

Abstract
Genome editing therapies represent a significant advancement in next-generation, precision medicine for the management of haematological diseases, and CRISPR/Cas9 has to date been the most successful implementation platform. From discovery in bacteria and archaea over 3 decades ago, through intensive basic research and pre-clinical development phases involving the modification of therapeutically relevant cell types, CRISPR/Cas9 genome editing is now being investigated in ongoing clinic trials. Despite the widespread enthusiasm brought by this new technology, significant challenges remain before genome editing can be routinely recommended and implemented in the clinic. These include risks of genotoxicity resulting from off-target DNA cleavage or chromosomal rearrangement, and suboptimal efficacy of homology-directed repair editing strategies, which thus limit therapeutic options. Practical hurdles such as high costs and inaccessibility to patients outside specialised centres must also be addressed. Future improvements in this rapidly developing field should circumvent current limitations with novel editing platforms and with the simplification of clinical protocols using in vivo delivery of editing reagents.
Keywords: CRISPR/Cas9, Genome editing, Haematopoietic stem cells, T lymphocytes, Clinical trial
Milestones of CRISPR/Cas9 research: from discovery to human therapeutics
Genome editing is now offering new hope for the cure of various congenital and acquired conditions through precise correction of aberrant genetic sequences, or by the site-specific replacement of defective genes. Biological applications of these technologies are expanding exponentially but are all based on the introduction of a double-stranded DNA break (DSB) at a given chromosomal locus by site-specific nucleases. Several genome editing platforms have been developed to date, including zinc finger nuclease (ZFN) and transcription activator-like nuclease (TALEN), as reviewed in (Porteus 2016), but the most widely employed is the Clustered Regularly Interspaced Short Palindromic Repeat (CRISPR)/ CRISPR-associated protein 9 (Cas9) platform. This RNA-guided endonuclease has received widespread public attention, probably due to its relative ease of use. By introducing a DNA DSB at a customised genetic location and enlisting the cellular repair machinery (described in further detail in the next section), CRISPR/Cas9 is a versatile tool that can be applied to a variety of biological contexts.
DNA sequences encoding this CRISPR system were first identified in several bacterial and archaeal species in the late 1980s-early 1990s (Ishino, et al 1987), and it is now evident that over 40% of bacteria and 90% of archaea contain genomic information for this system (Mojica, et al 2005) and reviewed in (Li and Peng 2019)). In these organisms, CRISPR functions as an element of the adaptive immune system against invading phages, but this system, and particularly type II CRISPR system that only requires one protein (Cas9) for the recognition and cleavage of the target site, was subsequently adapted for use in mammalian cells. In 2012, Doudna and Charpentier showed that CRISPR/Cas9 introduces DSBs in a target sequence (Gasiunas, et al 2012, Jinek, et al 2012), and a few months later, pioneering work demonstrated that CRISPR/Cas9 could be repurposed to gene-edit human cells (Cho, et al 2013, Cong, et al 2013, Mali, et al 2013a, Mali, et al 2013b) (Figure 1). In this context, the system was reconfigured and simplified by fusing the CRISPR RNA sequence (crRNA) with the transactivating crRNA (tracrRNA) into a single 100 nucleotide molecule, named single guide RNA (sgRNA), which complexes with the Cas9 protein and guides it to the target site.
Figure 1. Timeline describing the important chapters of CRISPR/Cas9 development and other editing platform development.
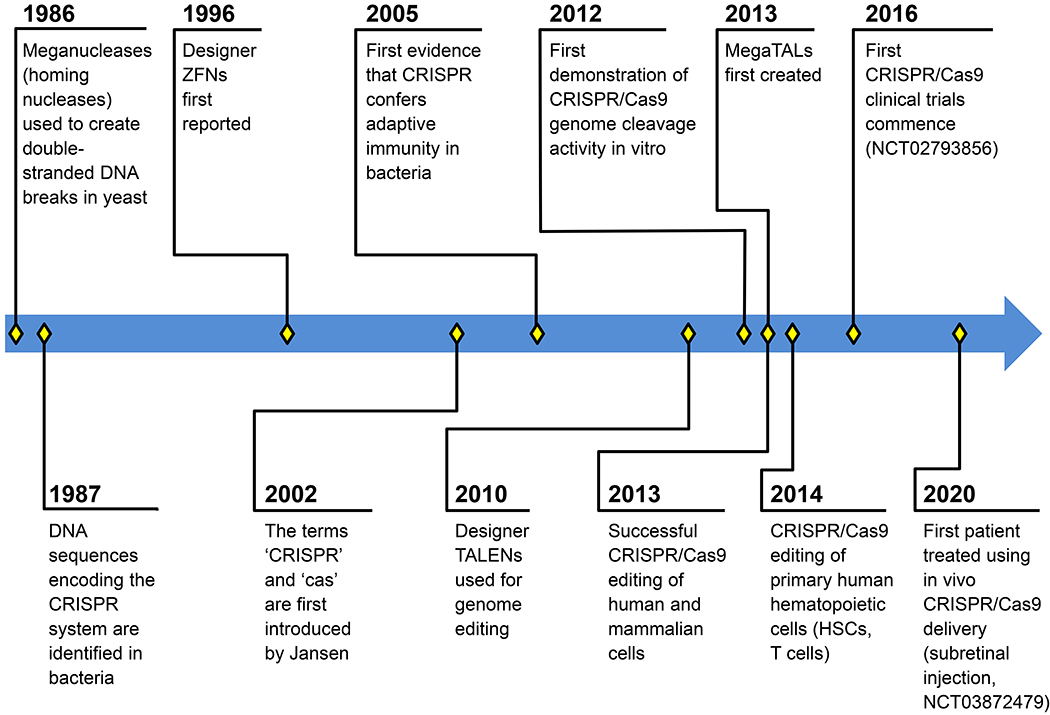
Notable publications associated with time events: 1986: (Kolodkin, et al 1986); 1987: (Ishino, et al 1987); 1996: (Kim, et al 1996); 2002: (Jansen, et al 2002); 2005: (Bolotin, et al 2005, Mojica, et al 2005, Pourcel, et al 2005); 2010: (Christian, et al 2010, Li, et al 2011); 2013: (Boissel, et al 2014, Cho, et al 2013, Cong, et al 2013, Mali, et al 2013a, Mali, et al 2013b); 2014: (Hendel, et al 2015, Mandal, et al 2014).
The ability of CRISPR/Cas9 to introduce a DSB in DNA at a given genomic site has opened the door to the manipulation of genes in a variety of ways, following repair of the lesion by the cellular repair machinery. The two major DNA repair pathways in mammalian cells are known as non-homologous end joining (NHEJ) or homologous recombination (commonly known as homology-directed repair, HDR) (Figure 2). Of note, a third repair pathway known as microhomology-mediated end-joining (MMEJ) also contributes to the generation of edited alleles after CRISPR/Cas9 treatment and shares components of both the NHEJ and HDR pathways (Bae, et al 2014). NHEJ is the default repair pathway in human cells and is an error-prone process because it involves the re-ligation of the two DNA ends with frequent nucleotide insertion or deletion (INDELS) at the cut site. This pathway is predominant in most cells types and can be harnessed to knock out a gene, disrupt a DNA regulatory motif (such as the binding site for a transcription factor), or create new splicing variants. One drawback of NHEJ is the production of an heterogenous population of edited cells: since a large variety of INDELS are generated, some may be undesirable for a given therapeutic application. The other major repair pathway, known as HDR, relies on a homologous donor DNA template, generally supplied along with the CRISPR/Cas9, to introduce more controlled genetic changes to the chromosomal target. This pathway involves different mechanisms that have been covered in detail elsewhere (Yeh, et al 2019) depending on whether a double- or single-stranded DNA is used, for example, with a single-stranded oligonucleotide (ssODN). HDR-editing allows for the introduction of a variety of changes in a target sequence, such as the substitution of one or multiple nucleotides, the insertion of novel coding information spanning several hundred base-pairs, or the deletion of genetic information. Adeno-associated virus (AAV) have generally been used for the delivery of large donor DNA templates (up to 4.4kbp) whereas donor ssODN have mainly been used for the introduction of small nucleotide variants within a target sequence. As mentioned previously, while HDR is attractive for its ability to encode precise edits, it is limited by low HDR/NHEJ ratios observed in most cells, particularly in long term engrafting haematopoietic stem cells.
Figure 2. Simplified mechanism of CRISPR/Cas9 genome editing.
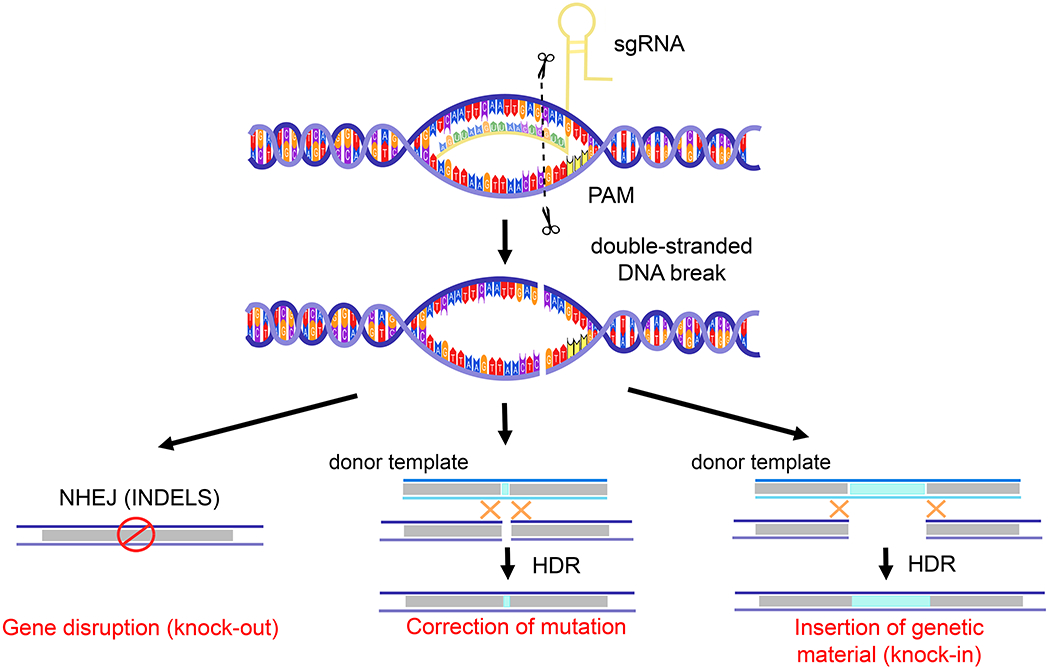
Single guide RNA (sgRNA)-mediated recognition of the chromosomal target sequence via the protospacer adjacent motif (PAM) sequence. A double-stranded cut occurs 3 nucleotides upstream of the PAM sequence and enlists the non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathways. INDELS= insertions and deletions.
Together, this plethora of genomic modifications enabled by CRISPR/Cas9 makes it a versatile tool for many valuable therapeutic applications. In this review, we particularly focus on haematological diseases, first by describing pre-clinical development, then by outlining active and completed clinical trials.
CRISPR/Cas9 genome editing of therapeutically relevant target cells
Two cell types with key roles in haematological disorders and which have therefore been prominent targets for CRISPR/Cas9-editing are human haematopoietic stem and progenitor cells (HSPCs) and T lymphocytes. Ideally, treatment should result in efficient, precise, non-toxic genetic alteration without perturbation of normal physiology of these cells. This is particularly important for HSPCs, which have self-renewal properties, multipotent differentiation potential (Cornu, et al 2017, Gonzalez-Romero, et al 2019), and for which the engineered footprint is expected to propagate across all blood lineages and persist for an entire lifetime. To fulfil these goals, several delivery vehicles for transient CRISPR/Cas9 expression have been tested and optimised, including plasmid DNA, RNA and ribonucleoprotein (RNPs), consisting of the Cas9 purified protein coupled with the sgRNA. RNPs have proven to be the preferred delivery method for ex vivo manipulation of HSPCs and T cells due to high editing efficiency and low toxicity (Bak, et al 2018a, Lattanzi, et al 2019), likely because they do not induce an innate immune response such as that seen with mRNA or double-stranded DNA (Cromer, et al 2018). The addition of chemical modifications such as 2’-O-methyl-3’phosphorothiate on the extremities of the sgRNA have further increased editing efficiency by enhancing stability after delivery to the target cells (Hendel, et al 2015). Importantly, both the Cas9 protein and the chemically modified sgRNA are commercially manufactured and can be purchased as “off-the-shelf” reagents with stringent release criteria to ensure consistency in quality across different lots and reproducibility in editing efficiency across experiments. Below, we describe therapeutic strategies that include the correction of disease-causing mutations in monogenic disorders, the introduction of beneficial mutations for malignant and non-malignant haematological disorders, and the inactivation of viral pathogens.
HSPC-based CRISPR/Cas9 therapeutic strategies
Therapeutic genome editing of HSPCs is still in its infancy, but these cells have been targeted ex vivo using retroviral vectors in gene therapy applications since the late 1980s. Despite early setbacks caused by insertional mutagenesis and oncogenesis of the provirus (Hacein-Bey-Abina, et al 2008), gene replacement therapies using optimised retroviral vectors are now being successfully applied in multiple clinical contexts that include β-haemoglobinopathies (Ribeil, et al 2017, Thompson, et al 2018), inherited bone marrow disorders such as Fanconi anemia (Rio, et al 2019) and primary immunodeficiencies (Mamcarz, et al 2019, Shaw, et al 2017). These vector-based approaches, however, are limited by the uncontrolled chromosomal integration of the therapeutic transgene, by the use of non-physiological (often suboptimal) promoters to drive expression of the transgene, and by challenges associated with large-scale, clinical-grade retroviral vector production. Genome editing can potentially offer a more precise and therefore safer alternative, and may be applicable to a wider array of diseases. Below, we focus on diseases for which HSPC CRISPR/Cas9 therapies have reached advanced stages following validation in preclinical experiments.
β-haemoglobinopathies are the most common monogenic disorder worldwide and affect the normal production of adult haemoglobin due to mutations in the β-globin gene (HBB). CRISPR/Cas9 editing approaches are being avidly investigated as a source of cure in these diseases. The two most common diseases are β-thalassaemia with low or absent β-globin production leading to dyserythropoiesis and severe anaemia, and sickle cell disease (SCD) with the production of a mutant form of β-globin causing polymerization of globin molecules and sickling of red blood cells in the circulation. Two main CRISPR/Cas9 strategies have been developed as attempts to treat these diseases: direct correction of the underlying mutation, or indirect correction of disease phenotype by reactivation of fetal haemoglobin (HbF). HbF has long been known to ameliorate clinical outcome in patients suffering from β-haemoglobinopathies from the study of individuals born with the benign condition known as hereditary persistence of fetal haemoglobin (HPFH) (Forget 1998).
In both approaches, HSPCs are the targets of editing to achieve long-term generation of modified erythrocytes that produce the therapeutic form of haemoglobin, thereby delivering a durable, one-time treatment that will last for an entire lifetime. In the first strategy, CRISPR/Cas9 is delivered along with a ssODN for the HDR-mediated correction of the SCD mutation (DeWitt, et al 2016, Pattabhi, et al 2019) or with an AAV encoding the entire HBB complementary DNA that serves as donor template for the site-specific replacement of the defective HBB gene (Dever, et al 2016). This universal approach is applicable to most patients’ mutations and uses the endogenous promoter to drive expression of the transgene (unlike vector-based gene therapy approaches). In comparison to these HDR-dependent strategies that are currently limited by low efficiency in long-term repopulating stem cells, CRISPR/Cas9-mediated HbF reactivation is more efficient in HSPCs since it is achieved by the prevalent NHEJ pathway. Proof of concept experiments have established that HbF reactivation can be accomplished either by inactivation of the gene encoding the HbF transcriptional repressor protein BCL11A, specifically by mutating an erythroid-enhancer motif located in the gene’s intronic sequence (Chang, et al 2017, Wu, et al 2019), or by disruption of its binding site located in the promoters of the gamma globin genes (Lux, et al 2019, Traxler, et al 2016, Ye, et al 2016). In the majority of studies, the humanised mouse transplantation model (reviewed in (Radtke, et al 2019)) was used to verify adequate engraftment and differentiation of engineered HSPCs. This model, however, is limited by the short-term monitoring of engraftment (< 6 months) and by the inability to measure haemoglobin output since peripheral erythroid differentiation is not supported in these animals. A recent mouse model generated by engineering a homozygous KitW41 allele was found to be more permissive to the production of human erythroid cells, but these are exclusively located in the bone marrow and are absent from peripheral blood (McIntosh, et al 2015). To circumvent these challenges, our group used the non-human primate transplantation model to document long-term (>2 years) engraftment of HSPCs edited by the NHEJ pathway to disrupt the BCL11A binding site, and peripheral blood HbF reactivation that correlated with editing achieved in vivo in nucleated cells (Humbert, et al 2019b). Together, these preclinical studies have enabled the initiation of several clinical trials that we discuss in the next section.
Genome editing is also actively being investigated as a novel treatment against human immunodeficiency virus (HIV). The description of a patient, known as the ‘Berlin Patient’, who was functionally cured of HIV following transplantation of allogeneic stem cells from a donor with a homozygous CCR5 allele containing a 32-bp deletion (CCR5Δ32) (Hutter, et al 2009), was a strong impetus for the investigation of novel HSPC editing strategies. More recently, HIV remission was reported in a second case known as the ‘London Patient’ who received a similar transplantation with CCR5Δ32 HSPCs but with a milder conditioning regimen (Gupta, et al 2019). CCR5 acts as a major coreceptor for HIV infection, and the generation of edited, CCR5-null, haematopoietic cells is expected to confer resistance to viral entry. Although initial work has aimed at specifically editing CD4+ T cells, being the major cell type targeted by the HIV virus, focus has recently switched toward the modification of HPSCs for a more durable response and a protection that also includes myeloid cells. Several studies have provided proof of concept results that NHEJ-mediated CRISPR/Cas9 inactivation of CCR5 in HSPCs does not impair engraftment in a mouse transplantation model and confers partial HIV resistance (Xu, et al 2017). These pre-clinical data served as a launching platform for testing in human patients as described in the next paragraph. While it is not entirely clear how much editing in nucleated cells will be required to achieve a complete cure, it is predicted that HIV infection will cause a gradual expansion in the number of edited and resistant cells (Perez, et al 2008, Peterson, et al 2018), due to their relative survival benefit, ultimately limiting the infection burden. Furthermore, this CCR5-editing approach may be combined with other editing strategies aimed at excising the integrated HIV proviral DNA from the host genome to eliminate the latent reservoir (Dash, et al 2019).
CRISPR/Cas9-editing has also been used as a powerful tool in cancer research, particularly for the rapid development of cancer immunotherapy. As we describe in the next paragraph, the vast majority of immunotherapy studies have focused on the modification of T-lymphocytes, for example, to express a chimeric antigen receptor (CAR) specific for a tumour antigen, but there are also some applications involving HSPC modification instead of T cells. CARs have been expressed in HSPCs to circumvent some of the limitations of CAR T cells, such as the short duration of the response or the exhaustion of the modified T cells (Larson and De Oliveira 2014). This strategy has so far employed retroviral vectors for the expression of the CAR, but current clinical CAR-T studies are using CRISPR/Cas9-editing to optimise CAR-T efficacy, and preclinical investigations into the use of CRISPR/Cas9-editing for CAR expression itself are ongoing.
Another approach using HSPCs aims to artificially create cancer-specific antigens by removing this same antigen from healthy cells to avoid toxicity. For example, specific treatment for myeloid malignancies, including acute myeloid leukaemia (AML), often focuses on CD33, a sialic acid binding receptor displayed on leukaemic cells but also on normal myeloid progenitors. For this reason, application of CD33-directed drugs such as the antibody-drug conjugate gemtuzumab ozogamicin (GO) results in significant on-target/off-leukaemia effects due to toxicity caused to healthy haematopoietic cells. Since allogeneic stem cell transplantation is already used to treat AML, we and others have demonstrated that CRISPR/Cas9 can safely remove CD33 from HSPCs and their derived progeny without altering engraftment capacity or biological function of these cells (Borot, et al 2019, Humbert, et al 2019a, Kim, et al 2018). Importantly, these studies demonstrated that haematopoietic cells generated from the edited and engrafted HSPCs were protected from the killing activity of GO, CD33/CD3 bispecific antibodies, or CD33 CAR-T cells, thus improving the outcome of AML therapies. This gene editing strategy also is likely to be applicable to other AML antigens such as CD123 or CD244 (Haubner, et al 2019) and to other forms of malignancies.
Beyond the diseases described here, HSPC editing has been applied to the treatment of other monogenic disorders such as Fanconi anemia, a hereditary defect of DNA repair in which causative mutations can be corrected by NHEJ-mediated repair of CRISPR/Cas9-induced DSBs (Roman-Rodriguez, et al 2019), or by using ZFN and the HDR pathway (Diez, et al 2017). X-linked severe combined immunodeficiency (SCID-X1) was also treated with a CRISPR/AAV-based strategy for targeted integration of a cDNA to functionally correct patient-derived HSPCs with as much as 45% efficiency (Pavel-Dinu, et al 2019). Similar to that described above for haemoglobinopathies, this strategy provides universal correction of all haematopoietic cell lineages by targeting the HSPC population, and allows for expression of the corrected gene under the control of the endogenous promoter. These are some examples for the most advanced functions using HSPC-directed CRISPR/Cas9-editing, but the repertoire of applications is expected to expand exponentially in the coming years.
T-cell based CRISPR/Cas9 therapeutic strategies
Beyond HSPCs, T cells have also been the target for genome editing mainly for the treatment of malignant conditions, by way of optimizing CAR-T cell efficacy, redirecting the T cell receptor (TCR), or by removing immune checkpoint inhibitors. Beyond the general advantages of genome editing over vector-based gene transfer that we highlighted earlier, it has become apparent that the site of CAR integration highly influences anti-tumour potency of the CAR-T cells, and this can be directed towards sites offering maximal efficacy with the use of genome editing strategies. CARs are engineered receptors consisting of an extracellular domain that recognises the antigen and an intracellular signal-activating domain. As an attempt to improve CAR-T cell performance, the Sadelain group used CRISPR/Cas9 and the HDR repair pathway to generate a uniform population of CD19-specific CAR T cells, in which the CAR transgene was precisely inserted at the TCR alpha locus (TRAC) (Eyquem, et al 2017). These T-cells showed CAR expression under the control of the native TCR promoter, and exhibited reduced exhaustion and increased activity in a mouse model of acute lymphoblastic leukaemia. Similarly, CRISPR/Cas9-mediated deletion and knockout of the endogenous TCR has been employed to facilitate subsequent TCR redirection by delivery of a specifically targeted TCR addition by retroviral vector. T cells modified with this approach exhibited greater tumour antigen sensitivity in a mouse model as compared to cells modified by standard TCR gene transfer, because the native TCR was no longer competing with the newly inserted receptor (Legut, et al 2018).
Beyond the use of autologous T cells that have to be tailored to individual patients, and that are costly and time consuming to produce at therapeutic scale, there is significant benefit to the availability of universal allogeneic T cells that can serve as “off-the-shelf” ready-to-use agents (reviewed in (Graham, et al 2018)). The ability to multiplex CRISPR/Cas9 gene editing through the usage of several sgRNAs simultaneously in the same cell has been harnessed to overcome immunological incompatibility by inactivation of the endogenous TCR and beta 2 microglobulin gene (B2M, a component of MHC class I molecules, Figure 3) (Ren, et al 2017). Further investigation will determine whether expression of HLA-E in these cells is required to avoid NK cell-mediated lysis (Gornalusse, et al 2017). In turn, these cells can be modified further to express a CAR for anti-tumour activity and enhanced by the simultaneous knockout of immune checkpoint inhibitors such as programmed cell death protein (PD-1) (Ren, et al 2017). T cells can also be further engineered by inactivating CD52, a protein targeted by the monoclonal antibody alemtuzumab, to promote engraftment of modified cells through lymphodepletion/ immunosuppression (Poirot, et al 2015). Despite the great potential of engineering T cells by multiplex gene editing, caution must be taken to avoid the generation of chromosomal translocations and off-target mutations that can increase risk for oncogenic transformation. As we describe in the next section, these different flavours of engineered T cells are currently being tested in patients with various malignancies.
Figure 3. Schematic of engineering of universal T cells by CRISPR/Cas9-knockout of different receptor molecules or by knockin of a CAR using lentiviral vector or CRISPR/Cas9-knockin.
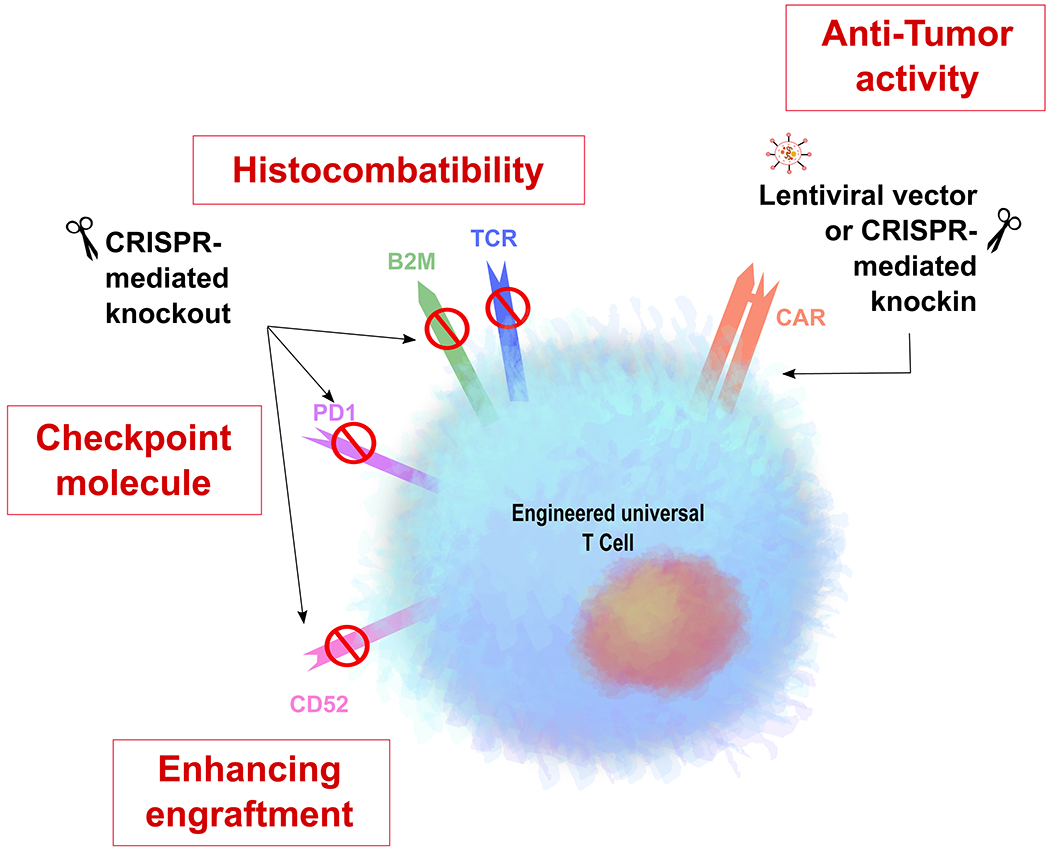
B2M= Beta 2 microglobulin; CD52=cluster of differentiation 52; CAR=chimeric antigen receptor; PD1= Programmed cell death protein 1; TCR= T cell receptor.
CRISPR/Cas as diagnostic and discovery tool for new targets in haematological malignancies
CRISPR/Cas9 editing is also a valuable tool in the discovery and validation of new therapeutic targets in haematological malignancies such as leukaemia, lymphoma and myeloma. Genome-wide CRISPR/Cas9 screens can be undertaken in malignant cell lines, to identify points at which disruption modulates growth or survival of the malignant clone, and have been described in detail elsewhere (Luo 2016). Postulated genes can be specifically interrogated in subsequent experiments to identify potential therapeutic targets such as FOXP1 in diffuse large B-cell lymphoma, DOT1L in multiple myeloma, MCL-1 in mantle cell lymphoma or several molecular effectors required for the regulation of the checkpoint molecule PD-1 in anaplastic lymphoma kinase-positive anaplastic large-cell lymphoma (Dafflon, et al 2020, Dengler, et al 2020, Felce, et al 2020, Zhang, et al 2019).
Modulators of cancer sensitivity to standard therapies can also be investigated, to provide insights into sensitivity and resistance mechanisms. For example, a CRISPR library screen in a Hodgkin lymphoma cell line has revealed two ubiquitin-editing enzymes as key regulators of sensitivity to brentuximab-vedotin (Wei, et al 2020).
CRISPR/Cas9 knockout provides more durable depletion than short interfering RNA applications, which have been previously used in such settings. This permits longer-term tracking of the effects of targeted knockouts, including in vivo effects following transplantation of edited cancer cell lines into mice. In situations where DSB-mediated gene knockout are not adequate, other options include loss-of-function through transcriptional repression (CRISPRi) or gain-of-function through transcriptional activation (CRISPRa) (Gilbert, et al 2014). In case of CRISPRi, a catalytically dead Cas9 (dCas9) protein is fused to a repressor domain such as Krüppel-associated box (KRAB) protein to silence genes by epigenetic mechanisms. In contrast, CRISPRa consists of a dCas9 fused to one or several activator domains such as the general control protein (GCN4) or virus protein 16 (VP16).
The CRISPR/Cas platform has also been harnessed as a sensitive tool for the detection of nucleic acid at very low concentration and with single-nucleotide specificity. This technology, named SHERLOCK (Gootenberg, et al 2017) (Specific High-Sensitivity Enzymatic Reporter UnLOCKing) is based on the promiscuous ribonuclease activity documented in some Cas proteins (not Cas9) upon DNA target recognition, which serves to activate a reporter and emit a signal upon detection of the target. This diagnostic tool has obvious applications in infectious diseases for the detection and quantification of viral and bacterial pathogens, but it has also proven useful in the detection of cancer-associated mutations from circulating cell-free DNA such as in non-small cell lung cancer (Gootenberg, et al 2018) or mutations in the EGFR or BRAF genes with allelic fractions as low as 0.1% (Gootenberg, et al 2017). We can even envision disease contexts where SHERLOCK will be used both for diagnosis and therapy using the Cas RNA editing/knockdown capability of this system.
Testing of CRISPR/Cas9-based therapies in clinical trials
Over 40 interventional trials of genome editing are currently registered with the United States (Clinicaltrials.gov) or European Union (EU Clinical Trials Register, EudraCT) clinical trials registries, targeting a multitude of benign or malignant and inherited or acquired conditions (summarized in Table I). The first trials involving ex vivo editing of T lymphocytes or HSPCs commenced in 2009, while trials investigating the direct in vivo delivery of editing agents were introduced later, since 2016. ZFNs and TALENs were used initially as the editing platform, but CRISPR/Cas9 has recently become the most common technology employed. For completeness, we describe clinical trials utilising all editing platforms and specify the type of nuclease used for each study (Table I).
Table I.
Summary of currently registered clinical trials
| Condition | Delivery and target cells | Genome editing platform(s) and target gene | Phase | Age | Dates (estimated study completion dates given where not complete) | Trial registration number (ClinicalTrials.gov or EU Clinical Trials Register) |
|---|---|---|---|---|---|---|
| Completed/status unknown | ||||||
| HIV | Ex vivo: autologous T-cells | ZFN: NHEJ editing of CCR5 | 1, 1/2 | Adult | Jan 2009-Jan 2013 | NCT00842634 |
| Dec 2009-Dec 2014 | NCT01044654 | |||||
| Dec 2011-Jul 2017 | NCT01543152 | |||||
| Aug 2014-Jun 2018 | NCT02225665 | |||||
| Apr 2015-Mar 2019 | NCT02388594 | |||||
| Oesophageal cancer | Ex vivo: autologous T-cells: PD-1 knockout | CRISPR/Cas9: NHEJ editing of PD-1 | 1 | Adult | Mar 2017-Feb 2018 |
NCT03081715 |
| Human papillomavirus (HPV) related; cervical Intraepithelial; neoplasia | In vivo: Topical gel / pessary targeting cervical epithelium | TALEN, CRISPR/Cas9, ZFN: NHEJ editing at HPV E6 / E7 | 1 | Adult | Dec 2017-July 2017 | NCT02800369 |
| Jan 2018-Jan 2019 | NCT03057912 | |||||
| Jan 2018-Jan 2019 | NCT03226470 | |||||
| Blastic plasmacytoid dendritic cell neoplasm | Ex vivo: autologous CD123 CAR T-cells | TALEN: NHEJ deletion of TCR, disruption of CD52 gene | 1 | Adult | Jun 2017-Jun 2019 |
NCT03203369 |
| Currently active | ||||||
| Sickle cell disease | Ex vivo: autologous HSPCs | CRISPR/Cas9, ZFN: NHEJ disruption of BCL11A enhancer |
1/2 | Adult | Nov 2018-May 2022 | NCT03745287 |
| Jun 2019-Apr 2023 | NCT03653247 | |||||
| Transfusion-dependent β-thalassaemia | Ex vivo: autologous HSPCs | CRISPR/Cas9, ZFN: NHEJ disruption of BCL11A enhancer |
1/2 | Adult | Mar 2018-Dec 2022 |
NCT03432364 |
| Sept 2018-May 2022 | NCT03655678 | |||||
| Sickle cell disease and transfusion-dependent β-thalassaemia |
(Long-term follow-up study in subjects who received CTX001 on studies NCT03655678 or NCT03745287) | Feb 2021-Sept 2039 | NCT04208529 | |||
| HIV | Ex vivo: autologous/allogeneic HSPCs | CRISPR/Cas9, ZFN: NHEJ disruption of CCR5 | 1 | Adult | Jul 2015-Apr 2022 |
NCT02500849 |
| May 2017-May 2021 |
NCT03164135 | |||||
| Non-small cell lung cancer; Epstein-Barr virus-associated malignancies | Ex vivo: autologous T cells | CRISPR/Cas9: NHEJ disruption of PD-1 | 1/2 | Adult | Aug 2016-Jan 2020 | NCT02793856 |
| Apr 2017-Mar 2022 | NCT03044743 | |||||
| Multiple myeloma; malignant melanoma, synovial sarcoma, myxoid / round cell liposarcoma | Ex vivo: autologous T cells | CRISPR/Cas9: NHEJ disruption of endogenous TCR and PD-1 | 1 | Adult | Sept 2018-Jan 2033 | NCT03399448 |
| B cell acute lymphoblastic leukaemia or lymphoma; non-Hodgkin lymphoma; T cell acute lymphoblastic leukaemia or lymphoma; T cell lymphoma; multiple myeloma; acute myeloid leukaemia; mesothelin-positive solid tumours; HIV | Ex vivo: autologous / allogeneic T-cells: CAR-T cells | CRISPR/Cas9, ZFN or TALEN: NHEJ disruption of multiple targets including endogenous PD-1, HPK1, CD52 | 1, 1/2 | Paediatric, adult | Jun 2016-July 2020 |
NCT02808442 |
| Aug 2016-July 2020 | NCT02746952 | |||||
| Jun 2017-May 2022 | NCT03166878 | |||||
| Jun 2017-Jun 2021 | NCT03190278 | |||||
| Jan 2018-May 2022 | NCT03398967 | |||||
| Jun 2018-Jun 2020 | NCT03545815 | |||||
| Nov 2018-May 2020 | NCT03747965 | |||||
| Jul 2019-Dec 2025 | NCT03617198 | |||||
| Jul 2019-Aug 2026 | NCT04035434 | |||||
| Aug 2019-Aug 2024 | NCT04037566 | |||||
| Oct 2019-Oct 2021 | NCT04150497 | |||||
| Nov 2019-Nov 2022 | NCT04142619 | |||||
| Mar 2020-May 2038 | NCT03690011 | |||||
| May 2016- | 2015-004293-15 | |||||
| May 2016- | 2016-000296-24 | |||||
| Information unavailable | 2019-003462-40 | |||||
| Multiple myeloma | Ex vivo: allogeneic T-cells: | CRISPR/Cas9 | 1 | Adult | Jan 2020-Jan 2027 | NCT04244656 |
| Mucopolysaccharidosis (I, II) | In vivo: Intravenous infusion targeting hepatocytes | AAV-delivered ZFN: HDR insertion of IDS or IDUA genes |
1/2 | Paediatric, adult | May 2017-Jan 2022 |
NCT02702115 |
| May 2017-Feb 2022 | NCT03041324 | |||||
| Severe haemophilia B | In vivo: Intravenous infusion targeting hepatocytes | AAV-delivered ZFN: HDR editing | 1 | Paediatric, adult | Nov 2016-Jan 2021 | NCT02695160 |
| Leber congenital amaurosis type 10 | In vivo: Subretinal injection | AAV-delivered CRISPR/Cas9: NHEJ to restore activity of CEP290 gene |
1/2 | Paediatric, adult | Sept 2019-Mar 2024 | NCT03872479 |
| Transfusion-dependent β-thalassaemia | Ex vivo: autologous HSPCs | CRISPR/Cas9: HDR correction of HBB | 1 | Paediatric, adult | Jan 2019-Jan 2021 | NCT03728322 |
Ex vivo editing of autologous HSPCs sourced from bone marrow or mobilised peripheral blood are the subjects of NHEJ editing for the treatment of β-haemoglobinopathies and HIV. Targeting of the BCL11A enhancer region with the aim of reversing the fetal-to-adult haemoglobin switch and increasing HbF levels is being investigated in adults for safety, tolerability and efficacy by CRISPR therapeutics and Vertex Pharmaceuticals (CTX001), and by Sanofi and Sangamo Therapeutics (BIVV003, ST400) as a treatment for both sickle cell disease and transfusion-dependent β-thalassaemia. Early results from the CTX001 study suggest successful amelioration of disease in first patients, but longer-term follow-up is required for more definitive results.
ZFNs were used for the generation of CCR5-knockout autologous CD4+ T cells, which were subsequently re-administered to HIV-positive patients in multiple clinical trials (Sangamo Therapeutics, University of Pennsylvania and National Institute of Allergy and Infectious Diseases). Interestingly, one study reported a significant increase in circulating T cells post-treatment, consistent with protection from HIV infection (Tebas, et al 2014). Medium-term persistence of CCR5-modified cells post-infusion was reported in this study, and, since then, other investigators have considered the use of repeated administration of similar cells to provide longer-term efficacy (NCT02225665, Sangamo Therapeutics). The application of HSPCs as an alternate editing target has recently become a more favoured approach to the maintenance of a long-lasting HIV-resistant population. Disruption of the CCR5 locus in autologous or allogeneic HSPCs prior to transplantation has therefore been studied as a method to confer HIV resistance to the mature progeny of these modified stem cells. Results from trial NCT03164135 were recently reported for one HIV patient suffering from acute lymphoblastic leukaemia who received donor stem cells from an HLA-compatible donor (Xu, et al 2019). CCR5-edited HSPCs stably engrafted, as represented by 5 to 8% editing frequencies detected in bone marrow and blood at 19 months post-transplant, spanning multiple lineages. Importantly, no adverse effects or off-target activity related to gene editing was reported. While the efficiency of post-transplantation editing in this patient was not adequate to achieve a cure for HIV infection, these data confirm that CCR5-edited cells can stably engraft and persist long-term without complication.
The most advanced clinical trials testing gene edited T cells as cancer immunotherapeutic agents used TALENs to knock out the endogenous TCR and CD52 genes, and a lentiviral vector that expressed CD19-directed CAR to treat B-cell acute lymphoblastic leukaemia (Qasim, et al 2017). This initial study lead to the launch of two further clinical trials investigating the use of the CD19-directed CAR-T cells in both children (NCT02808442) and adults (NCT02746952). In 2018 two separate CD19-directed autologous CAR-T products (tisagenlecleucel or “Kymriah” from Novartis and axicabtagene ciloleucel or “Yescarta” from Kite Pharma) were granted a license by the EMA and FDA to treat aggressive B-cell malignancies in specific patient populations. The CD19 CAR Lisocabtagene maraleucel developed and tested by Juno (now Bristol-Myers Squibb) for the treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL) is currently undergoing priority review by FDA (Abramson, et al 2019).
A similar editing approach is also being tested by Cellectis for AML (NCT03190278) and blastic plasmacytoid dendritic cell neoplasms (NCT03203369) using CD123-modified CAR-T cells. Clinical trials investigating CAR-T cell engineering currently involve lentiviral transduction of the CAR transgene, and adjuvant strategies based on gene editing are expected to improve CAR-T specificity, efficacy and longevity. Examples include increasing activity by PD-1 or HPK1 gene knock-out, or disruption of CD52 to confer alemtuzumab resistance on the modified CAR-T cells.
PD-1 checkpoint inhibitor expression has been disrupted ex vivo in autologous T cells prior to expansion and reinfusion, with the aim of increasing the anti-cancer potency of edited cells, a strategy recently trialed for the treatment of oesophageal (NCT02800369, NCT03081715), non-small cell lung and EBV-driven malignancies (NCT02793856, NCT03044743). CRISPR/Cas9 knock-out of the endogenous TCR has been employed to facilitate subsequent T-cell redirection by delivering specifically targeted TCR additions. Cancers targeted in trials of such redirected T cells include multiple myeloma and numerous solid tumours (NCT03399448). Results from three patients treated in this trial demonstrated safety and feasibility of engineering T cells with multiplex CRISPR/Cas9 to remove both the endogenous TCR and the checkpoint molecule PD-1, and with a lentiviral vector expressing a specific TCR to redirect these cells to target the cancer-testis antigens NY-ESO-1 and LAGE-1 (Stadtmauer, et al 2020). As was hypothesised, edited T cells showed a much more durable response as compared to results from other trials in which T cells retaining endogenous TCR and PD-1 expression were employed. However, the efficacy of these engineered T cells was ambiguous as clinical progression occurred post treatment in all patients. Residual tumour present after T cell infusion showed a reduction in target antigen expression, demonstrating a possible mechanism for tumour evasion. While minimal off-target CRISPR/Cas9 activity was documented in engineered cells, chromosomal translocations were observed at a frequency of less than 1% (discussed further below).
Considerable efforts are being made to improve the accessibility and reduce the cost of these gene editing therapies. This can be accomplished in principle by direct administration of the editing reagents to patients, bypassing the need to modify cells ex vivo. Intravenous delivery of AAV-packaged ZFN editing reagents to enlist the HDR repair pathway and deliver a functional copy of the therapeutic gene at the albumin locus is currently being investigated by Sangamo Therapeutics for the treatment of several genetic disorders. These include treatment of severe haemophilia B by delivery of the factor IX gene (NCT02695160) and mucopolysaccharidosis types I and II by targeted insertion of a corrected α-L-iduronidase (NCT02702115) or iduronate 2-Sulfatase gene (NCT03041324), respectively, into hepatocytes. Direct delivery of CRISPR/Cas9 editing reagents into the eye is also being studied for the control of visual loss caused by one subtype of Leber congenital amaurosis. This study, known as EDIT-101 (Editas Medicine and Allergan), is designed to remove an aberrant splicing site caused by a point mutation in the CEP290 gene, which results in production of a dysfunctional CEP290 protein. The AAV5 vector employed in EDIT-101 contains two guide RNA molecules and the Cas9 protein, and aims to remove the aberrant splice site created by the mutation by using the NHEJ pathway (Maeder, et al 2019). Treatment of the first patient in that study was announced in March 2020.
While many trials are still in early phase 1 stages, others include progression to phase 2 studies, and therefore robust data on therapeutic efficacy as well as safety is expected to emerge. It is encouraging to note that many investigators recognise the importance of long-term follow-up, with ongoing monitoring of up to 15 years integrated into the treatment protocol of some studies and a separate long-term follow-up study now registered for β-haemoglobinopathy patients who have received CTX001 on trial (NCT04208529).
Challenges and limitations to therapeutic CRISPR/Cas9 genome editing
Significant challenges to the clinical application of CRISPR/Cas9 therapies still remain to be overcome. These include genotoxicity caused both by the editing procedure and cytotoxic conditioning regimens often administered alongside; subtherapeutic levels of edited cells persisting post-treatment; and notable practical limitations such as cost and feasibility issues. It remains to be seen how effectively these will be tackled in current clinical trials, and it will be some time before data are mature enough to answer questions regarding long-term efficacy and toxicities.
Potential harms of therapeutic genome editing
There are still issues of safety as well as efficacy that must be addressed before routine clinical application of CRISPR/Cas9 editing strategies can be recommended (Demirci, et al 2018). Potential harms include off-target DNA cleavage at sites similar to the intended nuclease target, and chromosomal rearrangement events. Off-target activity may, for example, result in silencing of tumour suppressor genes or introduce gain-of-function mutations affecting oncogenes (Baylis 2018, Romero, et al 2019, Schaefer, et al 2017). Genomic rearrangements such as translocations and large deletions may result from on- or off-target DSBs and may have similar effects (Bak, et al 2018b). Even though these events are expected to occur at very low frequency, they will be a major concern for clinical translation if they induce oncogenic transformation of targeted cells (Gonzalez-Romero, et al 2019, Jeong, et al 2019). This is particularly a concern where a multiplexing CRISPR/Cas9 editing approach is applied. Autologous T cells engineered at multiple loci for the treatment of advanced myeloma or sarcoma showed multiple translocations events that were not only detectable in the infusion product but also persisted in vivo for up to 170 days after infusion in all three treated patients. Some reassurance was inferred from a decline in frequency of these events over time suggesting that they did not confer a selective growth advantage, but the outcome may be different with alternative multiplex targets or off-target DSBs (Stadtmauer, et al 2020).
These risks are especially concerning where HSPCs are the intended target of genome editing, given their high proliferation and differentiation capacity (Cornu, et al 2017, Gonzalez-Romero, et al 2019). While oncogenic risk is predicted to be significantly lower than with gene transfer therapy delivered by integrating lentiviral vectors (Braun, et al 2014, Howe, et al 2008), this possibility remains a realistic concern. Given that many of the currently active genome editing trials target non-malignant conditions such as haemoglobinopathies, even a small increase in risk of malignancy must be weighed carefully. The development of better tools to predict such adverse genetic events, and of more sensitive and standardised assays to detect them, still requires more work (Cornu, et al 2017). Unbiased, genome-wide tools allowing the long-term biological consequences of such events to be predicted are also needed (Tsai and Joung 2016).
Protocols for ex vivo modification of HSPCs followed by autologous re-transplantation still require the application of cytotoxic conditioning regimens. These are associated with acute complications such as myelosuppression and organ dysfunction, and long-term risk of secondary malignancies (Baker, et al 2003, Lidonnici and Ferrari 2018), thus amplifying risks for oncogenesis in such treatment protocols. An important challenge for clinical translation of gene therapy or genome editing approaches will be to use reduced-intensity conditioning regimens to 19tilizin toxicity without compromising engraftment, such as non-genotoxic antibody-drug conjugates (ADCs).
As investigators increasingly focus on in vivo administration of CRISPR reagents, the potential for unintended editing of the germline must be ethically considered. The possibility that gametes may be affected during in vivo genome editing is being investigated in current trials, although the consequences of such editing on any future progeny are largely unknown. Following the recent CRISPR/Cas9 editing of human embryos that were subsequently implanted and allowed to progress to delivery, the genome editing community and regulatory bodies have united in condemning any form of editing that would directly affect the germline. Substantially more research is needed to determine potential side-effects of germline editing including ethical and societal implications before this could be acceptable (Cyranoski and Ledford 2018).
Together, there must be careful consideration of the risk versus benefit balance of each CRISPR/Cas9 editing therapy for each patient. The risks of the disease itself and the toxicities of other potential treatments must be weighed against the known and unknown disadvantages associated with genome editing therapies.
Subtherapeutic levels of edited cells in vivo
Achieving and sustaining adequate numbers of edited cells in vivo to confer therapeutic benefit remains a major limitation to the clinical application of this technology to many diseases. This is particularly pertinent where HDR editing is applied to HSPCs, where a rapid fall-off is observed after transplantation and engraftment in many preclinical studies, even when respectable editing efficiency was initially obtained in infused cells (Dever, et al 2016, Genovese, et al 2014, Gomez-Ospina, et al 2019, Pattabhi, et al 2019). In comparison, HDR rates reported in T cells to insert a CAR within the TRAC locus were more encouraging. Very recently, αβ T cells treated with CRISPR/Cas9 editing and an AAV donor template resulted in >75% expression of the CAR along with >90% TCR loss (Wiebking, et al 2020). These unprecedented rates of targeted integration of large gene cassettes bode well for improved HDR efficiencies in other T-cell settings but additional optimization will be required before such impressive results are seen in long term engrafting stem cells. Although the clinical trial of CRISPR/Cas9 editing of HSPCs for ablating CCR5 expression as a treatment for HIV showed promising results in one patient (NCT03164135, (Xu, et al 2019)), the numbers of modified cells achieved post-transplant were sub-therapeutic, illustrating the ongoing challenges in this area. Stem cells with long term repopulating potential are one of the most challenging cell types to modify in this regard, due at least in part to their quiescent nature and therefore minimally active intracellular HDR gene-repair pathways. Apoptosis or cell cycle arrest mediated by p53 induction occurs in the majority of edited HSPCs, providing, at least in part, an explanation for their resistance to editing and high rates of cell death (Haapaniemi, et al 2018, Ihry, et al 2018). HDR-editing with an added AAV donor template resulted in a cumulative effect on p53 pathway activation but, interestingly, the defect seen in colony forming potential and engraftment of treated cells could be improved by transient p53 inhibition (Schiroli, et al 2019).
Pre-existing immunity to genome editing agents or delivery vectors and immunogenicity of the treatment itself has the potential to render in vivo treatment ineffective or trigger adverse reactions. Pre-existing anti-Cas9 antibodies and antigen-specific T cells are present in a significant proportion of the population, presumably stimulated by prior exposure to Cas9-producing bacteria (Charlesworth, et al 2019, Simhadri, et al 2018). The development of adverse immune responses to novel antigens presented by CRISPR/Cas9-edited cells must thus also be considered (Baylis 2018, Kang, et al 2018).
Practical limitations
Beyond the issues of efficacy and genetic risk, there are notable practical challenges involved in the delivery of CRISPR editing therapies to patients who may benefit from them. Access to the target cells for editing, costs, and feasibility of delivery are also potentially constraining factors. Accessing adequate numbers of HSPCs for ex vivo modification is not straightforward. Cells must either be harvested directly from the bone marrow in an invasive and painful procedure (Ho 2019, Siddiq, et al 2009), or mobilised into the peripheral blood prior to collection by apheresis. While the latter is less invasive, failure to collect adequate HSPCs is not uncommon (Duong, et al 2014), and mobilization is particularly difficult in patients with sickle cell disease where G-CSF administration risks precipitating life-threatening vaso-occlusive crises (Fitzhugh, et al 2009). Achieving access to tissue stem cells for CRISPR editing, such as in muscular dystrophies or cystic fibrosis, is even more challenging (Azvolinsky 2019).
The expenses involved in developing and delivering CRISPR genome editing therapies impose further limitations on this field. There is a degree of mismatch between the long-term investment required for the successful development of genome editing treatments and the appetite for immediate results and short-term rewards often demonstrated by industry investors (Sheridan 2018). Even post-development, the high costs of the genome editing procedure, and the necessary clinical interventions required to deliver such therapies, are likely to make access to this new genre of next-generation treatment unattainable for the vast majority of patients who may otherwise stand to benefit from them (Wilson and Carroll 2019).
In developed countries there must be open dialogue between patient and carer groups, health care leaders and the public, regarding the resource allocation implications of bringing genome editing therapies into the clinic. In the developing world there are even more hurdles to be overcome. Cell processing and manufacture of genome editing products must be to Good Manufacturing Practice (GMP) standards and in approved institutions. The clinical facilities required to deliver such treatments are also unavailable to most of the global population. The majority of patients with sickle cell disease, for example, are situated in Sub-Saharan Africa, where only a tiny minority have access to a modified autologous haematopoietic stem cell transplant procedure. Research efforts must focus on ensuring that genome editing therapies are deliverable to the patients who need them most, to avoid further exacerbating health inequalities between those of different socioeconomic circumstances (Baylis 2018, Wilson and Carroll 2019).
Finally, regulation of this rapidly progressive clinical field is a major challenge facing regulatory authorities and governments, as the speed of scientific progress, including broadening of the potential applications for such technology, outstrips the ability of many such bodies to keep pace. Genome editing therapies are being investigated for a multitude of diseases and cell types, utilising numerous editing strategies. A ‘one size fits all’ approach to the assessment of efficacy or safety of these applications is not realistic nor, therefore, to their regulation. Follow-up post-treatment is a particularly important area to be addressed. Lifelong follow-up to detect adverse events would be ideal; however, in many cases the resource and logistical implications would be prohibitive, and there is currently no consensus amongst governing bodies as to the required length of surveillance (Abou-El-Enein, et al 2017). Laws and regulations will inevitably reflect ongoing debate regarding social and ethical consequences of genome editing as well as biological effect, adding further complexity to this area (Abou-El-Enein, et al 2017, Baylis and McLeod 2017).
The future of CRISPR genome editing in the clinic
Upcoming developments predicted to facilitate further expansion and success of CRISPR/Cas9 editing therapies include methods by which levels of edited cells may be increased in vivo to more reliably allow for therapeutic benefit and strategies to further improve the safety profile of such treatments. In vivo delivery of therapeutic CRISPR reagents is likely to overtake ex vivo cellular modification strategies in the future, at least for some applications, and is already in use for the treatment of Leber congenital amaurosis type 10 (Table I), but this modality is not yet being applied to HSPC or T-lymphocyte modification. The potential applications for CRISPR editing are already expanding rapidly, and as the safety and efficacy of these technologies improves, this growth is expected to continue.
Improving in vivo editing efficiency
The challenge of achieving therapeutic levels of edited cells post-transplantation may be tackled in various ways. Other than increasing editing efficiency at the point of initial application, providing an engraftment or survival advantage to edited cells or enabling positive selection of cells harbouring the desired genetic modification would be expected to increase the proportion of modified cells post treatment. In vivo selection for modified cells may happen naturally for some disorders such as SCID-X1 or Fanconi Anemia where edited/corrected cells have a survival advantages over uncorrected cells, or it may have to be engineered, for example to increase resistance to cytotoxic drugs (Nagree, et al 2015).
In vivo positive selection of CRISPR/Cas9 edited cells is an attractive possibility, and one that is already under investigation in pre-clinical studies. An example of such an approach is the introduction of a chemoresistance gene cassette to edited cells, conferring resistance to a subsequently-applied chemotherapeutic agent and thereby resulting in positive selection pressure that favours the edited cells (Beard, et al 2010, Falahati, et al 2012, Paul, et al 2018, Wang, et al 2019). While further study is required to develop the use of non-genotoxic selection agents, these reports provide proof of concept that such an approach may allow for in vivo enhancement of subtherapeutic editing levels.
Other positive selection strategies to increase the proportion of CRISPR/Cas9 edited cells while still ex vivo are under investigation, by conferring a survival advantage to modified cells or via an alternative selection method such as flow cytometric purification of marked edited cells (Agudelo, et al 2017, Dever, et al 2016). Another method to improve in vivo editing levels where HSPCs are the targets of modification is to improve engraftment and survival capacity of edited cells post transplantation, thereby giving them a competitive advantage. One example of this strategy is the enhancement of CXCR4 expression on edited HSPCs, which has been shown to improve homing to and engraftment in the bone marrow niche (Arai, et al 2018, Brenner, et al 2004, Kahn, et al 2004).
Due to the requirement of CRISPR/Cas9 systems for a short sequence motif adjacent to target sites known as protospacer adjacent motif (PAM), a significant portion of the genome has not been amenable to editing. Recent efforts focused on the generation of “PAMless” Cas9 variants, which widely expanded the therapeutic applicability of this nuclease platform to chromosomal regions that were previously inaccessible to editing (Walton, et al 2020). These variants were generally found to have comparable activity to the original Cas9 enzyme in human cell lines but specificity remains to be evaluated in T cells and HSPCs.
Safety
There are many proposed methods for reducing genotoxic risk in CRISPR/Cas9 editing. The most established strategies under investigation are the use of bioinformatic prediction and design tools to produce editing compounds less likely to induce on- or off-target adverse effects (reviewed in (Liu, et al 2020)) and higher fidelity Cas9 preparations to reduce off-target mutagenesis (Chen, et al 2019, Kleinstiver, et al 2019, Vakulskas, et al 2018). Targeting integration of new genetic material by directing the HDR machinery to ‘safe harbour’ loci, where the risks of DNA DSBs or undesired INDELS would not affect functional DNA sequences, is another way by which the safety of CRISPR/Cas9 editing can be improved (Gomez-Ospina, et al 2019, Hong, et al 2017, Papapetrou, et al 2011, Yada, et al 2017). Anti-CRISPR/Cas proteins known as “Acr” could also be used as a tunable mechanism to limit off-target editing. Experiments conducted in CD34+ HSPCs showed feasibility of this approach when CRISPR/Cas9 was delivered as RNPs (Shin, et al 2017) or via adenoviral vectors (Li, et al 2018). It is encouraging to note that no off-target edits were reported in the liver of mice after in vivo delivery of CRISPR/Cas9 targeting the PCSK9 gene (Akcakaya, et al 2018), demonstrating that this system can be highly specific for carefully tested sgRNA sequences, particularly for in vivo genome editing applications.
Novel editing platforms recently reported employing modified CRISPR/Cas9 coupled to deaminase domains to catalyze precise A-to-G or C-to-T changes in a target sequence without the requirement for dsDNA breaks. These so-called base editors allow for the substitution of a single nucleotide for an alternate one to correct a disease-causing mutation or to disable function of a gene through the introduction of a stop codon or the disruption of conserved splicing sites (Gaudelli, et al 2017, Komor, et al 2016). The versatility of possible genetic alterations introduced at a given target was also recently enhanced with a new platform known as prime editors, which consist of CRISPR/Cas9 linked to a reverse-transcriptase. Interestingly, these enzymes can introduce novel genetic material into chromosomal DNA without the need for DNA DSBs and bypassing the HDR pathway; this method decreases genotoxicity, although this risk is not entirely eliminated (Anzalone, et al 2019, Gaudelli, et al 2017, Komor, et al 2016). Importantly, base editors have already been validated in both T cells (Webber, et al 2019) and in HSPCs (Zeng, et al 2020), even in the context of multiplex genome editing.
Finally, current protocols for modified autologous HSPC transplantation all require administration of genotoxic conditioning chemotherapy. A further improvement to the safety of such procedures will be the introduction of non-genotoxic targeted conditioning regimens such as ADCs. These antibodies can specifically target stem cells in the BM niche, and recent studies using anti-CD117 ADCs have demonstrated feasibility of this approach (Kwon, et al 2019, Li, et al 2019, Palchaudhuri, et al 2016, Srikanthan, et al 2020).
Improving cost and accessibility of CRISPR/Cas9 genome editing therapies
The majority of currently active CRISPR/Cas9 clinical trials rely on ex vivo editing procedures, but the first in vivo editing trials are already underway. Alternative methods of packaging and conveying CRISPR reagents, such as non-viral gold or lipid nanoparticle delivery, may improve options for in vivo delivery, and potentially also reduce the cytotoxicity and immunogenicity of such treatments (Kulkarni, et al 2018, Shahbazi, et al 2019).
In vivo delivery of CRISPR/Cas9 editing therapies have significant advantages in such that the ex vivo cellular processing and modification steps are avoided. This is likely to reduce both risk and cost for many patients, as the procedures associated with cell collection and re-transplantation are circumvented. In vivo delivery of CRISPR/Cas9 therapies is also expected to facilitate application in regions where GMP and transplantation facilities are unavailable.
Another method by which treatment costs may be reduced is by improving the accuracy with which true target cells are selected for editing. Where HSPCs are the target of CRISPR/Cas9 editing, it is possible to select a population of cells enriched for true, long-term repopulating haematopoietic stem cells based on the immunophenotype CD34+/CD90+/CD45RA-, reducing the number of cells submitted to CRISPR editing and, therefore, associated reagent costs by up to 90%, while still providing adequate long-term engraftment and differentiation post-modified transplant in preclinical studies (Humbert, et al 2019b, Radtke, et al 2017). A parallel strategy may be possible for other cell types.
In conclusion, the application of CRISPR/Cas9 editing to the cure of human disease is already delivering promising results, and its breadth and efficacy of usage is expected to continue advancing. While challenges facing the clinical application of genome editing therapies are recognised, many strategies to overcome each of these are already in development. It may in the end be practical issues of cost and delivery that are the last remaining limitations to the true potential of this technology to combat human disease.
Acknowledgements
We thank Helen Crawford for help with manuscript and figure preparation and Margaret Cui for generating the figures. This work was supported by grants from the National Heart, Lung, and Blood Institute (R01 HL136135 and R01 HL147324), from the National Institute of Allergy and Infectious Diseases (R01 AI135953-01, U19 AI096111 and UM1 AI126623), NIH, Bethesda, MD, and by charitable support to C.S. from the British Society for Haematology, the UK Thalassaemia Society and Sheffield Hospitals Charity. H.-P.K. is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Fred Hutch Endowed Chair for Cell and Gene Therapy.
Footnotes
Conflicts of interest
H.-P.K is consulting for Rocket Pharma, Homology Medicines, CSL Behring, Vor Biopharma, and Magenta Therapeutics. Other authors have no competing interests.
References
- Abou-El-Enein M, Cathomen T, Ivics Z, June CH, Renner M, Schneider CK & Bauer G (2017) Human genome editing in the clinic: New challenges in regulatory benefit-risk assessment. Cell Stem Cell, 21, 427–430. [DOI] [PubMed] [Google Scholar]
- Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang ML, Arnason JE, Mehta A, Purev E, Maloney DG, Andreadis C, Sehgal AR, Solomon SR, Ghosh N, Albertson T, Garcia J, Kostic A, Li D, Kim Y & Siddiqi T (2019) Pivotal safety and efficacy results from Transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas (Abstract). Blood, 134, 241–241 (doi: 10.1182/blood-2019-127508). [DOI] [Google Scholar]
- Agudelo D, Duringer A, Bozoyan L, Huard CC, Carter S, Loehr J, Synodinou D, Drouin M, Salsman J, Dellaire G, Laganiere J & Doyon Y (2017) Marker-free coselection for CRISPR-driven genome editing in human cells. Nature Methods, 14, 615–620. [DOI] [PubMed] [Google Scholar]
- Akcakaya P, Bobbin ML, Guo JA, Malagon-Lopez J, Clement K, Garcia SP, Fellows MD, Porritt MJ, Firth MA, Carreras A, Baccega T, Seeliger F, Bjursell M, Tsai SQ, Nguyen NT, Nitsch R, Mayr LM, Pinello L, Bohlooly YM, Aryee MJ, Maresca M & Joung JK (2018) In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature, 561, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A & Liu DR (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature, 576, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Choi U, Corsino CI, Koontz SM, Tajima M, Sweeney CL, Black MA, Feldman SA, Dinauer MC & Malech HL (2018) Myeloid conditioning with c-kit targeted CAR-T cells enables donor stem cell engraftment. Molecular Therapy, 26, 1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azvolinsky A (2019) Molecular scissors cut in on stem cells. Nature Medicine, 25, 864–866. [DOI] [PubMed] [Google Scholar]
- Bae S, Kweon J, Kim HS & Kim JS (2014) Microhomology-based choice of Cas9 nuclease target sites. Nature Methods, 11, 705–706. [DOI] [PubMed] [Google Scholar]
- Bak RO, Dever DP & Porteus MH (2018a) CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nature Protocols, 13, 358–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak RO, Dever DP, Reinisch A, Cruz Hernandez D, Majeti R & Porteus MH (2018b) Correction: Multiplexed genetic engineering of human hematopoietic stem and progenitor cells using CRISPR/Cas9 and AAV6. eLife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KS, Defor TE, Burns LJ, Ramsay NK, Neglia JP & Robison LL (2003) New malignancies after blood or marrow stem-cell transplantation in children and adults: incidence and risk factors [erratum appears in J Clin Oncol. 2003 Aug 15;21(16):3181]. Journal of Clinical Oncology, 21, 1352–1358. [DOI] [PubMed] [Google Scholar]
- Baylis F (2018) Counterpoint: The potential harms of human gene editing using CRISPR-Cas9. Clinical Chemistry, 64, 489–491. [DOI] [PubMed] [Google Scholar]
- Baylis F & McLeod M (2017) First-in-human Phase 1 CRISPR gene editing cancer trials: Are we ready? Current Gene Therapy, 17, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard BC, Trobridge GD, Ironside C, McCune JS, Adair JE & Kiem HP (2010) Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. Journal of Clinical Investigation, 120, 2345–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, Duchateau P, Shendure J, Stoddard BL, Certo MT, Baker D & Scharenberg AM (2014) megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Research, 42, 2591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin A, Quinquis B, Sorokin A & Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology, 151, 2551–2561. [DOI] [PubMed] [Google Scholar]
- Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM & Mukherjee S (2019) Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proceedings of the National Academy of Sciences of the United States of America, 116, 11978–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun CJ, Witzel M, Paruzynski A, Boztug K, von Kalle C, Schmidt M & Klein C (2014) Gene therapy for Wiskott-Aldrich Syndrome-Long-term reconstitution and clinical benefits, but increased risk for leukemogenesis. Rare Diseases, 2, e947749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, Whiting-Theobald N, Kawai T, Linton GF, Rudikoff AG, Choi U, Ryser MF, Murphy PM, Sechler JM & Malech HL (2004) CXCR4-transgene expression significantly improves marrow engraftment of cultured hematopoietic stem cells. Stem Cells, 22, 1128–1133. [DOI] [PubMed] [Google Scholar]
- Chang KH, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, Liu M, Liu Y, Vieira BF, Sun C, Hong VP, Zhang M, Yang X, Reik A, Urnov FD, Rebar EJ, Holmes MC, Danos O, Jiang H & Tan S (2017) Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Molecular Therapy. Methods & Clinical Development, 4, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth CT, Deshpande PS, Dever DP, Camarena J, Lemgart VT, Cromer MK, Vakulskas CA, Collingwood MA, Zhang L, Bode NM, Behlke MA, Dejene B, Cieniewicz B, Romano R, Lesch BJ, Gomez-Ospina N, Mantri S, Pavel-Dinu M, Weinberg KI & Porteus MH (2019) Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nature Medicine, 25, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhang H, Zhang Y, Wang Y, Gan J & Ji Q (2019) Molecular basis for the PAM expansion and fidelity enhancement of an evolved Cas9 nuclease. PLoS Biol, 17, e3000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM & Kim JS (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnology, 31, 230–232. [DOI] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ & Voytas DF (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics, 186, 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinicaltrials.gov: NIH U.S. National Library of Medicine. National Institutes of Health, Bethesda, MD. [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA & Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu TI, Mussolino C & Cathomen T (2017) Refining strategies to translate genome editing to the clinic. Nature Medicine, 23, 415–423. [DOI] [PubMed] [Google Scholar]
- Cromer MK, Vaidyanathan S, Ryan DE, Curry B, Lucas AB, Camarena J, Kaushik M, Hay SR, Martin RM, Steinfeld I, Bak RO, Dever DP, Hendel A, Bruhn L & Porteus MH (2018) Global Transcriptional Response to CRISPR/Cas9-AAV6-Based Genome Editing in CD34(+) Hematopoietic Stem and Progenitor Cells. Molecular Therapy, 26, 2431–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyranoski D & Ledford H (2018) Genome-edited baby claim provokes international outcry. Nature, 563, 607–608. [DOI] [PubMed] [Google Scholar]
- Dafflon C, Gaulis S, Barys L, Kapur K, Cornacchione V, Schukur L, Bergling S, Traggiai E, Jansky S, Hellmann L, Engstler BS, Kerr G, de Weck A, Ruddy DA, Naumann U, Stauffer F, Gaul C, Lin Y, Billy E, Weiss A, Hofmann F, Ito M & Tiedt R (2020) DOT1L inhibition is lethal for multiple myeloma due to perturbation of the endoplasmic reticulum stress pathway. Oncotarget, 11, 956–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, Chen C, Mancuso P, Sariyer R, Ferrante P, Donadoni M, Robinson JA, Sillman B, Lin Z, Hilaire JR, Banoub M, Elango M, Gautam N, Mosley RL, Poluektova LY, McMillan J, Bade AN, Gorantla S, Sariyer IK, Burdo TH, Young WB, Amini S, Gordon J, Jacobson JM, Edagwa B, Khalili K & Gendelman HE (2019) Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nature Communications, 10, 2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci S, Uchida N & Tisdale JF (2018) Gene therapy for sickle cell disease: An update. Cytotherapy, 20, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler MA, Teh CE, Thijssen R, Gangoda L, Lan P, Herold MJ, Gray DH, Kelly GL, Roberts AW & Adams JM (2020) Potent efficacy of MCL-1 inhibitor-based therapies in preclinical models of mantle cell lymphoma. Oncogene, 39, 2009–2023. [DOI] [PubMed] [Google Scholar]
- Dever DP, Bak RO, Reinisch A, Camarena J, Washington G, Nicolas CE, Pavel-Dinu M, Saxena N, Wilkens AB, Mantri S, Uchida N, Hendel A, Narla A, Majeti R, Weinberg KI & Porteus MH (2016) CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature, 539, 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWitt MA, Magis W, Bray NL, Wang T, Berman JR, Urbinati F, Heo SJ, Mitros T, Munoz DP, Boffelli D, Kohn DB, Walters MC, Carroll D, Martin DI & Corn JE (2016) Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Science Translational Medicine, 8, 360ra134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez B, Genovese P, Roman-Rodriguez FJ, Alvarez L, Schiroli G, Ugalde L, Rodriguez-Perales S, Sevilla J, Diaz de Heredia C, Holmes MC, Lombardo A, Naldini L, Bueren JA & Rio P (2017) Therapeutic gene editing in CD34(+) hematopoietic progenitors from Fanconi anemia patients. EMBO Mol Med, 9, 1574–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HK, Savani BN, Copelan E, Devine S, Costa LJ, Wingard JR, Shaughnessy P, Majhail N, Perales MA, Cutler CS, Bensinger W, Litzow MR, Mohty M, Champlin RE, Leather H, Giralt S & Carpenter PA (2014) Peripheral blood progenitor cell mobilization for autologous and allogeneic hematopoietic cell transplantation: guidelines from the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant, 20, 1262–1273. [DOI] [PubMed] [Google Scholar]
- EU Clinical Trials Register. European Medicines Agency, Amsterdam. [Google Scholar]
- Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M & Sadelain M (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature, 543, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falahati R, Zhang J, Flebbe-Rehwaldt L, Shi Y, Gerson SL & Gaensler KM (2012) Chemoselection of allogeneic HSC after murine neonatal transplantation without myeloablation or post-transplant immunosuppression. Molecular Therapy, 20, 2180–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felce SL, Anderson AP, Maguire S, Gascoyne DM, Armstrong RN, Wong KK, Li D & Banham AH (2020) CRISPR/Cas9-mediated Foxp1 silencing restores immune surveillance in an immunocompetent A20 lymphoma model. Front Oncol, 10, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzhugh CD, Hsieh MM, Bolan CD, Saenz C & Tisdale JF (2009) Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: time for a moratorium? Cytotherapy, 11, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forget BG (1998) Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci, 850, 38–44. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P & Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America, 109, E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI & Liu DR (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature, 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese P, Schiroli G, Escobar G, Tomaso TD, Firrito C, Calabria A, Moi D, Mazzieri R, Bonini C, Holmes MC, Gregory PD, van der Burg M, Gentner B, Montini E, Lombardo A & Naldini L (2014) Targeted genome editing in human repopulating haematopoietic stem cells. Nature, 510, 235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M & Weissman JS (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell, 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Ospina N, Scharenberg SG, Mostrel N, Bak RO, Mantri S, Quadros RM, Gurumurthy CB, Lee C, Bao G, Suarez CJ, Khan S, Sawamoto K, Tomatsu S, Raj N, Attardi LD, Aurelian L & Porteus MH (2019) Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nature Communications, 10, 4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Romero E, Martinez-Valiente C, Garcia-Ruiz C, Vazquez-Manrique RP, Cervera J & Sanjuan-Pla A (2019) CRISPR to fix bad blood: a new tool in basic and clinical hematology. Haematologica, 104, 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ & Zhang F (2018) Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science, 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, Myhrvold C, Bhattacharyya RP, Livny J, Regev A, Koonin EV, Hung DT, Sabeti PC, Collins JJ & Zhang F (2017) Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, Prunkard D, Colunga AG, Hanafi LA, Clegg DO, Turtle C & Russell DW (2017) HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nature Biotechnology, 35, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham C, Jozwik A, Pepper A & Benjamin R (2018) Allogeneic CAR-T Cells: More than Ease of Access? Cells, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, Martinez-Picado J, Nijhuis M, Wensing AMJ, Lee H, Grant P, Nastouli E, Lambert J, Pace M, Salasc F, Monit C, Innes AJ, Muir L, Waters L, Frater J, Lever AML, Edwards SG, Gabriel IH & Olavarria E (2019) HIV-1 remission following CCR5Delta32/Delta32 haematopoietic stem-cell transplantation. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J (2018) CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nature Medicine, 24, 927–930. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, Macintyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A & Cavazzana-Calvo M (2008) Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. Journal of Clinical Investigation, 118, 3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubner S, Perna F, Kohnke T, Schmidt C, Berman S, Augsberger C, Schnorfeil FM, Krupka C, Lichtenegger FS, Liu X, Kerbs P, Schneider S, Metzeler KH, Spiekermann K, Hiddemann W, Greif PA, Herold T, Sadelain M & Subklewe M (2019) Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia, 33, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, Bacchetta R, Tsalenko A, Dellinger D, Bruhn L & Porteus MH (2015) Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nature Biotechnology, 33, 985–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AD (2019) Evolution of peripheral blood stem cell transplantation. Methods Mol Biol, 2017, 1–10. [DOI] [PubMed] [Google Scholar]
- Hong SG, Yada RC, Choi K, Carpentier A, Liang TJ, Merling RK, Sweeney CL, Malech HL, Jung M, Corat MAF, AlJanahi AA, Lin Y, Liu H, Tunc I, Wang X, Palisoc M, Pittaluga S, Boehm M, Winkler T, Zou J & Dunbar CE (2017) Rhesus iPSC Safe Harbor Gene-Editing Platform for Stable Expression of Transgenes in Differentiated Cells of All Germ Layers. Molecular Therapy, 25, 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJ, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB & Thrasher AJ (2008) Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. Journal of Clinical Investigation, 118, 3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM, Beddoe ME, Carrillo RR, Kiem HP & Walter RB (2019a) Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia, 33, 762–808. [DOI] [PubMed] [Google Scholar]
- Humbert O, Radtke S, Samuelson C, Carrillo RR, Perez AM, Reddy SS, Lux C, Pattabhi S, Schefter LE, Negre O, Lee CM, Bao G, Adair JE, Peterson CW, Rawlings DJ, Scharenberg AM & Kiem HP (2019b) Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Science Translational Medicine, 11, eaaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, Blau IW, Hofmann WK & Thiel E (2009) Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. The New England Journal of Medicine, 360, 692–698. [DOI] [PubMed] [Google Scholar]
- Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, Randhawa R, Kulkarni T, Yang Z, McAllister G, Russ C, Reece-Hoyes J, Forrester W, Hoffman GR, Dolmetsch R & Kaykas A (2018) p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nature Medicine, 24, 939–946. [DOI] [PubMed] [Google Scholar]
- Ishino Y, Shinagawa H, Makino K, Amemura M & Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol, 169, 5429–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Embden JD, Gaastra W & Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol, 43, 1565–1575. [DOI] [PubMed] [Google Scholar]
- Jeong J, Jager A, Domizi P, Pavel-Dinu M, Gojenola L, Iwasaki M, Wei MC, Pan F, Zehnder JL, Porteus MH, Davis KL & Cleary ML (2019) High-efficiency CRISPR induction of t(9;11) chromosomal translocations and acute leukemias in human blood stem cells. Blood Adv, 3, 2825–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA & Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J, Byk T, Jansson-Sjostrand L, Petit I, Shivtiel S, Nagler A, Hardan I, Deutsch V, Gazit Z, Gazit D, Karlsson S & Lapidot T (2004) Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood, 103, 2942–2949. [DOI] [PubMed] [Google Scholar]
- Kang R, Zhu S, Zeh H & Tang D (2018) The STING-STAT6 pathway drives Cas9-induced host response in human monocytes. Biochem Biophys Res Commun, 506, 278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, Aljanahi AA, Schreeder D, Klichinsky M, Shestova O, Kozlowski MS, Cummins KD, Shan X, Shestov M, Bagg A, Morrissette JJD, Sekhri P, Lazzarotto CR, Calvo KR, Kuhns DB, Donahue RE, Behbehani GK, Tsai SQ, Dunbar CE & Gill S (2018) Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell, 173, 1439–1453 e1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Cha J & Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences of the United States of America, 93, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfo I, Maus MV, Pinello L, Aryee MJ & Joung JK (2019) Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nature Biotechnology, 37, 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin AL, Klar AJ & Stahl FW (1986) Double-strand breaks can initiate meiotic recombination in S. cerevisiae. Cell, 46, 733–740. [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature, 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni JA, Cullis PR & van der Meel R (2018) Lipid nanoparticles enabling gene therapies: From concepts to clinical utility. Nucleic Acid Ther, 28, 146–157. [DOI] [PubMed] [Google Scholar]
- Kwon HS, Logan AC, Chhabra A, Pang WW, Czechowicz A, Tate K, Le A, Poyser J, Hollis R, Kelly BV, Kohn DB, Weissman IL, Prohaska SS & Shizuru JA (2019) Anti-human CD117 antibody-mediated bone marrow niche clearance in non-human primates and humanized NSG mice. Blood, 133, 2104–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson S & De Oliveira SN (2014) Gene-modified hematopoietic stem cells for cancer immunotherapy. Hum Vaccin Immunother, 10, 982–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattanzi A, Meneghini V, Pavani G, Amor F, Ramadier S, Felix T, Antoniani C, Masson C, Alibeu O, Lee C, Porteus MH, Bao G, Amendola M, Mavilio F & Miccio A (2019) Optimization of CRISPR/Cas9 Delivery to Human Hematopoietic Stem and Progenitor Cells for Therapeutic Genomic Rearrangements. Molecular Therapy, 27, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legut M, Dolton G, Mian AA, Ottmann OG & Sewell AK (2018) CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood, 131, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Psatha N, Gil S, Wang H, Papayannopoulou T & Lieber A (2018) HDAd5/35(++) adenovirus vector expressing anti-CRISPR peptides decreases CRISPR/Cas9 toxicity in human hematopoietic stem cells. Molecular Therapy. Methods & Clinical Development, 9, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP & Yang B (2011) TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Research, 39, 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y & Peng N (2019) Endogenous CRISPR-Cas System-Based Genome Editing and Antimicrobials: Review and Prospects. Frontiers in Microbiology, 10, 2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Czechowicz A, Scheck A, Rossi DJ & Murphy PM (2019) Hematopoietic chimerism and donor-specific skin allograft tolerance after non-genotoxic CD117 antibody-drug-conjugate conditioning in MHC-mismatched allotransplantation. Nature Communications, 10, 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidonnici MR & Ferrari G (2018) Gene therapy and gene editing strategies for hemoglobinopathies. Blood cells, Molecules & Diseases, 70, 87–101. [DOI] [PubMed] [Google Scholar]
- Liu G, Zhang Y & Zhang T (2020) Computational approaches for effective CRISPR guide RNA design and evaluation. Computational and Structural Biotechnology Journal, 18, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J (2016) CRISPR/Cas9: From genome engineering to cancer drug discovery. Trends in Cancer, 2, 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lux CT, Pattabhi S, Berger M, Nourigat C, Flowers DA, Negre O, Humbert O, Yang JG, Lee C, Jacoby K, Bernstein I, Kiem HP, Scharenberg A & Rawlings DJ (2019) TALEN-Mediated Gene Editing of HBG in Human Hematopoietic Stem Cells Leads to Therapeutic Fetal Hemoglobin Induction. Molecular Therapy. Methods & Clinical Development, 12, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, Bumcrot D, Chao H, Ciulla DM, DaSilva JA, Dass A, Dhanapal V, Fennell TJ, Friedland AE, Giannoukos G, Gloskowski SW, Glucksmann A, Gotta GM, Jayaram H, Haskett SJ, Hopkins B, Horng JE, Joshi S, Marco E, Mepani R, Reyon D, Ta T, Tabbaa DG, Samuelsson SJ, Shen S, Skor MN, Stetkiewicz P, Wang T, Yudkoff C, Myer VE, Albright CF & Jiang H (2019) Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature Medicine, 25, 229–233. [DOI] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L & Church GM (2013a) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology, 31, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE & Church GM (2013b) RNA-guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamcarz E, Zhou S, Lockey T, Abdelsamed H, Cross SJ, Kang G, Ma Z, Condori J, Dowdy J, Triplett B, Li C, Maron G, Aldave Becerra JC, Church JA, Dokmeci E, Love JT, da Matta Ain AC, van der Watt H, Tang X, Janssen W, Ryu BY, De Ravin SS, Weiss MJ, Youngblood B, Long-Boyle JR, Gottschalk S, Meagher MM, Malech HL, Puck JM, Cowan MJ & Sorrentino BP (2019) Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. The New England Journal of Medicine, 380, 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, Musunuru K, Brand H, Tager AM, Allen TM, Talkowski ME, Rossi DJ & Cowan CA (2014) Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell, 15, 643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh BE, Brown ME, Duffin BM, Maufort JP, Vereide DT, Slukvin II & Thomson JA (2015) Nonirradiated NOD,B6.SCID Il2rgamma-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Reports, 4, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJ, Diez-Villasenor C, Garcia-Martinez J & Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of Molecular Evolution, 60, 174–182. [DOI] [PubMed] [Google Scholar]
- Nagree MS, Lopez-Vasquez L & Medin JA (2015) Towards in vivo amplification: Overcoming hurdles in the use of hematopoietic stem cells in transplantation and gene therapy. World Journal of Stem Cells, 7, 1233–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palchaudhuri R, Saez B, Hoggatt J, Schajnovitz A, Sykes DB, Tate TA, Czechowicz A, Kfoury Y, Ruchika F, Rossi DJ, Verdine GL, Mansour MK & Scadden DT (2016) Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nature Biotechnology, 34, 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, Kadota K, Roth SL, Giardina P, Viale A, Leslie C, Bushman FD, Studer L & Sadelain M (2011) Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nature Biotechnology, 29, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabhi S, Lotti SN, Berger MP, Singh S, Lux CT, Jacoby K, Lee C, Negre O, Scharenberg AM & Rawlings DJ (2019) In vivo outcome of homology-directed repair at the HBB gene in HSC using alternative donor template delivery methods. Molecular Therapy. Nucleic Acids, 17, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul B, Ibarra GSR, Hubbard N, Einhaus T, Astrakhan A, Rawlings DJ, Kiem HP & Peterson CW (2018) Efficient enrichment of gene-modified primary T cells via CCR5-targeted integration of mutant dihydrofolate reductase. Molecular Therapy - Methods & Clinical Development, 9, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavel-Dinu M, Wiebking V, Dejene BT, Srifa W, Mantri S, Nicolas CE, Lee C, Bao G, Kildebeck EJ, Punjya N, Sindhu C, Inlay MA, Saxena N, DeRavin SS, Malech H, Roncarolo MG, Weinberg KI & Porteus MH (2019) Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nature Communications, 10, 1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC & June CH (2008) Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nature Biotechnology, 26, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CW, Wang J, Deleage C, Reddy S, Kaur J, Polacino P, Reik A, Huang ML, Jerome KR, Hu SL, Holmes MC, Estes JD & Kiem HP (2018) Differential impact of transplantation on peripheral and tissue-associated viral reservoirs: Implications for HIV gene therapy. PLoS Pathogens, 14, e1006956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, Potrel P, Bas C, Lemaire L, Galetto R, Lebuhotel C, Eyquem J, Cheung GW, Duclert A, Gouble A, Arnould S, Peggs K, Pule M, Scharenberg AM & Smith J (2015) Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Research, 75, 3853–3864. [DOI] [PubMed] [Google Scholar]
- Porteus M (2016) Genome Editing: A New Approach to Human Therapeutics. Annual Review of Pharmacology and Toxicology, 56, 163–190. [DOI] [PubMed] [Google Scholar]
- Pourcel C, Salvignol G & Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology, 151, 653–663. [DOI] [PubMed] [Google Scholar]
- Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K, Rivat C, Wright G, Somana K, Ghorashian S, Pinner D, Ahsan G, Gilmour K, Lucchini G, Inglott S, Mifsud W, Chiesa R, Peggs KS, Chan L, Farzeneh F, Thrasher AJ, Vora A, Pule M & Veys P (2017) Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science Translational Medicine, 9. [DOI] [PubMed] [Google Scholar]
- Radtke S, Adair JE, Giese MA, Chan YY, Norgaard ZK, Enstrom M, Haworth KG, Schefter LE & Kiem HP (2017) A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Science Translational Medicine, 9, [ 10.1126/scitranslmed.aan1145]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke S, Humbert O & Kiem HP (2019) Mouse models in hematopoietic stem cell gene therapy and genome editing. Biochemical Pharmacology, 113692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu X, Fang C, Jiang S, June CH & Zhao Y (2017) Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clinical Cancer Research, 23, 2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet JF, Grevent D, Beuzard Y, Chretien S, Lefebvre T, Ross RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S, Leboulch P & Cavazzana M (2017) Gene therapy in a patient with sickle cell disease. The New England Journal of Medicine, 376, 848–855. [DOI] [PubMed] [Google Scholar]
- Rio P, Navarro S, Wang W, Sanchez-Dominguez R, Pujol RM, Segovia JC, Bogliolo M, Merino E, Wu N, Salgado R, Lamana ML, Yanez RM, Casado JA, Gimenez Y, Roman-Rodriguez FJ, Alvarez L, Alberquilla O, Raimbault A, Guenechea G, Lozano ML, Cerrato L, Hernando M, Galvez E, Hladun R, Giralt I, Barquinero J, Galy A, Garcia de Andoin N, Lopez R, Catala A, Schwartz JD, Surralles J, Soulier J, Schmidt M, Diaz de Heredia C, Sevilla J & Bueren JA (2019) Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nature Medicine, 25, 1396–1401. [DOI] [PubMed] [Google Scholar]
- Roman-Rodriguez FJ, Ugalde L, Alvarez L, Diez B, Ramirez MJ, Risueno C, Corton M, Bogliolo M, Bernal S, March F, Ayuso C, Hanenberg H, Sevilla J, Rodriguez-Perales S, Torres-Ruiz R, Surralles J, Bueren JA & Rio P (2019) NHEJ-Mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell, 25, 607–621 e607. [DOI] [PubMed] [Google Scholar]
- Romero Z, Lomova A, Said S, Miggelbrink A, Kuo CY, Campo-Fernandez B, Hoban MD, Masiuk KE, Clark DN, Long J, Sanchez JM, Velez M, Miyahira E, Zhang R, Brown D, Wang X, Kurmangaliyev YZ, Hollis RP & Kohn DB (2019) Editing the sickle cell disease mutation in human hematopoietic stem cells: Comparison of endonucleases and homologous donor templates. Molecular Therapy, 27, 1389–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer KA, Wu WH, Colgan DF, Tsang SH, Bassuk AG & Mahajan VB (2017) Unexpected mutations after CRISPR-Cas9 editing in vivo. Nature Methods, 14, 547–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiroli G, Conti A, Ferrari S, Della Volpe L, Jacob A, Albano L, Beretta S, Calabria A, Vavassori V, Gasparini P, Salataj E, Ndiaye-Lobry D, Brombin C, Chaumeil J, Montini E, Merelli I, Genovese P, Naldini L & Di Micco R (2019) Precise gene editing preserves hematopoietic stem cell function following transient p53-mediated DNA damage response. Cell Stem Cell, 24, 551–565 e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazi R, Sghia-Hughes G, Reid JL, Kubek S, Haworth KG, Humbert O, Kiem HP & Adair JE (2019) Targeted homology-directed repair in blood stem and progenitor cells with CRISPR nanoformulations. Nature Materials, 18, 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw KL, Garabedian E, Mishra S, Barman P, Davila A, Carbonaro D, Shupien S, Silvin C, Geiger S, Nowicki B, Smogorzewska EM, Brown B, Wang X, de Oliveira S, Choi Y, Ikeda A, Terrazas D, Fu PY, Yu A, Fernandez BC, Cooper AR, Engel B, Podsakoff G, Balamurugan A, Anderson S, Muul L, Jagadeesh GJ, Kapoor N, Tse J, Moore TB, Purdy K, Rishi R, Mohan K, Skoda-Smith S, Buchbinder D, Abraham RS, Scharenberg A, Yang OO, Cornetta K, Gjertson D, Hershfield M, Sokolic R, Candotti F & Kohn DB (2017) Clinical efficacy of gene-modified stem cells in adenosine deaminase-deficient immunodeficiency. The Journal of Clinical Investigation, 127, 1689–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C (2018) Sangamo’s landmark genome editing trial gets mixed reception. Nature Biotechnology, 36, 907–908. [DOI] [PubMed] [Google Scholar]
- Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, Baik SH, Nogales E, Bondy-Denomy J, Corn JE & Doudna JA (2017) Disabling Cas9 by an anti-CRISPR DNA mimic. Science Advances, 3, e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq S, Pamphilon D, Brunskill S, Doree C, Hyde C & Stanworth S (2009) Bone marrow harvest versus peripheral stem cell collection for haemopoietic stem cell donation in healthy donors. The Cochrane Database of Systematic Reviews,CD006406. [DOI] [PubMed] [Google Scholar]
- Simhadri VL, McGill J, McMahon S, Wang J, Jiang H & Sauna ZE (2018) Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Molecular Therapy. Methods & Clinical Development, 10, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanthan MA, Humbert O, Haworth KG, Ironside C, Rajawat YS, Blazar BR, Palchaudhuri R, Boitano AE, Cooke MP, Scadden DT & Kiem HP (2020) Effective multi-lineage engraftment in a mouse model of Fanconi anemia using non-genotoxic antibody-based conditioning. Molecular Therapy. Methods & Clinical Development, 17, 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA, Kulikovskaya I, Gupta M, Chen F, Tian L, Gonzalez VE, Xu J, Jung IY, Melenhorst JJ, Plesa G, Shea J, Matlawski T, Cervini A, Gaymon AL, Desjardins S, Lamontagne A, Salas-Mckee J, Fesnak A, Siegel DL, Levine BL, Jadlowsky JK, Young RM, Chew A, Hwang WT, Hexner EO, Carreno BM, Nobles CL, Bushman FD, Parker KR, Qi Y, Satpathy AT, Chang HY, Zhao Y, Lacey SF & June CH (2020) CRISPR-engineered T cells in patients with refractory cancer. Science, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL & June CH (2014) Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. The New England Journal of Medicine, 370, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M, Moshous D, Lefrere F, Puy H, Bourget P, Magnani A, Caccavelli L, Diana JS, Suarez F, Monpoux F, Brousse V, Poirot C, Brouzes C, Meritet JF, Pondarre C, Beuzard Y, Chretien S, Lefebvre T, Teachey DT, Anurathapan U, Ho PJ, von Kalle C, Kletzel M, Vichinsky E, Soni S, Veres G, Negre O, Ross RW, Davidson D, Petrusich A, Sandler L, Asmal M, Hermine O, De Montalembert M, Hacein-Bey-Abina S, Blanche S, Leboulch P & Cavazzana M (2018) Gene therapy in patients with transfusion-dependent beta-thalassemia. The New England Journal of Medicine, 378, 1479–1493. [DOI] [PubMed] [Google Scholar]
- Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C & Weiss MJ (2016) A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nature Medicine, 22, 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ & Joung JK (2016) Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nature Reviews. Genetics, 17, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, Bode NM, McNeill MS, Yan S, Camarena J, Lee CM, Park SH, Wiebking V, Bak RO, Gomez-Ospina N, Pavel-Dinu M, Sun W, Bao G, Porteus MH & Behlke MA (2018) A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nature Medicine, 24, 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton RT, Christie KA, Whittaker MN & Kleinstiver BP (2020) Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science, 368, 290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Georgakopoulou A, Psatha N, Li C, Capsali C, Samal HB, Anagnostopoulos A, Ehrhardt A, Izsvak Z, Papayannopoulou T, Yannaki E & Lieber A (2019) In vivo hematopoietic stem cell gene therapy ameliorates murine thalassemia intermedia. The Journal of Clinical Investigation, 129, 598–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber BR, Lonetree CL, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, Lahr WS, Draper GM, Slipek NJ, Smeester BA, Lovendahl KN, McElroy AN, Gordon WR, Osborn MJ & Moriarity BS (2019) Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nature Communications, 10, 5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Lin Y, Song Z, Xiao W, Chen L, Yin J, Zhou Y, Barta SK, Petrus M, Waldmann TA & Yang Y (2020) A20 and RBX1 regulate brentuximab vedotin sensitivity in Hodgkin lymphoma models. Clinical Cancer Research, 10.1158/1078-0432.CCR-19-4137]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiebking V, Lee CM, Mostrel N, Lahiri P, Bak R, Bao G, Roncarolo MG, Bertaina A & Porteus MH (2020) Genome editing of donor-derived T-cells to generate allogenic chimeric antigen receptor-modified T cells: Optimizing alphabeta T cell-depleted haploidentical hematopoietic stem cell transplantation. Haematologica, 10.3324/haematol.2019.233882]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RC & Carroll D (2019) The daunting economics of therapeutic genome editing. The CRISPR Journal, 2, 280–284. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren C, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA & Bauer DE (2019) Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nature Medicine, 25, 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, Wang L, Liu T, Wang X, Zhang B, Zhao L, Hu L, Ning H, Zhang Y, Deng K, Liu L, Lu X, Zhang T, Xu J, Li C, Wu H, Deng H & Chen H (2019) CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. The New England Journal of Medicine, 381, 1240–1247. [DOI] [PubMed] [Google Scholar]
- Xu L, Yang H, Gao Y, Chen Z, Xie L, Liu Y, Liu Y, Wang X, Li H, Lai W, He Y, Yao A, Ma L, Shao Y, Zhang B, Wang C, Chen H & Deng H (2017) CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Molecular Therapy, 25, 1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada RC, Ostrominski JW, Tunc I, Hong SG, Zou J & Dunbar CE (2017) CRISPR/Cas9-Based Safe-Harbor Gene Editing in Rhesus iPSCs. Current Protocols in Stem Cell Biology, 43, 5A 11 11–15A 11 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO & Kan YW (2016) Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and beta-thalassemia. Proceedings of the National Academy of Sciences of the United States of America, 113, 10661–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CD, Richardson CD & Corn JE (2019) Advances in genome editing through control of DNA repair pathways. Nature Cell Biology, 21, 1468–1478. [DOI] [PubMed] [Google Scholar]
- Zeng J, Wu Y, Ren C, Bonanno J, Shen AH, Shea D, Gehrke JM, Clement K, Luk K, Yao Q, Kim R, Wolfe SA, Manis JP, Pinello L, Joung JK & Bauer DE (2020) Therapeutic base editing of human hematopoietic stem cells. Nature Medicine, 26, 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Song Z, Wang HB, Lang L, Yang YZ, Xiao W, Webster DE, Wei W, Barta SK, Kadin ME, Staudt LM, Nakagawa M & Yang Y (2019) A novel model of controlling PD-L1 expression in ALK(+) anaplastic large cell lymphoma revealed by CRISPR screening. Blood, 134, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]