

Abstract
New and provocative insights into the relationships between iron and cancer have been uncovered in recent years. These include delineation of connections that link cellular iron to DNA repair, genomic integrity, and oncogenic signaling as well as the discovery of ferroptosis, a novel iron-dependent form of cell death. In parallel, new molecules and pathways that regulate iron influx, intracellular iron trafficking and egress in normal cells, and their perturbations in cancer have been discovered. In addition,, insights into the unique properties of iron handling in tumor initiating cells (cancer stem cells), novel contributions of the tumor microenvironment to the uptake and regulation of iron in cancer cells, and new therapeutic modalities that leverage the iron dependence of cancer have emerged.
Introduction
The field of iron biology, particularly aspects related to cancer, has evolved substantially since we last reviewed this topic for Cancer Research in 2011. New connections as well as mechanistic underpinnings of previously described relationships between iron and cancer have been uncovered, and novel interventions for cancer therapy that involve iron perturbations have been proposed. Iron trafficking is now understood to be considerably more complex and nuanced than envisioned 10 years ago, involving previously unknown mechanisms of uptake and efflux as well as intracellular and intercellular redistribution of iron that impacts tumor behavior and drug response. Further, iron is now known to influence not only tumor growth, but metastatic potential. In counterpoint to these pro-tumorigenic activities, iron has also been recognized as an essential element in ferroptosis, the eponymous mechanism of cell death that may ultimately allow us to target the iron addiction of cancer cells. To adequately review these topics, we have focused on recent discoveries of molecules and pathways that link iron homeostasis and cancer. We refer the reader to additional reviews to fill out the complete picture of iron dysregulation and cancer, including population-based studies, the role of mitochondrial iron, and early experimental work linking iron and cancer(1–4).
Iron Uptake, Redistribution and Egress
Cellular iron uptake
Transferrin receptor 1 (TFR1)/transferrin.
A major pathway of iron intake utilizes transferrin receptor 1 (TFR1) (Figure 1). This well-studied pathway(5) involves binding of diferric transferrin (TF) to TFR1, endocytosis of the TFR1/TF-Fe complex, release of iron from TF within the acidic endosome, iron reduction by STEAP3, and subsequent exit of iron from the endosome to the cytosol via DMT1. Following release of iron, the TFR1/TF complex is recycled to the cell surface, where apo-TF dissociates from TFR1 to participate in multiple additional rounds of iron delivery. As early as the 1980’s, it was shown that transferrin receptors are up-regulated in breast and other cancers(6,7), suggesting that tumor cells exhibit an enhanced demand for iron. These early findings have been corroborated by hundreds of subsequent studies, establishing TFR1 as a tumor marker (albeit not a specific marker, since non-cancer cells, especially rapidly proliferating cells, also express abundant transferrin receptors), and stimulating strategies to exploit the enhanced expression of TFR1 in anti-tumor therapy. However, TFR1 upregulation is only one of many strategies employed by tumor cells to acquire iron.
Figure 1. Intracellular and intercellular iron trafficking in the tumor microenvironment.
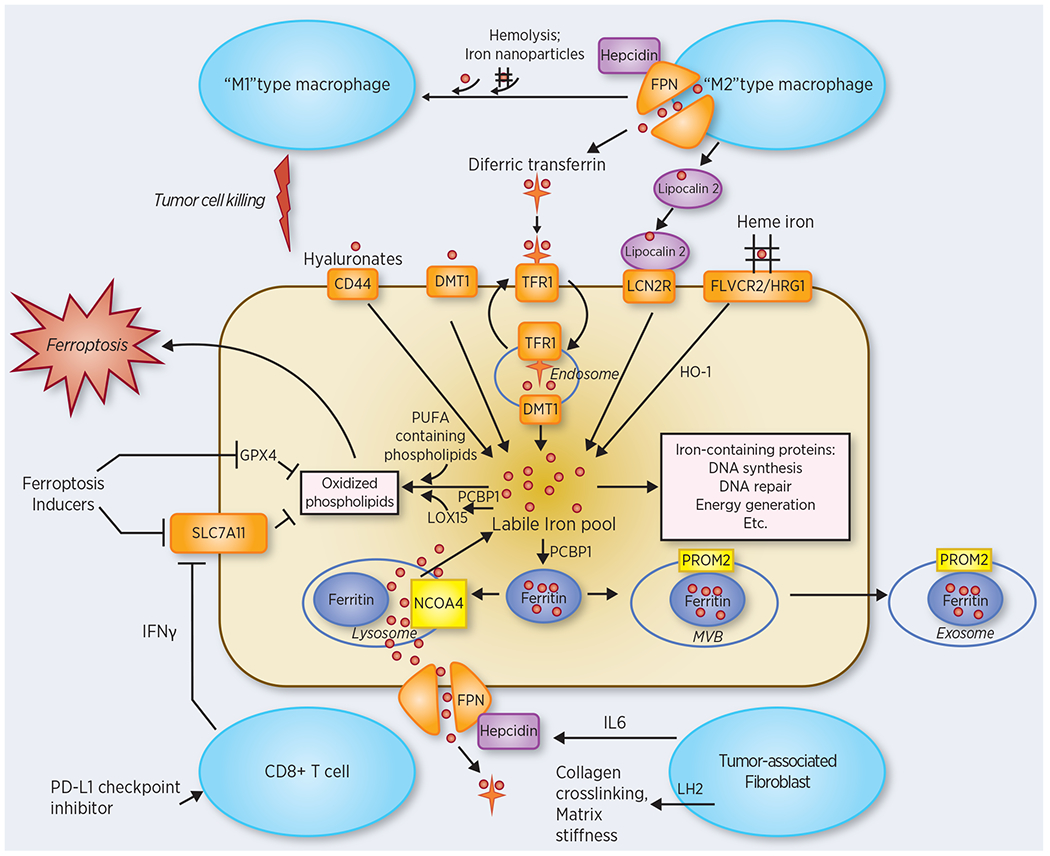
Intracellular trafficking: Plasma membrane proteins involved in iron uptake are heme transporters FLVCR2 and HRG1; lipocalin 2 receptor LCN2R (also called 24p3R), which binds a lipocalin 2-siderophore-iron complex; DMT1 (which is also involved in endosomal iron efflux), which transports Fe2+; CD44, which binds an iron-hyaluronate complex; and TFR1, which binds diferric transferrin. Iron taken up by these pathways is delivered to a pool of redox-active iron termed the labile iron pool to supply iron to iron-containing proteins. The chaperone PCBP1 delivers iron to ferritin and LOX15, among other targets. Ferritin stores excess iron in a non-toxic bioavailable form. Iron in ferritin can also be redistributed following binding to NCOA4 and lysosomal degradation (ferritinophagy), or ferritin-bound iron can be effluxed from the cell via a multivesicular body (MVB)/exosome pathway. The major route of iron efflux is ferroportin (FPN), which is degraded by the secreted peptide hepcidin. Iron participates in producing the oxidized membrane lipids that mediate ferroptotic cell death. Ferroptosis inducers disable endogenous pathways that reduce oxidized lipids, including the peroxidase GPX4 and the cystine/glutamate antiporter SLC7A11. Intercellular trafficking: Pro-tumorigenic effects are mediated by tumor-associated macrophages, which provide iron to tumor cells, as well as by tumor-associated fibroblasts (TAFs). TAFs express the iron-dependent enzyme lysyl hydroxylase 2 (LH2) an enzyme that enhances matrix stiffness; TAFs also secrete IL6, which upregulates hepcidin, degrades ferroportin, and limits iron efflux from tumor cells. Anti-tumorigenic effects are exerted by PD-L1-stimulated CD8+ T cells, which promote ferroptosis by secreting interferon gamma and inhibiting SLC7A11. Excess exogenous iron resulting from hemolysis or provided by iron-containing nanoparticles can trigger a shift of M2-type tumor associated macrophages to a tumoricidal M1 phenotype. Note that this figure consolidates findings from multiple different tumor types; not all pathways are operant in all cancers.
Lipocalin 2 (NGAL, LCN2), a member of the lipocalin superfamily, is a 25 kD secreted protein that binds bacterial siderophores or siderophore-like catechols of mammalian origin(8). Siderophores are small molecules that bind iron. Lipocalin 2 can therefore serve as an iron delivery vehicle following binding to its high affinity cell surface receptor, LCN2R (24p3R, SLC22A17), or megalin(9). Overexpression of LCN2 is associated with decreased survival of breast cancer patients(10), and depletion of lipocalin 2 inhibits tumor formation in mammary(11) as well as prostate(12,13) tumor models . Consistent with a role of iron in the pro-tumorigenic effects of lipocalin 2 in breast cancer(14), tumor tissue from renal clear cell carcinoma contained approximately 10-fold more lipocalin 2-bound iron than non-tumor tissue, and mutation of amino acid residues critical to the iron-binding function of lipocalin 2 or treatment of cells with apo-lipocalin 2 abolished its pro-tumorigenic activity in experimental models(15). Strikingly, cancer cells from leptomeningeal metastases of patients with breast or lung cancer express LCN2 and its receptor, enabling them to acquire iron in this nutrient-limited microenvironment(16). Accordingly, loss of LCN2 inhibited tumor growth in the leptomeninges and conferred a survival benefit in three mouse models(16). In addition to the ability of lipocalin 2 to promote tumor growth, invasiveness, and the epithelial to mesenchymal transition (EMT) in cancer cells, the role of this secreted protein in iron trafficking in the microenvironment likely contributes substantially to its pro-tumorigenic effects(17,18).
DMT1 (divalent metal transporter 1). In addition to its role in the transit of iron from the endosome to the cytosol, DMT1 serves as an apical iron transporter involved in the uptake of dietary iron by duodenal cells of the small intestine(19). Expression of DMT1 is increased in patients with colorectal carcinoma(20), and colon-selective disruption of DMT1 decreases sporadic and colitis-associated colon tumors in mice(21). The pro-tumorigenic activity of DMT1 is related to its iron transport activity: a high iron diet increased tumor burden in mice, and this effect was attenuated following DMT1 disruption. Effects of DMT1 are linked to its ability to stimulate JAK-STAT3 signaling(21) (see Iron and Signaling section).
CD44 is a transmembrane glycoprotein and cancer stem cell marker(22). A recent publication suggests that CD44 can function to transport iron-containing hyaluronates in breast and other cancer cells, and that this pathway of iron import may be particularly important in the mesenchymal phase of the EMT transition, when epithelial cells assume the more migratory and invasive phenotype that contributes to metastatic spread(23). Iron taken up by this CD44-dependent pathway sustains EMT by supporting the catalytic activity of PHF8, an iron-dependent histone demethylase that promotes demethylation of H3K9me2, a repressive histone mark(24). The implication that alternative pathways of iron uptake, such as that mediated by CD44, may acquire increased importance in mesenchymal cells is in accord with previous work showing that CD133 (prominin-1), a mesenchymal stem cell marker, downregulates TFR1-mediated endocytosis of diferric transferrin(25).
Heme uptake.
Heme iron refers to iron coordinated within a porphyrin ring, as found in hemoglobin. Because red meat serves as the principal dietary source of heme iron in humans and intake of red meat is associated with increased cancer risk(26), a role for heme iron in malignant processes has been suggested. Pathways of heme iron uptake are upregulated in cancer, as are pathways of heme export and heme catabolism via heme oxygenase 1(27), suggesting that an increased flux of heme is important to cancer cell growth or survival(28). However, identification of critical role(s) that heme iron plays in cancer cell metabolism has remained elusive: maintenance of the function of enzymes involved in histone demethylation, microRNA processing, or a role in cataplerosis have all been proposed (28).
NTBI.
Non-transferrin-bound iron (NTBI) refers to iron present in the circulation that exceeds the binding capacity of transferrin. Although NTBI is generally not present in cancer patients, NTBI is taken up by transporters that also transport other metals (e.g. zinc, calcium) such as ZIP14, L-type and T-type calcium channels, DMT1, ZIP8, and TRPC6(29), some of which are upregulated in cancer(21) or suppressed by mutated p53(30).
Iron Redistribution.
Ferritin is an intracellular iron storage protein that can store up to 4500 atoms of iron in a non-toxic but bioavailable form(31). Ferritin is regulated post-transcriptionally by iron, but can also be regulated transcriptionally by both oxidant and oncogenic stimuli (32–36). The discovery of NCOA4 (nuclear receptor coactivator 4) as a cargo receptor for ferritin uncovered a mechanism by which iron can be dynamically reallocated among intracellular compartments(37,38). NCOA4 mediates delivery of ferritin to the lysosome for degradation and subsequent release of ferritin-bound iron to the cytosol and mitochondria. This process, termed “ferritinophagy”, is regulated by iron through HERC2, a ubiquitin ligase that targets NCOA4 for degradation under iron-replete conditions (when the iron liberated by ferritin degradation is presumably not needed)(39).
Ferritinophagy and the consequent release of iron supports heme synthesis during erythropoesis(40), and also has important consequences in non-hematopoietic cells, including cancer cells. For example, interference with ferritin degradation by knockdown of NCOA4 inhibits ferroptosis, a death pathway dependent on iron, and overexpression of NCOA4 sensitizes pancreatic cancer cells to ferroptosis(41). Intriguingly, an oncogenic NCOA4-RET fusion protein has been identified in a small fraction of thyroid, breast and other cancers(42). Although NCOA4-RET is not predicted to retain the ferritin binding domain of the intact NCOA4 protein, cells expressing this fusion protein are sensitive to sorafinib(43), a multikinase inhibitor that induces ferroptosis. The effect of NCOA4-RET on iron metabolism may merit investigation.
Mitochondria are essential to intracellular iron metabolism, serving both as a site of FeS cluster biogenesis and heme synthesis. Mitochondrial iron accumulation has also been linked to cancer. For example, the NEET proteins mitoNEET (CISD1) and NAF-1 (CISD2) are Fe-S cluster-containing proteins located in the mitochondrial outer membrane that modulate mitochondrial iron homeostasis and oxidative stress; upregulation of NEET proteins have been implicated in breast and other cancers(44). PINK1 and PRKN, proteins that play a role in mitophagy, suppress KRAS-driven pancreatic tumor growth by inhibiting the mitochondrial iron importers mitoferrin-1 (SLC25A37) and mitoferrin-1 (SLC25A28) and attenuating mitochondrial iron accumulation(45,46). Inhibition of PINK1 and PRKN results in mitochondrial iron overload, chronic inflammation, and tumorigenesis in a mouse model of spontaneous pancreatic cancer. The iron chelator deferiprone reduced these pancreatic tumors, suggesting that mitochondrial iron homeostasis may constitute a therapeutic target.
Intracellular iron regulation.
IRP1 and IRP2 are a post-transcriptional regulators of intracellular iron metabolism that bind to IREs located in mRNAs encoding several critical proteins of iron metabolism (reviewed in (47)). Binding of IRP1 or IRP2 represses ferritin and the iron efflux pump ferroportin, and stabilizes TFR1, thus acting to increase intracellular iron. IRP2 is regulated by FBXL5, a ubiquitin ligase that degrades IRP2 under iron replete conditions. Inappropriate activation of IRP2 has been linked to cancer(48–50), perhaps in part by activating MDM2 (51) or suppressing TAp63(52). Dysregulation of FBXL5 is also associated with poor prognosis in human hepatocellular carcinoma (53). Further, in the first genetically engineered model of liver cancer promoted by iron overload (53), tissue-specific FBXL5 ablation in mouse liver was shown to result in iron overload, oxidative stress and inflammation, and promote tumor formation. Unexpectedly, the anti-cancer drug cisplatin may also act in part by disrupting IRP2-mediated iron regulation: recent work showed that cisplatin binds directly to cys512 and cys516 in human IRP2, disrupting IRP2 function and depleting the LIP(54). Accordingly, combining cisplatin with the iron chelator desferoxamine (DFO) led to enhanced iron depletion and reduced tumor growth in a mouse xenograft model of colon adenocarcinoma(54).
Iron efflux.
In 2010, our laboratory observed that ferroportin (FPN), a cellular iron efflux pump, is downregulated in breast cancer(55). We subsequently reported similar results in prostate(56) and ovarian(57) cancer. Overexpression of ferroportin reduced intracellular iron and slowed growth of cancer cells in vitro and in vivo. Ferroportin expression predicted outcome for women with breast cancer(55,57), implicating intracellular iron as a major driver of cancer cell growth, and suggesting that perturbation of this efflux pathway is sufficient to drive the biological behavior of cancers. Evaluation of additional iron regulatory genes revealed that gene combinations that minimized intracellular iron content conferred favorable prognosis regardless of whether they reduced iron import [anti-import TFR1(Low)/HFE(High)]; or increased iron export [SLC40A1 (ferroportin)(High)/HAMP(Low)] (58). Others have confirmed and expanded these results in other malignancies, including lung cancer(59), multiple myeloma(60) adrenocortical carcinoma(61), and a subset of patients with AML(62). Highlighting the crucial importance of iron to malignant processes, an iron-regulatory gene signature (IRGS) composed of 16 “iron” genes strongly predicted clinical outcome in women with breast cancer(58).
An additional mechanism of enhanced iron retention through downregulation of iron efflux was recently described(63). G9a, a H3K9 methyltransferase, is overexpressed in breast cancer and correlated with poor patient survival. Hephaestin, a ferroxidase involved in basolateral intestinal iron transport that facilitates iron egress from cells, was identified as an important target that is repressed by G9a. Blockade of iron efflux through silencing of hephaestin in cancer cells may thus represent an additional mechanism of iron retention.
Cells may also control intracellular iron by eliminating ferritin and its associated iron by secretion into the bloodstream (64,65). Additionally, breast cancer cells can activate a MVB-mediated pathway of ferritin secretion following detachment from the cell surface; this pathway is dependent on prominin-2(66). Ferritin released through this or other pathways may subsequently be taken by binding to TFR1 or other receptors(67–71).
Iron Addiction
Observations of enhanced iron uptake, redistribution and retention in cancer cells suggest that cancers are addicted to iron. What metabolic needs underpin this addiction? The answers are evolving, complex, and multifactorial. At a minimum, iron is a critical element in the proliferation, cell cycle control and genomic integrity of cancer cells.
Cellular proliferation:
Iron is essential for the catalytic function of ribonucleotide reductase, the enzyme that converts ribonucleotides to deoxyribonucleotides, the rate-limiting step in DNA synthesis and an obligate step in cell replication. Ribonucleotide reductase depends on a tyrosyl radical for ribonucleotide reduction that in turn requires a di ferric center coordinated with oxygen (Fe3+-O-Fe3+) for activity. Ribonucleotide reductase is highly regulated, and an imbalance in dNTP pools leads to increased DNA mutation rates, genome instability and replication anomalies. Iron chelators are a major class of ribonucleotide reductase inhibitors.
Cell cycle entry and progression:
Iron regulatory proteins (IRP1 and IRP2) are master regulators of intracellular iron. Both IRP2 knockdown (which results in iron depletion) and iron chelation induce the cell cycle regulators p15, p21 and p27, leading to cell cycle arrest with accumulation of cells in G0/G1(50). Mechanisms of p21 induction are still incompletely elucidated: in melanoma cells, SP1 and its interacting partners ER-α and c-Jun were implicated(72), whereas in prostate cancer cells, p53 and KLF6 appear to play a major role in iron-dependent regulation of p21(73). Through these effects on the cell cycle, iron directly impacts cell proliferation.
Genomic integrity:
Iron, in the form of iron-sulfur clusters, is required for the activity of virtually all replicative DNA polymerases as well as proteins involved in genome maintenance and repair(74). For example, iron-sulfur clusters are critical to the function of helicases such as XPD and FancJ, glycosylases involved in DNA repair, the primases Chir1 and DNA2 that are involved in DNA replication, RTEL1 that is involved in homologous recombination, and others(74). These pathways are used to initiate and maintain a highly replicative state.
For these and other reasons (for example, iron dysregulation also contributes to the activation of metastatic pathways [see Iron and Metastases section]), at least some cancer cells develop a dependence on iron that exceeds that of their non-malignant counterparts, a phenomenon we have termed “iron addiction”. As a result, these cancer cells are more sensitive to the anti-proliferative effects of a range of chemically distinct iron chelators, including desferoxamine, tachpyridine, the di-2-pyridylketone class of thiosemicarbazones (e.g. Dp44mT and DpC), than non-cancer cells(75,76). Similarly, forced expression of ferroportin or treatment with anti-hepcidin antibodies, which reduce intracellular iron by enhancing its efflux , preferentially reduces the rate of proliferation of breast, prostate and ovarian cancer cells(56,57,73) .
Iron and Metastases
With a few notable exceptions, most patients who die from cancer do so as the result of distant metastases rather than the growth of the primary tumor. Recent evidence has begun to link cancer cell iron to the ability of cancer cells to metastasize, although mechanisms are still uncertain. The observation that expression of ferroportin and TFR1 predict survival of breast cancer patients(58) and that disrupted regulation of these pathways is associated with poorer survival of patients with prostate cancer(56) implies a role for iron in human metastasis, since mortality from these cancers is primarily due to metastatic disease. Mouse models demonstrating that iron depletion inhibits metastases also support a role for iron in tumor dissemination. For example, treatment with the iron chelator Dp44mT reduced bone metastasis of human metastatic breast cancer cells (MDA-MB231-BoM) in intracardially injected mice(77). Dp44mT also inhibited the formation of lung tumors in a metastatic model created by injection of osteosarcoma cells in nude mice(78), and augmented the efficacy of a nanoparticle-delivered cisplatin prodrug in reducing mammary metastasis(79). The iron chelator desferiorox inhibited invasion of pancreatic cancer cells(80) and treatment with the iron chelator DFO inhibited cancer cell growth in the leptomeningeal metastatic site(16). In studies using induction of ferroportin rather than a chelator to deplete iron, our group observed a decrease in the number of tumors in a mouse model of ovarian cancer metastasis(57). In this model, ovarian cancer cells were injected directly into the peritoneum (a major site of ovarian cancer metastases); a reduction in both tumor number and mass was observed with tumor cells that overexpressed ferroportin. Similarly, ferroportin overexpression impeded not only tumor growth but EMT and the metastasis of 4T1 breast cancer cells to lung and liver in mice(81). Supporting these findings, global knockout of hepcidin, a regulator of ferroportin that hinders iron egress by promoting ferroportin degradation, decreased tumor formation and increased survival following tail vein injection of Lewis lung cancer cells in mice(81).
Effects of iron deprivation on metastasis have been linked to the upregulation of the metastasis suppressor NDRG1 as well as STAT3 signaling. NDRG1 is a metastasis suppressor that is downregulated in multiple cancer types(77). NDRG1 is induced transcriptionally following treatment with iron chelators(82). In ovarian cancer, STAT3 signaling was an important link between changes in cellular iron status and cancer: a FPN-mediated decrease in ovarian peritoneal tumors was associated with a decrease in invasion of ovarian cancer stem cells mediated by a decrease in IL6 and phosphorylated STAT3(57).
Iron and Molecular Signaling Pathways
Iron both regulates and is regulated by oncogenic and tumor suppressor signaling pathways.
Iron-regulated signaling.
WNT:
One of the strongest epidemiological associations between high dietary iron and cancer is in colorectal carcinoma (CRC)(26,83). The expression of multiple iron metabolism genes is altered in CRC leading to an accumulation of intracellular iron(20,84). Several studies have implicated Wnt signaling in this process. For example, iron augments Wnt signaling in cells with aberrant APC (a tumor suppressor frequently inactivated in CRC(20,84)). Further, high throughput screening for Wnt inhibitors identified iron chelators as top hits (85,86).
STAT3.
Genetic or pharmacological inhibition of the iron importer DMT1 reduced sporadic and colitis-associated colon tumors in mice(21). Unexpectedly, STAT3 rather than Wnt appeared critical to this process. Elevated levels of iron or DMT1 activated STAT3, and inhibition of this pathway reduced tumor burden, implying a critical role for activation of DMT1-mediated JAK/STAT signaling in iron-mediated enhancement of colonic tumorigenesis. Mechanistic studies revealed that iron binds directly to CDK1 kinase and potentiates STAT3 activity(21). A study in lung cancer similarly implicated iron-dependent activation of CDK1 and STAT3 signaling(87). The relative contributions of Wnt and STAT3 to iron signaling may depend on tissue type and requires further evaluation. Since hepcidin is downstream of the IL6/JAK/STAT3 pathway, it will be of interest to determine whether DMT1-mediated activation of STAT3 also contributes to iron retention in tumor cells by upregulating hepcidin.
EGFR.
EGFR binds to and regulates the cell surface distribution of TFR1 in lung adenocarcinoma cells and thereby influences iron import, illustrating an additional mechanism by which cancer cells may increase iron uptake(88) and linking EGFR to iron proteins in cancer.
NDRG1, a metastasis suppressor, is upregulated by iron chelation, implying a role for iron in regulating NDRG1. NDRG1 acts by multiple mechanisms(89), including attenuating signaling of EGFR family members by inhibiting the formation of HER2/EGFR and HER2/HER3 heterodimers and by stimulating degradation of EGFR(90).
ERK1/2 and Akt.
In head and neck carcinoma, iron stimulates ERK1/2 and AKT pathways, activates AP1, and increases expression of the metalloprotease MMP9(91), potentially contributing to invasion.
HIF1α and HIF2α.
Iron exerts a major influence on signaling in cancer cells by regulating the activity of HIF1α and HIF2α. These transcription factors are frequently upregulated in cancer and are associated with poor prognosis(92). Both HIF1α and HIF2α can complex with constitutively active HIF1β to transcriptionally induce many genes important to survival of tumor cells in a hypoxic environment, including VEGFA, EPO, GLUT1 and others. Levels of the alpha subunits of HIF1 are post-translationally controlled by prolyl hydroxylases, which are iron-, 2-oxoglutarate- and oxygen-dependent enzymes that modify HIF1 and target it for proteasomal degradation. Among other activities, HIF1 induces genes that increase iron uptake and intracellular iron release, notably TFRC, HO1 and ceruloplasmin(93–95). Thus, HIF1α and HIF2α, which are held in check by iron in normoxic cells, may be co-opted to increase iron availability in the hypoxic tumor environment. Further, studies in IRP1 knockout mice showed that HIF2α is repressed by IRP1, linking iron regulation to oxygen sensing, and suggesting that IRP1 may be a target for controlling HIF2 activity(96).
Ferritin.
Best known for its role as an intracellular iron storage protein, ferritin is also secreted into the serum, where it may have signaling activity(64,65,97). In hepatic stellate cells, exogenous ferritin was observed to induce NFKB (98), a transcription factor that induces multiple tumor-promoting cytokines including IL6. These observations are consistent with the association between elevated ferritin and poor outcome observed in a number of cancers, including AML (99).
Regulation of iron by oncogenic and tumor suppressive signaling pathways.
c-myc.
The earliest evidence of an association between oncogenic pathways and iron was the observation that adenoviral early gene product, E1A, a viral oncogene with immortalizing properties similar to those of the oncogene c-myc, represses ferritin at the level of transcription(35). Subsequently, it was shown in EBV-immortalized B cells that c-myc also represses transcription of ferritin and in addition transcriptionally induces IRP2 (100); induction of IRP2 further decreased ferritin and increased transferrin receptor post-transcriptionally. These effects of c-myc increased intracellular iron and were required for cell transformation by c-myc; IRP2 was later demonstrated to exert pro-tumorigenic activity through mechanisms in addition to its role in iron metabolism(48). C-myc was further shown to activate TFRC to enhance proliferation and tumorigenesis in in vitro and in vivo models of B cell lymphoma(101).
p53.
Iron influences the activity of the p53 tumor suppressor. Iron in the form of heme directly binds to p53, stimulating its degradation and interfering with its interaction with DNA, thus reducing the anti-tumor effects of p53(102,103). However, other iron regulatory pathways appear to have opposite effects on p53, suggesting that the final p53 status of cancer cells is influenced by multiple iron-regulatory interactions. Thus, supplementation of cell cultures with exogenous inorganic iron reduces levels of MDM2, the ubiquitin ligase responsible for p53 degradation(51,104), which would be expected to increase levels of p53, and in fact did so in one report(104). The iron-mediated decrease in MDM2 was shown to occur by an IRP2-dependent mechanism, in which stabilization of MDM2 mRNA by IRP2 binding to its 3’ UTR was disrupted by excess iron(51). Whether iron increases or decreases p53 may depend on the form of iron (inorganic iron versus heme), the relative rates of these apparently competing pathways, or the particular cell types involved.
There is also evidence that p53 in turn regulates iron: p53 translationally increases ferritin, reducing the availability of intracellular iron during p53-mediated cell cycle arrest(105). The effects of p53 on ferritin may occur through direct transcriptional induction of ISCU2 (iron sulfur cluster assembly enzyme) by p53 (106). Upregulation of ISCU2 favors the formation of iron-sulfur clusters in IRP1, diminishing the activity of IRP1 as an RNA binding molecule (i.e. reducing its activity as a translational repressor of ferritin)(106).
NRF2.
Iron signaling in cancer is also influenced by NRF2, a transcription factor that drives the chemopreventive response but also exhibits paradoxical oncogenic activity(107,108). NRF2 is upregulated and associated with poor prognosis in several cancers, including non-small cell carcinoma, papillary renal cell carcinoma, papillary thyroid cancer, esophageal and serous ovarian cancer (109). Among its targets, NRF2 induces HMOX1 (heme oxygenase 1), an enzyme that catabolizes heme into carbon monoxide, biliverdin and ferrous iron. NRF2 also reduces sensitivity to ferroptosis by transcriptionally inducing ferritin(108), which sequesters intracellular iron and limits its ability to catalyze formation of lipid radicals.
H-RAS.
Early studies demonstrated that cell cycling and proliferation in cells expressing oncogenic H-RAS was dependent on labile iron and could be increased by diverting iron from intracellular iron storage in ferritin to the labile iron pool(110,111). Highlighting the exquisite dependence of cancer cells on maintaining iron balance, excess iron was later shown to induce cell death in ovarian cancer cells expressing mutant H-ras, at least in part via a MAPK signaling pathway(112). Parallels between effects of iron and those of oncogenic H-RAS have also been suggested by the observation that in combination with MYC, H-RAS induced genetic changes in ovarian cancer precursor cells similar to those induced by chronic iron exposure, including increased expression of EVI1 (ecotropic virus integration site 1), a transcription regulator located at 3q26.2, a locus frequently amplified in ovarian cancer(113). These intriguing but limited results require further mechanistic study.
Iron and cancer stem cells
Tumor-initiating cells (often termed cancer stem cells (CSCs)), constitute a small but biologically important fraction of tumor cells. Their properties include unlimited self-renewal, the ability to differentiate into cells that constitute the bulk of the tumor, mesenchymal characteristics, drug- and radiation-resistance, and the ability to seed metastases(114). Evidence from a variety of cancer types suggests that addiction to iron may be particularly evident in cancer stem cells. It should be noted that many studies use cells grown as spheroids (tumorspheres) as a surrogate for cancer stem cells due to technical challenges in isolating and propagating cancer stem cells; conclusions drawn may reflect the limitations of this experimental model.
Tumor-initiating cells and iron have been linked in many cancer types. RNA sequencing and enhancer mapping revealed that gliobastoma stem-like cells upregulate transferrin (TF), and preferentially require TFR1 and ferritin to form tumors in vivo(115). In these cells, increased TF-mediated influx of iron upregulated ferritin and activated STAT3 and FOXM1 signaling, fostering cell cycle progression. Similarly, proteomic profiling of CSCs derived from tumors of HER2/neu transgenic mice identified H-ferritin as upregulated in CSCs compared to non-cancer stem cells(116). In contrast, in SKOV3 ovarian cancer cells, silencing of H-ferritin promoted EMT and spheroid formation(117). Silencing H-ferritin similarly promoted EMT in breast cancer stem cells, possibly due to increased ROS (expected due to the decreased iron sequestration and increased labile iron in cells with lowered ferritin), or to mechanisms unrelated to the function of ferritin in iron storage, such as control of miRNA profile(118).
Cholangiocarcinoma stem-like cells similarly exhibited dysregulated iron metabolism, with a profile of iron retention possibly tied to low expression of ferroportin(119). Accordingly, expression of CSC markers was reduced by iron chelation. Iron was also found to induce CSC in human lung cancer cells by an unexpected mechanism involving induction of the transcription factor SOX9(120). In concert with SOX2, SOX9 is a key participant in orchestrating and maintaining the breast cancer stem cell state(121).
A consistent theme in studies of iron and cancer stem cells has been the sensitivity of CSCs to iron deprivation induced either by exposure to iron chelators or by ferroportin overexpression. The sensitivity of cancer stem cells to iron chelation has been observed in virtually every instance that it has been studied. For example our laboratory observed that a genetic model of ovarian cancer stem cells exhibited an iron-retention profile, with upregulation of TfR and downregulation of FPN relative to non-cancer stem cells(57). These cells exhibited enhanced sensitivity to iron chelation. In a CSC model derived from induced pluripotent stem cells, iron chelation suppressed proliferation and decreased expression of stemness markers such as NANOG and SOX2 in vitro and in vivo(122). Other examples exist(119,123,124).
Breast cancer tumor initiating cells exhibit increased labile iron; along with prostate cancer tumor initiating cells they also exhibit a unique iron gene expression signature that distinguishes them from bulk tumor cells(125). Of the 10 genes identified in this signature, 4 (TFRC, CYBRD1, EPAS1, HFE) overlapped with the 16 member IRGS (iron regulatory gene signature) that predicts prognosis in breast cancer patients(58). Although specific genes in these two signatures are not identical, they exhibit substantial functional overlap, with roles in iron uptake, Fe-S cluster formation, and the hypoxic response(125), suggesting parallels in the iron dependencies of bulk cancer cells and the tumor initiating cell population.
Three-dimensional tumor models and the tumor microenvironment.
It is now commonly accepted that the tumor microenvironment affects the behavior of cancer cells. The tumor microenvironment includes the extracellular matrix (ECM) as well as non-tumor cells, including immune cells (such as macrophages and T cells), fibroblasts, endothelial cells, and others(126). These cells not only supply iron to tumor cells, but depend on iron to maintain extracellular matrix stiffness, as well as influence macrophage polarization and T-cell promoted tumor cell death (Figure 1).
Iron and immune cells in the tumor microenvironment.
Monocytes can be polarized into classically activated, proinflammatory (M1) macrophages and alternatively activated (M2) macrophages, although this dichotomy is likely an oversimplification. The M1 macrophage phenotype is characterized by the production of high levels of pro-inflammatory cytokines, production of reactive nitrogen and oxygen intermediates, and promotion of Th1 responses. In contrast, M2 macrophages are characterized by efficient phagocytic activity and a role in tumor promotion(127). However, macrophage differentiation is plastic, and tumor-associated macrophage switching to an M1 phenotype has been proposed as an anti-cancer strategy(127).
M1 and M2 macrophages differ in their iron phenotype: M2 exhibit an “iron donor” phenotype with enhanced expression of the iron efflux pump ferroportin, and the ability to deliver iron to cancer cells(128,129). The in vivo relevance of this finding is supported by the analysis of human samples that demonstrate high levels of both ferroportin and iron in macrophages in breast tumor tissue(130). Iron-laden tumor-associated macrophages (TAMs) were observed in human breast cancer samples using FeMRI; depletion of these iron-laden cells was associated with tumor reduction following treatment with anti-colony stimulating factor 1 receptor (CSF1R) in murine tumor models, supporting the concept that macrophage iron can play a tumor supportive role(131).
Delivery of iron to tumor cells may be accomplished by ferroportin-mediated efflux from macrophages followed by TFR1-dependent uptake by tumor cells. Alternatively or additionally, lipocalin-2 secreted by macrophages may serve to deliver iron to breast and other cancer cells(132). Ferritin, an iron-containing protein secreted by macrophages(64), may also serve as a mechanism of iron delivery, as noted previously. Further, because ferritin inhibits cleavage of kininogen to HKa, an endogenous inhibitor of angiogenesis, ferritin secretion may also affect endothelial cells and promote angiogenesis in the tumor microenvironment(133,134).
Iron itself may modulate the interplay between immune and cancer cells in the tumor microenvironment by influencing macrophage polarization. For example, a high iron diet promoted M2 macrophage polarization in vivo and dampened the inflammatory response(135). However, opposite results were observed in a mouse macrophage cell line, where iron induced polarization to a proinflammatory (M1) phenotype by increasing ROS(136). In line with these observations, tumor-associated macrophages (which have an M2-like phenotype) exposed to hemolytic RBCs or iron-containing nanoparticles were converted to pro-inflammatory macrophages capable of killing lung cancer cells in vitro and in vivo(137). The shift to an M1 phenotype was frequently observed in hemorrhagic areas within tumors. Thus, macrophage polarization and consequent effects on tumor behavior may vary within subdomains of the tumor.
In addition to providing tumor cells with iron, immune cells in the tumor microenvironment may also modulate susceptibility to ferroptosis, an iron dependent form of cell death (see Iron and Ferroptosis section). CD8+ tumor-killing T cells activated by treatment with PD-L1, an immune checkpoint inhibitor, were observed to drive ferroptosis of ovarian or melanoma cancer cells by secreting interferon gamma, which downregulates SLC7A11, a component of an anti-ferroptotic pathway(138).
Iron and fibroblasts in the tumor microenvironment.
Our work with breast cancer spheroids demonstrated that fibroblasts up-regulate synthesis of hepcidin in breast cancer cells by secreting IL6(139). Since hepcidin triggers ferroportin degradation, this pathway increases intracellular iron in tumor cells.
Fibroblasts present in the tumor microenvironment also produce a collagenous matrix that stiffens the tumor stroma and creates a hypoxic environment that activates tumor cell invasion(140). Iron-dependent enzymes play a critical role in this process: matrix stiffening is mediated by collagen crosslinking, a process driven by lysyl hydroxylase LH2, an Fe2+- and 2-oxoglutarate-dependent oxygenase. Fe2+ is required not only for the catalytic activity but for the stability of prometastaic collagen lysyl hydroxylase(141).
Iron and oxidative damage
The ability of iron to redox cycle between Fe2+ and Fe3+ oxidation states is central to its function in catalyzing biological reactions(142). In this regard iron differs from most biologically important transition metals except copper (e.g. zinc, magnesium, manganese), which do not redox cycle under physiological conditions. However the redox property of iron also underlies its mutagenic potential, since it enables iron to participate in the Fenton reaction, in which iron transfers an electron to hydrogen peroxide to generate highly reactive radical species (the hydroxyl radical OH., and/or FeO2+ (143)). These radical species can directly damage DNA by producing oxidatively modified bases, abasic sites, and DNA strand breaks. Aberrant repair of such lesions leads to mutations. The ability of iron to directly damage DNA establishes a direct link between iron and cancer, a disease characterized by the accumulation of oncogenic and tumor suppressor mutations. Iron can also oxidatively damage lipids, a property whose significance in cancer has grown with the discovery of ferroptosis.
Iron and Ferroptosis
In 2012, Stockwell and colleagues coined the term ferroptosis to describe an iron-dependent form of cell death characterized by the accumulation of oxidized lipids in membranes(144–146). Ferroptosis was so named due to its iron dependence: iron chelators inhibit ferroptosis, and high levels of iron promote ferroptosis(144,147). (Iron has been implicated in other forms of cell death such as apoptosis and necrosis through its participation in the formation of ROS(148); however, iron is not an essential constituent of the mechanism of these cell death pathways, as it is in ferroptosis). Ferroptosis is fundamentally different from other regulated cell death pathways such as apoptosis, necrosis, autophagy, and pyroptosis (149), and is most similar to oxytosis, a form of cell death largely studied in neuronal cells that is characterized by the accumulation of reactive oxygen species (ROS)(150).
Because an essential feature of ferroptosis is the accumulation of peroxidized membrane phospholipids, small molecules that inactivate glutathione peroxidase 4 (GPX4), an enzyme that detoxifies oxidized lipids, also induce ferroptosis(151); changes in membrane lipid composition similarly modulate sensitivity ferroptosis(152,153). In addition to candidate small molecule drugs, approved chemotherapeutic drugs, such as cisplatin and sorafinib, induce ferroptosis as part of their mechanism of action(154). Notably, cancer stem cells appear particularly vulnerable to ferroptosis, at least in part due to the activity of YAP, which antagonizes Hippo signaling to upregulate expression of ferroptosis modulators such as TFR1 and ACSL4 (155). These observations have stimulated an ongoing search for ferroptosis inducers for cancer treatment.
Although the precise role of mitochondria in ferroptosis remains unclear, alterations in mitochondrial morphology and membrane potential are observed in ferroptosis, and iron management in this organelle may play a role in ferroptosis. For example, recent work suggests that NEET proteins may contribute to protection from ferroptotic cell death: mitoferrin-1 and mitoferrin-2 reduced mitochondrial lipid peroxidation and ferroptosis in cultured hepatocellular carcinoma cells(156) and head and neck cancer cells(157). Conversely, inhibition of NEET proteins sensitized cells to ferroptosis by increasing lipid ROS and disrupting mitochondrial iron. Similarly, mitochondrial ferritin, which inhibits tumor growth by sequestering mitochondrial iron and causing cytosolic iron depletion(158), protects against ferroptosis in cultured neuroblastoma cells(159).
The discovery of ferroptosis as an iron-dependent form of cell death coupled with its potential role in cancer therapy have led to two critical questions: (1) what is the role of iron in ferroptosis? (2) given that non-cancer cells also contain iron, can ferroptosis inducers be selectively cytotoxic to cancer cells?
The role of iron in ferroptosis.
Does the ability of iron to redox cycle in Fenton chemistry explain the contribution of iron to ferroptosis? Since the redox activity of iron renders it capable of oxidizing lipids, it is reasonable to posit that non-specific iron-catalyzed lipid peroxidation through Fenton chemistry may underlie the dependence of ferroptosis on iron. Although this presumption is widely supported by the literature, the picture may be more nuanced.
The critical role of lipid peroxides in precipitating ferroptosis is documented by multiple independent lines of evidence(153,160–162). Current debate focuses on whether ferroptosis is attributable to a specific lipid (in particular hydroperoxy derivatives of arachidonoyl phosphatidyl ethanolamine (HOO-AA-PE) or adrenoyl phosphatidylethanolamine), or rather reflects the accumulation of numerous different oxidized lipid species (see (160,163) for review). As recently noted(160), these possibilities imply two different roles for iron. If a specific lipid is key, then the major role of iron may be as a cofactor in the enzyme(s) that produces it. There is evidence for this, since LOX15 (product of the ALOX15 gene), a lipoxygenase implicated in ferroptosis that catalyzes the peroxidation of arachidonic acid(164) contains a non-heme mono iron center that is required for its enzymatic activity(165). In addition, fatty acid desaturases, which contribute to the formation of readily oxidizable PUFA-containing lipids, contain iron in a diiron center(166). On the other hand, genetic studies demonstrating the inability of ALOX15 knockout to protect mice from ferroptosis suggests that LOX15 is not absolutely required for ferroptosis(167). Further, the demonstration of multiple oxidized lipid species in cells undergoing ferroptosis(161) suggests a less specific mechanism – i.e., iron-catalyzed Fenton reaction-mediated lipid oxidation. To date, technical challenges in characterizing and quantifying temporal changes in oxidized lipid species have limited the ability to distinguish between these mechanisms, and it remains possible, and perhaps likely, that both are involved.
What intracellular source of iron is involved in ferroptosis? Cellular redox active iron -- generally referred to as the labile iron pool (LIP) -- is a small, metabolically available fraction of total intracellular iron that is the likely form of iron for pro-ferroptotic processes, either directly or indirectly. LIP is defined functionally by its ability to be bound by permeable low molecular weight iron chelators(168). The LIP is believed to exist in a Fe2+ oxidation state that is coordinated by small intracellular ligands (possibly glutathione(169,170), although debate as to the nature of the intracellular ligand(s) that coordinate “free” iron has raged for decades). As such, the LIP can participate directly in the generation of oxygen and lipid radicals through Fenton chemistry. Iron in the LIP is also available to PCBP1, a chaperone that delivers iron to client proteins such as ferritin, prolyl hydroxylases, and others(171,172). Notably, PCBP1 can transfer iron from the LIP to enzymes containing mono-iron centers such as that found in LOX15(171,172), supporting a connection between this pool of iron and ferroptotic processes.
Iron redistribution into and out of the labile iron pool profoundly affects ferroptosis. Early studies documenting the general dependence of ferroptosis on iron using iron chelators to limit overall iron availability(144) were subsequently supported by experiments that manipulated levels of intracellular iron using genetic rather than pharmacologic methods: overexpression of ferroportin, an iron efflux pump, sensitized ovarian cancer cells to ferroptosis(57), and knockdown of ferroportin accelerated erastin-induced ferroptosis in neuroblastoma(173). More recent work has provided additional mechanistic detail by demonstrating that ferritin repartitions iron among intracellular compartments to modulate iron’s ferroptotic activity. Specifically, liberation of iron from ferritin, an intracellular iron storage protein, is associated with ferroptosis: (1) treatment of breast cancer stem cells with salinomycin or a synthetic derivative ironomycin leads to lysosomal degradation ferritin, liberation of iron and ferroptosis(174); (2) inhibition of NCOA4-mediated ferritin degradation reduces sensitivity of cancer cells to ferroptosis (41); (3) elimination of ferritin from breast cancer cells by the prominin-2 dependent, MVB-mediated, pathway protects them from ferroptosis(66). Underlining the link between iron and ferroptosis, TFR1 emerged as a candidate marker to identify ferroptotic tissue in vivo (175).
Can ferroptosis inducers be selectively cytotoxic to cancer cells?
Ferroptosis is not restricted to cancer cells. For example, ferroptosis has been implicated in the pathophysiology of neurodegenerative diseases and acute brain injury(176). Further, all cells have iron stores and labile iron pools that can potentially contribute to ferroptosis. However, several factors weight the scales towards preferential cytotoxicity of ferroptosis inducers to cancer cells. First, because the levels of labile iron are dependent on iron uptake and efflux and the proteins that regulate these properties are often expressed in cancer cells in a way that increases labile iron, the pool of metabolically available iron is frequently higher in malignant cells than non-malignant cells of the same histologic type. Second, oxidative stress is enhanced in cancer cells relative to their non-malignant counterparts, rendering them less tolerant to the increased oxidative burden imposed by ferroptosis inducers. Third, reduced levels of lipid radical detoxifying enzymes in some cancers augment their susceptibility to ferroptosis-inducing agents(177).
Nevertheless, some caution will be required in deploying ferroptosis inducers for cancer treatment, since increased autophagy, particularly selective autophagy, can contribute to ferroptotic cell death(178). In one report, it was demonstrated that pancreatic cancer cells with mutant KRASs released KRAS in exosomes, and that this release was fostered by ferroptosis(179). Uptake of released exosomes by macrophages promoted macrophage polarization to the M2 phenotype and accelerated tumor growth, suggesting that in selected contexts, induction of ferroptosis may foster rather than inhibit tumor growth. Thus additional research will be required to identify specific genetic characteristics that render tumors potentially susceptible to ferroptosis inducers, as has been suggested more broadly regarding targeting autophagy in cancer(180).
Iron and Cancer Therapeutics
It is rather extraordinary that approaches to target cancer by modulating intracellular iron involve two diametrically opposed strategies: iron depletion, including chelation, on the one hand, and purposeful use of excess iron to generate toxic free radicals on the other. In yet a third strategy, iron transporters are used to deliver cytotoxic payloads preferentially to tumor cells. Below we provide a brief overview and some current examples of these strategies; more detailed information can be found in recent reviews(181,182).
Early studies aimed at targeting iron for cancer therapy took advantage of an existing, clinically approved iron chelator used to treat iron overload, desferoxamine. This highly effective and specific iron chelator is used to treat patients with thalassemia and other conditions of iron overload. In small numbers of patients with hepatocellular carcinoma and leukemia(183,184), modest therapeutic success was attained, encouraging the search for new iron chelators with improved activity. Some of the more successful anti-cancer iron chelators have been thiosemicarbazone derivatives, such as Dp44mT(185). Mechanistic studies revealed that this tridentate chelator not only binds intracellular iron but does so in a way that permits iron to redox cycle, generating ROS that contribute to tumor cell killing. Dp44mT has entered clinical trials(181).
Gallium salts also represent a variant of an iron depletion strategy for cancer therapeutics. Due to the chemical similarly between iron and gallium, gallium incorporates into proteins and enzymes that use iron as a cofactor(186), inactivating enzymes that require iron for their function, such as ribonucleotide reductase. The anti-cancer activity of gallium maltolate or tris(8-quinolinolato)gallium III (KP46), which may be more effective than the nitrate salt used in the original clinical formulation of gallium, is currently being explored(187).
In a different approach, apolactoferrin, an Fe3+-binding protein with bactericidal properties present in mammalian milk, inhibited the growth of squamous cell carcinoma cell lines in vitro, and inhibited the growth of head and neck tumors following oral administration in mice(188). Whether the therapeutic benefit of lactoferrin depends on its iron-binding activity remains uncertain(188); however, in patients with colorectal cancer receiving chemotherapy, oral lactoferrin improved a number of clinical parameters, although survival benefit was not evaluated(189).
The opposite approach to iron-based cancer therapeutics is to provide excess iron. This supplemental iron, coupled with the high levels of labile iron already present in tumor cells, can produce sufficient oxidative stress to eliminate tumor cells, which are oxidatively stressed at baseline(190). A recent study with ferumoxytol (Feraheme), a clinically approved iron oxide nanoparticle used to treat iron deficiency, suggested that this approach may hold promise in AML. In this study, Feraheme induced oxidative stress and reduced tumor burden in cells from patients and in a murine leukemia model(62,191).
A novel variation on this approach has been to use supra-physiological levels of ascorbate to trigger iron dependent oxidative stress, an approach proposed for patients with non-small cell lung cancer and glioblastoma (192) as well as other malignancies. This approach is currently being pursued in multiple clinical trials(193) (e.g. NCT03508726, NCT02905578, NCT02420314, NCT02344355, NCT02905591, NCT03602235, and NCT03799094 in clinicaltrials.gov).
The use of ferroptosis-inducing agents is based on a similar rationale of leveraging excess iron in cancer therapy. Early evidence suggests that ferroptosis inducers may be effective anti-cancer drugs, and that they target cancer stem cells to reduce not only tumor growth but metastasis. Studies in our laboratory using a model of ovarian cancer metastasis driven by cancer stem cells demonstrated that erastin, a ferroptosis-inducing agent, effectively reduced tumor number and mass in the peritoneal cavity, the major site of metastatic dissemination in ovarian cancer(57). In another example, salinomycin triggered a ferroptotic cell death pathway in cancer stem cells by causing lysosomal degradation of ferritin, the release of ferritin-bound iron, and ensuing liberation of iron into the cytosol(174). Pursuing this observation, inhibitors of DMT1 that preferentially targeted CSCs were developed to block the translocation of iron out of the lysosome, leading to lysosomal iron accumulation, production of ROS, and ferroptotic cell death(194).
Apart from using small molecules to directly target iron, other approaches that use overexpression of TFR1 in cancer cells either as a target or as a portal for the delivery of anti-cancer therapeutics have been explored. For example, chemotherapeutic drugs such as doxorubicin and cisplatin have been conjugated to transferrin to preferentially deliver these drugs to cancers. Further, cellular toxins such as diphtheria toxin and ricin, as well as anti-neoplastic nucleic acids(195), have been linked to transferrin to deliver the toxin preferentially to malignant tissues. Because toxicity to normal tissues was observed in early trials, recent attention has focused on the development of new antibodies that may improve the therapeutic index(196,197). In addition, conjugation to lactoferrin (LF) enhances dendrimer-mediated gene delivery to tumors in mice, suggesting that like TF LF may have a role in cancer targeting(198).
Conclusions, limitations, unanswered questions, and future directions
The last ten years have seen extraordinary developments in understanding the biology of iron homeostasis in normal cells and the pathobiology of iron metabolism in cancer. Nonetheless, some fundamental questions remain. Can iron-dependent enzymes and pathways be prioritized in terms of their necessity for cancer cell growth? Can the level at which iron switches from nutrient to oxidative toxicant be quantified? Is the level different in different cell types? Can the amount of iron required by tumors and normal tissues be quantified, and can this information facilitate the more effective deployment of iron-targeting agents in cancer? Answers to these questions have been hindered in part because metabolically available iron (the labile iron pool) is a small fraction of total cellular iron and remains difficult to quantify. Levels of labile iron in specific intracellular compartments are even more difficult to measure but may be of crucial importance. Developing robust quantitative methods to measure metabolically available iron in the cytosol and in organelles as well as developing organelle-targeted iron chelators will help to address these issues. It also appears that tumors can deploy multiple strategies to extract iron from their environment; thus, tumor profiling may be necessary to identify tumor-specific iron targets. Systems biology approaches also hold substantial promise in identifying critical nodal points in tumor iron metabolism (199–201).
Despite the converging evidence that implicates iron in cancer, it should be remembered that experimental systems have their limits. Notably, individual reports generally focus on one type of cancer; the generality of results across malignancies cannot be assumed without confirmation. Additionally, the specifics of experimental systems can shape results: for example, we observed that mechanisms of hepcidin regulation are different when breast cancer cells are cultured in three dimensional spheroids when compared to two dimensional plates(139). However, in general there has been remarkable concordance between experimental results and predictions derived from databases of human gene expression; such databases have had the added benefit of focusing enquiry in new and often unexpected directions, and will likely continue to serve as important resources for future discovery.
Overall, the fundamental concept that iron is a critical nutrient that is essential for cancer cell survival appears well established. The next years promise not only continued discovery, but the successful leveraging of cancer cell iron addiction into effective cancer therapies. There are abundant possibilities: bringing ferroptosis-inducing drugs to the clinic, fostering oxidative cytotoxicity in already oxidatively fragile cancer cells, turning the unique iron metabolic properties of cancer stem cells to therapeutic advantage, developing better targeted and more effective anti-cancer iron chelators, or using recent insights into the contributions of the tumor microenvironment to limit the growth and metastatic potential of cancer.
Acknowledgements:
Supported by NCI grants R01 CA233636 (F.M.T), R01 CA188025 (S.V.T).
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References
- 1.Torti SV, Torti FM. Ironing out cancer. Cancer Res 2011;71:1511–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer 2013;13:342–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and Cancer. Annu Rev Nutr 2018;38:97–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu Y, Dean AE, Horikoshi N, Heer C, Spitz DR, Gius D. Emerging evidence for targeting mitochondrial metabolic dysfunction in cancer therapy. J Clin Invest 2018;128:3682–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004;117:285–97 [DOI] [PubMed] [Google Scholar]
- 6.Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet 1980;2:390–2 [DOI] [PubMed] [Google Scholar]
- 7.Galbraith GM, Galbraith RM, Faulk WP. Transferrin binding by human lymphoblastoid cell lines and other transformed cells. Cell Immunol 1980;49:215–22 [DOI] [PubMed] [Google Scholar]
- 8.Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, et al. Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol 2010;6:602–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asimakopoulou A, Weiskirchen S, Weiskirchen R. Lipocalin 2 (LCN2) Expression in Hepatic Malfunction and Therapy. Front Physiol 2016;7:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res Treat 2008;108:389–97 [DOI] [PubMed] [Google Scholar]
- 11.Berger T, Cheung CC, Elia AJ, Mak TW. Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc Natl Acad Sci U S A 2010;107:2995–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tung MC, Hsieh SC, Yang SF, Cheng CW, Tsai RT, Wang SC, et al. Knockdown of lipocalin-2 suppresses the growth and invasion of prostate cancer cells. Prostate 2013;73:1281–90 [DOI] [PubMed] [Google Scholar]
- 13.Ding G, Wang J, Feng C, Jiang H, Xu J, Ding Q. Lipocalin 2 over-expression facilitates progress of castration-resistant prostate cancer via improving androgen receptor transcriptional activity. Oncotarget 2016;7:64309–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005;123:1293–305 [DOI] [PubMed] [Google Scholar]
- 15.Rehwald C, Schnetz M, Urbschat A, Mertens C, Meier JK, Bauer R, et al. The iron load of lipocalin-2 (LCN-2) defines its pro-tumour function in clear-cell renal cell carcinoma. Br J Cancer 2020;122:421–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chi Y, Remsik J, Kiseliovas V, Derderian C, Sener U, Alghader M, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science 2020;369:276–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung M, Oren B, Mora J, Mertens C, Dziumbla S, Popp R, et al. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal 2016;9:ra64. [DOI] [PubMed] [Google Scholar]
- 18.Oren B, Urosevic J, Mertens C, Mora J, Guiu M, Gomis RR, et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J Pathol 2016;239:274–85 [DOI] [PubMed] [Google Scholar]
- 19.Garrick MD, Dolan KG, Horbinski C, Ghio AJ, Higgins D, Porubcin M, et al. DMT1: a mammalian transporter for multiple metals. Biometals 2003;16:41–54 [DOI] [PubMed] [Google Scholar]
- 20.Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006;55:1449–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab 2016;24:447–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Senbanjo LT, Chellaiah MA. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front Cell Dev Biol 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller S, Sindikubwabo F, Cañeque T, Lafon A, Versini A, Lombard B, et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nature Chemistry 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller S, Sindikubwabo F, Cañeque T, Lafon A, Versini A, Lombard B, et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. bioRxiv 2019:693424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourseau-Guilmain E, Griveau A, Benoit JP, Garcion E. The importance of the stem cell marker prominin-1/CD133 in the uptake of transferrin and in iron metabolism in human colon cancer Caco-2 cells. PLoS One 2011;6:e25515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cross AJ, Leitzmann MF, Gail MH, Hollenbeck AR, Schatzkin A, Sinha R. A prospective study of red and processed meat intake in relation to cancer risk. PLoS Med 2007;4:e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loboda A, Jozkowicz A, Dulak J. HO-1/CO system in tumor growth, angiogenesis and metabolism - Targeting HO-1 as an anti-tumor therapy. Vascul Pharmacol 2015;74:11–22 [DOI] [PubMed] [Google Scholar]
- 28.Fiorito V, Chiabrando D, Petrillo S, Bertino F, Tolosano E. The Multifaceted Role of Heme in Cancer. Front Oncol 2019;9:1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knutson MD. Non-transferrin-bound iron transporters. Free Radic Biol Med 2019;133:101–11 [DOI] [PubMed] [Google Scholar]
- 30.Zhao N, Zhang AS, Wortham AM, Jue S, Knutson MD, Enns CA. The Tumor Suppressor, P53, Decreases the Metal Transporter, ZIP14. Nutrients 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knovich MA, Storey JA, Coffman LG, Torti SV, Torti FM. Ferritin for the clinician. Blood Rev 2009;23:95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Ferritin and the response to oxidative stress. Biochem J 2001;357:241–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol Cell Biol 2000;20:5818–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuji Y, Akebi N, Lam TK, Nakabeppu Y, Torti SV, Torti FM. FER-1, an enhancer of the ferritin H gene and a target of E1A-mediated transcriptional repression. Mol Cell Biol 1995;15:5152–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuji Y, Kwak E, Saika T, Torti SV, Torti FM. Preferential repression of the H subunit of ferritin by adenovirus E1A in NIH-3T3 mouse fibroblasts. J Biol Chem 1993;268:7270–5 [PubMed] [Google Scholar]
- 36.Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci U S A 1991;88:4946–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014;509:105–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 2014;16:1069–79 [DOI] [PubMed] [Google Scholar]
- 39.Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santana-Codina N, Mancias JD. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals (Basel) 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016;12:1425–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paratala BS, Chung JH, Williams CB, Yilmazel B, Petrosky W, Williams K, et al. RET rearrangements are actionable alterations in breast cancer. Nat Commun 2018;9:4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levinson S, Cagan RL. Drosophila Cancer Models Identify Functional Differences between Ret Fusions. Cell Rep 2016;16:3052–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, et al. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc Natl Acad Sci U S A 2013;110:14676–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev Cell 2018;46:441–55 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang R, Xie Y, Zeh HJ, Klionsky DJ, Tang D. Mitochondrial quality control mediated by PINK1 and PRKN: links to iron metabolism and tumor immunity. Autophagy 2019;15:172–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Katsarou A, Pantopoulos K. Basics and principles of cellular and systemic iron homeostasis. Mol Aspects Med 2020:100866. [DOI] [PubMed] [Google Scholar]
- 48.Maffettone C, Chen G, Drozdov I, Ouzounis C, Pantopoulos K. Tumorigenic properties of iron regulatory protein 2 (IRP2) mediated by its specific 73-amino acids insert. PLoS One 2010;5:e10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang W, Deng Z, Hatcher H, Miller LD, Di X, Tesfay L, et al. IRP2 regulates breast tumor growth. Cancer Res 2014;74:497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng Z, Manz DH, Torti SV, Torti FM. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget 2017;8:82231–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Kong X, Zhang Y, Sun W, Xu E, Chen X. Mdm2 is a target and mediator of IRP2 in cell growth control. FASEB J 2020;34:2301–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Feng X, Zhang J, Chen X. Iron Regulatory Protein 2 Exerts its Oncogenic Activities by Suppressing TAp63 Expression. Mol Cancer Res 2020;18:1039–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muto Y, Moroishi T, Ichihara K, Nishiyama M, Shimizu H, Eguchi H, et al. Disruption of FBXL5-mediated cellular iron homeostasis promotes liver carcinogenesis. J Exp Med 2019;216:950–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyazawa M, Bogdan AR, Tsuji Y. Perturbation of Iron Metabolism by Cisplatin through Inhibition of Iron Regulatory Protein 2. Cell Chem Biol 2019;26:85–97 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinnix ZK, Miller LD, Wang W, D’Agostino R Jr., Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med 2010;2:43ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res 2015;75:2254–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 2017;36:4089–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller LD, Coffman LG, Chou JW, Black MA, Bergh J, D’Agostino R Jr., et al. An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res 2011;71:6728–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Babu KR, Muckenthaler MU. miR-20a regulates expression of the iron exporter ferroportin in lung cancer. J Mol Med (Berl) 2016;94:347–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong Y, Hu L, Lu K, Wang Y, Xie Y, Gao L, et al. Ferroportin downregulation promotes cell proliferation by modulating the Nrf2-miR-17–5p axis in multiple myeloma. Cell Death Dis 2019;10:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu B, Zhi Q, Xie Q, Wu X, Gao Y, Chen X, et al. Reduced expression of ferroportin1 and ceruloplasmin predicts poor prognosis in adrenocortical carcinoma. J Trace Elem Med Biol 2019;56:52–9 [DOI] [PubMed] [Google Scholar]
- 62.Trujillo-Alonso V, Pratt EC, Zong H, Lara-Martinez A, Kaittanis C, Rabie MO, et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat Nanotechnol 2019;14:616–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang YF, Zhang J, Su Y, Shen YY, Jiang DX, Hou YY, et al. G9a regulates breast cancer growth by modulating iron homeostasis through the repression of ferroxidase hephaestin. Nat Commun 2017;8:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010;116:1574–84 [DOI] [PubMed] [Google Scholar]
- 65.Truman-Rosentsvit M, Berenbaum D, Spektor L, Cohen LA, Belizowsky-Moshe S, Lifshitz L, et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018;131:342–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell 2019;51:575–86 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fisher J, Devraj K, Ingram J, Slagle-Webb B, Madhankumar AB, Liu X, et al. Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol Cell Physiol 2007;293:C641–9 [DOI] [PubMed] [Google Scholar]
- 68.Han J, Seaman WE, Di X, Wang W, Willingham M, Torti FM, et al. Iron uptake mediated by binding of H-ferritin to the TIM-2 receptor in mouse cells. PLoS One 2011;6:e23800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L, Fang CJ, Ryan JC, Niemi EC, Lebron JA, Bjorkman PJ, et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc Natl Acad Sci U S A 2010;107:3505–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Todorich B, Zhang X, Slagle-Webb B, Seaman WE, Connor JR. Tim-2 is the receptor for H-ferritin on oligodendrocytes. J Neurochem 2008;107:1495–505 [DOI] [PubMed] [Google Scholar]
- 71.Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med 2005;202:955–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moussa RS, Kovacevic Z, Bae DH, Lane DJR, Richardson DR. Transcriptional regulation of the cyclin-dependent kinase inhibitor, p21(CIP1/WAF1), by the chelator, Dp44mT. Biochim Biophys Acta Gen Subj 2018;1862:761–74 [DOI] [PubMed] [Google Scholar]
- 73.Deng Z, Manz DH, Torti SV, Torti FM. Effects of Ferroportin-Mediated Iron Depletion in Cells Representative of Different Histological Subtypes of Prostate Cancer. Antioxid Redox Signal 2019;30:1043–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fuss JO, Tsai CL, Ishida JP, Tainer JA. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim Biophys Acta 2015;1853:1253–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abeysinghe RD, Greene BT, Haynes R, Willingham MC, Turner J, Planalp RP, et al. p53-independent apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Carcinogenesis 2001;22:1607–14 [DOI] [PubMed] [Google Scholar]
- 76.Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood 2004;104:1450–8 [DOI] [PubMed] [Google Scholar]
- 77.Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK, et al. N-myc downstream regulated gene 1 modulates Wnt-beta-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol Med 2012;4:93–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li P, Zheng X, Shou K, Niu Y, Jian C, Zhao Y, et al. The iron chelator Dp44mT suppresses osteosarcoma’s proliferation, invasion and migration: in vitro and in vivo. Am J Transl Res 2016;8:5370–85 [PMC free article] [PubMed] [Google Scholar]
- 79.Ding F, Zhang L, Chen H, Song H, Chen S, Xiao H. Enhancing the chemotherapeutic efficacy of platinum prodrug nanoparticles and inhibiting cancer metastasis by targeting iron homeostasis. Nanoscale Horiz 2020 [DOI] [PubMed] [Google Scholar]
- 80.Amano S, Kaino S, Shinoda S, Harima H, Matsumoto T, Fujisawa K, et al. Invasion inhibition in pancreatic cancer using the oral iron chelating agent deferasirox. BMC Cancer 2020;20:681. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Guo W, Zhang S, Chen Y, Zhang D, Yuan L, Cong H, et al. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim Biophys Sin (Shanghai) 2015;47:703–15 [DOI] [PubMed] [Google Scholar]
- 82.Le NT, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood 2004;104:2967–75 [DOI] [PubMed] [Google Scholar]
- 83.Nelson RL. Iron and colorectal cancer risk: human studies. Nutr Rev 2001;59:140–8 [DOI] [PubMed] [Google Scholar]
- 84.Brookes MJ, Boult J, Roberts K, Cooper BT, Hotchin NA, Matthews G, et al. A role for iron in Wnt signalling. Oncogene 2008;27:966–75 [DOI] [PubMed] [Google Scholar]
- 85.Song S, Christova T, Perusini S, Alizadeh S, Bao RY, Miller BW, et al. Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res 2011;71:7628–39 [DOI] [PubMed] [Google Scholar]
- 86.Coombs GS, Schmitt AA, Canning CA, Alok A, Low IC, Banerjee N, et al. Modulation of Wnt/beta-catenin signaling and proliferation by a ferrous iron chelator with therapeutic efficacy in genetically engineered mouse models of cancer. Oncogene 2012;31:213–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuang Y, Guo W, Ling J, Xu D, Liao Y, Zhao H, et al. Iron-dependent CDK1 activity promotes lung carcinogenesis via activation of the GP130/STAT3 signaling pathway. Cell Death Dis 2019;10:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang B, Zhang J, Song F, Tian M, Shi B, Jiang H, et al. EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett 2016;381:331–40 [DOI] [PubMed] [Google Scholar]
- 89.Park KC, Paluncic J, Kovacevic Z, Richardson DR. Pharmacological targeting and the diverse functions of the metastasis suppressor, NDRG1, in cancer. Free Radic Biol Med 2019 [DOI] [PubMed] [Google Scholar]
- 90.Menezes SV, Sahni S, Kovacevic Z, Richardson DR. Interplay of the iron-regulated metastasis suppressor NDRG1 with epidermal growth factor receptor (EGFR) and oncogenic signaling. J Biol Chem 2017;292:12772–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kaomongkolgit R, Cheepsunthorn P, Pavasant P, Sanchavanakit N. Iron increases MMP-9 expression through activation of AP-1 via ERK/Akt pathway in human head and neck squamous carcinoma cells. Oral Oncol 2008;44:587–94 [DOI] [PubMed] [Google Scholar]
- 92.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 2011;12:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee PJ, Jiang BH, Chin BY, Iyer NV, Alam J, Semenza GL, et al. Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J Biol Chem 1997;272:5375–81 [PubMed] [Google Scholar]
- 94.Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem 2000;275:21048–54 [DOI] [PubMed] [Google Scholar]
- 95.Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem 1999;274:24142–6 [DOI] [PubMed] [Google Scholar]
- 96.Anderson SA, Nizzi CP, Chang YI, Deck KM, Schmidt PJ, Galy B, et al. The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab 2013;17:282–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV. Serum ferritin: Past, present and future. Biochim Biophys Acta 2010;1800:760–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ruddell RG, Hoang-Le D, Barwood JM, Rutherford PS, Piva TJ, Watters DJ, et al. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 2009;49:887–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bertoli S, Paubelle E, Berard E, Saland E, Thomas X, Tavitian S, et al. Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur J Haematol 2019;102:131–42 [DOI] [PubMed] [Google Scholar]
- 100.Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 1999;283:676–9 [DOI] [PubMed] [Google Scholar]
- 101.O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol 2006;26:2373–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep 2014;7:180–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shen J, Sheng X, Chang Z, Wu Q, Xie D, Wang F, et al. The heme-p53 interaction: Linking iron metabolism to p53 signaling and tumorigenesis. Mol Cell Oncol 2016;3:e965642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dongiovanni P, Fracanzani AL, Cairo G, Megazzini CP, Gatti S, Rametta R, et al. Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am J Pathol 2010;176:1006–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J Biol Chem 2008;283:33911–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Funauchi Y, Tanikawa C, Yi Lo PH, Mori J, Daigo Y, Takano A, et al. Regulation of iron homeostasis by the p53-ISCU pathway. Sci Rep 2015;5:16497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilkinson Jt, Pietsch EC, Torti SV, Torti FM. Ferritin regulation by oxidants and chemopreventive xenobiotics. Adv Enzyme Regul 2003;43:135–51 [DOI] [PubMed] [Google Scholar]
- 108.Pietsch EC, Chan JY, Torti FM, Torti SV. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J Biol Chem 2003;278:2361–9 [DOI] [PubMed] [Google Scholar]
- 109.Kerins MJ, Ooi A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep 2018;8:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Repression of ferritin expression modulates cell responsiveness to H-ras-induced growth. Biochem Soc Trans 2002;30:777–80 [DOI] [PubMed] [Google Scholar]
- 111.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Ferritin expression modulates cell cycle dynamics and cell responsiveness to H-ras-induced growth via expansion of the labile iron pool. Biochem J 2002;363:431–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bauckman KA, Haller E, Flores I, Nanjundan M. Iron modulates cell survival in a Ras- and MAPK-dependent manner in ovarian cells. Cell Death Dis 2013;4:e592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rockfield S, Kee Y, Nanjundan M. Chronic iron exposure and c-Myc/H-ras-mediated transformation in fallopian tube cells alter the expression of EVI1, amplified at 3q26.2 in ovarian cancer. Oncogenesis 2019;8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol 2017;14:611–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS, et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015;28:441–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kanojia D, Zhou W, Zhang J, Jie C, Lo PK, Wang Q, et al. Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics 2012;12:3407–15 [DOI] [PubMed] [Google Scholar]
- 117.Lobello N, Biamonte F, Pisanu ME, Faniello MC, Jakopin Z, Chiarella E, et al. Ferritin heavy chain is a negative regulator of ovarian cancer stem cell expansion and epithelial to mesenchymal transition. Oncotarget 2016;7:62019–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aversa I, Zolea F, Ierano C, Bulotta S, Trotta AM, Faniello MC, et al. Epithelial-to-mesenchymal transition in FHC-silenced cells: the role of CXCR4/CXCL12 axis. J Exp Clin Cancer Res 2017;36:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Raggi C, Gammella E, Correnti M, Buratti P, Forti E, Andersen JB, et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci Rep 2017;7:17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chanvorachote P, Luanpitpong S. Iron induces cancer stem cells and aggressive phenotypes in human lung cancer cells. Am J Physiol Cell Physiol 2016;310:C728–39 [DOI] [PubMed] [Google Scholar]
- 121.Domenici G, Aurrekoetxea-Rodriguez I, Simoes BM, Rabano M, Lee SY, Millan JS, et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019;38:3151–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ninomiya T, Ohara T, Noma K, Katsura Y, Katsube R, Kashima H, et al. Iron depletion is a novel therapeutic strategy to target cancer stem cells. Oncotarget 2017;8:98405–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Recalcati S, Gammella E, Cairo G. Dysregulation of iron metabolism in cancer stem cells. Free Radic Biol Med 2019;133:216–20 [DOI] [PubMed] [Google Scholar]
- 124.Katsura Y, Ohara T, Noma K, Ninomiya T, Kashima H, Kato T, et al. A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rychtarcikova Z, Lettlova S, Tomkova V, Korenkova V, Langerova L, Simonova E, et al. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017;8:6376–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Laplagne C, Domagala M, Le Naour A, Quemerais C, Hamel D, Fournie JJ, et al. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mills CD, Lenz LL, Harris RA. A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res 2016;76:513–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Recalcati S, Gammella E, Cairo G. Ironing out Macrophage Immunometabolism. Pharmaceuticals (Basel) 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol 2010;40:824–35 [DOI] [PubMed] [Google Scholar]
- 130.Marques O, Porto G, Rema A, Faria F, Cruz Paula A, Gomez-Lazaro M, et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer 2016;16:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Leftin A, Ben-Chetrit N, Joyce JA, Koutcher JA. Imaging endogenous macrophage iron deposits reveals a metabolic biomarker of polarized tumor macrophage infiltration and response to CSF1R breast cancer immunotherapy. Sci Rep 2019;9:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Duan X, He K, Li J, Cheng M, Song H, Liu J, et al. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int J Physiol Pathophysiol Pharmacol 2018;10:105–14 [PMC free article] [PubMed] [Google Scholar]
- 133.Coffman LG, Parsonage D, D’Agostino R Jr., Torti FM, Torti SV. Regulatory effects of ferritin on angiogenesis. Proc Natl Acad Sci U S A 2009;106:570–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Coffman LG, Brown JC, Johnson DA, Parthasarathy N, D’Agostino RB Jr., Lively MO, et al. Cleavage of high-molecular-weight kininogen by elastase and tryptase is inhibited by ferritin. Am J Physiol Lung Cell Mol Physiol 2008;294:L505–15 [DOI] [PubMed] [Google Scholar]
- 135.Agoro R, Taleb M, Quesniaux VFJ, Mura C. Cell iron status influences macrophage polarization. PLoS One 2018;13:e0196921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med 2018;7:4012–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front Immunol 2017;8:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019;569:270–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P, et al. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 2018;37:4013–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401 [DOI] [PubMed] [Google Scholar]
- 141.Guo HF, Tsai CL, Terajima M, Tan X, Banerjee P, Miller MD, et al. Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe(2+)-binding. Nat Commun 2018;9:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.R. C. Iron Metabolism: from Molecular Mechanisms to Clinical Consequences. John Wiley and Sons; 2009. p 17–58. [Google Scholar]
- 143.Koppenol WH, Hider RH. Iron and redox cycling. Do’s and donťs. Free Radic Biol Med 2019;133:3–10 [DOI] [PubMed] [Google Scholar]
- 144.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hirschhorn T, Stockwell BR. The development of the concept of ferroptosis. Free Radic Biol Med 2019;133:130–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dixon SJ, Stockwell BR. The Hallmarks of Ferroptosis. Annual Review of Cancer Biology 2019;3:35–54 [Google Scholar]
- 147.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008;15:234–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Morris G, Walker AJ, Berk M, Maes M, Puri BK. Cell Death Pathways: a Novel Therapeutic Approach for Neuroscientists. Mol Neurobiol 2018;55:5767–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017;171:273–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lewerenz J, Ates G, Methner A, Conrad M, Maher P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front Neurosci 2018;12:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 2019;26:420–32 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res 2019;79:5355–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis 2020;11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019;572:402–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun 2016;478:838–44 [DOI] [PubMed] [Google Scholar]
- 157.Kim EH, Shin D, Lee J, Jung AR, Roh JL. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett 2018;432:180–90 [DOI] [PubMed] [Google Scholar]
- 158.Nie G, Chen G, Sheftel AD, Pantopoulos K, Ponka P. In vivo tumor growth is inhibited by cytosolic iron deprivation caused by the expression of mitochondrial ferritin. Blood 2006;108:2428–34 [DOI] [PubMed] [Google Scholar]
- 159.Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front Aging Neurosci 2016;8:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bayir H, Anthonymuthu TS, Tyurina YY, Patel SJ, Amoscato AA, Lamade AM, et al. Achieving Life through Death: Redox Biology of Lipid Peroxidation in Ferroptosis. Cell Chem Biol 2020;27:387–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Forcina GC, Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019;19:e1800311. [DOI] [PubMed] [Google Scholar]
- 163.Stoyanovsky DA, Tyurina YY, Shrivastava I, Bahar I, Tyurin VA, Protchenko O, et al. Iron catalysis of lipid peroxidation in ferroptosis: Regulated enzymatic or random free radical reaction? Free Radic Biol Med 2019;133:153–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017;171:628–41 e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boyington JC, Gaffney BJ, Amzel LM. The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science 1993;260:1482–6 [DOI] [PubMed] [Google Scholar]
- 166.Lee JM, Lee H, Kang S, Park WJ. Fatty Acid Desaturases, Polyunsaturated Fatty Acid Regulation, and Biotechnological Advances. Nutrients 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 2014;16:1180–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik I. Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem 1997;248:31–40 [DOI] [PubMed] [Google Scholar]
- 169.Hider RC, Kong XL. Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 2011;24:1179–87 [DOI] [PubMed] [Google Scholar]
- 170.Patel SJ, Frey AG, Palenchar DJ, Achar S, Bullough KZ, Vashisht A, et al. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat Chem Biol 2019;15:872–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Philpott CC, Jadhav S. The ins and outs of iron: Escorting iron through the mammalian cytosol. Free Radic Biol Med 2019;133:112–7 [DOI] [PubMed] [Google Scholar]
- 172.Philpott CC, Ryu MS, Frey A, Patel S. Cytosolic iron chaperones: Proteins delivering iron cofactors in the cytosol of mammalian cells. J Biol Chem 2017;292:12764–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci 2018;22:3826–36 [DOI] [PubMed] [Google Scholar]
- 174.Mai TT, Hamai A, Hienzsch A, Caneque T, Muller S, Wicinski J, et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem 2017;9:1025–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep 2020;30:3411–23 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Weiland A, Wang Y, Wu W, Lan X, Han X, Li Q, et al. Ferroptosis and Its Role in Diverse Brain Diseases. Mol Neurobiol 2019;56:4880–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem Biol 2020;27:420–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer 2017;17:528–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Merlot AM, Kalinowski DS, Kovacevic Z, Jansson PJ, Sahni S, Huang ML, et al. Exploiting Cancer Metal Metabolism using Anti-Cancer Metal- Binding Agents. Curr Med Chem 2019;26:302–22 [DOI] [PubMed] [Google Scholar]
- 182.Crielaard BJ, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov 2017;16:400–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Estrov Z, Tawa A, Wang XH, Dube ID, Sulh H, Cohen A, et al. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood 1987;69:757–61 [PubMed] [Google Scholar]
- 184.Yamasaki T, Terai S, Sakaida I. Deferoxamine for advanced hepatocellular carcinoma. N Engl J Med 2011;365:576–8 [DOI] [PubMed] [Google Scholar]
- 185.Noulsri E, Richardson DR, Lerdwana S, Fucharoen S, Yamagishi T, Kalinowski DS, et al. Antitumor activity and mechanism of action of the iron chelator, Dp44mT, against leukemic cells. Am J Hematol 2009;84:170–6 [DOI] [PubMed] [Google Scholar]
- 186.Chitambar CR. Gallium Complexes as Anticancer Drugs. Met Ions Life Sci 2018;18. [DOI] [PubMed] [Google Scholar]
- 187.Chitambar CR. The therapeutic potential of iron-targeting gallium compounds in human disease: From basic research to clinical application. Pharmacol Res 2017;115:56–64 [DOI] [PubMed] [Google Scholar]
- 188.Wolf JS, Li G, Varadhachary A, Petrak K, Schneyer M, Li D, et al. Oral lactoferrin results in T cell-dependent tumor inhibition of head and neck squamous cell carcinoma in vivo. Clin Cancer Res 2007;13:1601–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Moastafa TM, El-Sissy Ael D, El-Saeed GK, Koura MS. Study on the Therapeutic Benefit on Lactoferrin in Patients with Colorectal Cancer Receiving Chemotherapy. Int Sch Res Notices 2014;2014:184278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 2010;49:1603–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Torti SV, Torti FM. Winning the war with iron. Nat Nanotechnol 2019;14:499–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, et al. O2(-) and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017;32:268. [DOI] [PubMed] [Google Scholar]
- 193.Allen BG, Bodeker KL, Smith MC, Monga V, Sandhu S, Hohl R, et al. First-in-Human Phase I Clinical Trial of Pharmacologic Ascorbate Combined with Radiation and Temozolomide for Newly Diagnosed Glioblastoma. Clin Cancer Res 2019;25:6590–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Turcu AL, Versini A, Khene N, Gaillet C, Caneque T, Muller S, et al. DMT1 Inhibitors Kill Cancer Stem Cells by Blocking Lysosomal Iron Translocation. Chemistry 2020 [DOI] [PubMed] [Google Scholar]
- 195.Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta 2012;1820:291–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Daniels-Wells TR, Penichet ML. Transferrin receptor 1: a target for antibody-mediated cancer therapy. Immunotherapy 2016;8:991–4 [DOI] [PubMed] [Google Scholar]
- 197.Kim SS, Harford JB, Moghe M, Rait A, Pirollo KF, Chang EH. Targeted nanocomplex carrying siRNA against MALAT1 sensitizes glioblastoma to temozolomide. Nucleic Acids Res 2018;46:1424–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lim LY, Koh PY, Somani S, Al Robaian M, Karim R, Yean YL, et al. Tumor regression following intravenous administration of lactoferrin- and lactoferricin-bearing dendriplexes. Nanomedicine 2015;11:1445–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chifman J, Arat S, Deng Z, Lemler E, Pino JC, Harris LA, et al. Activated Oncogenic Pathway Modifies Iron Network in Breast Epithelial Cells: A Dynamic Modeling Perspective. PLoS Comput Biol 2017;13:e1005352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Konstorum A, Lynch ML, Torti SV, Torti FM, Laubenbacher RC. A Systems Biology Approach to Understanding the Pathophysiology of High-Grade Serous Ovarian Cancer: Focus on Iron and Fatty Acid Metabolism. OMICS 2018;22:502–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Konstorum A, Tesfay L, Paul BT, Torti FM, Laubenbacher RC, Torti SV. Systems biology of ferroptosis: A modeling approach. J Theor Biol 2020;493:110222. [DOI] [PMC free article] [PubMed] [Google Scholar]