

Abstract
The lymphoid tyrosine phosphatase (LYP), encoded by the PTPN22 gene, has recently been identified as a promising drug target for human autoimmunity diseases. Like the majority of protein-tyrosine phosphatases LYP can adopt two functionally distinct forms determined by the conformation of the WPD-loop. The WPD-loop plays an important role in the catalytic dephosphorylation by protein-tyrosine phosphatases. Here we investigate the binding modes of two chemotypes of small molecule LYP inhibitors with respect to both protein conformations using computational modeling. To evaluate binding in the active form, we built a LYP protein structure model of high quality. Our results suggest that the two different compound classes investigated, bind to different conformations of the LYP phosphatase domain. Binding to the closed form is facilitated by an interaction with Asp195 in the WPD-loop, presumably stabilizing the active conformation. The analysis presented here is relevant for the design of inhibitors that specifically target either the closed or the open conformation of LYP in order to achieve better selectivity over phosphatases with similar binding sites.
Keywords: Protein tyrosine phosphorylation, PTPN22, LYP, Drug design, Docking, Homology modeling
Introduction
Protein tyrosine phosphorylation [1] is a key process in many essential physiological functions including cell growth, proliferation and differentiation, metabolism, cell cycle regulation, cell–cell interactions, neuronal development, gene transcription, and the immune response [2-4]. Dysregulated tyrosine phosphorylation is associated with many human diseases such as cancer, diabetes, hypertension, obesity, and inflammatory disorders [3-5]. Tyrosine phosphorylation levels are controlled by the coordinated actions of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) [3-6]. PTKs catalyze the phosphorylation of specific substrates at the 4-hydroxy group of tyrosyl moieties by ATP while PTPs remove the phosphoryl group from the phosphorylated tyrosines [4]. The importance of PTKs has been established for the last two decades with many clinical kinase drug targets. In contrast, only recently PTPs have equally been recognized as critical regulators of signal transduction and are no longer seen as passive housekeeping enzymes [6-8].
PTPs are characterized by the active site signature (H/V)C(X)5R(S/T) motif in the conserved PTP catalytic domain. This motif is responsible for PTPs’ catalytic activity, which is initiated by the cysteine thiol nucleophile attacking the phosphate ester [9]. The formed intermediate is then hydrolyzed with assistance of the catalytic aspartic acid located in the conserved WPD-loop followed by release of the dephosphorylated substrate. PTPs’ catalytic activity requires the WPD-loop in the active conformation—in vicinity to the catalytic cysteine. This active conformation is often called closed conformation referring to the binding site geometry changing from a shallow and open pocket to a deeper and closed pocket. The closed (active) conformation occurs in the presence of a substrate ligand (ligand-induced conformational change). Accordingly, the inactive form of PTPs is referred to as the open conformation [10, 11].
The lymphoid-specific phosphatase (LYP) encoded by the PTPN22 gene is a 105-kDa protein consisting of an N-terminal phosphatase domain and a noncatalytic C terminus with several proline-rich motifs [12, 13]. It belongs to the classical non-receptor PTPs. LYP is expressed exclusively in the cells of hematopoietic origin, predominantly in T cells where it acts as an inhibitor to downregulate T cell activation through dephosphorylation of the T cell receptor (TCR)-associated kinases LCK and ZAP70, as well as immunoreceptor tyrosine-based activation motifs (ITAMs) of the TCR/CD3 complex [13-15]. It also binds to the C-terminal Src tyrosine kinase (CSK) [16], which is an important suppressor of kinases that mediate T-cell activation [12]. It has also been demonstrated that LYP binds to the adaptor molecule GRB2 (growth factor receptor-bound protein 2), and this interaction is also thought to play a negative regulatory role in T-cell signaling [17].
Genetic studies have shown that a single-nucleotide polymorphism in PTPN22 at nucleotide 1,858 (C-to-T substitution) is associated with a number of autoimmune diseases including type 1 diabetes [16], rheumatoid arthritis [18], Graves disease [19], and systemic lupus erythematosus [20]. The autoimmune-associated allele changes the amino acid at position 620 from arginine to tryptophan, disrupting the proline-rich binding motif that is important for LYP to bind to both CSK and GRB2 [16-18]. Additionally it has been shown that the W620 variant of LYP is a gain-of-function mutation, generating a more active enzyme that inhibits T cell signaling to a higher extent than the R620 LYP variant [21]. Given the strong association of the C1858T polymorphism with various autoimmunity disorders and the elevated phosphatase activity associated with the R620W protein variant, LYP is currently considered a promising drug target for a broad spectrum of autoimmune diseases.
In order to identify small molecule inhibitors of LYP, a series of high throughput screens were performed at Columbia University as part of the Molecular Library Screening Center Network (MLSCN) of the NIH Molecular Libraries Initiative. Active series were identified and further optimized in a medicinal chemistry program. Two main chemotypes emerged, a series of thiazolidinedione-derived [22] and a series of 6-hydroxybenzofuran-5-carboxylic acid [23] inhibitors. Here we employed structure-based computational modeling to evaluate and analyze the binding modes of these two different classes of compounds. We wanted to understand the distinctions of their protein–ligand interactions with respect to the different conformations of LYP. Our hypothesis was that thiazolidinedione derivatives bind to the closed LYP conformation while 6-hydroxybenzofuran-5-carboxylic acid inhibitors bind to the open LYP conformation. This hypothesis is illustrated in Fig. 1.
Fig. 1.

Hypothesis: (a) thiazolidinedione type inhibitors bind to the closed, and (b) benzofuran salicylic acid inhibitors bind to the open LYP conformation
Materials and methods
Small molecule ligands
Several high throughput screens to identify small molecule LYP inhibitors were performed at Columbia University as part of the Molecular Library Screening Center Network (MLSCN) of the NIH Roadmap for Medical Research. Assay description and screening results were deposited to PubChem (AIDs 606, 640, 1253, and 1338). From the NIH compound libraries, one active series with several hits that share a thiazolidinedione motif was identified. These compounds were further optimized by a fragment-based approach with a total of 25 thiazolidinedione analogs being synthesized and tested. Seventeen of them showed potency toward LYP (IC50 < 44 μM) [22]. Their structures and activities (IC50 in μM units) are given in Table 1.
Table 1.
Set of 17 thiazolidinedione-derived LYP inhibitors and experimental IC50 values [22]
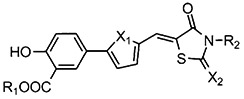 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | X1 | X2 | IC50, μM |
| 408 | CH3 | CH2CO2H | O | S | 21.0 |
| 409 | CH3 | (CH2)2CO2CH3 | O | O | 44.0 |
| 412 | H | CH2CO2H | O | S | 8.7 |
| 413 | H | (CH2)2CO2H | O | O | 5.1 |
| 414 | H | (CH2)2CO2H | S | O | 5.9 |
| 415 | H | CH2CO2H | S | O | 2.8 |
| 416 | H | CH2CO2H | S | S | 5.4 |
| 417 | CH3 | CH2CO2H | S | S | 4.3 |
| 422 | CH3 | CH2CO2H | S | O | 18 |
| 431 | H | CH2CO2H | O | O | 5.6 |
| 443 | CH3 | (CH2)2CO2H | O | O | 4.3 |
| 444 | CH3 | (CH2)2CO2H | S | O | 0.39 |
| 445 | H | CH2CO2CH3 | O | O | 4.8 |
| 446 | H | CH2CO2CH3 | S | O | 1.8 |
| 448 | H | H | O | S | 1.4 |
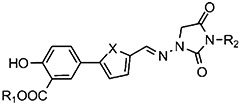 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | X | IC50, μM |
| 430 | H | H | S | 34 |
| 450 | H | CH2CO2H | O | 36 |
The activities were determined in the presence of Tween 20. Tween 20 is the common name for polyoxyethylene (20) sorbiton-monolaurate; it is a widely used nonionic surfactant preventing aggregate formation.
The thiazolidinedione substructure occurs among selective inhibitors of several proteins [24-27]. In addition, a number of experimental drugs contain this substructure (DrugBank [28] ID: DB04769, DB07503, DB07531, DB07838, DB08177). Based on this prior art and our SAR against LYP (Table 1) as well as the experimental assay conditions (detergent) we assume that the detected inhibition results from the reversible binding rather than an alternative (artifactual) mechanism such as compound aggregation, assay interference, or chemical reactivity (irreversible binding).
The other series of LYP inhibitors investigated here had been developed based on the chemical structure of the previously reported small-molecule LYP inhibitor, I-C11 [29]. A 34 member library of 6-hydroxybenzofuran-5-carboxylic acid-based ligands (I-C11 analogs) was synthesized and subsequently evaluated in a LYP enzyme activity assay [23]. Interestingly, most of the I-C11 analogs showed higher potency than I-C11 itself. Chemical structures of these compounds are given in Table 2.
Table 2.
Set of 35 6-hydroxybenzofuran-5-carboxylic acid inhibitors (34 new analogs and I-C11 as 478) [23]
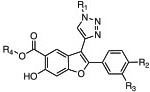 | |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | IC50, μM |
| 478 | H | H | H | 4.50 | |
| 479 | H | H | H | 3.22 | |
| 480 | H | H | H | 1.49 | |
| 481 | H | H | H | 0.56 | |
| 482 | H | H | H | 0.71 | |
| 483 | H | H | H | 3.84 | |
| 486 | H | H | Me | 4.86 | |
| 491 | H | H | H | 3.10 | |
| 492 | H | H | H | 0.77 | |
| 495 | H | H | H | 0.91 | |
| 496 | H | H | H | 2.21 | |
| 499 | H | H | H | 2.64 | |
| 522 | Me | H | H | 0.41 | |
| 523 | Me | H | H | 1.11 | |
| 524 | Me | H | H | 0.64 | |
| 525 | Me | H | H | 0.38 | |
| 526 | Me | H | H | 0.27 | |
| 527 | Me | H | H | 3.44 | |
| 528 | Me | H | H | 0.90 | |
| 529 | Me | H | H | 1.18 | |
| 530 | Me | H | H | 1.35 | |
| 531 | Me | H | H | 2.01 | |
| 532 | Me | H | H | 1.80 | |
| 569 | H | Me | H | 6.20 | |
| 577 | H | Me | H | 2.35 | |
| 584 | MeO | H | H | 0.34 | |
| 586 | MeO | H | H | 0.34 | |
| 604 | H | Me | H | 5.02 | |
| 609 | H | Me | H | 0.65 | |
| 610 | Me | H | H | 0.97 | |
| 611 | H | H | H | 0.74 | |
| 612 | H | Me | H | 0.65 | |
| 619 | Me | H | H | 0.48 | |
| 630 | H | Me | H | 0.52 | |
| 636 | H | Me | H | 0.61 | |
Ligand preparation
All ligands were prepared using LigPrep (Schrödinger LLC) [30] prior to docking. Tautomers were enumerated and protonation states within a pH range of 6 ± 2 were generated. Starting conformations for all ligands were minimized using the OPLS 2005 force field [31] implemented in LigPrep.
LYP structure prediction and receptor preparation
An isothiazolidinone inhibitor (IZD) of protein tyrosine phosphatase 1B (PTP1B) [10] was previously reported and co-crystalized (PDB entry 2cm7, resolution 2.10 Å) [32]. The inhibitor IZD binds to the active form of PTP1B. Our hypothesis was that thiazolidinedione analogs evaluated in the LYP assays bind in a similar mode, i.e. to the active conformation of LYP, because they can interact similarly in the active site. As no structure of the active LYP form was available at the beginning of this study, we built a model for the LYP PTP domain in the closed conformation. Specifically, we developed a hybrid model, combining the available LYP crystal structure in the inactive form (PDB entry 2p6x, resolution 1.90 Å) and the PTP1B structure in the active form (PDB code 2cm7). The template was constructed using the open conformation LYP structure for the PTP domain except the WPD-loop, and the closed conformation PTP1B structure for the region of the WPD-loop. The model of LYP in the active conformation was then generated by aligning the LYP PTP protein sequence to this template using Prime (Schrödinger LLC) [33, 34]. Interestingly a crystal structure of the active form of the LYP phosphatase domain was recently resolved and deposited to the PDB (PDB entry 3brh, 2.20 Å). However, this C227S-mutation structure is missing the peptide bond between His196 and Asp197 and does not resolve the Asp195 side chain in the WPD (the residue that plays a crucial role in the catalytic reaction). Hence, we used our model for the docking studies.
In order to investigate ligand binding to the open conformation LYP PTP domain we used a co-crystal structure of LYP with the inhibitor I-C11 (PDB entry 2qct, resolution 2.80 Å), which binds to the inactive form. We selected this PDB structure over the better resolved 2p6x LYP structure, because the studied compounds are analogs of the co-crystallized ligand in 2qct. Both structures have very similar conformations. Rather than performing ensemble docking by using the two protein structures, we accounted for protein flexibility by induced-fit docking (IFD) as further explained below.
Prior to performing docking experiments we corrected several deficiencies of this structure. After removing the ligand to obtain an apo-structure the missing atoms of the Lys32 and Lys42 side chains were predicted using Prime. The resulting (corrected) protein structure was then processed using the protein preparation facility in Maestro [35]. The I-C11 ligand structure was corrected to add the missing phenyl ring and prepared for re-docking using LigPrep as described under Ligand Preparation. The processed ligand was re-docked into the corrected LYP structure and the new protein–ligand complex was minimized using Macromodel with the OPLS-2005 force field. The LYP structure derived from the minimized complex was then used in the docking studies.
Docking
To generate possible binding modes of the two chemical series in the open and closed conformation of the LYP PTP domain, we performed molecular docking studies. All docking simulations were carried out in Glide (Schrödinger LLC) [36, 37]. Glide can generate ligand conformations (rotamers and ring conformations) internally and filter them through a series of calculations. Glide performs grid-based ligand docking and searches for favorable interactions between a ligand and a protein. To direct ligand docking to the catalytic pocket we defined hydrogen bonding constraints to residues around the catalytic cysteine Cys227. The residues included Ser228, Cys231, and Arg233. We required at least one hydrogen bond interaction in the final docking results, because we wanted to focus the analysis on binding poses interacting in the catalytic site while also allowing some degree of binding flexibility. We performed docking using both the Glide standard precision (SP) and extra precision (XP) protocols.
Prior to constraint docking we probed unconstraint docking (no required interactions within the catalytic pocket). However, the resulting ligand poses did not have a direct interaction with the residues in the catalytic pocket leaving the catalytic site exposed to the solvent and potentially to the phosphatase substrates. In addition, in many cases the obtained poses had a lower predicted binding affinity (worse docking scores) than the corresponding results derived from constraint docking, suggesting that they are less favorable. As the binding pocket is relatively open and flat, unconstrained docking may not have exhaustively sampled the conformational space likely missing energetically favorable poses. By introducing specific protein–ligand interaction constraints, which were based on knowledge of PTP—small molecule interactions, the sampling space was considerably reduced. The better docking scores indicated that these poses are more relevant compared to unconstraint docking.
To account for protein (receptor) flexibility we also applied constraint induced fit docking (IFD). IFD is implemented as a workflow in Schrödinger Maestro. It combines the Glide docking algorithm and Prime protein structure optimization to allow the receptor to relax in the presence of the docked ligands, followed by re-docking against the optimized receptor. We defined flexible residues as those within 12 Å around the docked ligand.
Protein–ligand interactions visualization
PyMol [38] was used for three-dimensional visualization of the protein–ligand interactions.
Results and discussion
Closed-conformation LYP model
Because no suitable structure was available when we initiated this study, we generated a hybrid homology model for the LYP closed conformation by combining the available open-form LYP structure with the closed conformation of the PTP1B WPD-loop as described in Materials and Methods. When compared to the recently published crystal structure of LYP in the closed conformation, our model performed very well with a RMSD of 0.506 Å (Cα-atoms aligned) indicating a high quality model. Importantly, our model is complete, comprising all atoms of the LYP PTP domain, in contrast to the experimental structure, which lacks several residues in the WPD-loop that are crucial for our study.
Figure 2 compares our model and the experimental structure (PDB entry 3brh) with focus on the WPD-loop and the signature (H/V)C(X)5R(S/T) motif that are most relevant for docking.
Fig. 2.

Comparison of the closed-conformation LYP PTP domain model (in cyan) and the experimental structure (PDB 3brh, in green); RMSD is 0.506 Å. The PTP signature (H/V)C(X)5R(S/T) motif residues are shown as sticks and the remaining residues as ribbon. The missing residues between His196 and Asp197 (ends shown in pink) of the experimental structure are located in the WPD-loop
Docking of thiazolidinedione inhibitors
To generate and rank plausible binding modes of LYP inhibitors on an atomic resolution and to evaluate our hypothesis of the thiazolidinedione class of ligands binding to the closed LYP conformation, we performed two series of Glide docking runs—to the closed and the open LYP conformations respectively. The series of 17 thiazolidinedione-derived ligands (shown in Table 1) were prepared and docked independently to the two LYP PTP domain structures.
We first performed non-constraint docking (with no preset required interactions). However, no plausible binding modes were obtained. The poses lacked key interactions with the catalytic residues leaving the catalytic site partially or entirely exposed to solvent or phosphatase substrates. Moreover, in many cases the docking scores indicated less favorable protein–ligand interactions than in case of subsequent constraint docking. This is likely due to incomplete conformational sampling in a relatively open and shallow binding site (compare Materials and methods). Because of the better docking scores and more plausible poses, we preferred the constraint docking procedure to rationalize the inhibitory mechanism of the reported LYP ligands. In this procedure, we required at least one hydrogen bond interaction to generate ensembles of relevant binding poses that interact at the catalytic site and are not located entirely outside the catalytic region. The backbone amides of Ser228 and Cys231 and the side chain hydrogen atoms of Arg233 were selected as candidates for hydrogen bond donors in both LYP conformations. These residues are part of the catalytic site, but are not part of the WPD-loop and therefore do not induce a bias towards a ligand’s preferential binding to one over the other form of LYP.
All experiments were run using two docking/scoring protocols, SP (standard precision) and XP (extra precision), and in addition using the induced fit docking protocol. Correlation coefficients between the corresponding docking scores and experimental pIC50 values are collected in Table 3 for both the closed and the open LYP conformations.
Table 3.
Squares of the correlation coefficients (R2) of the linear regression of docking scores and experimental pIC50 values from 17 thiazolidinedione-type inhibitors; using different docking protocols
| Docking protocol | Closed LYP conformation |
Open LYP conformation |
|---|---|---|
| SP docking | 0.052 | 0.014 |
| XP docking | 0.027 | 5 10E-6 |
| SP induced-fit docking | 0.399 | 0.001 |
The most informative results are shown in bold
As shown in Table 3, the estimated binding affinities by SP and XP docking scores do not correlate with the experimental pIC50 values. XP docking into the open conformation failed entirely for two compounds (408 and 417), producing no valid pose and resulted in very poor scores for several other compounds. However, the IFD results were much more encouraging. This could perhaps be expected, because IFD allows for receptor flexibility (see Materials and methods). For the closed conformation of LYP, IFD resulted in a quantitative improvement of the correlation between docking scores and the experimental pIC50 values (Fig. 3) while there was no correlation for the open conformation. The correlation for the SP induced-fit docking with the correlation coefficient R2 = 0.399 is considerably good given the small range of the IC50 values and the shallow binding site. Furthermore, we tested statistical significance for the presented linear model and found the p value of 0.0065. In comparison, the corresponding regression model of the IFD docking scores to the open LYP form (which we assume is not the conformation to which this chemotype binds) has a p value of 0.39.
Fig. 3.
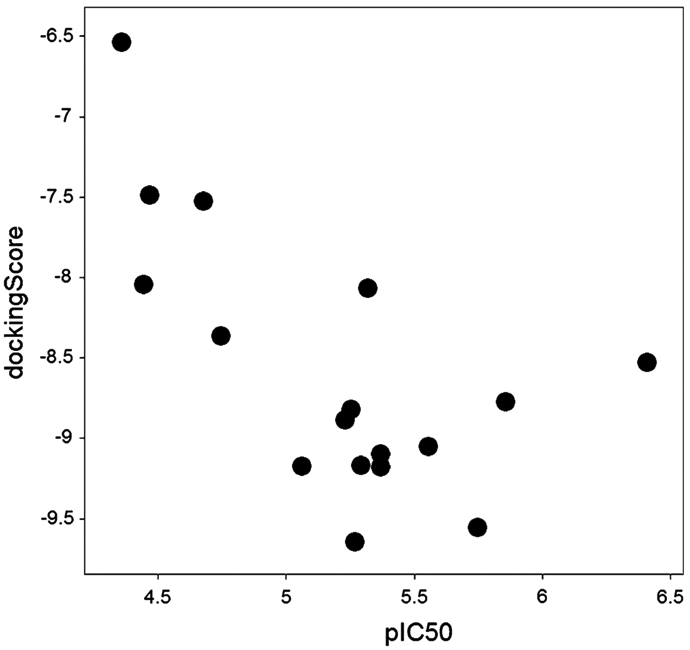
Induced fit docking scores versus experimental pIC50 values for the 17 thiazolidinedione inhibitors in the closed-conformation LYP model
The significant correlation of the experimental pIC50 values and the estimated binding affinities obtained by IFD with the closed conformation, in contrast to no correlation with the open form of LYP, suggests that the thiazolidinedione analogs preferentially bind to the closed LYP conformation, in a similar manner as IZD binds to PTP1B. This is also supported by the fact that several of the active thiazolidinedione analogs produced very poor scores or failed entirely to dock to the open conformation in case of the extra precision (XP) docking procedure.
To further investigate this preposition, we analyzed the best docking poses in more detail. Most of the ligands are two-headed (acid moieties at both ends). It was therefore reasonable to investigate if they could bind in two different orientations. However, the docking studies indicated that they bind via the salicylic acid moiety to the catalytic site. This was the binding mode for all ligands (best pose) in the closed conformation while the docked ligands’ orientations were not consistent in the open conformation.
An overlay of the best docking poses is shown in Fig. 4a, b for the active (closed) and inactive (open) form, respectively.
Fig. 4.

Best docking poses of the thiazolidinedione series of ligands in the active (closed) (a) and inactive (open) (b) LYP conformation. (a) The salicylic acid moieties interact in the catalytic site, including a hydrogen bond interaction to Asp195 in the WPD-loop in the closed conformation. (b) The docked ligands’ orientations are not consistent in the open form
Our results also indicated key residues interacting with the alkyl carboxylic acid moiety of the thiazolidinedione core: Lys32, Lys61, and Lys136 in both LYP conformations. However, the major difference in the binding modes between the two conformations was observed in the presence of an additional hydrogen bond interaction with Asp195 in the WPD-loop, which is in a suitable orientation when the WPD-loop adopts the closed conformation. This interaction is likely one of the determining contributors to binding affinity as indicated by the significant correlation of experimental activities to docking results with the closed, in contrast to the open conformation of LYP.
Docking of benzofuran salicylic acid inhibitors
We applied the same type of analysis to the 6-hydroxybenzofuran-5-carboxylic acid derivatives. In the case of non-constraint docking and similar to our results obtained with the thiazolidinedione compounds, we found that in many cases the generated ligand poses were outside the catalytic site. We therefore performed constraint docking employing both the SP and XP protocols. As described above, we required at least one hydrogen bond constraint within the catalytic site. To avoid potential biased docking of ligands in the active LYP conformation we did not select Asp195 (part of the WPD-loop) as a constraint (see Materials and methods). We also performed induced fit docking. Correlations of the docking scores and experimental pIC50 values for the respective docking protocols are collected in the Table 4 for both the closed and open LYP conformations.
Table 4.
Square of the correlation coefficients (R2) of the linear regression of docking scores and experimental pIC50 values from 35 benzofuran salicylic acid inhibitors; using different docking protocols
| Docking protocol | Closed LYP conformation |
Open LYP conformation |
|---|---|---|
| SP docking | 0.052 | 0.015 |
| XP docking | 0 (docking failed) | 0.006 |
| Induced-fit docking | 0.089 | 0.045 |
The most informative results are shown in bold
As shown in Table 4, there is no correlation of the SP docking scores and experimental results for either the active or the open LYP conformation. In contrast to the thiazolidinedione compounds, neither did IFD produce any significant correlation between predicted affinity of the generated poses and the experimental data. However, the XP docking protocol gave some indication of how the benzofuran salicylic acid compounds may bind. Although the XP docking protocol did not result in any improvement of correlation, we found no valid pose for the majority of the 6-hydroxybenzofuran-5-carboxylic acid inhibitors in the active LYP form, suggesting that they bind preferentially to the open form of LYP. This is also supported by the co-crystal structure of I-C11 (478) and LYP in the open conformation.
We analyzed the best docking poses in the open and closed confirmations of LYP in more detail (Fig. 5).
Fig. 5.

Best docking poses of 35 6-hydroxybenzofuran-5-carboxylic acid-derived ligands in the closed (a) and open (b) LYP conformation. Interactions outside the catalytic side include residues Lys32, Lys61, and Lys136
Docking poses of the 6-hydroxybenzofuran-5-carboxylic acid derivatives in the open conformation (Fig. 5b) resembled the co-crystal pose of the original benzofuran salicylic acid inhibitor 478 (I-C11, PDB code 2qct). Although this was expected, it confirmed that the docking protocol generated realistic poses that are at least qualitatively correct. In addition, the docking results suggested an interaction with Lys32, which is missing in the co-crystal structure. Ligand poses obtained after docking in the closed conformation primarily were oriented towards Lys136, presumably due to the restricted flexibility of the bulky benzofuran ring in the closed-conformation binding pocket. However, poor docking scores and missing interactions in the active site indicated that these are probably not realistic compared to binding in the open conformation.
Binding mode comparison
In order to better understand differences in the binding modes of the thiazolidinedione and the benzofuran salicylic acid series of inhibitors, we compared the best docking poses of the most active thiazolidinedione inhibitor, compound 444 (depicted in Fig. 6a), docked into the closed LYP conformation and compound 526 (the best inhibitor of the benzofuran salicylic acid series) docked to the open form (shown in Fig. 6b).
Fig. 6.

Protein-ligand interactions between (a) closed LYP conformation and thiazolidinedione 444 and (b) open LYP conformation and compound 526
Figure 6a illustrates that in the closed conformation residue Asp195 of the WPD-loop is in position to form a hydrogen bond with the phenol hydrogen of the salicylic acid moiety of the (thiazolidinedione) inhibitor—in contrast to the open conformation. It also illustrates the interaction of the alkyl carboxylic acid bound to the thiazolidinedione core with Lys61.
Summary and conclusions
In this report we used in-silico protein–ligand modeling to systematically generate and analyze the binding modes of two classes of small molecule LYP inhibitors—a thiazolidinedione and a benzofuran salicylic acid series—in the context of two functionally relevant conformations of the LYP catalytic PTP domain. The goal was to understand and rationalize how each chemotype interacts in the active or inactive form of LYP.
In the cause of this study we developed a high-quality model of LYP in the active LYP conformation. We also refined and corrected the existing crystal structure of the open LYP conformation (missing and unresolved segments and an incorrect ligand structure). We developed docking models of both forms of LYP and docked both series into each model using several docking protocols.
Correlating predicted binding affinities and experimental inhibitory activities (pIC50) is challenging for a number of reasons. PTP binding sites are rather flat, making them harder to target by small molecule inhibitors and also complicating automated docking procedures. As a consequence the number of available LYP inhibitors is still rather small and their activities are moderate (typically micromolar or higher IC50s). Moreover, the range of activities is also low covering only one to two orders of magnitude. Nevertheless, a combination of the different in-silico methods suggested that the two investigated series of inhibitors bind to two different LYP conformations. Furthermore, we consider the method fairly sensitive since it could generate distinct binding modes corresponding to two chemotypes that share a key (salicylic acid) binding element.
Despite the challenges above, in case of the thiazolidinedione series of inhibitors we achieved a reasonable correlation of predicted to experimental binding affinities using an induced fit docking protocol for the active LYP conformation. In contrast, no correlation was seen when this class of inhibitors was docked into the open form. Analysis of the consensus binding modes of the thiazolidinedione series in the closed conformation indicated a potentially critical hydrogen bond interaction with Asp195 in the WPD-loop. This interaction may contribute to stabilizing the WPD-loop in the closed conformation. It could therefore be one of the determinants selecting the active (closed) conformation for an inhibitor chemotype that can interact at this position.
A similar analysis of the benzofuran salicylic acid series of inhibitors showed that they are generally too bulky to fit into the binding pocket of the closed LYP conformation. In particular the XP docking results suggested that they bind to the open LYP conformation, which is also in agreement with the co-crystal structure of I-C11 bound to the open form of LYP [29].
Analysis of the binding modes of the thiazolidinedione class of inhibitors in the closed LYP conformation further revealed that the ligands with two potential head group moieties preferentially bind to the catalytic site via the salicylic acid moiety. In addition to the specific hydrogen bond interactions in the active site, we also identified additional residues that can interact with the ligand outside the catalytic pocket. These residues can potentially be targeted to increase potency and possibly improving selectivity.
In summary, we have illustrated how different inhibitor chemotypes interact with conformationally and functionally distinct forms of LYP. Insight into preferential binding of chemical classes of LYP ligands with respect to the conformation of the PTP domain is relevant in the context of developing novel inhibitors. There is currently only limited knowledge of how LYP inhibitors interact in the catalytic PTP domain and our studies provide new insights. More importantly, a method of predicting which LYP conformation is the primary target for a given chemical series of inhibitors is valuable. Because the binding sites of the active and inactive form of LYP are different with respect to shape and physicochemical characteristics determined by the types and orientations of active site residues, the protein–ligand interactions and ligand orientations in the binding pocket differ significantly among the two PTP conformations. This leads to the possibility of designing and optimizing inhibitors that specifically target one over the other form of LYP with implications for potency and selectivity. Such design considerations are particularly relevant given the high sequence and 3D binding site similarities among PTPs [39], and especially within the classic PTPs [40] where residues within the catalytic site and the WPD-loop are conserved. Inhibitor scaffold preferences towards one or the other conformation likely translate across the most similar PTPs. Selecting a suitable inhibitor chemotype may therefore be guided in part by how a desired selectivity profile across a set of relevant PTPs can best be related to the binding site similarities in the conformation that corresponds to the chemotypes under consideration.
Acknowledgment
This work was in part supported by the grant MLSCN U54 HG003914 and 1R21CA132121 (to L.T.) from the National Institutes of Health. We also acknowledge resources of the University of Miami Center for Computational Science (CCS).
Abbreviations
- LYP
Lymphoid tyrosine phosphatase
- PTKs
Protein tyrosine kinases
- PTPs
Protein tyrosine phosphatases
- TCR
T-cell receptor
- CSK
C-terminal Src tyrosine kinase
- GRB2
Growth factor receptor-bound protein
- MLSCN
Molecular Library Screening Center Network
- NIH
National Institutes of Health
- IZD
Izothiazolidinone inhibitor
- PTP1B
Protein tyrosine phosphatase 1B
- SAR
Structure activity relationship
- SP
Standard precision
- XP
Extra precision
- IFD
Induced-fit docking
Contributor Information
Dušica Vidović, Center for Computational Science, University of Miami, Miami, FL 33136, USA.
Yuli Xie, Department of Medicine, Columbia University, New York, NY 10032, USA.
Alison Rinderspacher, Department of Medicine, Columbia University, New York, NY 10032, USA.
Shi-Xian Deng, Department of Medicine, Columbia University, New York, NY 10032, USA.
Donald W. Landry, Department of Medicine, Columbia University, New York, NY 10032, USA
Caty Chung, Center for Computational Science, University of Miami, Miami, FL 33136, USA.
Deborah H. Smith, Department of Physiology and Cellular Biophysics, Columbia University, New York, NY 10032, USA
Lutz Tautz, Infectious and Inflammatory Disease Center, Sanford-Burnham Medical Research Institute, La Jolla, CA 92037, USA.
Stephan C. Schürer, Center for Computational Science, University of Miami, Miami, FL 33136, USA; Department of Pharmacology, Miller School of Medicine, University of Miami, Miami, FL 33136, USA
References
- 1.Hunter T, Sefton BM (1980) Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA 77(3):1311–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunter T (2000) Signaling—2000 and beyond. Cell 100(1):113–127 [DOI] [PubMed] [Google Scholar]
- 3.Tonks NK (1993) Introduction: protein tyrosine phosphatases. Semin Cell Biol 4(6):373–377 [DOI] [PubMed] [Google Scholar]
- 4.Zhang ZY (1998) Protein-tyrosine phosphatases: biological function, structural characteristics, and mechanism of catalysis. Crit Rev Biochem Mol Biol 33(1):1–52 [DOI] [PubMed] [Google Scholar]
- 5.Cohen P (2002) Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov 1(4):309–315 [DOI] [PubMed] [Google Scholar]
- 6.Hunter T (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80(2):225–236 [DOI] [PubMed] [Google Scholar]
- 7.Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7(11):833–846 [DOI] [PubMed] [Google Scholar]
- 8.Tautz L, Pellecchia M, Mustelin T (2006) Targeting the PTPome in human disease. Expert Opin Ther Targets 10(1):157–177 [DOI] [PubMed] [Google Scholar]
- 9.Guan KL, Dixon JE (1991) Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J Biol Chem 266(26):17026–17030 [PubMed] [Google Scholar]
- 10.Barford D, Flint AJ, Tonks NK (1994) Crystal structure of human protein tyrosine phosphatase 1B. Science 263(5152):1397–1404 [PubMed] [Google Scholar]
- 11.Jia Z, Barford D, Flint AJ, Tonks NK (1995) Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268(5218):1754–1758 [DOI] [PubMed] [Google Scholar]
- 12.Cohen S, Dadi H, Shaoul E, Sharfe N, Roifman CM (1999) Cloning and characterization of a lymphoid-specific, inducible human protein tyrosine phosphatase, Lyp. Blood 93(6):2013–2024 [PubMed] [Google Scholar]
- 13.Gjorloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T (1999) Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol 29(12):3845–3854 [DOI] [PubMed] [Google Scholar]
- 14.Cloutier JF, Veillette A (1999) Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med 189(1):111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, Tang J, Jeffery D, Mortara K, Sampang J, Williams SR, Buggy J, Clark JM (2006) Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem 281(16):11002–11010 [DOI] [PubMed] [Google Scholar]
- 16.Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, Eisenbarth GS, Comings D, Mustelin T (2004) A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36(4):337–338 [DOI] [PubMed] [Google Scholar]
- 17.Hill RJ, Zozulya S, Lu YL, Ward K, Gishizky M, Jallal B (2002) The lymphoid protein tyrosine phosphatase Lyp interacts with the adaptor molecule Grb2 and functions as a negative regulator of T-cell activation. Exp Hematol 30(3):237–244 [DOI] [PubMed] [Google Scholar]
- 18.Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batliwalla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75(2):330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, Vella A, Nutland S, Rance HE, Maier L, Barratt BJ, Guja C, Ionescu-Tirgoviste C, Savage DA, Dunger DB, Widmer B, Strachan DP, Ring SM, Walker N, Clayton DG, Twells RC, Gough SC, Todd JA (2004) Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53(11):3020–3023 [DOI] [PubMed] [Google Scholar]
- 20.Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM, Novitzke J, Williams AH, Gillett C, Rodine P, Graham RR, Ardlie KG, Gaffney PM, Moser KL, Petri M, Begovich AB, Gregersen PK, Behrens TW (2004) Genetic association of the R620 W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75(3):504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, Nika K, Tautz L, Tasken K, Cucca F, Mustelin T, Bottini N (2005) Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet 37(12):1317–1319 [DOI] [PubMed] [Google Scholar]
- 22.Xie Y, Liu Y, Gong G, Rinderspacher A, Deng SX, Smith DH, Toebben U, Tzilianos E, Branden L, Vidovic D, Chung C, Schurer S, Tautz L, Landry DW (2008) Discovery of a novel submicromolar inhibitor of the lymphoid specific tyrosine phosphatase. Bioorg Med Chem Lett 18(9):2840–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vang T, Xie Y, Liu WH, Vidovic D, Liu Y, Wu S, Smith DH, Rinderspacher A, Chung C, Gong G, Mustelin T, Landry DW, Rickert RC, Schurer SC, Deng SX, Tautz L (2011) Inhibition of lymphoid tyrosine phosphatase by benzofuran salicylic acids. J Med Chem 54(2):562–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J (2001) Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol 3(2):173–182 [DOI] [PubMed] [Google Scholar]
- 25.Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B, Martin T, Gretener D, Perrin D, Leroy D, Vitte PA, Hirsch E, Wymann MP, Cirillo R, Schwarz MK, Rommel C (2005) Blockade of PI3K-gamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med 11(9):936–943 [DOI] [PubMed] [Google Scholar]
- 26.Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, Schuurman K, van Tellingen O, Morris AJ, Smyth SS, Moolenaar WH, Ovaa H (2010) Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc Natl Acad Sci USA 107(16):7257–7262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wolf MC, Freiberg AN, Zhang T, Akyol-Ataman Z, Grock A, Hong PW, Li J, Watson NF, Fang AQ, Aguilar HC, Porotto M, Honko AN, Damoiseaux R, Miller JP, Woodson SE, Chantasirivisal S, Fontanes V, Negrete OA, Krogstad P, Dasgupta A, Moscona A, Hensley LE, Whelan SP, Faull KF, Holbrook MR, Jung ME, Lee B (2010) A broad-spectrum antiviral targeting entry of enveloped viruses. Proc Natl Acad Sci USA 107(7):3157–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS (2011) DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res 39(Database issue):D1035–D1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY (2007) Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci USA 104(50):19767–19772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LigPrep (2009) Schrödinger LLC. Portland, OR [Google Scholar]
- 31.Jorgensen W, Tirado-Rives J (1988) The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc 110(6):1657–1666 [DOI] [PubMed] [Google Scholar]
- 32.Ala PJ, Gonneville L, Hillman MC, Becker-Pasha M, Wei M, Reid BG, Klabe R, Yue EW, Wayland B, Douty B, Polam P, Wasserman Z, Bower M, Combs AP, Burn TC, Hollis GF, Wynn R (2006) Structural basis for inhibition of protein-tyrosine phosphatase 1B by isothiazolidinone heterocyclic phosphonate mimetics. J Biol Chem 281(43):32784–32795 [DOI] [PubMed] [Google Scholar]
- 33.Andrec M, Harano Y, Jacobson MP, Friesner RA, Levy RM (2002) Complete protein structure determination using backbone residual dipolar couplings and sidechain rotamer prediction. J Struct Funct Genomics 2(2):103–111 [DOI] [PubMed] [Google Scholar]
- 34.Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55(2):351–367 [DOI] [PubMed] [Google Scholar]
- 35.Maestro (2009) Schrödinger LLC. Portland, OR [Google Scholar]
- 36.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749 [DOI] [PubMed] [Google Scholar]
- 37.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47(7):1750–1759 [DOI] [PubMed] [Google Scholar]
- 38.DeLano WL (2009) PyMOL. Schrödinger LLC, Portland, OR [Google Scholar]
- 39.Vidovic D, Schurer SC (2009) Knowledge-based characterization of similarity relationships in the human protein-tyrosine phosphatase family for rational inhibitor design. J Med Chem 52(21):6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barr AJ, Ugochukwu E, Lee WH, King ON, Filippakopoulos P, Alfano I, Savitsky P, Burgess-Brown NA, Muller S, Knapp S (2009) Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 136(2):352–363 [DOI] [PMC free article] [PubMed] [Google Scholar]