

Abstract
Cardiovascular disease is characterized by lipid accumulation, inflammatory response, cell death, and fibrosis in the arterial wall and is the leading cause of morbidity and mortality worldwide. Cholesterol gallstone disease is caused by complex genetic and environmental factors and is one of the most prevalent and costly digestive diseases in the USA and Europe. Although sitosterolemia is a rare inherited lipid storage disease, its genetic studies led to identification of the sterol efflux transporters ABCG5/G8 that are located on chromosome 2p21 in humans and chromosome 17 in mice. Human and animal studies have clearly demonstrated that ABCG5/G8 play a critical role in regulating hepatic secretion and intestinal absorption of cholesterol and plant sterols. Sitosterolemia is caused by a mutation in either the ABCG5 or the ABCG8 gene alone, but not in both simultaneously. Polymorphisms in the ABCG5/G8 genes are associated with abnormal plasma cholesterol metabolism and may play a key role in the genetic determination of plasma cholesterol concentrations. Moreover, ABCG5/G8 is a new gallstone gene, LITH9. Gallstone-associated variants in ABCG5/G8 are involved in the pathogenesis of cholesterol gallstones in European, Asian, and South American populations. In this chapter, we summarize the latest advances in the critical role of the sterol efflux transporters ABCG5/G8 in regulating hepatic secretion of biliary cholesterol, intestinal absorption of cholesterol and plant sterols, the classical reverse cholesterol transport, and the newly established transintestinal cholesterol excretion, as well as in the pathogenesis and pathophysiology of ABCG5/G8-related metabolic diseases such as sitosterolemia, cardiovascular disease, and cholesterol gallstone disease.
Keywords: Bile flow, Bile salts, Biliary lipid secretion, Gallstones, Cardiovascular disease, Cholesterol-lowering drugs, Coronary heart disease, Intestinal lipid absorption, Lith gene, Lithogenic bile, Reverse cholesterol transport, Statins, Stroke
8.1. Introduction
It is well-known that cholesterol is essential for all cells in the body because it is widely used as a key structural component for cell membranes and as a central substrate for the synthesis of other steroids, including bile salts, vitamin D, and sex hormones such as estradiol, progesterone, androsterone, and testosterone, as well as adrenocortical hormones such as cortisone and aldosterone [1]. It has been found that the liver and small intestine are two major organs for cholesterol biosynthesis. Furthermore, high cholesterol biosynthesis in the liver leads to more very-low-density lipoprotein (VLDL) secreted into plasma, which has a significant impact on plasma total and low-density lipoprotein (LDL) cholesterol concentrations. High dietary cholesterol also could contribute an increase in plasma cholesterol concentrations in most individuals. Elevated plasma total and LDL cholesterol levels are an important risk factor for the development of cardiovascular disease in humans [2].
Clinical studies and epidemiological investigations have clearly demonstrated that cardiovascular disease is a leading cause of death and disability not only in the USA but also in European and Asian countries. Therefore, the National Cholesterol Education Program Adult Treatment Panel III guidelines [3] along with the 2012 update and the American Heart Association and American College of Cardiology recommendations [4–7] have proposed a much lower target for plasma LDL cholesterol concentrations (i.e., <100 mg/dL) for individuals at high risk for adverse cardiovascular events. As a result, the total number of patients requiring more aggressive cholesterol-lowering treatment has significantly increased. Because the cholesterol carried in LDL particles is derived mainly from both de novo biosynthesis in the liver and intestinal absorption from the diet, a better understanding of the cellular and molecular mechanisms of elucidating the regulation of hepatic cholesterol biosynthesis and intestinal cholesterol absorption should lead to novel approaches to the treatment and the prevention of cardiovascular disease. Despite significant advances in the treatment of cardiovascular disease, a large number of residual risks in these patients are still being fully studied. Based on the genetic studies on patients with sitosterolemia [8–10], the ATP-binding cassette (ABC) sterol efflux transporters ABCG5 and ABCG8, encoded by the ABCG5 and ABCG8 genes, have been identified, which are located primarily on the canalicular membrane of hepatocytes and the apical membrane of enterocytes and play a key role in hepatic secretion and intestinal absorption of cholesterol and plant sterols [9, 11–13].
Cholesterol gallstone disease is caused by complex genetic and environmental factors. It is one of the most common and costly digestive diseases worldwide. In Western countries, 15–20% of the populations suffer from gallstones. At least 20 million Americans (~12% of adults) have gallstones, leading to a considerable financial and social burden in the USA [14–19]. The prevalence of gallstones appears to be rising due to the epidemic of obesity that is associated with insulin resistance and the metabolic syndrome [16]. It is estimated that there are approximately 1 million new cases diagnosed each year [20–22]. Although most patients with gallstones are asymptomatic, one third of patients eventually develop clinical symptoms with or without complications [20]. The estimated 1,000,000 cholecystectomies are performed for gallstone disease every year. The annual medical cost of treating gallstones exceeded $6 billion in 2004 and even higher in 2019 [23]. The burden of gallstone disease is exacerbated by the fact that laparoscopic cholecystectomy remains the standard treatment for symptomatic gallstones worldwide [24]. In addition, unavoidable complications of gallstones result in 3000 deaths (~0.12% of all deaths) per year in the USA [14]. In general, persons with gallstone disease have increased overall, cardiovascular disease, and cancer mortality [18]. Most importantly, the prevalence of gallstones is increasing year by year because of the epidemic of obesity that is associated with insulin resistance, hyperlipidemia, and the metabolic syndrome.
To reduce the morbidity, mortality, and costs of health care associated with this disease, it is imperative to decipher the pathophysiology of cholesterol gallstone disease. This would facilitate the development of a novel, effective, and noninvasive therapy for patients with gallstone disease. Compelling evidence from the physical-chemical, pathophysiological, and genetic studies shows that cholesterol gallstone disease is determined by multiple Lith genes, which is a dominant trait. The principal pathogenic factor is persistent hepatic hypersecretion of cholesterol into bile, thereby contributing to the formation of cholesterol-supersaturated gallbladder bile. Clinical studies have found that cholesterol-supersaturated bile is an essential prerequisite for the precipitation of solid cholesterol monohydrate crystals and the formation of cholesterol gallstones [23]. Although it has been established that ABCG5/G8 play a key role in hepatic secretion and intestinal absorption of cholesterol and plant sterols [9, 11–13] and in the pathogenesis of sitosterolemia in patients [8–10], the Abcg5/g8 has also been identified as the mouse gallstone gene, Lith9, on chromosome 17 by quantitative trait locus (QTL) linkage analysis [25–28]. Subsequently, the ABCG5/G8 was found to be associated with cholesterol gallstone disease in patients, and two gallstone-associated variants in ABCG5/G8 (ABCG5-R50C and ABCG8-D19H) were identified not only in Germans and Chileans but also in Chinese and Indians [29–34]. These findings indicate the importance of ABCG5/G8 as LITH9 in the pathogenesis of gallstones not only in mice but also in humans [14].
In this chapter, we summarize the latest advances in the critical role of the sterol efflux transporters ABCG5/G8 in regulating hepatic secretion of biliary cholesterol, intestinal absorption of cholesterol and plant sterols, and reverse cholesterol transport, as well as in the pathogenesis and pathophysiology of ABCG5/G8-related metabolic diseases such as sitosterolemia, cardiovascular disease, and cholesterol gallstone disease.
8.2. Chemistry of Cholesterol and Plant Sterols
By definition, a steroid is a biologically active organic compound with four rings arranged in a specific molecular configuration, including the sterols, hormones (such as anabolic steroids or corticosteroids), and glycosides. The steroid core structure is typically composed of 17 carbon atoms, bonded in 4 “fused” rings: 3 6-member cyclohexane rings, called the A, B, and C rings, and 1 5-member cyclopentane ring, named the D ring [1]. It is well-known that the basic chemical structure of steroids has a nucleus containing the four-ringed carbon skeleton of cyclopentenophenanthrene and the numbering of the carbon atoms in steroids [1]. Furthermore, sterols are various solid steroid alcohols that are widely distributed in human, animal, and plant lipids. It is often called cholesterol in humans and animals, as well as phytosterols, or plant sterols, in plants.
As shown in Fig. 8.1, the basic chemical structure of the cholesterol molecule includes (i) the perhydrocyclopentenophenanthrene nucleus with its four fused rings, (ii) a single hydroxyl group at C-3, (iii) a double bond between C-5 and C-6, (iv) an eight-membered branched hydrocarbon chain attached to C-17 in the D ring, and (v) a methyl group (C-19) attached to C-10, and a second methyl group (C-18) attached to C-13. Furthermore, in the esterified form, a long-chain fatty acid, usually linoleic acid, is attached by ester linkage to the hydroxyl group at C-3 in the A ring. Similar to cholesterol in humans and animals, phytosterols, which encompass plant sterols and stanols, are phytosteroids, which occur in plants and vary only in carbon side chains and/or presence or absence of a double bond. Stanols are saturated sterols, having no double bonds in the sterol ring structure (Fig. 8.1).
Fig. 8.1.
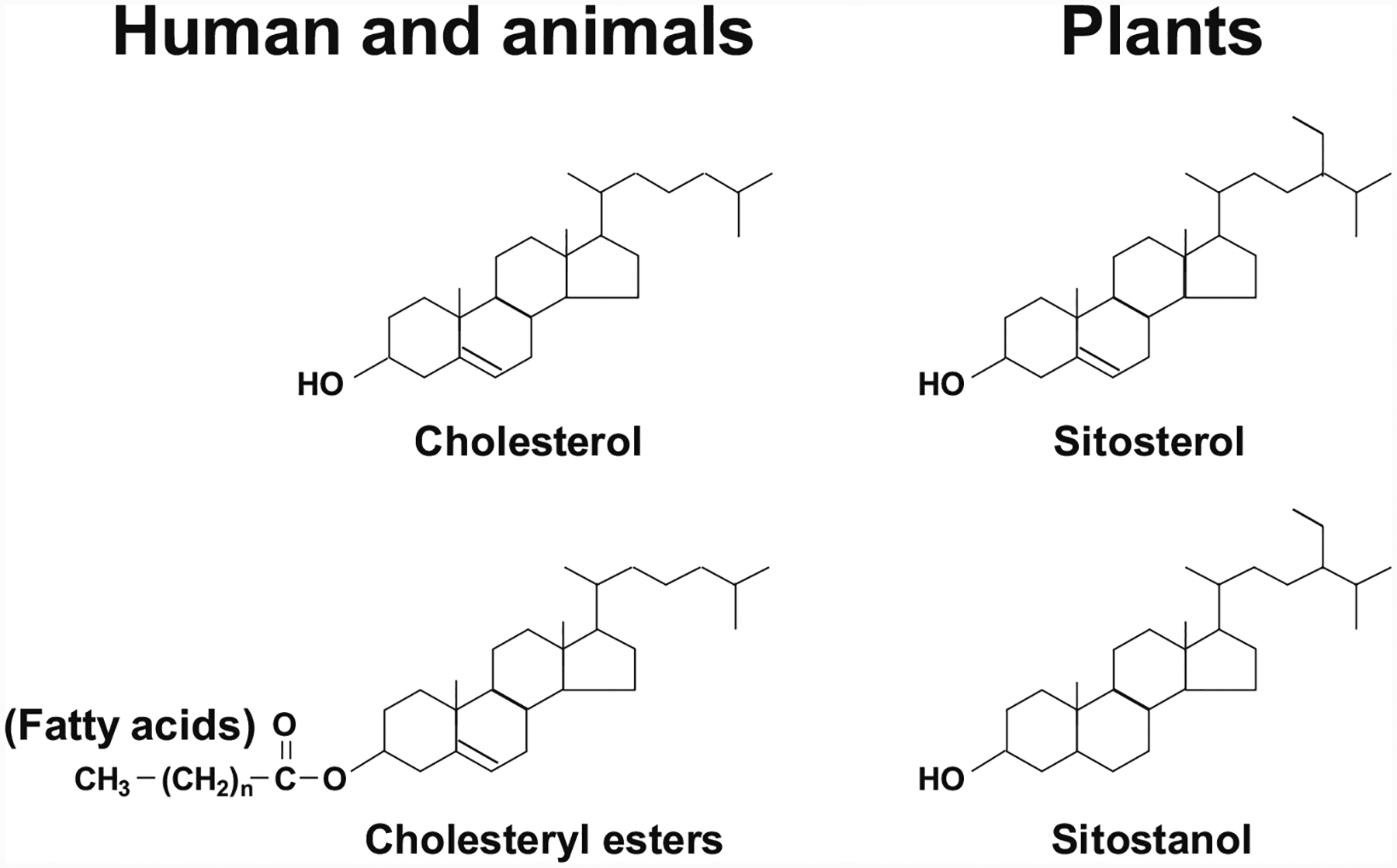
All these substances have a nucleus containing the four-ringed carbon skeleton of cyclopentenophenanthrene and are known as steroids. The sterols are one of the steroids and they are widely distributed in humans, animals, and plants. It is often called cholesterol in humans and animals and phytosterols (also called plant sterols) in plants. Notably, the general structural formula for the sterols includes the designation of the four rings with a side chain at C-17 and two methyl groups at C-18 and C-19. Cholesterol is one of the most abundant steroids in bile. Its hydroxyl group on the third carbon can react with the COOH group of a fatty acid molecule to form a cholesteryl ester. Plant sterols (e.g., β-sitosterol and β-sitostanol) are naturally occurring. Their chemical structures are very similar to cholesterol but with structural modifications of the side chain. In addition, stanols are saturated sterols, having no double bonds in the sterol ring structure, e.g., β-sitostanol
8.3. Discovery of the Sterol Efflux Transporters ABCG5/G8
The ATP-binding cassette (ABC) transporters are a family of large proteins in cell membranes. Using the energy from the ATP hydrolysis, these ABC transporters can make an active transport of various compounds crossing the cell membranes against steep concentration gradients [35]. Hitherto, 48 ABC genes have been found in the human genome [36]. The major physiological functions of these ABC transporters are involved in an active transport of a wide variety of substrates across extracellular and intracellular membranes, which include lipids, amino acids, sugars, vitamins, metals, drugs (xenotoxins) and drug conjugates, and peptides for antigen presentation or other purposes [37]. Of the 48 human ABC proteins, a significant number are known to mediate the extrusion of lipids from membranes or the flipping of membrane lipids across the bilayer to generate and maintain membrane lipid asymmetry [38]. For example, the bile salt export pump, ABCB11, is responsible for hepatic secretion of biliary bile salts. Other members of the subfamily of ABC transporters such as ABCB4, ABCG1, ABCC2, and ABCA1 implicated in lipid transport play important roles in diverse biological processes involving hepatic phospholipid secretion, cell signaling, membrane lipid asymmetry, removal of potentially toxic compounds and metabolites, and apoptosis [39]. The importance of the ABC lipid transporters in cell physiology is revealed based on the finding that mutations in the genes encoding many of these proteins are responsible for severe inherited diseases. At least 14 ABC genes have been found to be associated with a defined human disease due to genetic defects [40]. Especially, several ABC transporters are involved in inborn errors relevant to metabolic disorders [41]. For example, Tangier disease is caused by mutations in the ABCA1 gene, which is associated with defective efflux of cholesterol and phosphatidylcholine from the plasma membrane to the lipid acceptor protein, apolipoprotein A-I (apoA-I) [42]. In addition, relative phospholipid deficiency is caused mostly by missense mutations in the ABC subfamily B member 4 (ABCB4) gene, also known as the multidrug resistance protein 3 (MDR3) gene. The ABCB4 gene encodes for an energy-dependent phospholipid efflux translocator at the canalicular membrane of the hepatocytes, which facilitates the transport of phospholipids from the inner to the outer canalicular membrane of hepatocytes for hepatic secretion into canalicular bile [43–46].
The half-transporters, ABCG5 and ABCGG8, are found to heterodimerize into a functional transport. The genes, ABCG5 and ABCGG8, encoding these transporters are highly expressed in the liver and small intestine of both humans and mice [47–49]. The ABCG5/G8 genes are located on chromosome 2p21 in humans and chromosome 17 in mice. The two proteins form heterodimers in the endoplasmic reticulum and then traffic to the canalicular membrane of hepatocytes and the apical membrane of enterocytes where they transport neutral sterols into bile and into the gut lumen, respectively [48]. Further cellular and molecular studies found that ABCG5/G8 play a critical role in regulating hepatic secretion and intestinal absorption of cholesterol and plant sterols. Mutations in either ABCG5 or ABCG8 cause sitosterolemia [8–10], which is an autosomal recessive disorder characterized by phytosterolemia, hypercholesterolemia, and premature coronary heart disease [50].
8.4. Physiological Functions of ABCG5/G8
Many studies have found that almost all the cells in the body need a continuous supply of cholesterol. As a result, a series of complex and sophisticated transport, biosynthetic, and regulatory mechanisms have developed in humans and animals [51, 52]. Under normal physiological conditions, cholesterol is obtained from the intestinal absorption of dietary and biliary cholesterol, as well as the newly synthesized de novo from acetyl CoA in the body. However, because human and animal tissues do not possess enzymes that can degrade the ring structure of this sterol, cholesterol cannot be metabolized to CO2 and water in the body. Therefore, to prevent a potentially hazardous accumulation of cholesterol in the body, excess cholesterol must be metabolized to other compounds and/or excreted in the feces. This challenging task is usually accomplished by chemical modifications of certain substituent groups on the hydrocarbon tail or on the ring structure of the cholesterol molecule. Subsequently, excess cholesterol is excreted from the body essentially either as the unaltered molecule (i.e., in both unesterified and esterified forms) or after structural modifications to other sterol products such as bile salts and steroid hormones.
It has been recognized that the cholesterol molecule is a key lipid component of mostly all the cell membranes, as well as is the precursor of various steroid hormones such as the sex hormones (estrogen, progesterone, and testosterone) and corticosteroids (cortisone, corticosterone, cortisol, and aldosterone) [53–56]. Moreover, during the biosynthesis of bile salts in the liver, cholesterol is mainly converted into bile salts. As a result, large amounts of biliary cholesterol and bile salts are simultaneously secreted to bile. This dramatically reduces plasma cholesterol concentrations and greatly enhances removal of excess amounts of cholesterol from the body.
Because cholesterol is virtually insoluble in an aqueous solution, e.g., water, the mechanisms for cholesterol solubilization in plasma and bile are complex. It is well-known that cholesterol is mainly carried by lipoproteins in plasma and by micelles and vesicles in bile. If excess cholesterol is accumulated in the artery wall, it leads to atherosclerosis and causes cardiovascular disease. If excess cholesterol cannot be dissolved in bile by bile salts and/or phospholipids, it precipitates as plate-like solid cholesterol monohydrate crystals, thus leading to the formation of cholesterol gallstones in the gallbladder and/or the bile duct.
Based on animal studies [57–59], several pathways have been identified for elucidating the net flow of cholesterol through the major tissue compartments of the human, which explains how the cholesterol pool in the body is kept essentially constant. New cholesterol is added to the pool mainly from two sources: the absorbed cholesterol from dietary and biliary origins across the epithelial cells of small intestinal tract and the newly synthesized cholesterol in a variety of different tissues in the body, predominantly in the liver and small intestine. The availability of dietary and biliary cholesterol to the body varies tremendously in different individuals, and the consumed amounts of dietary cholesterol also vary dramatically from day to day [57–68]. The total amount of cholesterol from the small intestine to the body also depends mainly on the absorption efficiency of intestinal cholesterol and the amount of cholesterol that is consumed daily. Additionally, bile cholesterol is reabsorbed by the small intestine, which provides about two thirds of the total daily amount of cholesterol originating from the intestine [2]. The rate of cholesterol biosynthesis in the liver varies extremely in different individuals. The absorbed cholesterol from the small intestine can regulate hepatic cholesterol synthesis, depending on the amount of daily food intake, through a negative regulatory mechanism.
Taken together, the regulatory mechanisms on cholesterol metabolism must be operative, which can accurately and sophisticatedly adjust the rate of cholesterol biosynthesis in the body and the rate of cholesterol excretion from the body to accommodate the varying amounts of cholesterol that are absorbed by the small intestine at different times. Basically, these regulatory mechanisms on cholesterol metabolism work well. Therefore, there is little net accumulation of excess cholesterol in the body, and yet sufficient cholesterol is always available to meet the metabolic needs of the various cells. However, delicate imbalances lead to an increase in plasma cholesterol concentration and/or hepatic cholesterol hypersecretion in humans [69–72]. In the cardiovascular system, this metabolic abnormality often causes an accumulation of excess cholesteryl esters within the wall of arteries, leading to clinically apparent atherosclerosis mainly in the heart and brain and causing cardiovascular disease [73–80]. In the biliary system, when an imbalance of cholesterol metabolism in bile occurs, gallbladder bile becomes supersaturated with cholesterol, thereby promoting the precipitation of plate-like solid cholesterol monohydrate crystals and, eventually, leading to clinically apparent cholesterol gallstone formation [81–91].
Because the sterol efflux transporters ABCG5/G8 play a key role in the regulation of cholesterol metabolism in bile and plasma and in the maintenance of cholesterol homeostasis in the body, we will discuss the regulatory mechanisms of ABCG5/G8 in (i) hepatic secretion of biliary cholesterol; (ii) intestinal absorption of cholesterol and plant sterols; (iii) reverse cholesterol transport; and (iv) transintestinal cholesterol excretion.
(a). Hepatic secretion of biliary cholesterol
Bile is composed mainly of water, organic solutes, and inorganic electrolytes. Cholesterol, phospholipids, and bile salts are three major lipid species in bile, which account for approximate 99% of total lipids by weight. Bilirubin is a minor solute and represents less than 1% of biliary lipids. Hepatic secretion of biliary cholesterol and its degradation product, bile salts, represents the major route for elimination of cholesterol from the liver and, eventually, from the body. After entering the intestinal lumen, bile salts play an important role in the emulsification of dietary lipids and the breakdown of large lipid globules into a suspension of droplets for intestinal absorption. In addition, bile salts promote the intestinal absorption of cholesterol, fatty acids, fat-soluble vitamins (A, D, E, and K), and certain drugs.
Hepatic secretion of biliary lipids is determined by four members of the family of ABC transporters on the canalicular membrane of hepatocytes: ABCB4 for phospholipids, ABCB11 for bile salts, ABCG5/G8 for cholesterol, and ABCC2 for bilirubin (Fig. 8.2). Most, if not all, bile salts enter the canalicular space as monomers, whereas biliary phospholipids and cholesterol could enter together as unilamellar vesicles. Bile salts play a critical role in promoting hepatic secretion of vesicles that are always found in hepatic bile by quasi-elastic light-scattering spectroscopy and electronic microscopy with rapid fixation techniques. These imaging studies have provided clear morphologic evidence of the vesicle formation and secretion on the outer surface of the canalicular membrane of hepatocytes during the bile formation.
Fig. 8.2.
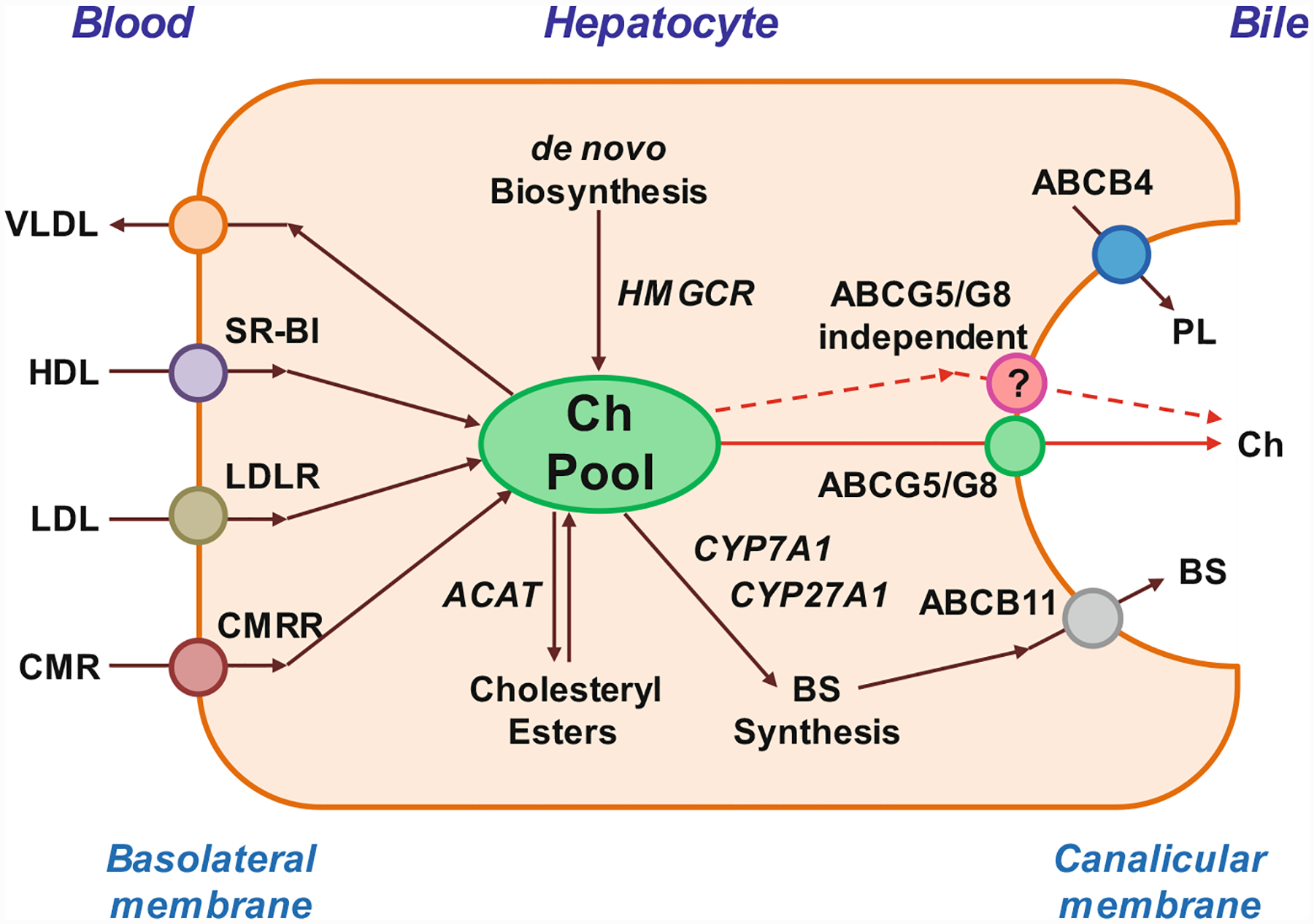
This diagram of the hepatocyte shows the ABCG5/G8-dependent (red solid lines) and the ABCG5/G8-independent (red dashed lines) pathways for biliary cholesterol (Ch) secretion, as well as the ABCB4 and ABCB11 transporters for biliary phospholipid (PL) and bile salt (BS) secretion, respectively. Abbreviations: ABC ATP-binding cassette (transporter), ACAT acyl-coenzyme A:cholesterol acyltransferase, CMR chylomicron remnants, CMRR CMR receptor, CYP7A1 cholesterol 7-α-hydroxylase, CYP27A1 sterol 27-hydroxylase, HDL high-density lipoprotein, HMGCR 3-hydroxy-3-methylglutaryl-coenzyme A reductase, LDL low-density lipoprotein, LDLR LDL receptor, SR-BI scavenger receptor class B type I, i.e., HDL receptor, VLDL very-low-density lipoprotein
Although biliary phospholipids are derived possibly from the cell membranes of hepatocytes, their compositions differ significantly. The cell membranes of hepatocytes contain a large amount of phosphatidylcholine (i.e., lecithin), phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine, and sphingomyelin. The major source of the phosphatidylcholine molecules destined for hepatic secretion into bile is its synthesis in the liver. However, a fraction of biliary phosphatidylcholines may also originate from the surface phospholipid coat of HDL particles. In the early 1990s, it was first reported that hepatic phospholipid secretion is a protein-mediated process because deletion of the Abcb4 gene completely inhibits hepatic phospholipid secretion in mice [43]. This important study provided clear evidence for the first time that a P-glycoprotein member of the multidrug resistance gene family, ABCB4, plays a key role in regulating hepatic secretion of biliary phospholipids [43]. Studies from cryoelectron microscopy with rapid fixation techniques found that the knockout of the Abcb4 gene in mice dramatically reduces the formation and secretion of vesicles on the outer surface of the canalicular membrane of hepatocytes [92–94]. It is highly likely that ABCB4 could be responsible for the translocation or “flip” of phosphatidylcholines from the endoplasmic (inner) to ectoplasmic (outer) leaflet of the canalicular membrane bilayer, and the action of ABCB4 may form phosphatidylcholine-rich microdomains within the outer membrane leaflet [95–99]. Notably, the ectoplasmic leaflet of the canalicular membrane is composed mainly with cholesterol and sphingomyelin. However, such chemical structure is quite resistant to penetration by bile salts. Thus, bile salts may interact with the canalicular membrane of hepatocytes and partition preferentially into these areas, enhancing biliary secretion of phosphatidylcholine-rich vesicles by destabilizing the membrane because of detergent-like properties of bile salts. Furthermore, mutations in the ABCB4 gene are the molecular defect underlying progressive familial intrahepatic cholestasis, type III in humans [99–104]. In addition, biliary phospholipids can dramatically solubilize excess cholesterol in bile through a vesicle mechanism. Low phospholipid-associated cholelithiasis is characterized mainly by the occurrence of gallbladder and intrahepatic microlithiasis in young adults associated with mutations in the ABCB4 gene [105–107]. To study the pathogenesis of low phospholipid-associated cholelithiasis, gallstone phenotypes have been systematically investigated in the ABCB4 knockout mouse model. It is interesting to find that even on the chow diet containing trace amounts of (<0.02%) cholesterol, ABCB4 knockout mice can spontaneously develop gallstones that are composed mainly of needle-shaped anhydrous cholesterol crystals [98]. These anhydrous cholesterol crystals and gallstones are formed in phospholipid-deficient gallbladder bile with its relative biliary lipid composition that is located in the far left crystallization region of the phase diagram [108]. These studies support the concept that this gene is a monogenic risk factor for this “peculiar” form of cholesterol gallstones and a target for novel therapeutic strategies in humans.
After bile salts are secreted into bile and enter the intestine, approximately 95% of the secreted bile salts are absorbed through an active transport mechanism by a specific bile salt transporter, apical sodium-dependent bile salt transporter expressed predominantly in the distal ileum [109–111]. These absorbed bile salts return to the liver through the enterohepatic circulation. As a result, the newly synthesized bile salts in the liver contribute only a small fraction (less than 5%) to biliary secretion, which compensate for bile salts that escape intestinal absorption and are lost in the feces. Therefore, there are two sources for hepatic bile salt secretion, which consist of those that are newly synthesized in the liver and those undergoing enterohepatic cycling [109, 112, 113]. In the late 1990s, the transporter ABCB11, also called the bile salt export pump, on the canalicular membrane of hepatocytes, was discovered to play a key role in hepatic secretion of biliary bile salts [114–118]. Deletion of the Abcb11 gene in mice completely impedes hepatic bile salt secretion. The cellular and molecular mechanisms by which bile salt secretion is coupled to cholesterol and phospholipid secretion are still under extensive investigations. Notably, hepatic secretion of bile salts could directly affect phospholipid vesicle secretion [119–122]. The relationship between bile salt secretion and cholesterol secretion has been found to be curvilinear. At a low hepatic bile salt secretion rate (<10 μmol/h/kg), more biliary cholesterol is secreted per molecule of bile salts compared to that at a higher rate. Although biliary bile salt secretion is not often low in healthy individuals, it could be reduced during prolonged fasting, during the overnight period, and with substantial bile salt losses such as with a biliary fistula or ileal resection when the liver cannot sufficiently compensate with increased bile salt synthesis. In contrast, at a high bile salt secretion rate, for example, during and after eating, biliary saturation is less compared to that during the interprandial period. Although biliary organic anion secretion does not influence bile acid secretion, it inhibits hepatic secretion of biliary phospholipids and cholesterol because organic anions can bind bile salts within the bile canaliculi and curtail interactions with the canalicular membrane of hepatocytes.
Many animal and human studies have found that bile salts promote vesicle secretion by the hepatocytes, and these unilamellar vesicles are always found in freshly collected hepatic bile [123–128]. In the early 2000s, genetic studies in patients with sitosterolemia revealed that the efflux of biliary cholesterol from the canalicular membrane of hepatocytes to bile is a protein-mediated process [8, 9, 129–139], which is determined by the sterol efflux transporters ABCG5/G8. Mutations in either ABCG5 or ABCG8 significantly reduce biliary cholesterol secretion in patients with sitosterolemia. The key role of ABCG5/G8 in hepatic cholesterol secretion has been investigated in genetically modified mice [11, 12, 140–142]. Overexpression of the human ABCG5/G8 gene in the liver increases the cholesterol content of gallbladder bile in transgenic mice. In contrast, hepatic secretion of biliary cholesterol is dramatically reduced in ABCG5/G8 double knockout mice, as well as in ABCG5 or ABCG8 single gene knockout mice. More interestingly, clinical studies found that sitosterolemia is caused by a mutation in either the ABCG5 or the ABCG8 gene alone, but not in both simultaneously, and hepatic cholesterol secretion is dramatically reduced, but not completely eliminated in these patients [50, 135, 143, 144]. To further study the cellular and molecular mechanisms underlying the key role of ABCG5/G8 in biliary sterol secretion, biliary cholesterol and sitostanol secretion is quantified for 6 h in ABCG8 knockout mice. Mass transport rate of [3H]sitostanol from plasma HDL into bile is significantly faster than that of [14C]cholesterol in wild-type mice; however, reduced amounts of [14C]cholesterol and no [3H]sitostanol are detected in bile of ABCG8 knockout mice [141]. These results indicate that knockout of the Abcg8 gene alone significantly reduces but does not eliminate hepatic cholesterol secretion. In addition, biliary cholesterol secretion studies uncovered that hepatic cholesterol output is dramatically diminished, but cholesterol is still secreted into bile in mice with the targeted deletion of either Abcg5 or Abcg8 alone, or both [11–13, 141, 145]. In agreement with the human data, these mouse results imply that an ABCG5/G8-independent pathway could also be involved in the regulation of hepatic cholesterol secretion in both humans and mice. In addition, scavenger receptor class B type I (SR-BI), the HDL receptor, is expressed mainly in the sinusoidal and, perhaps, in the canalicular membrane of hepatocytes. In transgenic and knockout mice, biliary secretion of cholesterol varies in proportion to the hepatic expression of SR-BI, and the established contribution of SR-BI to sinusoidal uptake of HDL cholesterol is destined for secretion into bile [146–148]. These studies indicate that SR-BI could play a critical role in the reverse cholesterol transport in the body.
(b). Intestinal absorption of cholesterol and plant sterols
Cholesterol is the most abundant steroid in human and animal tissues and in the intestinal lumen. It is poorly soluble in an aqueous environment. In addition to a double bond at C-5 and C-6 nucleus and a hydroxyl group on the third carbon of the cholestene nucleus (Fig. 8.1), the β-configuration is connected with the angular methyl groups at C-10 and C-13, the hydrogen atom at C-8, and the side chain at C-17. The hydrogen atoms at C-9 and C-14 are in the α-configuration [149].
Phytosterols, also called plant sterols, refer to sterols that originate from plants. These are abundant in the intestine, but not in human and animal tissues. As shown in Fig. 8.1, plant sterols are naturally occurring, and their chemical structures are very similar to cholesterol, i.e., a Δ5 double bond and a 3β-hydroxyl group, but with structural modifications of the side chain. Plant sterols have the same basic importance in plants as cholesterol in animals, playing a vital role in cell membrane function. Sitosterol and campesterol, which are 24-ethyl and the 24-methyl substituted variants of cholesterol, respectively, are the most abundant plant sterols [149]. They are consumed in the diet and may be absorbed in the intestine. However, they are often present only at very low plasma concentrations in human and animal tissues due to a poor (<5%) net absorption rate by the small intestine. Other sterols such as brassicasterol and isofucosterol may also originate from shellfish.
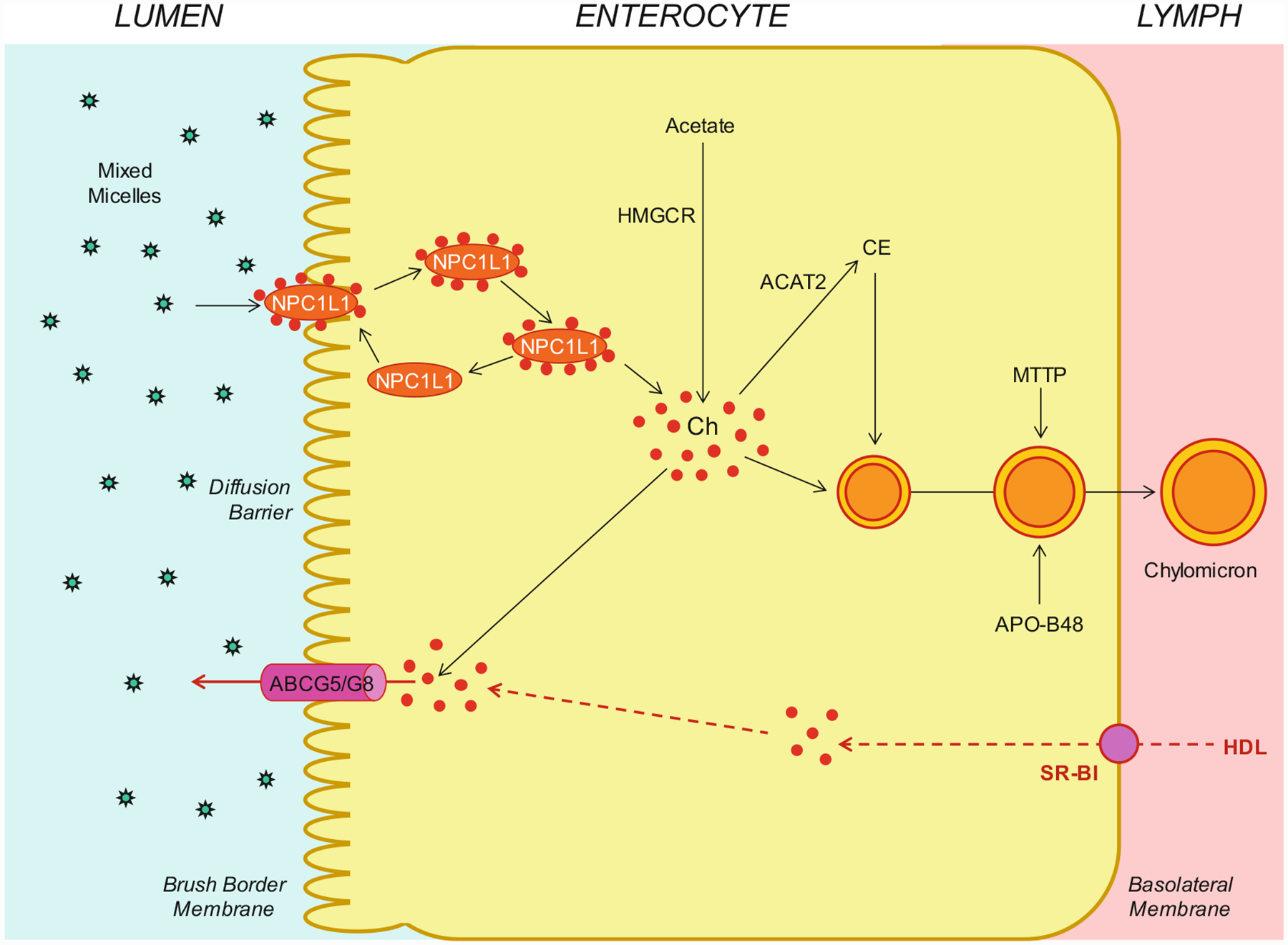
As shown in Fig. 8.3, within the intestinal lumen, the micellar solubilization of cholesterol and fatty acids facilitates movement through the diffusion barrier overlying the surface of the absorptive cells. In the presence of bile salts, mixed micelles deliver large amounts of the cholesterol molecules to the aqueous-membrane interface so that the uptake rate is greatly increased. Human and animal studies have found that the Niemann-Pick C1 like 1 (NPC1L1) protein, a sterol influx transporter, is expressed at the apical membrane of the enterocytes and can actively facilitate the uptake of cholesterol by promoting the passage of cholesterol across the brush border membrane of the enterocytes. Moreover, NPC1L1 plays a key role in the ezetimibe-sensitive cholesterol absorption pathway [150–154], which is highly likely to make the influx of cholesterol and likely plant sterols from the intestinal lumen into the cytoplasm of enterocytes. NPC1L1 could mediate cholesterol uptake via vesicular endocytosis, and ezetimibe may inhibit cholesterol absorption by suppressing the internalization of NPC1L1/cholesterol complex. In contrast, ABCG5/G8 are apical sterol export pumps promoting active efflux of cholesterol and plant sterols from the enterocytes back into the intestinal lumen for fecal excretion [8, 9, 12, 47, 48, 131, 141, 155–158]. The combined regulatory actions of NPC1L1 and ABCG5/G8 play a pivotal role in modulating the amount of cholesterol that reaches the lymph from the intestinal lumen. These findings imply that intestinal cholesterol absorption is a multistep process that is regulated by multiple genes at the enterocyte level and that the efficiency of cholesterol absorption is determined by the net effect between influx and efflux of intraluminal cholesterol molecules crossing the brush border membrane of the enterocyte [159]. In addition, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) is the rate-limiting enzyme for cholesterol biosynthesis in the body [160–165]. Cholesterol that is synthesized de novo from acetyl CoA in different organs (i.e., the liver and small intestine) is the second major source to the body [166–173]. The absorbed cholesterol molecules, as well as some that are newly synthesized from acetate by HMGCR within the enterocytes, are esterified to fatty acids by acyl-CoA:cholesterol acyltransferase isoform 2 (ACAT2) to form cholesteryl esters. Furthermore, there are three putative pathways for uptake of fatty acids and their transport across the apical membranes of enterocytes, either by simple passive diffusion mostly for short-chain fatty acids or by multiple transporters and proteins such as fatty acid transport protein 4 (FATP4), CD36 (also referred to as fatty acid translocase), and plasma membrane-associated fatty acid-binding protein (FABPpm; 43 kDa) largely for medium- and long-chain fatty acids. Finally, all of these lipids are used for the assembly of chylomicrons, which also requires the synthesis of apoB-48 and the activity of microsomal triglyceride transfer protein (MTTP). The core of chylomicrons secreted in lymph contains triglycerides and cholesteryl esters, and their surface is a monolayer containing phospholipids (mainly phosphatidylcholine), unesterified cholesterol, and apolipoproteins such as apoB-48, apoA-I, and apoA-IV [149].
Fig. 8.3.
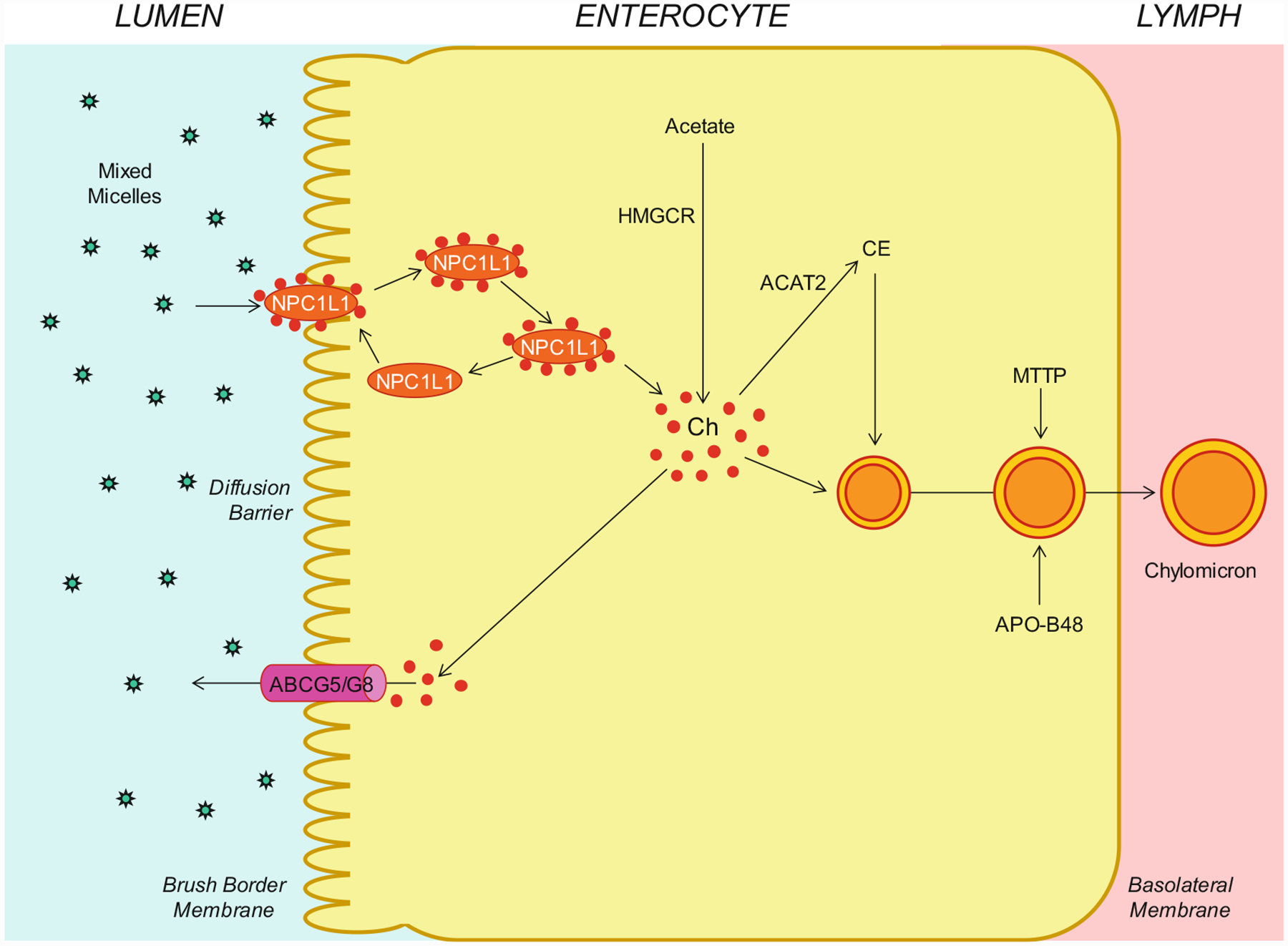
Molecular and cellular mechanisms of intestinal cholesterol absorption. Within the intestinal lumen, the micellar solubilization of sterols facilitates movement through the diffusion barrier overlying the surface of the absorptive cells in the small intestine. In the presence of bile salts, mixed micelles deliver large amounts of the cholesterol (Ch) molecules to the aqueous-membrane interface so that the uptake rate is greatly increased. The Niemann-Pick C1 like 1 (NPC1L1) protein, a sterol influx transporter, is located at the apical membrane of the enterocyte and can actively facilitate the uptake of cholesterol by promoting the passage of cholesterol across the brush border membrane of the enterocyte. NPC1L1 appears to mediate cholesterol uptake via vesicular endocytosis, and ezetimibe may inhibit cholesterol absorption by suppressing the internalization of NPC1L1/cholesterol complex. In contrast, ABCG5/G8 promote active efflux of cholesterol from the enterocyte back into the intestinal lumen for fecal excretion. The combined regulatory effects of NPC1L1 and ABCG5/G8 play a critical role in modulating the amount of cholesterol that reaches the lymph from the intestinal lumen. The absorbed cholesterol molecules, as well as some that are newly synthesized from acetate by 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) within the enterocytes, are esterified to fatty acids by acyl-CoA:cholesterol acyltransferase isoform 2 (ACAT2) to form cholesteryl esters (CE). All of these lipids are involved in the assembly of chylomicrons, which also requires the synthesis of apolipoprotein B-48 (apoB-48) and the activity of microsomal triglyceride transfer protein (MTTP). The core of chylomicrons secreted in lymph contains triglycerides and cholesteryl esters, and their surface is a monolayer containing phospholipids (mainly phosphatidylcholine), unesterified cholesterol, and apolipoproteins such as apoB-48, apoA-I, and apoA-IV
Although daily intake of cholesterol and plant sterols from the diet is almost equal, the intestinal absorption efficiency is significantly lower in the latter compared to the former. For example, the absorption efficiency of sitosterol and campesterol is 5–8% and 9–18%, respectively [174], compared with 30–60% of intestinal cholesterol absorption in humans [175–179]. It is highly likely that most of the plant sterols that do enter the enterocyte are rapidly pumped back into the intestinal lumen for excretion, as done by the actions of ABCG5/G8. In addition to poor net absorption, plant sterols are more efficiently secreted into bile compared to cholesterol. These combined mechanisms maintain plasma plant sterol concentrations at less than 1 mg/dL in humans. Because plant sterols are insoluble, they must be esterified and incorporated into triglycerides in margarines in order to achieve high concentrations within the intestine [180]. It has been found that large amounts of plant sterols could interfere with intestinal cholesterol absorption. The basic mechanism of inhibitory action of these compounds could be that plant sterols are efficiently incorporated into micelles in the intestinal lumen, displace the cholesterol, and lead to its precipitation with other, non-solubilized plant sterols [131, 158, 181–183]. Furthermore, competition between cholesterol and plant sterols for incorporation into micelles and for transfer into the brush border membrane could partly explain the inhibitory effect of large amounts of plant sterols on intestinal cholesterol absorption. This reduces both hepatic cholesterol and triglyceride contents by reducing delivery of intestinal cholesterol to the liver via chylomicrons. Because cholesterol absorption from dietary and biliary sources is reduced in the presence of plant sterols, the unabsorbed cholesterol excreted in the feces is substantially increased. Overall, plasma total and LDL cholesterol concentrations are lowered by two different mechanisms: reduced availability of cholesterol for incorporations into VLDL particles and increased expression of LDL receptor in the liver.
The identification of defective structures in the sterol efflux transporters ABCG5/G8 in patients with sitosterolemia indicates that cholesterol absorption is a selective process; also the activities of ABCG5/G8 provide an explanation for the selectivity against plant sterols so that plant sterols are absorbed poorly [159]. The NPC1L1 is also expressed at the apical membrane of enterocytes and plays a decisive role in the ezetimibe-sensitive cholesterol absorption pathway. As discussed above, intestinal cholesterol absorption is a multistep process that is regulated by multiple genes at the enterocyte level. The significant inter-individual differences in cholesterol absorption efficiency found in humans and the variations observed in inbred strains of mice strongly suggest that many additional genes may be involved in the regulation of intestinal cholesterol absorption. These differences also provide opportunities to apply advanced genetic techniques to identify the responsible genes that contribute to the regulation of intestinal lipid absorption. A better understanding of the cellular and molecular mechanisms whereby cholesterol and plant sterols are absorbed in the small intestine may provide more molecular targets for patients who require aggressive cholesterol-lowering therapy [149].
(c). Reverse cholesterol transport
Many clinical and animal studies have revealed that there are two major pathways for the removal of cholesterol from the body [184]. In humans and animals, hepatic secretion of biliary cholesterol across the canalicular membrane of hepatocytes is an important route for removing cholesterol from the body. Moreover, the cholesterol molecule can be metabolized to other compounds such as bile salts, which, in turn, are excreted from the body through the intestinal tract and eventually in the feces. Notably, the sterol efflux transporters ABCG5/G8 on the canalicular membrane of hepatocytes are responsible for regulating hepatic secretion of biliary cholesterol [11, 12, 140, 142, 185], and the bile salt export pump, ABCB11, plays a critical role in hepatic secretion of biliary bile salts [186]. These transporters in the liver has a vital impact on determining excretion of excess cholesterol from the body, either as unesterified cholesterol or as its metabolic products, bile salts.
In the mid-1960, the definition of the reverse cholesterol transport and the speculated role of HDL in promoting this process were first proposed [187]. Classically, the reverse cholesterol transport is a process involved in the removal of excess cholesterol that is accumulated in the peripheral tissues (e.g., macrophages in the aortae) by HDL, transporting it to the liver for excretion into the feces via the bile (Fig. 8.4). In the 1980s and 1990s, many results from animal studies strongly supported the concept that HDL could play a critical role in protecting against cardiovascular disease [188–191]. Subsequently, numerous clinical studies found that plasma HDL is the smallest lipoprotein particles and contains the highest proportion of apolipoproteins to lipids compared to LDL, VLDL, and chylomicrons. Although the molecular and genetic mechanisms underlying its beneficial properties in humans are not fully understood, HDL is most widely recognized for its ability to promote cholesterol efflux from the macrophages and other cells in the extrahepatic tissues and transport cholesterol from the periphery to the liver for hepatic secretion and, subsequently, fecal excretion. Obviously, during the process of the reverse cholesterol transport, the deposition of cholesterol in the peripheral tissues, including the aortae, is greatly reduced [192–194]. Many animal studies have consistently found that HDL is protective on several processes that are involved in preventing atherosclerosis, at least in part by mediating the removal of cholesterol from lipid-laden macrophages through the reverse cholesterol transport [189, 195, 196].
Fig. 8.4.
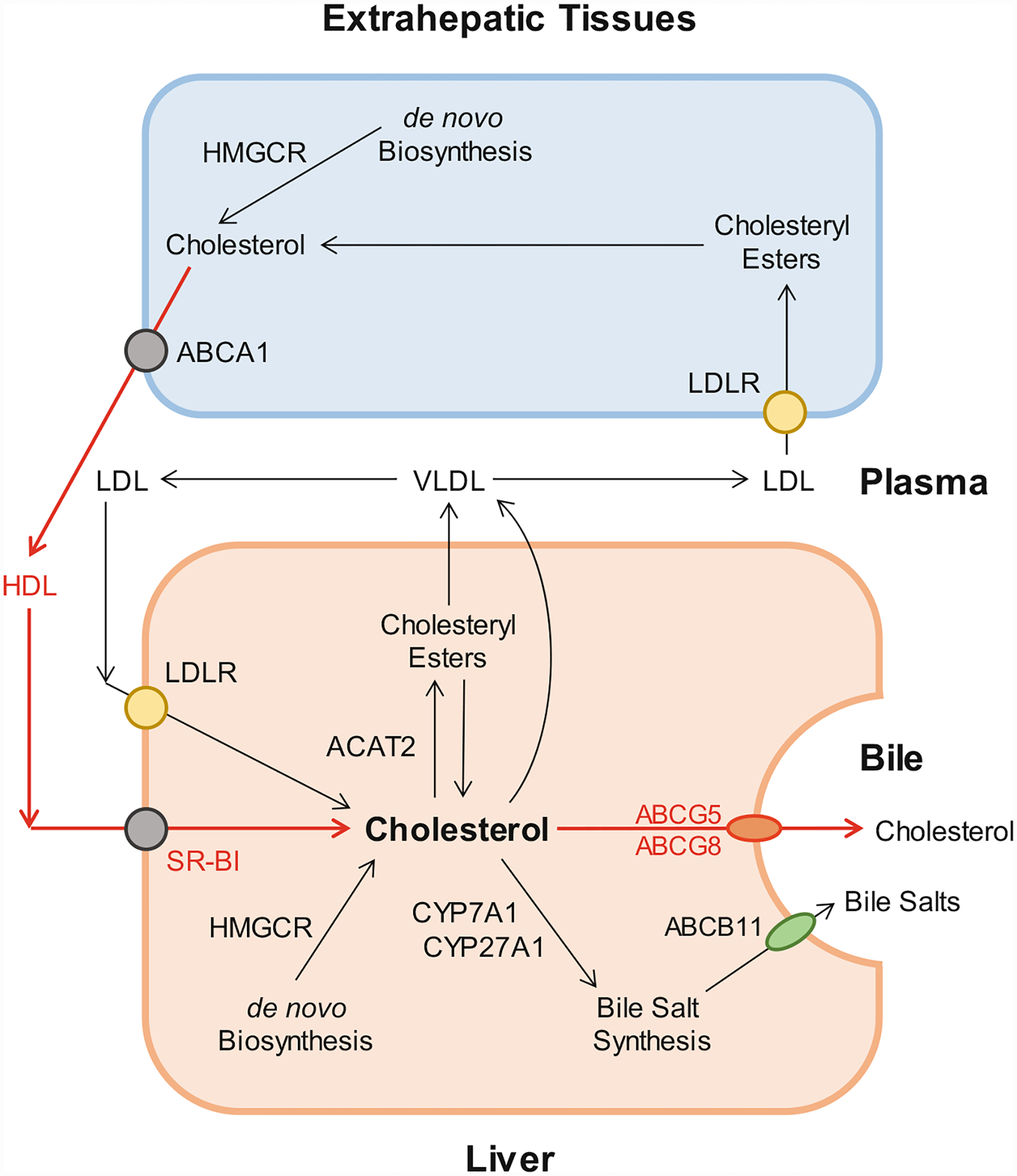
The reverse cholesterol transport through the classical biliary route to the bile as secreted by ABCG5/G8 on the canalicular membrane of hepatocytes. The reverse cholesterol transport in the hepatocytes is shown in red lines with arrows indicating the direction of transport. Abbreviations: ABC ATP-binding cassette (transporter), ACAT2 acyl-coenzyme A:cholesterol acyltransferase isoform 2, CYP7A1 cholesterol 7-α-hydroxylase, CYP27A1 sterol 27-hydroxylase, HDL high-density lipoprotein, HMGCR 3-hydroxy-3-methylglutaryl-CoA reductase, LDL low-density lipoprotein, SR-BI scavenger receptor class B type I, i.e., HDL receptor, VLDL very-low-density lipoprotein
The major HDL-associated apoA-I and apoA-II are secreted into plasma by the liver and intestine, where they are lipidated to form lipid-poor, discoidal, nascent HDL. Nascent HDL takes up cholesterol from cell membranes and other lipoproteins. Many studies have been performed to investigate whether an increase in plasma HDL cholesterol concentrations reduces the risk of developing cardiovascular disease. Substantial evidence from epidemiological investigations and clinical studies has clearly demonstrated that the level of plasma HDL cholesterol, especially at average to slightly above average concentrations, is inversely related to the incidence of cardiovascular disease and its thrombotic complications. Prospective population studies have found that humans with HDL cholesterol levels of 6–7 mg/dL, i.e., higher than average, have a 20–27% decrease in the risk of developing cardiovascular disease, and increasing HDL cholesterol levels by 1 mg/dL may reduce the risk of cardiovascular disease by 2% in men and 3% in women. Increasing plasma HDL cholesterol concentrations has been found to prevent atherogenesis and protect against atherosclerosis in mice, rabbits, and humans. When reconstituted HDL or apoA-I is provided exogenously, regressive changes in atherosclerotic plaques are found in human studies. Transgenic expression of the human APOAI gene increases HDL and suppresses atherosclerosis in APOE knockout mice, and genetic lowering of plasma HDL in mice reduces the appearance of macrophage-derived cholesterol in feces. Collectively, these results from human and animal studies have led to the idea that increasing plasma HDL may be a new strategy for the treatment and the prevention of cardiovascular disease.
Although most published studies attribute the atheroprotective properties of HDL to HDL2, a lot of results also reveal that HDL3 may be inversely related to the risk of developing cardiovascular disease. More recently, clinical studies of HDL metabolism have focused mainly on plasma total HDL cholesterol concentrations, but not on each HDL subclass. In addition, cardiovascular risk associated with HDL cholesterol levels is independent of plasma LDL cholesterol concentrations, as well as other lipid parameters (e.g., triglycerides and total cholesterol), and non-lipid risk factors. Although the concept has been proposed for many years that therapeutic interventions of increasing plasma HDL cholesterol levels could potentially reduce cardiovascular mortality [197], pharmacologic interventions to augment HDL cholesterol concentrations by delaying HDL catabolism do not translate into a marked reduction in cardiovascular risk. Therefore, the inability of therapies of increasing HDL cholesterol concentrations and new insights into the complexity of HDL composition and function have prompted researchers to further explore whether and how HDL exerts its cardioprotective functions [198–200]. Nevertheless, systematic interpretation of HDL metabolism could help identify therapeutic targets that may increase plasma HDL cholesterol concentrations and reduce the risk of developing cardiovascular disease.
(d). Transintestinal cholesterol excretion (TICE)
For many years, the reverse cholesterol transport is considered as an important route for transporting excess cholesterol that is accumulated within peripheral tissues back to the liver for hepatic secretion into bile and, eventually, to intestine for excretion in the feces. Some studies on patients with hepatobiliary and/or pancreatic disorders and several animal models with obstruction of the bile duct or cholestasis have found a novel non-biliary transport route likely for reverse cholesterol transport, independent of classical pathway of the reverse cholesterol transport through the liver. In the late 1950s, a secondary, non-biliary pathway was proposed, which was defined as the transintestinal cholesterol excretion (TICE) [201]. It is suggested that the TICE may contribute a new way to the reverse cholesterol transport. However, these studies were greatly criticized about the selection of patients and animal models because dramatic diminution or discontinuation of bile flow entering the small intestine could damage the normal physiological function of the epithelial cells of small intestine. Moreover, these alterations could lead to a remarkable reduction in intestinal lipid absorption because of a lack of bile salts. Such results with a striking increase in fecal neutral sterols were questioned because these studies were performed under conditions of severe hepatobiliary disease and inappropriate experimental approaches. Consequently, the TICE was not accepted even though this new concept challenged the classical view of the reverse cholesterol transport by showing that the small intestine is also highly likely to be involved in mass fecal neutral sterol excretion, independent of the biliary cholesterol excretion route. In the mid-2000s, using different mouse models with new experimental methods, some exciting data were reported that direct transintestinal excretion of plasma-derived cholesterol might contribute to the reverse cholesterol transport in mice [202, 203]. Based on the results from these mouse experiments, it is estimated that this non-biliary route may account for ~30% of total fecal neutral sterol excretion under basal conditions and could be regulated by several nuclear receptors such as liver X receptor (LXR), peroxisome proliferator-activated receptor-delta (PPAR-δ), and farnesoid X receptor (FXR) [204, 205]. Moreover, some results from animal studies suggest that this non-biliary route may be a novel therapeutic target to increase reverse cholesterol transport and, in this manner, confer protection against cardiovascular disease [205]. Although in vitro studies for examining the activity of this transintestinal route have been reported in explants from human small intestine mounted in Ussing chambers [206], the existence and importance of the TICE route in humans have not been established because of some difficult technical issues and methodology.
Interestingly, the contribution of TICE to total fecal neutral sterol excretion is recently studied in a small number of subjects [207]. Combining a cholesterol balance approach with stable isotopes that label cholesterol and bile salt molecules, the body cholesterol fluxes are analyzed in subjects with mild hypercholesterolemia. After 4 weeks of ezetimibe (10 mg/day) treatment for inhibiting the intestinal cholesterol influx transporter NPC1L1, the same studies are performed in the subjects eating a regular meal. Under basal conditions, the classical reverse cholesterol transport could contribute approximately 65% of daily fecal neutral sterol excretion, and it is likely that the TICE accounts for the remainder (~35%), as shown in Fig. 8.5. More interestingly, ezetimibe-treated subjects display a fourfold increase in total fecal neutral sterol excretion most likely through the TICE. To further confirm the results reported from human studies, chow-fed ABCG8 knockout and wild-type mice are treated with ezetimibe at 0 or 8 mg/kg/day for 2 weeks. As a result, most of the ezetimibe-modulated TICE flux is likely to be determined by the intestinal sterol efflux transporters ABCG5/G8. These studies suggest that TICE may exist in humans, and most of the ezetimibe-modulated TICE flux may be regulated by ABCG5/G8. For that reason, the TICE may be a new therapeutic target to enhance the removal of excess cholesterol from the body in patients at risk for cardiovascular disease. It is highly likely that the TICE may be an alternative route to the biliary route of the reverse cholesterol transport. However, it is imperative to explore the cellular and molecular mechanisms underlying the pivotal role of the TICE alone in the regulation of reverse cholesterol transport in humans [208]. More importantly, it is crucial to decipher whether the TICE could excrete more cholesterol from the body in patients with hypercholesterolemia, as well as how the TICE works together with the classical biliary route and whether it is fully independent from the latter. In addition, it is critical to elucidate whether there is a striking difference between the fasting state and the fed condition for the TICE to regulate plasma cholesterol, HDL, and LDL metabolism. More studies are also needed to investigate how the TICE is regulated in the normal physiological state, as well as under conditions of high plasma total and LDL cholesterol concentrations. With new experimental techniques, it is crucial for exploring whether the TICE is associated with the absorption efficiency of intestinal cholesterol because it is well-known that ABCG5/G8 is actively involved in regulating both the TICE and intestinal cholesterol absorption. Definitely, it is interesting to study whether abnormality in the molecular and genetic regulation of the TICE is associated with the prevalence of cardiovascular disease in humans [208]. Taken together, the TICE might provide a new target for the prevention and the treatment of cardiovascular disease.
Fig. 8.5.
Schematic diagram of the proposed transintestinal cholesterol excretion (TICE) pathway in the enterocytes, as showed in red dashed lines with arrows indicating the direction of transport. Abbreviations: HDL high-density lipoprotein, SR-BI scavenger receptor class B type I, i.e., HDL receptor. See Fig. 8.3 for other abbreviations
8.5. Roles of ABCG5/G8 in Pathophysiology of Lipid-Related Metabolic Disorders
(a). Sitosterolemia
Sitosterolemia was first reported by Bhattacharyya and Connor in 1974 based on a clinical study on two sisters with tendon xanthomas and with elevated plant sterol concentrations in plasma [129]. Sitosterolemia is a rare inherited lipid storage disease characterized chemically by the accumulation of plant sterols and 5α-saturated stanols in plasma and tissues [134]. As analyzed by the sterol balance method, a large amount of dietary sitosterol is absorbed from the small intestine, thereby leading to the plant sterol accumulation in the body of patients with sitosterolemia. Further genetic studies find that sitosterolemia is a rare autosomal, recessively inherited lipid metabolic disorder [209]. However, the majority of heterozygous subjects are clinically and biochemically normal, and some heterozygotes display a slight, but not significant, increase in plasma sitosterol concentrations compared to normal healthy subjects [210]. Nevertheless, plasma sitosterol concentrations are 10- to 20-fold higher in homozygotes than in heterozygotes [211]. Therefore, the diagnosis of sitosterolemia is based mainly on a significant increase in the concentrations of plant sterols (sitosterol, campesterol, stigmasterol, and avenosterol) and 5α-stanols in plasma and tissues [212]. The clinical presentation in these patients includes tendon xanthomas, accelerated atherosclerosis particularly affecting males at a young age, hemolytic episodes, arthritis, and arthralgia [134]. The risk of premature atherosclerosis has been found in some young male patients who died because of acute myocardial infarctions associated with extensive coronary and aortic arteriosclerosis [139, 213].
Sitosterolemia is caused by a mutation in either the ABCG5 or the ABCG8 gene alone, but not in both simultaneously [8, 9, 136, 137, 214]. It is characterized mainly by increased intestinal absorption of cholesterol and sitosterol and diminished hepatic secretion of these sterols into bile [129, 209, 215]. In patients with sitosterolemia, the intestinal absorption of cholesterol is augmented by ~30%, from ~46% to ~60%; however, the intestinal absorption of sitosterol is dramatically increased by ~800%, from <5% to ~45% [50, 135, 143, 144]. Therefore, more cholesterol of intestinal origin, through the chylomicron pathway, reaches the liver for VLDL secretion into plasma, thereby increasing risk of developing cardiovascular disease in patients with sitosterolemia. Indeed, intestinal cholesterol absorption efficiency is also significantly increased in ABCG5/G8, ABCG5, and ABCG8 knockout mice [11–13, 141, 145].
Notably, several human studies on biliary lipid secretion have found that hepatic cholesterol secretion is markedly reduced and hepatic secretion of sitosterol and other plant sterols is almost totally inhibited [50, 135, 143, 144]. As a result, these patients often display hypercholesterolemia, tendon and tuberous xanthomas, premature development of atherosclerosis, and abnormal hematologic and liver function test results [134]. Further animal studies show that hepatic cholesterol output is dramatically reduced, but cholesterol is still secreted into bile in mice with the deletion of either Abcg5 or Abcg8 alone, or both [11–13, 141, 145]. These results clearly support the concept that the deletion of the Abcg5/g8 double genes and Abcg5 or Abcg8 single gene significantly reduces, but does not eliminate, hepatic cholesterol secretion. In addition, consistent with the human results, these mouse data imply that an ABCG5/G8-independent pathway is also involved in hepatic cholesterol secretion, as discussed above.
The cholesterol molecules derived from HDL, but not LDL or VLDL, are an important source for hepatic secretion into bile because HDL promotes reverse cholesterol transport from peripheral tissues to the liver where the HDL-derived cholesterol is secreted preferentially into the bile [216]. After intravenous injection, HDL-derived [14C]cholesterol, but not [3H] sitostanol, is recovered in hepatic bile of ABCG5/G8 and ABCG8 knockout mice. This indicates that the ABCG5/G8-independent pathway is also able to regulate hepatic secretion of HDL-derived cholesterol, but not sitostanol. By contrast, ABCG5/G8 is involved in the regulation of hepatic secretion of both cholesterol and plant sterols. These results are consistent with the finding in sitosterolemic patients in whom only reduced amounts of cholesterol are found in bile and hepatic secretion of plant sterols is completely inhibited, leading to a significant increase in plasma plant sterol concentrations [135].
The treatment of sitosterolemia includes bile salt sequestrants such as cholestyramine, colestipol, and colesevelam in combination with the low-sterol diet [217–220]. Bile salt sequestrants bind bile salts in the intestine and increase the excretion of bile salts in the feces. This greatly diminishes the amount of bile salts returning to the liver and forces the liver to produce more bile salts to replace the bile salts lost in the feces. To synthetize more bile salts, the liver must convert more cholesterol into bile salts, thus leading to a reduction in plasma total and LDL cholesterol concentrations in sitosterolemic patients [221]. Moreover, ezetimibe, a potent intestinal cholesterol absorption inhibitor, has been used to treat patients with sitosterolemia [222–224] because ezetimibe can diminish plasma LDL cholesterol levels in patients with hypercholesterolemia by inhibiting the function of intestinal NPC1L1, the cholesterol influx transport protein [150, 153, 225–228].
(b). Cardiovascular disease
Atherosclerosis is characterized by lipid accumulation, inflammatory response, cell death, and fibrosis in the arterial wall, which is the pathological basis for cardiovascular disease, and the leading cause of morbidity and mortality in the USA and other industrialized nations [229]. Major risk factors for atherosclerosis include high plasma levels of LDL cholesterol and lipoprotein(a), as well as low plasma concentrations of HDL cholesterol [230]. Although genetic mechanisms underlying the pathogenesis of cardiovascular disease are largely unknown, accumulated evidence from human and animal studies has clearly demonstrated that cardiovascular disease may be determined by multiple genes disrupting cholesterol and lipoprotein metabolism [231–236]. Because mutations in either ABCG5 or ABCG8 cause phytosterolemia, hypercholesterolemia, and premature coronary heart disease in patients with sitosterolemia, this strongly suggests that defect or reduction in the ABCG5/G8 expression and function may be an important risk factor for the development of cardiovascular disease [141, 237–240]. Increased expression of Abcg5/g8 attenuates Western-diet-induced hypercholesterolemia and atherosclerosis in LDL receptor knockout mice [241]. However, overexpression of Abcg5/g8 in the liver, but not in the small intestine, does not reduce atherosclerosis development in LDL receptor or ApoE knockout mice fed the Western diet for 6 months [242]. This suggests that the increased hepatic secretion of biliary cholesterol could be absorbed back into the body, thus leading to unaltered atherosclerosis in these knockout mice. When these mice are fed ezetimibe, the potent intestinal cholesterol absorption inhibitor, total plasma cholesterol concentrations, and atherosclerosis are dramatically reduced in LDL receptor knockout mice overexpressing the human ABCG5/G8 genes in the liver alone compared to LDL receptor knockout mice [243]. These mouse studies indicate that deletion of Abcg5/g8 could play a determinant role in the development of hypercholesterolemia and atherosclerosis in mice fed the Western diet. In contrast, this suggests that ABCG5/G8 may be a novel target for the prevention and the treatment of cardiovascular disease. Furthermore, more studies are needed to explore whether dysfunction of ABCG5/G8 in the liver, or small intestine, or both sites is responsible for increased risk for the development of hypercholesterolemia and atherosclerosis in mice fed the Western diet.
In addition, it is interesting to investigate whether polymorphisms in the ABCG5 and ABCG8 genes are associated with plasma total and LDL cholesterol concentrations, increasing susceptibility to cardiovascular disease. Various polymorphisms (A632V, T400K, D19H, M429V, and C54Y) in the ABCG8 and ABCG5 (Q604E) genes have been found to be associated with several facets of cholesterol metabolism, including baseline cholesterol level, cholesterol kinetics, and individual responsiveness of plasma cholesterol to dietary and pharmaceutical interventions for hypercholesterolemia. For example, Tyr54Cys and Thr400Lys variations in the ABCG8 gene may play a role in the genetic determination of plasma cholesterol levels and could influence the gender-specific response of plasma cholesterol levels after dietary changes [244]. More interestingly, low serum cholesterol concentrations and intestinal cholesterol absorption are found to be linked to the D19H polymorphism of the ABCG8 gene, and characteristics of the insulin resistance syndrome in men are linked with the Q604E polymorphism of the ABCG5 gene [245]. However, an association study between five common ABCG5/G8 polymorphisms (p.Q604E, p.D19H, p.Y54C, p. T400K, and p.A632V) and plasma sterol levels was performed in 245 patients with hypercholesterolemia, and no significant associations were found [246]. Thus, most, but not all, studies reported that polymorphisms in the ABCG5 and ABCG8 genes may be associated with increased total and LDL-cholesterol concentrations [32]. Furthermore, a meta-analysis that comprised 3,364 subjects from 16 studies was carried out [246]. This study found that the ABCG8 632V variant is associated with a clinically irrelevant LDL-cholesterol reduction, whereas the 19H allele correlates with decreased cholesterol absorption and increased synthesis without affecting the lipid profile [246]. However, it is largely unknown whether small amounts of phytosterol exposure over a lifetime cause pathology in healthy humans with polymorphic variants in the ABCG5 and ABCG8 genes. Taken together, polymorphic variants in the ABCG5 and ABCG8 genes could increase or reduce the risk of these phenotypes, and loss of ABCG5/G8 function could cause more significant phenotypes, including premature atherosclerosis, platelet dysfunction, and thrombocytopenia, and perhaps, increased endocrine disruption and liver dysfunction [239]. Obviously, more studies are strongly needed to investigate how specific polymorphisms of the ABCG5 and ABCG8 genes confer to higher risk of these diseases.
Because elevated LDL cholesterol levels are a major causal factor for cardiovascular disease and have been a primary target of therapy for more than 30 years, the potent HMGCR inhibitors, statins, have been developed to lower plasma LDL cholesterol levels and reduce the risk of adverse cardiovascular events [247]. Moreover, reducing LDL cholesterol levels to below current guideline targets further inhibits atherogenesis and decreases adverse coronary events [4, 5, 248]. Many clinical studies have found that statins can reduce new adverse cardiovascular events and cardiovascular disease mortality by ~35%, but even aggressive statin therapy can not completely eliminate cardiovascular risk. Approximately 65% of the patients treated with statins still develop adverse cardiovascular events. Therefore, additional therapeutic interventions beyond statins are strongly needed to further reduce the risk of developing cardiovascular disease [249]. Overall, ABCG5/G8 may be an attractive target for the prevention and the treatment of hypercholesterolemia, and increasing their expression may reduce the risk of developing cardiovascular disease in humans.
(c). Cholesterol gallstone disease
Clinical investigations and animal studies have clearly established that hepatic hypersecretion of biliary cholesterol is the primary defect in the pathogenesis of cholesterol gallstone disease [14]. Hepatic cholesterol hypersecretion into bile may or may not be accompanied by normal, high, or low hepatic secretion rates of biliary bile salts and/or phospholipids [250]. Cholesterol-supersaturated bile is often defined as a state in which cholesterol cannot be dissolved in bile by biliary bile salts or phospholipids at equilibrium [70]. Therefore, the formation of supersaturated bile is often caused by (i) hepatic hypersecretion of biliary cholesterol; (ii) reduced hepatic bile salt and/or phospholipid secretion with normal biliary cholesterol secretion; or (iii) a combination of hepatic cholesterol hypersecretion with hyposecretion of these solubilizing lipids [251].
Genetic studies have been performed to investigate Lith genes in different strains of inbred mice fed the lithogenic diet for 8 weeks [26]. As shown in Fig. 8.6, Lith9 is localized on mouse chromosome 17 and is co-localized with a genetic biomarker D17Mit155 at approximately 55 centimorgans (cM). Genotyping and phenotyping studies have found that in the Lith9 QTL region, Abcg5/g8 is a strong candidate for this gallstone gene. Subsequently, Abcg5/g8 is identified as a new gallstone gene, Lith9, by QTL studies in mice [25, 252, 253]. Based on mouse genetic analysis of the Lith genes, a genome-wide association study in a large cohort of patients with gallstones and a linkage study in affected sibling pairs have identified a common variant (D19H) of the sterol efflux transporters ABCG5/G8 as a key risk factor for cholesterol gallstone disease [29]. Indeed, ABCG5/G8 is found to be associated with gallstones in patients, proving that it is human LITH9. Other ABCG8 variants (T400K, D19H, A632V, M429V, and C54Y) and ABCG5 variants (Q604E) have also been found to be associated with cholesterol gallstone disease in humans. Furthermore, many research groups have reported that two gallstone-associated variants in ABCG5/G8, i.e., ABCG5-R50C and ABCG8-D19H, are involved in the pathogenesis of gallstones not only in Germans and Chileans but also in Chinese and Indians [29–34, 254, 255]. These studies strongly imply that ABCG5-R50C and ABCG8-D19H could play a central role in hepatic cholesterol hypersecretion, thereby leading to the formation of cholesterol-supersaturated bile in humans.
Fig. 8.6.
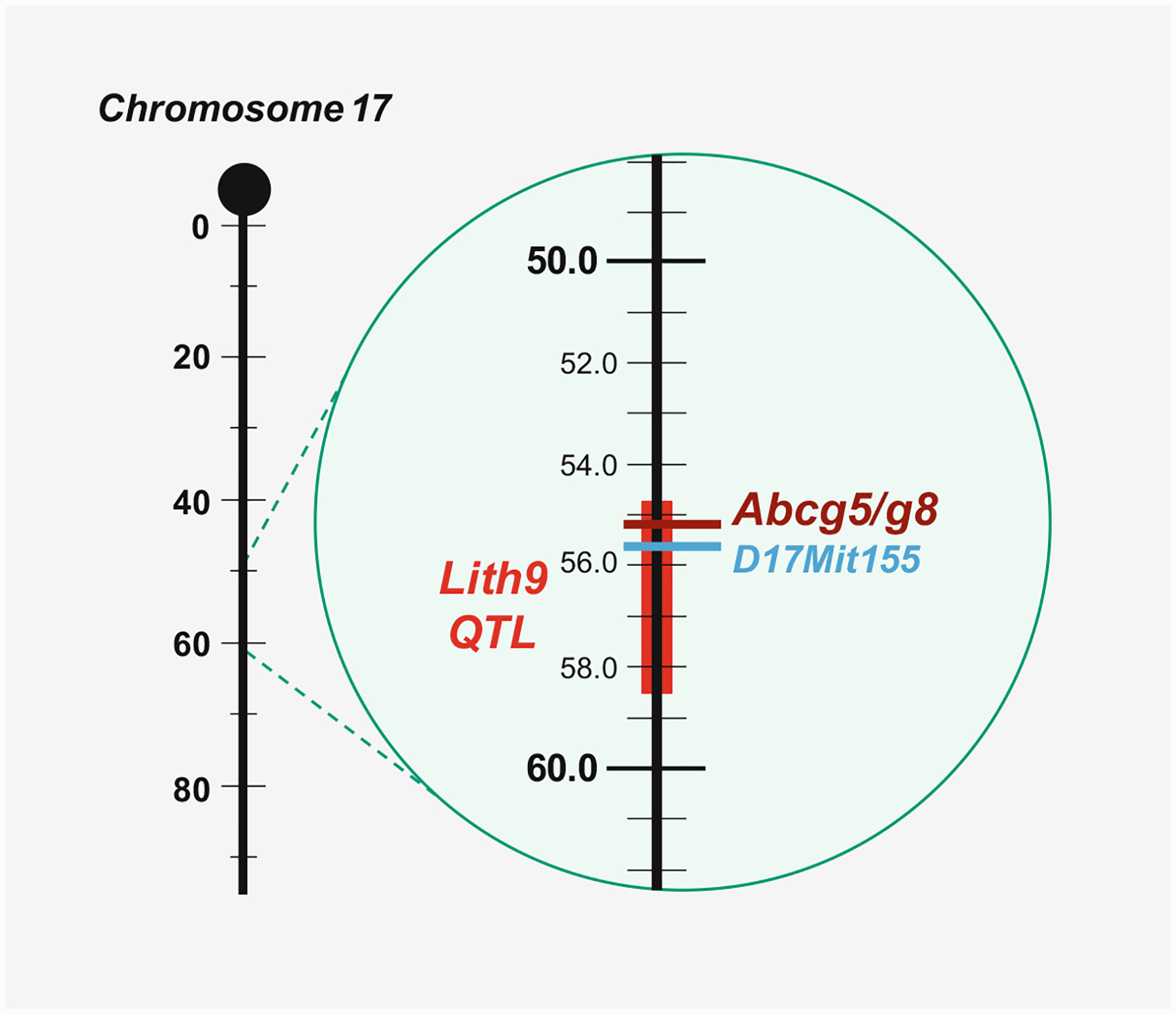
As shown in the composite map, the quantitative trait locus (QTL) region of the Lith9 gene is localized on chromosome 17 in mice. A vertical line represents chromosome 17, with the centromere at the top; genetic distances from the centromere (horizontal black lines) are indicated to the left of the chromosomes in centimorgans (cM). Chromosomes are drawn to scale, based on the estimated cM position of the most distally mapped locus taken from Mouse Genome Database. The gallstone gene, the Lith9, QTL region is represented by a vertical red bar, and the Abcg5/g8 gene location is indicated by a horizontal borrow line. A genetic biomarker, D17Mit155, that is co-localized with Lith9 is indicated by a horizontal blue line with the marker symbol to the right
Because Abcg5/g8 is Lith9 in mice and two gallstone-associated variants in ABCG5/G8 have been identified in humans, it is important to further investigate whether targeted disruption of the Abcg5/g8 double genes or the Abcg8 single gene protects against the formation of cholesterol gallstones in gallstone-susceptible C57BL/6J mice fed the lithogenic diet for 8 weeks [256]. It is surprising to find that despite a significant reduction in gallstone prevalence in ABCG5/G8 and ABCG8 knockout mice, classical parallelogram-shaped cholesterol monohydrate crystals and gallstones are still formed in these mice during the 8-week period of feeding the lithogenic diet. As discussed above, although sitosterolemia is caused by mutations in either the ABCG5 or the ABCG8 gene alone, but not in both simultaneously, hepatic cholesterol secretion is reduced, but not completely eliminated, in these patients [50, 135, 143, 144]. To explore the mechanism underlying the effect of ABCG5/G8 on hepatic cholesterol and plant sterol secretion, biliary cholesterol and sitostanol secretion is quantified for 6 h in ABCG8 knockout mice. Mass transport rate of [3H]sitostanol from plasma HDL into bile is significantly faster than that of [14C]cholesterol in wild-type mice; however, reduced amounts of [14C]cholesterol and no [3H] sitostanol are found in bile of ABCG8 knockout mice [141]. These results clearly exhibit that the deletion of the Abcg8 gene alone significantly reduces, but does not eliminate, hepatic cholesterol secretion. In addition, biliary cholesterol studies show that hepatic cholesterol output is significantly reduced, but cholesterol is still secreted into bile in mice with the deletion of either Abcg5 or Abcg8 alone, or both [11–13, 141, 145].
Although ABCG5/G8 display a striking impact on hepatic cholesterol and plant sterol secretion, cholesterol is still secreted to bile in sitosterolemic patients with a defect in either ABCG5 or ABCG8 and in either ABCG5/G8 double or single gene knockout mice. This strongly suggests that in the defect of ABCG5/G8, an ABCG5/G8-independent pathway is essential for regulating hepatic secretion of biliary cholesterol, which is independent of the lithogenic mechanism of the ABCG5/G8 pathway. To decipher the effect of the ABCG5/G8-independent pathway on cholelithogenesis, the biliary and gallstone characteristics are investigated in wild-type as well as ABCG5/G8 and ABCG8 knockout mice fed the lithogenic diet or varying amounts of cholesterol, or injected intravenously with [3H]sitostanol- and [14C]cholesterol-labeled HDL. These studies show that ABCG5/G8 and ABCG8 knockout mice display the same biliary and gallstone phenotypes. Although both groups of knockout mice show a significant reduction in hepatic cholesterol output compared to wild-type mice, they still form gallstones. Especially, the ABCG5/G8-independent pathway plays an important role in the regulation of biliary cholesterol secretion, the transport of HDL-derived cholesterol from plasma to bile, and the formation of cholesterol gallstones, which works independently of the ABCG5/G8 pathway.
It is well-known that the LXR agonist T0901317 activates hepatic LXR, promoting biliary cholesterol secretion by stimulating hepatic Abcg5/g8 expression in mice [145, 257, 258]. Additionally, LXR activation by T0901317 greatly promotes cholesterol crystallization and gallstone formation in mice fed the lithogenic diet [259]. However, this is not the case in ABCG5/G8 or ABCG8 knockout mice. This clearly implies that the hepatic LXR does not have an effect on the ABCG5/G8-independent pathway for regulating biliary cholesterol secretion, which is distinct from the ABCG5/G8 pathway that is effectively regulated by the hepatic LXR through a transcriptional mechanism. The LXR agonist dramatically increases biliary cholesterol secretion and gallstones in wild-type, but not ABCG5/G8 or ABCG8 knockout, mice. Taken together, these studies [256] provide clear evidence in support of the concepts that (i) the ABCG5/G8-independent pathway accounts for 30% to 40% of hepatic cholesterol output in the lithogenic state and plays a critical role in the regulation of biliary cholesterol secretion in response to high dietary cholesterol; (ii) in the absence of ABCG5/G8, it determines biliary cholesterol secretion and the formation of cholesterol gallstones; (iii) it modulates hepatic secretion of HDL-derived cholesterol, but not sitostanol; and (iv) its activity in the liver is not regulated by the LXR agonist through the LXR signaling cascade. These findings strongly support the existence of an ABCG5/G8-independent pathway for regulating hepatic cholesterol secretion. Moreover, these results imply that in the absence of ABCG5/G8, the ABCG5/G8-independent pathway is essential for the regulation of hepatic cholesterol secretion and also plays a vital role in determining the susceptibility to cholesterol gallstones, working independently of the ABCG5/G8 pathway in mice. However, further studies are strongly needed to observe if this pathway is also operational in humans. Nevertheless, both ABCG5/G8-dependent and ABCG5/G8-independent pathways could be potential therapeutic targets for cholesterol gallstone disease.
8.6. Conclusions and Future Directions
Accumulated evidence has clearly demonstrated that ABCG5/G8 play a key role not only in hepatic secretion of biliary cholesterol and plant sterols but also intestinal absorption of these two sterols. Moreover, ABCG5/G8 have an important impact on the classical reverse cholesterol transport and the TICE pathway. Obviously, mutations in either ABCG5 or ABCG8 are the major genetic mechanisms causing sitosterolemia. It is highly likely that gene therapy is a better option for curing this genetic disorder by repairing ABCG5 or ABCG8 gene mutations. Lowering plasma total and LDL cholesterol concentrations is also crucial to reduce the risk of cardiovascular disease in patients with sitosterolemia.
Many clinical studies have shown that statins can reduce the risk of developing cardiovascular disease; however, other lipid-lowering therapies are often used adjunctively when statin therapy is inadequate or as an alternative for patients who are intolerant of statins. More importantly, intensive lipid and pharmaceutical studies have led to significant development of new agents that could work on novel targets in the metabolic pathways of lipids and lipoproteins and that have the potential to serve as new alternative or adjunctive agents to the existing cholesterol-lowering drugs such as statins. Clinical trials in patients receiving these new classes of lipid-lowering agents, especially in individuals with monogenic disorders of lipid and/or lipoprotein metabolism, will certainly increase a great opportunity to identify the genotype that predicts response to lipid-lowering therapy and thus guides the choice of drug and dose for high-risk patients and, especially, for patients with the hardest-to-treat elevated plasma cholesterol concentrations due to intolerance to any statins and severe side effects of these drugs.
Although the pharmacogenomics of lipid-lowering drugs have greatly advanced and a few consistent trends on the therapy of cardiovascular disease have emerged, mainly relating to the genetic determinants of response to statins, many new cellular, molecular, genetic, and biochemical studies on lipid and lipoprotein metabolism are being extensively explored. Therefore, it is more interesting to investigate the cellular and molecular mechanisms of deciphering how ABCG5/G8 regulate cholesterol and lipoprotein metabolism in the plasma, liver, and intestine. In addition, the potential mechanisms underlying the removal of cholesterol from the body through the classical reverse cholesterol transport, i.e., the biliary route, and the TICE, i.e., the non-biliary routes, are desired to be revealed. Advances in the elucidation of lipid and lipoprotein metabolism, as well as the biliary and the non-biliary routes for removal of cholesterol and plant sterols from the body, will provide a great opportunity of finding new lipid-lowering strategies and proving that they are more effective in the prevention and therapeutic intervention of cardiovascular disease that affects millions worldwide.
The gallstone (Lith) gene map has been updated, which lists all known genetic loci that confer gallstone susceptibility, as well as candidate genes in inbred strains of mice. This would greatly help identify human LITH genes because genetic analysis of Lith genes in mouse models open the way for searching for the orthologous human LITH genes and for exploring their cholelithogenic effects in humans. Given that the ABCG5/G8-dependent and the ABCG5/G8-independent pathways are essential in the regulation of hepatic cholesterol secretion, both routes could be potential therapeutic targets for the prevention and the treatment of cholesterol gallstone disease. Deciphering the molecular and cellular mechanisms on the formation of cholesterol-supersaturated bile could be very helpful for exploring novel therapeutic approaches through modulating both the ABCG5/G8-dependent and the ABCG5/G8-independent pathways, thus greatly reducing the risk of developing gallstones.
More importantly, there should be a great development of the personalized medicine for the prevention and the treatment of cardiovascular disease and cholesterol gallstone disease because they are highly prevalent not only in the USA but also in European and Asian countries. The ideal application of lipid-lowering drugs and bilecholesterol-desaturating drugs would be to identify patients at risk for either a suboptimal response with respect to efficacy or a marked adverse response to either a drug class or a specific drug. For that reason, individuals who would be predicted to have an unfavorable benefit-to-risk ratio can be identified and might be obtained from alternative methods more expeditiously and without the trial-and-error process that typically accompanies initiation and maintenance of such commonly used treatment. Obviously, it is imperative to understand the cellular and molecular mechanisms underlying the key role of ABCG5/G8 in regulating hepatic secretion of biliary cholesterol and plant sterols and intestinal absorption of these two sterols, as well as in modulating the classical reverse cholesterol transport and the TICE pathway, because it could provide novel insights into strategies for the prevention and the treatment of sitosterolemia, cardiovascular disease, and cholesterol gallstone disease.
Acknowledgments
This work was supported in part by research grants DK101793, DK106249, DK114516, and AA025737 (to DQ-HW), as well as P30 DK041296 (to Marion Bessin Liver Research Center), all from the National Institutes of Health (US Public Health Service). This chapter was modified from the paper published by our group in Annals of Hepatology (Helen H. Wang, Gabriella Garruti, Min Liu, Piero Portincasa, David Q.-H. Wang. 2017; 16 (Suppl. 1): s28-s43). The related contents are re-used with the permission.
Abbreviations
- ABC
ATP-binding cassette (transporter)
- ACAT2
Acyl-CoA: cholesterol acyltransferase isoform 2
- APO
Apolipoprotein
- CSI
Cholesterol saturation index
- CYP7A1
Cholesterol 7α-hydroxylase
- CYP27A1
Sterol 27-hydroxylase
- FABPpm
Plasma membrane-associated fatty acid-binding protein
- FATP4
Fatty acid transport protein 4
- FXR
Farnesoid X receptor
- HDL
High-density lipoprotein
- HMGCR
3-Hydroxy-3-methylglutaryl coenzyme A reductase
- LDL
Low-density lipoprotein
- LXR
Liver X receptor
- MTTP
Microsomal triglyceride transfer protein
- NPC1L1
Niemann-Pick C1 like 1 (protein)
- PPAR-δ
Peroxisome proliferator-activated receptor-delta
- QTL
Quantitative trait locus
- SR-BI
Scavenger receptor class B type I
- TICE
Transintestinal cholesterol excretion
- VLDL
Very-low-density lipoprotein
Footnotes
Conflict of Interest There is no conflict of interest to disclose for all authors.
Contributor Information
Helen H. Wang, Department of Medicine and Genetics, Division of Gastroenterology and Liver Diseases, Marion Bessin Liver Research Center, Einstein-Mount Sinai Diabetes Research Center, Albert Einstein College of Medicine, Bronx, NY, USA
Min Liu, Department of Pathology and Laboratory Medicine, University of Cincinnati College of Medicine, Cincinnati, OH, USA.
Piero Portincasa, Department of Biomedical Sciences and Human Oncology, Clinica Medica “A. Murri”, University of Bari Medical School, Bari, Italy.
David Q.-H. Wang, Department of Medicine and Genetics, Division of Gastroenterology and Liver Diseases, Marion Bessin Liver Research Center, Einstein-Mount Sinai Diabetes Research Center, Albert Einstein College of Medicine, Bronx, NY, USA
References
- 1.Wang DQ, Neuschwander-Tetri BA, Portincasa P (2012) The biliary system. Morgan & Claypool Life Sciences, Princeton [Google Scholar]
- 2.Wang DQ (2007) Regulation of intestinal cholesterol absorption. Annu Rev Physiol 69:221–248 [DOI] [PubMed] [Google Scholar]
- 3.Panel TNCEPE (2001) Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 285 (19):2486–2497 [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC Jr, Stone NJ (2004) Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol 44(3):720–732 [DOI] [PubMed] [Google Scholar]
- 5.Smith SC Jr, Allen J, Blair SN, Bonow RO, Brass LM, Fonarow GC, Grundy SM, Hiratzka L, Jones D, Krumholz HM et al. (2006) AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update: endorsed by the National Heart, Lung, and Blood Institute. Circulation 113(19):2363–2372 [DOI] [PubMed] [Google Scholar]
- 6.Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fayad ZA, Foster E, Hlatky MA, Hodgson JM, Kushner FG et al. (2010) 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 56(25):e50–103 [DOI] [PubMed] [Google Scholar]
- 7.Martin SS, Metkus TS, Horne A, Blaha MJ, Hasan R, Campbell CY, Yousuf O, Joshi P, Kaul S, Miller M et al. (2012) Waiting for the National Cholesterol Education Program Adult Treatment Panel IV Guidelines, and in the meantime, some challenges and recommendations. Am J Cardiol 110 (2):307–313 [DOI] [PubMed] [Google Scholar]
- 8.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH (2000) Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290(5497):1771–1775 [DOI] [PubMed] [Google Scholar]
- 9.Lee MH, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Kojima H, Allikmets R, Sakuma N, Pegoraro R et al. (2001) Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet 27(1):79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee MH, Lu K, Patel SB (2001) Genetic basis of sitosterolemia. Curr Opin Lipidol 12(2):141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2002) Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci U S A 99(25):16237–16242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu L, Li-Hawkins J, Hammer RE, Berge KE, Horton JD, Cohen JC, Hobbs HH (2002) Overexpression of ABCG5 and ABCG8 promotes biliary cholesterol secretion and reduces fractional absorption of dietary cholesterol. J Clin Invest 110(5):671–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Mitsche MA, Lutjohann D, Cohen JC, Xie XS, Hobbs HH (2015) Relative roles of ABCG5/ABCG8 in liver and intestine. J Lipid Res 56(2):319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang DQ, Portincasa P (2017) Gallstones: recent advances in epidemiology, pathogenesis, diagnosis and management. Nova Biomedical, New York [Google Scholar]
- 15.Lammert F, Gurusamy K, Ko CW, Miquel JF, Mendez-Sanchez N, Portincasa P, van Erpecum KJ, van Laarhoven CJ, Wang DQ (2016) Gallstones. Nat Rev Dis Primers 2:16024. [DOI] [PubMed] [Google Scholar]
- 16.Wang DQ, Cohen DE, Carey MC (2009) Biliary lipids and cholesterol gallstone disease. J Lipid Res 50(Suppl):S406–S411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruhl CE, Everhart JE (2011) Gallstone disease is associated with increased mortality in the United States. Gastroenterology 140(2):508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peery AF, Crockett SD, Barritt AS, Dellon ES, Eluri S, Gangarosa LM, Jensen ET, Lund JL, Pasricha S, Runge T et al. (2015) Burden of gastrointestinal, liver, and pancreatic diseases in the United States. Gastroenterology 149(7):1731–41 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Portincasa P, Wang DQ (2016) In: Podolsky DK, Camilleri M, Fitz JG, Kalloo AN, Shanahan F, Wang TC (eds) Yamada’s Atlas of gastroenterology. Wiley-Blackwell, Hoboken, pp 335–353 [Google Scholar]
- 20.Portincasa P, Di Ciaula A, de Bari O, Garruti G, Palmieri VO, Wang DQ (2016) Management of gallstones and its related complications. Expert Rev Gastroenterol Hepatol 10(1):93–112 [DOI] [PubMed] [Google Scholar]
- 21.Di Ciaula A, Wang DQ, Portincasa P (2018) An update on the pathogenesis of cholesterol gallstone disease. Curr Opin Gastroenterol 34(2):71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Ciaula A, Wang DQ, Garruti G, Wang HH, Grattagliano I, de Bari O, Portincasa P (2014) Therapeutic reflections in cholesterol homeostasis and gallstone disease: a review. Curr Med Chem 21(12):1435–1447 [DOI] [PubMed] [Google Scholar]
- 23.Wang DQ, Afdhal NH (2014) In: Feldman M, Friedman LS, Brandt L (eds) Sleisenger and Fordtran’s gastrointestinal and liver disease. Elsevier Saunders, Philadelphia, pp 1100–1133 [Google Scholar]
- 24.Portincasa P, Ciaula AD, Bonfrate L, Wang DQH (2012) Therapy of gallstone disease: what it was, what it is, what it will be. World J Gastrointest Pharmacol Ther 3(2):7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wittenburg H, Lyons MA, Li R, Churchill GA, Carey MC, Paigen B (2003) FXR and ABCG5/ABCG8 as determinants of cholesterol gallstone formation from quantitative trait locus mapping in mice. Gastroenterology 125(3):868–881 [DOI] [PubMed] [Google Scholar]
- 26.Wang TY, Portincasa P, Liu M, Tso P, Wang DQ (2018) Mouse models of gallstone disease. Curr Opin Gastroenterol 34(2):59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang HH, Portincasa P, Afdhal NH, Wang DQ (2010) Lith genes and genetic analysis of cholesterol gallstone formation. Gastroenterol Clin N Am 39(2):185–207. vii–viii [DOI] [PubMed] [Google Scholar]
- 28.Wang DQ, Afdhal NH (2004) Genetic analysis of cholesterol gallstone formation: searching for Lith (gallstone) genes. Curr Gastroenterol Rep 6(2):140–150 [DOI] [PubMed] [Google Scholar]
- 29.Grunhage F, Acalovschi M, Tirziu S, Walier M, Wienker TF, Ciocan A, Mosteanu O, Sauerbruch T, Lammert F (2007) Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology 46(3):793–801 [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Jiang ZY, Fei J, Xin L, Cai Q, Jiang ZH, Zhu ZG, Han TQ, Zhang SD (2007) ATP binding cassette G8 T400K polymorphism may affect the risk of gallstone disease among Chinese males. Clinica Chimica Acta Int J Clin Chem 384(1–2):80–85 [DOI] [PubMed] [Google Scholar]
- 31.Kuo KK, Shin SJ, Chen ZC, Yang YH, Yang JF, Hsiao PJ (2008) Significant association of ABCG5 604Q and ABCG8 D19H polymorphisms with gallstone disease. Br J Surg 95(8):1005–1011 [DOI] [PubMed] [Google Scholar]
- 32.Rudkowska I, Jones PJ (2008) Polymorphisms in ABCG5/G8 transporters linked to hypercholesterolemia and gallstone disease. Nutr Rev 66(6):343–348 [DOI] [PubMed] [Google Scholar]
- 33.Katsika D, Magnusson P, Krawczyk M, Grunhage F, Lichtenstein P, Einarsson C, Lammert F, Marschall HU (2010) Gallstone disease in Swedish twins: risk is associated with ABCG8 D19H genotype. J Intern Med 268(3):279–285 [DOI] [PubMed] [Google Scholar]
- 34.von Kampen O, Buch S, Nothnagel M, Azocar L, Molina H, Brosch M, Erhart W, von Schonfels W, Egberts J, Seeger M et al. (2013) Genetic and functional identification of the likely causative variant for cholesterol gallstone disease at the ABCG5/8 lithogenic locus. Hepatology 57(6):2407–2417 [DOI] [PubMed] [Google Scholar]
- 35.Borst P, Elferink RO (2002) Mammalian ABC transporters in health and disease. Annu Rev Biochem 71:537–592 [DOI] [PubMed] [Google Scholar]
- 36.Dean M, Allikmets R (1995) Evolution of ATP-binding cassette transporter genes. Curr Opin Genet Dev 5(6):779–785 [DOI] [PubMed] [Google Scholar]
- 37.Allikmets R, Gerrard B, Glavac D, Ravnik-Glavac M, Jenkins NA, Gilbert DJ, Copeland NG, Modi W, Dean M (1995) Characterization and mapping of three new mammalian ATP-binding transporter genes from an EST database. Mamm Genome 6(2):114–117 [DOI] [PubMed] [Google Scholar]
- 38.Quazi F, Molday RS (2011) Lipid transport by mammalian ABC proteins. Essays Biochem 50(1):265–290 [DOI] [PubMed] [Google Scholar]
- 39.Iida A, Saito S, Sekine A, Mishima C, Kitamura Y, Kondo K, Harigae S, Osawa S, Nakamura Y (2002) Catalog of 605 single-nucleotide polymorphisms (SNPs) among 13 genes encoding human ATP-binding cassette transporters: ABCA4, ABCA7, ABCA8, ABCD1, ABCD3, ABCD4, ABCE1, ABCF1, ABCG1, ABCG2, ABCG4, ABCG5, and ABCG8. J Hum Genet 47(6):285–310 [DOI] [PubMed] [Google Scholar]
- 40.van Meer G, Halter D, Sprong H, Somerharju P, Egmond MR (2006) ABC lipid transporters: extruders, flippases, or flopless activators? FEBS Lett 580(4):1171–1177 [DOI] [PubMed] [Google Scholar]
- 41.Stefkova J, Poledne R, Hubacek JA (2004) ATP-binding cassette (ABC) transporters in human metabolism and diseases. Physiol Res/Academia Scientiarum Bohemoslovaca 53(3):235–243 [PubMed] [Google Scholar]
- 42.Marcil M, Brooks-Wilson A, Clee SM, Roomp K, Zhang LH, Yu L, Collins JA, van Dam M, Molhuizen HO, Loubster O et al. (1999) Mutations in the ABC1 gene in familial HDL deficiency with defective cholesterol efflux. Lancet 354(9187):1341–1346 [DOI] [PubMed] [Google Scholar]
- 43.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, Mol CA, Ottenhoff R, van der Lugt NM, van Roon MA et al. (1993) Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 75(3):451–462 [DOI] [PubMed] [Google Scholar]
- 44.Oude Elferink RP, Ottenhoff R, van Wijland M, Smit JJ, Schinkel AH, Groen AK (1995) Regulation of biliary lipid secretion by mdr2 P-glycoprotein in the mouse. J Clin Invest 95(1):31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groen AK, Van Wijland MJ, Frederiks WM, Smit JJ, Schinkel AH, Oude Elferink RP (1995) Regulation of protein secretion into bile: studies in mice with a disrupted mdr2 p-glycoprotein gene. Gastroenterology 109(6):1997–2006 [DOI] [PubMed] [Google Scholar]
- 46.Oude Elferink RP, Ottenhoff R, van Wijland M, Frijters CM, van Nieuwkerk C, Groen AK (1996) Uncoupling of biliary phospholipid and cholesterol secretion in mice with reduced expression of mdr2 P-glycoprotein. J Lipid Res 37(5):1065–1075 [PubMed] [Google Scholar]
- 47.Graf GA, Li WP, Gerard RD, Gelissen I, White A, Cohen JC, Hobbs HH (2002) Coexpression of ATP-binding cassette proteins ABCG5 and ABCG8 permits their transport to the apical surface. J Clin Invest 110(5):659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Graf GA, Yu L, Li WP, Gerard R, Tuma PL, Cohen JC, Hobbs HH (2003) ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J Biol Chem 278(48):48275–48282 [DOI] [PubMed] [Google Scholar]
- 49.Zhang DW, Graf GA, Gerard RD, Cohen JC, Hobbs HH (2006) Functional asymmetry of nucleotide-binding domains in ABCG5 and ABCG8. J Biol Chem 281(7):4507–4516 [DOI] [PubMed] [Google Scholar]
- 50.Miettinen TA (1980) Phytosterolaemia, xanthomatosis and premature atherosclerotic arterial disease: a case with high plant sterol absorption, impaired sterol elimination and low cholesterol synthesis. Eur J Clin Investig 10(1):27–35 [DOI] [PubMed] [Google Scholar]
- 51.Turley SD, Dietschy JM (1988) In: Arias IM, Jakoby WB, Popper H, Schachter D, Shafritz DA (eds) The liver: biology and pathobiology. Raven Press, New York, pp 617–641 [Google Scholar]
- 52.Cohen DE (2009) In: Arias IM, Alter HJ, Boyer JL, Cohen DE, Fausto N, Shafritz DA, Wolkoff AW (eds) The liver: biology and pathobiology. Wiley-Blackwell, Chichester, pp 271–285 [Google Scholar]
- 53.Andersen JM, Dietschy JM (1977) Regulation of sterol synthesis in 16 tissues of rat. I. Effect of diurnal light cycling, fasting, stress, manipulation of enterohepatic circulation, and administration of chylomicrons and triton. J Biol Chem 252(11):3646–3651 [PubMed] [Google Scholar]
- 54.Andersen JM, Dietschy JM (1977) Regulation of sterol synthesis in 15 tissues of rat. II. Role of rat and human high and low density plasma lipoproteins and of rat chylomicron remnants. J Biol Chem 252(11):3652–3659 [PubMed] [Google Scholar]
- 55.Dietschy JM (1984) Regulation of cholesterol metabolism in man and in other species. Klin Wochenschr 62(8):338–345 [DOI] [PubMed] [Google Scholar]
- 56.Spady DK, Dietschy JM (1985) Rates of cholesterol synthesis and low-density lipoprotein uptake in the adrenal glands of the rat, hamster and rabbit in vivo. Biochim Biophys Acta 836(2):167–175 [DOI] [PubMed] [Google Scholar]
- 57.Dietschy JM, Turley SD (2002) Control of cholesterol turnover in the mouse. J Biol Chem 277(6):3801–3804 [DOI] [PubMed] [Google Scholar]
- 58.Spady DK, Turley SD, Dietschy JM (1985) Rates of low density lipoprotein uptake and cholesterol synthesis are regulated independently in the liver. J Lipid Res 26(4):465–472 [PubMed] [Google Scholar]
- 59.Spady DK, Turley SD, Dietschy JM (1985) Receptor-independent low density lipoprotein transport in the rat in vivo. Quantitation, characterization, and metabolic consequences. J Clin Invest 76(3):1113–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turley SD, Dietschy JM (2003) Sterol absorption by the small intestine. Curr Opin Lipidol 14(3):233–240 [DOI] [PubMed] [Google Scholar]
- 61.Wang DQ, Cohen DE (2008) In: Ballantyne CM (ed) Lipidology in the treatment and prevention of cardiovascular disease (Clinical lipidology: a companion to Braunwald’s heart disease). Elsevier Saunders, Philadelphia, pp 26–44 [Google Scholar]
- 62.Tso P, Fujimoto K (1991) The absorption and transport of lipids by the small intestine. Brain Res Bull 27 (3–4):477–482 [DOI] [PubMed] [Google Scholar]
- 63.Wang DQ, Lee SP (2008) Physical chemistry of intestinal absorption of biliary cholesterol in mice. Hepatology 48(1):177–185 [DOI] [PubMed] [Google Scholar]
- 64.Wang DQ, Paigen B, Carey MC (2001) Genetic factors at the enterocyte level account for variations in intestinal cholesterol absorption efficiency among inbred strains of mice. J Lipid Res 42(11):1820–1830 [PubMed] [Google Scholar]
- 65.Bhattacharyya AK, Eggen DA (1987) Relationships between dietary cholesterol, cholesterol absorption, cholesterol synthesis, and plasma cholesterol in rhesus monkeys. Atherosclerosis 67(1):33–39 [DOI] [PubMed] [Google Scholar]
- 66.Trautwein EA, Forgbert K, Rieckhoff D, Erbersdobler HF (1999) Impact of beta-cyclodextrin and resistant starch on bile acid metabolism and fecal steroid excretion in regard to their hypolipidemic action in hamsters. Biochim Biophys Acta 1437(1):1–12 [DOI] [PubMed] [Google Scholar]
- 67.Turley SD, Daggy BP, Dietschy JM (1996) Effect of feeding psyllium and cholestyramine in combination on low density lipoprotein metabolism and fecal bile acid excretion in hamsters with dietary-induced hypercholesterolemia. J Cardiovasc Pharmacol 27(1):71–79 [DOI] [PubMed] [Google Scholar]
- 68.Turley SD, Daggy BP, Dietschy JM (1991) Cholesterol-lowering action of psyllium mucilloid in the hamster: sites and possible mechanisms of action. Metab Clin Exp 40(10):1063–1073 [DOI] [PubMed] [Google Scholar]
- 69.Admirand WH, Small DM (1968) The physicochemical basis of cholesterol gallstone formation in man. J Clin Invest 47(5):1043–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carey MC, Small DM (1978) The physical chemistry of cholesterol solubility in bile. Relationship to gallstone formation and dissolution in man. J Clin Invest 61(4):998–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang DQ, Carey MC (1996) Characterization of crystallization pathways during cholesterol precipitation from human gallbladder biles: identical pathways to corresponding model biles with three predominating sequences. J Lipid Res 37(12):2539–2549 [PubMed] [Google Scholar]
- 72.Wang DQ, Cohen DE, Lammert F, Carey MC (1999) No pathophysiologic relationship of soluble biliary proteins to cholesterol crystallization in human bile. J Lipid Res 40(3):415–425 [PubMed] [Google Scholar]
- 73.Goldstein JL, Brown MS (1975) Lipoprotein receptors, cholesterol metabolism, and atherosclerosis. Arch Pathol 99(4):181–184 [PubMed] [Google Scholar]
- 74.Brown MS, Goldstein JL (1984) How LDL receptors influence cholesterol and atherosclerosis. Sci Am 251(5):58–66 [DOI] [PubMed] [Google Scholar]
- 75.Brown MS, Goldstein JL (1983) Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem 52:223–261 [DOI] [PubMed] [Google Scholar]
- 76.Brown MS, Kovanen PT, Goldstein JL (1981) Regulation of plasma cholesterol by lipoprotein receptors. Science 212(4495):628–635 [DOI] [PubMed] [Google Scholar]
- 77.Small DM (1977) Cellular mechanisms for lipid deposition in atherosclerosis (first of two parts). N Engl J Med 297(16):873–877 [DOI] [PubMed] [Google Scholar]
- 78.Small DM (1988) George Lyman Duff memorial lecture. Progression and regression of atherosclerotic lesions. Insights from lipid physical biochemistry. Arteriosclerosis 8(2):103–129 [DOI] [PubMed] [Google Scholar]
- 79.Small DM, Shipley GG (1974) Physical-chemical basis of lipid deposition in atherosclerosis. Science 185(147):222–229 [DOI] [PubMed] [Google Scholar]
- 80.Goldstein JL, Brown MS (1987) Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation 76(3):504–507 [DOI] [PubMed] [Google Scholar]
- 81.Portincasa P, Moschetta A, Di Ciaula A, Pontrelli D, Sasso RC, Wang HH, Wang DQ (2008) In: Borzellino G, Cordiano C (eds) Biliary lithiasis: basic science, current diagnosis and management. Springer, Milano, pp 19–49 [Google Scholar]
- 82.Portincasa P, Moschetta A, Palasciano G (2006) Cholesterol gallstone disease. Lancet 368(9531):230–239 [DOI] [PubMed] [Google Scholar]
- 83.Portincasa P, Moschetta A, Palasciano G (2002) From lipid secretion to cholesterol crystallization in bile. Relevance in cholesterol gallstone disease. Ann Hepatol 1(3):121–128 [PubMed] [Google Scholar]
- 84.Portincasa P, Moschetta A, van Erpecum KJ, Calamita G, Margari A, vanBerge-Henegouwen GP, Palasciano G (2003) Pathways of cholesterol crystallization in model bile and native bile. Dig Liver Dis 35(2):118–126 [DOI] [PubMed] [Google Scholar]
- 85.Afdhal NH, Smith BF (1990) Cholesterol crystal nucleation: a decade-long search for the missing link in gallstone pathogenesis. Hepatology 11(4):699–702 [DOI] [PubMed] [Google Scholar]
- 86.Holan KR, Holzbach RT, Hermann RE, Cooperman AM, Claffey WJ (1979) Nucleation time: a key factor in the pathogenesis of cholesterol gallstone disease. Gastroenterology 77(4 Pt 1):611–617 [PubMed] [Google Scholar]
- 87.Holzbach RT (1995) Cholesterol nucleation in bile. Ital J Gastroenterol 27(2):101–105 [PubMed] [Google Scholar]
- 88.Holzbach RT (1990) Nucleation of cholesterol crystals in native bile. Hepatology 12(3 Pt 2):155S–159S. discussion 9S–61S [PubMed] [Google Scholar]
- 89.Holzbach RT (1986) Recent progress in understanding cholesterol crystal nucleation as a precursor to human gallstone formation. Hepatology 6(6):1403–1406 [DOI] [PubMed] [Google Scholar]
- 90.Holzbach RT (1984) Factors influencing cholesterol nucleation in bile. Hepatology 4(5 Suppl):173S–176S [DOI] [PubMed] [Google Scholar]
- 91.Holzbach RT, Busch N (1991) Nucleation and growth of cholesterol crystals. Kinetic determinants in supersaturated native bile. Gastroenterol Clin N Am 20(1):67–84 [PubMed] [Google Scholar]
- 92.Crawford JM, Mockel GM, Crawford AR, Hagen SJ, Hatch VC, Barnes S, Godleski JJ, Carey MC (1995) Imaging biliary lipid secretion in the rat: ultrastructural evidence for vesiculation of the hepatocyte canalicular membrane. J Lipid Res 36(10):2147–2163 [PubMed] [Google Scholar]
- 93.Crawford AR, Smith AJ, Hatch VC, Oude Elferink RP, Borst P, Crawford JM (1997) Hepatic secretion of phospholipid vesicles in the mouse critically depends on mdr2 or MDR3 P-glycoprotein expression. Visualization by electron microscopy. J Clin Invest 100(10):2562–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crawford JM (1996) Role of vesicle-mediated transport pathways in hepatocellular bile secretion. Semin Liver Dis 16(2):169–189 [DOI] [PubMed] [Google Scholar]
- 95.Oude Elferink RP, Beuers U (2011) Targeting the ABCB4 gene to control cholesterol homeostasis. Expert Opin Ther Targets 15(10):1173–1182 [DOI] [PubMed] [Google Scholar]
- 96.Langheim S, Yu L, von Bergmann K, Lutjohann D, Xu F, Hobbs HH, Cohen JC (2005) ABCG5 and ABCG8 require MDR2 for secretion of cholesterol into bile. J Lipid Res 46(8):1732–1738 [DOI] [PubMed] [Google Scholar]
- 97.Dikkers A, Tietge UJ (2010) Biliary cholesterol secretion: more than a simple ABC. World J Gastroenterol 16(47):5936–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lammert F, Wang DQ, Hillebrandt S, Geier A, Fickert P, Trauner M, Matern S, Paigen B, Carey MC (2004) Spontaneous cholecysto- and hepatolithiasis in Mdr2−/− mice: a model for low phospholipid-associated cholelithiasis. Hepatology 39(1):117–128 [DOI] [PubMed] [Google Scholar]
- 99.Oude Elferink RP, Paulusma CC (2007) Function and pathophysiological importance of ABCB4 (MDR3 P-glycoprotein). Pflugers Arch – Eur J Physiol 453(5):601–610 [DOI] [PubMed] [Google Scholar]
- 100.Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E (2010) The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis 30(2):134–146 [DOI] [PubMed] [Google Scholar]
- 101.Gonzales E, Davit-Spraul A, Baussan C, Buffet C, Maurice M, Jacquemin E (2009) Liver diseases related to MDR3 (ABCB4) gene deficiency. Front Biosci 14:4242–4256 [DOI] [PubMed] [Google Scholar]
- 102.Stapelbroek JM, van Erpecum KJ, Klomp LW, Houwen RH (2010) Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol 52(2):258–271 [DOI] [PubMed] [Google Scholar]
- 103.Paulusma CC, Elferink RP, Jansen PL (2010) Progressive familial intrahepatic cholestasis type 1. Semin Liver Dis 30(2):117–124 [DOI] [PubMed] [Google Scholar]
- 104.Jansen PL, Sturm E (2003) Genetic cholestasis, causes and consequences for hepatobiliary transport. Liver Int 23(5):315–322 [DOI] [PubMed] [Google Scholar]
- 105.Rosmorduc O, Hermelin B, Boelle PY, Parc R, Taboury J, Poupon R (2003) ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 125(2):452–459 [DOI] [PubMed] [Google Scholar]
- 106.Rosmorduc O, Hermelin B, Poupon R (2001) MDR3 gene defect in adults with symptomatic intrahepatic and gallbladder cholesterol cholelithiasis. Gastroenterology 120(6):1459–1467 [DOI] [PubMed] [Google Scholar]
- 107.Rosmorduc O, Poupon R (2007) Low phospholipid associated cholelithiasis: association with mutation in the MDR3/ABCB4 gene. Orphanet J Rare Dis 2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang DQ, Carey MC (1996) Complete mapping of crystallization pathways during cholesterol precipitation from model bile: influence of physical-chemical variables of pathophysiologic relevance and identification of a stable liquid crystalline state in cold, dilute and hydrophilic bile salt-containing systems. J Lipid Res 37(3):606–630 [PubMed] [Google Scholar]
- 109.Carey MC, Cahalane MJ (1988) In: Arias IM, Jakoby WB, Popper H, Schachter D, Shafritz DA (eds) The liver: biology and pathobiology. Raven Press, New York, pp 573–616 [Google Scholar]
- 110.Hofmann AF, Hagey LR (2008) Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci 65(16):2461–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chiang JY (2009) Bile acids: regulation of synthesis. J Lipid Res 50(10):1955–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hofmann AF (2009) The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci 14:2584–2598 [DOI] [PubMed] [Google Scholar]
- 113.Hofmann AF (2009) In: Arias IM, Alter HJ, Boyer JL, Cohen DE, Fausto N, Shafritz DA, Wolkoff AW (eds) The liver: biology and pathobiology. Wiley-Blackwell, Chichester, pp 290–304 [Google Scholar]
- 114.Suchy FJ, Ananthanarayanan M (2006) Bile salt excretory pump: biology and pathobiology. J Pediatr Gastroenterol Nutr 43(Suppl 1):S10–S16 [DOI] [PubMed] [Google Scholar]
- 115.Trauner M, Boyer JL (2003) Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 83(2):633–671 [DOI] [PubMed] [Google Scholar]
- 116.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, Meier PJ (1998) The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem 273(16):10046–10050 [DOI] [PubMed] [Google Scholar]
- 117.Wang R, Salem M, Yousef IM, Tuchweber B, Lam P, Childs SJ, Helgason CD, Ackerley C, Phillips MJ, Ling V (2001) Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci U S A 98(4):2011–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang R, Lam P, Liu L, Forrest D, Yousef IM, Mignault D, Phillips MJ, Ling V (2003) Severe cholestasis induced by cholic acid feeding in knockout mice of sister of P-glycoprotein. Hepatology 38(6):1489–1499 [DOI] [PubMed] [Google Scholar]
- 119.Cohen DE, Leighton LS, Carey MC (1992) Bile salt hydrophobicity controls vesicle secretion rates and transformations in native bile. Am J Physiol 263 (3 Pt 1):G386–G395 [DOI] [PubMed] [Google Scholar]
- 120.Cohen DE, Leonard MR, Carey MC (1994) In vitro evidence that phospholipid secretion into bile may be coordinated intracellularly by the combined actions of bile salts and the specific phosphatidylcholine transfer protein of liver. Biochemistry 33(33):9975–9980 [DOI] [PubMed] [Google Scholar]
- 121.Fuchs M, Carey MC, Cohen DE (1997) Evidence for an ATP-independent long-chain phosphatidylcholine translocator in hepatocyte membranes. Am J Physiol 273(6 Pt 1):G1312–G1319 [DOI] [PubMed] [Google Scholar]
- 122.Carey MC, Cohen DE (1995) Update on physical state of bile. Ital J Gastroenterol 27(2):92–100 [PubMed] [Google Scholar]
- 123.Somjen GJ, Marikovsky Y, Lelkes P, Gilat T (1986) Cholesterol-phospholipid vesicles in human bile: an ultrastructural study. Biochim Biophys Acta 879(1):14–21 [DOI] [PubMed] [Google Scholar]
- 124.Mazer NA, Carey MC, Kwasnick RF, Benedek GB (1979) Quasielastic light scattering studies of aqueous biliary lipid systems. Size, shape, and thermodynamics of bile salt micelles. Biochemistry 18 (14):3064–3075 [DOI] [PubMed] [Google Scholar]
- 125.Mazer NA, Benedek GB, Carey MC (1980) Quasielastic light-scattering studies of aqueous biliary lipid systems. Mixed micelle formation in bile salt-lecithin solutions. Biochemistry 19(4):601–615 [DOI] [PubMed] [Google Scholar]
- 126.Mazer NA, Schurtenberg P, Carey MC, Preisig R, Weigand K, Kanzig W (1984) Quasi-elastic light scattering studies of native hepatic bile from the dog: comparison with aggregative behavior of model biliary lipid systems. Biochemistry 23(9):1994–2005 [DOI] [PubMed] [Google Scholar]
- 127.Somjen GJ, Gilat T (1985) Contribution of vesicular and micellar carriers to cholesterol transport in human bile. J Lipid Res 26(6):699–704 [PubMed] [Google Scholar]
- 128.Somjen GJ, Gilat T (1983) A non-micellar mode of cholesterol transport in human bile. FEBS Lett 156(2):265–268 [DOI] [PubMed] [Google Scholar]
- 129.Bhattacharyya AK, Connor WE (1974) Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest 53(4):1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Berge KE (2003) Sitosterolemia: a gateway to new knowledge about cholesterol metabolism. Ann Med 35(7):502–511 [DOI] [PubMed] [Google Scholar]
- 131.Berge KE, von Bergmann K, Lutjohann D, Guerra R, Grundy SM, Hobbs HH, Cohen JC (2002) Heritability of plasma noncholesterol sterols and relationship to DNA sequence polymorphism in ABCG5 and ABCG8. J Lipid Res 43(3):486–494 [PubMed] [Google Scholar]
- 132.Salen G, Patel S, Batta AK (2002) Sitosterolemia. Cardiovas Drug Rev 20(4):255–270 [DOI] [PubMed] [Google Scholar]
- 133.Salen G, Shefer S, Nguyen L, Ness GC, Tint GS, Batta AK (1997) Sitosterolemia. Subcell Biochem 28:453–476 [DOI] [PubMed] [Google Scholar]
- 134.Salen G, Shefer S, Nguyen L, Ness GC, Tint GS, Shore V (1992) Sitosterolemia. J Lipid Res 33(7):945–955 [PubMed] [Google Scholar]
- 135.Salen G, Shore V, Tint GS, Forte T, Shefer S, Horak I, Horak E, Dayal B, Nguyen L, Batta AK et al. (1989) Increased sitosterol absorption, decreased removal, and expanded body pools compensate for reduced cholesterol synthesis in sitosterolemia with xanthomatosis. J Lipid Res 30(9):1319–1330 [PubMed] [Google Scholar]
- 136.Lu K, Lee MH, Yu H, Zhou Y, Sandell SA, Salen G, Patel SB (2002) Molecular cloning, genomic organization, genetic variations, and characterization of murine sterolin genes Abcg5 and Abcg8. J Lipid Res 43(4):565–578 [PMC free article] [PubMed] [Google Scholar]
- 137.Lu K, Lee MH, Hazard S, Brooks-Wilson A, Hidaka H, Kojima H, Ose L, Stalenhoef AF, Mietinnen T, Bjorkhem I et al. (2001) Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am J Hum Genet 69(2):278–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nguyen LB, Shefer S, Salen G, Ness GC, Tint GS, Zaki FG, Rani I (1990) A molecular defect in hepatic cholesterol biosynthesis in sitosterolemia with xanthomatosis. J Clin Invest 86(3):923–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Salen G, Horak I, Rothkopf M, Cohen JL, Speck J, Tint GS, Shore V, Dayal B, Chen T, Shefer S (1985) Lethal atherosclerosis associated with abnormal plasma and tissue sterol composition in sitosterolemia with xanthomatosis. J Lipid Res 26(9):1126–1133 [PubMed] [Google Scholar]
- 140.Yu L, Gupta S, Xu F, Liverman AD, Moschetta A, Mangelsdorf DJ, Repa JJ, Hobbs HH, Cohen JC (2005) Expression of ABCG5 and ABCG8 is required for regulation of biliary cholesterol secretion. J Biol Chem 280(10):8742–8747 [DOI] [PubMed] [Google Scholar]
- 141.Wang HH, Patel SB, Carey MC, Wang DQ (2007) Quantifying anomalous intestinal sterol uptake, lymphatic transport, and biliary secretion in Abcg8(−/−) mice. Hepatology 45(4):998–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Klett EL, Lu K, Kosters A, Vink E, Lee MH, Altenburg M, Shefer S, Batta AK, Yu H, Chen J et al. (2004) A mouse model of sitosterolemia: absence of Abcg8/sterolin-2 results in failure to secrete biliary cholesterol. BMC Med 2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lutjohann D, Bjorkhem I, Beil UF, von Bergmann K (1995) Sterol absorption and sterol balance in phytosterolemia evaluated by deuterium-labeled sterols: effect of sitostanol treatment. J Lipid Res 36(8):1763–1773 [PubMed] [Google Scholar]
- 144.Gould RG, Jones RJ, LeRoy GV, Wissler RW, Taylor CB (1969) Absorbability of beta-sitosterol in humans. Metab Clin Exp 18(8):652–662 [DOI] [PubMed] [Google Scholar]
- 145.Plosch T, Bloks VW, Terasawa Y, Berdy S, Siegler K, Van Der Sluijs F, Kema IP, Groen AK, Shan B, Kuipers F et al. (2004) Sitosterolemia in ABC-transporter G5-deficient mice is aggravated on activation of the liver-X receptor. Gastroenterology 126(1):290–300 [DOI] [PubMed] [Google Scholar]
- 146.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271(5248):518–520 [DOI] [PubMed] [Google Scholar]
- 147.Wang DQ, Carey MC (2002) Susceptibility to murine cholesterol gallstone formation is not affected by partial disruption of the HDL receptor SR-BI. Biochim Biophys Acta 1583(2):141–150 [DOI] [PubMed] [Google Scholar]
- 148.Kozarsky KF, Donahee MH, Rigotti A, Iqbal SN, Edelman ER, Krieger M (1997) Overexpression of the HDL receptor SR-BI alters plasma HDL and bile cholesterol levels. Nature 387(6631):414–417 [DOI] [PubMed] [Google Scholar]
- 149.Wang DQ, Cohen DE (2015) In: Ballantyne CM (ed) Clinical lipidology: a companion to Braunwald’s heart disease. Elsevier Saunders, Philadephia, pp 25–42 [Google Scholar]
- 150.Altmann SW, Davis HR Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Iyer SP, Maguire M, Golovko A, Zeng M et al. (2004) Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 303(5661):1201–1204 [DOI] [PubMed] [Google Scholar]
- 151.Davis HR Jr, Altmann SW (2009) Niemann-Pick C1 Like 1 (NPC1L1) an intestinal sterol transporter. Biochim Biophys Acta 1791(7):679–683 [DOI] [PubMed] [Google Scholar]
- 152.Davis HR Jr, Basso F, Hoos LM, Tetzloff G, Lally SM, Altmann SW (2008) Cholesterol homeostasis by the intestine: lessons from Niemann-Pick C1 Like 1 [NPC1L1]. Atheroscler Suppl 9(2):77–81 [DOI] [PubMed] [Google Scholar]
- 153.Davis HR Jr, Zhu LJ, Hoos LM, Tetzloff G, Maguire M, Liu J, Yao X, Iyer SP, Lam MH, Lund EG et al. (2004) Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem 279(32):33586–33592 [DOI] [PubMed] [Google Scholar]
- 154.Garcia-Calvo M, Lisnock J, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HR Jr, Dean DC, Detmers PA et al. (2005) The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc Natl Acad Sci U S A 102(23):8132–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Graf GA, Cohen JC, Hobbs HH (2004) Missense mutations in ABCG5 and ABCG8 disrupt heterodimerization and trafficking. J Biol Chem 279(23):24881–24888 [DOI] [PubMed] [Google Scholar]
- 156.Hubacek JA, Berge KE, Cohen JC, Hobbs HH (2001) Mutations in ATP-cassette binding proteins G5 (ABCG5) and G8 (ABCG8) causing sitosterolemia. Hum Mutat 18(4):359–360 [DOI] [PubMed] [Google Scholar]
- 157.Duan LP, Wang HH, Ohashi A, Wang DQ (2006) Role of intestinal sterol transporters Abcg5, Abcg8, and Npc1l1 in cholesterol absorption in mice: gender and age effects. Am J Physiol Gastrointest Liver Physiol 290(2):G269–G276 [DOI] [PubMed] [Google Scholar]
- 158.Duan LP, Wang HH, Wang DQ (2004) Cholesterol absorption is mainly regulated by the jejunal and ileal ATP-binding cassette sterol efflux transporters Abcg5 and Abcg8 in mice. J Lipid Res 45(7):1312–1323 [DOI] [PubMed] [Google Scholar]
- 159.Lammert F, Wang DQ (2005) New insights into the genetic regulation of intestinal cholesterol absorption. Gastroenterology 129(2):718–734 [DOI] [PubMed] [Google Scholar]
- 160.Brown MS, Brannan PG, Bohmfalk HA, Brunschede GY, Dana SE, Helgeson J, Goldstein JL (1975) Use of mutant fibroblasts in the analysis of the regulation of cholesterol metabolism in human cells. J Cell Physiol 85(2 Pt 2 Suppl 1):425–436 [DOI] [PubMed] [Google Scholar]
- 161.Brown MS, Dana SE, Dietschy JM, Siperstein MD (1973) 3-Hydroxy-3-methylglutaryl coenzyme A reductase. Solubilization and purification of a cold-sensitive microsomal enzyme. J Biol Chem 248(13):4731–4738 [PubMed] [Google Scholar]
- 162.Brown MS, Dana SE, Goldstein JL (1975) Receptor-dependent hydrolysis of cholesteryl esters contained in plasma low density lipoprotein. Proc Natl Acad Sci U S A 72(8):2925–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brown MS, Dana SE, Goldstein JL (1975) Cholesterol ester formation in cultured human fibroblasts. Stimulation by oxygenated sterols. J Biol Chem 250(10):4025–4027 [PubMed] [Google Scholar]
- 164.Brown MS, Dana SE, Goldstein JL (1974) Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem 249(3):789–796 [PubMed] [Google Scholar]
- 165.Brown MS, Dana SE, Goldstein JL (1973) Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts by lipoproteins. Proc Natl Acad Sci U S A 70(7):2162–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Dietschy JM, Spady DK (1984) Measurement of rates of cholesterol synthesis using tritiated water. J Lipid Res 25(13):1469–1476 [PubMed] [Google Scholar]
- 167.Dietschy JM, Spady DK (1984) Regulation of low density lipoprotein uptake and degradation in different animals species. Agents Actions Suppl 16:177–190 [DOI] [PubMed] [Google Scholar]
- 168.Dietschy JM, Spady DK, Stange EF (1983) Quantitative importance of different organs for cholesterol synthesis and low-density-lipoprotein degradation. Biochem Soc Trans 11(6):639–641 [DOI] [PubMed] [Google Scholar]
- 169.Dietschy JM, Turley SD, Spady DK (1993) Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res 34(10):1637–1659 [PubMed] [Google Scholar]
- 170.Turley SD, Spady DK, Dietschy JM (1997) Identification of a metabolic difference accounting for the hyper- and hyporesponder phenotypes of cynomolgus monkey. J Lipid Res 38(8):1598–1611 [PubMed] [Google Scholar]
- 171.Turley SD, Spady DK, Dietschy JM (1997) Regulation of fecal bile acid excretion in male golden Syrian hamsters fed a cereal-based diet with and without added cholesterol. Hepatology 25(4):797–803 [DOI] [PubMed] [Google Scholar]
- 172.Turley SD, Spady DK, Dietschy JM (1995) Role of liver in the synthesis of cholesterol and the clearance of low density lipoproteins in the cynomolgus monkey. J Lipid Res 36(1):67–79 [PubMed] [Google Scholar]
- 173.Spady DK, Dietschy JM (1983) Sterol synthesis in vivo in 18 tissues of the squirrel monkey, guinea pig, rabbit, hamster, and rat. J Lipid Res 24(3):303–315 [PubMed] [Google Scholar]
- 174.Heinemann T, Axtmann G, von Bergmann K (1993) Comparison of intestinal absorption of cholesterol with different plant sterols in man. Eur J Clin Investig 23(12):827–831 [DOI] [PubMed] [Google Scholar]
- 175.McNamara DJ, Kolb R, Parker TS, Batwin H, Samuel P, Brown CD, Ahrens EH Jr (1987) Heterogeneity of cholesterol homeostasis in man. Response to changes in dietary fat quality and cholesterol quantity. J Clin Invest 79(6):1729–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kesaniemi YA, Miettinen TA (1987) Cholesterol absorption efficiency regulates plasma cholesterol level in the Finnish population. Eur J Clin Investig 17(5):391–395 [DOI] [PubMed] [Google Scholar]
- 177.Sehayek E, Nath C, Heinemann T, McGee M, Seidman CE, Samuel P, Breslow JL (1998) U-shape relationship between change in dietary cholesterol absorption and plasma lipoprotein responsiveness and evidence for extreme interindividual variation in dietary cholesterol absorption in humans. J Lipid Res 39(12):2415–2422 [PubMed] [Google Scholar]
- 178.Miettinen TA, Tilvis RS, Kesaniemi YA (1990) Serum plant sterols and cholesterol precursors reflect cholesterol absorption and synthesis in volunteers of a randomly selected male population. Am J Epidemiol 131(1):20–31 [DOI] [PubMed] [Google Scholar]
- 179.Bosner MS, Lange LG, Stenson WF, Ostlund RE Jr (1999) Percent cholesterol absorption in normal women and men quantified with dual stable isotopic tracers and negative ion mass spectrometry. J Lipid Res 40(2):302–308 [PubMed] [Google Scholar]
- 180.Hallikainen MA, Sarkkinen ES, Gylling H, Erkkila AT, Uusitupa MI (2000) Comparison of the effects of plant sterol ester and plant stanol ester-enriched margarines in lowering serum cholesterol concentrations in hypercholesterolaemic subjects on a low-fat diet. Eur J Clin Nutr 54(9):715–725 [DOI] [PubMed] [Google Scholar]
- 181.Gremaud G, Dalan E, Piguet C, Baumgartner M, Ballabeni P, Decarli B, Leser ME, Berger A, Fay LB (2002) Effects of non-esterified stanols in a liquid emulsion on cholesterol absorption and synthesis in hypercholesterolemic men. Eur J Nutr 41(2):54–60 [DOI] [PubMed] [Google Scholar]
- 182.Maki KC, Davidson MH, Umporowicz DM, Schaefer EJ, Dicklin MR, Ingram KA, Chen S, McNamara JR, Gebhart BW, Ribaya-Mercado JD et al. (2001) Lipid responses to plant-sterol-enriched reduced-fat spreads incorporated into a National Cholesterol Education Program Step I diet. Am J Clin Nutr 74(1):33–43 [DOI] [PubMed] [Google Scholar]
- 183.Miettinen TA, Puska P, Gylling H, Vanhanen H, Vartiainen E (1995) Reduction of serum cholesterol with sitostanol-ester margarine in a mildly hypercholesterolemic population. N Engl J Med 333(20):1308–1312 [DOI] [PubMed] [Google Scholar]
- 184.Wang DQ, Neuschwander-Tetri BA, Portincasa P (2017) The biliary system. Morgan & Claypool Life Sciences, Princeton [Google Scholar]
- 185.Kosters A, Frijters RJ, Schaap FG, Vink E, Plosch T, Ottenhoff R, Jirsa M, De Cuyper IM, Kuipers F, Groen AK (2003) Relation between hepatic expression of ATP-binding cassette transporters G5 and G8 and biliary cholesterol secretion in mice. J Hepatol 38(6):710–716 [DOI] [PubMed] [Google Scholar]
- 186.Wang HH, Lammert F, Schmitz A, Wang DQ (2010) Transgenic overexpression of Abcb11 enhances biliary bile salt outputs, but does not affect cholesterol cholelithogenesis in mice. Eur J Clin Investig 40(6):541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Glomset JA, Janssen ET, Kennedy R, Dobbins J (1966) Role of plasma lecithin: cholesterol acyltransferase in the metabolism of high density lipoproteins. J Lipid Res 7(5):638–648 [PubMed] [Google Scholar]
- 188.Rader DJ (2014) New therapies for coronary artery disease: genetics provides a blueprint. Sci Transl Med 6(239):239ps4. [DOI] [PubMed] [Google Scholar]
- 189.Tuteja S, Rader DJ (2014) High-density lipoproteins in the prevention of cardiovascular disease: changing the paradigm. Clin Pharmacol Ther 96(1):48–56 [DOI] [PubMed] [Google Scholar]
- 190.Rader DJ, Hovingh GK (2014) HDL and cardiovascular disease. Lancet 384(9943):618–625 [DOI] [PubMed] [Google Scholar]
- 191.Rader DJ (2014) Spotlight on HDL biology: new insights in metabolism, function, and translation. Cardiovasc Res 103(3):337–340 [DOI] [PubMed] [Google Scholar]
- 192.Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ et al. (2012) Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 125(15):1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lewis GF, Rader DJ (2005) New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res 96(12):1221–1232 [DOI] [PubMed] [Google Scholar]
- 194.Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH (2009) The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res 50(Suppl):S189–S194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Khera AV, Rader DJ (2010) Future therapeutic directions in reverse cholesterol transport. Curr Atheroscler Rep 12(1):73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Degoma EM, Rader DJ (2011) Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol 8(5):266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rader DJ (2006) Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest 116(12):3090–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rosenson RS, Brewer HB Jr, Ansell B, Barter P, Chapman MJ, Heinecke JW, Kontush A, Tall AR, Webb NR (2013) Translation of high-density lipoprotein function into clinical practice: current prospects and future challenges. Circulation 128(11):1256–1267 [DOI] [PubMed] [Google Scholar]
- 199.Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, D’Agostino RB Sr, Davidson MH, Davidson WS, Heinecke JW et al. (2013) High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol 7(5):484–525 [DOI] [PubMed] [Google Scholar]
- 200.Birner-Gruenberger R, Schittmayer M, Holzer M, Marsche G (2014) Understanding high-density lipoprotein function in disease: recent advances in proteomics unravel the complexity of its composition and biology. Prog Lipid Res 56C:36–46 [DOI] [PubMed] [Google Scholar]
- 201.Stanley MM, Pineda EP, Cheng SH (1959) Serum cholesterol esters and intestinal cholesterol secretion and absorption in obstructive jaundice due to cancer. N Engl J Med 261:368–373 [DOI] [PubMed] [Google Scholar]
- 202.Kruit JK, Plosch T, Havinga R, Boverhof R, Groot PH, Groen AK, Kuipers F (2005) Increased fecal neutral sterol loss upon liver X receptor activation is independent of biliary sterol secretion in mice. Gastroenterology 128(1):147–156 [DOI] [PubMed] [Google Scholar]
- 203.Temel RE, Sawyer JK, Yu L, Lord C, Degirolamo C, McDaniel A, Marshall S, Wang N, Shah R, Rudel LL et al. (2010) Biliary sterol secretion is not required for macrophage reverse cholesterol transport. Cell Metab 12(1):96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.de Boer JF, Schonewille M, Boesjes M, Wolters H, Bloks VW, Bos T, van Dijk TH, Jurdzinski A, Boverhof R, Wolters JC et al. (2017) Intestinal farnesoid X receptor controls transintestinal cholesterol excretion in mice. Gastroenterology 152(5):1126–1138. e6 [DOI] [PubMed] [Google Scholar]
- 205.de Boer JF, Schonewille M, Dikkers A, Koehorst M, Havinga R, Kuipers F, Tietge UJ, Groen AK (2017) Transintestinal and biliary cholesterol secretion both contribute to macrophage reverse cholesterol transport in rats-brief report. Arterioscler Thromb Vasc Biol 37(4):643–646 [DOI] [PubMed] [Google Scholar]
- 206.Le May C, Berger JM, Lespine A, Pillot B, Prieur X, Letessier E, Hussain MM, Collet X, Cariou B, Costet P (2013) Transintestinal cholesterol excretion is an active metabolic process modulated by PCSK9 and statin involving ABCB1. Arterioscler Thromb Vasc Biol 33(7):1484–1493 [DOI] [PubMed] [Google Scholar]
- 207.Jakulj L, van Dijk TH, de Boer JF, Kootte RS, Schonewille M, Paalvast Y, Boer T, Bloks VW, Boverhof R, Nieuwdorp M et al. (2016) Transintestinal cholesterol transport is active in mice and humans and controls ezetimibe-induced fecal neutral sterol excretion. Cell Metab 24(6):783–794 [DOI] [PubMed] [Google Scholar]
- 208.Wang DQ, Portincasa P, Tso P (2017) Transintestinal cholesterol excretion (TICE): a secondary, non-biliary pathway contributing to reverse cholesterol transport. Hepatology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Shulman RS, Bhattacharyya AK, Connor WE, and Fredrickson DS. Beta-sitosterolemia and xanthomatosis. N Engl J Med 1976;294(9):482–3. [DOI] [PubMed] [Google Scholar]
- 210.Hidaka H, Nakamura T, Aoki T, Kojima H, Nakajima Y, Kosugi K, Hatanaka I, Harada M, Kobayashi M, Tamura A et al. (1990) Increased plasma plant sterol levels in heterozygotes with sitosterolemia and xanthomatosis. J Lipid Res 31(5):881–888 [PubMed] [Google Scholar]
- 211.Cobb MM, Salen G, Tint GS (1997) Comparative effect of dietary sitosterol on plasma sterols and cholesterol and bile acid synthesis in a sitosterolemic homozygote and heterozygote subject. J Am Coll Nutr 16(6):605–613 [PubMed] [Google Scholar]
- 212.Connor WE, Lin DS, Pappu AS, Frohlich J, Gerhard G (2005) Dietary sitostanol and campestanol: accumulation in the blood of humans with sitosterolemia and xanthomatosis and in rat tissues. Lipids 40(9):919–923 [DOI] [PubMed] [Google Scholar]
- 213.Salen G, Kwiterovich PO Jr, Shefer S, Tint GS, Horak I, Shore V, Dayal B, Horak E (1985) Increased plasma cholestanol and 5 alpha-saturated plant sterol derivatives in subjects with sitosterolemia and xanthomatosis. J Lipid Res 26(2):203–209 [PubMed] [Google Scholar]
- 214.Patel SB, Salen G, Hidaka H, Kwiterovich PO, Stalenhoef AF, Miettinen TA, Grundy SM, Lee MH, Rubenstein JS, Polymeropoulos MH et al. (1998) Mapping a gene involved in regulating dietary cholesterol absorption. The sitosterolemia locus is found at chromosome 2p21. J Clin Invest 102(5):1041–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Salen G, Tint GS, Shefer S, Shore V, Nguyen L (1992) Increased sitosterol absorption is offset by rapid elimination to prevent accumulation in heterozygotes with sitosterolemia. Arterioscler Thromb 12(5):563–568 [DOI] [PubMed] [Google Scholar]
- 216.Robins SJ, Fasulo JM (1997) High density lipoproteins, but not other lipoproteins, provide a vehicle for sterol transport to bile. J Clin Invest 99(3):380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Belamarich PF, Deckelbaum RJ, Starc TJ, Dobrin BE, Tint GS, Salen G (1990) Response to diet and cholestyramine in a patient with sitosterolemia. Pediatrics 86(6):977–981 [PubMed] [Google Scholar]
- 218.Nguyen LB, Cobb M, Shefer S, Salen G, Ness GC, Tint GS (1991) Regulation of cholesterol biosynthesis in sitosterolemia: effects of lovastatin, cholestyramine, and dietary sterol restriction. J Lipid Res 32(12):1941–1948 [PubMed] [Google Scholar]
- 219.Cobb MM, Salen G, Tint GS, Greenspan J, Nguyen LB (1996) Sitosterolemia: opposing effects of cholestyramine and lovastatin on plasma sterol levels in a homozygous girl and her heterozygous father. Metab Clin Exp 45(6):673–679 [DOI] [PubMed] [Google Scholar]
- 220.Parsons HG, Jamal R, Baylis B, Dias VC, Roncari D (1995) A marked and sustained reduction in LDL sterols by diet and cholestyramine in beta-sitosterolemia. Clin Invest Med 18(5):389–400 [PubMed] [Google Scholar]
- 221.Hidaka H, Kojima H, Kawabata T, Nakamura T, Konaka K, Kashiwagi A, Kikkawa R, Shigeta Y (1995) Effects of an HMG-CoA reductase inhibitor, pravastatin, and bile sequestering resin, cholestyramine, on plasma plant sterol levels in hypercholesterolemic subjects. J Atheroscler Thromb 2(1):60–65 [DOI] [PubMed] [Google Scholar]
- 222.Salen G, Starc T, Sisk CM, Patel SB (2006) Intestinal cholesterol absorption inhibitor ezetimibe added to cholestyramine for sitosterolemia and xanthomatosis. Gastroenterology 130(6):1853–1857 [DOI] [PubMed] [Google Scholar]
- 223.Salen G, von Bergmann K, Lutjohann D, Kwiterovich P, Kane J, Patel SB, Musliner T, Stein P, Musser B (2004) Ezetimibe effectively reduces plasma plant sterols in patients with sitosterolemia. Circulation 109(8):966–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Tsubakio-Yamamoto K, Nishida M, Nakagawa- Toyama Y, Masuda D, Ohama T, Yamashita S (2010) Current therapy for patients with sitosterolemia – effect of ezetimibe on plant sterol metabolism. J Atheroscler Thromb 17(9):891–900 [DOI] [PubMed] [Google Scholar]
- 225.Iyer SP, Yao X, Crona JH, Hoos LM, Tetzloff G, Davis HR Jr, Graziano MP, Altmann SW (2005) Characterization of the putative native and recombinant rat sterol transporter Niemann-Pick C1 Like 1 (NPC1L1) protein. Biochim Biophys Acta 1722(3):282–292 [DOI] [PubMed] [Google Scholar]
- 226.Sudhop T, Lutjohann D, Kodal A, Igel M, Tribble DL, Shah S, Perevozskaya I, von Bergmann K (2002) Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation 106(15):1943–1948 [DOI] [PubMed] [Google Scholar]
- 227.Sudhop T, von Bergmann K (2002) Cholesterol absorption inhibitors for the treatment of hypercholesterolaemia. Drugs 62(16):2333–2347 [DOI] [PubMed] [Google Scholar]
- 228.Pandor A, Ara RM, Tumur I, Wilkinson AJ, Paisley S, Duenas A, Durrington PN, Chilcott J (2009) Ezetimibe monotherapy for cholesterol lowering in 2,722 people: systematic review and meta-analysis of randomized controlled trials. J Intern Med 265(5):568–580 [DOI] [PubMed] [Google Scholar]
- 229.Lusis AJ (2000) Atherosclerosis. Nature 407 (6801):233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Emerging Risk Factors Collaboration, Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N et al. (2009) Major lipids, apolipoproteins, and risk of vascular disease. JAMA 302(18):1993–2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Allen JK (2000) Genetics and cardiovascular disease. Nurs Clin North Am 35(3):653–662 [PubMed] [Google Scholar]
- 232.Chico TJ, Milo M, Crossman DC (2010) The genetics of cardiovascular disease: new insights from emerging approaches. J Pathol 220(2):186–197 [DOI] [PubMed] [Google Scholar]
- 233.Cymet T (2010) Genetics of cardiovascular disease. Compr Ther 36:18–19 [PubMed] [Google Scholar]
- 234.Hajar R (2020) Genetics in cardiovascular disease. Heart Views 21(1):55–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kathiresan S, Srivastava D (2012) Genetics of human cardiovascular disease. Cell 148(6):1242–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Katsuya T, Harrap SB, Ogihara T (2011) Genetics of hypertension and cardiovascular disease. Int J Hypertens 2010:951254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sabeva NS, Liu J, Graf GA (2009) The ABCG5 ABCG8 sterol transporter and phytosterols: implications for cardiometabolic disease. Curr Opin Endocrinol Diabetes Obes 16(2):172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Klett EL, Patel S (2003) Genetic defenses against noncholesterol sterols. Curr Opin Lipidol 14(4):341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Patel SB, Graf GA, Temel RE (2018) ABCG5 and ABCG8: more than a defense against xenosterols. J Lipid Res 59(7):1103–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Patel SB (2014) Recent advances in understanding the STSL locus and ABCG5/ABCG8 biology. Curr Opin Lipidol 25(3):169–175 [DOI] [PubMed] [Google Scholar]
- 241.Wilund KR, Yu L, Xu F, Hobbs HH, Cohen JC (2004) High-level expression of ABCG5 and ABCG8 attenuates diet-induced hypercholesterolemia and atherosclerosis in Ldlr−/− mice. J Lipid Res 45(8):1429–1436 [DOI] [PubMed] [Google Scholar]
- 242.Wu JE, Basso F, Shamburek RD, Amar MJ, Vaisman B, Szakacs G, Joyce C, Tansey T, Freeman L, Paigen BJ et al. (2004) Hepatic ABCG5 and ABCG8 overexpression increases hepatobiliary sterol transport but does not alter aortic atherosclerosis in transgenic mice. J Biol Chem 279 (22):22913–22925 [DOI] [PubMed] [Google Scholar]
- 243.Basso F, Freeman LA, Ko C, Joyce C, Amar MJ, Shamburek RD, Tansey T, Thomas F, Wu J, Paigen B et al. (2007) Hepatic ABCG5/G8 overexpression reduces apoB-lipoproteins and atherosclerosis when cholesterol absorption is inhibited. J Lipid Res 48(1):114–126 [DOI] [PubMed] [Google Scholar]
- 244.Hubacek JA, Berge KE, Stefkova J, Pitha J, Skodova Z, Lanska V, Poledne R (2004) Polymorphisms in ABCG5 and ABCG8 transporters and plasma cholesterol levels. Physiol Res/Academia Scientiarum Bohemoslovaca 53(4):395–401 [PubMed] [Google Scholar]
- 245.Gylling H, Hallikainen M, Pihlajamaki J, Agren J, Laakso M, Rajaratnam RA, Rauramaa R, Miettinen TA (2004) Polymorphisms in the ABCG5 and ABCG8 genes associate with cholesterol absorption and insulin sensitivity. J Lipid Res 45(9):1660–1665 [DOI] [PubMed] [Google Scholar]
- 246.Jakulj L, Vissers MN, Tanck MW, Hutten BA, Stellaard F, Kastelein JJ, Dallinga-Thie GM (2010) ABCG5/G8 polymorphisms and markers of cholesterol metabolism: systematic review and meta-analysis. J Lipid Res 51(10):3016–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cholesterol Treatment Trialists’ (CTT) Collaboration, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A et al. (2010) Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet 376(9753):1670–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, Pasternak RC, Smith SC Jr, Stone NJ (2004) Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 110(2):227–239 [DOI] [PubMed] [Google Scholar]
- 249.Barter PJ, Brewer HB Jr, Chapman MJ, Hennekens CH, Rader DJ, Tall AR (2003) Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol 23(2):160–167 [DOI] [PubMed] [Google Scholar]
- 250.Wang HH, Portincasa P, Wang DQ (2008) Molecular pathophysiology and physical chemistry of cholesterol gallstones. Front Biosci 13:401–423 [DOI] [PubMed] [Google Scholar]
- 251.Wang HH, Li T, Portincasa P, Ford DA, Neuschwander-Tetri BA, Tso P, Wang DQ (2017) New insights into the role of Lith genes in the formation of cholesterol-supersaturated bile. Liver Res 1(1):42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Wittenburg H, Lyons MA, Li R, Kurtz U, Mossner J, Churchill GA, Carey MC, Paigen B (2005) Association of a lithogenic Abcg5/Abcg8 allele on Chromosome 17 (Lith9) with cholesterol gallstone formation in PERA/EiJ mice. Mamm Genome 16(7):495–504 [DOI] [PubMed] [Google Scholar]
- 253.Wittenburg H, Lyons MA, Paigen B, Carey MC (2003) Mapping cholesterol gallstone susceptibility (Lith) genes in inbred mice. Dig Liver Dis 35(Suppl 3):S2–S7 [DOI] [PubMed] [Google Scholar]
- 254.Jiang ZY, Parini P, Eggertsen G, Davis MA, Hu H, Suo GJ, Zhang SD, Rudel LL, Han TQ, Einarsson C (2008) Increased expression of LXR alpha, ABCG5, ABCG8, and SR-BI in the liver from normolipidemic, nonobese Chinese gallstone patients. J Lipid Res 49(2):464–472 [DOI] [PubMed] [Google Scholar]
- 255.von Schonfels W, Buch S, Wolk M, Aselmann H, Egberts JH, Schreiber S, Krawczak M, Becker T, Hampe J, Schafmayer C (2013) Recurrence of gallstones after cholecystectomy is associated with ABCG5/8 genotype. J Gastroenterol 48(3):391–396 [DOI] [PubMed] [Google Scholar]
- 256.Wang HH, Li X, Patel SB, Wang DQ (2016) Evidence that the adenosine triphosphate-binding cassette G5/G8-independent pathway plays a determinant role in cholesterol gallstone formation in mice. Hepatology 64(3):853–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ (2002) Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J Biol Chem 277(21):18793–18800 [DOI] [PubMed] [Google Scholar]
- 258.Yu L, York J, von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2003) Stimulation of cholesterol excretion by the liver X receptor agonist requires ATP-binding cassette transporters G5 and G8. J Biol Chem 278(18):15565–15570 [DOI] [PubMed] [Google Scholar]
- 259.Uppal H, Zhai Y, Gangopadhyay A, Khadem S, Ren S, Moser JA, Xie W (2008) Activation of liver X receptor sensitizes mice to gallbladder cholesterol crystallization. Hepatology 47(4):1331–1342 [DOI] [PubMed] [Google Scholar]