

Abstract
Banked CAR T-cells immediately available for off-the-shelf (OTS) application can solve key limitations of patient-specific CAR T-cell products while retaining their potency. The allogeneic nature of OTS cell therapies requires additional measures to minimize graft-versus-host disease and host-versus-graft immune rejection in immunocompetent recipients. In this review, we discuss engineering and manufacturing strategies aimed at minimizing unwanted interactions between allogeneic CAR T-cells and the host. Overcoming these limitations will improve safety and anti-tumor potency of OTS CAR T-cells and facilitate their wider use in cancer therapy.
Introduction
Autologous T-cell products modified to express chimeric antigen receptors (CARs) have produced tremendous clinical benefit in patients with some treatment-resistant cancers. Expanded studies of CAR T-cells targeting the CD19 antigen resulted in three FDA-approved autologous cell therapies for B-cell leukemia and lymphoma1–3, with more products expected to obtain regulatory approval soon. However, these highly potent cell therapies have to be produced for each patient individually from autologous material in cell manufacturing facilities. Overall, patient-specific manufacturing is expensive, time-consuming, and yields cell products with varying potency. Producing CAR T-cells requires complex logistics for centralized or distributed manufacturing and ample time to generate, test, and release the product4–7. These requirements delay treatment, often allowing disease to progress. Further, variation in the quality and quantity of patient blood cells results in highly heterogenous cell products and potential manufacturing failures. Finally, for certain cancers, there is a risk of genetically modifying malignant cells and thereby contaminate starting cell material, with the potential consequence of creating CAR T-cell resistant tumor mutants.
Many hurdles associated with autologous CAR T-cells can be overcome with banked cell products pre-manufactured from highly potent T-cell sources, such as selected healthy donors or induced pluripotent stem cells (iPSC)8,9. These cells would be extensively characterized and tested to ensure potent and predictable anti-tumor activity in all patients. The streamlined manufacturing and release testing of banked T cells has the potential to dramatically reduce the cost and simplify logistics, while off-the-shelf availability of these cell products would facilitate prompt treatment10.
While off-the-shelf (OTS) CAR T-cells have many obvious advantages over autologous cell products, two major steps must be taken before these therapies become standard. First, an appropriate cell platform must be chosen. Conventional polyclonal αβT-cells, which are commonly used for autologous CAR T-cell products, can recognize mismatched major and minor histocompatibility antigens on host cells and promote off-tumor graft-versus-host activity. Therefore, the cell platform must be devoid of significant alloreactivity in order to maximize CAR-directed anti-tumor activity and minimize off-tumor toxicities. Second, the OTS CAR T-cells must resist recognition and elimination by the host immune system.
Immunogenicity of allogenic CAR T-cells may elicit rapid rejection via cellular and humoral immunity, which would limit expansion and persistence of infused OTS CAR T-cells and minimize the therapeutic benefit of re-dosing. Both the graft-versus-host and host-versus-graft activities must be minimized for the OTS CAR T-cell therapy to produce maximal clinical benefit.
Mitigating graft-versus-host alloreactivity in OTS CAR T-cells
The phenomenon of graft-versus-host disease (GvHD) is common in patients undergoing allogeneic hematopoietic stem cell transplant where donor T-cells mount deleterious cytotoxicity against normal recipient tissues and organs11–13. This process depends chiefly on the TCR-mediated recognition of host antigens by rare alloreactive donor αβT-cells that contaminate donor marrow. Therefore, either disrupting TCR expression or using non-alloreactive T-cell subsets should abrogate the unwanted off-tumor alloreactivity in OTS CAR T-cells.
Removing surface TCR in αβT-cells
Engineered T-cells utilize the CAR to recognize and eliminate tumor cells, bypassing the need for the TCR-mediated recognition. Multiple independent studies have shown surface TCR expression is not necessary for CAR-mediated tumor cytotoxicity in αβT-cells, at least in the short term14–18. Therefore, disrupting TCR expression would minimize GvHD potential in αβT-cells while preserving CAR-mediated cytotoxicity. Because proper assembly of the TCR complex requires the presence of all six subunits (two variable chains and four CD3 chains), disrupting expression of any of those subunits is sufficient to prevent surface TCR expression. For example, knocking out TRAC, the gene encoding the constant region of TCRα chain, using genome editing tools completely eliminates surface TCR expression and prevents alloreactivity by αβT-cells19,20. Similar outcomes were observed when genes encoding TCRβ chain or the CD3ε subunit were disrupted19–21. Furthermore, combining TRAC genome editing with homology-directed repair allows for the CAR cassette to be integrated into the TRAC gene locus, generating TCR-edited CAR T-cells in a single step.
Inserting a CAR under the endogenous TRAC promoter results in a relatively narrow range of CAR expression on the cell surface compared to CARs driven by gammaretroviral or lentiviral promoters14–16. Thus, these T-cells have a more calibrated response to CAR antigen stimulation. The CAR transgene located in the TRAC locus is also under transcriptional regulation of TCR gene expression, resulting in physiologic oscillations of CAR transcript levels in response to CAR-embedded CD3ζ chain signaling14. While genetic ablation of TCR genes permanently disables alloreactivity in OTS CAR T-cells, the resulting TCR-edited T-cells must be purified for clinical application, as recent studies showed that even ~99% pure TCR-edited CD19 CAR T-cells produced clinically significant GvHD in patients with B-ALL22,23. Additionally, multiplexed genome editing tools may induce genotoxicity producing additional mutations and chromosomal aberrations23,24 and reducing T-cell viability. A recent study showed evidence of TCR-edited CAR T-cells hypofunctionality in mouse xenograft models of human cancer, suggesting TCR gene editing can be toxic to T-cells25. It is also possible that the observed effects were amplified in the xenogeneic environment, where human T-cells receive limited cytokine support and may require homeostatic TCR stimulation to persist in murine hosts.
TCR function in αβT-cells also can be disrupted without genome editing. For example, trapping the CD3ε subunit intracellularly with a protein expression blocker (PEBL) anchored in the endoplasmic reticulum prevents proper TCR assembly, resulting in a loss of surface TCR expression26. Alternatively, TCR signaling can be disrupted with an engineered TCR inhibitory molecule (TIM) that displaces the CD3ζ chain from the TCR, unlinking the antigen recognition part of the main signaling subunit27. These strategies switch off TCR function and minimize alloreactivity in engineered T-cells by simply incorporating another transgene in the CAR expression cassette. Indeed, recent results from a Phase I study of allogeneic T-cells modified with NKG2D-based CAR and TIM demonstrated absence of GvHD after repeated dosing28. A potential limitation of these approaches is the risk of regaining TCR function in allogeneic T-cells if transgene expression is eventually downregulated or lost (e.g., via epigenetic silencing).
Utilizing non-alloreactive CAR T-cells as a therapeutic platform
A minority of circulating T-cells are alloreactive, and most are phenotypically naïve αβT-cells. Therefore, the risk of GvHD can be substantially reduced or eliminated by depleting from the CAR T-cell product the host-reactive population or selecting cells with defined TCR specificity. Expressing a CAR on polyclonal virus-specific T-cells (VST) expanded from peripheral or cord blood by stimulation with viral peptides is one strategy to reduce the risk of GvHD. Clinical studies showed allogeneic VST lack significant alloreactivity and do not produce severe GvHD29–31. Similarly, host-reactive T-cells can be eliminated prior to manufacture by depleting CD45RA+ naïve T-cells and enriching for the non-alloreactive memory TCR repertoire32,33. Allogeneic αβT-cells host reactivity can be further attenuated by expressing a CAR with a CD28 costimulatory endodomain, which leads to overstimulation and deletion of T-cells that receive both TCR and CAR stimulation34.
To further streamline manufacturing and banking of OTS cell therapies, T-cells with non-alloreactive TCRs can be reprogrammed into induced pluripotent stem cells (iPSC) to create a master cell bank of genetically identical progenitors that can be de-differentiated back to mature cytotoxic T-cell subsets. These iPSC can be modified with CARs, either through retroviral transduction or targeted integration into a desired gene locus (e.g., TRAC) and undergo extensive expansion thus providing a virtually limitless and homogenous source of engineered T-cells for clinical application35,36.
Instead of minimizing αβT-cell host reactivity through additional selection or engineering, one could utilize naturally non-alloreactive subsets of innate-like T-cells, including γδT-cells, NKT-cells, and mucosal-associated invariant T-cells (MAITs)37–40. These cytotoxic T-cell subsets express TCRs with defined specificity to metabolites and bacterial antigens in the context of invariant MHC-like molecules that are conserved in humans. Innate-like T-cells also often employ effector mechanisms commonly found in NK-cells thus effectively combining cancer-targeting capabilities of both adaptive and innate killer cells without producing undesired alloreactivity.
γδT-cells comprise 0.5–10% of peripheral blood mononuclear cells. The majority of γδT cells found in the peripheral blood express Vγ9 and Vδ2 TCR chains, which recognize phospho-antigens overproduced in cancer cells like mevalonate pathway intermediate isopentenyl pyrophosphate (IPP)37,41. Unlike αβT cells, Vγ9Vδ2 T cells express cytotoxic receptors typically found in NK-cells, such as NKG2D42, which recognizes MHC class I polypeptide-related sequence A and B (MICA/B) and other stress-induced ligands upregulated on tumor cells, and CD16 (FcγRIII). Thus, these cells can mediate antibody-dependent cellular cytotoxicity in an MHC-independent manner43. Vγ9Vδ2 T cells can be expanded ex vivo by stimulating PBMC with aminobisphosphonates, such as zoledronate or pamidronate, and easily transduced with CARs and other transgenes44. Engineered γδT cells can mediate multivalent cytotoxicity against tumor cells via both the CAR and TCR, as well as through NKG2D and FcR. Adoptively transferred allogeneic Vγ9Vδ2 T cells have demonstrated safety in several independent clinical studies with more than a hundred cancer patients45. Limitations of Vγ9Vδ2 T cells as a platform for OTS CAR-T cell therapy include limited survival and proliferation, due in part to low production of supporting cytokines and high sensitivity to activation-induced cell death46. Administering exogenous cytokines enhanced Vγ9Vδ2 T cell persistence in preclinical models47, suggesting the anti-tumor activity of these cells can be increased through additional engineering.
The subset of γδT cells that expresses Vδ1 TCR paired with various Vγ chains also holds promise as an OTS cell platform. These cells recognize antigens in the context of non-polymorphic receptors, such as CD1c and CD1d and therefore are non-alloreactive48,49. Compared to Vγ9Vδ2 T cells, Vδ1 T-cells are more difficult to selectively expand and may exhibit regulatory function by secreting immunomodulatory cytokines IL-10, TGF-b and IL-1750. On the other hand, Vδ1 T-cells exert additional anti-tumor activity via NK-cell cytotoxic receptors (NCRs)51 such as NKp30, NKp44, and NKp46 and primarily home to mucosal tissues, which may be advantageous when targeting malignancies like colorectal cancer52. To date, no clinical results have been reported from the registered clinical trials utilizing CAR-modified γδT cells.
Invariant NKT (iNKT) cells are a minor subset of circulating T-cells that recognize foreign lipid antigens in the context of CD1d, a non-polymorphic molecule mainly expressed on antigen-presenting cells37. Additionally, iNKT cells can mediate indirect antitumor activity by targeting immunosuppressive myeloid cells in tumor stroma53. iNKT cells modified with CARs elicited both CAR- and TCR-mediated activity against tumor cells in preclinical models of human cancer54,55 and demonstrated anti-tumor activity in patients with neuroblastoma56. iNKT cells can be selectively stimulated and expanded from PBMC using α-Galactosylceramide (and its derivatives)54,56,57, although this process is often lengthy due to the low frequency of circulating iNKT (<1% of total T-cells). Compared to polyclonal αβT-cells, expansion and persistence of CAR iNKT cells in mouse xenograft models of human cancers was abbreviated, owing to their terminal differentiation and decreased production of homeostatic cytokines54,55. However, recent studies showed the CD62L+ subset of iNKT cells has superior persistence and anti-tumor function in vivo, suggesting this population can be selectively expanded for optimal therapeutic activity57,58. Clinical evaluation of OTS CAR-modified iNKT cells is currently ongoing59.
MAITs recognize bacterial metabolites presented by MHC-like receptor 1 (MR1), a ubiquitously expressed monomorphic molecule60,61. Activated MAITs produce inflammatory cytokines and cytotoxic mediators and functionally resemble iNKT cells62,63. Highly prevalent in liver, lungs, and gastrointestinal tract, MAITs comprise 1–10% of circulating T-cells and can be selectively expanded to achieve high purity64,65. While more studies are needed to demonstrate feasibility and activity of CAR-modified MAITs, this cell subset is an attractive alternative to conventional T-cell platforms due its lack of alloreactivity and ability to home to common sites of primary and metastatic tumors.
Engineering OTS CAR T-cells to resist host immune rejection
Allogeneic CAR T-cells may precipitate an immune response directed at mismatched antigens, such as HLA class I and class II alleles and minor histocompatibility antigens. Unlike graft-versus-host activity, which has one central mechanism (TCR-driven alloreactivity) and several effective mitigating solutions, host-versus-graft immune rejection is more difficult to overcome as there are multiple mechanisms by which the host immune system can recognize and eliminate allogeneic CAR T-cells. The key mediators of allogeneic rejection include host CD8+ and CD4+ T-cells that recognize mismatched HLA antigens as foreign, NK-cells that detect the absence of host histocompatibility molecules, and B-cells that may elicit antibody response against foreign MHC or engineered surface proteins.
Broad immunosuppression
In general, treatment-induced lymphopenia and decreased metabolic fitness of host lymphocytes can decrease the recognition and elimination of allogeneic cells by the adaptive immune system in patients after multiple rounds of chemotherapy. Conventional lymphodepletive conditioning with fludarabine and cyclophosphamide prior to the infusion of OTS CAR T-cells temporarily reduces the numbers of circulating lymphocytes to create favorable conditions for the engraftment and expansion of allogeneic cells. Host immune activation can be further inhibited by administering OTS CAR T-cells in combination with lymphoablative agents like CD52-specific cytotoxic antibody alemtuzumab. To impart OTS CAR T-cells with resistance to alemtuzumab-mediated lymphoablation, the CD52 gene in is commonly disrupted using genome editing tools66,67. A recent report demonstrated incorporation of alemtuzumab into the cyclophosphamide/fludarabine-based lymphodepletive conditioning enabled expansion of allogeneic TCR/CD52-edited CD19 CAR T-cells (UCART19) in 15 out of 17 patients with B-ALL whereas no expansion was noted in patients following lymphodepletion without alemtuzumab23. Expanded UCART19 cells produced complete responses or complete responses with incomplete hematologic recovery in 14 of 17 patients (82%) and 10 patients went on to receive allogeneic stem cell transplant23. Expansion and persistence of UCART19 cells correlated with responses but was limited to 28 days in most patients, which is shorter than that of autologous 4–1BB.ζ CD19 CAR T-cells suggesting eventual immune rejection of allogeneic cells. A limitation of CD52-directed conditioning is that broad depletion of CD52+ lymphocytes, including T-, NK-, and B-cells increases susceptibility to infections caused by environmental and latent pathogens. An alternative strategy is to target CD38-positive activated lymphocytes with daratumumab, a CD38-specific cytotoxic antibody with a more favorable safety profile. As with CD52, CD38 can be genetically ablated in immune effector cells to provide resistance to CD38-targeted conditioning68. Finally, OTS CAR T-cells can be engineered to resist lymphodepletive conditioning with purine nucleotide analogs (e.g. clofarabine or fludarabine) by disrupting expression of the deoxycytidine kinase gene69. Unlike the CD52-directed lymphoablation, the latter two strategies are still in preclinical development.
Passive and active inhibition of T- and NK-cell cytotoxicity
Removing surface HLA in OTS CAR T-cells is another effective way to suppress T-cell mediated rejection. While activated T-cells express both Class I and Class II HLA alleles, reducing expression of only Class I alleles may be sufficient to minimize cytotoxic alloimmune responses to the infused cells. Most commonly, Class I HLA proteins can be ablated by genetic disruption of the beta-2 microglobulin (β2m) gene, which is required for their surface expression17,20. This approach minimizes the expression of all Class I alleles by targeting a single gene. Similarly, editing genes encoding the class II transactivator (CIITA) or regulatory factor X ankyrin repeat-containing protein (RFXANK), which are required to initiate transcription of all MHC Class II genes, can decrease the expression of HLA Class II alleles in T-cells70–72.
T-cell cytotoxicity can also be inhibited by engaging checkpoint inhibitory receptors, such as CTLA-4 and PD-1. This mechanism, which many tumors use to suppress unwanted activation and degranulation of T-cells in the microenvironment, can be adapted to enable OTS CAR T-cells evade responses by allogeneic immune cells. Indeed, forced expression of CTLA4-Ig and PD-L1 protects human embryonic stem cells from alloimmune recognition by T-cells73. However, this approach may require additional modifications in the OTS context to minimize engagement of inhibitory checkpoint receptors on CAR T-cells themselves, which could attenuate their anti-tumor activity.
Deleting surface HLA molecules is a straightforward method to inhibit host T-cell responses to allogeneic OTS CAR T-cells. However, NK-cell cytotoxicity triggered by lack of HLA molecules is a major consequence of this strategy, as HLA serve as potent ligands for NK-cell inhibitory receptors. This phenomenon has been demonstrated in several experimental models where MHC-edited cells were eliminated by NK cells21,71,74. In some instances, NK-cell mediated rejection of OTS CAR T-cells may be even more robust than T-cell mediated responses. NK-cells possess strong and immediate cytotoxicity and, unlike most T cells, do not require prior priming. Furthermore, NK-cells rebound quickly after cyclophosphamide/fludarabine-mediated lymphodepletion75,76, as NK cell lymphopoiesis in the bone marrow occurs more rapidly than T-cell selection in the thymus and does not decline with age77. Therefore, NK-cell mediated rejection is a powerful mechanism that can limit therapeutic benefit of MHC-mismatched or MHC-edited OTS CAR T-cells in pediatric and adult patients.
A few strategies exist to overcome NK-cell rejection of allogeneic cells by overexpressing ligands for inhibitory NK-cell receptors. One approach uses expression of HLA-E, which is a minimally polymorphic non-classical MHC class I allele that inhibits NK-cell degranulation by binding to inhibitory CD94/NKG2A and NKG2B receptors. Indeed, ectopic expression of the HLA-E/β2m fusion protein protected MHC-edited iPSC and B-cell lymphoblasts from elimination by NK-cell lines and, to some extent, by primary NK cells74. A similar effect can be achieved by overexpression of HLA-G78, another non-classical MHC I allele that binds to the inhibitory receptor ILT2 (LIR-1). These mechanisms can suppress NK-cell cytotoxicity against OTS CAR T-cells. However, because both HLA-G and HLA-E can also bind to activating NK-cell receptors (KIR2DL4 and CD94/NKG2C, respectively) and thus trigger NK-cell cytotoxicity, it is unclear whether expression of these alleles alone or in combination will be sufficient for uniform protection against all NK-cell subsets.
To further reduce NK-cell activation, only highly polymorphic HLA-A and -B alleles can be removed, leaving HLA-C and non-classical HLA-G/-E/-F alleles intact. Because non-classical HLA alleles are relatively homogenous in humans, a bank containing 12 cell lines expressing main HLA-C alleles individually was estimated to be immunologically compatible with >90% of human recipients70. Evaluating whether these strategies can be combined with disrupting expression of ligands for major NK-cell activating receptors like NKG2D or 2B4 is of interest. Finally, a recent study suggested NK-cell activity against HLA-edited iPSC can be suppressed71 by forced expression of CD47 — a “don’t eat me” signal for phagocytes. CD47 is often overexpressed in tumor cells to suppress phagocytosis by macrophages but the mechanism of NK-cell inhibition remains to be elucidated and NK-cell subsets that are inhibited by CD47 need to be characterized.
There is a possibility that both T- and NK-cell responses can be attenuated by reducing, rather than eliminating, expression of MHC alleles. Downregulating MHC I expression using shRNA-mediated knockdown of the β2m gene may reduce expression enough to suppress T-cell recognition while remaining sufficient to engage NK-cell inhibitory receptors. Similar effects can be achieved by expressing viral genes that inhibit HLA expression after infection as a part of the immune evasion mechanism. For example, overexpression in T-cells of the E3 ubiquitin ligases K3 and K5 from human herpes virus-8 (HHV-8) significantly decreases HLA molecules on the cell surface and protects these cells from allorejection79.
Active targeting of alloreactive lymphocytes
Our group recently developed an alternative strategy to impart resistance to host immune rejection. We engineered a cytotoxic alloimmune defense receptor (ADR) that recognizes 4–1BB/CD137 on activated T- and NK-cells21. Expression of the ADR enables OTS CAR T-cells to target and eliminate host activated alloreactive lymphocytes while sparing resting subsets, which do not express the 4–1BB activation marker. ADR-expressing T-cells were completely protected from rejection by both T- and NK-cells in mixed lymphocyte reaction assays. Arming OTS CAR T-cells with the ADR allowed them to resist alloimmune rejection in mouse models of allogeneic cell therapy of cancer and produce sustained CAR-mediated anti-tumor activity. Importantly, ADR expression on T-cells protected them from 4–1BB directed self-targeting upon antigen stimulation by binding the 4–1BB receptor in cis and thus masking it from external recognition. This simple genetic modification effectively protects OTS CAR T-cells from alloimmune rejection without the need for gene editing or systemic lymphodepletion. IN addition to 4–1BB, other T-cell activation markers like OX40, CD30, or CD38 can be explored as potential targets. However, the inability of ADR T-cells to distinguish between pathogenic and beneficial activated T-cells may result in some collateral damage to tumor- and pathogen-specific T-cell subsets, while also promoting depletion of activated Tregs in the tumor microenvironment. The extent and the outcome of this on-target activity is difficult to predict in preclinical models but is expected to be less damaging than broad immunosuppressive conditioning (e.g. a CD52-directed lymphoablation). It is worth noting that, to date, there is no evidence that engineered CAR T-cells targeting CD5 or CD30 — antigens more broadly expressed on T-cells than 4–1BB – produce any significant damage to systemic T-cell immunity in patients80,81.
Evading humoral responses
Presence of chimeric receptors or mismatched MHC molecules on the surface of OTS T-cells may activate alloreactive B-cells and induce antibody responses. While B-cell responses would likely be inhibited when targeting CD19 and other B-lineage antigens, it is possible that humoral responses may play an active role in host immune rejection of OTS CAR T-cells with other specificities. Immunogenicity of engineered receptors can be minimized through their humanization (e.g., grafting complementarity-determining regions of an antibody onto human framework regions or generating antigen binders using a library of human antibodies). Some strategies discussed above, including removal of surface HLA on OTS CAR T-cells or CD52-mediated lymphoablation of the host, would also help suppress alloreactive B-cell responses.
Summary
Transition from patient-specific CAR T-cell products to pre-manufactured, immediately available banks of engineered T-cells will democratize these life-saving therapies. While both host-versus-graft and graft-versus-host mechanisms remain to be the main hurdles limiting broader implementation of the allogeneic cell platforms, there is a growing list of manufacturing and engineering strategies to mitigate those unwanted activities (Figure 1). The requirement to minimize GvHD boosted efforts in gene editing of αβT-cells and promoted exploration of alternative, non-alloreactive effector cell subsets. Abrogating host immune rejection remains a more challenging problem to solve. Recent clinical studies utilizing broad lymphoablation have demonstrated the critical importance of suppressing or evading host immune responses in facilitating engraftment and sustaining the activity of allogeneic CAR T-cell therapies. Development and clinical evaluation of alternative approaches is warranted to obviate the need for the pan-lymphocyte ablation and decrease the risks associated with broad immunosuppression. Exploring combinations of “cloaking” and active suppression strategies may be required to ensure maximal protection against multiple mechanisms of host immune rejection.
Figure 1.
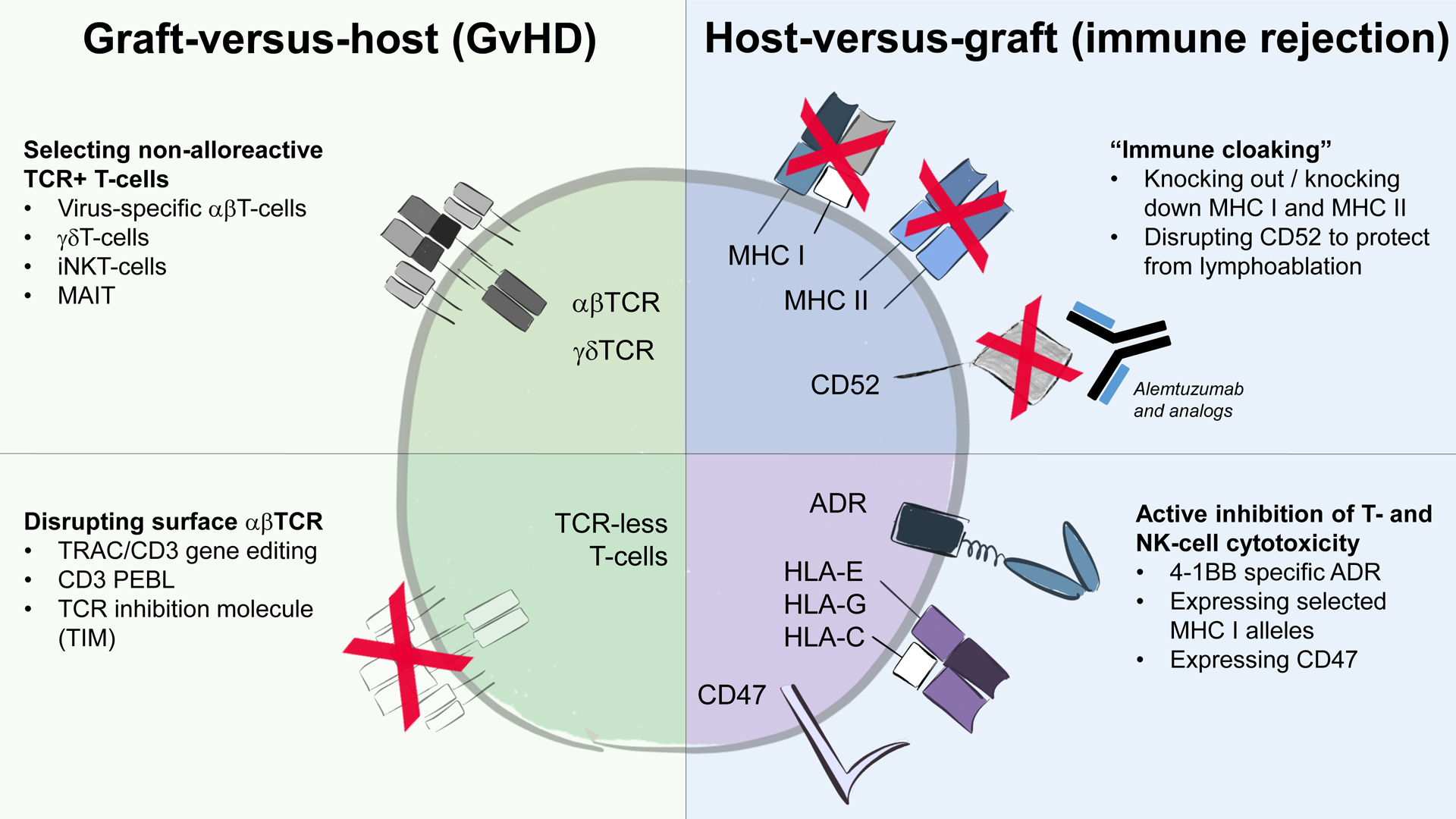
Main strategies to minimize unwanted alloreactivity between OTS CAR T-cells and the recipient.
Limited availability (and often quality) of autologous CAR T-cells requires highly optimized manufacturing processes and CAR constructs that potentiate robust expansion and persistence of therapeutic T-cells to produce durable anti-tumor activity after a single injection. Establishing banks of OTS CAR T-cells available ad libitum for repeated infusions affords a change in the treatment paradigm where the long-term persistence of therapeutic T-cells becomes less important - or even unwanted. Repeated administration of OTS CAR T-cells programmed to undergo predictable expansion and inevitable contraction would minimize the risk of long-term off-tumor toxicities and make cell therapies behave more like conventional drugs with defined pharmacokinetics and pharmacodynamics. Importantly, these OTS cell therapies must still resist host immune rejection which would otherwise limit the activity of repeatedly administered allogeneic CAR T-cells. Therefore, tuning CAR signaling and/or utilizing highly cytotoxic but shorter-lived effector cell platforms may be desirable to generate safe and effective OTS cell therapies with predictable activity.
Acknowledgements
NIH SPORE in Lymphoma 5P50CA126752, LLS SCOR award, LLS Translational Research Project Award #6566, Stand Up to Cancer Dream Team in Lymphoma, CPRIT Award RP150611. The authors thank Catherine Gillespie for editing the manuscript
Footnotes
Conflict of Interest: MM has a patent application in the field of allogeneic CAR T-cell therapies and licensed technology to Fate Therapeutics and Allogene Therapeutics
References
- 1.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine 2017;377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New England Journal of Medicine Massachusetts Medical Society, 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. New England Journal of Medicine Massachusetts Medical Society, 2020;382:1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin JK, Lerman BJ, Barnes JI, Boursiquot BC, Tan YJ, Robinson AQL, et al. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory Pediatric B-Cell Acute Lymphoblastic Leukemia. Journal of Clinical Oncology American Society of Clinical Oncology, 2018; [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Molecular Therapy - Oncolytics Elsevier, 2016;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Current Opinion in Biotechnology 2018;53:164–181. [DOI] [PubMed] [Google Scholar]
- 7.Hay AE, Cheung MC. CAR T-cells: costs, comparisons, and commentary. Journal of Medical Economics Taylor & Francis, 2019;22:613–615. [DOI] [PubMed] [Google Scholar]
- 8.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nature Reviews Drug Discovery Nature Publishing Group, 2020;19:185–199. [DOI] [PubMed] [Google Scholar]
- 9.Aftab BT, Sasu B, Krishnamurthy J, Gschweng E, Alcazer V, Depil S. Toward “off-the-shelf” allogeneic CAR T cells. ADVANCES IN CELL AND GENE THERAPY 2020;3:e86. [Google Scholar]
- 10.Harrison RP, Zylberberg E, Ellison S, Levine BL. Chimeric antigen receptor–T cell therapy manufacturing: modelling the effect of offshore production on aggregate cost of goods. Cytotherapy 2019;21:224–233. [DOI] [PubMed] [Google Scholar]
- 11.Zeiser R, Blazar BR. Acute Graft-versus-Host Disease — Biologic Process, Prevention, and Therapy. Longo DL, ed. New England Journal of Medicine 2017;377:2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perkey E, Maillard I. New Insights into Graft-Versus-Host Disease and Graft Rejection. Annual Review of Pathology: Mechanisms of Disease 2018;13:219–245. [DOI] [PubMed] [Google Scholar]
- 13.Toubai T, Magenau J. Immunopathology and biology-based treatment of steroid-refractory graft-versus-host disease. Blood 2020;136:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature Nature Publishing Group, 2018;559:405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacLeod DT, Antony J, Martin AJ, Moser RJ, Hekele A, Wetzel KJ, et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Molecular Therapy Elsevier, 2017;25:949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clinical Cancer Research American Association for Cancer Research, 2017;23:2255–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An ‘off-the-shelf’ fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nature Medicine Nature Publishing Group, 2012;18:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torikai H, Reik A, Liu P-Q, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012;119:5697–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nature Biotechnology Nature Publishing Group, 2020;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science Translational Medicine 2017;9:eaaj2013. [DOI] [PubMed] [Google Scholar]
- 23.Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. The Lancet Elsevier, 2020;396:1885–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science American Association for the Advancement of Science, 2020;367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020;136:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamiya T, Wong D, Png YT, Campana D. A novel method to generate T-cell receptor–deficient chimeric antigen receptor T cells. Blood Advances 2018;2:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilham DE, Michaux A, Breman E, Mauen S, Bolsée J, Huberty F, et al. TCR inhibitory molecule as a promising allogeneic NKG2D CAR-t cell approach. Journal of Clinical Oncology American Society of Clinical Oncology, 2018;36:e15042–e15042. [Google Scholar]
- 28.Prenen H, Dekervel J, Anguille S, Hendlisz A, Michaux A, Sotiropoulou PA, et al. CYAD-101: An innovative non-gene edited allogeneic CAR-T for solid tumor cancer therapy. Journal of Clinical Oncology American Society of Clinical Oncology, 2020;38:3032–3032.32552276 [Google Scholar]
- 29.Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. Journal of Clinical Oncology American Society of Clinical Oncology, 2017;35:3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, et al. Allogeneic Virus-Specific T Cells with HLA Alloreactivity Do Not Produce Graft-Versus-Host Disease In Human Subjects. Blood American Society of Hematology, 2010;116:1252–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houghtelin A, Bollard CM. Virus-Specific T Cells for the Immunocompromised Patient. Frontiers in Immunology 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bleakley M, Heimfeld S, Jones L, Chaney C, Turtle C, Gooley T, et al. Depletion of Naïve T Cells From Peripheral Blood Stem Cell Grafts for GVHD Prevention. Biology of Blood and Marrow Transplantation 2013;19:S318. [Google Scholar]
- 33.Müller N, Landwehr K, Langeveld K, Stenzel J, Pouwels W, van der Hoorn MAWG, et al. Generation of alloreactivity-reduced donor lymphocyte products retaining memory function by fully automatic depletion of CD45RA-positive cells. Cytotherapy 2018;20:532–542. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nature Medicine 2017;23:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nature Biotechnology 2013;31:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iriguchi S, Kaneko S. Toward the development of true “off-the-shelf” synthetic T-cell immunotherapy. Cancer Science 2019;110:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mori L, Lepore M, De Libero G. The Immunology of CD1- and MR1-Restricted T Cells. Annual Review of Immunology 2016;34:479–510. [DOI] [PubMed] [Google Scholar]
- 38.Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018;48:453–473. [DOI] [PubMed] [Google Scholar]
- 39.Consonni M, Dellabona P, Casorati G. Potential advantages of CD1-restricted T cell immunotherapy in cancer. Molecular Immunology 2018;103:200–208. [DOI] [PubMed] [Google Scholar]
- 40.Perez C, Gruber I, Arber C. Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring “Universal” Donor T Cells. Frontiers in Immunology Frontiers, 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human γδ T cells. Nature Nature Publishing Group, 1995;375:155–158. [DOI] [PubMed] [Google Scholar]
- 42.Das H, Groh V, Kuijl C, Sugita M, Morita CT, Spies T, et al. MICA Engagement by Human Vγ2Vδ2 T Cells Enhances Their Antigen-Dependent Effector Function. Immunity Elsevier, 2001;15:83–93. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Freedman MS. CD16+ γδ T cells mediate antibody dependent cellular cytotoxicity: Potential mechanism in the pathogenesis of multiple sclerosis. Clinical Immunology 2008;128:219–227. [DOI] [PubMed] [Google Scholar]
- 44.Rischer M, Pscherer S, Duwe S, Vormoor J, Jürgens H, Rossig C. Human γδ T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. British Journal of Haematology 2004;126:583–592. [DOI] [PubMed] [Google Scholar]
- 45.Xu Y, Xiang Z, Alnaggar M, Kouakanou L, Li J, He J, et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cellular & Molecular Immunology Nature Publishing Group, 2020;1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrarini M, Delfanti F, Gianolini M, Rizzi C, Alfano M, Lazzarin A, et al. NF-κB Modulates Sensitivity to Apoptosis, Proinflammatory and Migratory Potential in Short- versus Long-Term Cultured Human γδ Lymphocytes. The Journal of Immunology American Association of Immunologists, 2008;181:5857–5864. [DOI] [PubMed] [Google Scholar]
- 47.Devaud C, Bilhere E, Loizon S, Pitard V, Behr C, Moreau J-F, et al. Antitumor Activity of γδ T Cells Reactive against Cytomegalovirus-Infected Cells in a Mouse Xenograft Tumor Model. Cancer Research American Association for Cancer Research, 2009;69:3971–3978. [DOI] [PubMed] [Google Scholar]
- 48.Faure F, Jitsukawa S, Miossec C, Hercend T. CD1c as a target recognition structure for human T lymphocytes: Analysis with peripheral blood γ/δ cells. European Journal of Immunology 1990;20:703–706. [DOI] [PubMed] [Google Scholar]
- 49.Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vδ1 TCR. European Journal of Immunology 2012;42:2505–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chabab G, Barjon C, Abdellaoui N, Salvador-Prince L, Dejou C, Michaud H-A, et al. Identification of a regulatory Vδ1 gamma delta T cell subpopulation expressing CD73 in human breast cancer. Journal of Leukocyte Biology 2020;107:1057–1067. [DOI] [PubMed] [Google Scholar]
- 51.Correia DV, Fogli M, Hudspeth K, da Silva MG, Mavilio D, Silva-Santos B. Differentiation of human peripheral blood Vδ1+ T cells expressing the natural cytotoxicity receptor NKp30 for recognition of lymphoid leukemia cells. Blood 2011;118:992–1001. [DOI] [PubMed] [Google Scholar]
- 52.Mikulak J, Oriolo F, Bruni E, Roberto A, Colombo FS, Villa A, et al. NKp46-expressing human gut-resident intraepithelial Vδ1 T cell subpopulation exhibits high antitumor activity against colorectal cancer. JCI Insight American Society for Clinical Investigation, 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song L, Asgharzadeh S, Salo J, Engell K, Wu H, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. The Journal of Clinical Investigation 2009;119:1524–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014;124:2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell Elsevier, 2018;34:596–610.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nature Medicine 2020;26:1686–1690. [DOI] [PubMed] [Google Scholar]
- 57.Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. The Journal of Clinical Investigation American Society for Clinical Investigation, 2016;126:2341–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ngai H, Tian G, Courtney AN, Ravari SB, Guo L, Liu B, et al. IL-21 Selectively Protects CD62L+ NKT Cells and Enhances Their Effector Functions for Adoptive Immunotherapy. Journal of Immunology (Baltimore, Md.: 1950) 2018;201:2141–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin J, Liu B, Guo L, Di Pierro EJ, Balzeau J, Courtney A, et al. Development of an Allogeneic Universally Tolerated NKT Cell Platform for Off-the-Shelf Cancer Immunotherapy. CELL PRESS 50 HAMPSHIRE ST, FLOOR 5, CAMBRIDGE, MA 02139 USA, 2019:321–321. [Google Scholar]
- 60.Tilloy F, Treiner E, Park S-H, Garcia C, Lemonnier F, de la Salle H, et al. An Invariant T Cell Receptor α Chain Defines a Novel TAP-independent Major Histocompatibility Complex Class Ib–restricted α/β T Cell Subpopulation in Mammals. Journal of Experimental Medicine 1999;189:1907–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature Nature Publishing Group, 2003;422:164–169. [DOI] [PubMed] [Google Scholar]
- 62.Dusseaux M, Martin E, Serriari N, Péguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17–secreting T cells. Blood 2011;117:1250–1259. [DOI] [PubMed] [Google Scholar]
- 63.Sundström P, Szeponik L, Ahlmanner F, Sundquist M, Wong JSB, Lindskog EB, et al. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget Impact Journals, 2019;10:2810–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gherardin NA, Souter MN, Koay H, Mangas KM, Seemann T, Stinear TP, et al. Human blood MAIT cell subsets defined using MR1 tetramers. Immunology and Cell Biology 2018;96:507–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Y, Wang W, Wu X, Weng X. Detection, Expansion, and Isolation of Human MAIT Cells. In: Liu C, ed. T-Cell Receptor Signaling: Methods and Protocols New York, NY: Springer US, 2020:285–293. [DOI] [PubMed] [Google Scholar]
- 66.Poirot L, Philip B, Schiffer-Mannioui C, Clerre DL, Chion-Sotinel I, Derniame S, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Research American Association for Cancer Research, 2015;75:3853–3864. [DOI] [PubMed] [Google Scholar]
- 67.Sommer C, Boldajipour B, Kuo TC, Bentley T, Sutton J, Chen A, et al. Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma. Molecular Therapy 2019;27:1126–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bjordahl R, Gaidarova S, Woan K, Cichocki F, Bonello G, Robinson M, et al. FT538: Preclinical Development of an Off-the-Shelf Adoptive NK Cell Immunotherapy with Targeted Disruption of CD38 to Prevent Anti-CD38 Antibody-Mediated Fratricide and Enhance ADCC in Multiple Myeloma When Combined with Daratumumab. Blood 2019;134:133–133. [Google Scholar]
- 69.Valton J, Guyot V, Marechal A, Filhol J-M, Juillerat A, Duclert A, et al. A Multidrug-resistant Engineered CAR T Cell for Allogeneic Combination Immunotherapy. Molecular Therapy 2015;23:1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu H, Wang B, Ono M, Kagita A, Fujii K, Sasakawa N, et al. Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell Elsevier, 2019;24:566–578.e7. [DOI] [PubMed] [Google Scholar]
- 71.Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nature Biotechnology Nature Publishing Group, 2019;37:252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kagoya Y, Guo T, Yeung B, Saso K, Anczurowski M, Wang C-H, et al. Genetic Ablation of HLA Class I, Class II, and the T-cell Receptor Enables Allogeneic T Cells to Be Used for Adoptive T-cell Therapy. Cancer Immunology Research American Association for Cancer Research, 2020;8:926–936. [DOI] [PubMed] [Google Scholar]
- 73.Rong Z, Wang M, Hu Z, Stradner M, Zhu S, Kong H, et al. An Effective Approach to Prevent Immune Rejection of Human ESC-Derived Allografts. Cell Stem Cell 2014;14:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nature Biotechnology Nature Publishing Group, 2017;35:765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Storek J, Geddes M, Khan F, Huard B, Helg C, Chalandon Y, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Seminars in Immunopathology 2008;30:425. [DOI] [PubMed] [Google Scholar]
- 76.Ysebaert L, Gross E, Kühlein E, Blanc A, Corre J, Fournié JJ, et al. Immune recovery after fludarabine–cyclophosphamide–rituximab treatment in B-chronic lymphocytic leukemia: implication for maintenance immunotherapy. Leukemia 2010;24:1310–1316. [DOI] [PubMed] [Google Scholar]
- 77.Hazeldine J, Lord JM. The impact of ageing on natural killer cell function and potential consequences for health in older adults. Ageing Research Reviews 2013;12:1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pazmany L, Mandelboim O, Valés-Gómez M, Davis DM, Reyburn HT, Strominger JL. Protection from natural killer cell-mediated lysis by HLA-G expression on target cells. Science (New York, N.Y.) 1996;274:792–795. [DOI] [PubMed] [Google Scholar]
- 79.Wang X, Cabrera FG, Sharp KL, Spencer DM, Foster AE, Bayle JH. Engineering Tolerance toward Allogeneic CAR-T Cells by Regulation of MHC Surface Expression with Human Herpes Virus-8 Proteins. Molecular Therapy Elsevier, 2020;0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hill LC, Rouce RH, Smith TS, Yang L, Srinivasan M, Zhang H, et al. Safety and Anti-Tumor Activity of CD5 CAR T-Cells in Patients with Relapsed/Refractory T-Cell Malignancies. Blood 2019;134:199–199.31064751 [Google Scholar]
- 81.Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu M-F, Ivanova A, et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. Journal of Clinical Oncology: Official Journal of the American Society of Clinical Oncology 2020;38:3794–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]