

Abstract
Chromatin-associated condensates are implicated in many nuclear processes, but the underlying mechanisms remain elusive. This protocol describes a chemically-induced protein dimerization system to create condensates on telomeres. The chemical dimerizer consists of two linked ligands that can each bind to a protein: Halo ligand to Halo-enzyme and trimethoprim (TMP) to E. coli dihydrofolate reductase (eDHFR), respectively. Fusion of Halo enzyme to a telomere protein anchors dimerizers to telomeres through covalent Halo ligand-enzyme binding. Binding of TMP to eDHFR recruits eDHFR-fused phase separating proteins to telomeres and induces condensate formation. Because TMP-eDHFR interaction is non-covalent, condensation can be reversed by using excess free TMP to compete with the dimerizer for eDHFR binding. An example of inducing promyelocytic leukemia (PML) nuclear body formation on telomeres and determining condensate growth, dissolution, localization and composition is shown. This method can be easily adapted to induce condensates at other genomic locations by fusing Halo to a protein that directly binds to the local chromatin or to dCas9 that is targeted to the genomic locus with a guide RNA. By offering the temporal resolution required for single cell live imaging while maintaining phase separation in a population of cells for biochemical assays, this method is suitable for probing both the formation and function of chromatin-associated condensates.
Keywords: Condensates, Liquid-liquid phase separation, Chemical dimerizer, Local condensation, Telomeres, PML nuclear body
SUMMARY:
This protocol illustrates a chemically induced protein dimerization system to create condensates on chromatin. The formation of promyelocytic leukemia (PML) nuclear body on telomeres with chemical dimerizers is demonstrated. Droplet growth, dissolution, localization and composition are monitored with live cell imaging, immunofluorescence (IF) and fluorescence in situ hybridization (FISH).
INTRODUCTION:
Many proteins and nucleic acids undergo liquid-liquid phase separation (LLPS) and self-assemble into biomolecular condensates to organize biochemistry in cells1, 2. LLPS of chromatin-binding proteins leads to the formation of condensates that are associated with specific genomic loci and are implicated in various local chromatin functions3. For example, LLPS of HP1 protein underlies the formation of heterochromatin domains to organize the genome4, 5, LLPS of transcription factors forms transcription centers to regulate transcription6, LLPS of nascent mRNAs and multi-sex combs protein generates histone locus bodies to regulate the transcription and processing of histone mRNAs7. However, despite many examples of chromatin-associated condensates being discovered, the underlying mechanisms of condensate formation, regulation and function remain poorly understood. In particular, not all chromatin-associated condensates are formed through LLPS and careful evaluations of condensate formation in live cells are still needed8, 9. For example, HP1 protein in mouse is shown to have only a weak capacity to form liquid droplets in live cells and heterochromatin foci behave as collapsed polymer globules10. Therefore, tools to induce de novo condensates on chromatin in living cells are desirable, particularly those that allow the use of live imaging and biochemical assays to monitor the kinetics of condensate formation, the physical and chemical properties of the resulting condensates, and the cellular consequences of condensate formation.
This protocol reports a chemical dimerization system to induce protein condensates on chromatin11 (Figure 1A). The dimerizer consists of two linked protein-interacting ligands: trimethoprim (TMP) and Halo ligand and can dimerize proteins fused to the cognate receptors: Escherichia coli dihydrofolate reductase (eDHFR) and a bacterial alkyldehalogenase enzyme (Halo enzyme), respectively12. The interaction between Halo ligand and Halo enzyme is covalent, allowing Halo enzyme to be used as an anchor by fusing it to a chromatin-binding protein to recruit a phase-separating protein fused to eDHFR to chromatin. After the initial recruitment, increased local concentration of the phase separating protein passes the critical concentration needed for phase separation and thus nucleates a condensate at the anchor (Figure 1B). By fusing fluorescent proteins (e.g. mCherry and eGFP) to eDHFR and Halo, nucleation and growth of condensates can be visualized in real time with fluorescence microscopy. Because the interaction between eDHFR and TMP is non-covalent, excess free TMP can be added to compete with the dimerizer for eDHFR binding. This will then release the phase separation protein from the anchor and dissolve the chromatin-associated condensate.
Figure 1: Chemical dimerization to induce chromatin-associated condensates.
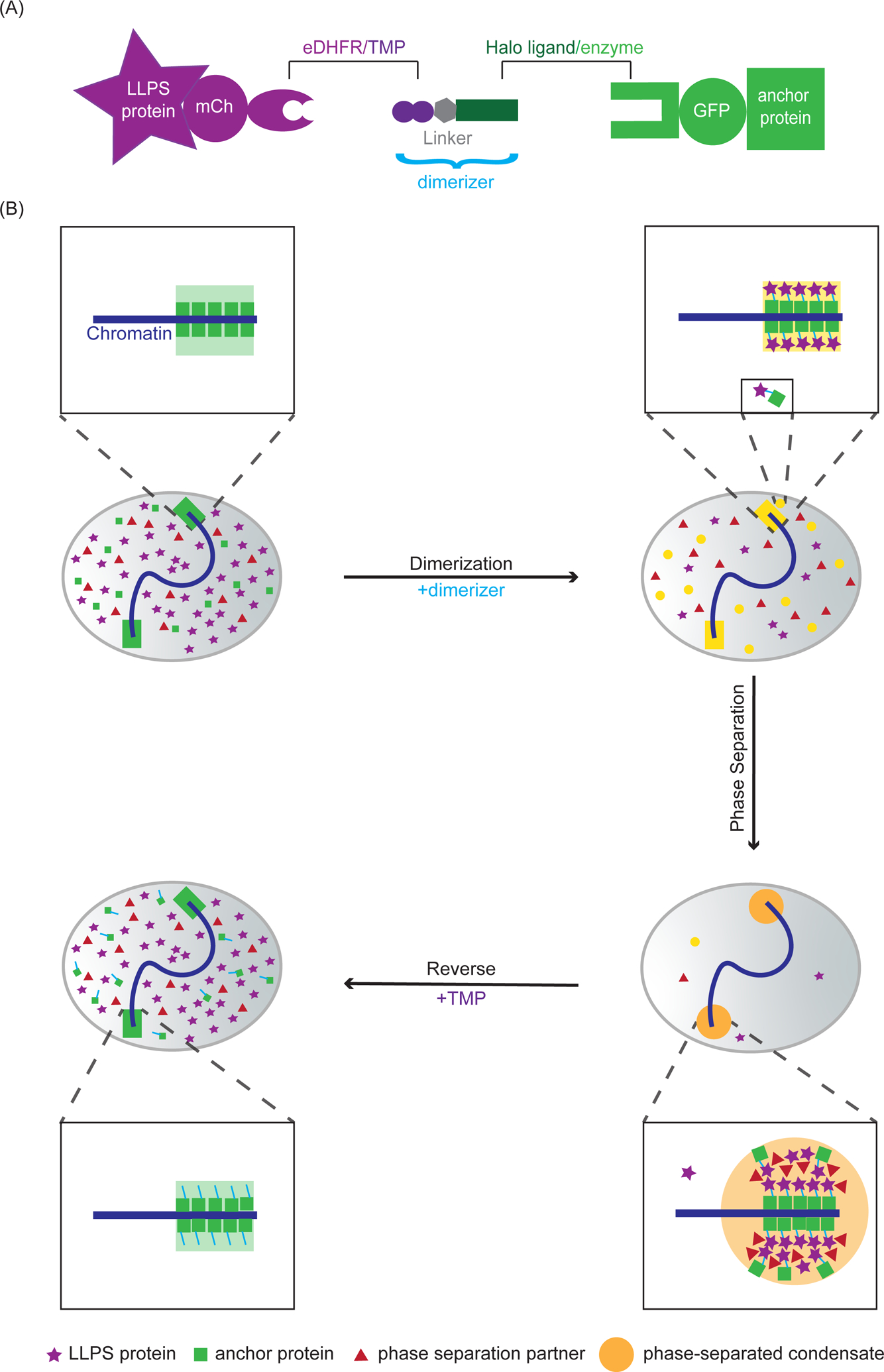
(A) Dimerization schematic: The dimerizer consists of two linked ligands, TMP and Halo that interact with eDHFR and Haloenzyme, respectively. The phase separating protein is fused to mCherry and eDHFR, and the chromosome anchor protein is fused to Halo and GFP. (B) Before adding dimerizer (top left nucleus), the majority of chromosome anchor proteins (green squares) are localized to the chromosomes and a small amount of anchor proteins are diffusely localized in the nucleoplasm. Phase separating proteins to be recruited (purple stars) and phase separating partners (proteins that will condense with the phase separating protein, red triangles) are diffusely localized in the nucleoplasm. After adding dimerizers (top right nucleus), phase separating proteins are dimerized to the anchor protein on the chromosomes and in the nucleoplasm. There could be some excess phase separating proteins in the nucleoplasm, depending on the relative concentration of the anchor protein, phase separating protein and the dimerizer used. After dimerization (bottom right nucleus), increased local concentration of the phase separating proteins at the anchor leads to phase separation and the formation of chromatin-associated condensates. Phase separating partners are enriched at the anchor because of co-condensation with the eDHFR-fused phase separating protein. Anchor proteins that are not directly bound to the chromatin can be enriched at the anchor because of dimerization to the phase separating protein. After adding excess free TMP to compete with the dimerizer for eDHFR binding (bottom left nucleus), the phase separating protein is released from the chromatin and the condensate is dissolved.
We used this tool to induce de novo promyelocytic leukemia (PML) nuclear body formation on telomeres in telomerase-negative cancer cells that use an alternative lengthening of telomeres pathway (ALT) pathway for telomere maintenance13, 14. PML nuclear bodies are membrane-less compartments involved in many nuclear processes15, 16 and are uniquely localized to ALT telomeres to form APBs, for ALT telomere-associated PML bodies17, 18. Telomeres cluster within APBs, presumably to provide repair templates for homology-directed telomere DNA synthesis in ALT19. Indeed, telomere DNA synthesis has been detected in APBs and APBs play essential roles in enriching DNA repair factors on telomeres20, 21. However, the mechanisms underlying APB assembly and telomere clustering within APBs were unknown. Since telomere proteins in ALT cells are uniquely modified by small ubiquitin-like modifier (SUMOs)22, many APB components contain sumoylation sites22–25 and/or SUMO-interacting motifs (SIMs)26, 27 and SUMO-SIM interactions drive phase separation28, we hypothesized that sumoylation on telomeres leads to enrichment of SUMO/SIM containing proteins and SUMO-SIM interactions between those proteins lead to phase separation. PML protein, which has three sumoylation sites and one SIM site, can be recruited to sumoylated telomeres to form APBs and coalescence of liquid APBs leads to telomeres clustering. To test this hypothesis, we used the chemical dimerization system to mimic sumoylation-induced APB formation by recruiting SIM to telomeres (Figure 2A)11. GFP is fused to Haloenzyme for visualization and to the telomere-binding protein TRF1 to anchor the dimerizer to telomeres. SIM is fused to eDHFR and mCherry. Kinetics of condensate formation and droplet fusion-induced telomere clustering are followed with live cell imaging. Phase separation is reversed by adding excess free TMP to compete with eDHFR binding. Immunofluorescence (IF) and fluorescence in situ hybridization (FISH) are used to determine condensate composition and telomeric association. Recruiting SIM enriches SUMO on telomeres and the induced condensates contain PML and therefore are APBs. Recruiting a SIM mutant that cannot interact with SUMO does not enrich SUMO on telomeres or induce phase separation, indicating that the fundamental driving force for APB condensation is SUMO-SIM interaction. Agreeing with this observation, polySUMO-polySIM polymers that fused to a TRF2 binding factor RAP1 can also induce APB formation29. Compared to the polySUMO-polySIM fusion system where phase separation occurs as long as enough proteins are produced, the chemical dimerization approach presented here induces phase separation on demand and thus offers better temporal resolution to monitor the kinetics of phase separation and telomere clustering process. In addition, this chemical dimerization system permits the recruitment of other proteins to assess their ability in inducing phase separation and telomere clustering. For example, a disordered protein recruited to telomeres can also form droplets and cluster telomeres without inducing APB formation, suggesting telomere clustering is independent of APB chemistry and only relies on APB liquid property11.
Figure 2: SUMO is enriched after recruiting SIM to telomeres with dimerizers.
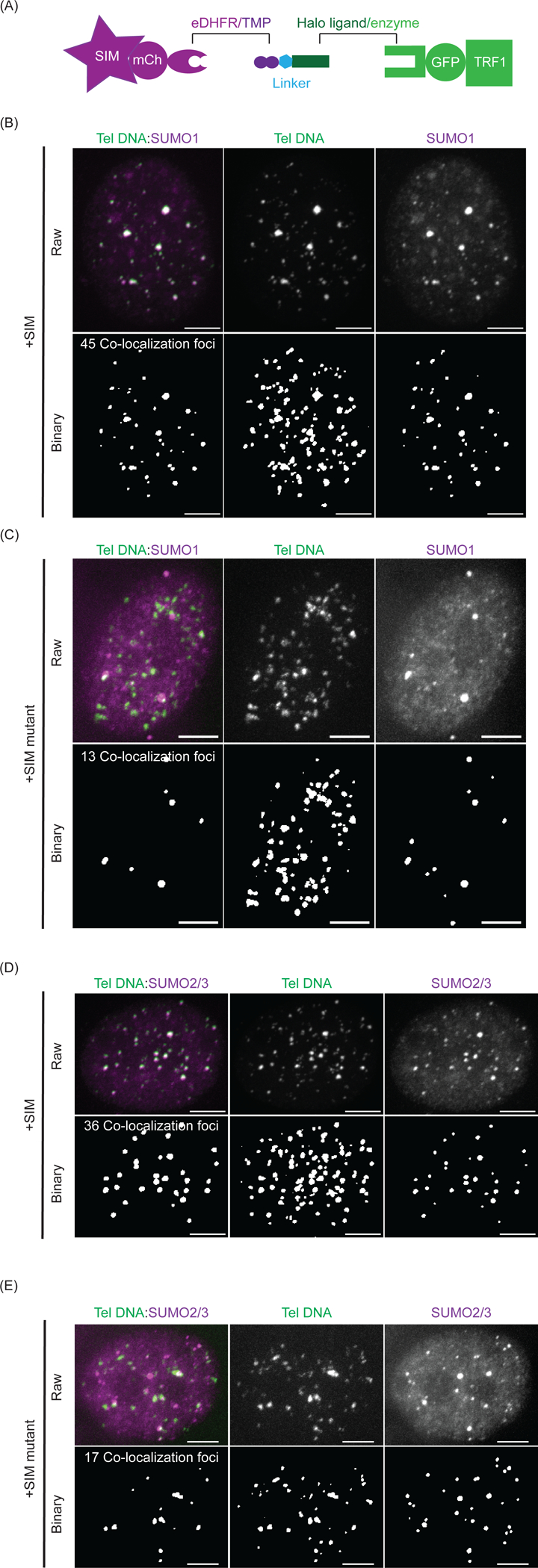
(A) Dimerization schematic in this experiment: SIM (or SIM mutant) is fused to mCherry and eDHFR, and TRF1 is fused to Halo and GFP. (B) A representative cell for telomere DNA FISH and SUMO1 IF after recruiting SIM. Bottom is binary layer identifying telomeres, SUMO1 and number of colocalized SUMO1 and telomere DNA foci. Scale bars, 5 μm. (C) A representative cell for telomere DNA FISH and SUMO1 IF after recruiting SIM mutant. At the bottom is the binary layer of the images used to identify the number of colocalized SUMO1 and telomere DNA foci. Scale bars, 5 μm. (D) A representative cell for telomere DNA FISH and SUMO2/3 IF after recruiting SIM. At the bottom is the binary layer identifying telomeres, SUMO2/3, and the number of colocalized SUMO2/3 and telomere DNA foci. Scale bars, 5 μm. (E) A representative cell for telomere DNA FISH and SUMO2/3 IF after recruiting SIM mutant. At the bottom is the binary layer of the images used to identify the number of colocalized SUMO2/3 and telomere DNA foci. Scale bars, 5 μm.
PROTOCOLS:
1. Production of transient cell lines
-
1.1
Culture U2OS acceptor cells on 22 × 22 mm glass coverslips (for live imaging) or 12 mm diameter circular covterslips (for IF or FISH) coated with poly-D-lysine in 6-well plate with growth medium (10% fetal bovine serum and 1% Penicillin-Streptomycin solution in DMEM) until they reach 60–70% confluency.
-
1.2
Replace growth medium with 1 mL transfection medium (growth medium without Penicillin-Streptomycin solution) prior to transfection.
-
1.3
For each transfection well, add 4 μL transfection reagent to 150 μL reduced serum media, vortex for 10 seconds and then incubate for 5 mins.
-
1.4
For each well, add Halo construct plasmid (Halo-GFP-TRF1 or Halo-TRF1) and eDHFR construct plasmid (mCherry-eDHFR-SIM or mCherry-eDHFR-SIM mutant) at a 1:1 mass ratio (0.5 μg Halo construct plasmid with 0.5 μg eDHFR construct plasmid) to 150 μL reduced serum media, dropwise, mix by pipetting.
NOTE: The tags are relatively small (Halo 33 kD, eDHFR 28 kD, mCherry 30 kD, eGFP 27 kD) and no effect on phase separation was observed. However, using mutants such as SIM mutant used here to make sure that the phase behavior is sensitive to the mutations not the tags is advised. SIM is from PIASx28,30. SIM sequence is AAAGTCGATGTAATTGACTTAACGATCGAATCTAGCAGCGATGAAGAAGAAGATCCACCGGCTAAACGT. SIM mutant is generated by mutating SIM amino acids VIDL to VADA28, and the sequence is AAAGTCGATGTAGCCGACGCCACGATCGAATCTAGCAGCGATGAAGAAGAAGATCCACCGGCTAAACGT. We deposited our plasmids to addgene: #164644 3XHalo-GFP-TRF1; #164646 mCherry-eDHFR-SIM; #164649 mCherry-eDHFR-SIM mutant.
-
1.5
Add 150 μL of transfection reagent -reduced serum media mixture to 150 μL reduced serum media with DNA, dropwise, mix by pipetting, incubate for 5 mins.
-
1.6
Add the 300 μL of transfection reagent-DNA mixture to cells, dropwise and then place cells back to incubator.
-
1.7
Wait 24–48 hours before live imaging, immunofluorescence (IF) or fluorescence in situ hybridization (FISH).
2. Dimerization on telomeres
-
2.1
Dissolve dimerizers in dimethyl sulphoxide at 10 mM and store in plastic microcentrifuge tubes at −80 °C for long term storage.
NOTE: Instead of using the dimerizer with TMP directly linked to Halo (TMP-Halo, TH), dimerizer TMP-NVOC-Halo (TNH) that has a photosensitive linker, 6-nitroveratryl oxycarbonyl (NVOC), between TMP and Haloligand is used31. This is because it takes less time for TNH (~5 mins) to diffuse into the cell than TH (~20 mins). Results shown here can also be obtained using TH. When using TNH, be careful not to expose the dimerizer to light to avoid NOVC cleavage. Handle TNH in a dark room with a dim red-light lamp, store the dimerizer in amber plastic tubes and wrap the container containing TNH or treated cells with aluminum foil. Imaging TNH with DIC is safe.
-
2.2
Take an aliquot of 10 mM dimerizer from −80 °C and dilute in imaging medium to a stock concentration of 10 μM and store at −20 °C.
-
2.3
When ready to use, dilute dimerizers from 10 μM stock solution to a final working concentration of 100 nM in growth medium (for fixed imaging) or imaging medium. Treat cells with 100 nM dimerizers (final concentration) on stage for live imaging or incubate for 4–5 hours for immunofluorescence (IF) and fluorescence in situ hybridization (FISH).
NOTE: The concentration of dimerizers used affects dimerization efficiency and thus phase separation. The dimerizer concentration allowing maximum dimerization efficiency depends on cell type and anchor protein concentration, so it will need to be determined for different experiments. One simple way is to incubate cells expressing the anchor protein and mCherry-eDHFR (without fusing to the phase separating protein or fused to a non-phase separating mutant) and identify the dimerizer concentration at which the highest mCherry intensity at the anchor is achieved. A more systematic approach is to incubate cells expressing the anchor only with different concentrations of dimerizers and then with a Halo binding dye to help determine the dimerizer concentration at which the Halo binding dye intensity starts to plateau (i.e., all Halo-fused anchors are occupied by the dimerizer and no more left for the Halo binding dye)32.
-
2.4
To reverse dimerization, incubate cells with dimerizers for 2–5 hours (with or without live imaging) or until desired droplet size is achieved and add 100 μM free TMP (final concentration) diluted in imaging medium to cells.
3. Immunofluorescence (IF)
-
3.1
Seed 105 cells on 12 mm diameter circular cover glasses coated with poly-D-lysine in 6-well plate. Then transfect the two plasmids (Halo-GFP-TRF1 with mCherry-eDHFR-SIM or mCherry-eDHFR-SIM mutant) and wait for 24–48 hours before proceeding to immunofluorescence.
NOTE: Wait more than one day after transfection can obtain higher expression.
-
3.2
Dilute dimerizers with growth medium to reach a final concentration of 100 nM, add diluted dimerizers to cells and incubate at 37 °C for 4–5 hours.
NOTE: Phase separation is quickly induced after adding dimerizers (< 30 mins). Longer incubation helps droplets coarsen into larger sizes. Following droplet growth with live imaging can be used to determine the time it takes for droplets to reach a desired state.
-
3.3
Fix cells in PBS solution containing 4% formaldehyde and 0.1% Triton X-100 for 10 mins at room temperature to permeabilize cells. Wash cells 3 times with PBS.
NOTE: After this step, it can be paused and cells can be stored at 4 °C for up to a week.
Caution: Formaldehyde is harmful by inhalation and if swallowed, it also irritates eyes, respiratory system and skin and is a possible cancer hazard. Need to wear personal protective equipment, use only in a chemical fume hood. Also put it in a waste container after use, do not dispose it in the sink.
-
3.4
Wash coverslips two times with 50 μL TBS-Tx and once with 50 μL Antibody Dilution Buffer (AbDil). TBS-Tx was made by TBS, 0.1% Triton X-100 and 0.05% Na-azide. AbDil was made by TBS-Tx, 2% BSA and 0.05% Na-azide.
-
3.5
Incubate each coverslip with 50 μL primary anti-PML (1:50 dilution in AbDil) / anti-SUMO1 (1:200 dilution in AbDil) / anti-SUMO2/3 antibody (1:200 dilution in AbDil) at 4 °C in a humidified chamber overnight. mCherry antibody can also be used (1:200 dilution in AbDil) to help detect mCherry signal for FISH.
NOTE: FISH quenches the mCherry fluorescent signal, which makes it difficult to differentiate cells transfected with mCherry plasmids from those not transfected in FISH experiments. Using mCherry antibody is advised. Alternatively, one can make a stable cell line expressing eDHFR containing protein.
-
3.6
Wash coverslips 3 times with AbDil to remove unbound primary antibody.
-
3.7
Incubate cells with secondary antibody [anti-mouse IgG (H+L) secondary antibody conjugated with Alexa Fluor 647 for PML and SUMO, anti-rabbit IgG (H+L) secondary antibody conjugated with Alexa Fluor 555 for mCherry, both at 1:1000 dilution in AbDil] for 1 hour in dark box at room temperature.
-
3.8
Wash coverslips 3 times with TBS-Tx.
-
3.9
Label slides, dilute DAPI in mounting media to reach DAPI final concentration of 1 μg/mL. Then put 2 μL diluted DAPI on the slide. Flip the coverslips over and place them onto DAPI drop, aspire extra fluid from the edge of the coverslip.
-
3.10
Seal with nail polish, let it dry and rinse from top of the coverslip with water. Save in freezer for imaging.
4. Fluorescence in situ hybridization (FISH)
-
4.1
Seed 105 cells on 12 mm diameter circular cover glasses coated with poly-D-lysine in 6-well plate. Transfected cells with Halo-TRF1 and mCherry-eDHFR-SIM or mCherry-eDHFR-SIM mutant plasmids and wait for 24–48 h before proceeding to FISH. The dimerization step is the same as described in 3.2.
NOTE: Here TRF1 is not fused with GFP to free the green channel up for telomere DNA probe.
-
4.2
Fix cells with 4% formaldehyde for 10 mins at room temperature and wash 4 times with PBS. For IF-FISH, proceed to IF protocol from here and after washing off secondary antibody in IF (3.8), refix cells with 4% formaldehyde for 10 mins at room temperature and wash three times with PBS.
-
4.3
Dehydrate coverslips in an ethanol series (70%, 80%, 90%, 2 mins each).
-
4.4
Incubate coverslips with 488-telC PNA probe (1:2000 ratio) in 5 μL hybridization solution at 75 °C for 5 mins. Hybridization solution contains 70% deionized formamide, 10 mM Tris (pH 7.4), 0.5% blocking reagent. Then incubate overnight in a humidified chamber at room temperature.
-
4.5
Wash coverslips with wash buffer (70% formamide, 10 mM Tris) 2 mins for 3 times at room temperature and mount with 1 μg/mL DAPI in mounting media for imaging.
5. Live imaging
-
5.1
When cells are ready for imaging, mount coverslips in magnetic chambers with cells maintained in 1 mL imaging medium without phenol red on a heated stage in an environmental chamber.
-
5.2
Set up microscope and environmental control apparatus. Images were acquired with a spinning disk confocal microscope with a 100× 1.4 NA objective, a Piezo Z-Drive, an electron multiplying charge-coupled device (EMCCD) camera CCD camera, and a laser merge module equipped with 455, 488, 561, 594 and 647 nm lasers controlled by imaging software. Output powers for all lasers are 20 mW measured at the fiber end.
-
5.3
Locate cells with bright GFP signal on telomeres and diffusively localized mCherry signal in the nucleoplasm. Find around 20 cells, memorize each position with x, y, z information and set up parameters for time lapse imaging with 0.5 μm spacing for a total of 8 μm in Z and 5-minute time interval for 2–4 hours for both GFP and mCherry channels. Use 30% of 594 nm and 50% of 488 nm power intensity, with exposure times of 200 ms and camera gain 300.
NOTE: The output power of laser units is 20 mW. Bright GFP foci indicate larger anchor size which can nucleate condensates more easily. Find cells with a wide range of mCherry signal because phase separation depends on SIM concentration in the cell. Cells with too dim or too bright mCherry signal may not phase separate. Do not use too much laser power or too long exposure time to avoid photobleaching.
-
5.4
Start imaging and take one-time loop as pre-dimerization. Pause imaging, add 0.5 mL imaging media containing 15 μL of 10 μM dimerizer to the imaging chamber on the stage so that the final dimerizer concentration is 100 nM. Resume imaging.
-
5.5
When ready to reverse dimerization, pause imaging, add 0.5 mL imaging media containing 2 μL of 100 mM stock TMP to the imaging chamber on the stage to get 100 μM TMP final concentration. Continue imaging cells for 1–2 hours.
6. Fixed imaging
-
6.1
Same microscope set up as live imaging, stage heating is not needed. Use 488 nm to image telomere FISH, 561 nm for mCherry IF, and 647 nm for PML or SUMO IF.
NOTE: If not using a mCherry antibody, just directly image mCherry protein but signal maybe dim because of quenching in FISH. Still use 561 nm rather than 594 nm laser to image mCherry to avoid signal bleed-through of Cy5 to mCherry.
-
6.2
Locate around 30–50 cells with red signal (mCherry or mCherry IF) to select for transfected cells.
-
6.3
Images were taken with 0.3 μm spacing for a total of 8 μm in Z for collecting more signals. Use 80% of 647 nm, 80% of 561 nm, and 70% of 488 nm power intensity, with exposure times of 600 ms and camera gain 300.
7. Process time-lapse images
-
7.1
Define binary for telomeres
Choose one cell with all time and z-stack information, choose only the GFP channel and create a binary layer by defining threshold. Adjust the lower and upper values of the threshold and use functions such as “Smooth”, “Clean” and “Fill holes” to see how well the threshold picks up the desired objects through all time points.
-
7.2
Subtract background
Choose all channels, draw rectangle Region of Interest (ROI) on the background (aside from the cell). Define this ROI as background and then subtract background intensity.
-
7.3
Link telomere binary to telomere intensity
Choose telomere binary and link it to GFP channel for calculating GFP intensity in the binary objects as telomere intensity.
-
7.4
Calculate telomere number and intensity over time
Specify which information to be exported, such as time, object ID, mean intensity, sum intensity, and export data. Use figure plotting software to read the exported table and generate figures of telomere number and telomere intensity (summarize intensity over the volume in each telomere and then average over all telomeres in a cell) over time.
8. Process fixed-cell images
-
8.1
Define binary for APBs
Choose transfected cells for analysis by looking for signal in 561nm channel. Following the procedure in 7.1, define threshold in both GFP (telomere DNA FISH) and Cy5 (PML or SUMO IF) channel to generate binary for telomeres and PML bodies or SUMO, respectively. Merge GFP and Cy5 binary layers and create a new layer containing particles that both have GFP and Cy5 signal to represent co-localization of PML body on telomeres, thus APBs.
-
8.2
Calculate APB/SUMO number and intensity
Subtract image background following 7.2. Link APB/SUMO binary layer to Cy5 channel following 7.3. Calculate APB/SUMO number and intensity. Export and plot data following 7.4.
REPRESENTATIVE RESULTS:
Representative images of telomeric localization of SUMO identified by telomere DNA FISH and SUMO protein IF are shown in Figure 2. Cells with SIM recruitment enriched SUMO1 and SUMO 2/3 on telomeres compared to cells with SIM mutant recruitment. This indicates SIM dimerization induced SUMO enrichment on telomeres depends on SUMO-SIM interactions.
A representative time lapse movie of TRF1 and SIM after dimerization is shown in Video 1. Snapshots at four time points are shown in Figure 3A. SIM was successfully recruited to telomeres and both SIM and TRF1 foci became larger and brighter, as predicted for liquid droplet formation and growth (Figure 1B). In addition, fusion of TRF1 foci was observed (Figure 3B), which led to telomere clustering as shown in the reduced telomere number (Figure 3E) and increased telomere intensity over time (Figure 3D). In contrast, SIM mutant was recruited to telomeres after dimerization but did not induce any droplet formation or telomere clustering, as telomere intensity did not grow and telomere number did not reduce (Figure 3C, D, E, Video 2). This indicates that phase separation and thus telomere clustering is driven by SUMO-SIM interactions.
Figure 3: Dimerization-induced phase separation drives telomere clustering.
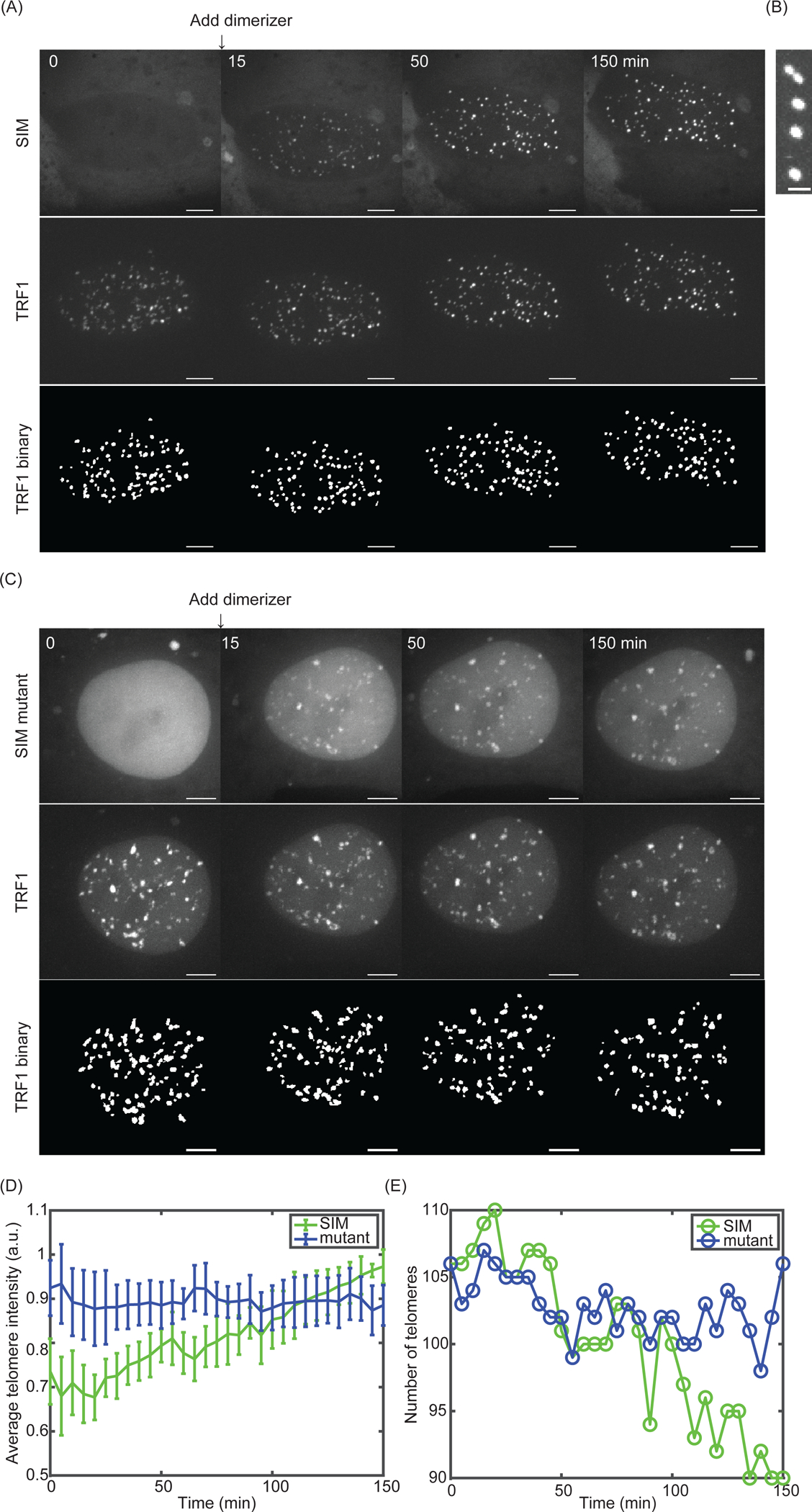
(A) Snapshots of TRF1-GFP and SIM-mCherry before and after adding 100 nM dimerizer (final concentration). AT the bottom is the telomere binary layer identified from TRF1-GFP. Scale bars, 5 μm. (B) A fusion event after recruiting SIM to telomeres. Scale bars, 2 μm. Time interval, 5 mins. (C) Snapshots of TRF1-GFP and SIM mutant-mCherry before and after adding 100 nM dimerizer (final concentration). At the bottom is the telomere binary layer identified from TRF1-GFP. Scale bars, 5 μm. (D) Average telomere intensity (summarize intensity over the volume in each telomere and then average over all telomeres in a cell) over time after recruiting SIM (green, for cell in Figure 3A) and SIM mutant (blue, for cell in Figure 3C). (E) Telomere number over time after recruiting SIM (green, for cell in Figure 3A) and SIM mutant (blue, for cell in Figure 3C).
The reversal of phase separation and telomere clustering after adding excess free TMP is shown in Video 3. Snapshots at four time points are shown in Figure 4A. Agreeing with the predicted condensate dissolution and de-clustering of telomeres, telomere number increased, and telomere intensity decreased over time (Figure 4B, C).
Figure 4: Reversal of condensation and telomere clustering.
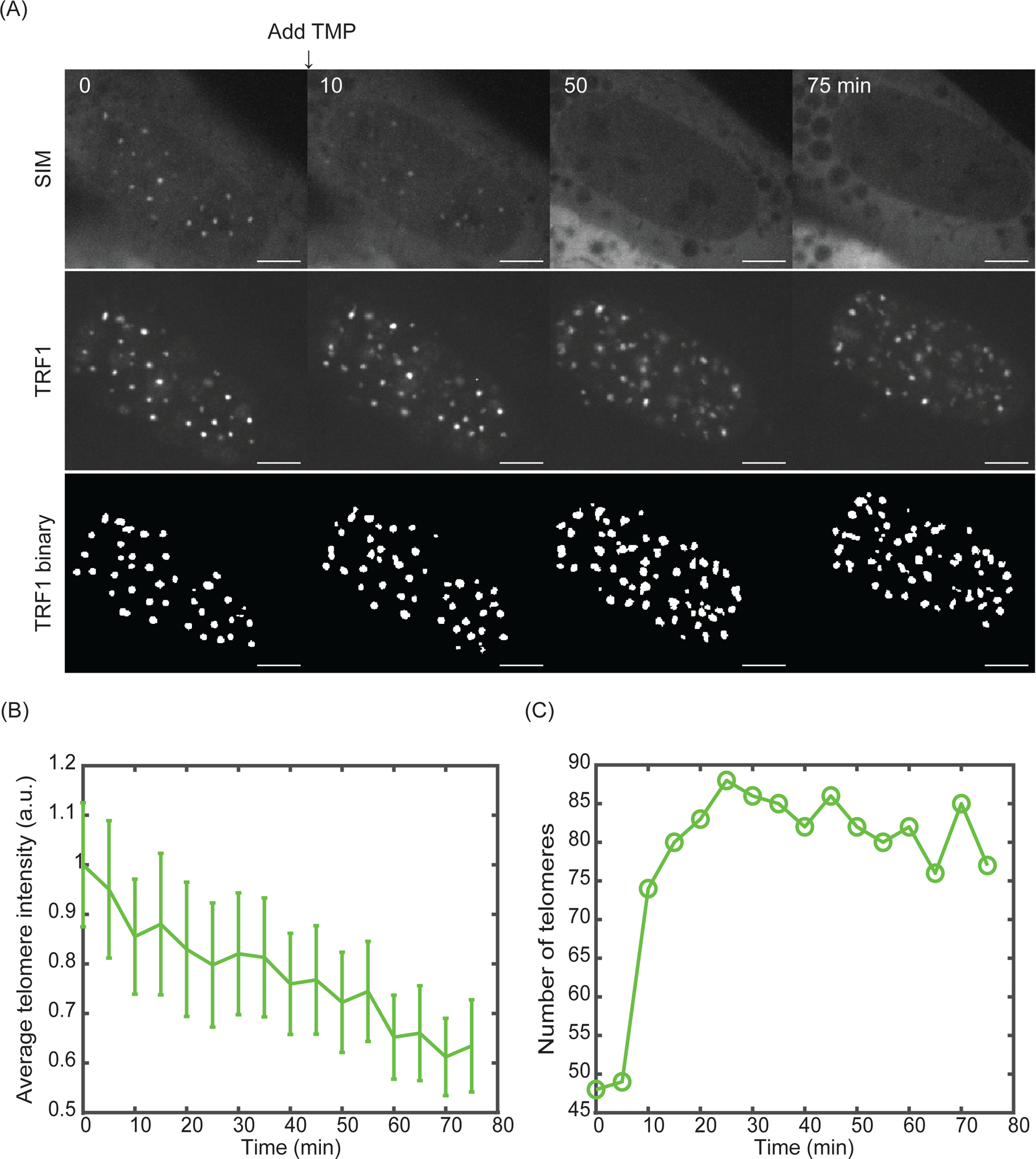
(A) Snapshots of TRF1-GFP and SIM-mCherry after adding 100 μM TMP (final concentration) to cells with condensates formed for 3 hours. At the bottom is the telomere binary layer identified from TRF1-GFP. Scale bars, 5 μm. (B) Average telomere intensity (summarize intensity over the volume in each telomere and then average over all telomeres in a cell) over time for cell in Figure 4A. (C) Telomere number over time for cell in Figure 4A.
Representative images of APBs identified by telomere DNA FISH and PML protein IF are shown in Figure 5. Cells with SIM recruited have more APBs than cells with SIM mutant recruited, suggesting dimerization-induced condensates are indeed APBs.
Figure 5: Dimerization-induced condensates are APBs.
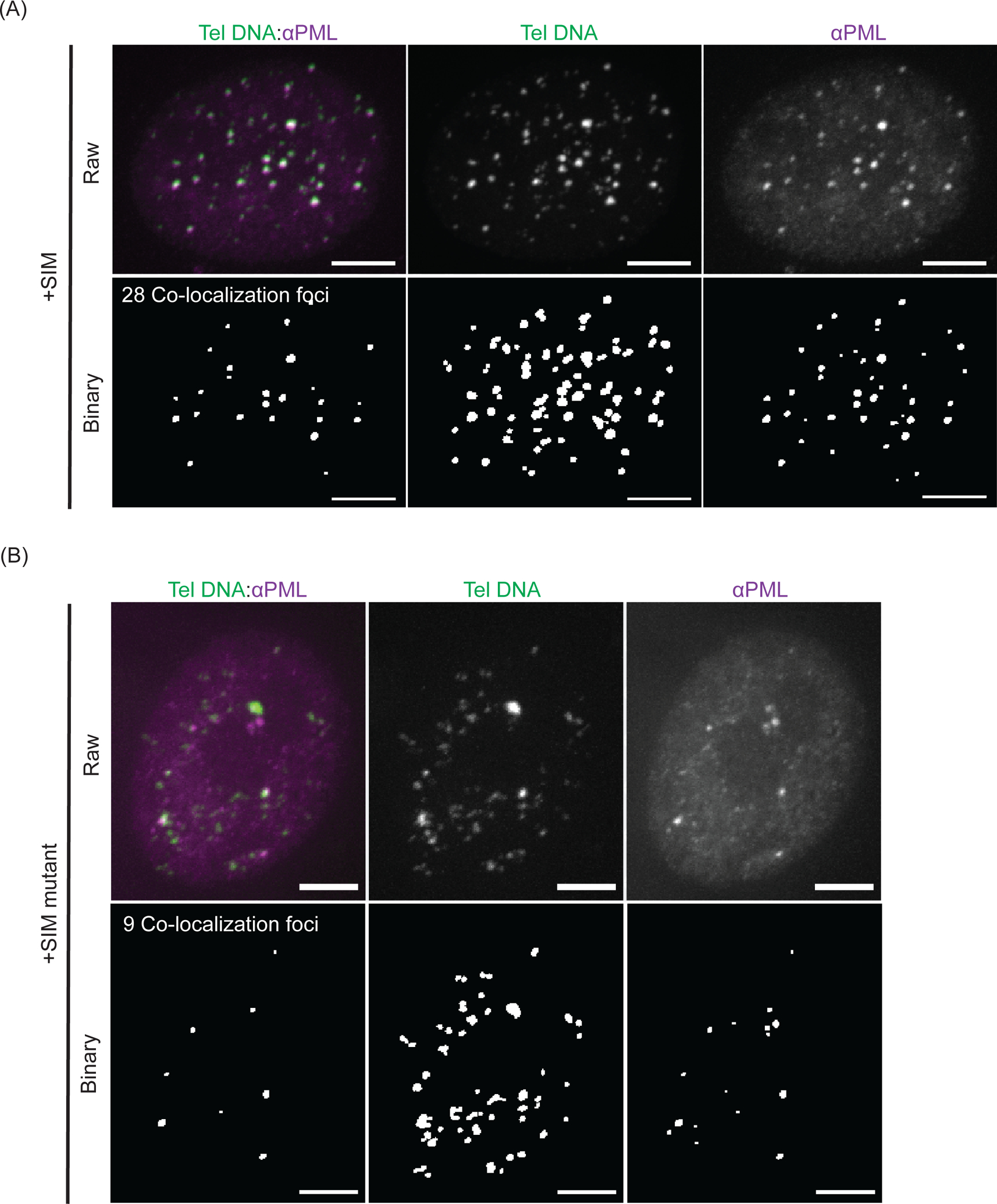
(A) A representative cell for telomere DNA FISH and PML IF after recruiting SIM. Bottom is binary layer identifying telomeres, PML bodies and the number of colocalized PML and telomere DNA foci, i.e., number of APBs. Scale bars, 5 μm. (B) A representative cell for telomere DNA FISH and PML IF after recruiting SIM mutant. At the bottom is the binary layer of the images used to identify the number of colocalized PML and telomere DNA foci, i.e., number of APBs. Scale bars, 5 μm.
The figures here show representative images. For statistical analysis with more cells, please refer to Zhang et. al., 202011.
DISCUSSION:
This protocol demonstrated the formation and dissolution of condensates on telomeres with a chemical dimerization system. Kinetics of phase separation and droplet-fusion-induced telomere clustering are monitored with live imaging. Condensate localization and composition are determined with DNA FISH and protein IF.
There are two critical steps in this protocol. The first is to determine protein and dimerizer concentration. The success in inducing local phase separation at a genomic locus relies on the increase in local concentration of the phase separating protein above the critical concentration for phase separation (Figure 1B). The global concentration of the phase separating protein needs to be high enough so that there are enough proteins to be concentrated locally. The concentration of the phase separating protein cannot be too high so that global phase separation has occurred or can be easily induced. The anchor DNA length (or size of the modified chromatin that the anchor protein binds to) and concentration of Halo-fused anchor protein determine the size of the nucleation center at maximum dimerization efficiency. The larger the anchor size the easy it is to nucleate condensates. The dimerization efficiency is affected by the amount of dimerizers relative to the amount of anchor proteins. Too few dimerizers cannot occupy all the available anchor proteins while too many dimerizers result in non-productive binding of eDHFR to the excess dimerizers rather than to the ones on the anchor protein. Dimerizer concentration, along with the anchor DNA length and concentration of Halo-fused anchor protein, can be used to determine the critical concentration required for nucleating local phase separation. A systematic approach to vary those parameters (anchor DNA length, anchor protein concentration, phase separation protein concentration and dimerizer concentration) can be used to map a multi-dimensional phase diagram. However, if the interest is not in mapping phase diagram but forming chromatin associated-condensates like demonstrated here, it is very easy to simply pick cells with bright Halo-GFP signal (larger anchor size) and cells with a wide range of brightness for mCherry-eDHFR (various phase separating protein concentration) to image with the dimerizer concentration for maximum dimerization determined in Protocol 2.3. The second critical step is to avoid photobleaching in live imaging. Different from global phase separation where droplets (bright mCherry foci labeling the phase separating protein) will emerge after phase separation, local condensation at genomic locations cannot be easily spotted by judging the presence of mCherry foci. This is because recruitment of the protein alone, without phase separation, to genomic loci will result in formation of mCherry local foci. Phase separation occurs after recruitment, so mCherry foci continue to become bigger and brighter after initial recruitment. The phase separation-induced enrichment can occur in GFP channel (the anchor protein) as well, due to the dimerization of the anchor protein to the phase separation protein. Therefore, change of physical properties (size and intensity) of the foci over time rather than the presence of foci should be used to judge phase separation. While it might be difficult to differentiate dimerization or phase separation-induced enrichment of mCherry (prey protein) foci, enrichment of GFP (anchor protein) foci only occurs if there is phase separation (Figure 3D). Therefore, the enrichment of anchor protein can be used to easily judge phase separation. Photobleaching resulted from high laser power or long exposure time during imaging makes it more difficult to judge phase separation from live imaging and therefore should be avoided as much as possible by adjusting imaging conditions. Note that the increases in foci intensity and size over time are characteristics of LLPS but cannot be used as the sole evidence for LLPS. In the case presented here, droplet fusion was used as evidence for the formation of liquid droplets, which may not occur for smaller number of anchors or fewer mobile anchors. Without droplet fusion, other methods such as diffusion of condensate components and sensitivity to small molecule perturbation can be used to further confirm condensate formation8, 9,11.
Though this chemical dimerization system renders temporal resolution required for monitoring phase separation in live cells, it lacks spatial resolution at the cellular and subcellular level. Thanks to the modular design of the dimerizers, it is possible to make light-sensitive dimerizers by attaching a photocage to TMP, making the linker photosensitive or both12, 32, 35. By simply switching dimerizers within the same engineered cell background for different applications, high spatial and temporal control of the dimerization, reversal of dimerization, or both with light can be achieved. We envision with those light-sensitive dimerizers, it will be able to control phase separation with high spatial and temporal precision. Compared to the available optogenetic tools to control phase separation through light sensitive proteins36, 37, a disadvantage of the chemical dimerization system is that it can only reverse phase separation once. However, this system can maintain sustained recruitment and thus phase separation without light, which makes it more suitable for long term live imaging applications such as to follow droplet growth or cellular consequences of phase separation. In addition, the ability to treat a population of cells without light makes it convenient for biochemical assays such as those needed to determine condensate composition or changes in genome organization.
This method can be easily adapted to induce condensates at other locations on the genome. One can simply identify a protein that binds to the genomic location of interest and fuse it to Halo to use it as an anchor (Figure 1B). Alternatively, one can combine this with CRISPR and fuse dCas9 to Halo and use guide RNAs to anchor Halo to the genomic loci of interest38. In addition, one can anchor Halo to an ectopic DNA array (e.g. LacO) integrated into the genome by fusing Halo to the targeting protein (e.g. LacI). One can then use a bottom-up approach to assess the ability of a protein to phase separate locally on chromatin, how its phase separation ability is affected by protein truncations, mutations or post-translational modifications, or how the condensate affects local functions such as chromatin modification, replication or transcription. To summarize, this chemical dimerization system can be used to induce a wide range of condensates on various chromatin locations and is particularly suitable for investigating how the material properties and chemical composition of chromatin-associated condensates contribute to chromatin functions by combining long-term live imaging with biochemical assays.
Supplementary Material
ACKNOWLEDGEMENTS:
This work was supported by US National Institutes of Health (1K22CA23763201 to H.Z., GM118510 to D.M.C.) and Charles E. Kaufman foundation to H.Z.. The authors thank Jason Tones for helping proofread the manuscript.
Footnotes
DISCLOSURES:
The authors have nothing to disclose.
REFERENCES:
- 1.Shin Y, Brangwynne CP Liquid phase condensation in cell physiology and disease. Science. 357 (6357), doi: 10.1126/science.aaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Banani SF, Lee HO, Hyman AA, Rosen MK Biomolecular condensates: Organizers of cellular biochemistry. Nature Reviews Molecular Cell Biology. 18 (5), 285–298, doi: 10.1038/nrm.2017.7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabari BR, Dall’Agnese A, Young RA Biomolecular Condensates in the Nucleus. Trends in Biochemical Sciences. 1–17, doi: 10.1016/j.tibs.2020.06.007 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH Phase separation drives heterochromatin domain formation. Nature. 547 (7662), 241–245, doi: 10.1038/nature22989 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larson AG et al. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 547 (7662), 236–240, doi: 10.1038/nature22822 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boija A et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell. 175 (7), 1842–1855.e16, doi: 10.1016/j.cell.2018.10.042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hur W et al. CDK-Regulated Phase Separation Seeded by Histone Genes Ensures Precise Growth and Function of Histone Locus Bodies. Developmental Cell. 54 (3), 379–394.e6, doi: 10.1016/j.devcel.2020.06.003 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McSwiggen DT, Mir M, Darzacq X, Tjian R Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes & development. 33 (23–24), 1619–1634, doi: 10.1101/gad.331520.119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng A, Weber SC Evidence for and against liquid-liquid phase separation in the nucleus. Non-coding RNA. 5 (4), doi: 10.3390/ncrna5040050 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erdel F et al. Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Molecular Cell. 78 (2), 236–249.e7, doi: 10.1016/j.molcel.2020.02.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang H et al. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Molecular biology of the cell. 31 (18), 2048–2056, doi: 10.1091/mbc.E19-10-0589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ballister ER, Aonbangkhen C, Mayo AM, Lampson MA, Chenoweth DM Localized light-induced protein dimerization in living cells using a photocaged dimerizer. Nature Communications. 5, 1–9, doi: 10.1038/ncomms6475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Research. 59 (17), 4175–4179 (1999). [PubMed] [Google Scholar]
- 14.Zhang JM, Zou L Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell and Bioscience. 10 (1), 1–9, doi: 10.1186/s13578-020-00391-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corpet A et al. PML nuclear bodies and chromatin dynamics: catch me if you can! Nucleic Acids Research. 1–23, doi: 10.1093/nar/gkaa828 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Ma X, Wu W, Chen Z, Meng G PML Nuclear Body Biogenesis, Carcinogenesis, and Targeted Therapy. Trends in Cancer. 6 (10), 889–906, doi: 10.1016/j.trecan.2020.05.005 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Dilley RL, Greenberg RA ALTernative Telomere Maintenance and Cancer. Trends in Cancer. 1 (2), 145–156, doi: 10.1016/j.trecan.2015.07.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobinoff AP, Pickett HA Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends in Genetics. 33 (12), 921–932, doi: 10.1016/j.tig.2017.09.003 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londoño-Vallejo A Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proceedings of the National Academy of Sciences. 106 (37), 15726 LP–15731, doi: 10.1073/pnas.0907689106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang JM, Yadav T, Ouyang J, Lan L, Zou L Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Reports. 26 (4), 955–968.e3, doi: 10.1016/j.celrep.2018.12.102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loe TK, Zhou Li JS, Zhang Y, Azeroglu B, Boddy MN, Denchi EL Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes and Development. 34 (9–10), 650–662, doi: 10.1101/gad.333963.119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potts PR, Yu H The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nature Structural and Molecular Biology. 14 (7), 581–590, doi: 10.1038/nsmb1259 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Chung I, Leonhardt H, Rippe K De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. Journal of Cell Science. 124 (21), 3603–3618, doi: 10.1242/jcs.084681 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP The Mechanisms of PML-Nuclear Body Formation. Molecular Cell. 24 (3), 331–339, doi: 10.1016/j.molcel.2006.09.013 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shima H et al. Activation of the SUMO modification system is required for the accumulation of RAD51 at sites of DNA damage. Journal of Cell Science. 126 (22), 5284–5292, doi: 10.1242/jcs.133744 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Yalçin Z, Selenz C, Jacobs JJL Ubiquitination and SUMOylation in telomere maintenance and dysfunction. Frontiers in Genetics. 8 (MAY), 1–15, doi: 10.3389/fgene.2017.00067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarangi P, Zhao X SUMO-mediated regulation of DNA damage repair and responses. Trends in Biochemical Sciences. 40 (4), 233–242, doi: 10.1016/j.tibs.2015.02.006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banani SF et al. Compositional Control of Phase-Separated Cellular Bodies. Cell. 166 (3), 651–663, doi: 10.1016/j.cell.2016.06.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Min J, Wright WE, Shay JW Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes and Development. 33 (13–14), 814–827, doi: 10.1101/gad.324905.119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proceedings of the National Academy of Sciences of the United States of America. 101 (40), 14373–14378, doi: 10.1073/pnas.0403498101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Chenoweth DM, Lampson MA Chapter 7 - Optogenetic control of mitosis with photocaged chemical dimerizers. Mitosis and Meiosis Part A. 144, 157–164, doi: 10.1016/bs.mcb.2018.03.006 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Aonbangkhen C, Tarasovetc EV, Ballister ER, Chenoweth DM, Lampson MA Optogenetic control of kinetochore function. Nature Chemical Biology. 13 (10), 1096–1101, doi: 10.1038/nchembio.2456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho NW, Dilley RL, Lampson MA, Greenberg RA Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 159 (1), 108–121, doi: 10.1016/j.cell.2014.08.030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA Breakinduced telomere synthesis underlies alternative telomere maintenance. Nature. 539 (7627), 54–58, doi: 10.1038/nature20099 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aonbangkhen C, Zhang H, Wu DZ, Lampson MA, Chenoweth DM Reversible Control of Protein Localization in Living Cells Using a Photocaged-Photocleavable Chemical Dimerizer. Journal of the American Chemical Society. 140 (38), 11926–11930, doi: 10.1021/jacs.8b07753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, Brangwynne CP Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell. 168 (1–2), 159–171.e14, doi: 10.1016/j.cell.2016.11.054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin Y et al. Liquid Nuclear Condensates Mechanically Sense and Restructure the Genome. Cell. 175 (6), 1481–1491.e13, doi: 10.1016/j.cell.2018.10.057 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiang X et al. CRISPR/Cas9-Mediated Gene Tagging: A Step-by-Step Protocol. Methods in Molecular Biology. 1961, 255–269, doi: 10.1007/978-1-4939-9170-9_16 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.