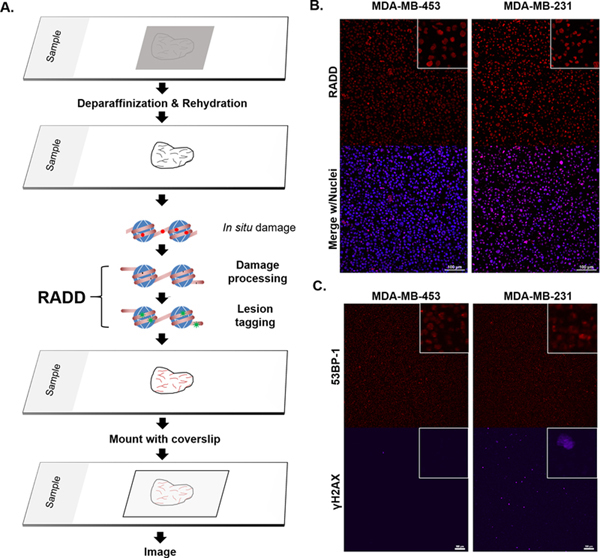
A. RADD workflow. FFPE tissue samples are deparaffinized and rehydrated. The RADD assay is performed with a cocktail of DNA repair enzymes that detects and removes damaged DNA leaving gaps where damaged bases were encountered. Lesion filling occurs with a DNA polymerase and a tagged dUTP molecule allows for fluorescent detection of the damage site. B. RADD detection of DNA lesions within FFPE immortalized cell lines. C. Strand break markers for γH2AX and 53BP-1 confirm presence of DNA damage within the FFPE samples. Scale bars 100 μm.
We have utilized this system to monitor the induction of oxidized adducts and ultraviolet (UV) induced DNA adducts within cells and isolated DNA [13,14]. We have also demonstrated the ability of the method to monitor the dynamics of DNA repair by inducing DNA damage and measure DNA adduct levels over time [13]. Altogether, these assays demonstrate the ability of the RADD assay to detect a broad-spectrum of DNA lesions in a variety of sample formats. Here, we further extend the applications of the RADD assay by detecting DNA damage within formalin-fixed paraffin-embedded (FFPE) tissue samples. With this application, the RADD assay is the only DNA damage assessment method capable of detecting a broad-spectrum of DNA lesions across biological sample formats.
2. Material and methods
2.1. Formalin-fixed paraffin-embedded (FFPE) samples
Immortalized breast cancer cell lines, MDA-MB-231 and MDA-MB-453, embedded in paraffin were purchased from AMSBio. Tissues used for analysis are described in detail in Table 2. Archival xenograft tumors of the ovarian cancer cell line TOV112D cells from two separate mice were provided by Drs. Scalici and Madeira da Silva (acquired under IACUC protocol #742025). Normal lung from a Balb/c mouse was acquired under IACUC protocol #1400753. Archival B16 mouse tumors harvested from C57BL/6 mice were acquired under IACUC protocol #933458 and were a kind gift of Dr. Gary Piazza, University of South Alabama Mitchell Cancer Institute. De-identified ovarian tumor samples from patients who had undergone treatment with at the Mitchell Cancer Institute were acquired from the University of South Alabama Department of Pathology under IRB protocol #1223907. Ovarian patient samples are divided into pre- and post- treatment group. Pre-treatment tumors were acquired during a de-bulking surgery before any drug treatment regime was started. The post-treatment tumor samples were collected after three to six rounds of combination therapy with carboplatin and paclitaxel when the tumor recurrence was detected. Post-treatment tumor samples were collected during a second de-bulking surgery.
Table 2.
Tissues used for analysis
| Tissue | Pathological Diagnosis | Tumor/Normal | Age/Sex | Specimen Label |
|---|---|---|---|---|
| Lung | Squamous Cell Carcinoma, Poorly Differentiated | Tumor | 72/M | T8235152-PT |
| Lung | Normal Adjacent to Tumor | Normal | 72/M | T8235152-PN |
| Ovary | Serous Adenocarcinoma | Tumor | 59/F | 27856 |
| Ovary | Normal Adjacent to Tumor | Normal | 59/F | 27853 |
| Breast | Ductal Infiltrative Carcinoma | Tumor | 52/F | 30355 |
| Breast | Normal Adjacent to Tumor | Normal | 52/F | 30353 |
| Breast | Ductal Infiltrative Carcinoma | Tumor | 53/F | 1340 |
| Breast | Normal Adjacent to Tumor | Normal | 53/F | 1340–1 |
| Breast | Ductal Infiltrative Carcinoma | Tumor | 70/F | 2180 |
| Breast | Normal Adjacent to Tumor | Normal | 70/F | 2180–1 |
2.2. Tissue RADD
FFPE samples are sectioned in 5 μm thick slices and mounted on poly-lysine functionalized glass coverslides. Slides are placed on a heat block set for 65°C and incubated for 8 min to melt the paraffin. Slides are then placed directly in 100% xylene and incubated twice for 10 min each. Slides are rehydrated to water through sequential incubations in ethanol and water mixtures. Specifically, slides are incubated for 5 min each in sequential order of 100% ethanol-0% water; 70% ethanol-30% water; 50% ethanol-50% water; 30% ethanol-70% water; 0% ethanol-100% water. Rehydrated slides are then placed in glass coplin jars with 200 mL of 10 mM sodium citrate in water and microwaved twice for 2.5 min at 120 watts until solution reaches 47°C for antigen retrieval [16,17]. Slides are allowed to cool in water for 2 min. Slides are briefly dried, and tissue samples are outlined with a PAP pen. The lesion removal cocktail (Table 3) is added to each tissue sample and incubated for 1 h at 37°C. For the full RADD lesion removal cocktail, all enzymes in Table 3 are included. For FPG+EndoIV only, T4 PDG, UDG, and AAG are removed and replaced with water. For EndoIV, all enzymes except EndoIV are removed and replaced with water. For Klenow only, all enzymes are removed and replaced with water. The gap filling solution (Table 3) added directly on top of the lesion removal solution and incubated for 1 h at 37°C.
Table 3. RADD sequential reaction conditions.
RADD is performed in two sequential reactions without aspirating reagents between reactions. The lesion processing mix (Left) is placed on prepared tissues and placed in a humidified incubator. The gap filling mix (Right) is added directly to the lesion processing mix and incubated for an additional hour. The reagents are then aspirated and the cells are washed and incubated with anti-digoxigenin antibody.
| Lesion processing mix |
100 μL total reaction volume |
Gap filling mix | 100 μL total reaction volume |
|---|---|---|---|
| UDG (NEB M0280s) | 2.5 U | Klenow exo- (Thermo Fisher EP0422) | 5 U |
| FPG (NEB M0240L) | 4 U | Digoxigenin dUTP (Sigma Aldrich 11573152910) | 1 μM |
| T4PDG (NEB M0308S) | 5 U | Therm Pol buffer (NEB B9004S) | 10 μL |
| EndoIV (NEB M0304L) | 5 U | ||
| hAAG (NEBM0313S) | 5 U | ||
| NAD+ (100x, NEB B9007S) | 500 μM | ||
| BSA (Sigma Aldrich) | 200 μg/mL | ||
| Therm Pol buffer (NEB B9004S) | 10 μL |
Slides are then washed three times in phosphate-buffered saline (PBS) for 5 min each and blocked in 2% BSA in PBS for 30 min at room temperature (RT, ~24°C). Anti-Digoxigenin (Dig) antibody (abcam; #ab420 clone 21H8) is incubated at a dilution of 1:250 in 2% BSA in PBS at 4°C overnight. For IgG control, one extra slide processed with the full RADD enzyme cocktail is incubated with mouse IgG isotype control antibody (Cell Signaling 5415, clone G3A1) at a dilution of 1:625 at 4°C overnight. This dilution factor matched the μg of anti-Dig antibody used per 100 μL. The next day slides are washed three times in PBS for 5 min each, and Alexa Fluor 546 goat anti-mouse secondary (Invitrogen) is incubated at a dilution of 1:400 in 2% BSA in PBS for 1 h at RT. Hoescht 33342 is added at a final dilution of 1:1000 for 15 min at RT to stain the nuclei. Slides are washed three times in PBS for 5 min each, briefly dried, and mounted with coverslips using ProLong Gold Antifade reagent. Slides are allowed to dry overnight in the dark at RT and visualized using a Nikon A1R confocal microscope or stored at 4°C until analysis. Where applicable, Ki67 (abcam 16667) and γH2AX (Millipore JWB301) antibodies are co-incubated with the anti-Dig antibody overnight at 4°C, before being counterstained with Alexa Fluor 647 secondary (1:400, Invitrogen).
2.3. Exogenous DNA damage induction
MDA-MB-231 cells were acquired from American Type Culture Collection (ATCC). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) + Glutamax (Life Technologies) supplemented with sodium pyruvate and 10% FBS (Atlantic Biologicals). Cells were plated on glass bottomed coverslides coated with EmbryoMax at 40,000 cells/plate and incubated for 48 h at 37°C and 10% CO2 in a humidified incubator. FFPE normal mouse lung tissues were sectioned into 5 μm thick sections. Ovarian carcinoma cells were also sectioned into 5 μm thick sections. Cells and normal mouse lung tissues were irradiated by XRAD320 (dose rate 117 cGy/min, Precision X-ray) for doses indicated.
MDA-MB-231 cells were immediately fixed, washed three times with PBS, then permeabilized with Biotium permeabilization buffer. Once permeabilized, the cells were processed with the complete RADD damage processing and lesion tagging mixtures (Table 3). Irradiated normal mouse lung tissues were then deparaffinized, rehydrated, antigen retrieved, and RADD processed as described above (Table 3).
Restriction enzyme digest of mouse lung tissue or ovarian tissue with SalI (ThermoFisher, FD0644), Vsp1 (AseI, ThermoFisher FD0914), EcoRI (ThermoFisher FD0274), VspI +SalI, or VspI + EcoRI was conducted after antigen retrieval and before the damage processing cocktail. Restriction recognition sites are present in Table 4. Enzymes were diluted in 1X FastDigest buffer, and 5 U of indicated enzymes were added to tissues. After incubating for 1 h at 37°C, cells were washed three times in PBS. Then the RADD assay (Table 3) was applied as described above.
Table 4.
Restriction enzyme recognition sites.
| Restriction Enzyme | Recognition Site Cut site indicated with ‘ |
|---|---|
| EcoRI | 5’-G’AATTC-3’ 3’-CTTAA’G-5’ |
| SalI | 5’-G’TCGAC-3’ 3’-CAGCT’G-5’ |
| VspI | 5’-AT’TAAT-3’ 3’-TAAT’TA-5’ |
2.4. Immunohistochemistry (IHC)
Tissue slides were deparaffinized and rehydrated to water the same as for tissue RADD. For antigen retrieval, slides were placed in a decloaking chamber in a 1X solution of Nuclear Decloaker (Biocare Medical) until 15 psi was reached, roughly 20 min. Slides were gradually cooled with water using five buffer changes. Endogenous peroxidase activity was blocked using Peroxidazed 1 (Biocare Medical) for 40 min at RT. Tissues were then blocked in 2% BSA in PBS for 30 min at RT. Primary antibody for 53BP1 (Novus Biologicals; NB100–304) was incubated at a dilution of 1:400 in 2% BSA in PBS at 4°C overnight. Slides were then washed three times in PBS, and 53BP1 was counterstained with Alexa Fluor 546 secondary antibody. Next, slides were incubated with anti-γH2AX antibody (Millipore; clone JBW301 05–636-AF647) conjugated to Alexa Fluor 647 for 1 h at RT. Hoescht 33342 was used to stain nuclei at a dilution of 1:1000 for 15 min at RT. Slides were washed three times in PBS for 5 min each, briefly dried, and mounted with coverslips using ProLong Gold Antifade reagent. Slides are allowed to dry overnight in the dark at RT and visualized using a Nikon A1R confocal microscope or stored at 4°C until analysis.
2.5. Image acquisition
All images were acquired using a Nikon A1r scanning confocal microscope with a Plan-Apochromat 10x/0.5 or 20x/0.75 objective. Image acquisition settings were obtained by imaging the full RADD samples for tumor tissues and identifying gain settings that limited the number of saturated pixels. These imaging conditions were then used for all tissue imaging allowing for direct comparisons and analysis between tumor and normal tissues. For large tissue sections, images were first mapped using the Acquire Large Image acquisition tool in the Nikon Elements software (NIS-Elements AR 4.51.00), acquired using the 10x objective, and stitched post-acquisition. The tool maps the X-Y-Z positions of individual images within the tissue slice which are then acquired individually at 10x, 1024×1024 resolution for further analysis. For some images, the 20x objective was used to acquire better resolution images of heterogeneous regions or boundaries between stained and unstained tissues.
2.6. Image analysis
The individual images which make up the large stitched images, between 30 and 150 images depending on tissue section size, were used for analysis. The Nikon Elements software was used to create a binary mask of the RADD signal intensity, and the sum fluorescence intensity was exported. Gating for the binary mask was defined by the lowest intensity image, and these settings were used between all images for analysis. The sum intensity was used rather than the mean intensity to better reflect the heterogeneity of the staining. The sum intensity for each individual image making up the whole tissue slice are then reported in a scatter plot to show the heterogeneity of the entire tissue slice. The mean ± standard error of the mean (mean ± SEM) of the individual summed images are then compared between samples. Data were plotted using GraphPad Prism software and analyzed for significance using either a Student’s t-test (normal vs tumor) or one-way analysis of variance (ANOVA) with a Dunnett’s post-hoc analysis for mean comparisons (cocktail breakdown or treatments). For the patient samples, a two-tailed t-test is used to compare pre- and post-treatment samples for the same patient.
3. Results
3.1. Adaption of RADD for tissues
To develop RADD for cells, we modified existing protocols from TUNEL and immunohistochemistry (IHC) to allow a cocktail of repair enzymes (Table 1) to access the nucleus, detect and excise a broad-spectrum of DNA adducts from abasic sites to oxidative base lesions [13]. Once DNA adducts are removed, a DNA polymerase inserts a modified nucleotide into the gapped DNA for detection (Table 3) [13]. In experiments using monolayer cell culture, we demonstrated that the RADD assay could detect DNA adducts induced by the oxidizing agent potassium bromate and induced by ultraviolet (UV) radiation [13]. Here, we further expanded the detection capability of RADD by extending the assay into FFPE samples.
The workflow for FFPE samples is shown in Figure 1A. FFPE blocks are sectioned into 5 μm slices and mounted on poly-lysine functionalized coverslides. The tissue samples are then deparaffinized by heating at 65°C and rehydrated from 100% xylene to water. Heated sodium citrate is used for antigen retrieval. The RADD assay is then performed in two sequential steps. First, damage detection and processing is conducted with an enzyme cocktail of the bacterial and human glycosylases shown in Table 1. This enzyme cocktail detects alkylation, oxidation, UV, and deamination events. We have chosen FPG to detect oxidative base lesions, and T4PDG to detect UV-induced lesions. The specificities for each of these glycosylases does not cover all of the possible oxidative or UV-induced lesions that can be created. For example, oxidized pyrimidines would be more effectively detected by endonuclease III or endonuclease VIII, though FPG does have some activity for these lesions [18]. We have tailored our cocktail to be more focused on smaller base lesions or crosslinks, like 8-oxoguanine (8-oxodG) or cyclobutane pyrimidine dimers (CPD), which are known to be mutagenic. However, the RADD lesion processing cocktail could be created to detect different lesion classes with additional enzymes.
After lesions are removed, gaps in the DNA sequence where these adducts sites were located are left behind (Figure 1A) [13]. After the damage processing, gap-filling of damaged sites is accomplished by Klenow DNA polymerase I (exo-) and digoxigenin-labeled dUTP (Dig-dUTP). Klenow covalently attaches the modified dUTP to strand breaks and gaps within the genome, allowing downstream detection. Tissues are then washed to remove enzymes, blocked, and the incorporated Dig-dUTP detected by an anti-digoxigenin antibody. After antibody incubation, cell nuclei are labeled with Hoechst, and tissues are mounted (Figure 1A).
With the full cocktail described in Table 1 (also Table 3), we validated DNA damage detection using the procedure outline above with immortalized cell lines embedded in paraffin. MDA-MB-231 and MDA-MB-453 cells show specific nuclear staining for DNA adducts using RADD (Figure 1B). To confirm the presence of DNA damage within the immortalized cell lines, we also conducted IHC for the strand break signals phosphorylated H2AX (γH2AX) and 53BP-1 (Figure 1C). Damage levels for MDA-MB-231 observed with the paraffin are consistent with damage levels observed in monolayer cell culture (Supplemental Figure 1).
3.2. RADD detection of DNA damage within tumor xenografts
The RADD assay was then used to detect DNA damage within two orthotopic tumor xenografts of the ovarian cell line TOV112D (Figure 2). Archival FFPE tumors were sectioned, and the full RADD assay (Table 3) was performed. In parallel with the anti-digoxigenin antibody, an antibody specific for the proliferation marker Ki67 was also added to discriminate tumor tissue from normal mouse tissue. Again, the RADD signal is observed predominantly within the nucleus of cells within the tumor (Figure 2A, top). The RADD signal also corresponds to the Ki67 staining, discriminating the proliferating tumors from surrounding normal tissue (Figure 2A, middle). When a non-specific mouse Ig antibody is used in place of the specific anti-digoxigenin antibody, we observed little non-specific signal (Supplemental Figure 2). Unlike some DNA adduct measurement methods, RADD can be multiplexed with other antibodies, allowing simultaneous assessment of DNA damage with DNA damage response and repair proteins, cell cycle proteins or even oncogenes. Supplemental Figure 3 shows RADD multiplexed with the strand break marker γH2AX.
Figure 2. RADD signal in FFPE xenograft tumors correlates with higher proliferation.

TOV112D xenografts from two separate mice are co-stained for DNA damage using (A) RADD (red) and cell proliferation wth (B) Ki67 (purple). C. Merged image with nuclear staining. D. H&E stain of adjacent tumor slice.
In addition to detecting a broad-spectrum of DNA lesions with the full RADD assay damage processing cocktail (Table 3), a subset of enzymes, i.e., EndoIV, FPG+EndoIV, can be used to specifically detect classes of lesions, like abasic sites or oxidative lesions. Supplemental Figure 4 shows DNA adduct measurements by different glycosylase cocktails using RADD.
3.3. Detection of DNA damage within normal and tumor tissues
DNA damage detection within clinically relevant samples was then demonstrated by applying the RADD assay to human tumor samples. Matched pairs of lung squamous cell carcinoma tumor and adjacent normal lung tissue and breast ductal infiltrative carcinoma tumor and adjacent normal breast tissue in FFPE were purchased and the full RADD assay conducted (Table 3). Imaging conditions were identical across all samples, and the summed intensity for each section of the tumor recorded. As shown in Figure 3, RADD staining is higher in tumor tissues than in normal tissues for both lung (Figure 3A) and breast (Figure 3B). There are also significant differences in staining intensity between lung and breast tissues and tumors (Figure 3C). The stitched images and scatter plots also show significant heterogeneity in DNA adduct levels within each tissue and tumor slice. Additional RADD tumor analysis of ovarian adenocarcinoma and normal adjacent tissue is shown in Supplemental Figure 5.
Figure 3. RADD detection of DNA adducts within human tumors and adjacent normal tissues.
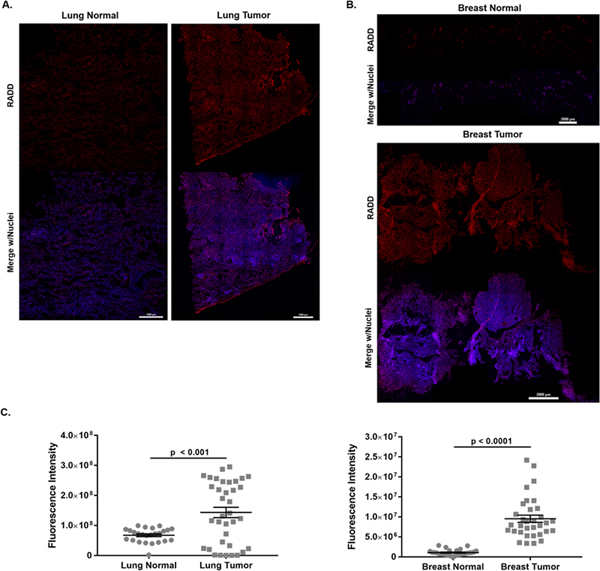
Stitched image of RADD performed on a single lung squamous cell carcinoma and adjacent normal tissue (A) and on a single breast ductal infiltrative carcinoma and adjacent normal tissue (B). Brightness and contrast are optimized for visualization in this image. C. Tissue slices were imaged using constant laser power and gain, and the fluorescent intensity of individual tissue images measured. Graphed is the summed fluorescent intensity from each individual tissue section imaged at 10x.
To further validate that the RADD assay is detecting DNA damage within FFPE sections, we induced DNA damage within fixed tissue sections from normal mouse lung using X-ray irradiation or restriction enzyme digest (Figure 4). X-ray irradiation will induce a mixture of single- and double-strand breaks along with base lesions, while the restriction enzymes will induce double-strand breaks with single-strand overhangs (detailed in Table 4). After inducing DNA damage, the complete RADD assay (Table 3) was performed as described in the Material and Methods. RADD detects increasing DNA damage induced by X-ray irradiation (Figure 4A) and by restriction digest (Figure 4B). Induction of damage by these agents may be limited by the chromatin compaction induced by fixation which prevents precise quantification of induced damage based on applied Gy or number of restriction enzyme sites within the genome [19,20]. However, both agents induce DNA damage within the tissues, and RADD detects this damage. Supplemental Figure 6 shows additional RADD detection of exogenously induced damage.
Figure 4. RADD measures DNA damage induced after fixation in normal mouse lung.

A. Fluorescent intensity of RADD staining after exposure to increasing amounts of X-ray irradiation. B. Fluorescent intensity of RADD staining after exposure to 5 units of the restriction enzymes VspI, EcoRI, VspI + EcoRI, or VspI + SalI for 1 h prior to performing the RADD assay.
3.4. RADD assessment of DNA damage in pre- and post-treatment clinical samples
In the final demonstration of the application of RADD for the detection of DNA damage within human tissue samples, we performed the RADD assay on matched pairs of de-identified ovarian tumors pre- and post-treatment. Initial tumors were collected during a de-bulking surgery before the initiation of chemotherapy. Patients then underwent three to six rounds of combination treatment with carboplatin and paclitaxel. When tumors recurred, post-treatment tumors were collected at a second de-bulking surgery. Figure 5 shows average RADD fluorescent intensity signal for pre- and post-treatment patient samples. Figure 5B shows a representative image from the tumor section for patients 1 and 4 pre- and post- treatment to show the heterogeneity in the sample staining within even one tumor section and to show the increase in staining observed for patient 4 in the post-treatment recurrent tumor (Figure 5A&B).
Figure 5. RADD measures DNA levels in ovarian patient tumor samples.

A. Basal levels of DNA damage were detected in tumor samples collected in a de-bulking surgery before the start of chemotherapy (pre) and at a second de-bulking surgery when the tumor recurred after treatment with carboplatin and paclitaxel (post). A two-tailed t-test is used to determine significance between pre- and post-treatment tumors for a single patient. B. Representative images of a tumor section imaged from patient 1 and patient 4 pre- and post-treatment are shown. C. Fluorescent intensity of DNA damage staining in the pre-treatment tumor is divided into low, moderate, and high staining categories, so the trend in DNA damage staining intensity pre- and post-treatment can be observed across the eight patient samples.
Different trends in initial levels of DNA damage and post-treatment levels of DNA damage are observed among the samples (Figure 5A). Patients 1, 3, 5, 6, and 8 show a significant decrease in DNA damage after treatment with a p value of 0.001 for patient 1 and p < 0.0001 for patients 3, 5, 6, and 8 compared to the matched pre-treatment tumor. Patient 4 shows a significant increase in DNA damage after treatment (p < 0.01), and patient 7 shows a non-significant increase in DNA damage after treatment. Patients 5 and 8 also showed the highest level of overall fluorescent staining, indicating high levels of DNA damage. To better examine the variation in the pre-treatment staining levels, we divided the samples in three categories based on the average fluorescent intensity observed (Figure 5C). We based the categories on the staining intensity observed for the exogenously induced DNA damage in Figure 4. A low (1 × 106 – 1 × 107), moderate (2 × 107 – 1 × 108), and high (>2 × 108) levels of staining and therefore DNA damage is observed in the pre-treatment tumor sample across the patients sampled. Larger sample populations are needed to understand the trends in DNA damage pre- and post-treatment and the variation in staining intensity of the pre-treatment tumors. However, RADD clearly detects difference in DNA damage levels among the patient population.
4. DISCUSSION
DNA damaging events are induced by normal metabolic processes and exposures to environmental toxins and toxicants. There is a real need to measure DNA adducts within target tissues like liver, kidney, breast, etc. to provide more specific epidemiological links between environmental exposures and their role in disease development and progression [21]. Similarly, being able to measure DNA adducts within tumors pre- and post-treatment could provide information about DNA repair defects, report on the efficacy of DNA damaging chemotherapies, and potentially help refine the selection of chemotherapies for individual patients [22–24].
The limitation is that there are currently no DNA damage detection methods easy to employ clinically compatible with small amounts of tissue and detect a broad range of DNA adducts. There have been recent developments in using FFPE tissues for LC-MS quantification of specific DNA adducts, but these methods have not yet been employed outside of the developing laboratory [2]. Currently, DNA adducts can be assessed within frozen and FFPE tissue samples using IHC for specific DNA lesions like CPD or 8-oxodG. Immunostaining for adducts works well in cultured cells where permeabilization and denaturing of the DNA are more easily accomplished. In tissues, permeabilization can be more difficult and variable, requiring the addition of proteinase K and stronger detergents to allow DNA lesions to be accessed [9–11,25,26]. These requirements, coupled with the limited number of highly specific antibodies for DNA adducts, has limited quantification of DNA adducts using these methods.
Given the limitations of current methodologies, the RADD assay was developed as a flexible and broad-spectrum method for detecting DNA damage across biological samples. A cocktail of DNA repair enzymes is employed to detect and excise DNA lesions from the DNA and DNA polymerase is used to tag the excised damaged site within the genome for analysis. Here, we have employed a cocktail of human and bacterial DNA glycosylases to detect alkylation, oxidation, UV, and deamination events. The selected cocktail here is focused on smaller base lesions that will be repaired by base excision repair (BER) and nucleotide excision repair (NER). It is not exhaustive in the types of BER and NER lesions it can detect, but it does detect a broad spectrum of adducts repaired by these pathways. We have previously demonstrated the detection of oxidative and UV-induced lesions [13]. Here, we utilize X-rays to induce base lesions and strand breaks to demonstrate the functionality of the RADD cocktail (Figure 4). Importantly, RADD also detects DNA strand breaks, which we demonstrated through the application of restriction enzymes to the FFPE samples (Figure 4). The restriction enzymes employed will generate double strand breaks with single strand sequence overhangs that could be repaired by non-homologous end joining or even homologous recombination, if encountered in a live cell. This is the first time we have demonstrated DNA strand breaks are also specifically detected by the assay (Figure 4 and Supplemental Figure 6).
With lesions detected from most major DNA repair pathways, the RADD assay exceeds the detection capabilities of current DNA damage assessment methods. However, the RADD assay is also flexible in that different DNA repair enzymes could be incorporated into the cocktail to allow for more DNA adduct classes to be detected, i.e., endonuclease III, endonuclease VIII, or SMUG1. Additionally, all human enzymes could be used to limit the substrate specificity and tailor detection to vary narrow lesion classes. Single enzymes can also be used to separate the adduct classes (Supplemental Figure 4) that are currently averaged by using the complete enzymatic cocktails. Altogether, RADD allows the user the freedom to tailor lesion detection, which is again a distinct advantage over current DNA damage detection methods.
The flexibility in the RADD assay also extends to the sample formats to which it can be applied. Here, we have demonstrated the application of this assay in a variety of fixed mouse and human tissues. This extends RADD beyond isolated DNA [12,14] and cells [13,15] to clinically relevant sample formats. The RADD assay also allows for the examination of DNA adducts and strand breaks at single-cell resolution in these tissues, outpacing current detection schemes. More importantly, RADD is compatible with small amounts of biological material acquired from tissue biopsies. With the RADD assay, DNA damage measurements can be made in target tissues of interest rather than in surrogate material such as peripheral blood mononuclear cells (PBMCs).
We also demonstrated that the RADD analysis could be multiplexed with antibodies for cell proliferation and DNA damage response within tissue sections (Figure 2 and Supplemental Figure 3). Finally, we demonstrated the applicability of the RADD assay to clinically relevant samples by examining DNA damage levels in ovarian tumors collected pre- and post-treatment.
DNA repair defects in homologous recombination, NER, and BER have been identified in numerous cancers, including ovarian cancer [15,27–30]. Exploiting DNA repair defects for therapeutic intervention is a growing area of research, but methods are needed to characterize DNA repair defects and understand their role in disease development and progression [24,27]. Gene expression changes are frequently measured in tumors, but linking gene expression changes in DNA repair proteins to changes in DNA repair capacity has been lacking. RADD assay application to patient tumor samples may provide the critical DNA damage measurements needed to understand the consequences of changes in the expression of DNA repair proteins in cancer.
The main limitations of this assay are the relative quantification between samples and the use of glycosylases that recognize multiple DNA adducts. The nature of analyzing DNA damage in situ means that chromatin or other cellular proteins could interfere with the efficiency of the detection, lesion removal, and subsequent gap filling [19,20]. Current comparable DNA damage detection methods within tissues, IHC or TUNEL, also only provide relative quantifications. There are emerging methods with LC-MS that could be more quantitative, but to date most cell-based methods lack precise quantitation [31–33]. While isolation of DNA can improve quantification, there is still a risk of introducing DNA lesions during isolation procedures. The experiments with X-ray irradiation and restriction enzymes demonstrate that it may be possible to refine the quantification of DNA damage with RADD in the future, or the RADD assay could be performed in parallel with isolated DNA from tissues to provide more precise quantification [12,14]. The relative quantifications performed here still demonstrate the value of DNA damage assessment within tissues using RADD, and the compatibility with small samples exceeds most DNA adduct detection methods. The limitation induced by the use of bacterial DNA glycosylases can be overcome by creating a DNA damage processing mixture with human enzymes that have more limited substrate specificity or even through protein engineering to create enzymes specific to lesion types. The format of the RADD assay is flexible enough to use any enzymes of interest for damage detection. Overall, the RADD assay offers a flexible, customizable method for examining DNA damage from isolated DNA to tissue samples.
Supplementary Material
Highlights.
Current DNA damage detection methods in tissues are limited
Methods for assessing strand breaks and adducts with FFPE tissues are needed
RADD detects both strand breaks and adducts in a broad-spectrum assay
Major lesion classes detected by nucleotide and base excision repair are measured
RADD can be customized to detect specific lesion classes within FFPE tissues
Acknowledgments
Imaging was performed in the Cellular and Biomolecular Imaging Core at the University of South Alabama Mitchell Cancer Institute with the assistance of Dr. Joel Andrews.
Funding
This work was supported by the National Institutes of Health [ES028015] and start-up funding from the University of South Alabama Mitchell Cancer Institute.
Footnotes
Declaration of competing interests
The authors declare no conflicts of interest.
References
- 1.Balbo S, Turesky RJ, Villalta PW: DNA adductomics. Chem Res Toxicol 2014, 27:356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo J, Yun BH, Upadhyaya P, Yao L, Krishnamachari S, Rosenquist TA, Grollman AP, Turesky RJ: Multiclass Carcinogenic DNA Adduct Quantification in Formalin-Fixed Paraffin-Embedded Tissues by Ultraperformance Liquid Chromatography-Tandem Mass Spectrometry. Anal Chem 2016, 88:4780–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dizdaroglu M, Jaruga P, Rodriguez H: Measurement of 8-hydroxy-2’-deoxyguanosine in DNA by high-performance liquid chromatography-mass spectrometry: comparison with measurement by gas chromatography-mass spectrometry. Nucleic acids research 2001, 29:E12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olive PL, Banáth JP: The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 2006, 1:23–29. [DOI] [PubMed] [Google Scholar]
- 5.Smith CC, O’Donovan MR, Martin EA: hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis 2006, 21:185–190. [DOI] [PubMed] [Google Scholar]
- 6.Braafladt S, Reipa V, Atha DH: The Comet Assay: Automated Imaging Methods for Improved Analysis and Reproducibility. Sci Rep 2016, 6:32162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins AR, El Yamani N, Lorenzo Y, Shaposhnikov S, Brunborg G, Azqueta A: Controlling variation in the comet assay. Front Genet 2014, 5:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Collins AR, Oscoz AA, Brunborg G, Gaivao I, Giovannelli L, Kruszewski M, Smith CC, Stetina R: The comet assay: topical issues. Mutagenesis 2008, 23:143–151. [DOI] [PubMed] [Google Scholar]
- 9.Didenko VV: In situ labeling of DNA breaks and apoptosis by T7 DNA polymerase. Methods Mol Biol 2011, 682:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hornsby PJ, Didenko VV: In situ ligation: a decade and a half of experience. Methods Mol Biol 2011, 682:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loo DT: TUNEL assay. An overview of techniques. Methods Mol Biol 2002, 203:21–30. [DOI] [PubMed] [Google Scholar]
- 12.Zirkin S, Fishman S, Sharim H, Michaeli Y, Don J, Ebenstein Y: Lighting up individual DNA damage sites by in vitro repair synthesis. J Am Chem Soc 2014, 136:7771–7776. [DOI] [PubMed] [Google Scholar]
- 13.Holton NW, Ebenstein Y, Gassman NR: Broad spectrum detection of DNA damage by Repair Assisted Damage Detection (RADD). DNA Repair (Amst) 2018, 66–67:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Torchinsky D, Michaeli Y, Gassman NR, Ebenstein Y: Simultaneous detection of multiple DNA damage types by multi-colour fluorescent labelling. Chem Commun (Camb) 2019, 55:11414–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KJ, Piett CG, Andrews JF, Mann E, Nagel ZD, Gassman NR: Defective base excision repair in the response to DNA damaging agents in triple negative breast cancer. PLoS One 2019, 14:e0223725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lucassen P, Chung W, Vermeulen J, Van Lookeren Campagne M, Van Dierendonck J, Swaab D: Microwave-enhanced in situ end-labeling of fragmented DNA: parametric studies in relation to postmortem delay and fixation of rat and human brain. J Histochem Cytochem 1995, 43:1163–1171. [DOI] [PubMed] [Google Scholar]
- 17.Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, Negoescu A: TUNEL Apoptotic Cell Detection in Tissue Sections: Critical Evaluation and Improvement. J Histochem Cytochem 1998, 46:327–334. [DOI] [PubMed] [Google Scholar]
- 18.Wallace SS, Harrison L, Jiang D, Blaisdell JO, Purmal AA, Hatahet Z: Processing and Consequences of Oxidative DNA Base Lesions. In Advances in DNA Damage and Repair: Oxygen Radical Effects, Cellular Protection, and Biological Consequences. Edited by Dizdaroglu M, Karakaya AE. Springer US; 1999:419–430. [Google Scholar]
- 19.Takata H, Hanafusa T, Mori T, Shimura M, Iida Y, Ishikawa K, Yoshikawa K, Yoshikawa Y, Maeshima K: Chromatin compaction protects genomic DNA from radiation damage. PLoS One 2013, 8:e75622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campalans A, Kortulewski T, Amouroux R, Menoni H, Vermeulen W, Radicella JP: Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res 2013, 41:3115–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rundle A: Carcinogen-DNA adducts as a biomarker for cancer risk. Mutat Res 2006, 600:23–36. [DOI] [PubMed] [Google Scholar]
- 22.da Cunha Colombo Bonadio RR, Fogace RN, Miranda VC, Diz M: Homologous recombination deficiency in ovarian cancer: a review of its epidemiology and management. Clinics (Sao Paulo) 2018, 73:e450s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ, et al. : Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first-in-class proof-of-concept phase 2 study. Lancet Oncol 2018, 19:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kristeleit RS, Miller RE, Kohn EC: Gynecologic Cancers: Emerging Novel Strategies for Targeting DNA Repair Deficiency. Am Soc Clin Oncol Educ Book 2016, 35:e259–268. [DOI] [PubMed] [Google Scholar]
- 25.Canene-Adams K: Preparation of formalin-fixed paraffin-embedded tissue for immunohistochemistry. Methods Enzymol 2013, 533:225–233. [DOI] [PubMed] [Google Scholar]
- 26.Liou GY, Storz P: Detecting reactive oxygen species by immunohistochemistry. Methods Mol Biol 2015, 1292:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li SX, Sjolund A, Harris L, Sweasy JB: DNA repair and personalized breast cancer therapy. Environ Mol Mutagen 2010, 51:897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gee ME, Faraahi Z, McCormick A, Edmondson RJ: DNA damage repair in ovarian cancer: unlocking the heterogeneity. J Ovarian Res 2018, 11:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broustas CG, Lieberman HB: DNA damage response genes and the development of cancer metastasis. Radiat Res 2014, 181:111–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurfurstova D, Bartkova J, Vrtel R, Mickova A, Burdova A, Majera D, Mistrik M, Kral M, Santer FR, Bouchal J, et al. : DNA damage signalling barrier, oxidative stress and treatment-relevant DNA repair factor alterations during progression of human prostate cancer. Mol Oncol 2016, 10:879–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown K: Methods for the detection of DNA adducts. Methods Mol Biol 2012, 817:207–230. [DOI] [PubMed] [Google Scholar]
- 32.Phillips DH, Farmer PB, Beland FA, Nath RG, Poirier MC, Reddy MV, Turteltaub KW: Methods of DNA adduct determination and their application to testing compounds for genotoxicity. Environ Mol Mutagen 2000, 35:222–233. [DOI] [PubMed] [Google Scholar]
- 33.Reddy MV: Methods for testing compounds for DNA adduct formation. Regul Toxicol Pharmacol 2000, 32:256–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.