

Abstract
Cholecystokinin (CCK) is an important neuro-intestinal peptide hormone produced by the enteroendocrine I-cells in the upper part of small intestine. Protein- and fat-enriched food plays an important role in triggering CCK secretion from the intestine. Carbohydrates stimulate only small amounts of CCK release. The CCK-1 receptor (CCK-1R) is largely localized in the gallbladder, sphincter of Oddi, pancreas, small intestine, gastric mucosa, and pyloric sphincter, where it is responsible for CCK to regulate multiple digestive processes including gallbladder contraction, pancreatic secretion, small intestinal transit, and gastric emptying. Accumulated evidence clearly demonstrates that CCK regulates gallbladder and small intestinal motility through CCK-1R signaling cascade and the effect of CCK-1R on small intestinal transit is a physiological response for regulating intestinal cholesterol absorption. Disruption of the Cck or the Cck-1r gene in mice significantly increases the formation of cholesterol gallstones by disrupting gallbladder emptying and biliary cholesterol metabolism, as well as promoting intestinal absorption of cholesterol. Abnormalities in gallbladder motility function in response to exogenously administered CCK are found primarily in patients with cholesterol gallstones. Patients with pigment gallstones display an intermediate degree of gallbladder motility defect without gallbladder inflammation and enlarged fasting gallbladder. Dysfunctional gallbladder contractility has been found under several conditions such as pregnancy, obesity, diabetes, celiac disease, and total parenteral nutrition although gallstones are not observed. The gallbladder-specific CCK-1R-selective agonist may lead to an efficacious novel way for preventing gallstone formation by promoting gallbladder emptying, particularly for pregnant women and subjects with dysfunctional gallbladder motility function such as celiac patients, as well as patients with total parenteral nutrition.
Keywords: Bile salt, biliary sludge, cholesterol crystallization, gallbladder motility, lithogenic bile, Cholecystokinin (CCK)
1. INTRODUCTION
Cholecystokinin (CCK) is an important neuro-intestinal peptide hormone that is produced by the enteroendocrine I-cells mainly in the upper part of small intestine. CCK plays multiple regulatory roles in many target organs and tissues [1]. Plasma CCK levels are significantly elevated after consumption of a meal containing a large amount of fat and protein [2, 3]. It is well established that the CCK-1 and the CCK-2 receptors (CCK-1R and CCK-2R) play a biological role predominantly in gallbladder smooth muscle and pancreatic acini, as well as at multiple levels, in the gastrointestinal tract, the enteric nervous system, and brain. A high level of plasma CCK has been found in some patients with chronic pancreatitis. It is highly likely that it is induced by reduced secretion of pancreatic enzymes and interrupted negative feedback regulation of CCK secretion. In contrast, a low level of plasma CCK has been found in patients with celiac disease, mostly due to reduced intestinal mucosal surface area and bulimia nervosa. Many clinical and animal studies have reported that impaired CCK release is a high risk for many digestive diseases such as gallstone formation and for metabolic disorders such as obesity [4–7]. Furthermore, a defective gallbladder motility plays a critical role in the pathogenesis of cholesterol gallstone disease [8], and abnormal gallbladder contractility has been found in patients with cholesterol gallstones [9]. Dysfunctional gallbladder motility is also observed under several conditions such as pregnancy, obesity, diabetes, total parenteral nutrition, and celiac disease although these subjects are free of gallstones [10–16], suggesting that it is a high risk factor for gallstone formation [17]. All these studies clearly demonstrate that CCK and its receptor CCK-1R play an important role in the pathophysiology of cholesterol gallstone disease. In this review article, we summarize the molecular biology and physiology of CCK and its receptors, and discuss recent progress in its physiological role in the digestive system, with emphasis on the pathogenesis of cholesterol gallstone formation at a molecular and cellular level.
2. MOLECULAR BIOLOGY OF CCK AND ITS RECEPTORS
In 1928, CCK was first reported to be a gallbladder contracting hormone in extracts of the small intestine [18]. CCK consists of different numbers of amino acids, which depends on post-translational reformation of the CCK gene product, i.e., preproCCK. Therefore, CCK is basically a family of hormones that are composed of varying numbers of amino acids, including CCK-58, CCK-33, CCK-22, and CCK-8. Fig. (1) shows the molecular structure of sulfated cholecystokinin octapeptide (CCK-8). Moreover, CCK-58 is a helix-turn-helix configuration [19]. The structure of CCK is very similar to that of gastrin, one of the gastrointestinal hormones. Although CCK and gastrin share the same five amino acids at their C-termini, most CCK peptides contain a sulfate-group connected to the tyrosine in the C-terminus. Such a structure plays an important role for CCK to stimulate CCK-1R. Although non-sulfated CCK peptides also exist, they consequently cannot stimulate CCK-1R. The human CCK gene is mapped to chromosome 3 (p22.1). The mouse and rat CCK genes are localized to chromosomes 9 (72.43 cM) and 8 (q32), respectively.
Fig. (1). Molecular structure of sulfated cholecystokinin octapeptide (CCK-8).
(A) The standard chemical formula (left panel), (B) the perspective formula (right top panel), (C) and the space-filling model (right bottom panel) are shown.
The transcription unit of the CCK gene is 7 kb interrupted by two introns. The CCK gene contains three exons, with the first one being small and noncoding. Of special note, there is the homology between the CCK gene and the gastrin gene in the coding region and in the 5’-untranslated region. Because there is only one CCK mRNA molecule, the CCK peptides must be fragments of the same mRNA product. The CCK mRNA has 750 bases, of which 345 are protein coding. The translational product, i.e., preproCCK, is composed of 115 amino acid residues. The bioactive parts of CCK are mainly originated from the subsequent 58 amino acid residues (i.e., CCK-58), and there is a very small species variation in this sequence. The affinity of CCK CCK for its receptors is determined by Tyr-77 that is O-sulfated. Of note, the cell-specific product with different numbers of amino acids is essential for the biological functions of CCK in diverse tissues of the body. For example, endocrine cells release a mixture of medium-sized CCKs and neurons principally synthesize CCK-8. The specific bioactive CCKs consist of varying numbers of amino acids, including CCK-83, CCK-58, CCK-39, CCK-33, CCK-22 and CCK-8 [19].
Wank and colleagues first cloned the CCK-1R gene from the rat pancreas [20]. Subsequently, Kopin and co-workers cloned a gastrin receptor, i.e., CCK-2R, from a canine parietal cell expression library [21]. The genomic structures of CCK-1R and CCK-2R in mice, rats, and humans have been reported and the physiological and clinical functions of CCK, CCK-1R, and CCK-1R have been studied extensively.
The CCK-1R gene is composed of five exons that are interrupted by four introns. The human CCK-1R gene is estimated to be 11.0 kb in length. The mouse Cck-1r gene and the rat CCK-1R gene are estimated to be approximately 9.0 kb and about 9.5 kb in length, respectively. The human CCK-1R gene is mapped to chromosome 4 (p15.2). The mouse Cck-1r gene and the rat CCK-1R gene are localized to chromosomes 5 (29.52 cM) and 14 (q11), respectively.
The CCK-2R gene encodes a G protein-coupled receptor for gastrin and CCK, regulatory peptides of the brain and gastrointestinal tract. This protein is a type B gastrin receptor, which has a high affinity for both sulfated and non-sulfated CCK analogs and is found principally in the central nervous system and the gastrointestinal tract. Similar to CCK-1R, CCK-2R displays seven hydrophobic segments that contain transmembrane helices and form a helical bundle domain, which is typical of Family A in sharing the signature sequences of such kind of the receptor family within these structural regions. The human CCK-2R gene is mapped to chromosome 11 (p15.4). The mouse and rat CCK-2R genes are localized to chromosomes 7 (55.86 cM) and 1 (q33), respectively.
3. PHYSIOLOGY OF CCK AND ITS RECEPTORS
As shown in (Fig. 2), plasma CCK comes predominantly from the intestinal endocrine I-cells. Protein- and fat-enriched food is the most important trigger for its secretion. Among the nutritional components, protein and L-amino acids, as well as digested fat significantly stimulate CCK secretion from the intestine (Table 1). Carbohydrates stimulate only small amounts of CCK release. CCK triggers bile release from the gallbladder and the secretion of digestive enzymes from the pancreas [1, 22–27].
Fig. (2). Effect of diet on the release of cholecystokinin (CCK) for the regulation of hepatobiliary and pancreatic functions and gastrointestinal tract motility.
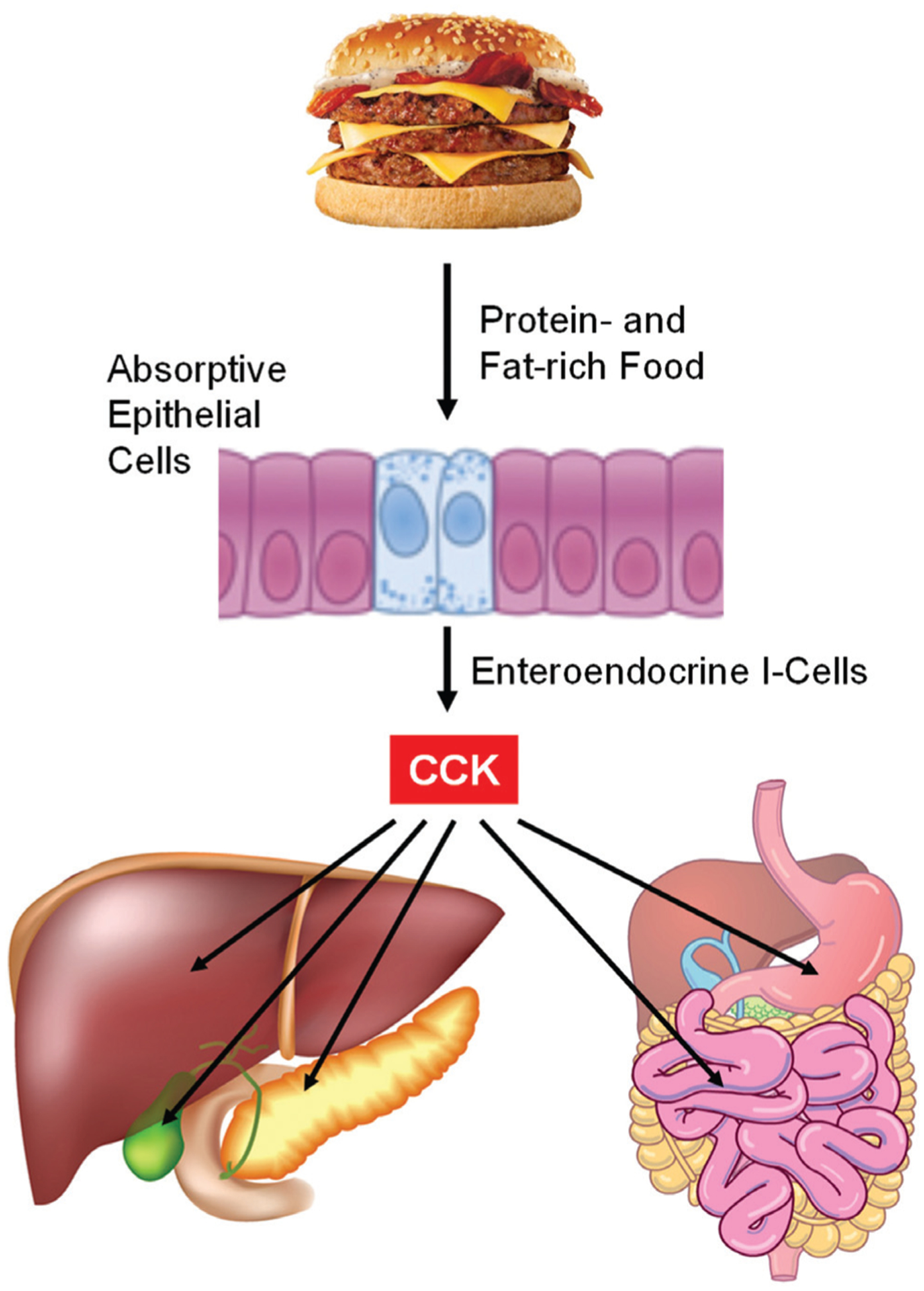
Among the nutritional components, protein- and fat-enriched food is the most important trigger stimulating CCK secretion from the intestinal endocrine I-cells. Carbohydrates stimulate only small amounts of CCK release. CCK causes gallbladder contraction by acting on gallbladder smooth muscles. CCK mainly stimulates hepatic secretion of bicarbonate from hepatic ductular cells. CCK promotes the secretion of pancreatic enzymes such as pancreatic amylase, chymotrypsinogen, and trypsinogen, as well as several small intestinal enzymes such as alkaline phosphatase, disaccharidase and enterokinase. CCK accelerates small intestinal transit through the CCK-1 receptor (CCK-1R) signaling cascade. In contrast, CCK inhibits gastric emptying. See text for more details.
Table 1.
Effect of dietary nutrients on CCK release.
| Nutrients | CCK Release |
|---|---|
| Fat | +++++ |
| Protein | ++++ |
| Carbohydrate | + |
In the human, CCK-22 and CCK-33 are present predominantly in the circulation, but CCK-8 and CCK-58 are also found in plasma [19]. Because the cholecystokinetic and pancreozymic potencies of CCK-33 and CCK-8 are basically similar, it may not be important what are secreted by the enteroendocrine I-cells during meals. Of special note, clearance rate of CCK-58, CCK-33 and CCK-22 from the circulation is markedly slower compared to that of CCK-8 [28].
It is well established that CCK exerts its biological and physiological actions through CCK-1R and CCK-2R, as summarized in Table 2. Accumulated evidence from animal experiments and human studies supports a role for CCK through the CCK-1R pathway in regulating many physiological processes such as gallbladder emptying, relaxation of the sphincter of Oddi, as well as increasing bile flow, accelerating small intestinal transit, stimulating pancreatic secretion, inhibiting gastric emptying and acid secretion, regulating satiety, relaxing lower esophageal sphincter tone, and slowing of colonic motility [1]. These are regulated by CCK-1R on gallbladder muscularis, pancreatic neurons (in humans, and also directly on pancreatic acinar cells in rodents), pyloric smooth muscle, enteric neurons, and central nervous system nuclei [1]. CCK-2R is distributed in the gastric oxyntic mucosa and is also present in the brain, with a high concentration expressing in the striatum, cerebral cortex, and limbic system [29]. Besides stimulating gastric acid secretion, CCK-2R plays a role in anxiety and nociception. Thereafter, we will focus mainly on the role of CCK in the regulation of gallbladder and small intestinal motility through the CCK-1R signaling pathway.
Table 2.
Role of CCK in health and disease.
| Target Organs | Physiological Functions | Pathophysiological Conditions |
|---|---|---|
| Gallbladder | Gallbladder contraction and emptying; bile emulsifies dietary fat and aids the digestion and absorption of cholesterol, fatty acids, and fat-soluble vitamins. | Gallbladder hypomotility; gallbladder stasis; biliary sludge; gallstone formation; impaired bile-induced emulsion of dietary fat and the digestion and absorption of cholesterol, fatty acids, and fat-soluble vitamins. |
| Biliary tract | Sphincter of Oddi relaxation. | Bile release into the intestine. |
| Pancreas | Secretion of pancreatic enzymes such as pancreatic amylase, chymotrypsinogen, and trypsinogen; pancreatic exocrine secretion. | Impaired digestion and absorption of dietary fat, protein, carbohydrate, cholesterol, bile salts, and fat-soluble vitamins; pancreatitis; reduced pancreatic exocrine secretion. |
| Liver | Bile flow and hepatic secretion mainly as bicarbonate from hepatic ductular cells. | Reduced bile formation. |
| Stomach | Gastric emptying and acid secretion; relaxation of the gastric corpus. | Delayed gastric emptying and delivery of food to the duodenum; increased resistance to flow of chyme across the pyloric sphincter of stomach; impaired digestion of dietary fat and protein. |
| Esophagus | Relaxation of lower esophageal sphincter tone. | Impaired esophageal motility. |
| Small intestine | Small intestinal transit. Secretion of several small intestinal enzymes such as alkaline phosphatase, disaccharidase and enterokinase. |
Slow small intestinal transit, impaired digestion and absorption of dietary fat, protein, carbohydrate, cholesterol, bile salts, and fat-soluble vitamins. |
| Colon | Colonic motility. | Slow colonic motility. |
| Brain | Regulation of satiety. | Food intake; obesity; anxiety; nociception. |
| Adipose tissue | Energy expenditure. | Obesity. |
4. EFFECTS OF CCK AND CCK-1R ON DIGESTIVE SYSTEM
CCK-1R is mainly localized in the gallbladder, sphincter of Oddi, pancreas, small intestine, gastric mucosa, and pyloric sphincter, where it is responsible for regulating multiple digestive processes, including gallbladder emptying, pancreatic secretion, small intestinal motility, and gastric emptying (Fig. 2). Intestinal CCK secretion is triggered by fat- or protein-enriched chyme entering the duodenum [30]. In the basal state, the concentration of CCK in the circulation is much lower (less than 1 pmol/L) than that of other pancreatic and gastrointestinal hormones [31]. As shown in Fig. (3), the concentration of CCK in plasma increases to 3–5 pmol/L within 20 minutes during maximal stimulation, and then reduces gradually to basal levels [31]. Of note, the circulating CCK concentrations are sufficient to stimulate the gallbladder emptying and pancreatic enzyme secretion during meals [32].
Fig. (3).
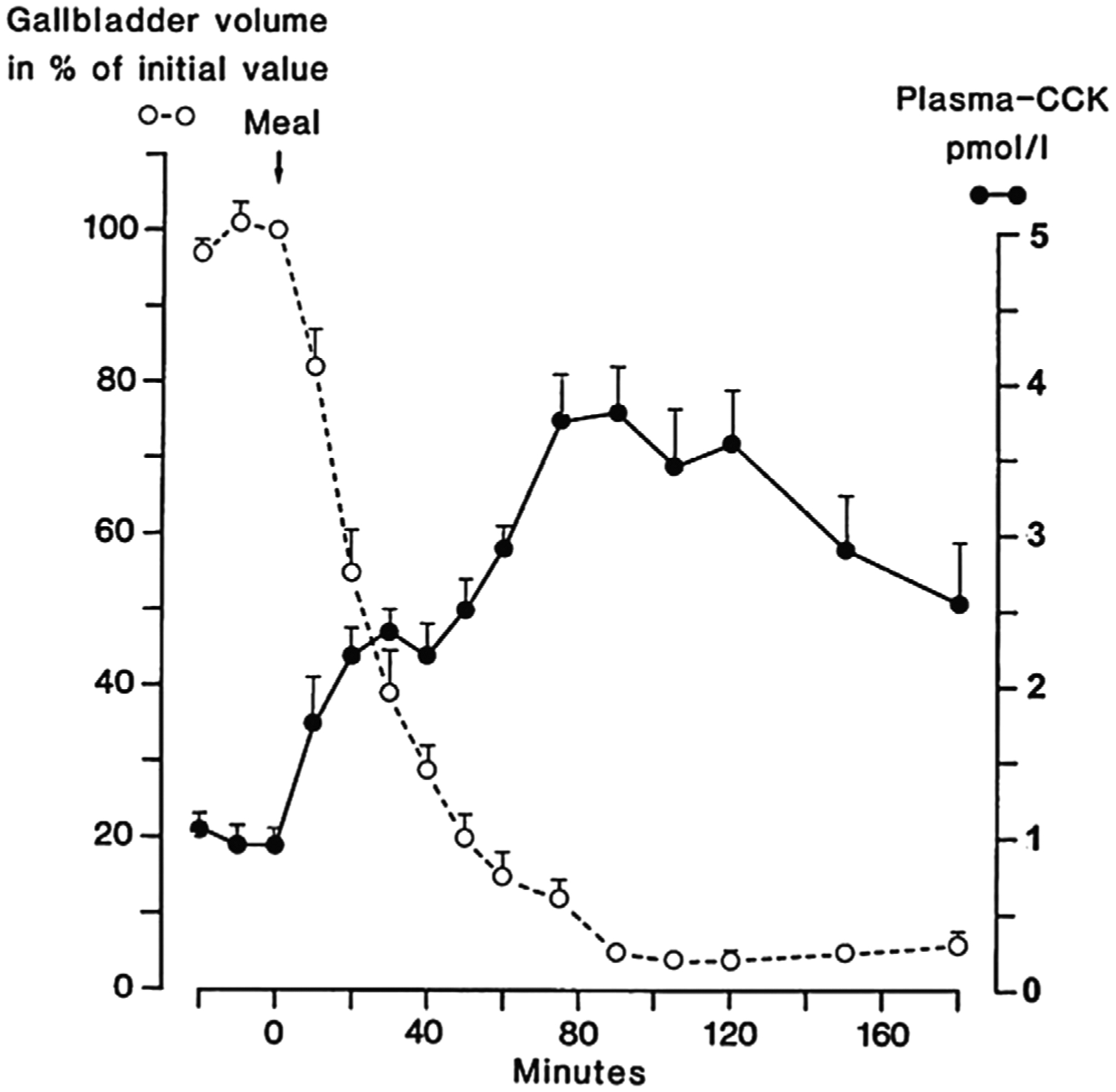
Plasma cholecystokinin (CCK) concentrations and gallbladder volume during a meal in normal human subjects. Reprinted, with permission, from Ref. [31].
The gallbladder is a pear-shaped organ located on the inferior surface of the liver at the junction of the right and left hepatic lobes, which typically hangs from the anterior inferior margin of the liver [33]. The volume of a moderately distended gallbladder is approximately between 30 and 50 mL. Furthermore, the gallbladder’s volume varies considerably, being large because of the storage of concentrated bile during fasting states and becoming small due to its postprandial emptying [34]. The gallbladder can be divided into four parts: the neck, body, infundibulum, and fundus. After being stored in the gallbladder between meals, gallbladder bile becomes more concentrated compared to hepatic bile, intensifying its action, by emulsification, on fat digestion and absorption. Notably, among solute components of bile, bile acids play a critical role in the digestion and absorption of lipids [35]. They are synthesized by the liver from cholesterol and are typically conjugated with glycine and taurine in a ratio of 3:1 [36–38]. Bile acids are secreted into hepatic bile by the liver. When bile is concentrated in the gallbladder, their concentrations are increased by 5 fold compared to those in hepatic bile [33]. When protein- and fat-rich food enters the small intestine and stimulates CCK secretion, CCK causes gallbladder contraction by acting on gallbladder smooth muscles with a potency correlated to the low plasma concentrations of sulfated CCK (Fig. 4). As a result, bile is released into the duodenum via CCK-mediated rhythmic contraction and relaxation of muscles in the common bile duct and the sphincter of Oddi. Thus, the gallbladder empties bile into the duodenum where bile emulsifies dietary fat and aids the digestion and absorption of cholesterol, fatty acids, and fat-soluble vitamins [39].
Fig. (4).
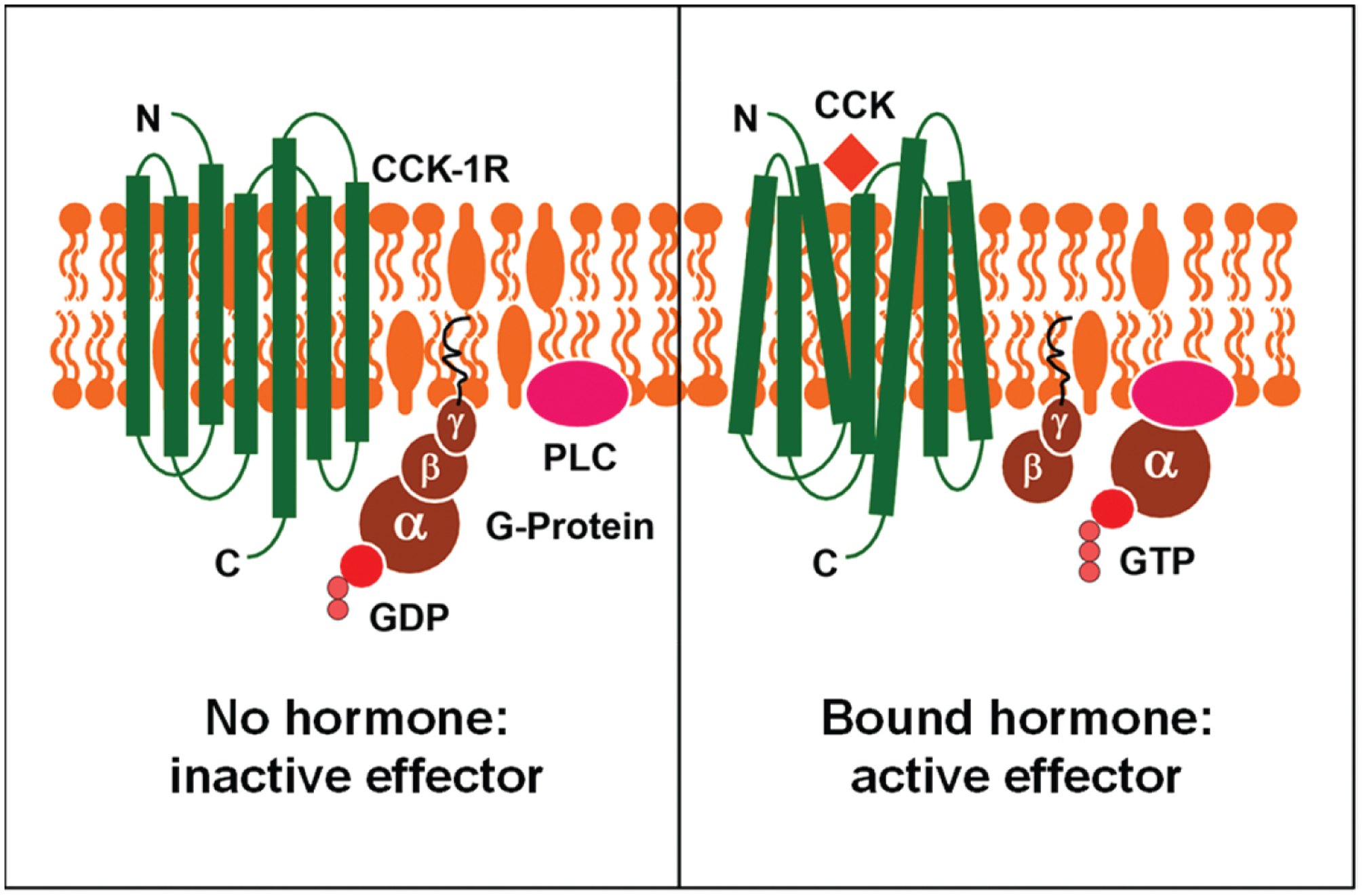
This diagram illustrates how cholecystokinin (CCK) stimulates gallbladder contraction by activating the CCK-1 receptor (CCK-1R) signaling pathway in the sarcolemmae of the gallbladder smooth muscle. The left panel shows the CCK-1R signaling cascade in the inactive state, whereas the right panel shows the CCK-1R activated by CCK, coupled with the stimulation of G proteins in the gallbladder smooth muscle.
CCK stimulates hepatic secretion mainly as bicarbonate from hepatic ductular cells. CCK promotes the secretion of pancreatic enzymes including pancreatic amylase, chymotrypsinogen, and trypsinogen, as well as several small intestinal enzymes such as alkaline phosphatase, disaccharidase and enterokinase [40]. Although there are species differences in the mechanism underlying the effect of CCK on pancreatic exocrine secretion, CCK stimulates pancreatic enzyme secretion through a cholinergic pathway in humans, which is considerably less significant in rodents [41–43].
CCK is involved in regulating the gastrointestinal tract motility. CCK markedly inhibits gastric emptying and food intake, as well as delays delivery of food to the duodenum possibly through stimulation of vagal afferent neurons that mainly express CCK-1R. Furthermore, gastric emptying is inhibited mostly through activation of vagovagal reflexes, thus inducing relaxation of the gastric corpus and increasing resistance to flow of chyme across the pyloric sphincter of stomach.
In addition, CCK accelerates small intestinal transit through the CCK-1R signaling cascade because there is a sluggish small intestinal motility in mice with disruption of either the Cck or the Cck-1r gene [44, 45]. Furthermore, the potent CCK-1R-selective antagonist devazepide not only impairs gallbladder emptying function, but also decelerates small intestinal transit in mice [46]. These abnormalities lead to gallbladder stasis, increased intestinal cholesterol absorption, and augmented hepatic secretion of biliary cholesterol, as well as rapid cholesterol crystallization, and accelerated the growth of solid cholesterol crystals and the formation of microlithiasis in mice treated with devazepide.
5. MOLECULAR MECHANISMS UNDERLYING THE CRITICAL ROLE OF CCK AND CCK-1R IN THE PATHOGENESIS OF GALLSTONE FORMATION
Abnormalities in gallbladder contractility in response to exogenously administered CCK are found primarily in patients with cholesterol gallstones [9, 47, 48]. Patients with pigment gallstones display an intermediate degree of gallbladder motility defect without enlarged fasting gallbladder and gallbladder inflammation [49]. Moreover, impaired gallbladder contractile function exists in some gallstone-free subjects under several conditions such as pregnancy, obesity, diabetes, celiac disease, and total parenteral nutrition [17, 50–56]. Increased fasting volume and slow emptying of the gallbladder, as measured by ultrasonography, are often found in women receiving oral contraceptives, pregnant women, and postmenopausal women with estrogen replacement therapy, all of which predispose to gallstone formation [57]. Furthermore, impaired gallbladder emptying in patients with cholesterol gallstones is not the result of the physical presence of gallstones per se, since the defect does not correlate with the size or number of stones, and lithotripsy-induced gallstone ablation does not restore impaired gallbladder motility [17, 58, 59]. In patients with cholesterol gallstones, gallbladder contraction in response to exogenous CCK is dampened compared with healthy subjects [60], implying that gallbladder CCK-1R function is impaired possibly due to reduced CCK-1R number and/or CCK binding capacity to CCK-1R’s in some gallstone patients [61–64].
The absence of endogenous CCK in mice impairs gallbladder emptying function and enlarges both fasting and postprandial gallbladder sizes (Fig. 5), and consequently these alterations promote gallstone formation by accelerating cholesterol crystallization and crystal growth. CCK knockout mouse model provides an opportunity to investigate the role of gallbladder hypomotility in cholesterol crystallization and crystal growth and to study the gallbladder stasis mechanism [44, 65]. Thus, whether impaired gallbladder emptying plays a critical role in the evolutionary sequences of cholesterol crystallization and gallstone formation has been investigated in CCK knockout mice. This is not easily achieved in patients although investigators have attempted to study the physical-chemical phase separation sequences in bile before and during gallstone formation in obese patients who ingest a very low calorie diet or are undergoing weight reduction after gastric bypass surgery [66–68].
Fig. (5).
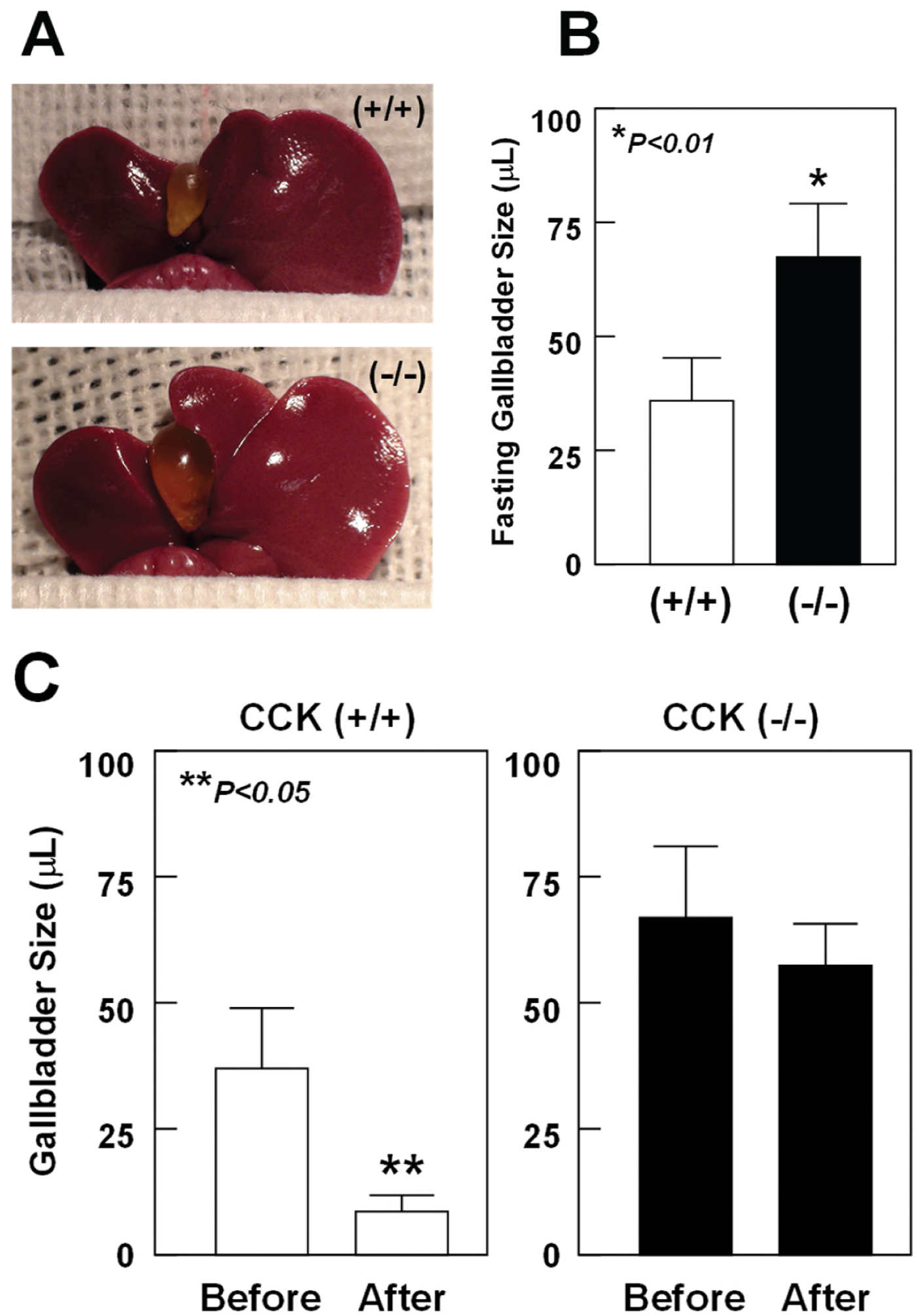
(A) Representative photographs and (B) gross observations of gallbladders exhibit that fasting gallbladder volumes are significantly enlarged in CCK knockout mice compared with CCK wild-type mice. (C) Postprandial gallbladder sizes in response to the high fat meal. Duodenal infusion of corn oil could stimulate the release of CCK from the upper part of small intestine. As a result, the secreted CCK induces gallbladder emptying in CCK wild-type mice but not in CCK knockout mice. These results indicate that gallbladder contractile function is impaired in CCK knockout mice. Reprinted, with permission, from Ref. [65].
Under lithogenic conditions, the evolutionary sequences of gallstone formation (Fig. 6) are characterized by the initial accumulation of mucin gel in the gallbladder, followed by the appearances of liquid crystals and/or anhydrous cholesterol crystals and classic plate-like cholesterol monohydrate crystals, and then agglomerated solid cholesterol crystals, sandy stones, and gallstones in CCK knockout mice fed the lithogenic diet for 8 weeks [44, 65]. These sequences are in agreement with findings in other animal models of cholesterol gallstones, such as gallstone-susceptible C57L mice [69] and prairie dogs [70, 71]. In addition to the well-known liquid crystal to classic plate-like cholesterol monohydrate crystal pathway [72–74], the anhydrous cholesterol crystal to solid cholesterol monohydrate crystal pathway [69, 75, 76] plays a crucial role in the early stage of cholesterol gallstone disease in CCK knockout mice with gallbladder stasis. Therefore, there are two cholesterol crystallization pathways in these mice fed the lithogenic diet, which are identical to those found in model bile with the same lipid composition and in human bile [69, 75–77]. Of note, the sequences of cholesterol crystallization and gallstone formation are more rapid in CCK knockout mice than in other mouse models of gallstones fed the same lithogenic diet [69], which underscores the importance of gallbladder stasis.
Fig. (6).
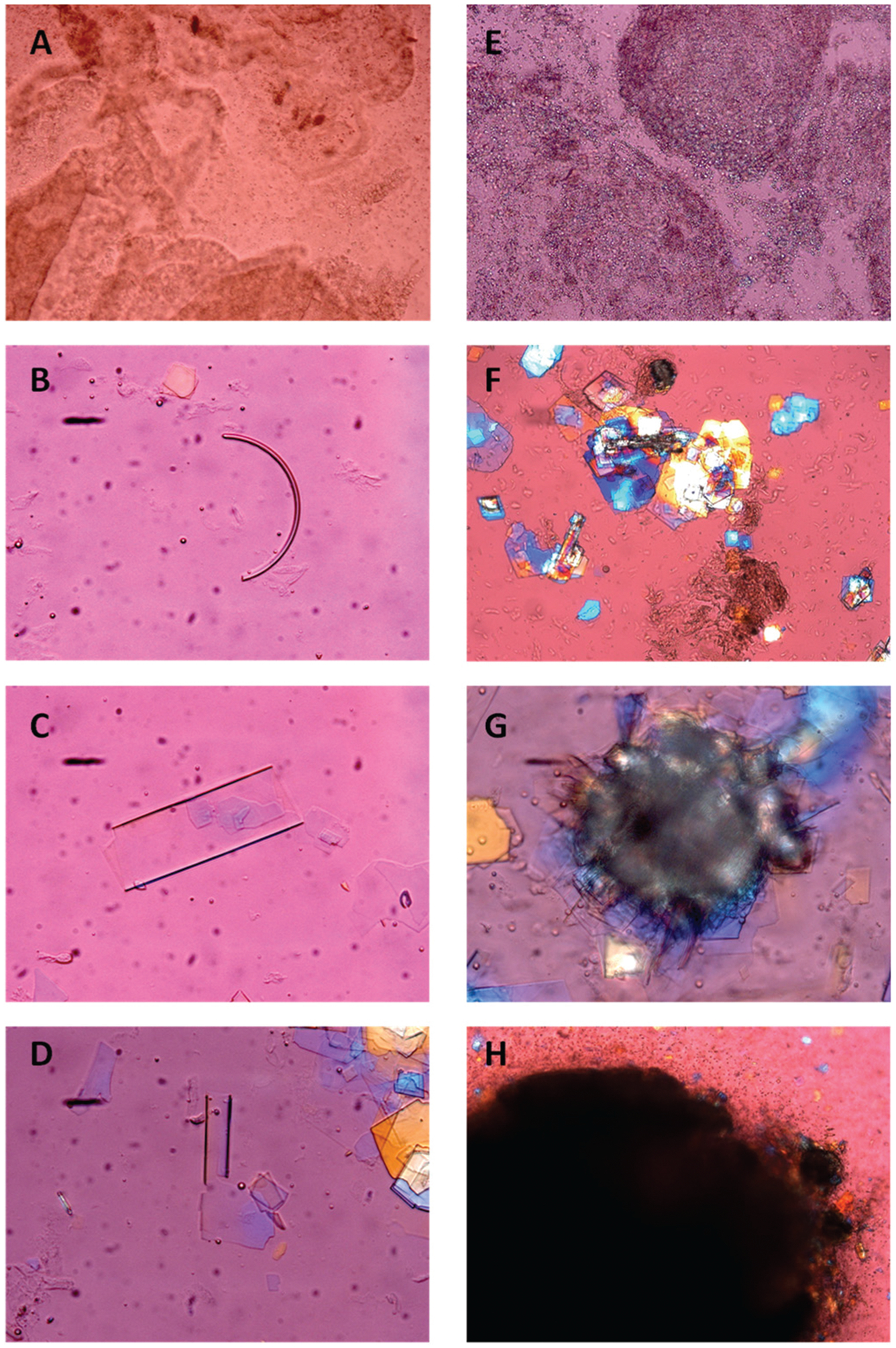
Representative photomicrographs of mucin gel as well as habits of liquid crystals, solid cholesterol crystals, and gallstones as observed in fresh gallbladder bile of CCK knockout mice by phase contrast and polarizing light microscopy: (A) non-birefringent amorphous mucin gel; (B) arc-like (possible anhydrous cholesterol) crystal; (C) tubular crystal; (D) tubular crystal fracturing at the end to produce plate-like cholesterol monohydrate crystals; (E) numerous aggregated non-birefringent liquid crystals and few fused liquid crystals; (F) agglomerates of typical cholesterol monohydrate crystals, with 79.2° and 100.8° angles, and often a notched corner; (G) disintegrable amorphous sandy stones surrounded by mucin gel, with individual plate-like cholesterol monohydrate crystals projecting from the edges; (H) true gallstones displaying rounded contours and black centers from light scattering/absorption. All magnifications are ×800, except Figure 6(F and G) ×400 and Figure 6H ×200, by polarizing light microscopy. Reprinted, with permission, from Ref. [65].
CCK knockout mice display a significant retardation of small intestinal transit, leading to augmented intestinal cholesterol absorption [44, 65]. This change partly elucidates why CCK knockout mice display earlier elevation of CSI values and more rapid cholesterol-supersaturated gallbladder bile compared to wild-type mice. Because of the absence of CCK-induced emptying, the resulting gallbladder stasis provides a microenvironment for the longer stay of excess cholesterol in the lumen. As a result, the increase in bile cholesterol, coupled with longer contact time of saturated mixed micelles with cholesterol transporters on the apical membrane of gallbladder epithelial cells, greatly facilitates gallbladder cholesterol absorption [78, 79]. Elevated cholesterol content in the gallbladder wall further impairs gallbladder emptying and promotes mucin hypersecretion and accumulation in the lumen. The gel-forming mucins secreted by specialized gallbladder mucin-producing cells, lead to the formation of a gel phase in higher concentrations because they form disulfide-stabilized oligo- or polymers with viscoelastic properties. In addition, hydrophobic domains in the mucin molecule (on the non-glycosylated regions of the polypeptide core) allow binding of mucin to lipids such as cholesterol, phospholipids, and bilirubin. The resulting water-insoluble complex of mucous glycoproteins and calcium bilirubinate provides a surface for rapid nucleation and crystallization of solid cholesterol crystals and a framework of matrix for the growth of crystals and stones [80, 81]. During the early stage of gallstone formation, very tiny cholesterol monohydrate crystals are often found within the mucin gel. A lot of evidence has indicated that gallbladder mucins are a strong pro-nucleating/crystallizing agent leading to rapid cholesterol crystallization in native and model bile and play a crucial role in the pathogenesis of gallstone formation [82, 83]. Gallbladder mucin hypersecretion is also a prerequisite for the formation of gallstones [69, 70, 84]. Mucins have been identified within gallstones where they could act as a matrix for accelerating the growth of crystals and stones [85, 86].
Although over the past years, the growth habits of solid cholesterol crystals have been extensively investigated mostly in model bile systems [87, 88], whether these pathophysiologically relevant phenomena take place in native gallbladder bile of humans or mice is still unknown. In mouse lithogenic bile, three modes of cholesterol crystal growth habits have been found in the early stage of cholesterol gallstone disease, all of which increase the size of solid cholesterol crystals. These findings are consistent with the results observed in model bile systems [87]. When the CSI values in bile are higher, there are two major crystal growth habits: the spiral dislocation growth and the twin crystal growth patterns [87]. When bile CSI values are lower, proportional enlargement patterns are found to be the third mode of crystal growth habits. Obviously, impaired gallbladder contractility is associated with larger sizes of solid cholesterol crystals probably because of more rapid crystal growth by way of these three crystal growth patterns in CCK knockout mice.
Clinical studies found that repeated gallbladder emptying induced by supra-physiological doses of CCK-8 restores gallbladder contractility, thereby preventing gallstones in subjects undergoing total parenteral nutrition [89]. Thus, the gallbladder-specific CCK-1R-selective agonist may provide an efficacious novel approach for the prevention of cholesterol gallstones by promoting gallbladder emptying, particularly for pregnant women and subjects with dysfunctional gallbladder contraction such as celiac patients, as well as patients undergoing total parenteral nutrition.
Expression of the Cck-1r gene has been found on the smooth muscle of the gallbladder and stomach, as well as of the small intestine [90]. CCK-1R knockout mice display a significantly slower small intestinal transit times compared with wild-type mice, regardless of whether chow or the lithogenic diet is fed (Fig. 7). Small intestinal transit time in mice is not influenced by feeding high levels of dietary cholesterol or cholic acid [91]. However, this alteration can be induced by fatty acids during the digestion of dairy fat contained in the lithogenic diet, implying that the large intake of fat could trigger maximal endogenous CCK release exaggerating differences between CCK-1R knockout and wild-type mice. This shows that a physiologically relevant mechanism regulated by CCK-1R mediates small intestinal motility. In addition, slow small intestinal transit time promotes cholesterol absorption from the small intestine most likely due to longer contact time of the cholesterol molecule in the small intestinal lumen. This would increase cholesterol’s incorporation into mixed micelles and also promote partitioning of cholesterol monomers out of micelles for uptake by intestinal cholesterol transporters on the apical membrane of enterocytes [39, 92, 93]. This indicates that the transit time of cholesterol molecules through the small intestine is an important factor for regulating cholesterol absorption [94]. As examined by a highly precise cholesterol balance method [95–97], intestinal cholesterol absorption efficiency is significantly higher in CCK-1R knockout mice than in wild-type mice, regardless of whether chow or the lithogenic diet is fed. Moreover, the cholesterol absorbed from the small intestine reaches the liver through the chylomicron remnant pathway which, in turn, enhances biliary cholesterol secretion and promotes biliary cholesterol supersaturation [98]. It has been found that the efficiency of intestinal cholesterol absorption correlates positively with the prevalence of cholesterol gallstones in inbred strains of mice [98]. These indicate that high intestinal cholesterol absorption efficiency is an independent risk factor for the formation of cholesterol gallstones in mice. Although there is a slow small and large intestinal transit rate in patients with cholesterol gallstones [99], this probably differs from the findings in CCK-1R knockout mice since these human studies found that both prolonged large intestinal transit time and increased numbers of anaerobes with elevated 7α-dehydroxylation activity greatly promotes the synthesis and absorption of deoxycholic acid in the large intestine [100–103]. However, in none of these investigations is the possibility of increased intestinal cholesterol absorption addressed or even alluded to. Increased levels of deoxycholate conjugates in bile could be prolithogenic in gallstone patients [104–106], enhancing biliary cholesterol hypersecretion and promoting cholesterol crystallization in gallbladder bile. However, there is no difference in biliary bile salt composition between CCK-1R knockout and wild-type mice. Moreover, biliary deoxycholate pools are increased by 27% in male C57L mice fed 0.5% deoxycholic acid compared to chow (3.4%), but small intestinal transit time remains similar [107].
Fig. (7).
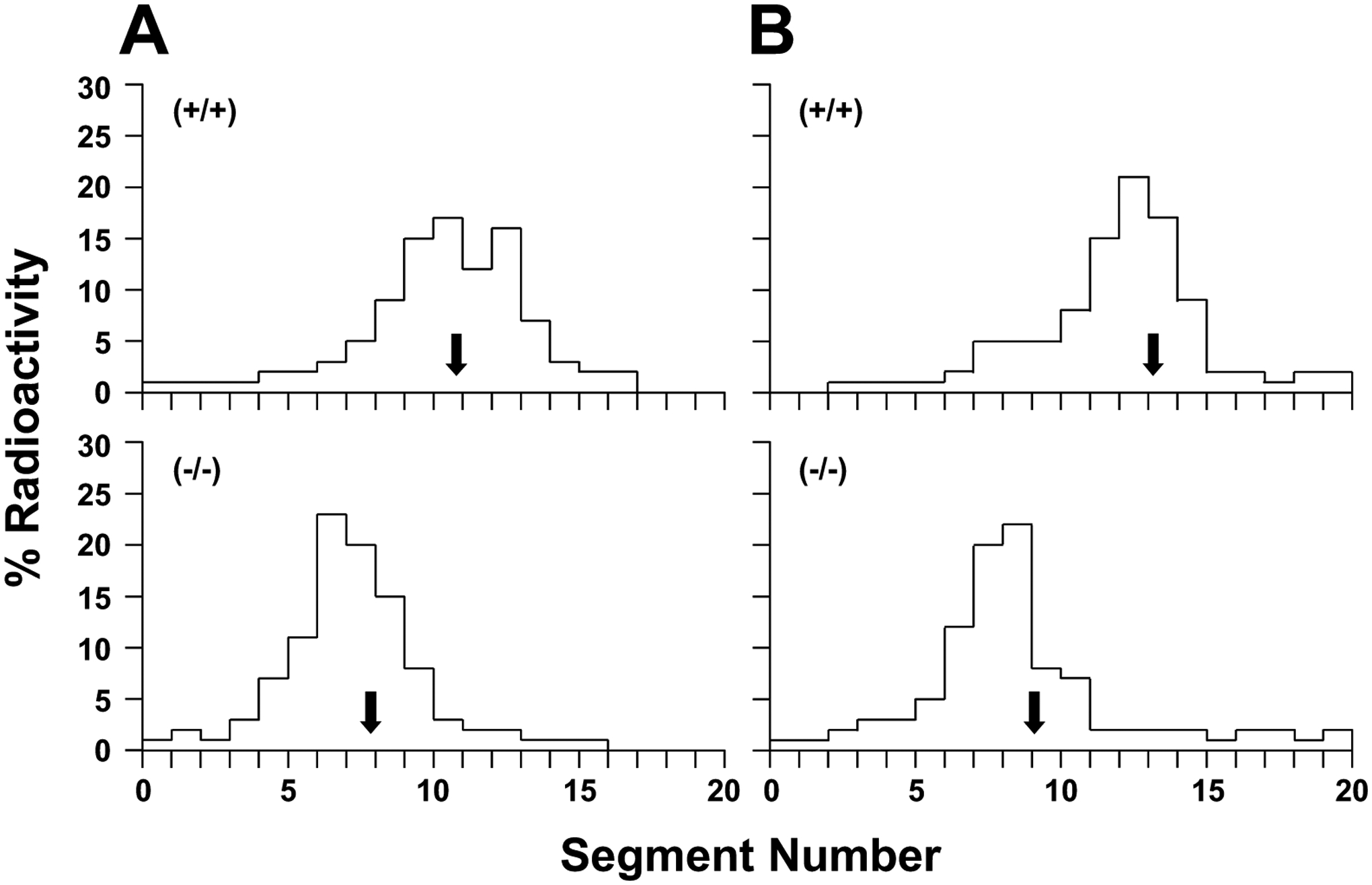
Small intestinal transit rates in CCK-1R wild-type mice (top panel) and knockout mice (bottom panel) on chow or fed the lithogenic diet for 14 days as analyzed by the distribution of radioactivity at 30 minutes along the small intestine following intraduodenal instillation of [3H]sitostanol dissolved in medium-chain triglyceride [96]. Each bar is the mean percentage of radioactivity in each segment. Segments 1 and 20 represent evenly divided segments from the most proximal to the distal part of the small intestine. (A) Arrows show the geometric center that is significantly (P<0.001) shorter in chow-fed CCK-1R knockout mice, indicating significantly slower small intestinal transit times (geometric center=7.8±0.8) compared with the wild-type mice (geometric center=10.8±1.0). (B) Under conditions of feeding lithogenic diet, CCK-1R knockout mice continue to display significantly slower small intestinal transit times (geometric center=9.1±1.5) than the wild-type mice (geometric center=13.3±2.0). Reprinted, with permission, from Ref. [45].
Because impaired smooth muscle contractility is found in cell isolates from human gallbladders and from animal models with cholesterol gallstones, Behar and co-workers [62, 108, 109] have suggested that the gallbladder contractile mechanisms involve CCK-1R mediated activation of phospholipase C, resulting in signal-transduction decoupling when the sarcolemmae of the gallbladder smooth muscle are enriched with cholesterol. This may be caused by a cholesterol-dependent reduction in the number of CCK-1R or its defects in the gallbladder smooth muscle [110], which impairs contractility mostbly from involvement of excess cholesterol within the caveolin rafts of sarcolemmal membranes. The critical CCK-1R signal-transduction pathway that mediates contraction of gallbladder smooth muscle becomes further dysfunctional by the absorbed cholesterol from bile in CCK-1R knockout mice and each, in turn, increases cholelithogenesis [62, 108, 109, 111]. Similar to the findings in humans, the deletion of CCK-1R in mice disrupts the physiological control of gallbladder contraction and small intestinal transit, thereby inducing muscle dysfunction in the gallbladder and small intestine. This scenario is propagated since bile is cholesterol-supersaturated from cholesterol absorption from the small intestine and gallbladder function is further compromised by the absorbed cholesterol from its lumen. Obviously, deletion of the Cck-1r gene augments intestinal cholesterol absorption by impairing small intestinal transit, which, in turn, promotes biliary cholesterol hypersecretion. Dysfunctional CCK-1R also enlarges gallbladder size and enhances cholesterol absorption from bile, which further impairs gallbladder contractility, thus resulting in remarkable gallbladder stasis [45]. Furthermore, gallstone prevalence is greatly increased in C57BL/6 mice treated with the potent CCK-1R antagonist devazepide by impairing gallbladder emptying, disrupting cholesterol metabolism in bile, and increasing intestinal cholesterol absorption [46]. These defects in gallbladder and small intestinal motility due to dysfunctional CCK or CCK-1R greatly contribute to the formation of cholesterol gallstones by promoting intestinal cholesterol absorption and biliary cholesterol hypersecretion, followed by rapid cholesterol crystallization and crystal growth, as well as agglomeration of solid cholesterol crystals in “large lax and lazy” gallbladder [17]. These results, taken together, clearly demonstrate that prolonged small intestinal transit time, augmented cholesterol absorption, and impaired gallbladder motility function are interdependent but conflating risk factors for the formation of cholesterol gallstones in mice deficient in CCK or CCK-1R [112].
Celiac disease is a chronic, small intestinal, immune-mediated enteropathy caused by a permanent intolerance to dietary gluten in genetically predisposed individuals. The major site of damage is localized on the proximal part of small intestine, which typically displays villus atrophy of the mucosa [113]. Clinical studies have found 40 years ago that gallbladder emptying in response to a fatty meal is dramatically decreased because of a defect in CCK secretion by the small intestine caused by enteropathy in celiac patients before they start the gluten-free diet [10]. Thus, celiac disease is an important risk factor for gallstone formation because defective intestinal CCK secretion markedly enhances susceptibility to cholesterol gallstones via a mechanism that is involved in hypomotility of both the gallbladder and the small intestine.
Compelling evidence has clearly demonstrated that postprandial gallbladder emptying in response to a fatty meal is impaired in untreated celiac patients because of a marked defect in CCK secretion from the atrophic small intestinal mucosa as found by low CCK concentrations in plasma and duodenal extracts [10–13, 114, 115]. Another possible explanation is that gallbladder responsiveness to CCK may also be impaired [14]. Further clinical studies found that in the fasting state, untreated celiac patients display dramatically lower CCK levels in the circulating compared to healthy control subjects [10]. These findings show that a fatty meal cannot effectively stimulate CCK secretion by the I-cells in celiac patients. This abnormality is highly attributed to severe villus atrophy, enterocyte disarray, crypt hyperplasia, epithelial cell layer, and intense inflammation of the lamina propria as found in celiac patients [116]. Obviously, celiac patients display gallbladder stasis before starting the gluten-free diet [16], and this may put them at a high risk of developing biliary sludge and gallstones.
Based on mouse studies, a possible relationship between abnormality of the CCK and/or CCK-1R genes and gallstone disease has suggested in humans. Miller and colleagues reported abnormal processing of the CCK-1R gene in patients with gallstones and obesity [117]. However, Nardone and co-workers failed to identify any mutations or sequence variants in patients with the same phenotype undergoing cholecystectomy [118]. Nevertheless, a further study is strongly needed to investigate whether there is mutation in the CCK and/or CCK-1R genes in gallstone patients and whether the CCK and/or CCK-1R gene variants are associated with increased gallstone prevalence in patients. It is also imperative to determine whether some of the single nucleotide polymorphisms (SNPs) in the CCK and/or CCK-1R genes are responsible for enhanced susceptibility to biliary sludge and gallstones in pregnant women and in subjects on total parenteral nutrition.
CONCLUSION
Cholesterol gallstone disease is induced by hepatic cholesterol supersaturation, rapid cholesterol nucleation and crystallization through several intermediate steps, and gallbladder stasis [112]. Recent progress in exploring the molecular, genetic, and physical-chemical basis of hepatic cholesterol hypersecretion and cholesterol crystallization has provided many new insights into the complex pathophysiological mechanisms underlying the pathogenesis of cholesterol gallstone disease in humans [119]. The CCK knockout and the CCK-1R knockout mouse models highlight two important issues: (i) gallbladder and small intestinal motility are, in part, regulated by CCK through the CCK-1R signaling cascade; and (ii) the role of CCK-1R in small intestinal motility is a physiological response that could be a significant factor in regulating intestinal cholesterol absorption. As impaired gallbladder emptying function is a high risk for developing gallbladder stasis, biliary sludge (a precursor of gallstones), and microlithiasis, as well as sluggish small intestinal transit increases cholesterol absorption for promoting hepatic cholesterol hypersecretion, two defects in the gallbladder and the intestine should be prevented by oral administration of the potent CCK-1R agonists. Thus, susceptibility to gallstone formation may be markedly reduced.
FUNDING
This work was supported in part by research grants DK101793 and DK106249 (to DQ-HW), both from the National Institutes of Health (US Public Health Service), and by European Joint Programming Initiative “A Healthy Diet for a Healthy Life (JPI HDHL), action DEterminants of DIet and Physical Activity Choice (DEDIPAC)-National Project Wellness, nutrItion, Sport and Exercise prevention (WISE) 2013-16 (to P.P.).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].Chandra R; Liddle RA Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes, 2007, 14(1), 63–67. [ 10.1097/MED.0b013e3280122850] [DOI] [PubMed] [Google Scholar]
- [2].Ivy AC; Oldberg E A hormone mechanism for gallbladder contraction and evacuation. Am. J. Physiol, 1928, 86, 599–613. [ 10.1152/ajplegacy.1928.86.3.599] [DOI] [Google Scholar]
- [3].Schjoldager BT Role of CCK in gallbladder function. Ann. N. Y. Acad. Sci, 1994, 713, 207–218. [ 10.1111/j.1749-6632.1994.tb44067.x] [DOI] [PubMed] [Google Scholar]
- [4].Spiegelman BM; Flier JS Obesity and the regulation of energy balance. Cell, 2001, 104(4), 531–543. [ 10.1016/S0092-8674(01)00240-9] [DOI] [PubMed] [Google Scholar]
- [5].Badman MK; Flier JS The gut and energy balance: visceral allies in the obesity wars. Science, 2005, 307(5717), 1909–1914. [ 10.1126/science.1109951] [DOI] [PubMed] [Google Scholar]
- [6].Guyenet SJ; Schwartz MW Clinical review: Regulation of food intake, energy balance, and body fat mass: implications for the pathogenesis and treatment of obesity. J. Clin. Endocrinol. Metab, 2012, 97(3), 745–755. [ 10.1210/jc.2011-2525] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martínez-González MA; Bes-Rastrollo M Nut consumption, weight gain and obesity: epidemiological evidence. Nutr. Metab. Cardiovasc. Dis, 2011, 21(Suppl. 1), S40–S45. [ 10.1016/j.numecd.2010.11.005] [DOI] [PubMed] [Google Scholar]
- [8].Portincasa P; Moschetta A; Palasciano G Cholesterol gallstone disease. Lancet, 2006, 368(9531), 230–239. [ 10.1016/S0140-6736(06)69044-2] [DOI] [PubMed] [Google Scholar]
- [9].Portincasa P; Di Ciaula A; Baldassarre G; Palmieri V; Gentile A; Cimmino A; Palasciano G Gallbladder motor function in gallstone patients: sonographic and in vitro studies on the role of gallstones, smooth muscle function and gallbladder wall inflammation. J. Hepatol, 1994, 21(3), 430–440. [ 10.1016/S0168-8278(05)80324-1] [DOI] [PubMed] [Google Scholar]
- [10].Low-Beer TS; Harvey RF; Davies ER; Read AF Abnormalities of serum cholecystokinin and gallbladder emptying in celiac disease. N. Engl. J. Med, 1975, 292(18), 961–963. [ 10.1056/NEJM197505012921807] [DOI] [PubMed] [Google Scholar]
- [11].Maton PN; Selden AC; Fitzpatrick ML; Chadwick VS Defective gallbladder emptying and cholecystokinin release in celiac disease. Reversal by gluten-free diet. Gastroenterology, 1985, 88(2), 391–396. [ 10.1016/0016-5085(85)90497-4] [DOI] [PubMed] [Google Scholar]
- [12].Hopman WP; Rosenbusch G; Hectors MP; Jansen JB Effect of predigested fat on intestinal stimulation of plasma cholecystokinin and gall bladder motility in coeliac disease. Gut, 1995, 36(1), 17–21. [ 10.1136/gut.36.1.17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fraquelli M; Bardella MT; Peracchi M; Cesana BM; Bianchi PA; Conte D Gallbladder emptying and somatostatin and cholecystokinin plasma levels in celiac disease. Am. J. Gastroenterol, 1999, 94(7), 1866–1870. [ 10.1111/j.1572-0241.1999.01221.x] [DOI] [PubMed] [Google Scholar]
- [14].Brown AM; Bradshaw MJ; Richardson R; Wheeler JG; Harvey RF Pathogenesis of the impaired gall bladder contraction of coeliac disease. Gut, 1987, 28(11), 1426–1432. [ 10.1136/gut.28.11.1426] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sjölund K; Alumets J; Berg NO; Håkanson R; Sundler F Duodenal endocrine cells in adult coeliac disease. Gut, 1979, 20(7), 547–552. [ 10.1136/gut.20.7.547] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Low-Beer TS; Heaton KW; Heaton ST; Read AE Gallbladder inertia and sluggish enterohepatic circulation of bile-salts in coeliac disease. Lancet, 1971, 1(7707), 991–994. [ 10.1016/S0140-6736(71)91387-0] [DOI] [PubMed] [Google Scholar]
- [17].Portincasa P; Di Ciaula A; Wang HH; Palasciano G; van Erpecum KJ; Moschetta A; Wang DQ Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology, 2008, 47(6), 2112–2126. [ 10.1002/hep.22204] [DOI] [PubMed] [Google Scholar]
- [18].Ivy AC; Oldberg E A hormone mechanism for gallbladder contraction and evacuation. Am. J. Physiol, 1928, 86, 559–613. [ 10.1152/ajplegacy.1928.86.3.599] [DOI] [Google Scholar]
- [19].Liddle RA; Goldfine ID; Rosen MS; Taplitz RA; Williams JA Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. J. Clin. Invest, 1985, 75(4), 1144–1152. [ 10.1172/JCI111809] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wank SA; Harkins R; Jensen RT; Shapira H; de Weerth A; Slattery T Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc. Natl. Acad. Sci. USA, 1992, 89(7), 3125–3129. [ 10.1073/pnas.89.7.3125] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kopin AS; Lee YM; McBride EW; Miller LJ; Lu M; Lin HY; Kolakowski LF Jr.; Beinborn M Expression cloning and characterization of the canine parietal cell gastrin receptor. Proc. Natl. Acad. Sci. USA, 1992, 89(8), 3605–3609. [ 10.1073/pnas.89.8.3605] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liddle RA Gastrointestinal hormones and neurotransmiters. In: Sleisenger and Fordtran’s gastrointestinal and liver disease. Edited by: Feldman M; Friedman LS; Brandt L Philadelphia: Elsevier Saunders; 2010, pp. 3–19. [ 10.1016/B978-1-4160-6189-2.00001-9] [DOI] [Google Scholar]
- [23].Martínez MA; Lajas AI; Yago MD; Redondo PC; Granados MP; González A; Rosado JA; Martínez-Victoria E; Mañas M; Pariente JA Dietary virgin olive oil enhances secretagogue-evoked calcium signaling in rat pancreatic acinar cells. Nutrition, 2004, 20(6), 536–541. [ 10.1016/j.nut.2004.03.018] [DOI] [PubMed] [Google Scholar]
- [24].Liddle RA Regulation of cholecystokinin secretion by intraluminal releasing factors. Am. J. Physiol, 1995, 269(3 Pt 1), G319–G327. [DOI] [PubMed] [Google Scholar]
- [25].Wang Y; Chandra R; Samsa LA; Gooch B; Fee BE; Cook JM; Vigna SR; Grant AO; Liddle RA Amino acids stimulate cholecystokinin release through the Ca2+-sensing receptor. Am. J. Physiol. Gastrointest. Liver Physiol, 2011, 300(4), G528–G537. [ 10.1152/ajpgi.00387.2010] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang Y; Prpic V; Green GM; Reeve JR Jr; Liddle RA Luminal CCK-releasing factor stimulates CCK release from human intestinal endocrine and STC-1 cells. Am. J. Physiol. Gastrointest. Liver Physiol, 2002, 282(1), G16–G22. [ 10.1152/ajpgi.2002.282.1.G16] [DOI] [PubMed] [Google Scholar]
- [27].Ohta H; Guan D; Tawil T; Liddle RA; Green GM Regulation of plasma cholecystokinin levels by bile and bile acids in the rat. Gastroenterology, 1990, 99(3), 819–825. [ 10.1016/0016-5085(90)90974-6] [DOI] [PubMed] [Google Scholar]
- [28].Maton PN; Selden AC; Chadwick VS Differential distribution of molecular forms of cholecystokinin in human and porcine small intestinal mucosa. Regul. Pept, 1984, 8(1), 9–19. [ 10.1016/0167-0115(84)90024-7] [DOI] [PubMed] [Google Scholar]
- [29].Noble F; Roques BP CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog. Neurobiol, 1999, 58(4), 349–379. [ 10.1016/S0301-0082(98)00090-2] [DOI] [PubMed] [Google Scholar]
- [30].Maton PN; Selden AC; FitzPatrick ML; Chadwick VS Infusion of cholecystokinin octapeptide in man: relation between plasma cholecystokinin concentrations and gallbladder emptying rates. Eur. J. Clin. Invest, 1984, 14(1), 37–41. [ 10.1111/j.1365-2362.1984.tb00701.x] [DOI] [PubMed] [Google Scholar]
- [31].Rehfeld JF; Friis-Hansen L; Goetze JP; Hansen TV The biology of cholecystokinin and gastrin peptides. Curr. Top. Med. Chem, 2007, 7(12), 1154–1165. [ 10.2174/156802607780960483] [DOI] [PubMed] [Google Scholar]
- [32].Rehfeld JF; Larsson LI; Goltermann NR; Schwartz TW; Holst JJ; Jensen SL; Morley JS Neural regulation of pancreatic hormone secretion by the C-terminal tetrapeptide of CCK. Nature, 1980, 284(5751), 33–38. [ 10.1038/284033a0] [DOI] [PubMed] [Google Scholar]
- [33].Wang DQ; Neuschwander-Tetri BA; Portincasa P The Biliary System, 2012, [ 10.4199/C00051ED1V01Y201202ISP033] [DOI]
- [34].Meilstrup JW; Hopper KD; Thieme GA Imaging of gallbladder variants. AJR Am. J. Roentgenol, 1991, 157(6), 1205–1208. [ 10.2214/ajr.157.6.1950867] [DOI] [PubMed] [Google Scholar]
- [35].Carey MC; Hernell O Digestion and absorption of fat. Semin. Gastrointest. Dis, 1992, 3, 189–208. [Google Scholar]
- [36].Hofmann AF; Hagey LR Bile acids: chemistry, patho-chemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci, 2008, 65(16), 2461–2483. [ 10.1007/s00018-008-7568-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hofmann AF Bile Acids and the Enterohepatic Circulation. In: The Liver: Biology and Pathobiology; Arias Irwin M. , Ed.; John Wiley & Sons, Ltd.:2009; pp. 290–304. [ 10.1002/9780470747919.ch20] [DOI] [Google Scholar]
- [38].Hay DW; Carey MC Chemical species of lipids in bile. Hepatology, 1990, 12(3 Pt 2), 6S–14S. [PubMed] [Google Scholar]
- [39].Wang DQ Regulation of intestinal cholesterol absorption. Annu. Rev. Physiol, 2007, 69, 221–248. [ 10.1146/annurev.physiol.69.031905.160725] [DOI] [PubMed] [Google Scholar]
- [40].Chandra R; Liddle RA Recent advances in pancreatic endocrine and exocrine secretion. Curr. Opin. Gastroenterol, 2011, 27(5), 439–443. [ 10.1097/MOG.0b013e328349e2e1] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen D; Nylander AG; Rehfeld JF; Axelson J; Ihse I; Håkanson R Does vagotomy affect the growth of the pancreas in the rat? Scand. J. Gastroenterol, 1992, 27(7), 606–608. [ 10.3109/00365529209000126] [DOI] [PubMed] [Google Scholar]
- [42].Chu M; Borch K; Lilja I; Blomqvist L; Rehfeld JF; Ihse I Endogenous hypercholecystokininemia model in the hamster: trophic effect on the exocrine pancreas. Pancreas, 1992, 7(2), 220–225. [ 10.1097/00006676-199203000-00014] [DOI] [PubMed] [Google Scholar]
- [43].Shetzline MA; Liddle RA Neurohumoral control of the exocrine pancreas. Curr. Opin. Gastroenterol, 1999, 15(5), 380–384. [ 10.1097/00001574-199909000-00002] [DOI] [PubMed] [Google Scholar]
- [44].Wang HH; Liu M; Portincasa P Lack of endogenous cholecystokinin promotes cholelithogenesis in mice. Neurogastroenterol. Motil, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang DQ; Schmitz F; Kopin AS; Carey MC Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J. Clin. Invest, 2004, 114(4), 521–528.[ 10.1172/JCI16801] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang HH; Portincasa P; Wang DQ The cholecystokinin-1 receptor antagonist devazepide increases cholesterol cholelithogenesis in mice. Eur. J. Clin. Invest, 2016, 46(2), 158–169. [ 10.1111/eci.12580] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Behar J; Lee KY; Thompson WR; Biancani P Gallbladder contraction in patients with pigment and cholesterol stones. Gastroenterology, 1989, 97(6), 1479–1484. [ 10.1016/0016-5085(89)90392-2] [DOI] [PubMed] [Google Scholar]
- [48].Pomeranz IS; Shaffer EA Abnormal gallbladder emptying in a subgroup of patients with gallstones. Gastroenterology, 1985, 88(3), 787–791. [ 10.1016/0016-5085(85)90152-0] [DOI] [PubMed] [Google Scholar]
- [49].Portincasa P; Di Ciaula A; Vendemiale G; Palmieri V; Moschetta A; Vanberge-Henegouwen GP; Palasciano G Gallbladder motility and cholesterol crystallization in bile from patients with pigment and cholesterol gallstones. Eur. J. Clin. Invest, 2000, 30(4), 317–324. [ 10.1046/j.1365-2362.2000.00639.x] [DOI] [PubMed] [Google Scholar]
- [50].Wang HH; Portincasa P; Wang DQ Molecular pathophysiology and physical chemistry of cholesterol gallstones. Front. Biosci, 2008, 13, 401–423. [ 10.2741/2688] [DOI] [PubMed] [Google Scholar]
- [51].Stampfer MJ; Maclure KM; Colditz GA; Manson JE; Willett WC Risk of symptomatic gallstones in women with severe obesity. Am. J. Clin. Nutr, 1992, 55(3), 652–658. [ 10.1093/ajcn/55.3.652] [DOI] [PubMed] [Google Scholar]
- [52].Kodama H; Kono S; Todoroki I; Honjo S; Sakurai Y; Wakabayashi K; Nishiwaki M; Hamada H; Nishikawa H; Koga H; Ogawa S; Nakagawa K Gallstone disease risk in relation to body mass index and waist-to-hip ratio in Japanese men. Int. J. Obes. Relat. Metab. Disord, 1999, 23(2), 211–216.[ 10.1038/sj.ijo.0800781] [DOI] [PubMed] [Google Scholar]
- [53].Vezina WC; Paradis RL; Grace DM; Zimmer RA; Lamont DD; Rycroft KM; King ME; Hutton LC; Chey WY Increased volume and decreased emptying of the gallbladder in large (morbidly obese, tall normal, and muscular normal) people. Gastroenterology, 1990, 98(4), 1000–1007. [ 10.1016/0016-5085(90)90025-V] [DOI] [PubMed] [Google Scholar]
- [54].Haber GB; Heaton KW Lipid composition of bile in diabetics and obesity-matched controls. Gut, 1979, 20(6), 518–522. [ 10.1136/gut.20.6.518] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pitt HA; King W III; Mann LL; Roslyn JJ; Berquist WE; Ament ME; DenBesten L Increased risk of cholelithiasis with prolonged total parenteral nutrition. Am. J. Surg, 1983, 145(1), 106–112. [ 10.1016/0002-9610(83)90175-7] [DOI] [PubMed] [Google Scholar]
- [56].Roslyn JJ; Pitt HA; Mann LL; Ament ME; DenBesten L Gallbladder disease in patients on long-term parenteral nutrition. Gastroenterology, 1983, 84(1), 148–154. [PubMed] [Google Scholar]
- [57].Ko CW; Beresford SA; Schulte SJ; Matsumoto AM; Lee SP Incidence, natural history, and risk factors for biliary sludge and stones during pregnancy. Hepatology, 2005, 41(2), 359–365.[ 10.1002/hep.20534] [DOI] [PubMed] [Google Scholar]
- [58].LaMorte WW; Schoetz DJ Jr; Birkett DH; Williams LF Jr. The role of the gallbladder in the pathogenesis of cholesterol gallstones. Gastroenterology, 1979, 77(3), 580–592. [ 10.1016/0016-5085(79)90027-1] [DOI] [PubMed] [Google Scholar]
- [59].Spengler U; Sackmann M; Sauerbruch T; Holl J; Paumgartner G Gallbladder motility before and after extracorporeal shock-wave lithotripsy. Gastroenterology, 1989, 96(3), 860–863. [ 10.1016/0016-5085(89)90913-X] [DOI] [PubMed] [Google Scholar]
- [60].Schneider H; Sänger P; Hanisch E In vitro effects of cholecystokinin fragments on human gallbladders. Evidence for an altered CCK-receptor structure in a subgroup of patients with gallstones. J. Hepatol, 1997, 26(5), 1063–1068.[ 10.1016/S0168-8278(97)80115-8] [DOI] [PubMed] [Google Scholar]
- [61].Yu P; Chen Q; Harnett KM; Amaral J; Biancani P; Behar J Direct G protein activation reverses impaired CCK signaling in human gallbladders with cholesterol stones. Am. J. Physiol, 1995, 269(5 Pt 1), G659–G665.[DOI: 10.1152/ajpgi.1995.269.5.G659] [DOI] [PubMed] [Google Scholar]
- [62].Yu P; Chen Q; Xiao Z; Harnett K; Biancani P; Behar J Signal transduction pathways mediating CCK-induced gallbladder muscle contraction. Am. J. Physiol, 1998, 275(2), G203–G211.[DOI: 10.1152/ajpgi.1998.275.2.G203] [DOI] [PubMed] [Google Scholar]
- [63].Yu P; De Petris G; Biancani P; Amaral J; Behar J Cholecystokinin-coupled intracellular signaling in human gallbladder muscle. Gastroenterology, 1994, 106(3), 763–770. [ 10.1016/0016-5085(94)90713-7] [DOI] [PubMed] [Google Scholar]
- [64].Yu P; Harnett KM; Biancani P; De Petris G; Behar J Interaction between signal transduction pathways contributing to gallbladder tonic contraction. Am. J. Physiol, 1993, 265(6 Pt 1), G1082–G1089. [DOI: 10.1152/ajpgi.1993.265.6.G1082] [DOI] [PubMed] [Google Scholar]
- [65].Wang HH; Portincasa P; Liu M; Tso P; Samuelson LC; Wang DQ Effect of gallbladder hypomotility on cholesterol crystallization and growth in CCK-deficient mice. Biochim. Biophys. Acta, 2010, 1801(2), 138–146. [ 10.1016/j.bbalip.2009.10.003] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Marks JW; Bonorris GG; Albers G; Schoenfield LJ The sequence of biliary events preceding the formation of gallstones in humans. Gastroenterology, 1992, 103(2), 566–570. [ 10.1016/0016-5085(92)90848-S] [DOI] [PubMed] [Google Scholar]
- [67].Shiffman ML; Sugerman HJ; Kellum JM; Brewer WH; Moore EW Gallstone formation after rapid weight loss: a prospective study in patients undergoing gastric bypass surgery for treatment of morbid obesity. Am. J. Gastroenterol, 1991, 86(8), 1000–1005. [PubMed] [Google Scholar]
- [68].Broomfield PH; Chopra R; Sheinbaum RC; Bonorris GG; Silverman A; Schoenfield LJ; Marks JW Effects of ursodeoxycholic acid and aspirin on the formation of lithogenic bile and gallstones during loss of weight. N. Engl. J. Med, 1988, 319(24), 1567–1572. [ 10.1056/NEJM198812153192403] [DOI] [PubMed] [Google Scholar]
- [69].Wang DQ; Paigen B; Carey MC Phenotypic characterization of Lith genes that determine susceptibility to cholesterol cholelithiasis in inbred mice: physical-chemistry of gallbladder bile. J. Lipid Res, 1997, 38(7), 1395–1411. [PubMed] [Google Scholar]
- [70].Lee SP; LaMont JT; Carey MC Role of gallbladder mucus hypersecretion in the evolution of cholesterol gallstones. J. Clin. Invest, 1981, 67(6), 1712–1723. [ 10.1172/JCI110209] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Holzbach RT; Corbusier C; Marsh M; Naito HK The process of cholesterol cholelithiasis induced by diet in the prairie dog: a physicochemical characterization. J. Lab. Clin. Med, 1976, 87(6), 987–998. [PubMed] [Google Scholar]
- [72].Mazer NA; Carey MC Quasi-elastic light-scattering studies of aqueous biliary lipid systems. Cholesterol solubilization and precipitation in model bile solutions. Biochemistry, 1983, 22(2), 426–442. [ 10.1021/bi00271a029] [DOI] [PubMed] [Google Scholar]
- [73].Halpern Z; Dudley MA; Lynn MP; Nader JM; Breuer AC; Holzbach RT Vesicle aggregation in model systems of supersaturated bile: relation to crystal nucleation and lipid composition of the vesicular phase. J. Lipid Res, 1986, 27(3), 295–306. [PubMed] [Google Scholar]
- [74].Halpern Z; Dudley MA; Kibe A; Lynn MP; Breuer AC; Holzbach RT Rapid vesicle formation and aggregation in abnormal human biles. A time-lapse video-enhanced contrast microscopy study. Gastroenterology, 1986, 90(4), 875–885. [ 10.1016/0016-5085(86)90863-2] [DOI] [PubMed] [Google Scholar]
- [75].Konikoff FM; Chung DS; Donovan JM; Small DM; Carey MC Filamentous, helical, and tubular microstructures during cholesterol crystallization from bile. Evidence that cholesterol does not nucleate classic monohydrate plates. J. Clin. Invest, 1992, 90(3), 1155–1160. [ 10.1172/JCI115935] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang DQ; Carey MC Complete mapping of crystallization pathways during cholesterol precipitation from model bile: influence of physical-chemical variables of pathophysiologic relevance and identification of a stable liquid crystalline state in cold, dilute and hydrophilic bile salt-containing systems. J. Lipid Res, 1996, 37(3), 606–630. [PubMed] [Google Scholar]
- [77].Wang DQ; Carey MC Characterization of crystallization pathways during cholesterol precipitation from human gallbladder biles: identical pathways to corresponding model biles with three predominating sequences. J. Lipid Res, 1996, 37(12), 2539–2549. [PubMed] [Google Scholar]
- [78].Neiderhiser DH; Harmon CK; Roth HP Absorption of cholesterol by the gallbladder. J. Lipid Res, 1976, 17(2), 117–124. [PubMed] [Google Scholar]
- [79].Ginanni Corradini S; Ripani C; Della Guardia P; Gio-vannelli L; Elisei W; Cantafora A; Codacci Pisanelli M; Tebala GD; Nuzzo G; Corsi A; Attili AF; Capo-caccia L; Ziparo V The human gallbladder increases cholesterol solubility in bile by differential lipid absorption: a study using a new in vitro model of isolated intra-arterially perfused gallbladder. Hepatology, 1998, 28(2), 314–322. [ 10.1002/hep.510280205] [DOI] [PubMed] [Google Scholar]
- [80].Lee SP; Nicholls JF Nature and composition of biliary sludge. Gastroenterology, 1986, 90(3), 677–686. [ 10.1016/0016-5085(86)91123-6] [DOI] [PubMed] [Google Scholar]
- [81].Lee SP; Maher K; Nicholls JF Origin and fate of biliary sludge. Gastroenterology, 1988, 94(1), 170–176. [ 10.1016/0016-5085(88)90626-9] [DOI] [PubMed] [Google Scholar]
- [82].Levy PF; Smith BF; LaMont JT Human gallbladder mucin accelerates nucleation of cholesterol in artificial bile. Gastroenterology, 1984, 87(2), 270–275. [ 10.1016/0016-5085(84)90700-5] [DOI] [PubMed] [Google Scholar]
- [83].Afdhal NH; Niu N; Gantz D; Small DM; Smith BF Bovine gallbladder mucin accelerates cholesterol monohydrate crystal growth in model bile. Gastroenterology, 1993, 104(5), 1515–1523. [ 10.1016/0016-5085(93)90364-I] [DOI] [PubMed] [Google Scholar]
- [84].Pemsingh RS; MacPherson BR; Scott GW Mucus hypersecretion in the gallbladder epithelium of ground squirrels fed a lithogenic diet for the induction of cholesterol gallstones. Hepatology, 1987, 7(6), 1267–1271. [ 10.1002/hep.1840070615] [DOI] [PubMed] [Google Scholar]
- [85].Womack NA The development of gallstones. Surg. Gynecol. Obstet, 1971, 133(6), 937–945. [PubMed] [Google Scholar]
- [86].Pearson JP; Foster SN Mucus glycoprotein content of human cholesterol gallstones. Digestion, 1987, 36(3), 132–140. [ 10.1159/000199410] [DOI] [PubMed] [Google Scholar]
- [87].Toor EW; Evans DF; Cussler EL Cholesterol monohydrate growth in model bile solutions. Proc. Natl. Acad. Sci. USA, 1978, 75(12), 6230–6234. [ 10.1073/pnas.75.12.6230] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Portincasa P; Venneman NG; Moschetta A; van den Berg A; Palasciano G; vanBerge-Henegouwen GP; van Erpecum KJ Quantitation of cholesterol crystallization from supersaturated model bile. J. Lipid Res, 2002, 43(4), 604–610. [PubMed] [Google Scholar]
- [89].Sitzmann JV; Pitt HA; Steinborn PA; Pasha ZR; Sanders RC Cholecystokinin prevents parenteral nutrition induced biliary sludge in humans. Surg. Gynecol. Obstet, 1990, 170(1), 25–31. [PubMed] [Google Scholar]
- [90].Kopin AS; Mathes WF; McBride EW; Nguyen M; Al-Haider W; Schmitz F; Bonner-Weir S; Kanarek R; Beinborn M The cholecystokinin-A receptor mediates inhibition of food intake yet is not essential for the maintenance of body weight. J. Clin. Invest, 1999, 103(3), 383–391.[ 10.1172/JCI4901] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wang DQ; Lammert F; Cohen DE; Paigen B; Carey MC Cholic acid aids absorption, biliary secretion, and phase transitions of cholesterol in murine cholelithogenesis. Am. J. Physiol, 1999, 276(3), G751–G760. [DOI] [PubMed] [Google Scholar]
- [92].Wang DQ; Cohen DE Absorption and Excretion of Cholesterol and Other Sterols. In: Lipidology in the Treatment and Prevention of Cardiovascular Disease (Clinical Lipidology: A Companion to Braunwald’s Heart Disease);Ballantyne CM, Ed.; Elsevier Inc.: Philadelphia, 2008; pp. 26–44. [ 10.1016/B978-141605469-6.50007-X] [DOI] [Google Scholar]
- [93].Wang TY; Liu M; Portincasa P; Wang DQ New insights into the molecular mechanism of intestinal fatty acid absorption. Eur. J. Clin. Invest, 2013, 43(11), 1203–1223. [ 10.1111/eci.12161] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Ponz de Leon M; Iori R; Barbolini G; Pompei G; Zaniol P; Carulli N Influence of small-bowel transit time on dietary cholesterol absorption in human beings. N. Engl. J. Med, 1982, 307(2), 102–103. [ 10.1056/NEJM198207083070207] [DOI] [PubMed] [Google Scholar]
- [95].Wang DQ; Carey MC Measurement of intestinal cholesterol absorption by plasma and fecal dual-isotope ratio, mass balance, and lymph fistula methods in the mouse: an analysis of direct versus indirect methodologies. J. Lipid Res, 2003, 44(5), 1042–1059. [ 10.1194/jlr.D200041-JLR200] [DOI] [PubMed] [Google Scholar]
- [96].Wang DQ; Paigen B; Carey MC Genetic factors at the enterocyte level account for variations in intestinal cholesterol absorption efficiency among inbred strains of mice. J. Lipid Res, 2001, 42(11), 1820–1830. [PubMed] [Google Scholar]
- [97].Duan LP; Wang HH; Wang DQ Cholesterol absorption is mainly regulated by the jejunal and ileal ATP-binding cassette sterol efflux transporters ABCg5 and ABCg8 in mice. J. Lipid Res, 2004, 45(7), 1312–1323. [ 10.1194/jlr.M400030-JLR200] [DOI] [PubMed] [Google Scholar]
- [98].Wang DQ; Zhang L; Wang HH High cholesterol absorption efficiency and rapid biliary secretion of chylomicron remnant cholesterol enhance cholelithogenesis in gallstone-susceptible mice. Biochim. Biophys. Acta, 2005, 1733(1), 90–99. [ 10.1016/j.bbalip.2004.12.005] [DOI] [PubMed] [Google Scholar]
- [99].Heaton KW; Emmett PM; Symes CL; Braddon FE An explanation for gallstones in normal-weight women: slow intestinal transit. Lancet, 1993, 341(8836), 8–10. [ 10.1016/0140-6736(93)92479-D] [DOI] [PubMed] [Google Scholar]
- [100].Marcus SN; Heaton KW Intestinal transit, deoxycholic acid and the cholesterol saturation of bile-three inter-related factors. Gut, 1986, 27(5), 550–558. [ 10.1136/gut.27.5.550] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Shoda J; He BF; Tanaka N; Matsuzaki Y; Osuga T; Yamamori S; Miyazaki H; Sjövall J Increase of deoxycholate in supersaturated bile of patients with cholesterol gallstone disease and its correlation with de novo syntheses of cholesterol and bile acids in liver, gallbladder emptying, and small intestinal transit. Hepatology, 1995, 21(5), 1291–1302. [ 10.1016/0270-9139(95)90050-0] [DOI] [PubMed] [Google Scholar]
- [102].Thomas LA; Veysey MJ; Murphy GM; Russell-Jones D; French GL; Wass JA; Dowling RH Octreotide induced prolongation of colonic transit increases faecal anaerobic bacteria, bile acid metabolising enzymes, and serum deoxycholic acid in patients with acromegaly. Gut, 2005, 54(5), 630–635. [ 10.1136/gut.2003.028431] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Azzaroli F; Mazzella G; Mazzeo C; Simoni P; Festi D; Colecchia A; Montagnani M; Martino C; Villanova N; Roda A; Roda E Sluggish small bowel motility is involved in determining increased biliary deoxycholic acid in cholesterol gallstone patients. Am. J. Gastroenterol, 1999, 94(9), 2453–2459. [ 10.1111/j.1572-0241.1999.01375.x] [DOI] [PubMed] [Google Scholar]
- [104].Pereira SP; Bain IM; Kumar D; Dowling RH Bile composition in inflammatory bowel disease: ileal disease and colectomy, but not colitis, induce lithogenic bile. Aliment. Pharmacol. Ther, 2003, 17(7), 923–933. [ 10.1046/j.1365-2036.2003.01529.x] [DOI] [PubMed] [Google Scholar]
- [105].Low-Beer TS; Nutter S Colonic bacterial activity, biliary cholesterol saturation, and pathogenesis of gallstones. Lancet, 1978, 2(8099), 1063–1065. [ 10.1016/S0140-6736(78)91800-7] [DOI] [PubMed] [Google Scholar]
- [106].Thomas LA; Veysey MJ; Bathgate T; King A; French G; Smeeton NC; Murphy GM; Dowling RH Mechanism for the transit-induced increase in colonic deoxycholic acid formation in cholesterol cholelithiasis. Gastroenterology, 2000, 119(3), 806–815. [ 10.1053/gast.2000.16495] [DOI] [PubMed] [Google Scholar]
- [107].Wang DQ; Tazuma S; Cohen DE; Carey MC Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol, 2003, 285(3), G494–G502. [ 10.1152/ajpgi.00156.2003] [DOI] [PubMed] [Google Scholar]
- [108].Chen Q; Chitinavis V; Xiao Z; Yu P; Oh S; Biancani P; Behar J Impaired G protein function in gallbladder muscle from progesterone-treated guinea pigs. Am. J. Physiol, 1998, 274(2), G283–G289. [DOI] [PubMed] [Google Scholar]
- [109].Chen Q; Amaral J; Biancani P; Behar J Excess membrane cholesterol alters human gallbladder muscle contractility and membrane fluidity. Gastroenterology, 1999, 116(3), 678–685. [ 10.1016/S0016-5085(99)70190-3] [DOI] [PubMed] [Google Scholar]
- [110].Suzuki S; Takiguchi S; Sato N; Kanai S; Kawanami T; Yoshida Y; Miyasaka K; Takata Y; Funakoshi A; Noda T Importance of CCK-A receptor for gallbladder contraction and pancreatic secretion: a study in CCK-A receptor knockout mice. Jpn. J. Physiol, 2001, 51(5), 585–590. [ 10.2170/jjphysiol.51.585] [DOI] [PubMed] [Google Scholar]
- [111].Shaffer EA Abnormalities in gallbladder function in cholesterol gallstone disease: bile and blood, mucosa and muscle--the list lengthens. Gastroenterology, 1992, 102(5), 1808–1812. [ 10.1016/0016-5085(92)91749-T] [DOI] [PubMed] [Google Scholar]
- [112].Wang DQ; Afdhal NH Gallstone Disease. In: Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Feldman M; Friedman LS; Brandt L; Ed.; Elsevier Inc.; Philadelphia: 2014; pp. 1100–1133. [Google Scholar]
- [113].Green PH; Cellier C Celiac disease. N. Engl. J. Med, 2007, 357(17), 1731–1743. [ 10.1056/NEJMra071600] [DOI] [PubMed] [Google Scholar]
- [114].Calam J; Ellis A; Dockray GJ Identification and measurement of molecular variants of cholecystokinin in duodenal mucosa and plasma. Diminished concentrations in patients with celiac disease. J. Clin. Invest, 1982, 69(1), 218–225. [ 10.1172/JCI110433] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Masclee AA; Jansen JB; Driessen WM; Geuskens LM; Lamers CB Gallbladder sensitivity to cholecystokinin in coeliac disease. Correlation of gallbladder contraction with plasma cholecystokinin-like immunoreactivity during infusion of cerulein. Scand. J. Gastroenterol, 1991, 26(12), 1279–1284. [ 10.3109/00365529108998625] [DOI] [PubMed] [Google Scholar]
- [116].Kelly CP Celiac Disease. In: Sleisenger and Fordtran’s Gastrointestinal and Liver Disease; Feldman M; Friedman LS; Brandt LJ, Ed.; Elsevier Inc.:2014, pp.1849–1872. [Google Scholar]
- [117].Miller LJ; Holicky EL; Ulrich CD; Wieben ED Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology, 1995, 109(4), 1375–1380. [ 10.1016/0016-5085(95)90601-0] [DOI] [PubMed] [Google Scholar]
- [118].Nardone G; Ferber IA; Miller LJ The integrity of the cholecystokinin receptor gene in gallbladder disease and obesity. Hepatology, 1995, 22(6), 1751–1753. [PubMed] [Google Scholar]
- [119].Lammert F; Gurusamy K; Ko CW; Miquel JF; Méndez-Sánchez N; Portincasa P; van Erpecum KJ; van Laarhoven CJ; Wang DQ Gallstones. Nat. Rev. Dis. Primers, 2016, 2, 16024. [ 10.1038/nrdp.2016.24] [DOI] [PubMed] [Google Scholar]