

Abstract
The bacterial cell wall, composed of peptidoglycan (PG), provides structural integrity for the cell and is responsible for cell shape in most bacteria. Here we present tools to study the cell wall using a clickable PG-specific sugar, MurNAc-alk, as a metabolic probe. Here we present a new reaction pathway for generating MurNAc-alk. We also include protocols for labeling PG synthesis in Helicobacter pylori, determining the identity of the labeled muropeptides using LC-MS/MS, sample preparation of cells labeled for a short fraction of the doubling time, and visualization using 3D structured illumination microscopy.
Keywords: N-acetylmuramic acid, Helicobacter pylori, cell wall, microscopy, metabolic labeling, LC-MS/MS identification
INTRODUCTION:
Cell shape was one of the first noted properties of bacteria, but how bacteria maintain their diverse shapes was a largely impenetrable question until recently. For most bacteria, the bag-shaped peptidoglycan (PG) sacculus (commonly referred to as the cell wall), a single mesh-like molecule surrounding the cytoplasmic membrane, maintains cell shape (Höltje, 1998). PG subunits, muropeptides, are composed of N-acetylglucosamine (GlcNAc) linked to N-acetylmuramic acid (MurNAc) with an attached short peptide stem on the lactyl moiety, and are released from the PG sacculus by a muramidase such as lysozyme (Vollmer, Blanot, & de Pedro, 2008). The PG precursor is synthesized stepwise in the cytoplasm before being flipped across the cytoplasmic membrane to the periplasmic side and incorporated into the sacculus (Barreteau et al., 2008). Strands of PG are generated by glycosyltransferases, which catalyze the formation of β 1-4 linkages between disaccharides. These strands are interlinked to form a meshwork by the action of transpeptidases, which crosslink stem peptide of nearby PG strands (Sauvage, Kerff, Terrak, Ayala, & Charlier, 2008).
Helicobacter pylori is a helical Gram-negative bacterium with minimal genetic redundancy that is a useful model system to study bacterial cell shape (Tomb et al., 1997). In the PG of H. pylori and many other Gram-negative bacteria, the short peptide stem is comprised of L-alanine, D-glutamic acid, meso-diaminopimelic acid, D-alanine, and D-alanine, from position 1 through 5 respectively (Vollmer et al., 2008). In H. pylori, the only detectable crosslinks are between position 3 on one stem and position 4 on another, though other bacteria can also generate crosslinks between position 3 and 3 (Costa et al., 1999; Sycuro et al., 2010).
Bacteria must faithfully maintain the characteristic shape of their cell wall, which is a dynamic structure. For bacteria to grow, PG crosslinks must be broken to make space for the insertion of new PG strands (Singh, SaiSree, Amrutha, & Reddy, 2012). One mechanism for preserving cell shape is regulating where PG precursors are incorporated in the sacculus (Caccamo & Brun, 2018; Shi, Bratton, Gitai, & Huang, 2018; Taylor, Sichel, & Salama, 2019). Partnered with advances in microscopy, the development of probes to metabolically label the cell wall has facilitated an increased understanding of and interest in bacterial cell shape maintenance.
Within the past decade, a handful of probes that allow metabolic fluorescent labeling of PG have revolutionized the field. The available probes for labeling PG are incorporated through different steps of PG metabolism, which impacts interpretation of these labels. The first of these probes developed were D-Ala functionalized with either a fluorescent or clickable moiety (Kuru et al., 2012; Siegrist et al., 2013). These probes can be incorporated by the action of transpeptidases (Kuru et al., 2019). Shortly thereafter, clickable D-Ala-D-Ala dipeptide probes that can be incorporated into PG were developed. These are likely incorporated through the cytoplasmic steps of PG precursor synthesis (Kuru et al., 2019; Liechti et al., 2014). While D-amino acid probes are very useful for studying PG metabolism in a wide range of bacteria, there are some key limitations to this class of probe. Since D-amino acid probes label the peptide stem of muropeptides, they can be removed as the PG is modified after synthesis even if the rest of the muropeptide remains. Also, the D-Ala probes that are incorporated at sites of crosslinking activity label both sites of new PG synthesis as well as sites of modification of established PG via crosslinking.
Recently, a MurNAc probe modified with a clickable handle was developed (Liang et al., 2017). Because this probe is incorporated through the cytoplasmic steps of muropeptide synthesis, it reports only on sites of new PG synthesis. Furthermore, since the label is on the sugar backbone of the PG, the label is only removed with removal of the whole muropeptide, making this a much more straightforward reporter of PG synthesis. Using this probe requires the presence of PG recycling enzymes that can add UDP to MurNAc, which is the substrate used for muropeptide biogenesis. The recycling enzymes AmgK and MurU from Pseudomonas putida can be expressed in the organism to be labeled if this recycling activity is not natively present.
This manuscript will focus primarily on MurNAc-alk, with some included comparison to D-Ala-alk. Prior to our work (Taylor et al., 2020), MurNAc-alk had been primarily used for sparse labeling of the whole cell wall to verify incorporation of the probe into PG and to serve as a method for generating labeled PG for downstream experiments (DeMeester et al., 2018; DeMeester et al., 2019; Liang et al., 2017).
Our interest has been in characterizing the pattern of new PG incorporation as H. pylori grows and to determine if H. pylori uses spatial regulation of growth to help maintain helical shape. Accomplishing this goal required appreciable modifications to previous MurNAc-alk labeling and synthesis protocols. Since PG is modified as it matures, we needed to label incorporation for only a small fraction of the doubling time.
We had to take both our experimental goals and H. pylori physiology into consideration when deciding how to best image labeled cells. Our question of where PG synthesis occurs in H. pylori is inherently a three-dimensional question, considering the helical shape of the organism. We needed to collect images of cells that were of sufficient quality to allow us to computationally identify geographic landmarks on the cell and to determine how the amount of PG incorporation relates to these landmarks. H. pylori cells are on average approximately 0.45 μm in diameter and 2.5 μm in length, with a helical diameter of 0.3 μm (Taylor et al., 2020). Given these dimensions and the resolution limits of light microscopy, we relied on super-resolution microscopy, specifically 3D structured illumination microscopy (SIM) to elucidate subcellular details of PG incorporation. SIM is amenable to the use of a wide variety of fluorescent dyes but works best when labeling is bright and photostable. Furthermore, because SIM imaging requires acquisition of multiple images per z-slice and because samples can drift slightly, it is prudent to aim for as bright a signal as possible to reduce imaging time and ensure optimal image quality. To achieve an acceptable signal to noise ratio with a short labeling pulse, we labeled cells with a much higher concentration of MurNAc-alk than had been used previously for whole-cell labeling.
There were several requirements for sample preparation that necessitated creation of a tool for carefully affixing fixed cells to clean glass coverslips. We needed to preserve 3D geometry for subcellular analysis, so it was essential to prevent cells from drying out at any step during sample preparation. Because SIM works best for imaging close to the coverslip and because we needed to image and analyze hundreds of individual, well-separated cells per slide, we also needed to reproducibly obtain an optimal density of cells on the coverslip. Drawing inspiration from cytocentrifuges, we used readily-accessible materials to generate a sealed chamber above the coverslip that could be centrifuged in a swinging-bucket tabletop centrifuge that can accommodate 96-well plates. Depositing fixed cells on the coverslip also allows us to minimize the starting culture volume and thus amount of probe required. Thus, we can achieve an acceptable density of cells on the coverslip from a dilute suspension of cells. Additionally, the number of centrifugations required can be reduced because labeling steps following fixation and permeabilization can be performed on the coverslip rather than in suspension.
These protocols can be used as a starting point for short-pulse labeling of PG in other organisms to image new PG synthesis. Basic Protocol 1 outlines the synthesis of highly-purified MurNAc-alk. Basic Protocol 2 demonstrates a procedure for the detection of muropeptides using LC-MS/MS and how this can be used to prove incorporation of MurNAc-alk into the PG. Support Protocol 1 is used to grow H. pylori cells in liquid culture. Support Protocol 2 is used to determine if a strain can use exogenous MurNAc. Support Protocol 3 is used to determine if SDS has been completely removed from the PG preparation before moving on to the next step of Basic Protocol 2. Support Protocol 4 describes how to create cytocentrifuge units for high-quality slide preparation where fixed cells need to be reliably affixed to a coverslip for imaging. Basic Protocol 3 is used to perform a short pulse labeling of cell wall synthesis using MurNAc-alk or D-Ala-alk and to make high-quality slides of these cells. Basic Protocol 4 instructs the user how to perform structured illumination microscopy on the DeltaVision OMX microscope using the slides from Basic Protocol 3.
BASIC PROTOCOL 1
BASIC PROTOCOL TITLE
Alternative Synthesis of MurNAc-alk (direct coupling)
Introductory paragraph:
2-Alkyne Muramic acid (MurNAc-alk hereafter, Figure 1C.) is one of the most convenient and relevant compounds used for in bacterial peptidoglycan (PG) labeling techniques (DeMeester et al., 2019; Zhang, Wang, Yang, & Hang, 2020).
Figure 1.
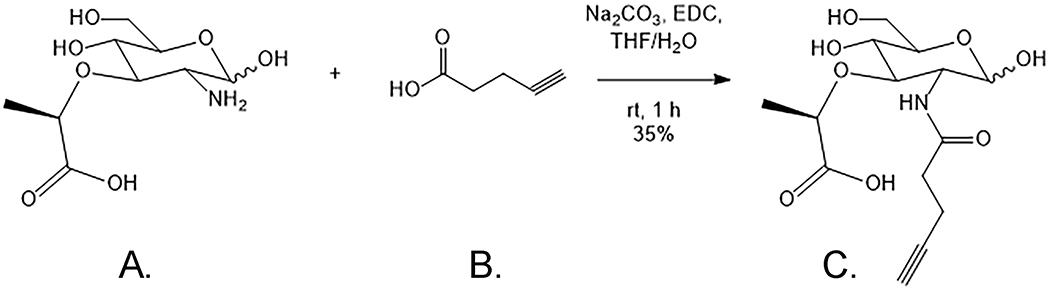
Alternative synthesis of MurNAc-alk (C.) from Muramic acid (A.) and 4-pentynoic acid (B.).
As a muramic acid analog, this probe can be effectively (metabolically) incorporated into the bacterial cell wall of a wide variety of microorganisms. Moreover, its key alkyne moiety allows a sensitive fluorescent visualization of the labeled structures through a simple and accessible click reaction.
Therefore, in addition to the previously reported synthesis protocol (DeMeester et al., 2019), we developed a second reaction pathway in order to generate this essential probe. (Figure 1). This alternative method, that also uses muramic acid (Figure 1A.) as substrate, consists of a coupling reaction with the free acid, 4-pentynoic acid (Figure 1B.), catalyzed by EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride). Basic conditions and a polar binary solvent system are also necessary. At the end of this protocol, the user will have generated purified MurNAc-alk that can be used to label PG in H. pylori and other bacteria.
Materials:
Muramic acid (Figure 1A.) (Prepared as in DeMeester et al. 2019, Liang et al. 2017 or as in Brown et al. 2021, or purchased from Sigma-Aldrich, #M2503)
4-pentynoic acid (Figure 1B., Sigma-Aldrich, #232211)
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, Chem-Impex International, Inc. #00050)
Sodium carbonate (Na2CO3) (Fisher Scientific #S263-500)
Tetrahydrofuran (THF) (Sigma, #186562)
Deionized H2O (dH2O)
Dichloromethane (DCM) (Fisher Scientific, #D37-20)Ethyl acetate (EtOAc) (Fisher Scientific, #124-20)
Methanol (MeOH) (Fisher Scientific, #A412-20)
p-anisaldehyde stain (see Reagents and Solutions)
Celite (Fisher Chemical, #C212-500)
Silica Gel, Standard Grade, 60 Å, 40-63 μm (Sorbtech, #30930M)
10-mL round bottom flasks
Stir bars
Stir plate
Pasteur pipettes
Rotary evaporator
Lyophilizer
5 mL collection tubes
Glass chromatographic column (or pipette with a cotton)
Tube rack
TLC plates
TLC Spotting Capillary Tubes
TLC Chamber
Tweezers
Hot plate
Additional reagents and equipment for LC/MS
Protocol steps — Step annotations:
Prepare Mixture A
Add 4-pentynoic acid (Figure 1B., 43.2 mg, 0.4404 mmol, 1.1 eq) to a 10-mL round-bottom flask.
Add EDC (92.8 mg, 0.4841 mmol, 1.2 eq).
Finally add 2 mL of THF to both solids.
-
Stir the reaction at room temperature for 0.5 h.
The appearance of This mixture will appear as either a cloudy solution or an off-white suspension
Prepare Mixture B
5. Add Muramic acid (Figure 1A., 100 mg, 0.3981 mmol) to a 10-mL round-bottom flask.
6. Add Na2CO3 (127.2 mg, 1.201 mmol, 3 eq).
7. Dissolve these solids in 2 mL of dH2O
-
8. Stir the reaction at room temperature for 0.5 h.
Due to the high solubility of these compounds in water, mixture B is always a yellowish solution
Perform the coupling reaction
9. Add Mixture B (Muramic acid aqueous basic solution) to Mixture A (activated 4-pentynoic acid). Stir for approximately 1h at room temperature.
10. Monitor reaction by LC/MS ESI pos [M+23]+ = 354 and by TLC (MeOH/EtOAc 4:5, Spot with Rf = 0.39, detection by p-anisaldehyde stain). Typically, after 1-2 h, the reaction is isolated to avoid probable product degradation. Once complete, evaporate the THF under reduced pressure without heat using a rotary evaporator.
-
11. Wash the resulting aqueous phase with DCM three times (3 x 1 ml).
This step is intended to extract the remaining 4-pentynoic acid and EDC from the reaction crude
12. Lyophilize the crude to obtain an off-white solid.
- 13. Purify the crude utilizing a silica gel chromatographic column, considering the following features.
- Use a dry load technique. Dissolve the crude in MeOH and use four times the weight of celite comparing to weight of the crude (x4 celite).
- Use an amount of silica gel at least 30 times the weight of crude material. In terms of volume, this ratio will be ml of silica = mass of crude x 60 ml.
- Apply a slow gradient of MeOH/EtOAc 2:8 through 4:5.
-
Collect the product based on TLC control (Spot with Rf = 0.39, detected by p-anisaldehyde stain).The excess 4-pentynoic acid elutes first (Rf = 0.8) and then the product (Rf = 0.39)
-
14. Lyophilize the collected fractions to obtain a white solid (46.3 mg, Yield = 35%, see NMR spectra on Figure 2).
We note that yields can be significantly improved by using larger amounts of starting material. Using 0.5 g of muramic acid, the yield reaches 40% +/- 5%. Since approximately 30% of the initial amount of muramic acid can be recovered for use in a second reaction, the overall yield will be even higher.
Figure 2.
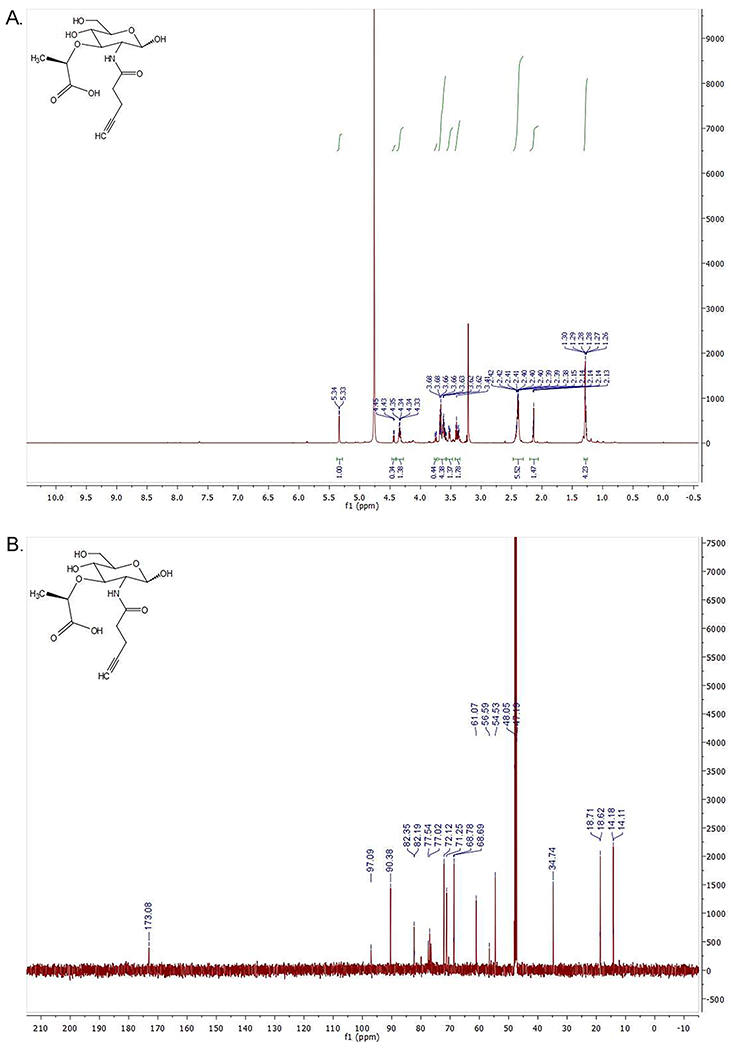
Spectral analysis of MurNAc-alk (Figure 1C.). A. (top): 1H NMR (600 MHz, Methanol-d4) δ 5.34 (d, J = 3.0 Hz, H1α), 4.45 (d, J=7.81 Hz, H1β), 4.36–4.32 (m, CH propionic), 3.76–3.74 (dd, J=2.44 Hz, H6a), 3.69–3.57 (m, H6b, H4, H3), 3.53–3.51 (dd, H2), 3.42–3.36 (m, H5), 2.42–2.38 (m, methylene adjacent to amide and methylene adjacent to alkyne), 2.15–2.13 (m, CH alkyne), 1.30–1.26 (bd, CH3 propionic acid). Ratio α/β=3. B. (bottom): 13C NMR (151 MHz, MeOD) δ 173.08 (carbonyl), 97.09 (C1β), 90.38 (C1α), 82.35 (C quaternary α), 82.19 (C quaternary β), 77.54 (CH propionic), 77.02 (C3), 72.12 (C4), 71.25 (C5), 68.78 (CH alkyne β), 68.69 (CH alkyne α), 61.07 (C6), 56.59 (C2β), 54.53 (C2α), 34.74 (CH2 adjacent to amide), 18.71 (CH3 propionic acid β), 18.62 (CH3 propionic acid α), 14.18 (CH2 adjacent to alkyne α), 14.11 (CH2 adjacent to alkyne β). Note: The upper green lines in A. are called integral lines and measure the area below the curve of the 1H NMR signal which is proportional to the number of protons that originate the signal (the larger the signal, the more protons).
Sample Data
SUPPORT PROTOCOL 1
SUPPORT PROTOCOL TITLE
Growing Helicobacter pylori in liquid culture
Introductory paragraph:
This protocol instructs the user how to reliably grow a robust culture of H. pylori starting from a frozen stock. The included calculations are helpful for planning experiments, as the user can inoculate cells to be ready at a desired time the following day. The user will likely need to go through several iterations to determine the precise doubling time of the strain grown under these conditions.
Materials:
Helicobacter pylori −80°C freezer stock (strain G27, or alternatively J99, ATCC® 700824™)
Horse blood plates (See Reagents and Solutions)
BB10 (See Reagents and Solutions)
Sterile wooden sticks
Inoculating loops
50 ml baffled culture flasks with lids
Trigas incubator with a shaker (Sanyo O2/CO2 incubator; 10% O2, 10% CO2, Nitrogen to balance)
spectrophotometer
cuvettes
Protocol steps—Step annotations:
Remove a small amount of the H. pylori freezer stock using a sterile wooden stick. Inoculate a horse blood agar plate from the freezer stock. Once the liquid has soaked into the plate, invert the plate and place it in a Trigas incubator set at 10% CO2, 10% O2, 37°C for 24-72 hours until growth occurs. If you are not ready to begin a liquid culture for labeling, passage the culture on horse blood plates daily.
To start a liquid culture of H. pylori, in the morning the day before using cells, pick a small patch of low-density culture growth from the horse blood plate and use it to inoculate 10 ml of BB10 in a small baffled culture flask. Grow the culture in the trigas incubator shaking at 150-200 RPM.
In the evening the day before using cells, pipette 1 ml of BB10 into one cuvette and 1 ml of the liquid culture into another. Blank the spectrophotometer using the cuvette of BB10 and then read the optical density at 600 nm (OD600) of the liquid culture. (Note if OD600 > 1.5, dilute the sample 1:10 and reread to ensure you are within the linear range of the spectrophotometer.)
-
Calculate the inoculum in μl:
(1000*V*ODlabel)/(ODinoculant*2T/DT)
where
V = overnight culture volume (10 ml)
ODlabel = target OD600/ml of labeling culture (0.4 OD600/ml)
ODinoculant = measured OD600/ml of liquid culture for inoculating (target 0.1-1.0)
T = number of hours desired between inoculating culture and having culture ready for labeling (target 12-18)
DT = doubling time in hours (approximately 2.1 for LSH100 under these conditions; your strain and growth conditions will likely vary)
Inoculate 10 ml BB10 in a small baffled culture flask with the volume of inoculant culture calculated in Step 4. Grow the flask overnight in the Trigas incubator shaking at 150-200 RPM.
Sample Data
After successfully following this protocol, the user should have a culture of exclusively rod-shaped (mostly helical rods, though there is cell-to-cell variability in morphological parameters) H. pylori cells without any coccoid cells (see Figure 3). Some swimming cells should be observed when looking at fresh cells in suspension using phase contrast microscopy (at least 400x magnification). There should be no contaminating organisms present. The culture should reach the target density at approximately the calculated time. Large deviations indicate a problem with the culture or growth conditions and the problem(s) should be diagnosed.
Figure 3:
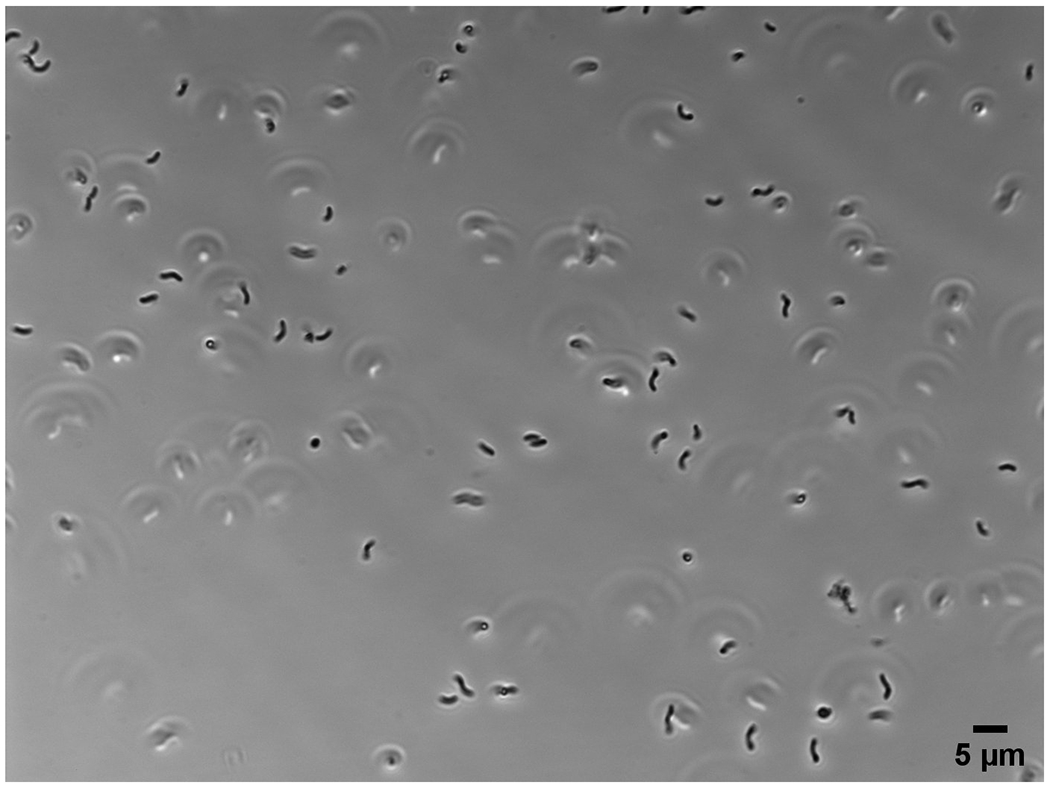
Image of a wet mount of a mid-log phase H. pylori culture using a 60x objective. Scale bar = 5 μm.
SUPPORT PROTOCOL 2
Fosfomycin rescue assay
Introductory paragraph:
This protocol is used to verify that a strain of interest is capable of using exogenously provided MurNAc. In this protocol, the user will be growing H. pylori in liquid culture under several different conditions to determine if adding MurNAc to cells treated with fosfomycin will help rescue growth. Serial dilutions of each culture will be plated to determine cell viability after 12 hours. Partial growth rescue indicates that the strain should be appropriate for labeling with MurNAc-alk (detailed in Basic Protocol 3).
Materials:
Liquid culture of wild-type Helicobacter pylori (from Support Protocol 1)
Liquid culture of Helicobacter pylori expressing AmgK and MurU (from Support Protocol 1)
BB10 (See Reagents and Solutions)
MurNAc (Sigma-Aldrich, #A3007), 200 mg/ml in ddH2O
Fosfomycin (Sigma-Aldrich, # 34089), 7.5 mg/ml in ddH2O
70% ethanol
50 ml baffled culture flasks
Trigas incubator [Sanyo O2/CO2 incubator; 10% O2, 10% CO2, Nitrogen to balance]
Spectrophotometer
Cuvettes
Sterile, flat-bottomed, untreated 96-well plates
5 ml polystyrene round-bottom tubes
Trigas incubator with shaker
Dan Kar Corp 48 Prong Frogger (ThermoFisher Scientific, #NC9843368)
Sterile reagent reservoir
Multichannel pipettes
Protocol steps—Step annotations:
As detailed in Support Protocol 1, prepare a liquid culture of H. pylori to reach 0.35-0.50 OD600/ml in the morning.
Check the culture density with the spectrophotometer. If the culture is between 0.35-0.5 OD600/ml, proceed with labeling. If OD600 > 0.5, dilute the culture and grow for at least 1 doubling to ensure the cells are in early/mid log phase.
Put the frogger tip side-down in a small open container of 70% ethanol.
Make 1 ml of each culture diluted to 0.002 OD600/ml in BB10.
-
Label eight 5 ml polystyrene round-bottom tubes 1-8. Add 200 μl of the diluted wild-type culture to tubes 1-4. Add 200 μl of the diluted AmgK MurU culture to tubes 5-8.
See Figure 4 for a schematic guide to the experimental setup in this protocol.
Add 1.33 μl of 7.5 mg/ml fosfomycin (final concentration = 50 μg/ml) to tubes 2, 4, 6, and 8.
Add 4 μl of 200 mg/ml MurNAc (final concentration = 4 mg/ml) to tubes 3, 4, 7, and 8.
Gently flick each tube 10 times to mix.
-
Take the frogger out of the ethanol, gently shake off the excess ethanol, and pass the frogger through a Bunsen burner flame to ignite any remaining ethanol. After the flames are extinguished, gently set the frogger on top of an open blood plate to mark faint indentations in a grid on the plate. No additional pressure beyond the weight of the frogger is required.
It is extremely important to make sure that no burning ethanol drips from the frogger and to ensure that the open container of ethanol is kept far from the flames. Make sure no loose papers or other flammable materials are above, below, or near the flame. The flame on the frogger may extend several inches vertically. Wait an extra 5 seconds after you think all the ethanol has burnt off to ensure the burn is indeed complete. Before igniting the ethanol, make sure you have a plan ready in case the ethanol vessel is accidentally set on fire.
First make a two-fold dilution in row A, columns 1-8 in a 96-well plate. To do this, pipette 20 μl of BB10 into columns 1-8 of row A. Then pipette 20 μl of each culture to the correspondingly numbered well in row A and pipette up and down 10 times to mix.
-
To make a series of 10-fold dilutions in the 96-well plate, use a multichannel pipette to add 180 μl of BB10 to row B-F, columns 1-8. Using a multichannel pipette, pipette up and down 10 times in column A to mix, then transfer 20 μl of each two-fold dilution in row A to the correspondingly numbered column in row B. Using a multichannel pipette set to 20 μl, pipette up and down 10 times in row B to mix, then transfer 20 μl from row B to row C. Pipette up and down 10 times to mix, then eject the tips. Using fresh tips, mix row C an additional 10 times, then transfer 20 μl from row C to row D. Repeat for each row, ending by adding 20 μl to row F and mixing.
It is crucial to change pipette tips between rows in the 10-fold dilution series. H. pylori sticks readily to pipette tips, so using the same tips for multiple dilution steps will not result in 10-fold dilutions.
Using the indentations on the blood plate made by the frogger as a guide, spot 2 μl of each dilution onto the plate, such that the dilutions from culture 1 are in the top row, with the most concentrated dilution on the left and down to the greatest dilution on the right. Repeat in rows 2-8 for cultures 2-8.
Allow all the spots to completely dry and then place the plate inverted in the Trigas incubator to grow for 3-5 days until distinct colonies are visible at the lower dilutions.
Incubate the tubes shaking in the Trigas incubator for 12 hours.
Repeat steps #9-13.
Once distinct colonies are clearly visible, photograph the plate to record the results.
Figure 4:
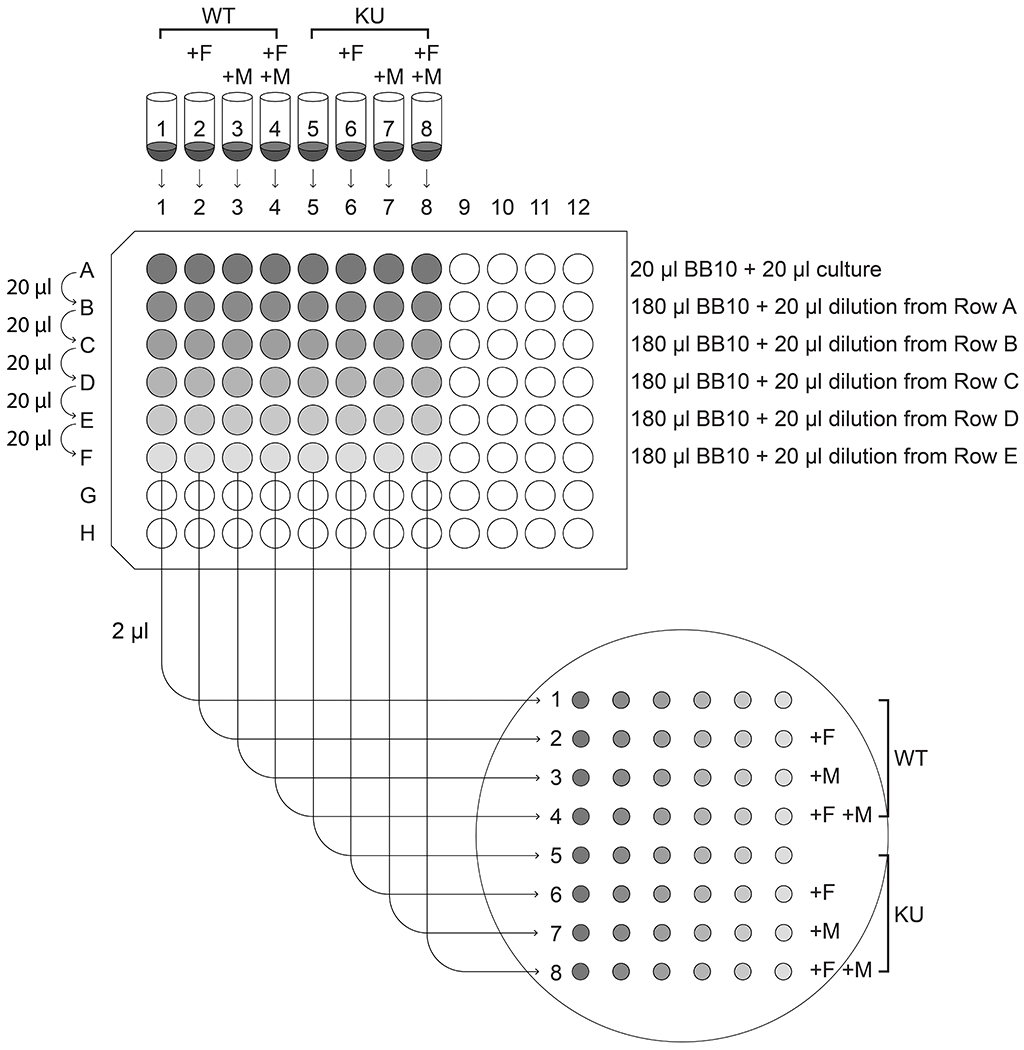
Schematic of the experimental setup for Support Protocol 2.
Sample Data
Figure 5:
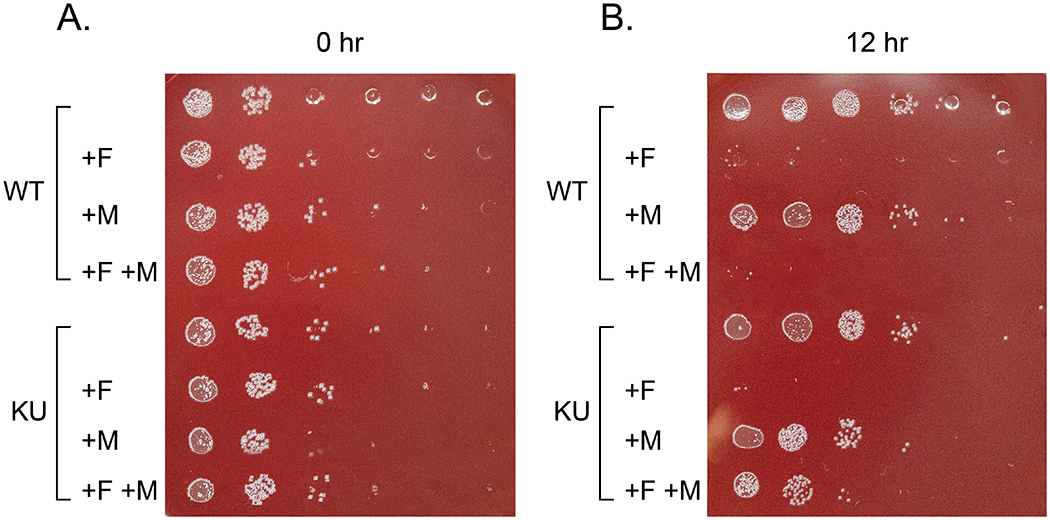
An example of how plates should look if your H. pylori strain can successfully use exogenous MurNAc. All conditions should grow well on the plate from the 0 hour timepoint (A.). For a strain similar to LSH100 (WT), there should be robust growth for all conditions on the two most concentrated spots, and a few colonies at higher dilutions. On the 12 hour timepoint plate (B.), there should be robust growth for untreated cultures on the three most concentrated spots and scattered colonies at higher dilutions. Cultures with just MurNAc added may have a small reduction in growth. If AmgK and MurU (KU) are successfully expressed, there should be more growth in row 8 (AmgK MurU strain + fosfomycin, + MurNAc) than in row 4 (wild-type strain + fosfomycin, + MurNAc). (Note the top row indentations appear as shiny circles, but the colonies are much smaller dots.)
BASIC PROTOCOL 2
BASIC PROTOCOL TITLE
Mass Spectrometry (MS) analysis to determine incorporation of MurNAc-alk within the peptidoglycan of H. pylori
Introductory paragraph:
This protocol instructs the user how to determine the PG subunits (muropeptides) that are labeled by MurNAc-alk. As MurNAc-alk is incorporated into PG precursors in the cytoplasm, the initial labeled muropeptide in the PG is the pentapeptide, but other labeled species can be generated by modification and/or crosslinking of these initial labeled pentapeptides. H. pylori cell walls labeled with MurNAc-alk are purified, digested, and analyzed using LC-MS/MS. This protocol provides both a means for determining which muropeptides are labeled within the cell over a given labeling period and unambiguous validation that MurNAc-alk successfully labels the PG.
Materials:
Starting liquid H. pylori culture of a known density (generated using Steps 1-3 of Support Protocol 1)
BB10 (see recipe in the Reagents and Solutions section)
MurNAc-alk (from Basic Protocol 1), 200 mg/ml stock in ddH2O or D-Ala-alk (ThermoFisher Scientific, #AC441221000), 100 mM stock in ddH2O
8% solution of HPLC electrophoresis-grade SDS (FisherScientific #BP166-500) in ddH2O
0.5% (w/v) methylene blue (Millipore Sigma # M9140)
0.2% sodium azide (w/v) (Millipore Sigma, #71289)
0.7 M sodium phosphate buffer pH 7.2 (see recipe in the Reagents and Solutions section)
10 mM Tris-HCl, 10 mM NaCl pH 7.0 (see recipe in the Reagents and Solutions section)
3.2 M Imidazole (see recipe in the Reagents and Solutions section)
10 mg/ml α-amylase in 10 mM Tris-HCl, 10 mM NaCl pH 7.0 (see recipe in the Reagents and Solutions section)
10 mg/ml Pronase E (Fisher Scientific, #50-146-956) in 10 mM Tris-HCl, 10 mM NaCl pH 7.0 (see recipe in the Reagents and Solutions section)
10% Formic acid (v/v) LC-MS grade (Millipore Sigma, #5.33002)
Water with 0.1% (v/v) Formic acid (VWR, #85960.320)
Acetonitrile with 0.1% (v/v) Formic acid (VWR, #84866.320)
50 mM Ammonium Formate Buffer pH 4.8 (see recipe in the Reagents and Solutions section)
0.5 mg/ml cellosyl (kind gift from Hoechst, Frankfurt am Main, Germany) in 10 mM Ammonium Formate Buffer pH 4.8 (see recipe in the Reagents and Solutions section)
50 ml baffled culture flasks with lids
Trigas incubator with a shaker [Sanyo O2/CO2 incubator; 10% O2, 10% CO2, Nitrogen to balance]
Spectrophotometer
Tabletop centrifuge
Two crystallizing dishes (FisherScientific #50-121-5014)
Two magnetic stirring hotplates
Small stir bars
50 ml flasks
Aluminum foil
Two round covered lead flask ring
50 ml conical tubes
Glass rods
Vortex mixer
Two P1000 pipettors
Glass Pasteur pipettes and bulb
15 ml conical vials
Ultracentrifuge
Ultracentrifuge tubes
2 ml microcentrifuge tubes
Speedvac vacuum concentrator (Thermo Scientific)
HPLC system (NanoACQUITY UPLC, Waters).
HPLC sample loop, 2 μL (Waters, # 430001264).
ACE Ultracore 2.5 super C18, 0.5 X 150 mm microbore column (VWR, #CORE-25A-15005).
Impact II QTOF LC-MS/MS system (Bruker).
Dry heat block (Dri-Block, Techne, Cole-Parmer).
Compass DataAnalysis™ software (Bruker).
ChemDraw™ software package (Perkin Elmer).
Protocol steps—Step annotations:
Digestion Protocol
-
Calculate the inoculum of the liquid H. pylori culture in ml using the following equation:
(V*ODlabel)/(ODinoculant*26)
where
V = overnight culture volume (330 ml)
ODlabel = target OD600/ml of labeling culture (1.0 OD600/ml)
ODinoculant = measured OD600/ml of liquid culture for inoculating (target 0.1-1.0)
Or more simply,
(5.16)/(ODinoculant)
-
Add 330 ml of BB10 to a sterile 500 ml bottle. Add in the calculated volume of liquid culture H. pylori calculated (in ml) above. Add 20.625 mg MurNAc-alk (103.1 μl of the 200 mg/ml stock; 62.5 μg/ml final concentration) or 33 mg D-Ala-alk (2.92 ml of the 100 mM stock; 100 μg/ml final concentration). Mix and aliquot into three 500-ml baffled culture flasks.
Note that in earlier cell shape publications, H. pylori cells for PG preps were harvested from agar plates rather than from liquid culture. One consequence of analyzing PG from cells grown on plates is that there is a greater distribution of growth phases represented in the population, including a notable proportion of cells near/at stationary phase. Older cultures of H. pylori will have a higher dipeptide composition (Costa et al., 1999), so PG composition results obtained from liquid cultures are best compared to more recent publications that also used liquid H. pylori cultures.
To make the negative control culture, add 330 ml of BB10 and the calculated volume of liquid culture H. pylori calculated (in ml) above to a second sterile 500 ml bottle. Mix and aliquot into three 500-ml baffled culture flasks.
-
Check the culture OD. Once the cultures reach 1 OD600/ml, put the flasks on ice.
A higher culture density is used here than in Basic Protocol 3 to strike a compromise between growth phase and amount of cells harvested per ml of culture, and thus amount of MurNAc-alk needed.
-
Add 6 ml of an 8% SDS solution to a 50 ml flask, add a small stir bar, and cover the top of the flask with aluminum foil. Poke a small hole in the top of the foil (Figure 6A.). Place a crystallizing dish on the hotplate, place the flask in the crystallizing dish, and weigh down the flask with a covered lead flask ring. Add water to the crystallizing dish to about midway up the culture flask (Figure 6B.). Add aluminum foil with a hole in the center over the top of the crystallizing dish to minimize steam escaping. The top of the flask should stick out above the aluminum foil (Figure 6C.). Turn on the hotplate and stirrer and to begin heating the water to boiling. Repeat to make a second setup for lysing the second set of cells.
Make sure to seal the top of the flask very tightly with the aluminum foil to minimize evaporation.
Begin harvesting the cells by filling a two 50-ml conical tubes per sample (4 in total) and centrifuging in a tabletop centrifuge at 2400 x g at 4°C for 10 min. Pour off the supernatant and repeat centrifugations until all of the culture volume for the labeled culture and experimental control have been processed.
-
Set aside one glass rod for the labeled cells and one glass rod for the negative control. Using the appropriate rod, resuspend each pellet in the residual media. To resuspend, hold the end of the rod gently between 2-3 fingers of one hand and hold the 50 ml conical on a running vortex mixer with the other hand. Move the rod up and down in the base of the tube and adjust the speed of the vortex mixer until the rod swirls readily about the sides of the tube. With the glass rod swirling, move the rod up and down the tube to distribute the pellet so that no discrete chunks of cells remain.
This may take some practice to get a feel for how best to hold the rod. This method will also be used to resuspend sacculi pellets following ultracentrifugation in later steps. For E. coli cells, harvested cell pellets can easily be resuspended by pipetting up and down. However, H. pylori cells tend to both clump to each other and to stick to pipette tips. Therefore, the glass rod resuspension method is much easier.
Begin consolidating the labeled cell pellets into one tube and the negative control cell pellets into one tube. Use two P1000 pipettors, one for pipetting PBS and one for transferring cell slurries. Transfer the cell suspension from one conical to the other. Then pipette 1 ml PBS into the emptied vial using the PBS pipette and wash the walls of the vial using the cell slurry pipette. Repeat until the volume of the collected cell suspension reaches 6 ml.
-
Keep the cell suspension on ice. Using a Pasteur pipette, slowly add the cell suspension to the boiling SDS. Once all of the cell culture has been pipetted into the flask with SDS, let the cells sit in the boiling SDS stirring for 30 minutes.
Take up only 1 ml of suspension at a time so the rest of the culture stays cold. Stick the end of the Pasteur pipette into the small hole in the foil on the top of the SDS flask. Add the suspension in tiny vigorous squirts (several squirts per ml) such that the cell suspension is rapidly dispersed into the SDS solution (each squirt dispersed within a few seconds). Wait until the previous addition is fully dispersed before adding more cells. At the end of this process, the liquid should be a golden color. Note the coloration varies based on the species of bacteria harvested but the final suspension should be clear regardless of color. If the SDS solution begins to bubble up, add a few drops of cold ddH2O, decrease the heat, and/or speed of the stir bar.
Repeat step 9 for the second set of cells with the second flask of SDS.
-
After each flask is finished boiling, remove the flask and cool at RT. Transfer each to a 15 ml conical vial and store at 4°C.
These samples can be stored for months at 4°C if necessary without degradation of the sample. The sacculi will eventually settle out and may be visible as a very faint clear pellet.
Repeat for the second set of cells with the second flask of SDS.
Place a bottle of ddH2O in a 60°C water bath.
Place the conical vials in an 80°C water bath for 30 minutes to redissolve SDS. Then cool to room temperature.
-
Pipette to mix each cell suspension then transfer each to an open-top ultracentrifuge tube. Balance the two tubes to within 0.01g of one another and fill tubes enough to prevent collapse (within 2-3 mm of top).
Nearly all if not all of the cell suspension should fit in the ultracentrifuge tube. If there is some left over, it can be discarded. Tubes can be topped off with ddH2O if more volume is necessary. Note that we use a larger ultracentrifuge (tube volume of approximately 12 ml) for the first set of washes and a smaller ultracentrifuge (tube volume of approximately 3 ml) for washes after digestion with α-amylase and Pronase E since pellets shrink notably after that step. It is acceptable to perform all washes in the smaller tubes, but this will increase the number of washes that need to be performed.
-
Ultracentrifuge for 60 minutes at 28°C at 133 907 RCF.
We use a Beckman-Coulter Optima L-90K ultracentrifuge with a SW41 rotor. Centrifugation steps should be performed warm to ensure that SDS does not precipitate. The size and appearance of the pellet will vary based on SDS concentration. The high concentration of SDS generally results in a much smaller pellet that will be larger after the following wash. Do not be alarmed. Also, prior to using an ultracentrifuge for the first time, seek out training to ensure you use this equipment properly.
-
After the first ultracentrifugation, pipette out the supernatant, leaving a small amount above the pellet.
PG pellets of H. pylori are very loose with 4% SDS, but will be much more robust in all subsequent wash steps in this protocol. E. coli pellets are robust even at 4% SDS. Supernatant should be pipetted, or in later steps decanted, into a small beaker to allow recovery in case the pellet is disrupted or lost during removal of the supernatant. If this occurs, ultracentrifuge the supernatant again. See Figure 7 for images of how the pellets should appear (A.) after removal of the supernatant, (B.) during resuspension with the glass rod, and (C.) After initial disruption of the pellet.
-
Resuspend the pellet using a clean glass rod. First vortex with the rod only (moving rod up and down while vortexing as in Step 7), then slowly add a drop or two of ddH2O (60°C) and vortex with rod as before. Continue adding and vortexing until a few ml have been added and no more water can be safely added without risk of losing some sample. Rinse the glass rod with water into the ultracentrifuge tube to avoid sample loss and return it to a labeled 15ml conical vial. Fill the rest of the centrifuge tube with water (to within 2-3 mm of the top) and balance to within 0.01g. Return the ddH2O to 60°C.
Note to pause for the day, add sodium azide (0.02% final concentration) and store at 4°C. Seal open-top ultracentrifuge tubes with parafilm. The next day, replace the parafilm with aluminum foil and reheat any samples still containing SDS in 80°C water bath for 30 minutes to redissolve SDS.
-
Repeat ultracentrifugation and wash steps approximately 3-4 times to completely remove SDS.
You can check for complete removal of SDS earlier; the number of wash steps required depends on wash volume (determined by ultracentrifuge and corresponding tube). After the first wash is complete, the supernatant can be decanted from rather than pipetted out of the tube in all following wash steps.
Use Support Protocol 3 (Hayashi Test) to check if all the SDS has been removed from the sample. Proceed to step 21 once the SDS has been removed from each sample.If one sample is SDS free before the second, use water in a blank tube to balance the remaining sample. Minimizing the number of wash steps for each sample minimizes the amount of PG loss.
-
Resuspend sacculi in 900 μl of 10 mM Tris HCl with 10 mM NaCl, pH 7.0.
To achieve quantitative transfer, resuspend the pellet by first vortexing the pellet alone (with a glass rod), then adding one to two drops of solution from a pipette tip containing 900 μL of solution and vortexing again with the glass rod. Carefully set the pipette with solution aside. Transfer the PG material to a 2 ml microcentrifuge tube using a different 1 ml pipette tip. Do not eject the tip. Carefully set this other pipette with tip aside. Using half of the remainder of the solution in the pipette tip, rinse the glass rod into the ultracentrifuge tube and again set the rest of the solution in the pipette aside. Place the glass rod back in its container. Using the pipette tip used to transfer the sacculi, pipette the solution down the sides of the ultracentrifuge tube to rinse them. Then pipette all this liquid into the 2 ml microcentrifuge tube. Still keep the pipette tip. Pipette the remainder of the solution into the ultracentrifuge tube. Then using the tip used for transfer, again rinse the walls of the ultracentrifuge tube and transfer this liquid to the microcentrifuge tube.
-
Add 100 μl of 3.2M imidazole, pH 7.0 and 15 μl α-amylase (10 mg/ml), vortex briefly, and incubate for 2 hours at 37°C.
This degrades the high molecular-weight glycogen that is trapped inside the sacculi.
-
Add 20 μl Pronase E (10 mg/ml) and incubate for 1 hour at 60°C.
This releases the covalently bound lipoproteins, which should not be present in H. pylori, but this step is necessary for other bacteria and is thus included for uniformity. Note that Pronase E needs to be pre-incubated for two hours at 60°C to degrade a contaminating enzyme that comes with it. You can pre-incubate aliquots and then store them at −20°C.
-
Add 500 μL 8% SDS and boil for 15 minutes on heating block.
Alternatively, samples can be incubated at 80°C for 30 minutes. Beware the caps popping off. At this stage, the PG prep can be stored over the night or weekend at 4°C without the addition of sodium azide.
-
Quantitatively transfer the material to ultracentrifuge tubes and remove SDS again by ultracentrifugation followed by washes with 60°C water as described above.
Using a tabletop ultracentrifuge, tube balancing can be performed by eyeballing equal volumes. We used a TLA 100.3 fixed angle rotor. Take care not to fill the tubes too full to ensure sample is not lost in the fixed angle rotor, but do include enough volume to support the tubes during ultracentrifugation (fill such that when angled, the side of the liquid closest to the tube rim is 3-4 mm below the rim).
Suspend isolated PG saccculi in 100 μl of 20 mM ammonium formate pH 4.8, add 10 μg of 0.5 mg/ml cellosyl, and incubate overnight at 37⁰C on a Thermomixer (Eppendorf) at 900 rpm.
Figure 6:

(A.) 50 ml flask with 6ml 8% SDS and a small stir bar. The top is tightly covered with aluminum foil with a small hole poked in the center and a lead weight is placed around the flask. (B.) The flask is placed in a crystallizing dish with water. (C.) The top of the crystallizing dish is covered with aluminum foil with a hole in the center to accommodate the top of the flask.
Figure 7:
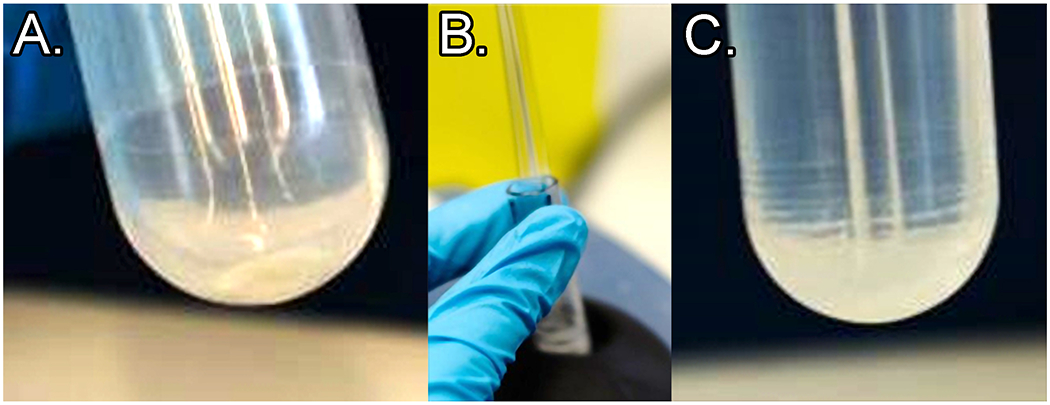
(A.) PG pellet at the tube bottom (material should be almost clear; sometimes a small amount of opaque material is present at the very bottom of the pellet – this is acceptable). (B.) Example of desired rod motion during proper vortexing. (C.) PG pellet after vortexing with glass rod: note rings of sacculus material up the side of the tube.
Sample Preparation for LC-MS
-
27. Following digestion, place samples in a dry heat block at 100⁰C for 10 min and then centrifuge at room temperature for 15 min at 15,000×g. Retrieve the supernatant and place in a clean tube.
Note: In conventional muropeptide preparation methods the released muropeptides are finally treated with a reducing agent (sodium borohydride/Tetramethyl ammonium borohydride) to remove anomerization by reducing the aldehyde group of the muramic acid (at sugar carbon position 1). Reduction is used in order to prevent the formation of anomer-related chromatographic artefacts such as peak splitting (see Figure 10D). Unfortunately, the reduction step (in our hands) seems to degrade or modify the MurNAc-alk label, and so this step is omitted from our protocol.
28. Dry supernatant samples using a vacuum concentrator.
-
29. Resuspend MurNAc-alk labeled digests in 1% formic acid (20 – 50 μL of distilled water plus 1/10th volume of 10% formic acid) with vortexing for 5 minutes.
Sometimes it is difficult to completely resuspend all of the muropeptide pellet. Samples may be cleared by centrifugation (next step).
30. Centrifuge at 15 000xg for 3 minutes. Carefully remove the supernatant to a clean microcentrifuge tube.
Figure 10.
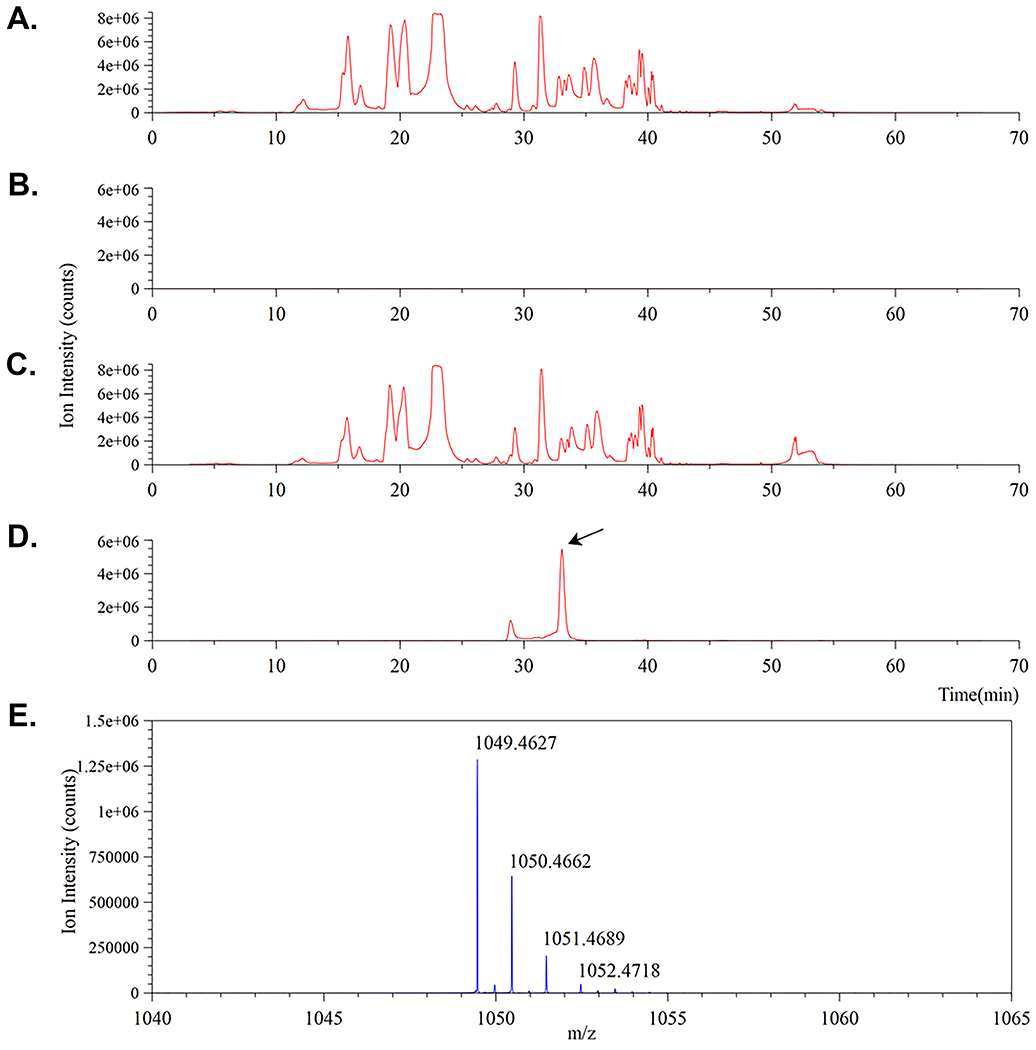
Confirmation of MurNAc-alk incorporation into H. pylori PG. A.-D. comprises four ion traces. A. and C. correspond to total ion chromatograms for the control and labeled muropeptide digests respectively. B. and D. correspond to mass extracted ion chromatograms for the same control and labeled muropeptide digests set to monitor for the predicted 1+ monomer ion mass (m/z = 1049.45) eluting around 33 min and shown in E. The presence of the MurNAc-alk pentapeptide muropeptide monomer (D.) is indicated by an arrow.
The samples are now ready for electrospray mass spectrometry analysis as described below
LC-MS Setup
For sensitive LC-MS/MS analysis, prepare a microbore HPLC resolving column arrangement based around a 6-port selection valve and a 2 μl injection loop. Any modern HPLC system equipped with such a switching valve may be configured in the arrangement shown in the schematic in Figure 8 (below). HPLC requires configuration of a 6-port valve for low/sub-microliter volume injections. To maintain the separation efficiency of the microbore column, target injection volumes of 0.1-0.2 μl are required. A HPLC system equipped with a 2 μl (or lower) injection loop is suitable. If such a setup is not available, adaption of the HPLC gradient and flow rate (below) to suit a larger diameter column and injection volume should be possible.
Figure 8:
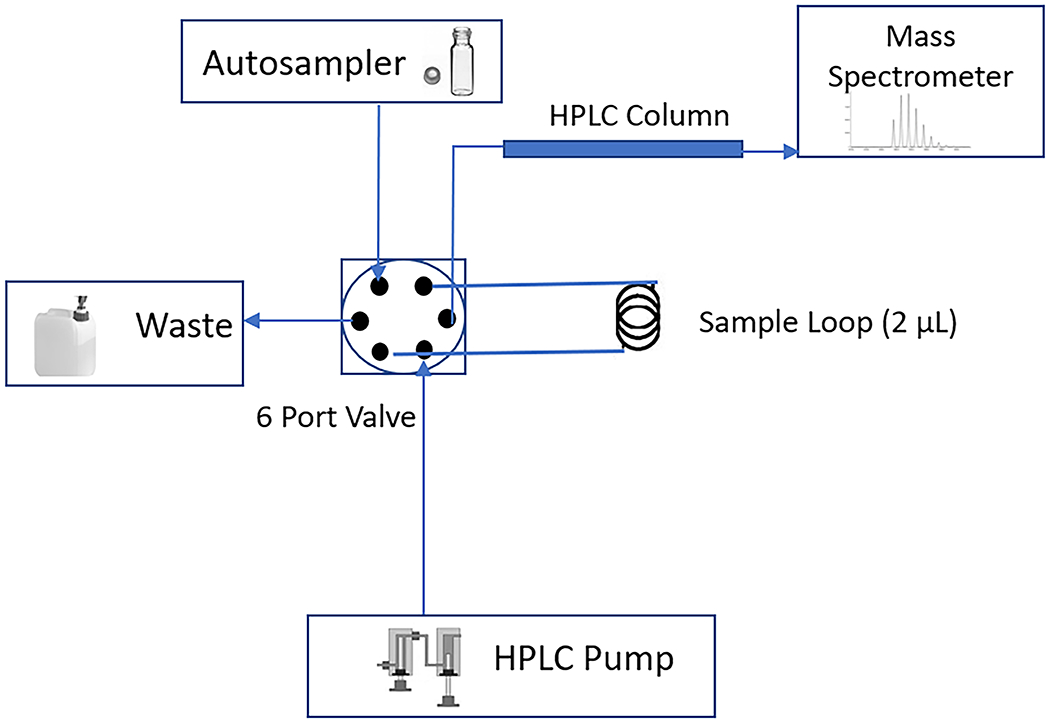
Schematic representation of the microbore column LC-MS/MS arrangement for muropeptide analysis.
HPLC Conditions
For buffer A, use 0.1% formic acid in water (VWR). For buffer B, use 0.1% formic acid in acetonitrile (VWR).
31. Use the following microbore RP-HPLC conditions: 0% buffer B for 3 min, rising to 1.5% B at 20 min, then on to 3.0% B at 35 min, ramp the gradient to 15% B at 45 min, then on to 50% B at 50 min, followed by 2 min at 85% B, and finally 15 min re-equilibration at 0% B.
32. Set the HPLC flow rate to 20 μl/min and the microbore column temperature to 35°C.
-
33. Dilute sample 1:5 (v/v) with buffer A and then place 10 μl in an autosampler vial. Inject directly (typically 0.2 μl) onto the microbore HPLC column configured as shown in Figure 8.
Muropeptides have quite hydrophilic properties. In the presence of a low percentage of organic solvent, they will elute in order of increasing hydrophobicity and size.
MS conditions
Mass spectrometers can be rather complicated instruments to program for LC-MS/MS analysis.For novice users we highly recommend that you consult an experienced operator or service provider. The instrument settings below should provide enough detail for data acquisition.
34. Direct the microbore column eluate to the mass spectrometer via an Apollo electrospray ion source (Bruker, Billerica, MA). Typical settings for the ion source are capillary voltage and temperature settings of 3200 V and 150oC respectively, together with a drying gas flow of 5 L/min and nebulizer pressure of 0.6 Bar.
35. Collect the MS data in positive ion mode over the range of 50 – 2000 m/z at a spectral rate of 2 Hz.
36. Perform MS/MS acquisition on the top 5 intense precursors in each scan (exclude these ions after two MS/MS events). The preferred charge states are set at +1 to +4 (undetermined charge states are excluded) and the sampling rate is varied from 0.5 Hz at low ion counts (10,000) to 5.0 Hz for intense ion counts (500,000).
37. The resulting MS spectral data can be opened for analysis using Compass DataAnalysis™ software (Bruker).
Data Analysis – Example data
This section presents an overview of how MS data files are manipulated to extract ion chromatogram traces and to interpret MS/MS data to confirm muropeptide structures. This is best explained with a few worked examples (below).
-
38. For the purpose of this protocol, we shall illustrate how to link accurate mass and fragmentation patterns to structure models in order to confirm identity for an exemplar monomer and dimer muropeptide.
Use the ChemDraw™ software package (Perkin Elmer) to model the structures of predicted MurNAc-alkyne labeled muropeptides as shown below in Figure 9. Then confirm structure models using MS data manipulation with the Compass DataAnalysis™ software (Bruker) as illustrated.
This example shows confirmation of the presence and identity of two suspected MurNAc-alkyne labelled muropeptides in cellosyl digest profiles of the H. pylori KU strain (rdxA::amgKmurU) cell wall grown in the presence of fosfomycin and supplemented with MurNAc-alkyne. The control digestion is grown without addition of MurNAc-alkyne.
39. Figure 9 illustrates the complete structures and MS/MS fragmentation points for the predicted modified MurNAc-alk pentapeptide (monomer) and MurNAc-alk pentapeptide-tetrapeptide (dimer) muropeptides. The calculated theoretical masses are 1048.44 and 1969.83 Da respectively, with expected singly [M+H]+ (monomer) and doubly [M+2H]2+ (dimer) charged ions to be observed at m/z = 1049.45 (1+) and 985.92 (2+) respectively (ChemDraw™).
-
40. The first exemplar mass spectral data is for the MurNAc-alk pentapeptide monomer whose structure is illustrated in Figure 9A. Load the labeled and non-labeled (control) MS data files into Compass DataAnalysis. Initial MS ion traces for the unlabeled cell wall digest and MurNAc-alk labelled cell wall digest are shown below in Figure 10A. and C., respectively.
Notice how the traces appear to be essentially identical. This is because the alkyne label is only incorporated as a small percentage of the total MurNAc during cell wall synthesis. Therefore, superficially all traces will look alike, hence the reason why we must ‘search’ for predicted modified muropeptide masses as determined in ChemDraw™.
41. Figure 10 (A.–D.) illustrates four ion chromatograms representing muropeptide LC-MS analysis performed following the digest and LC-MS conditions outlined in the previous sections. As stated, traces A. and C. represent total ion chromatogram traces (TICs) for the H. pylori KU strain PG digests that were grown in the absence (A.) and presence (C.) of MurNAc-alkyne.
-
42. Traces B. and D. represent ‘extracted’ ion chromatograms (XICs) taken from A. and C. respectively. set the DataAnalysis™ software to monitor for the expected 1+ ion of the MurNAc-alkyne pentapeptide at m/z = 1049.45 ± 0.1 (Figure 9A.).
Notice the absence of the expected ion signal in trace B. and the presence of a major ion peak around 33 min in D. (black arrow).
-
43. Figure 10E. shows the mass spectrum of the peak at approximately 33 min in D. confirming the presence of an ion at m/z = 1049.46 as expected.
Note: The presence of a second, lower intensity peak (here ~29 min) is something we commonly observe with non-reduced muropeptides. We suspect it to be a feature of the anomeric carbon because muropeptide reduction resolves such chromatograms to just a single peak.
-
44. In order to confirm that the ion at m/z = 1049.46 is indeed the expected MurNAc-alk pentapeptide muropeptide, refer to the MS/MS fragmentation spectrum for this ion. Figure 11 shows the MS/MS spectrum obtained from fragmentation of the parent ion at m/z = 1049.46 that eluted at around 33 min. We list abundant fragment ions (m/z labeled) that contributed to the positive identification of the MurNAc-alkyne pentapeptide in Table 1.
Fragment ion structures can be modeled and verified using the ChemDraw™ software package (Perkin Elmer).
-
45. Table 1 illustrates assignment of the top 10 ion peaks in Figure 11 (descending intensity) confirming the modified MurNAc-alk pentapeptide (monomer) structure as predicted in Figure 9A.
Notice how all the ion fragmentations have taken place across the peptide and glycosidic bonds that link the subunits. Loss of water (δ = 18) is not uncommon with peptidoglycan during MS/MS fragmentation.
-
46. The second exemplar mass spectral data is for the MurNAc-alk pentapeptide-tetrapeptide dimer muropeptide structure illustrated in Figure 9B. Again, load the two MS data files. The initial TIC ion traces for the unlabeled cell wall digest and MurNAc-alk labelled cell wall digest are shown below in Figure 12A. and C., respectively.
Notice once again how these TIC traces appear to be essentially identical for the same reason of low percentage incorporation of tag discussed previously.
47. Once more, Figure 12A.–D. shows four ion chromatograms representing DataAnalysis™ software analysis of muropeptide LC-MS data acquired following cell wall digest and muropeptide LC-MS separation. Traces A. and C. represent total ion chromatograms for the H. pylori KU strain PG digests that were grown in the absence (A.) and presence (C.) of MurNAc-alkyne.
-
48. Traces B. and D. are extracted ion chromatograms taken from A. and C. respectively, with the DataAnalysis™ software set to monitor for the expected 2+ ion of the MurNAc-alkyne pentapeptide-tetrapeptide at m/z = 985.92 ± 0.1 (Figure 9B.).
Again, note the absence of the expected ion signal in trace B. and the presence of a major ion peak around 40 min in D. (black arrow).
49. Figure 12E. shows the mass spectrum of the peak eluting around 40 min in D. confirming the presence of an ion at m/z = 985.93 as expected.
50. Once more use the MS/MS spectrum obtained from fragmentation of the parent ion at m/z = 985.93 (eluting around 40 min) to confirm identity, as shown in Figure 13. Abundant fragment ions (m/z labeled) that contributed to the positive identification of the MurNAc-alkyne pentapeptide-tetrapeptide are listed in Table 2 below.
-
51. Table 2 shows assignment of the top 10 ion peaks in Figure 13 (descending intensity) confirming the modified MurNAc-alk pentapeptide-tetrapeptide (dimer) structure as predicted in Figure 9B.
Again, note how the intense ions are comprised from fragmentations that have taken place across the peptide and glycosidic bonds that link the subunits, thereby confirming the structure as predicted in Figure 9B.
52. Lastly, repeat the above process of modelling predicted structures in ChemDraw™, using the calculated ion masses to search raw data files (labelled versus unlabeled control) with DataAnalysis™, and finally confirming all predicted ion identities via inspection of the MS/MS fragmentation spectrum of each and applying the rules for expected ion fragmentation across peptide and glycosidic bonds, until you have sufficient conclusive evidence to prove tag labelling.
Figure 9.
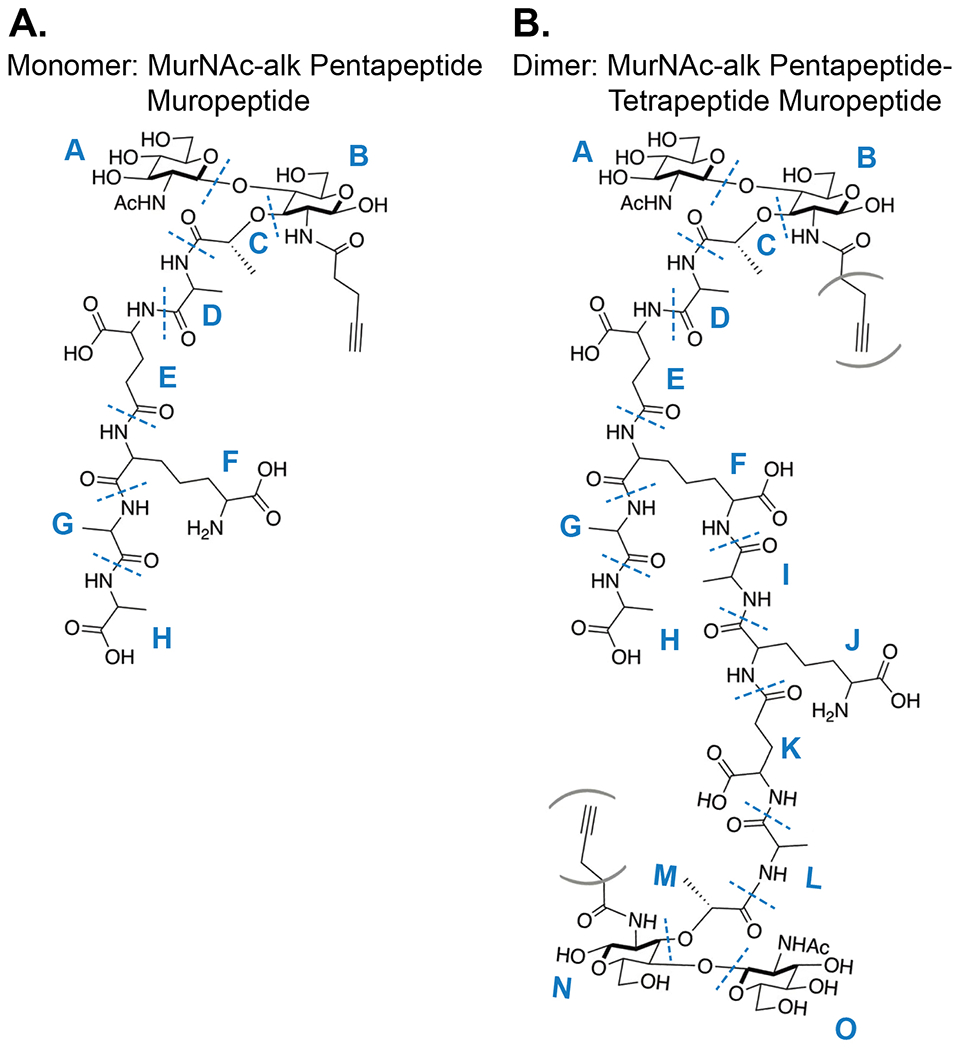
Target structures to test for incorporation of MurNAc-alk into H. pylori cell wall peptidoglycan. Bonds predisposed to MS/MS fragmentation are indicated with blue dotted lines. Subunit structures are labelled (A.) A – H (monomer) and (B.) A – O (dimer) respectively. Parentheses indicate that the Alk motif could be present at either position.
Figure 11.
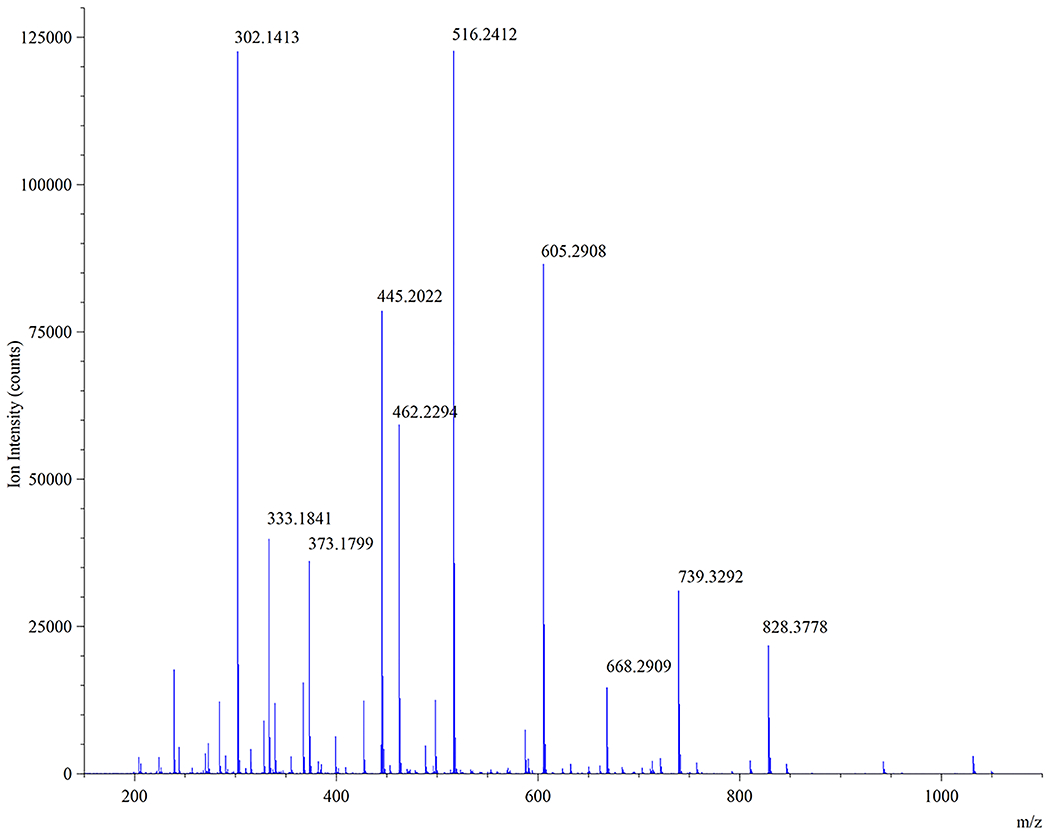
MS/MS spectrum of suspected MurNAc-alk pentapeptide muropeptide ion (m/z = 1049.46) eluting around 33 min in Fig. 9A.
Table 1.
Top 10 abundant ion fragment peaks in Figure 11 (MS/MS spectrum of the m/z = 1049.46 ion) confirming the MurNAc-alk pentapeptide muropeptide structure as depicted in Figure 9A.
| Ion Peak | Observed m/z | Ion Fragment Containing |
|---|---|---|
| 1 | 516.24 | [A to C]+ |
| 2 | 302.14 | [E to F]+ |
| 3 | 605.29 | [C to H]+ |
| 4 | 445.20 | [A + B]+ |
| 5 | 462.23 | [E to H]+ |
| 6 | 333.18 | [F to H]+ |
| 7 | 373.18 | [E to G]+ |
| 8 | 739.33 | [B to G – H2O]+ |
| 9 | 828.38 | [B to H – H2O]+ |
| 10 | 668.29 | [B to F – H2O]+ |
Figure 12.
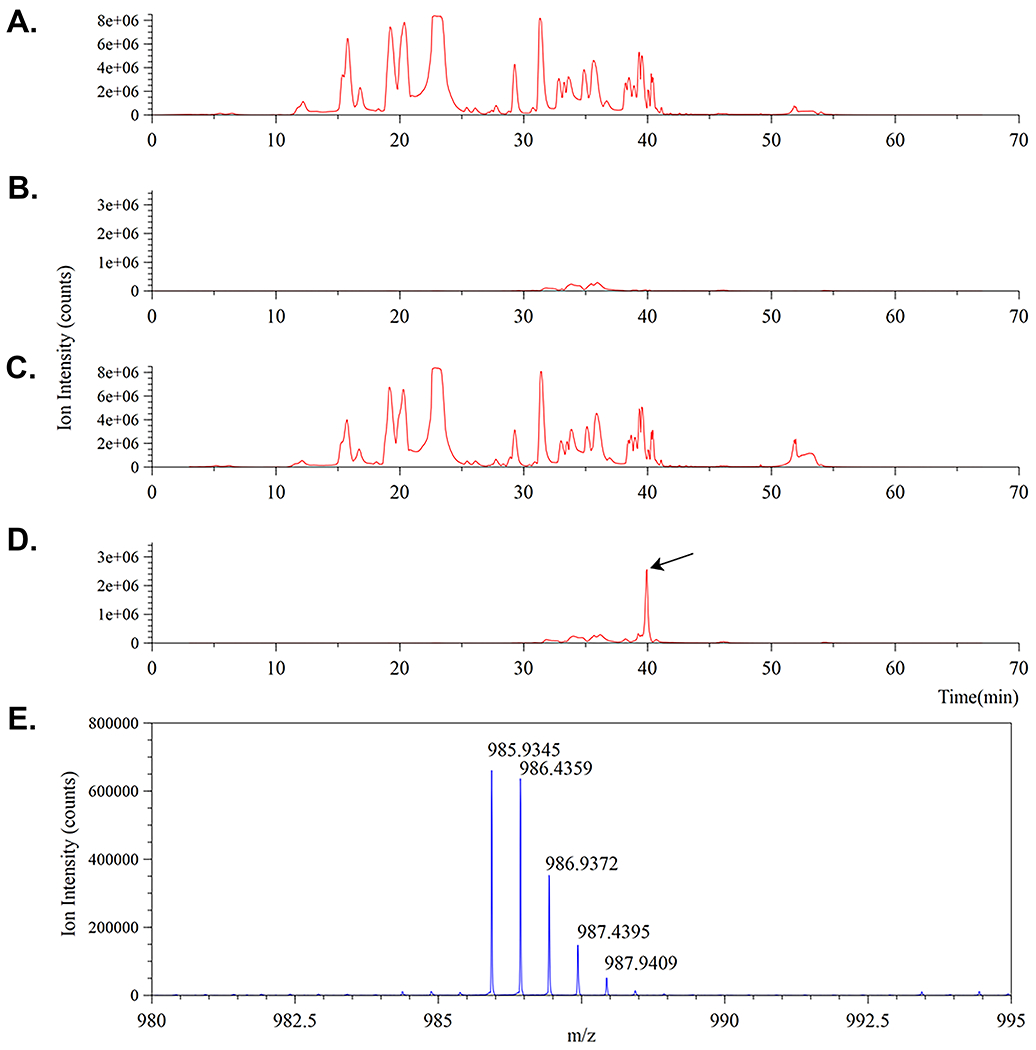
Confirmation of MurNAc-alk incorporation into H. pylori PG. A.-D. are four ion traces. A. and C. correspond to total ion chromatograms for the control and labeled muropeptide digests respectively. B. and D. correspond to mass extracted ion chromatograms for the same control and labeled muropeptide digests set to monitor for the predicted 2+ dimer ion mass (m/z = 985.92) eluting around 40 min and shown in E. The presence of the MurNAc-alk pentapeptide-tetrapeptide muropeptide dimer (D.) is indicated (black arrow).
Figure 13.
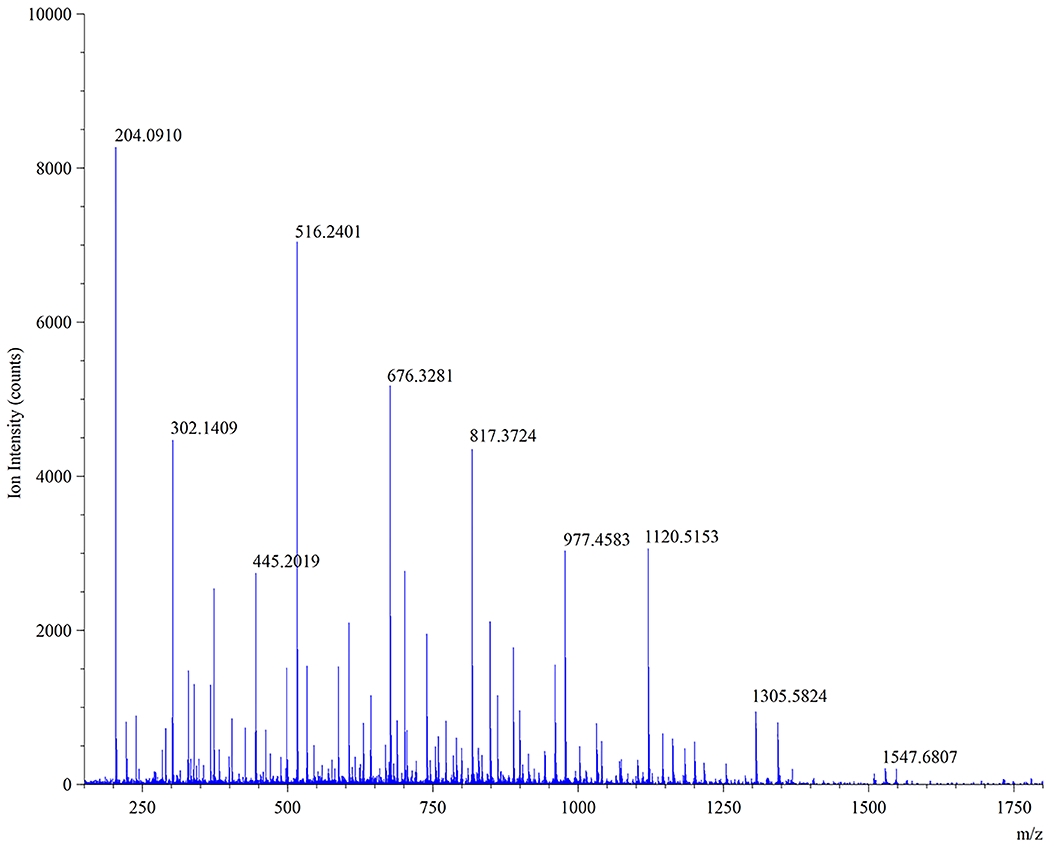
MS/MS spectrum of MurNAc-alk pentapeptide-tetrapeptide muropeptide ion (m/z = 985.93) eluting around 40 min in Fig. 12D.
Table 2.
Top 10 abundant ion fragment peaks in Figure 13 (MS/MS spectrum of the m/z = 985.93 ion) confirming the MurNAc-alk pentapeptide-tetrapeptide muropeptide structure as depicted in Figure 9B.
| Ion Peak | Observed m/z | Ion Fragment Containing |
|---|---|---|
| 1 | 204.09 | [A & O]+ |
| 2 | 516.24 | [C to F + I – H2O]+ |
| 3 | 676.33 | [C to I – H2O]+ |
| 4 | 302.14 | [E to F – H2O]+ |
| 5 | 817.37 | [C to F + I to K – H2O]+ |
| 6 | 977.46 | [C to K – H2O]+ |
| 7 | 1120.51 | [C to M – H2O]+ |
| 8 | 445.20 | [C to F – H2O]+ |
| 9 | 1305.58 | [C to N – H2O]+ |
| 10 | 1547.68 | [B to N – H2O]+ |
SUPPORT PROTOCOL 3
SUPPORT PROTOCOL TITLE
Hayashi test to determine if SDS is present in the supernatant of peptidoglycan preparations.
Introductory paragraph:
Here, the user performs a colorimetric assay on the supernatant of each peptidoglycan prep after washing to determine if all the SDS has been removed from the sample (Hayashi, 1975). The sample is SDS-free when the blue color is only present in the upper layer and the lower (chloroform) layer is clear or tinted slightly pink.
Materials:
335 μl sample of the supernatant after centrifugation
7 μl 0.5% methylene blue
170 μl 0.7 M sodium phosphate buffer pH 7.2
1 ml chloroform
2 ml microcentrifuge tube
vortexer
Protocol steps—Step annotations:
Pipette a 335 μl sample of supernatant after centrifugation, 7 μl 0.5% methylene blue, and 170 μl 0.7 M sodium phosphate buffer pH 7.2 into a 2 ml microcentrifuge tube and mix.
Add 1 ml chloroform, cap, and mix using a vortex mixer for 30 sec.Chloroform that has been stored in the dark must be used, otherwise the test may indicate that SDS is still present when it has already been successfully removed. You may need to invert/shake the tube by hand prior to vortexing to get layers to mix properly.
Let the tubes sit for 30 sec or centrifuge briefly to separate the phases.
-
Immediately look for coloration in the chloroform (lower) layer. Appearance of blue color in the chloroform layer indicates the presence of SDS. If the chloroform layer is clear or tinted pink, SDS has been successfully removed.
Eventually the lower phase will turn blue even if all the SDS has been removed from the sample. Reading the test too late may incorrectly indicate that SDS is still present.
Sample Data
At the end of this protocol, users will have determined whether SDS has been completely removed from their peptidoglycan prep. If so (Figure 14, Samples 1-3), the user may proceed to the next step of Basic Protocol 1. If not (Figure 14, Sample 4), repeat the wash step and subsequent Hayashi test until all of the SDS has been removed.
Figure 14:
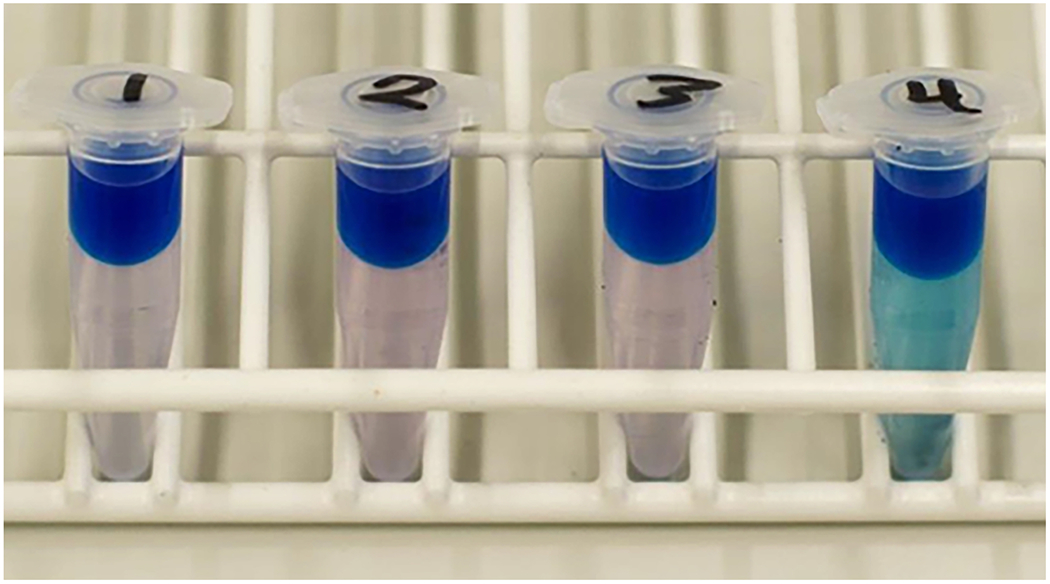
Samples 1–3 are SDS-free, while sample 4 still contains SDS; sample 4 must be washed again and re-tested.
SUPPORT PROTOCOL 4
SUPPORT PROTOCOL TITLE
Creating custom cytocentrifuge units for use in a swinging bucket tabletop centrifuge
Introductory paragraph:
Here, the user creates cytocentrifuge units that can be used in a standard tabletop swinging-bucket centrifuge that can accommodate 96-well plates. The user will cut out the necessary pieces using a provided template and will assemble four top and four bottom units using superglue. Wear gloves and take care not to glue skin. Use superglue in a well-ventilated area.
Materials:
Materials list to make four cytocentrifuge units:
two sheets of gasket rubber: 6” x 6” x 1/16”
Tenura anti-slip silicone roll
four 1.5 ml microcentrifuge tube
one sheet of 1.3 mm thick plexiglass
scissors
dissecting scissors
razor blade or exacto knife
safety glasses
straight edge for a cutting guide
disposable gloves
superglue
Templates 1 and 2 (included with this manuscript), printed at 100% size
Protocol steps—Step annotations:
Cut out the pieces from the rubber gasket (Figure 15A.). Using a razor blade, exacto knife, or scissors, cut four 1” x 3” rubber gasket base pieces (Template 1, Piece A).
Using a razor blade, exacto knife, or scissors, cut four 1” x 1” rubber gasket pieces. Using the dissecting scissors, cut one hole 5/16” in diameter in the center of each piece (Template 1, Piece B).
Using the dissecting scissors, cut 32 rubber gasket circles with a diameter of 5/8”. Then cut one hole 10.8 mm in diameter (or the outer diameter of the flat side of your 1.5 ml microcentrifuge tube) in the center of each of these pieces (Template 1, Piece C).
Using a razor blade, exacto knife, or scissors, cut 12 1” x 1” and eight 5/8” x 5/8” rubber gasket pieces (Template 1, Piece D and E, respectively).
-
Using a razor blade or exacto knife and a straight edge as a guide, cut four 1” x 1” squares out of the 1.3 mm-thick plexiglass (Template 2, Piece A) and remove protective coating, if present (Figure 15B.).
To cut the plexiglass, deeply score the plexiglass with the blade. This will take multiple passes of the blade. Score the outer edges of the rectangle of squares first. Once these edges are scored to a depth of at least half the thickness of the plexiglass, carefully bend the plexiglass at the score lines until it snaps. Wear safety glasses and gloves while snapping the plexiglass. If the plexiglass snaps at the wrong location, repeat the process and score more deeply. After snapping the rectangle from the sheet of plexiglass, repeat the scoring and snapping process for each of the squares within the rectangle. Be cautious both when scoring and snapping the plexiglass to prevent lacerations.
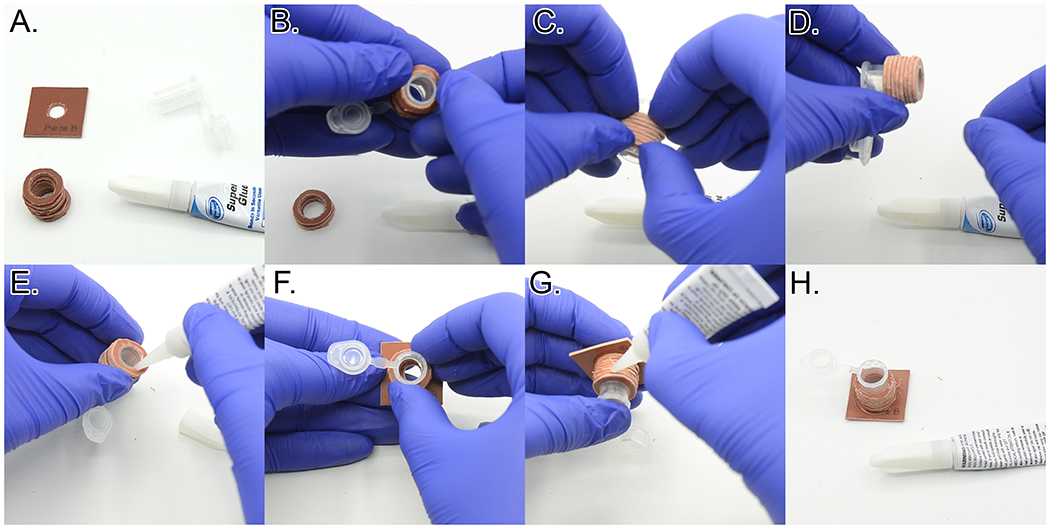
To make the tops (Video 1), use a razor blade to carefully cut four 1.5ml centrifuge tubes at the point where the walls begin to taper in to form the point at the bottom of the tube such that the straight-walled cylindrical part of the tube and the cap apparatus remain (Figure 15C.).
Thread eight rubber gasket rings (Template 1, Piece C) onto the base of each cut centrifuge tube. Squeeze the stack of rings together and the push the stack of rings to the base of the tube (Figure 16A.–D. and Video 1).
Wearing gloves, apply a small amount of superglue around the lowest ring (Template 1, Piece C) on the cytocentrifuge tube (Figure 16E.). Place the cut centrifuge tube cut-side down on top of the superglue, centered on the hole (Figure 16F.). If necessary, add a little more superglue around the outer base of the tube.
Before the superglue dries, carefully push the rings (Template 1, Piece C) around the centrifuge tube down to contact the rubber gasket base (Template 1, Piece B), such that the rings are tightly stacked. Add superglue to the exposed ring faces (sides and top) and let thoroughly dry (Figure 16G.–H.).
To make the bottoms (Video 2), wearing gloves, superglue one Template 1 Piece D to the center of each Template 1, Piece A. Superglue two additional Template 1, Piece D on to the first Piece D and then two Template 1, Piece E to create a stack.
Superglue one Template 2, Piece A to the top of each Template 1, Piece D stack and let dry.
Using scissors, cut four 1” x 1” silicone squares (Template 3, Piece A). Cut an additional four 1” x 1” silicone squares, and using dissecting scissors, cut out one 5/16” diameter hole in the center of each (Template 3, Piece B) (Figure 15D.).
Figure 15:

Rubber gasket pieces (A.), plexiglass pieces (B.), and cut microcentrifuge tops (C.) for assembling cytocentrifuge tops and bottoms. (D.) Silicone pieces for creating a liquid-tight seal in assembled cytocentrifuge units.
Figure 16:
Assembling the cytospin tops. A. Components needed to make one cytospin top. B. Adding a stack of rubber gasket rings to the cut microcentrifuge tube. C. Squeezing the rubber rings together to remove spaces between the rings. D. Final position of the stack of rubber rings. E. Applying superglue to the bottom of the ring stack. F. Gluing the cut microcentrifuge tube and ring stack to the bottom rubber gasket piece, centering the hole. G. Applying superglue to the side of the ring stack. H. Completed cytocentrifuge top. See Video 1.
Sample Data
At the end of this protocol, users should have successfully created four cytocentrifuge units, as pictured in Figure 17. Each unit consists of a base and a top. The base serves as a pedestal to support the coverslip during centrifugation. The top serves as a container for the cell suspension during centrifugation. The tops and bottoms should be interchangeable and should seal securely when assembled with the silicone pieces and the micro binder clips.
Figure 17:

Completed cytocentrifuge unit components
BASIC PROTOCOL 3
BASIC PROTOCOL TITLE
Labeling H. pylori with MurNAc-alk or D-Ala-alk
Introductory paragraph:
This protocol instructs the user how to create high-quality slides of H. pylori cells labeled for a short pulse with MurNAc-alk or D-Ala-alk. Cells are grown in liquid culture with the metabolic probe, fixed, and permeabilized. Cells are then spun onto coverslips using the cytospin units from Support Protocol 4. Click labeling and whole cell wall staining are performed on cells on the coverslips, and coverslips are mounted on slides and cured before imaging.
Materials:
Liquid Helicobacter pylori culture (from Support Protocol 1)
BB10 (see recipe in the Reagents and Solutions section)
D-Ala-alk (ThermoFisher Scientific, #AC441221000), 100 mM stock in ddH2O
MurNAc-alk (from Basic Protocol 1), 200 mg/ml stock in ddH2O
10% sodium azide stock in ddH2O
Brucella broth (see recipe in the Reagents and Solutions section)
20% paraformaldehyde (PFA) aqueous solution (Fisher Scientific, #50-980-492)
70% ethanol, aqueous solution
Alconox® detergent (Sigma-Aldrich, #Z273228-1PAK)
0.05% Tween-20 in Dulbecco’s Phosphate Buffered Saline (PBST) (PBS from ThermoFisher Scientific #14190144)
Click-iT Cell Reaction Buffer Kit (ThermoFisher Scientific #C10269)
Dimethyl sulfoxide >99.9% , Alfa Aesar (Fisher, #AA22914K2)
Alexa Fluor 555 Azide, Triethylammonium Salt (Thermofisher Scientific, #A20012)
Dulbecco’s Phosphate Buffered Saline (PBS)
Wheat Germ Agglutinin Alexa Fluor 488 Conjugate (ThermoFisher Scientific, #W11261)
ProLong™ Diamond Antifade Mountant (ThermoFisher Scientific, #P36961)
Trigas incubator with shaker [Sanyo O2/CO2 incubator; 10% O2, 10% CO2, Nitrogen to balance]
Water bath, set to 55°C
spectrophotometer
cuvettes
5 ml polystyrene round-bottom tubes
timer
ice and ice bucket
1.5 ml microcentrifuge tubes
circular sticker labels
fine-tipped permanent marker
microcentrifuge
Glass screw-cap test tube for storing excess PFA at −20°C
Rotisserie shaker
hemocytometer with cover glass
phase contrast microscope
Corning™ Square cover classes, No. 1.5 (Fisher Scientific, #12-553-453)
cytocentrifuge units (from Support Protocol 4)
micro binder clips
two 96-well plate lids
laboratory tape
swinging bucket centrifuge that can accommodate 96-well plates
high precision tweezers (Electron Microscopy Sciences, #78752-08)
parafilm
paper towels
opaque box lid
Plain glass slides (FisherScientific, #12-549-3)
microfiber towel
Protocol steps—Step annotations:
As detailed in Support Protocol 1, prepare a liquid culture of H. pylori to reach 0.35-0.50 OD600/ml in the morning.
In the morning, before beginning labeling, put ice in a bucket and place the 70% EtOH on ice to cool. If you are using a previously-used aliquot of PFA stored in a screw-top glass test tube at −20°C, place the test tube in a 55°C water bath to warm. Place a tube of ProLong Diamond Antifade Mountant cap-down in a tube rack to thaw.
Check the culture density with the spectrophotometer. If the culture is between 0.35-0.5 OD600/ml, proceed with labeling. If OD600 > 0.5, dilute the culture and grow for at least 1 doubling to ensure the cells are in early/mid log phase.
Label two round-bottomed 5 ml polystyrene tubes, one for the culture to be labeled and one for the unlabeled control.
Pipette 400 μl culture into each polystyrene tube and place the tube in the incubator for 15 minutes, shaking at an angle.
While the cultures are warming in the incubator, thaw the MurNAc-alk or D-Ala-alk stock.
After 15 minutes, add 8 μl of the 200 mg/ml MurNAc-alk stock or 4 μl of the 100 mM D-Ala-alk stock to the culture and put the culture back into the Trigas incubator, shaking at an angle for 18 minutes.
After 18 minutes, take the tubes out of the incubator, add 4 μl 10% sodium azide to suspend growth, swirl to mix, and place on ice for 5 minutes.
While the cells sit on ice, label one 1.5 microcentrifuge tube per sample using a circular sticker label.
Transfer each culture from the 5 ml round bottom polystyrene tube to a 1.5 ml microcentrifuge tube.
-
Centrifuge cultures for 5 minutes at 5000 rpm [2300 RCF].
Helical H. pylori cells tend to clump tightly, so gentle spins are important.
Resuspend each pellet in 1 ml Brucella Broth (without FBS). Add 250 μl 20% PFA (final concentration = 4% PFA) to each sample and invert to mix. Rotate at room temperature 45 minutes to fix cells. Store any unused 20% PFA in a screw cap glass test tube at −20°C.
Centrifuge cells for 5 minutes at 5000 rpm [2300 RCF] and remove supernatant.
Resuspend pellets in 1 ml ice cold 70% ethanol. Place tubes on ice for 30 minutes.
During this step, get the Click-iT® reaction buffer additive out of the freezer and place on the benchtop to thaw.
Centrifuge cells for 5 minutes at 2000 rpm [400 RCF] and remove supernatant.
-
Thoroughly and quickly resuspend pelleted cells in 200 μl PBS.
Note that cells will likely be pelleted up the side of the tube where the pellet formed, so make sure to pipette PBS over this surface multiple times to resuspend these cells.
Let the cell suspensions sit 10 minutes at room temperature.
-
Place the cover glass on top of the hemocytometer. Pipette 10 μl of one sample into the top wedge and 10 μl of the second sample into the bottom wedge. Observe the density of the cell suspension at 400x magnification with phase contrast. Within the smallest squares, the optimal density is approximately three cells per square per focal plane (Video 3).
Because of the small size of the cells and the extremely narrow depth of field, not all of the cells within the liquid volume above the square will be visible at any given focal point. Adjust the focus up and down between the cover glass and the hemocytometer to get a proper estimate of cell density.
If necessary, dilute the cell suspensions with PBS to achieve the appropriate cell density as visualized on the hemocytometer.
-
Clean two No. 1.5 glass coverslips by placing a small amount of Alconox™ detergent powder into one gloved hand. Add a few drops of distilled H2O to the powder and mix. Get a small amount of the detergent mix on the thumb and forefinger. Rub the coverslip thoroughly between these fingers and then rinse completely with distilled H2O, ensuring no detergent remains. Blot dry on paper towels.
Coverslips can be cleaned in advance. Keep clean coverslips in a Petri dish on top of a small piece of Kimwipe.
Assemble cytocentrifuge units by placing the base on the benchtop, then place one 1”x1” silicone square (Template 3, Piece A) on top of the center pedestal. While wearing gloves to prevent oil transfer from your fingers, carefully place one clean No. 1.5 coverslip on top of the silicone square. It is best to handle the coverslip by just touching the edges. Then place one 1”x1” silicone square with a hole in the center (Template 3, Piece B) on top of the coverslip. Next place the cytocentrifuge unit top on top of the stack. Holding the entire stack together between thumb and forefinger of your non-dominant hand, use your dominant hand to clip all four sides of the apparatus together using micro binder clips. See Figure 18A.–E. and Video 4 for a detailed assembly diagram.
Pipette 200 μl of the normalized density suspension into the modified 1.5 ml microcentrifuge tube and close the lid. Using a circular label sticker, label the base of the cytocentrifuge apparatus. Tape the base of each cytocentrifuge unit onto the inside surface of a 96-well plate. See Figure 18F.–H. for a detailed assembly diagram.
Place each 96-well plate cytocentrifuge apparatus into a tabletop swinging bucket centrifuge and spin at 500 rpm [59 RCF] for 5 minutes.
-
Remove from the centrifuge. Open the lid of each cytocentrifuge apparatus and pipette in 200 μl of PBST. Wait five minutes to disassemble.
This step helps allay issues with hydrophobicity of the coverslip and helps ensure cells remain hydrated throughout the whole protocol.
In the meantime, cut a strip of parafilm 0.25 m in length. Remove the paper backing. Lay the parafilm flat on the benchtop and tack it to the surface along the edges using your fingernail. Use a permanent marker to label columns on the parafilm for labeling. Pipette 200 μl PBST on top of the parafilm, one for each coverslip (Figure 19A.–B. and Video 5).
-
Carefully disassemble the cytocentrifuge units (Figure 19C.–H. and Video 5). Set the cytocentrifuge unit on top of a paper towel. Unclip all four micro binder clips. Peel off the top of the cytocentrifuge unit, and if necessary, the 1”x1” piece of blue silicone with a hole in the center (Template 3, Piece B). Make sure some liquid remains on the top surface of the coverslip. Exercising extreme care to avoid breaking the coverslip, pull a corner of the 1”x1” silicone square (Template 3, Piece A) parallel to the plane of the coverslip to help release the coverslip. While holding the corner of the silicone, gently slide one pincer of the tweezers between the coverslip and the silicone square and move pincer as necessary to release the seal between the coverslip and silicone. Pick the coverslip up using the tweezers (Figure 19C.–H.).
This step is the most likely time to accidentally break a coverslip. It may take some practice to reliably be successful with releasing the coverslip. If you do break a coverslip into two pieces, you can carefully carry both pieces through the labeling and mounting steps.
With the coverslip gripped securely in the tweezers, rotate the coverslip 90° and carefully wick the liquid off on a paper towel. Quickly place the coverslip cell side-down on the corresponding droplet of PBST on the parafilm. Repeat for any remaining coverslips.
Once all of the coverslips are cell side-down on PBST, make a Click-it reaction master mix: 396 μl ddH2O, 44 μl 10x buffer, 10 μl CuSO4, 50 μl reaction additive, and 4 μl AF555 azide (1 mg/ml).
-
Pipette 50 μl of the Click-it reaction master mix to the next position on the parafilm. Pick up each coverslip securely with the tweezers, rotate the coverslip 90°, wick it on paper towels, and quickly place it cell side-down on the drop of Click-it reaction master mix. Cover with a box lid to block light exposure and wait 30 minutes.
All wicking steps will be done with the coverslip approximately perpendicular to the paper towel and coverslips will always be placed onto liquid drops cell side-down. In all following steps, coverslips will be handled exclusively with tweezers and coverslips will be protected from light. After a drop has been used, you can aspirate or wipe it away.
Pipette two 200 μl drops of PBST per coverslip onto the parafilm. After 30 minutes of Click-it labeling, pick up each coverslip, wick it, and quickly place it cell side-down on the corresponding PBST drop. Once all coverslips are on PBST, you may pick up and place the coverslip back on the droplet to mix the solution to promote optimal washing.
Let the coverslip sit on the PBST droplet for 10 minutes, then wick and transfer it to the next PBST drop and let sit another 10 minutes.
While the coverslips are in the second wash step, make a master mix of fluorescent Wheat Germ Agglutinin (1.5 μl of a 1 mg/ml solution per 50 μl PBS). Pipette 50 μl of the WGA solution per coverslip onto the parafilm. Once the second wash is finished, wick and transfer each coverslip onto the corresponding WGA drop. Let coverslips label for 30 minutes.
Pipette 4 x 200 μl drops of PBST per coverslip onto parafilm. When the WGA labeling has been completed, wick each coverslip and place it cell side-down on the corresponding PBST drop. Proceed with four washes as described above in Steps 31-32.
While washing the coverslips, spray slides with 70% ethanol and rub dry with the microfiber towel to clean them. Label slides.
-
Once the final wash is finished, carefully add one drop of ProLong Diamond anti-fade mountant to the top, center of each slide. Ensure that there are no bubbles in the drop of mountant. If there are bubbles, fold a kimwipe several times and use the resulting corner to pop or move bubbles away from the drop. Pick up each coverslip, wick well on a paper towel, and mount on the slide (Figure 20 and Video 6).
To minimize the risk of generating bubbles under the coverslip, at a 60°angle from the coverslip, contact the coverslip bottom edge with the drop of mountant. Then, keeping the edge of the coverslip near the slide, move the coverslip edge along the long axis of the slide away from the drop of mountant to spread out the drop some. Then, slowly and gently decrease the angle of the coverslip until the coverslip is parallel to the slide.
-
Place the slides on a level surface under an opaque box lid to cure for one week.
Slides can be imaged immediately with great care after tacking the corners with VALP for screening purposes if necessary. It is preferable to wait 24 hours to screen slides; VALP is not necessary after the slide mountant has been able to cure for 24 hours.
Evaluate the quality of the slide using a basic widefield fluorescence microscope.
Figure 18:
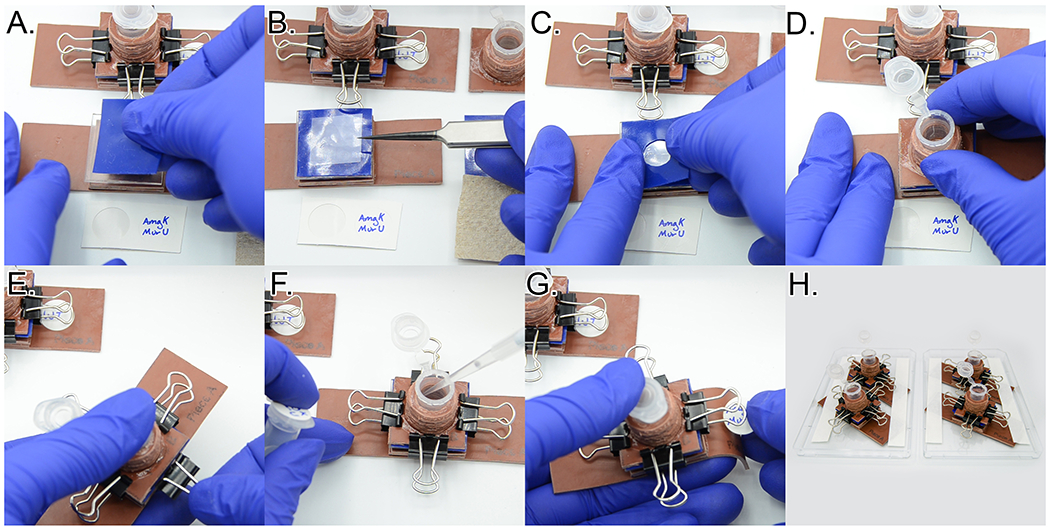
Assembling cytocentrifuge units. A. Placing a silicone square and B. clean coverslip on the cytocentrifuge base. C. Adding a silicone square with a hole cut out of the center. D. Adding the cytocentrifuge top. E. Creating a seal by clipping the cytocentrifuge unit together with four micro binder clips. F. Pipetting in 200 μl of cell suspension. G. Adding a label to the assembled cytocentrifuge unit. H. Fully assembled cytospin units attached to 96 well plate lids. See Video 4.
Figure 19:
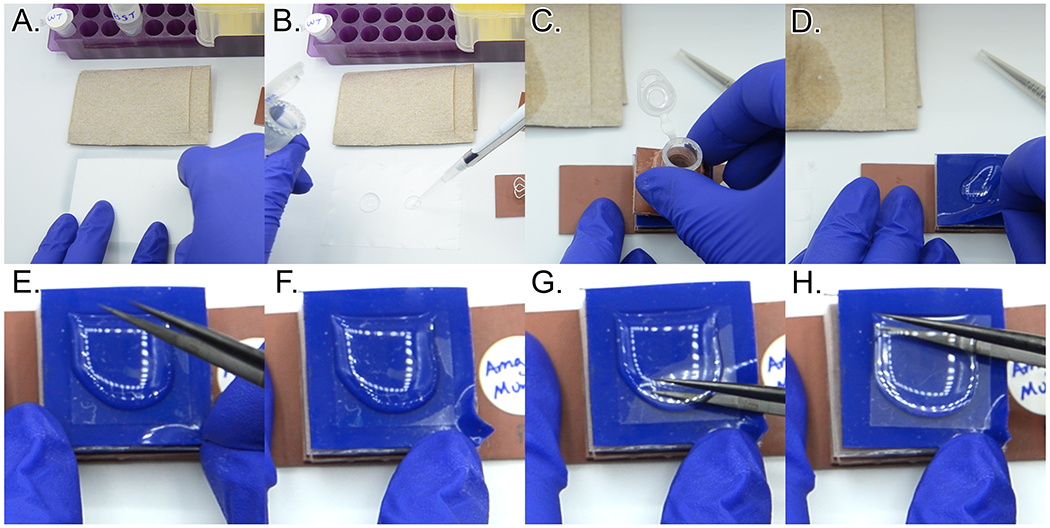
Disassembling cytocentrifuge units. A. Tacking parafilm to work surface. B. Pipetting 200 μl PBST onto the parafilm. C. Removing cytocentrifuge top after unclipping binder clips. D. Removing top piece of silicone. E-H. Removing the coverslip. E. Pulling a corner of the blue silicone down. F. Securing the silicone with a finger from the opposite hand. G-H. Slowly breaking the seal between the silicone and the coverslip. See Video 5.
Figure 20:

Mounting coverslips. A. Squeezing a small amount of the ProLong Diamond out of the tube. This often forms a bubble. B. Wiping off the bubble while holding the tube squeezed. C. Depositing a small drop of mounting medium onto a clean glass slide. D. Wicking the excess liquid off the coverslip. E-H. Mounting the coverslip onto the slide. E. Placing one edge into the drop of mounting medium. F. Dragging that edge slowly away from the drop of mounting medium. G. Slowly lowering the rest of the coverslip. H. Mounted coverslip without bubbles. See Video 6.
Sample Data
After successfully completing this protocol, the user will have at least two slides – one of labeled cells and one of a negative control. Coverslips should have remained hydrated throughout the protocol and should be mounted on the slide with no or minimal bubbles. Slides can be checked immediately after completion by tacking the corners only with a melted 1:1:1 mixture of vasaline:lanolin:paraffin by weight (VaLP). However, we recommend waiting at least 24 hours to check slides to prevent disruption of the mounting medium. The cell wall for both slides should be brightly labeled (green channel), while there should only be notable signal in the red channel for the labeled AmgK MurU-expressing cells. Figure 21 shows example images. Wild-type cells labeled with MurNAc-alk can be used as a negative control, though this may not be ideal if conservation of the MurNAc-alk is a priority.
Figure 21:
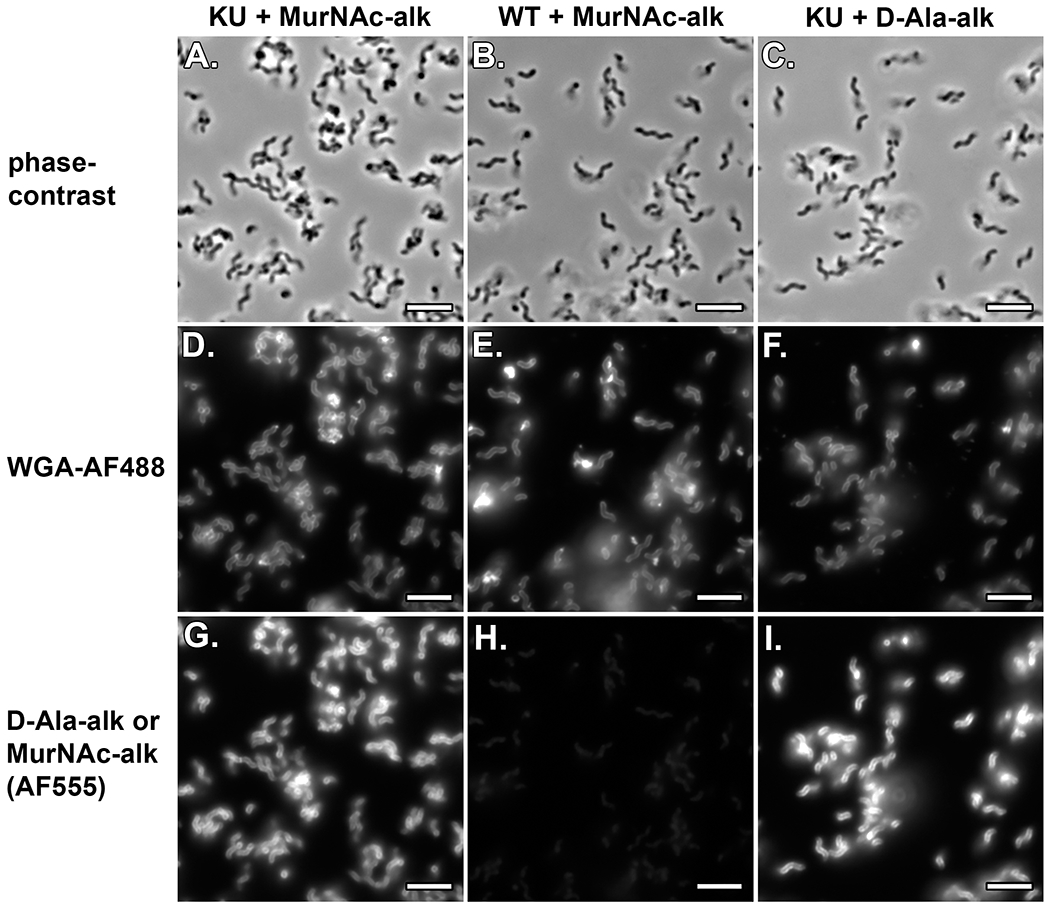
Phase contrast (A.-C.; 50 msec exposures), Fluorescent WGA-AF488 (D.-F.; 50 msec exposures), MurNAc-alk AF555 (G. and H.; 500 msec exposures), and D-Ala-alk AF555 (I.; 500 msec exposure). Images in column 1 (A., D., and G.) are from H. pylori KU labeled with MurNAc-alk; images in column 2 (B., E., and H.) are from wild-type H. pylori labeled with MurNAc-alk; and images in column 3 (C., F., and I.) are from H. pylori KU labeled with D-Ala-alk. As expected, there is minimal signal for KU labeled with MurNAc-alk (H.). Images A.-C., D.-F., and G.-I. are each scaled equally (within groups). Scale bar = 5 μm.
BASIC PROTOCOL 4
BASIC PROTOCOL TITLE
Structured Illumination Microscopy (SIM) imaging on the DeltaVision OMX
Introductory paragraph:
This protocol provides the user with a framework for performing structured illumination microscopy of the slides from Basic Protocol 2 using the DeltaVision OMX microscope. Do not attempt independent use of this microscope until you have completed training and have been cleared for use by the manager of the microscope. This protocol is intended to provide useful guidance for obtaining high-quality images, but it is expected that the user will need to optimize parameters.
Materials:
Chloroform
DeltaVision OMX microscope and accompanying immersion oil kit
cotton-tipped swabs
lens paper
slides from Basic Protocol 3
Protocol steps—Step annotations:
For more complete instructions on imaging with the OMX, please refer to documents provided by the manufacturer (See Internet Resources section).
INTERNET RESOURCES: (optional).
URLs for important sites relevant to the method. Each must be accompanied by a short description of the subject of the site.
https://cdn.cytivalifesciences.com/dmm3bwsv3/AssetStream.aspx?mediaformatid=10061&destinationid=10016&assetid=27720 – Cytivia’s guide to sample preparation for OMX SIM imaging.
https://microscopy.jhmi.edu/Learn/refman/GE/DVOMXSR_SI_UserGuide.PDF - GE Healthcare’s customer instructions for structured illumination imaging on the OMX.
https://microscopy.jhmi.edu/Learn/refman/GE/DVOMXSR_QRef1_Acq_29159151AB.pdf - GE Healthcare’s Quick Start Guide #1 for the OMX – Startup and Accquisition.
https://microscopy.jhmi.edu/Learn/refman/GE/DVOMX_AppGuide_OilOpt_29250659AA.pdf - GE Healthcare’s customer instructions for determining the correct oil for imaging your specimen.
https://microscopy.jhmi.edu/Learn/refman/GE/DVOMXSR_QRef2_Recon_29159154AB.pdf - GE Healthcare’s Quick Reference #2 for the OMX – SI Image Reconstruction.
https://microscopy.jhmi.edu/Learn/refman/GE/DVOMXSR_QRef4_SIM_Sample_Prep_29193027AB.pdf - GE Healthcare’s Quick Reference #4 for the OMX – SIM Sample Preparation.
https://microscopy.jhmi.edu/Learn/refman/GE/DVOMXSR_ImageAlignment_04-720165-000CC.pdf - GE Healthcare’s customer instructions for image alignment (for use by advanced users only when the color channels are not aligned).
At the beginning of each imaging session, the microscope will need to be initialized and the immersion oil and alignment will need to be checked using an image from your samples.
At the beginning of imaging a new set of slides that will be directly compared, you will need to empirically determine an optimal set of exposure time, laser power, and if applicable, camera gain.
At the beginning of imaging every slide, you will need to ensure that the sample has stopped drifting before you begin collecting images.
-
Each imaging session: turn on and initialize microscope as necessary.
Depending on the policies of the party in charge of microscope maintenance, the OMX may need certain components turned on prior to imaging.
Each imaging session: once the microscope is turned on, restart hardware and restart cameras to ensure that the microscope is properly initialized and ready for imaging.
-
Each slide: clean your coverslip thoroughly but gently using a clean cotton swab dipped in chloroform.
Take care not to contact cured mountant surrounding the coverslip with the swab.
-
Each imaging session: determine the appropriate immersion oil for imaging your sample. The immersion oil is matched well when the out of focus blur above and below an object is symmetrical. Add oil to the objective and then lower the stage until the oil contacts the slide. Looking at the fluorescent WGA channel, which should be brightest, use the software to incrementally lower the stage until the cells can be seen. The histogram values should increase as the sample comes closer to being in focus. Focus on the middle of a cell, then move the stage the same distance above and below the center to compare the out of focus blurring. When changing oils to optimize the index of refraction, make sure to thoroughly clean the oil off both the slide and the objective.
Note that the same sample may need a different immersion oil on a different OMX microscope. Also not that it is impossible to optimize the immersion oil for both color channels. A reasonable strategy is to compromise with the oil matching such that the chosen index of refraction is between the optimal index of refraction for each channel. To facilitate focusing with other slides, set a preset Z-height. Slides prepared as in Protocol 3 should all be relatively similar in z-height. In new imaging sessions to save time, you may add oil to the objective, center the coverslip, and use the preset z-height to quickly approach a height near the likely focal plane. It is a good idea to start with the oil selected during the previous imaging session and ensure that the image quality with that index of refraction oil is still acceptable. Start with 1.514-1.516 oil.
-
Once per slide set: after you have optimized the oil and the cells are in focus, optimize the image acquisition parameters for both channels. These imaging parameters should be established once for a given set of comparable slides using the D-Ala-alk or MurNAc-alk-labeled cells. The negative control slide and any other slides to be compared directly should be imaged using the same parameters for both channels. Ensure that you are using the structured illumination imaging mode and imaging a 512 x 512 pixel field of view. Acquire an image of a plane at the middle of the cells and check the provided histogram. Change the exposure times and laser intensities to obtain satisfactory imaging. Greater signal (below the maximum threshold value) can enhance image quality, but will also require longer exposure times and/or higher laser intensity. The maximum signal intensities should be greater than 3000 and less than 20000 (approximate value range). Laser intensity should be no greater than 30%.
Longer exposure times can substantially increase the amount of time required to acquire images and could exacerbate any issues with drift. Higher laser intensities could bleach your sample. Image one z-stack of a field of view twice and compare raw intensities at comparable z-slices to check if the sample has dimmed significantly. Depending on the installed camera, there may be options to increase the gain settings to boost signal intensity, which may be useful. Consult with the microscope manager before altering these settings. Begin with the following settings: 488- 20 msec at 5% laser power, and 568 – 100 msec at 15% laser power.
-
Each slide: check to make sure the sample is not drifting. Acquire a snapshot image of the current field of view with the cells in focus. Wait two minutes and capture another snapshot. If the cells have moved or the focus has shifted, refocus if necessary and repeat the process until the cells remain stationary and in focus.
You might not notice drift on the slide you have used to optimize the imaging parameters since it likely has been on the microscope long enough to equilibrate. Take care not to omit this step with subsequent slides.
-
Acquire a 3D-SIM image. To facilitate downstream analysis, collect images with the same z-height each time. Ensure that you are using the structured illumination imaging mode and imaging a 512 x 512 pixel field of view. Focus at the center of the image and ensure that the microscope is set up such that the current z-plane is the center of the z-stack to be acquired.
For slides of H. pylori with cells that retain their 3D geometry prepared as in Protocol 3, a z-stack of 3 μm is advised.
Each imaging session: check the X, Y, and Z alignment. Run the SIM reconstruction and alignment algorithms on the image and inspect it visually. The D-Ala-alk or MurNAc-alk signal should align completely with the WGA signal. There may be subtle alignment mismatches at the edges of the field of view, but they should be minimal. If the signals are not properly aligned, consult with the microscope manager to realign the scope and/or apply any necessary z-correction.
-
Each slide: proceed with collecting images as described in step 7. Depending on the density of the cells on the slide, the number of images you need to collect will differ. For quantitative analysis aim for imaging approximately 200 well-separated cells. Ensure that you are collecting images where cells are still 3-dimensional; avoid the edges of the cell deposition spot as they often dry out and cells flatten.
Because cells were spun gently onto the coverslip, it is likely that where cells were well-hydrated, only parts of the cell will be in contact with the coverslip and other parts will be at a different z-height. Looking for this feature will help ensure that you are imaging in a good area.
Sample Data
Figure 22:
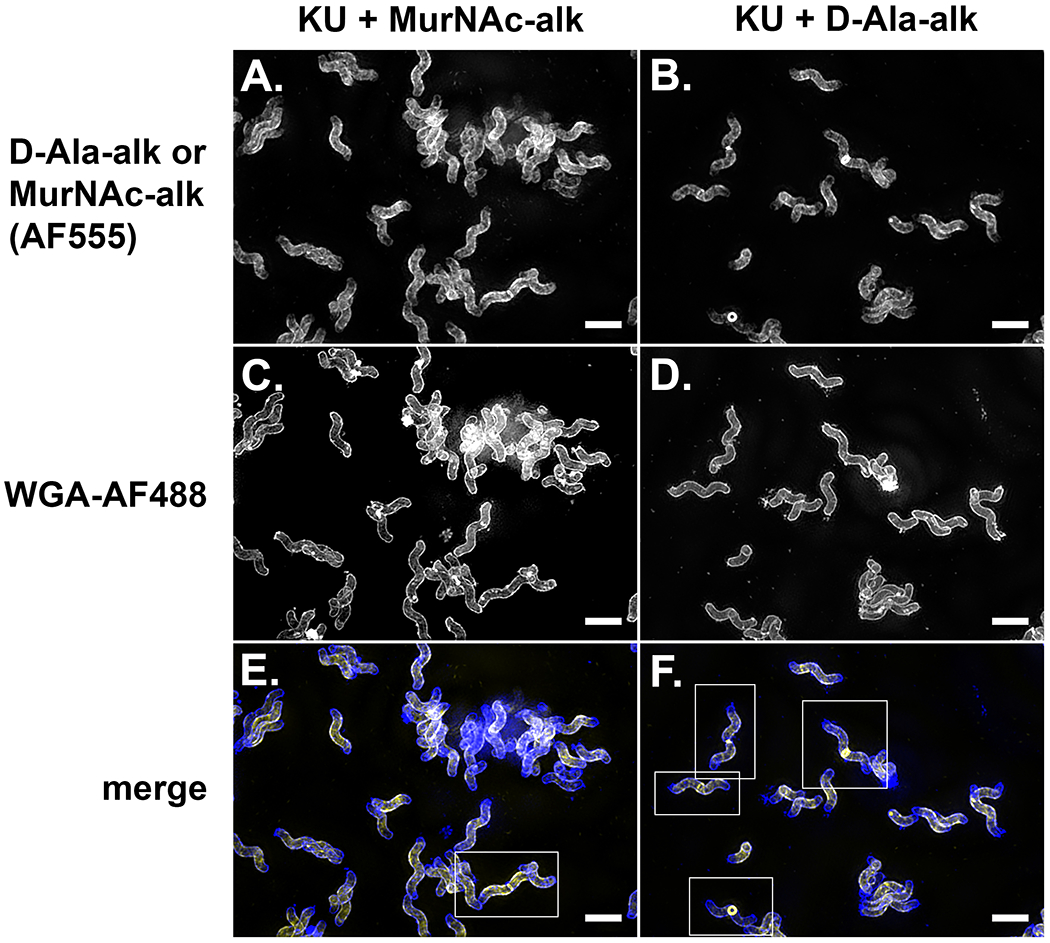
3D SIM images of the H. pylori KU strain labeled with WGA-AF488 (C., D., and blue – E., F.) and MurNAc-alk (A. and yellow – E.) or D-Ala-alk (B. and yellow – F.). Septating cells are denoted by white boxes in E. and F. Note the increased relative brightness of septal labeling vs. sidewall labeling for D-Ala-alk labeling in comparison to MurNAc-alk labeling.
REAGENTS AND SOLUTIONS:
0.5 mg/ml Cellosyl in 10mM ammonium formate Buffer pH 4.8
Dilute 1 ml of 50mM ammonium formate Buffer pH 4.8 (see recipe) to 5 ml with distilled water.
Add 2.5 mg Cellosyl (kind gift from Hoechst, Frankfurt am Main, Germany).
Stir until dissolved.
Aliquot 100 μl into microfuge tubes and store at −20°C until use (up to 1 year).
Note: The muramidase cellosyl is not commercially available. The commercially available muramidase mutanolysin (Merck-Sigma, #SAE0092) has the same amino acid sequence as cellosyl and can be used with the same conditions.
0.7 M sodium phosphate buffer pH 7.2
Add 2.8 g of sodium hydroxide (Honeywell, UK, cat. no. 306576) and add distilled water to 90 ml.
Stir until dissolved.
Adjust pH to 7.2 with dropwise addition of concentrated phosphoric acid (Honeywell, UK, #79606).
Adjust volume to 100 ml with distilled water.
Store up to 1 week at 4°C.
3.2 M imidazole pH 7.0
Add 21.8 grams of imidazole (Merck-Sigma, #56750) and add distilled water to 70 ml.
Stir until dissolved.
Adjust pH to 7.0 with dropwise addition of concentrated hydrochloric acid (Honeywell, UK, #30721).
Adjust volume to 100 ml with distilled water.
Store up to 1 week at 4°C.
10 mg/ml α-amylase in 10mM Tris-HCl,10mM NaCl pH 7.0
Add 50 mg α-amylase (Merck-Sigma, #86250) and add 10mM Tris-HCl, 10mM NaCl pH 7.0 (see recipe) to 5 ml.
Stir until dissolved.
Aliquot into microfuge tubes (100 μl).
Store at −20°C until use (up to 1 year).
10 mg/ml Pronase E in 10mM Tris-HCl,10mM NaCl pH 7.0
Add 50 mg Pronase E (Merck-Millipore, #1074330001) and add 10mM Tris-HCl, 10mM NaCl pH 7.0 (see recipe) to 5 ml. Stir until dissolved.
Transfer to a sterile falcon tube (15 ml).
Activate the enzyme by incubation at 60°C for 2 hours in a water bath.
Cool to room temperature and aliquot into microfuge tubes (100 μl).
Store at −20°C until use.
10 mM Tris-HCl, 10mM NaCl pH 7.0
Add 121 mg Trizma base (Merck-Sigma, #T6066), 58 mg sodium chloride (VWR, #27810.295), and add distilled water to 90 ml.
Stir until dissolved.
Adjust pH to 7.0 with dropwise addition of concentrated hydrochloric acid (Honeywell, UK, #30721).
Adjust volume to 100 ml with distilled water.
Store up to 1 week at 4°C.
20mM Ammonium formate Buffer pH 4.8
Add 126 mg ammonium formate (Honeywell-Fluka, UK, #17843) and add distilled water to 90 ml.
Stir until dissolved.
Adjust pH to 4.8 with dropwise addition of concentrated formic acid (Merck-Sigma, #33015).
Adjust volume to 100 ml with distilled water.
Store up to 1 week at 4°C.
Antibiotic 200X Stock
Materials:
Vancomycin (Fisher Scientific #AAJ6279006)
Polymixin B (Fisher Scientific #P1004)
ddH2O
Stericup vacuum filtration unit, 0.22 μm pore size (Millipore Sigma #S2GPU02RE)
15 ml conical tubes
Measure out 0.2 g of vancomycin and 0.0064 g of polymyxin B.
Dissolve the antibiotics into 100 ml of ddH2O
Sterile-filter the solution using the vacuum filtration unit.
Aliquot 7.5 ml into 15 ml conical tubes and store at −20°C; stable for >3 months.
Antibiotic 1000X Stock
Materials:
Amphotericin B (Sigma #A4888-5G)
DMSO
2 ml microcentrifuge tubes
Measure out 0.4 g of Amphotericin B and dissolve in 50 ml DMSO
Without filtering, aliquot 1.5 ml into 2 ml microcentrifuge tubes and store at −20°C; stable for >3 months.
Brucella Broth
Materials:
Dehydrated Brucella Broth culture media (Fisher Scientific #B11088)
ddH2O
1 L glass bottle
Magnetic stir bar
Magnetic stir plate
Autoclave
Measure out 28 g of Brucella Broth and add to a 1 L glass bottle with a magnetic stir bar.
Add 900 ml ddH2O and mix using a magnetic stir plate.Make sure there are no clumps of media remaining at the bottom of the bottle before autoclaving.
Ensure caps are loose then autoclave using a 30-45 minute liquid cycle.A longer cycle is necessary when more bottles are being autoclaved at one time.
Once the bottles have completely cooled, the caps may be tightened and the bottles can be stored at room temperature for >3 months.
BB10
Materials:
Brucella broth (see above)
BenchMark™ Fetal Bovine Serum (GeminiBio #100-106)
37°C water bath
56°C water bath
50 μl conical vials
Stericup vacuum filter system, 0.22 μm pore size, polyethersulfone membrane (Millipore Sigma #S2GPU02RE)
-
Thaw the serum for 10-20 minutes at room temperature and then in the 37°C water bath.
Gently mix the serum by inverting the bottle every 10 minutes and remove from the 37°C water bath when thawed.
-
Heat inactivate the serum for 30 minutes in the 56°C water bath.
Submerge the bottles to just below the cap; do not fully submerge. Gently mix the bottles every 5 minutes. After 30 minutes, remove the bottles from the water bath and allow them to cool at room temperature. Heat inactivated serum can be aliquoted into 50 μl conical vials, stored at −20°C for >6 months, and thawed at 37°C when needed. Note that the freezer should not be a frost-free freezer as defrost cycles will damage the FBS. FBS that has not been heat inactivated can inhibit H. pylori growth, particularly at low culture densities.
-
Filter sterilize 90% Brucella broth plus 10% heat-inactivated FBS, by volume. For 250 μl of BB10, add 225 μl Brucella broth plus 25 μl heat-inactivated FBS.
BB10 should be stored at 4°C for up to one month.
Horse blood plates
Materials:
β-Cyclodextrin hydrate (Fisher Scientific #AC227281000)
DMSO
Columbia Blood Agar Base (Fisher Scientific #DF0792-07-5)
ddH2O
Defibrinated horse blood (Hemostat Labs #DHB100)
Antibiotic 200X stock (see above)
Antibiotic 1000X stock (see above)
15 ml plastic conical tube
Rotisserie shaker
1 L glass bottle
Magnetic stir bar
Stir plate
Autoclave
Water bath set at 55°C
100 mm Petri dishes (Fisher Scientific #FB0875712)
Measure 1 g of β-Cyclodextrin in a 15 ml conical tube. Add 5 ml DMSO to make a 200 mg/ml stock and rotate on a rotisserie shaker at room temperature several hours to dissolve. This stock can be made the morning of media preparation and left at room temperature until use.
Set the horse blood, antibiotic 200X stock, and antibiotic 1000x stock on the bench to come to room temperature. The antibiotic stocks can take some time to thaw and adding cold blood to hot liquid media can damage the blood.
Measure out 19.5 g Columbia Blood Agar Base and add to a 1 L glass bottle. Add 500 ml ddH2O and a magnetic stir bar to the bottle. Briefly mix the contents of the bottle using a stir plate. Note that the agar mix will not dissolve at this step.
Loosely cap the bottle so steam can easily escape, then autoclave the bottle on an appropriate liquid cycle.
Once the autoclave cycle is complete, put the bottle into the 55°C water bath for 1-5 hours. The media needs to cool to 55°C so that the blood does not lyse when added. If the media sits too long in the water bath, chunks will begin to form at the bottom of the bottle.
-
Once the media has cooled to approximately 55°C, put it on a stir plate and stir at a medium speed. Do not stir the media so rapidly as to cause bubbles to form.
Add:
25 ml defibrinated horse blood
5 ml β-Cyclodextrin 200 mg/ml stock in DMSO
2.5 ml antibiotic 200X stock
500 μl antibiotic 1000X stock
Once the additions are mixed in, turn on a Bunsen burner and use aspetic technique to pour approximately 20-25 ml of the blood agar media into each Petri dish. One bottle should make approximately one sleeve of plates, depending on how full the plates are poured.
Let the plates set at room temperature. Leave the plates out (with lids on) overnight to dry excess moisture.
The following day, place the horse blood plates in a Petri dish sleeve and store at 4°C. Plates can be stored at least 1 month at 4°C.
P-anisaldhyde stain
While stirring add to770 ml Ethanol (Fisher Scientific, #A4094) 40 ml DI water, 30 ml concentrated sulfuric acid (Fisher Scientific, #SA213), and 9 ml of glacial acetic acid (Fisher Scientific, #18-602-917).
Cool to room temperature.
Add 22.2 ml P-anisaldhyde (Oakwood Chemical, #098960).
COMMENTARY
BACKGROUND INFORMATION:
The first tool employed for metabolic labeling of PG was D-cysteine, which can be incorporated into PG through the action of transpeptidases (de Pedro, Quintela, Holtje, & Schwarz, 1997; Horcajo, de Pedro, & Cava, 2012). New PG incorporation can be visualized by performing pulse-chase labeling followed by purification of the cell wall, biotinylation of the D-Cys, and immunolabeling with gold-coupled antibodies. Purified cell walls can be imaged using transmission electron microscopy; new PG incorporation is unlabeled. While helpful for identifying large-scale PG synthesis features, this labeling method is of limited utility for studying finer scale details and, because purified sacculi flatten when dried, obscures obscuring 3D information. Though not a metabolic probe, fluorescent vancomycin has been used as a proxy label of new cell wall material (Daniel & Errington, 2003; Tiyanont et al., 2006). Vancomycin binds the terminal D-Ala-D-Ala motif of PG pentapeptide stems. Since many bacteria rapidly trim the pentapeptide stem after new PG synthesis, fluorescent vancomycin labeling can provide a reasonable picture of sites of new PG synthesis. Use of this probe is limited to Gram-positive organisms and a few Gram-negative organisms that have an outer membrane that is permeable to vancomycin. Additionally, vancomycin provides a poor readout of new PG synthesis in bacteria that are pentapeptide-rich.
The development of metabolic probes to fluorescently label PG has greatly expanded the depth and breadth of our understanding of the dynamic bacterial cell wall. One of the advantages of these probes is that they can be successfully used in a diversity of bacteria, facilitating development of a better picture of the range of strategies bacteria can use to shape their cell wall. Moreover, different probes report on different metabolic activities. D-Ala can be incorporated both at sites of PG modification and sites of new PG strand incorporation (Kuru et al., 2019). In contrast, the MurNAc-alk probe described here and D-Ala-D-Ala dipeptide probes (Liechti et al., 2014) exclusively report on sites of new PG strand incorporation, though the dipeptide probes are still subject to removal during peptide stem trimming. With their different labeling specificities, these probes can be used in concert to query these different aspects of PG metabolism. Finally, different modifications can be placed onto the probes for different biochemical purposes. Similar to the alkyne probe described here, azide linked probes can be used in click reactions to add a variety of chemical moieties and could be used in combination with an alkyne modified probe for dual labeling (Liang et al., 2017). Finally, probes can be directly labeled with fluorophores, though these large functional groups could result in steric hinderance and can limit cell permeabilization, a particular problem for labeling Gram-negative bacteria.
Previous work in the Grimes laboratory led to the development of the synthesis of MurNac-alk and its successful application to study PG biosynthesis and cell wall recycling pathways (DeMeester et al., 2019; Liang et al., 2017). Herein we presented enhancements to the method to allow ease of production of starting materials, enhanced visualization techniques, and elaborated mass spectrometric protocols.
The new approach, proposed in Basic Protocol 1, avoids the generation of N-hydroxysuccinimide (NHS), an inconvenient sub product that complexes the purification step. According to our investigations, conventional organic synthesis purification techniques proved not to be sufficient in the task of refining a crude sample containing NHS. Column chromatography, either normal phase or reverse phase, preparative thin-layer chromatography, crystallization, and even repetitions and combinations of them, could not effectively separate the NHS from the carbohydrate product (Figure 23). Only use of preparatory HPLC (AutoPurification system) resulted in a pure sample of the probe.
Figure 23:
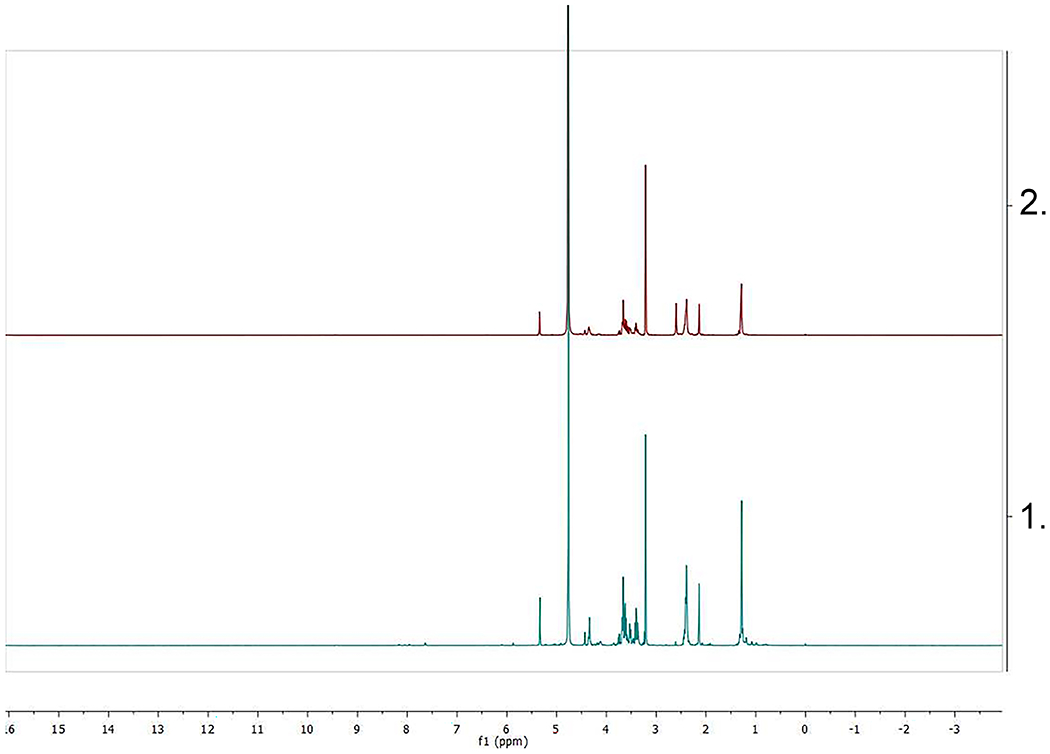
Spectra of two samples of MurNAc-alk obtained using the direct coupling method (1, lower spectrum) and with the NHS method (2, upper one), both purified through column chromatography. On (2), the signal at δ=2.60 ppm originated by the methylenes of the NHS shows that further purification is needed.
Template: This template should be printed at 100% size to cut the properly-sized components to make the cytocentrifuge units in Support Protocol 4.
The utilization of the coupling reagent EDC allows to use the 4-pentynoic acid directly, without previous esterification steps in order to activate this intermediate. Therefore, NHS contamination issues are avoided and the whole synthesis of the NHS-Alk ester (2,5-dioxopyrrolidin-1-yl pent-4-ynoate), an extra previous step, is no longer needed. In other words, using Basic Protocol 1, MurNac-alk can be prepared in a single synthetic step from commercially available reagents, whereas the one previously reported requires two synthetic steps.
Even though the small-scale yields of this option are lower (35% vs 62%), the current protocol is extremely straightforward since it can be performed in any basic organic synthesis laboratory. Furthermore, ongoing investigations suggest that this method might be suitable to access to other 2-substituted NAM probes.
The composition of bacterial peptidoglycan is an important parameter of the bacterial cell wall structure, informing about the cellular activities of cell wall synthases and hydrolases, and the presence or absence of modifications affecting pathogenesis. Traditionally, peptidoglycan composition is reflected as the quantification of muropeptides, the disaccharide peptide subunits released by a muramidase, and the resulting muropeptide pattern can be a characteristic signature of a bacterial species or strain (Vollmer et al., 2008). For several decades now the method published by Glauner in 1988 has been considered the de facto gold standard technique for the analysis of muropeptides (MPs) (Bernd Glauner, 1988). Typically, released MPs of varying size and crosslinking complexity are separated via C-18 reverse phase high performance liquid chromatography (RP-HPLC) on 4.6 mm ID columns using a linear separation gradient of ~pH 4 aqueous phosphate buffer versus phosphate buffer containing 15% methanol. MPs were detected with a UV detector measuring the response at 205 nm. Originally, Escherichia coli MPs isolated (from 400 ml culture) using this method were characterized using amino acid analysis, assigning MP identifications by their retention time on the same elution gradient (B. Glauner, Höltje, & Schwarz, 1988).
Amino acid analysis for identification was quickly superseded by the use of mass spectrometry (MS) analysis. In 1997, Xu et al. published details of a method that combined peak collection from a Glauner separation, followed by desalting and the use of matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) and post-source decay (PSD) fragmentation to unequivocally identify a range of monomer, and crosslinked dimer muropeptides from Staphylococcus aureus (Xu, Huang, de Jonge, & Gage, 1997). In 2009 an electrospray method was described in Bui et al. that involved an off-line desalting technique (RP-C18 tips) and the use of high-resolution mass spectrometry (HRMS). Desalted samples were accurately measured by offline-electrospray MS and fragmentation (MS/MS) was additionally performed to provide sequence information on a range of monomer, dimer and higher crosslinked species (Bui et al., 2009).
In more recent years researchers have moved to replicate Glauner separation coupled directly to the mass spectrometer in a LC-MS/MS configuration (Huang et al., 2017; Kühner, Stahl, Demircioglu, & Bertsche, 2014). Such setups typically involve the use of 2 mm ID columns and MS-compatible buffers, usually water versus acetonitrile in the presence of 0.1% formic acid as a modifier. Cell preparations analyzed have ranged between 2 ml and 100 ml.
Herein, we describe our muropeptide analysis method which represents a scaling-down from 2 mm ID (narrow bore) columns to 0.5 mm ID (microbore) columns. Our setup provides for a couple of benefits over prior methods.
The reduction in column diameter results in a highly sensitive method, permitting the generation of LC-MS/MS muropeptide profiles from as little as 300 μL of culture.
Previous methods contained upstream steps that were relatively unchanged from the Glauner method. We have made MS-friendly adaptations to the digestion (phosphate buffer), and the final acidification (phosphoric acid) steps prior to analysis. As a result, our modifications circumvent the need for desalting prior to LC-MS/MS analysis. In our experience, this pre-analysis C-18 desalt frequently results in the loss of shorter hydrophilic species (i.e., those of smaller mass eluting earlier than the main tetrapeptide monomer).
This scaled down LC-MS/MS method still delivers a separation profile that reasonably mimics our 4.6 mm ID column Glauner separations.
CRITICAL PARAMETERS:
Basic Protocol 1
Regarding the synthesis of MurNAc-alk, it is important to pre-form the two separate solutions before starting the reaction itself. In solution A the acid is activated by EDC, while in solution B the amino group of muramic acid is deprotonated for facilitating the nucleophilic attack on the now activated acid.
A one-pot strategy could lead to secondary products, likely affecting reaction yield. For instance, the EDC might react with the carboxyl group from the sugar instead of the one from the acid.
On the other hand, a larger base excess (more than 3 eq.) can result in problems since this could cause the loss of the anomeric hydrogen originating a new and reactive nucleophilic center in the reaction media.
Finally, it is recommended to store the MurNAc-alk probe properly. Inert atmosphere (argon or dry nitrogen), low temperatures (−20°C), and if possible, storage in solid form, are key parameters to maintain the quality of the compound and ensure the reproducibility of results.
Basic Protocol 2
Since there is no simple assay for muropeptide quantification and the amount often differs from preparation to preparation (depending upon experience and/or species), it can be tricky judging the optimum amount for LC-MS/MS injection. We recommend testing a range of dilutions (in 1% aqueous formic acid). Typically, a 1:5 (v/v) dilution is a useful starting point, but depending upon how successful your preps have been, you may need to experiment with such dilutions to obtain the desired muropeptide chromatogram profile.
Temporary (e.g., between run days) storage of uninjected muropeptides dissolved in 1% formic acid in the freezer can sometimes result in the appearance of precipitated material upon subsequent thawing.
Briefly heating the samples to 60oC with vortexing for around 5 min usually restores soluble samples.
For microbore/capillary LC-MS/MS analysis, injection of smaller volumes (e.g., 0.1 μL) from smaller loops (e.g., 1.0 μL) should result in the best chromatographic profiles. We have no doubts that the best analysis would be available via access to a modern UPLC setup.
When targeting modified muropeptides for identification, begin your modeling with simple monomer and dimer muropeptide structures. The reason for this is because these smaller muropeptide structures not only tend to be more abundant, but they are also detectable at lower ion charge states (1+ and 2+) and give much more complete MS/MS fragmentation ion patterns, thereby making verification easier.
Basic Protocols 3 and 4
For metabolic labeling of PG, MurNAc-alk and other MurNAc probes are the most straightforward to interpret since they are incorporated during muropeptide synthesis in the cytoplasm and are not removed when the peptide stem is modified. However, there are several considerations when applying this probe in a new bacterium or to a new biological question. First, is the bacterium capable of incorporating exogenous MurNAc into its PG? This can be determined by whether or not supplementation with MurNAc at least partially rescues growth during fosfomycin treatment. If, like H. pylori, the bacterium of interest is not natively capable of using exogenous MurNAc, some upfront genetic manipulation is required. Expression of the PG recycling enzymes AmgK and MurU from P. putida is sufficient in H. pylori to both partially rescue growth from fosfomycin treatment and to allow robust PG labeling with MurNAc-alk. In H. pylori, we expressed these genes chromosomally, but expression of amgK and murU from a plasmid in E. coli has also been sufficient to enable MurNAc-alk labeling (Taylor 2020, Liang 2017). The choice of expression system will have to be decided for the organism of interest and may require some optimization of expression levels.
Once the ability to use exogenous MurNAc has been achieved, the next step is to determine the concentration and duration of labeling. Depending on the scientific question of interest, a long or a short pulse may be appropriate. For determining where new PG is incorporated, an acceptable signal to noise ratio must be achieved. When using the OMX to acquire images, signal that is too low will result in poor image quality and noticeable honeycomb-like artifacts in the image. While increasing laser intensity and exposure time can help boost signal, the tradeoffs of sample bleaching and acquisition time limit how far these parameters can be extended.
Selection of the pulse length and concentration are interdependent. A longer pulse length will result in a higher signal but will also be more likely to be influenced by PG remodeling. We recommend a pulse length that is shorter than 15% of the organism’s doubling time. Shorter pulses require a higher concentration of MurNAc-alk to achieve sufficient signal. However, synthesis of the probe is costly, both in terms of time and money. Thus it is prudent to use only as much MurNAc-alk as necessary. For labeling PG, we suggest a pilot study to determine the combination of pulse length and concentration that enables the acquisition of high-quality 3D images.
For investigating questions such as PG turnover that require a long pulse to achieve uniform labeling of the cell wall, a much lower concentration of the probe will be necessary to achieve bright labeling. Note that if your application requires a pulse followed by a chase period without probe, it may be necessary to add unlabeled MurNAc to the culture. The conversion of MurNAc-alk to UDP-MurNAc-alk impedes its ability to diffuse out of the cell. The rate of conversion to UDP-MurNAc-alk may outpace the rate of its consumption, leading to continued PG labeling despite washing out MurNAc-alk following the labeling pulse.
TROUBLESHOOTING:
Basic Protocol 1
Muramic acid, the precursor of MurNAc-alk, usually turns into a deep yellow/ brownish solid by direct light exposure and aging (long time-storage). This coloring can be prevented by protecting the material from the light when storage and use, e.g. by covering containers with aluminum foil. There is no conclusive evidence that this coloration significantly affects the reaction yields of MurNAc-alk. However, if yield-related issues are detected, it is strongly recommended to first assure the purity of this substrate.
Purification of MurNAc-alk only requires a normal phase column and can be performed in any organic synthesis laboratory. Nonetheless, sometimes a second column may be necessary to obtain a truly pristine probe. The utilization of larger excess of reagents in the preparation step, a rushed elution or change between the two mobile phases, and the use of less than the recommended amount of silica gel on the purification column are conditions that can lead to the necessity of a second chromatographic procedure.
Support Protocol 1
If your H. pylori cells do not grow well, ensure that all traces of detergent have been removed from culture flasks as well as all glassware used to prepare media. H. pylori is extremely detergent-sensitive. If this does not resolve your issues, make sure that the FBS has been properly heat-inactivated, that the media is not too old, and that the proper growth conditions in the incubator have been maintained.
Basic Protocol 2
There can be numerous reasons behind a failure to obtain the desired LC-MS ion trace (TIC)/ profile. The most common point of failure is loss of pellet material during the preparation of sacculi. You can check this by running a portion of the undiluted material (typically half) using a standard Glauner setup on a conventional HPLC with UV detection. If no profile is visible, repeat the digestion protocol, paying particular attention to the washing and centrifugation steps. If peaks are clearly visible, then you can investigate the LC/MS/MS setup for problems. Common issues here include over-dilution of samples with 1% formic acid resulting in the production weak ion peaks. Reducing the dilution factor or concentrating the sample in vacuo to 5 – 10 μL often resolves this issue.
If you use a different LC-MS/MS setup to the one detailed here, you will certainly have to spend time optimizing the chromatographic separation using the gradient and mass spectrometer details given here as a starting point.
Sometimes the LC-MS/MS fails due to the presence of too much salt. Check all buffers, enzyme stocks (e.g., cellosyl) and material sources to ensure they do not introduce excessive amounts of non-volatile salt. On other occasions, problems with injector pickup of low volumes or non-optimal ion source spray and/or general MS parameters may be at fault. In this instance, you are advised to consult your local LC-MS specialist or service provider.
When all seems lost, simply going back to basics and performing an E. coli muropeptide preparation can help pinpoint the source of a problem.
Basic Protocol 3
Even after labeling parameters for metabolic labeling of PG have been optimized, difficulties may arise unexpectedly. The following discussion highlights potential issues and strategies for troubleshooting.
Several issues can arise with the cytospin apparatus. If the plexiglass piece (Template 2, Piece A) comes off the rubber gasket, simply dry the two pieces and reaffix with superglue. Allow this bond to cure overnight before continued use. If you have issues with the coverslips breaking when you remove it from the cytospin apparatus, significantly slow down how quickly you try to release the coverslip. Sometimes pulling each corner of the silicone below the coverslip can help (Video 5). When the silicone releases from parts of the coverslip, those areas will look less dark than before. Ensure that all of the coverslip has released from the silicone before attempting to lift it. Also, try to put the forceps at an angle nearly parallel to the coverslip and lift without changing the angle of the forceps. If the apparatus leaks, the seal was not made well. If the leak is minor, it will likely not be an issue during the short centrifugation step. However, you can pipette the solution back into the Eppendorf tube and reassemble the apparatus to try to obtain a more secure seal.
If you observe no MurNAc-alk fluorescent signal, several possibilities exist for troubleshooting. There is a possibility that the click reaction buffer was made without the all the necessary components. Using Alexa Fluor 555, the click reaction buffer is visibly pink, so it is unlikely that this reagent would have been omitted without notice. Repeat the labeling experiment once to ensure that the protocol was followed correctly. If trouble persists, verify that the strain can be rescued from fosfomycin treatment by the addition of exogenous MurNAc. Note, it is not necessary to use MurNAc-alk for this step. MurNAc in our hands is more efficient at rescuing H. pylori from fosfomycin treatment and use of MurNAc instead of MurNAc-alk preserves valuable reagent. If the strain can be rescued from fosfomycin treatment, the MurNAc-alk may have degraded during long-term storage or improper handling.
If you observe cytoplasmic MurNAc-alk fluorescent signal, this may indicate that there is still unreacted 4-pentynoic acid present in the MurNAc-alk prep. MurNAc-alk can be repurified with additional silica gel column. If silica gel column does not remove the excess 4-pentynoic acid, purification can be completed using a Waters preparative HPLC/MS.
Basic Protocol 4
If your images are of poor quality, there are a number of possible culprits. Ensure that the slide is well-cleaned, does not have bubbles in the mounting medium, and that the oil is properly matched to the sample. Additionally, verify that the slide is not drifting by collecting two snapshot single-plane images separated by a few minutes and checking that the image looks the same. If this does not resolve the issue, check the general quality of the slide on a widefield fluorescence microscope. If the slide still appears to be of poor quality, slide preparation is likely to blame. Ensure that the detergent is completely rinsed when cleaning coverslips and be careful to make sure that the cells do not dry out at any point during the procedure. Generally there will be a ring of cells at the outside of the deposition circle on the coverslip that will be dried and thus flattened. This is normal and you must take care not to image cells from this region. If you verify that the slide preparation is high quality, that slides are not drifting during imaging, and that you are imaging in the correct region with properly matched immersion oil without bubbles between the slide and objective, the problem could be a malfunction with the microscope. Contact the microscope manager for further assistance.
UNDERSTANDING RESULTS:
Basic Protocol 1
Basic Protocol 1 will provide MurNAc-alk, a muramic acid analog with an alkyne moiety that allows the user to perform the labeling experiments described in the following protocols. Also, through a simple modification of the reagents, this method can be used to obtain 2-Azide NAM, another muramic acid derivative with an azide group linked to C2. This interesting compound is another useful probe for click chemistry experiments.
Basic Protocol 2
Please see the thorough discussion in the Sample Data section at the end of Basic Protocol 2.
Basic Protocol 3
After performing a short MurNAc-alk pulse with H. pylori as described in Basic Protocol 3, you should have high-quality slides with a good density of cells on the slide such that in each field of view there should be at least 10 well-isolated single cells. All of the cells, except perhaps cells in a narrow band along the periphery of the deposition spot, should be clearly three-dimensional. Cells should be approximately parallel to the imaging plane, but since the centrifugation step is gentle, cells may only be stuck to the coverslip on one side of the cell. There will likely be some clumps of cells also present, but these are generally not problematic so long as the cell density is not too high. While less phase dark than intact cells in media, the mounted fixed cells will still be visible using phase contrast microscopy. For both the labeled and unlabeled control, the green WGA stain should be bright and clear around the periphery of the cell. The poles might stain brighter than the cell body and there may be some stained debris visible. For the unlabeled control, there should only be barely visible signal in the red channel. For the labeled slide, there should be clearly visible red staining at the cell periphery. There should be less staining at the cell poles, and diffuse staining along the sidewall. In cells that are actively dividing, a stained septal ring should be visible. There should only be negligible signal in the center of the cell.
Basic Protocol 4
OMX imaging of these slides as described in Basic Protocol 4 should yield clear images of staining at the cell periphery and minimal signal inside the cell. The 3D geometry of the cells should be clearly visible, though the resolution in Z will be noticeably lower than that in X-Y. Structured artifacts should be minimal and largely only present in out-of-focus regions. The MurNAc-alk staining should appear patchy with a roughly circumferential band-like patterning. The MurNAc-alk and fluorescent WGA signals should align in X, Y, and Z. The scope may need to be realigned to achieve optimal positional matching of the two channels, and alignment may be less precise at the periphery of each field of view. Additionally, image brightness will also be lower around the periphery.
TIME CONSIDERATIONS:
Basic Protocol 1
The full synthesis of MurNAc-alk from Basic Protocol 1 takes 3 weeks, approximately. However, this depends on the expertise of the researcher. Comparing the two presented last steps of the preparation, the NHS coupling has one more reaction (NHS Alk ester) and uses a reverse phase purification method (water as the major solvent), making the evaporation step longer. Therefore, slightly less time is required for the direct coupling method.
Support Protocol 1
Growing H. pylori on a plate from a freezer stock should take 1-3 days but could take longer depending on the strain used and the quality of the freezer stock. Once robust growth is achieved on plates, a liquid culture should be started the morning before labeling. In the evening, the starter culture should be back-diluted to reach the desired density the next day. There should be 12-17 hours between diluting the culture and labeling the next day.
Basic Protocol 2
Performing the PG preps takes 5-7 days from harvesting cells to having samples ready to run for LC-MS/MS. Each LC-MS/MS run takes just over 1.5 hour to complete (100 min). You may run blanks in-between all samples. We typically do not run blanks between replicate injections of the same sample.
Support Protocol 4
Cytocentrifuge apparatuses from Support Protocol 4 can be made in a few hours but should cure overnight before first being used. Unless more units are desired, there should be no need to repeat this step.
Basic Protocol 3
Once the labeling described in Basic Protocol 3 begins, the whole slide preparation protocol should be able to be completed in fewer than 8 hours. The fixation, permeabilization, and labeling steps provide good time windows for preparation and/or cleanup. After the slide preparation is complete, the slides should cure for one week to ensure the ideal refractive index of the mounting medium.
Basic Protocol 4
For Basic Protocol 4, if the user is new to using the OMX microscope, several hours should be first dedicated to training. It would be ideal to bring some prepared slides to the training to have assistance with optimizing oils, imaging, and checking the channel alignment. After the user is confident in independent use of the microscope, it should take less than 30 minutes to initialize the microscope, determine the proper oil, and ensure that the alignment looks correct. If the scope needs to be realigned, this can typically be performed by an experienced user in under 30 minutes. Once image acquisition is proceeding smoothly, the amount of time required to image each slide will depend on the density of the sample, the exposure time, and how many individual cells the user needs to image. Typically it will take 20-60 minutes to collect enough images to have several hundred individual cells, but this time period can vary greatly.
Supplementary Material
Videos 1: Creating cytocentrifuge tops. This video shows the reader how to assemble the cytocentrifuge top components created in Support Protocol 4.
Video 2: Creating cytocentrifuge bases. This video shows the reader how to assemble the cytocentrifuge bottom components created in Support Protocol 4.
Video 3: Optimal cell suspension density. This video shows the reader a view of the proper cell suspension density for loading into cytocentrifuge units. The view shows multiple focal planes of the cell suspension through a phase contrast microscope using a 40x objective.
Video 4: Assembling cytocentrifuge units. This video shows the reader how to assemble a complete cytocentrifuge unit to spin cells onto coverslips using the top and bottom created in Support Protocol 4.
Video 5: Disassembling cytocentrifuge units: This video shows the reader how to disassemble cytocentrifuge units and remove coverslips after spinning cells onto coverslips.
Video 6: Mounting coverslips. This video shows the reader how to carefully mount coverslips onto slides with ProLong Diamond while minimizing bubble formation.
Template:
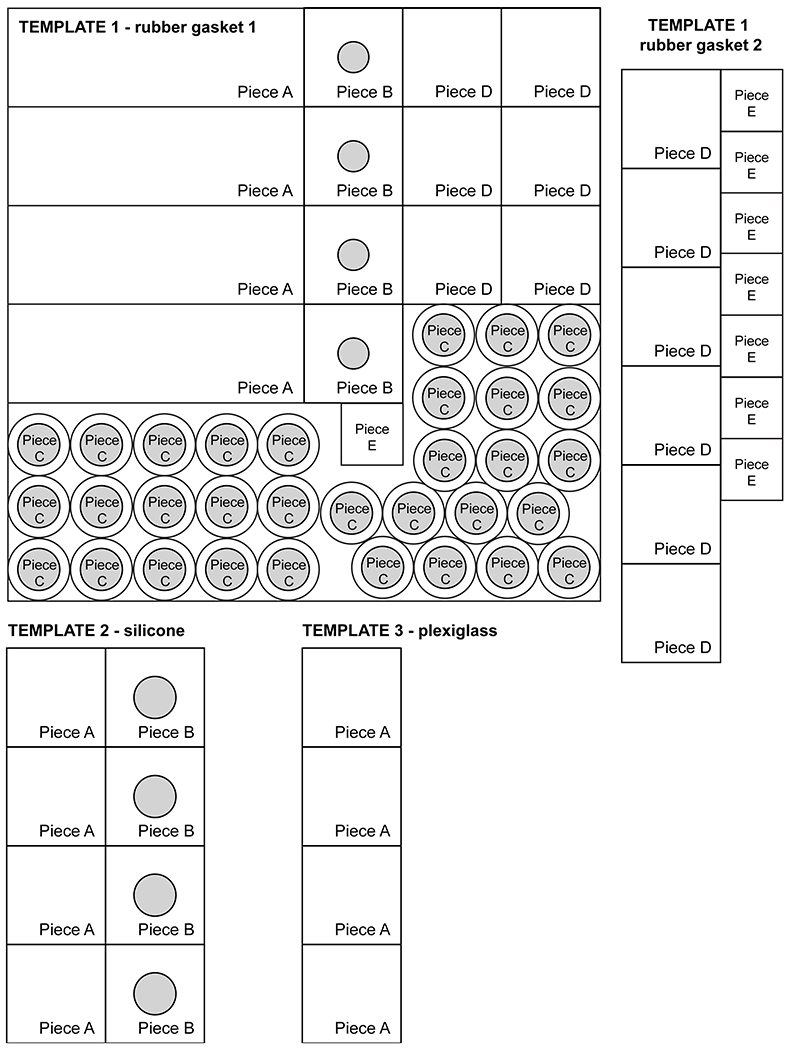
This template should be printed at 100% size to cut the properly-sized components to make the cytocentrifuge units in Support Protocol 4.
Table 3.
Troubleshooting table for bacterial labeling and imaging issues.
| Problem | Possible Cause | Solution |
|---|---|---|
| Degradation of MurNAc-alk | Light exposure/aging | Protect from light with aluminum foil |
| Poor H. pylori growth | Detergent residue in glassware | Completely clean glassware |
| FBS not heat-inactivated | Heat-inactivate FBS as described in the Reagents and Solutions section | |
| Improper growth conditions | Check incubator settings | |
| No labeling observed | Bacterial species does not have recycling machinery present | Genetic modification to include recycling enzymes AmgK and MurU |
| Click reaction buffer missing reagent | Remake click reaction buffer | |
| Cytoplasmic labeling reducing fluorescent quality of cell wall labelling | Excess 4-pentoynoic acid present with MurNAc-alk | Run additional column on MurNAc-alk; Run Waters preparative HPLC/MS |
| Poor quality microscopy images | Slide is dirty/not mounted properly (i.e. bubbles) | Clean slide and ensure oil is matched to sample; Remount slide to ensure no bubbles; Ensure detergent is completely rinsed when cleaning coverslips |
| Broken coverslips in cytospin apparatus | Improper removal from apparatus | Pull each corner of silicone below coverslip and ensure all of coverslip is released before attempting to remove; Release coverslip slower |
| Leaking cytospin apparatus | Improper seal formation | Pipette solution back into Eppendorf tube and reassemble apparatus |
| Failure to get desired LC-MS ion trace | Loss of PG pellet during preparation | Run undiluted sample to check; if no peaks, repeat PG preps |
| Over-dilution of samples | Reduce the dilution factor or concentrate the samples | |
| Excess salt | Check buffers, enzyme stocks, and material sources | |
| LC-MS equipment failure | Contact your service provider |
ACKNOWLEDGEMENTS: (mandatory for NIH, optional for all others)
We would like to thank Dr. Papa Nii Asare-Okai, Director of the MS facility at the University of Delaware, and Dr. Shi Bai, Director of the NMR Laboratory of the University of Delaware for their kind assistance. We also thank Dr. Jeffrey Caplan, Director of Bioimaging, and the Delaware Biotechnology Institute for assistance with super-resolution microscopy. We thank Alicia Meyer for the image in Figure 3 and Dr. David Baker, Department of Biochemistry, University of Washington for OMX SIM microscope access. This research was supported by the NIH U01 Common Fund program (grant number U01 CA221230), NIAID R01 AI136946, the Genomics Shared Resource of the Fred Hutch/University of Washington Cancer Consortium (P30 CA015704) and the Delaware COBRE program with a grant from the National Institute of General Medical Sciences (NIGMS P20 GM104316). Work in WV's lab was supported by a UKRI Strategic Priorities Fund (Grant no. EP/T002778/1).
C.L.G. is a Pew Biomedical Scholar, Sloan Scholar, and Cottrell Scholar and thanks the Pew Foundation, Sloan Foundation, and the Research Corporation for Science Advancement. K.E.D. and K.A.W. thank the NIH for support through a CBI training grant (T32 GM008550). K.E.D. also thanks the University of Delaware for support through the University Dissertation Fellowship program. J.A.T. thanks the NSF and DoD for support through the NSF Graduate Research Fellowship Program (DGE-0718124 and DGE-1256082) and the DoD National Defense Science and Engineering Graduate Fellowship.
The authors acknowledge the infrastructure and support of Delaware COBRE and INBRE programs, supported by a grant from the National Institute of General Medical Sciences (NIGMS, P30 GM110758, P20 GM104316, and P20 GM103446) of the National Institutes of Health.
LITERATURE CITED:
- 1.Barreteau H, Kovac A, Boniface A, Sova M, Gobec S, & Blanot D (2008). Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev, 32(2), 168–207. doi: 10.1111/j.1574-6976.2008.00104.x [DOI] [PubMed] [Google Scholar]
- 2.Brown AR et al. , 20201, manuscript in preparation.
- 3.Bui NK, Gray J, Schwarz H, Schumann P, Blanot D, & Vollmer W (2009). The peptidoglycan sacculus of Myxococcus xanthus has unusual structural features and is degraded during glycerol-induced myxospore development. J Bacteriol, 191(2), 494–505. doi: 10.1128/JB.00608-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caccamo PD, & Brun YV (2018). The Molecular Basis of Noncanonical Bacterial Morphology. Trends Microbiol, 26(3), 191–208. doi: 10.1016/j.tim.2017.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa K, Bacher G, Allmaier G, Dominguez-Bello MG, Engstrand L, Falk P, … García-del Portillo F (1999). The Morphological Transition of Helicobacter pylori Cells from Spiral to Coccoid Is Preceded by a Substantial Modification of the Cell Wall. Journal of Bacteriology, 181(12), 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniel RA, & Errington J (2003). Control of Cell Morphogenesis in Bacteria. Cell, 113(6), 767–776. doi: 10.1016/s0092-8674(03)00421-5 [DOI] [PubMed] [Google Scholar]
- 7.de Pedro MA, Quintela JC, Holtje JV, & Schwarz H (1997). Murein segregation in Escherichia coli. J Bacteriol, 179(9), 2823–2834. doi: 10.1128/jb.179.9.2823-2834.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeMeester KE, Liang H, Jensen MR, Jones ZS, D'Ambrosio EA, Scinto SL, … Grimes CL (2018). Synthesis of Functionalized N-Acetyl Muramic Acids To Probe Bacterial Cell Wall Recycling and Biosynthesis. J Am Chem Soc, 140(30), 9458–9465. doi: 10.1021/jacs.8b03304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeMeester KE, Liang H, Zhou J, Wodzanowski KA, Prather BL, Santiago CC, & Grimes CL (2019). Metabolic Incorporation of N-Acetyl Muramic Acid Probes into Bacterial Peptidoglycan. Curr Protoc Chem Biol, 11(4), e74. doi: 10.1002/cpch.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glauner B (1988). Separation and quantification of muropeptides with high-performance liquid chromatography. Analytical Biochemistry, 172(2), 451–464. doi: 10.1016/0003-2697(88)90468-x [DOI] [PubMed] [Google Scholar]
- 11.Glauner B, Höltje JV, & Schwarz U (1988). The composition of the murein of Escherichia coli. The Journal of biological chemistry, 263(21), 10088–10095. [PubMed] [Google Scholar]
- 12.Hayashi K (1975). A rapid determination of sodium dodecyl sulfate with methylene blue. Analytical Biochemistry, 67(2), 503–506. doi: 10.1016/0003-2697(75)90324-3 [DOI] [PubMed] [Google Scholar]
- 13.Höltje J-V (1998). Growth of the Stress-Bearing and Shape-Maintaining Murein Sacculus of Escherichia coli. Microbiology and Molecular Biology Reviews, 62(1), 181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horcajo P, de Pedro MA, & Cava F (2012). Peptidoglycan plasticity in bacteria: stress-induced peptidoglycan editing by noncanonical D-amino acids. Microb Drug Resist, 18(3), 306–313. doi: 10.1089/mdr.2012.0009 [DOI] [PubMed] [Google Scholar]
- 15.Huang YW, Wang Y, Lin Y, Lin C, Lin YT, Hsu CC, & Yang TC (2017). Impacts of Penicillin Binding Protein 2 Inactivation on beta-Lactamase Expression and Muropeptide Profile in Stenotrophomonas maltophilia. mSystems, 2(4). doi: 10.1128/mSystems.00077-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kühner D, Stahl M, Demircioglu DD, & Bertsche U (2014). From cells to muropeptide structures in 24 h: peptidoglycan mapping by UPLC-MS. Sci Rep, 4, 7494. doi: 10.1038/srep07494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, … VanNieuwenhze MS (2012). In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl, 51(50), 12519–12523. doi: 10.1002/anie.201206749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuru E, Radkov A, Meng X, Egan A, Alvarez L, Dowson A, … VanNieuwenhze MS (2019). Mechanisms of Incorporation for D-Amino Acid Probes That Target Peptidoglycan Biosynthesis. ACS Chem Biol, 14(12), 2745–2756. doi: 10.1021/acschembio.9b00664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang H, DeMeester KE, Hou CW, Parent MA, Caplan JL, & Grimes CL (2017). Metabolic labelling of the carbohydrate core in bacterial peptidoglycan and its applications. Nat Commun, 8, 15015. doi: 10.1038/ncomms15015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liechti GW, Kuru E, Hall E, Kalinda A, Brun YV, VanNieuwenhze M, & Maurelli AT (2014). A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis. Nature, 506(7489), 507–510. doi: 10.1038/nature12892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sauvage E, Kerff F, Terrak M, Ayala JA, & Charlier P (2008). The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev, 32(2), 234–258. doi: 10.1111/j.1574-6976.2008.00105.x [DOI] [PubMed] [Google Scholar]
- 22.Shi H, Bratton BP, Gitai Z, & Huang KC (2018). How to Build a Bacterial Cell: MreB as the Foreman of E. coli Construction. Cell, 172(6), 1294–1305. doi: 10.1016/j.cell.2018.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegrist MS, Whiteside S, Jewett JC, Aditham A, Cava F, & Bertozzi CR (2013). (D)-Amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol, 8(3), 500–505. doi: 10.1021/cb3004995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh SK, SaiSree L, Amrutha RN, & Reddy M (2012). Three redundant murein endopeptidases catalyse an essential cleavage step in peptidoglycan synthesis of Escherichia coli K12. Mol Microbiol, 86(5), 1036–1051. doi: 10.1111/mmi.12058 [DOI] [PubMed] [Google Scholar]
- 25.Sycuro LK, Pincus Z, Gutierrez KD, Biboy J, Stern CA, Vollmer W, & Salama NR (2010). Peptidoglycan crosslinking relaxation promotes Helicobacter pylori's helical shape and stomach colonization. Cell, 141(5), 822–833. doi: 10.1016/j.cell.2010.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor JA, Bratton BP, Sichel SR, Blair KM, Jacobs HM, DeMeester KE, … Salama NR (2020). Distinct cytoskeletal proteins define zones of enhanced cell wall synthesis in Helicobacter pylori. Elife, 9. doi: 10.7554/eLife.52482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor JA, Sichel SR, & Salama NR (2019). Bent Bacteria: A Comparison of Cell Shape Mechanisms in Proteobacteria. Annu Rev Microbiol, 73, 457–480. doi: 10.1146/annurev-micro-020518-115919 [DOI] [PubMed] [Google Scholar]
- 28.Tiyanont K, Doan T, Lazarus MB, Fang X, Rudner DZ, & Walker S (2006). Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc Natl Acad Sci U S A, 103(29), 11033–11038. doi: 10.1073/pnas.0600829103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomb J-F, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, … Venter JC (1997). The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature, 388(6642), 539–547. [DOI] [PubMed] [Google Scholar]
- 30.Vollmer W, Blanot D, & de Pedro MA (2008). Peptidoglycan structure and architecture. FEMS Microbiol Rev, 32(2), 149–167. doi: 10.1111/j.1574-6976.2007.00094.x [DOI] [PubMed] [Google Scholar]
- 31.Xu N, Huang ZH, de Jonge BL, & Gage DA (1997). Structural characterization of peptidoglycan muropeptides by matrix-assisted laser desorption ionization mass spectrometry and postsource decay analysis. Anal Biochem, 248(1), 7–14. doi: 10.1006/abio.1997.2073 [DOI] [PubMed] [Google Scholar]
- 32.Zhang ZJ, Wang YC, Yang X, & Hang HC (2020). Chemical Reporters for Exploring Microbiology and Microbiota Mechanisms. Chembiochem, 21(1–2), 19–32. doi: 10.1002/cbic.201900535 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Videos 1: Creating cytocentrifuge tops. This video shows the reader how to assemble the cytocentrifuge top components created in Support Protocol 4.
Video 2: Creating cytocentrifuge bases. This video shows the reader how to assemble the cytocentrifuge bottom components created in Support Protocol 4.
Video 3: Optimal cell suspension density. This video shows the reader a view of the proper cell suspension density for loading into cytocentrifuge units. The view shows multiple focal planes of the cell suspension through a phase contrast microscope using a 40x objective.
Video 4: Assembling cytocentrifuge units. This video shows the reader how to assemble a complete cytocentrifuge unit to spin cells onto coverslips using the top and bottom created in Support Protocol 4.
Video 5: Disassembling cytocentrifuge units: This video shows the reader how to disassemble cytocentrifuge units and remove coverslips after spinning cells onto coverslips.
Video 6: Mounting coverslips. This video shows the reader how to carefully mount coverslips onto slides with ProLong Diamond while minimizing bubble formation.