

Abstract
The minireview surveys the modification of native nucleosides as a result of which huge libraries of nucleoside analogs of various structures were synthesized. Particular attention is paid to the synthesis of the so-called prodrug forms of nucleoside analogs which ensure their penetration into the cell and metabolism to active 5'-triphosphate derivatives. All the best known antiviral cyclic nucleoside analogs approved for the treatment of HIV infections, hepatitis B, C, and influenza since the 1960s, as well as those in various stages of clinical trials in recent years, are listed. Nucleoside analogs that have shown the ability to inhibit the replication of SARS-CoV and MERS-CoV are discussed, including remdesivir, approved by the FDA for emergency use in the fight against COVID-19.
Keywords: nucleoside analogs, pronucleotides, antiviral activity
Discovering in the 1950s of DNA structure attracted the attention of chemists and biologists to nucleosides which are starting compounds for the synthesis of nucleotides, the building blocks of nucleic acids. In the early 1960s, the idea arose that analogs of native nucleosides of uridine (1), thymidine (2), cytidine (3), adenosine (4), guanosine (5), 2'-deoxycytidine (6), 2'-deoxyadenosine (7), and 2'-deoxyguanosine (8) (Fig. 1) would be able to change biochemical processes in cells infected with viruses, inhibiting viral replication, that is, to act as antiviral drugs.
Figure 1.
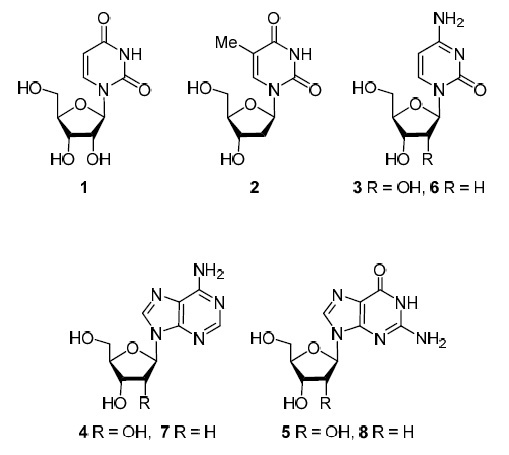
Native nucleosides.
Over the past more than 50 years, numerous libraries of various nucleoside analogs exhibiting antiviral activity have been synthesized. More than 90 compounds have been approved for use as pharmaceuticals.1 The synthesis of nucleoside analogs was carried out by modifying the structure of native nucleosides in three ways:2 1) modification of the nucleobase by functionalizing it with various substituents, replacing the nucleobase with another nitrogen-containing heterocycle; 2) modification of the D-ribofuranosyl fragment carried out by its functionalization with various substituents, elimination of one or both hydroxy groups at C-2' and C-3' atoms, removal of hydrogen atoms at C-2' and C-3' atoms, introduction of an oxygen and/or sulfur atom, replacing the oxygen atom with a sulfur atom; replacement of the D-ribofuranosyl moiety with a cyclopentane or cyclopentene ring; 3) phosphorylation of the hydroxy group at the C-5' atom.
In this minireview, only cyclic nucleoside analogs with antiviral activity are presented which are grouped in accordance with the specified structural features. In order not to violate the logic of the presentation of the material, several analogs of nucleosides in which both the nucleic base and the D-ribofuranosyl fragment are simultaneously modified are not separated into a special section but are mentioned in sections 2 and 3.
1. NUCLEOSIDE ANALOGS WITH A MODIFIED HETEROCYCLIC FRAGMENT
2'-Deoxy-5-iodouridine (9) (Fig. 2) was one of the first synthetically simplest analogs of nucleosides that showed antiviral activity. In 1963, it was approved by the Food and Drug Administration (FDA) for the treatment of herpes simplex and chickenpox under the name idoxuridine.1,2 Two other 5-substituted 2'-deoxyuridines 10 and 11, known as trifluridine and brivudine,1,2 also showed antiviral activity against herpes simplex and varicella zoster viruses (Table 1).
Figure 2.
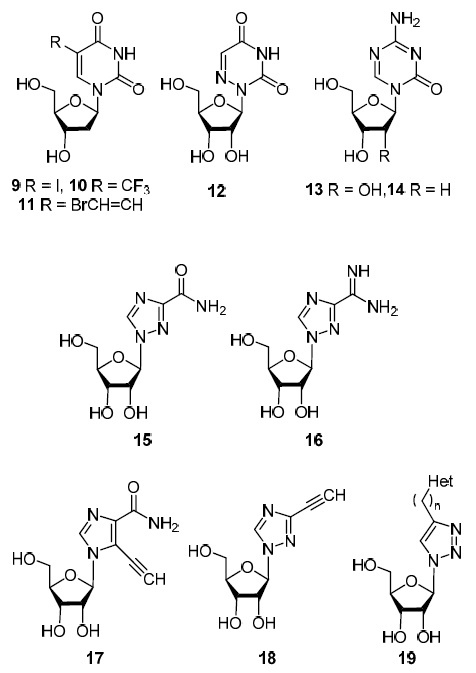
Nucleoside analogs with a modified heterocyclic fragment (Het = uracil-1-yl, 5-fluorouracil-1-yl, 5-bromouracil-1-yl, 5-iodouracil-1-yl, thymin-1-yl, cytisin-1-yl, adenin-9-yl, 2,4-dioxo- 3,4-dihydroquinazolin-1(2H)-yl, n = 1, 3, 4).
Table 1.
Antiviral activity of nucleoside analogs discussed in the review
| Compound | Trivial name, code, systematic name | Virus* | IC50 (EC50), μM | Reference |
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 |
| 9 | Idoxuridine, IdU, 5-iodo-2'-deoxyiodouridine | HSV | 0.37 | 66 |
| 10 | Trifluridine, TFT, 5-trifluoromethyl-2'-deoxyuridine | HSV | 0.88 | 67 |
| 11 | Brivudine, BvdU, (E)-5-(2-bromovinyl)-2'-deoxyuridine | VSV, HSV | 0.033 | 68 |
| 12 | 6-Azauridine |
DENV, WNV, YFV ZIKV HCoV-NL63 |
0.1–0.5 1.5 0.03 |
3 3 61 |
| 13 | 5-AZC, 5-azacytidine | HIV-1 | 57 | 69 |
| 14 | Decitabine, 5-aza-2'-deoxycytidine | HIV-1 | 0.354 | 70 |
| 15 | Ribavirin, RBN, 1-β-D-ribofuranosyl-1,2,4-triazol-3-carboxamide |
HBV VSV RSV H1N1 SARS-CoV MERS-CoV |
43.8 82 25 2.5 50–819 20 |
71 6 6 72 61 61 |
| 16 | Viramidine, taribavirin, 1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamidine |
H1N1 RSV |
8 65 |
72 73 |
| 17 | EICAR, 1-β-D-ribofuranosyl-5-ethynyl-4-carboxamide |
CMV H1N1 RSV VSV Coxsackie B3 |
1.5 3.4 0.75 15 7.5 |
6 6 6 6 |
| 18 | EICAR, 1-β-D-ribofuranosyl-5-ethynyl-4-carboxamide |
CMV H1N1 RSV VSV Coxsackie B3 |
1.5 3.4 0.75 15 7.5 |
6 6 6 6 |
| 17 | triazoleZidovudine, AZT, 3'-azido-3'-deoxythymidine |
ANDV DENV HTNV |
4.4 9.5 10 |
74 75 74 |
| 20 | Zidovudine, AZT, 3'-azido-3'-deoxythymidine |
HIV HBV |
0.03 >100 |
2 76 |
| 21 | 4'-Azidothymidine |
HIV-1 HBV |
0.01 0.63 |
13 13 |
| 22 | 4'-Azido-2'-deoxy-5-methylcytidine | HBV | 5.99 | 13 |
| 23 | 2'-Deoxy-2'-β-fluoro-4'-azidocytidine | HBV | 0.12 | 13 |
| 24 | Balapiravir, R-1626, 4'-azido-2',3',5'-tri-O-isopropanoylcytidine | HCV | 1.3 | 15 |
| 25 | Mericitabine, RGT-7128, 2'-deoxy-2'-α-fluoro-3',5'-di-O-isopropanoyl-2'-β-methylcytidine | HCV | 4.6 | 77 |
| 26 | AadA, 2-amino-4'-azido-2'-deoxyadenosine |
HIV-1 HBV |
0.003 0.006 |
17 17 |
| 27 | AdG, 4'-azido-2'-deoxyguanosine |
HIV-1 HBV |
0.0005 0.009 |
17 17 |
| 28 | CadA, 2-amino-4'-cyano-2'-deoxyadenosine |
HIV-1 HBV |
0.0003 0.0003 |
17 17 |
| 29 | CdG, 4'-cyano-2'-deoxyguanosine |
HIV-1 HBV |
0.0004 0.0004 |
17 17 |
| 30 | Festinavir, censavudine, BMS-986001, 4'-ethynyl-3'-deoxy-2',3'-didehydrothymidine | HIV-1 | 0.002–0.008 | 78 |
| 31 | Islatravir, EfdA, МK-8591, 4'-ethynyl-2-fluoro-2'-deoxyadenosine | HIV-1 | 0.003 | 79 |
| 32 | 2-Amino-4'-vinyl-2'-deoxyadenosine |
HIV-1 HBV |
0.004 0.005 |
17 17 |
| 33 | 2-Amino-4'-trifluoromethyl-2'-deoxyadenosine |
HIV-1 HBV |
0.003 0.004 |
17 17 |
| 34 | 2-Amino-4'-ethyl-2'-deoxyadenosine |
HIV-1 HBV |
0.009 0.025 |
17 17 |
| 35 | 4'-Vinyl-2'-deoxyguanosine |
HIV-1 HBV |
0.004 0.002 |
17 17 |
| 36 | 4'-Trifluoromethyl-2'-deoxyguanosine |
HIV-1 HBV |
0.027 0.005 |
17 17 |
| 37 | 4'-Ethyl-2'-deoxyguanosine |
HIV-1 HBV |
0.006 0.008 |
17 17 |
| 38 | Gemcitabine, 2',2'-difluoro-2'-deoxycytidine |
HIV-1 HCV MERS-CoV SARS-CoV |
0.016. 0.012 1.22 4.96 |
70 80 61 61 |
| 40 | 2'-Ethinyladenosine | DENV | 1.41 | 3 |
| 41 | NITD008, 2'-ethynyl-7-deazaadenosine |
DENV ZIKV |
0.46 0.241 |
3 3 |
| 42 | GS-441524, 1'-cyano-7,9-dideaza-4-azaadenosine |
YFV DENV HCV SARS-CoV EBOV RSV |
11 9.46 4.1 2.24 0.78 0.53 |
3 3 20 20 21 21 |
| 43 | 1'-Cyano-2'-fluoro-2'-deoxy-7,9-dideaza-4-azaadenosine | EBOV, RSV, HCV | >200 | 21 |
| 44 | Didanosine, ddI, 2',3'-dideoxyinosine | HIV | 0.049 | 2 |
| 45 | Zalcitabine, ddC, 2',3'-dideoxycytidine | HIV | 0.6 | 2 |
| 46 | Stavudine, d4T, 3'-deoxy-2',3'-didehydrothymidine |
HIV-1 HIV-1 HIV-2 HIV-1 HIV-1 |
0.009 0.43 0.6 0.4 0.02–0.05 |
22 45 47 57 78 |
| 47 | d4C, 2',3'-dideoxy-2',3'-didehydrocytidine | HIV-1 | 0.005 | 22 |
| 48 | D-Fd4C, 5-fluoro-2',3'-dideoxy-2',3'-didehydrocytidine | HIV | 0.046 | 23 |
| 49 | Elvucitabine, L-Fd4C, (–)-L-(1S,4R)-5-fluoro-2',3'-didehydro-2',3'-dideoxycytidine | HIV | 0.034 | 24 |
| 50 | Lamivudine, 3TC, (–)-L-(1S,4R)-2',3'-dideoxy-3'-thiacytidine |
HIV-1 HBV |
0.002 0.01 |
25 25 |
| 51 | (+)-D-BCH-189, (+)-(1R,4S)-2',3'-dideoxy-3'-thiacytidine |
HIV-1 HBV |
0.2 0.5 |
25 25 |
| 52 | Emtricitabine, (–)-L-FTC, (–)-L-(1S,4R)-5-fluoro-2',3'-dideoxy-3'-thiacytidine |
HIV-1 HBV |
0.008 0.01 |
25 25 |
| 53 | (+)-D-FTC, (+)-D-(1R,4S)-5-fluoro-2',3'-dideoxy-3'-thiacytidine |
HIV-1 HBV |
0.84 0.96 |
25 25 |
| 54 | Apricitabine, (–)-D-dOTC, ATC, (–)-D-(1R,4R)-2'-deoxy-3'-oxa-6'-thiocytidine |
HIV-1 HIV-1 |
0.2 0.36 |
26 27 |
| 55 | (+)-L-dOTC, (+)-L-(1S,4S)-2'-deoxy-3'-oxa-6'-thiocytidine | HIV-1 | 0.28 | 26 |
| 56 | (–)-D-β-Dioxolane-T, (–)-D-1-[(2R,4R)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]thymine | HIV | 0.39 | 28 |
| 57 | (+)-L-β-Dioxolane-T, (+)-L-1-[(2S,4S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]thymine | HIV | 4.81 | 28 |
| 58 | (+)-D-1-[(2R,4R)-2-(Hydroxymethyl)-1,3-dioxolan-4-yl]cytosine |
HIV HBV |
0.016 0.01 |
28 28 |
| 59 | Troxacitabine, BCH-4556, (−)-L-β-dioxolane-C, (–)-L-[(2S,4S)-2-(hydroxymethyl)-1,3-dioxolan-4-yl]cytosine |
HIV HBV |
0.002 0.0005 |
28 28 |
| 60 | Aristerоmycin, Ari, Ado, 6'-deoxyadenosine |
VV VSV Coxsackie B4 |
2.6 0.75 7.5 |
30 30 30 |
| 61 | Neplanocin A, 4',6'-didehydro-6'-deoxyadenosine |
VV VSV Coxsackie B4 |
0.11 0.04 0.15 |
30 30 30 |
| 62 | Abacavir, ABC, 2-amino-10-cyclopropyl-2',3'-dideoxy-2',3'-didehydro-6'-deoxyadenosine | HIV-1 | 4.0 | 81 |
| 63 | Carbovir, CBV, 2',3'-dideoxy-2',3'-didehydro-6'-deoxyguanosine | HIV-1 | 4.6 | 81 |
| 64 | Entecavir, ETV, 2'-deoxy-6'-methylidene-6'-deoxyguanosine |
HIV-1 HBV HBV |
1.0 0.0007 0.00018 |
76 76 82 |
| 74 | Nikavir, 3'-azido-3'-deoxythymidine-5'-phosphite | HIV | 1.0 | 41 |
| 80 | TriPPPro-d4TTP, 3'-deoxy-2',3'-didehydrothymidine-5'-[bis(4-aroyloxyphenyl)] triphosphate | HIV-1 | 0.03–0.07 | 45 |
| 81 | TriPPPro-CBVTP, 2',3'-dideoxy-2',3'-didehydro-6'-deoxyguanosine-5'-[bis(4-aroyloxyphenyl)] triphosphate | HIV-1 | 0.99 | 36 |
| 86 | cycloSal-d4T, (3'-deoxy-2',3'-didehydrothymidin-5'-yl)(cyclosaligenyl) monophosphate | HIV-1 | 0.15 | 47 |
| 87 | ddA, 2',3'-dideoxyadenosine | HIV-1 | 4.33 | 48 |
| 88 | cycloSal-ddA, (2',3'-dideoxyadenosin-5'-yl)(cyclosaligenyl) monophosphate | HIV-1 | 0.03 | 49 |
| 89 | d4A, 2',3'-dideoxy-2',3'-didehydroadenosine | HIV-1 | 30 | 49 |
| 90 | cycloSal-d4A, (2',3'-dideoxy-2',3'-didehydroadenosin-5'-yl)(cyclosaligenyl)-monophosphate | HIV-1 | 0.065 | 49 |
| 91 | cycloSal-ABC, {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-ylmethyl}(cyclosaligenyl) monophosphate | HIV-1 | 0.95 | 50 |
| 92 | cycloSal-CBC, [(1S,4R)-4-(2-amino-6-oxo-9H-purin-9-yl)-2-en-1-ylmethyl](cyclosaligenyl) monophosphate | HIV-1 | 0.15 | 50 |
| 93 | cycloSal-BvdU, [2'-deoxy-5-(2-bromovinyl)uridin-5'-yl](cyclosaligenyl) monophosphate | EBV | 1.8 | 46 |
| 101 |
ProTide-d4T, 3'-deoxy-5'-О-[4-(trifluoromethyl)phenoxy(О-methylalanino)phosphoryl]-2',3'-didehydrothymidine ProTide-d4T, 3'-deoxy-5'-О-[3-(trifluoromethyl)phenoxy(О-methylalanino)phosphoryl]-2',3'-didehydrothymidine |
HIV-1 HIV-1 |
0.037 0.041 |
57 57 |
| 108 | Sofosbuvir, isopropyl (S)-2-({[(2R,3R,4R,5R)-3-hydroxy-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-methyl-4-fluorotetrahydrofuran-2-yl]methoxy}(phenoxy)phosphoryl)aminopropionate |
HCV DENV ZIKV |
0.52 1.4 1.0–32 |
55 83 84 |
| 110 | Remdesivir, GS-5734, (2-ethylbutyl) (S)-[({[(2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-cyanotetrahydrofuran-2-yl]methoxy}(phenoxy)phosphoryl)amino]propionate |
SARS-CoV MERS-CoV EBOV RSV HCV |
0.07 0.03–0.07 0.066 0.021 0.18 |
61 61 85 85 86 |
*ANDV – Andes orthohantavirus; CMV – cytomegalovirus of the herpes virus family; Coxsackie virus – Coxsackie viruses of the enterovirus family; DENV – dengue virus; EBOV – Ebola virus; EBV – herpes virus type 4; H1N1 – one of the influenza A viruses; HBV – hepatitis B virus; HCoV-NL63 – human coronavirus of the Netherlands; HCV – hepatitis C virus; HIV – human immunodeficiency virus; HSV – herpes simplex virus; HTNV – Hantaan orthohantavirus (one of the causative agents of hemorrhagic fever); MERS-CoV – Middle Eastern coronavirus strain; RSV – human respiratory syncytial virus; SARS-CoV – the coronavirus strain that caused the COVID-19 epidemic; VSV – vesicular stomatitis virus; VV – vaccinia virus; WNV – West Nile virus; YFV – yellow fever virus; ZIKV – Zika virus.
The introduction of a nitrogen atom into the heterocyclic fragment of uridine (1) and cytidine (3) at position 5 or 6 led to a sharp change in the antiviral activity. If 5-substituted 2'-deoxyuridines 9 and 10 are active against DNA viruses of herpes simplex and varicella zoster, then 6-azauridine (12), 5-azacytidine (13), and 2'-deoxy-5-azacytidine (decitabine) (14) (Fig. 2) already inhibit the replication of RNA viruses. Thus, 6-azauridine (12) has a pronounced activity against dengue, Zika, yellow fever, and West Nile fever flaviviruses,3 whereas cytidine (3) analogs – compounds 13 and 14 – were approved by the FDA for the treatment of HIV in 20051 (Table 1 ).
Among the considered class of nucleoside analogs, ribavirin (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide) (15) is noteworthy (Fig. 2). Ribavirin (15) is the first nucleoside analog that showed activity against several RNA viruses4 (hepatitis C, influenza A viruses, respiratory syncytial virus, Table 1). Additionally, ribavirin (15) is the only nucleoside analog among all drugs approved by the FDA for the treatment of influenza.4 Moreover, ribavirin (15) is the first example of the search for antiviral agents among isosterics of native nucleosides (ribavirin is an isostere of guanosine (5)).
This approach has proven to be very productive. Viramidine (16) (Fig. 2), an isostere of adenosine (4), was active against influenza viruses, hepatitis C and B, yellow fever, and respiratory syncytial virus.5 Substitution of 1,2,4-triazole ring in ribavirin (15) to imidazole led to 1-(β-D-ribofuranosyl)-5-ethynylimidazole-4-carboxamide (EICAR) (17) which demonstrates a wide spectrum of activity against DNA and RNA viruses and significantly exceeds the activity of ribavirin (15).6 Replacement of the carboxamide group in ribavirin (15) with an ethynyl group resulted in 1-(β-D-ribofuranosyl)-3-ethynyl-1,2,4-triazole (ETAR) (18) which showed a high level of antiviral activity against hantaviruses and flaviviruses.7 Advances in the synthesis of antiviral agents based on the scaffold of ribavirin (15) led chemists to employ the 1,2,3-triazole ring as a bridge between the nucleic base and the D-ribofuranosyl fragment. A series of analogs of nucleosides 19 were synthesized (Fig. 2),8–12 in which compounds with antiviral activity against influenza A H1N1 virus and Coxsackie B3 virus were found.12
2. NUCLEOSIDE ANALOGS WITH A MODIFIED SUGAR FRAGMENT
2.1. Variation of substituents at the C-4' atom
The first nucleoside analog found to have antiHIV activity was zidovudine (3'-azido-3'-deoxythymidine) (20) (Fig. 3), approved in 1987 by the FDA for the treatment of HIV infection.4 Zidovudine (20) was the first nucleoside analog for which the target – HIV reverse transcriptase (RNA-dependent DNA polymerase) – was established.2,4
Figure 3.
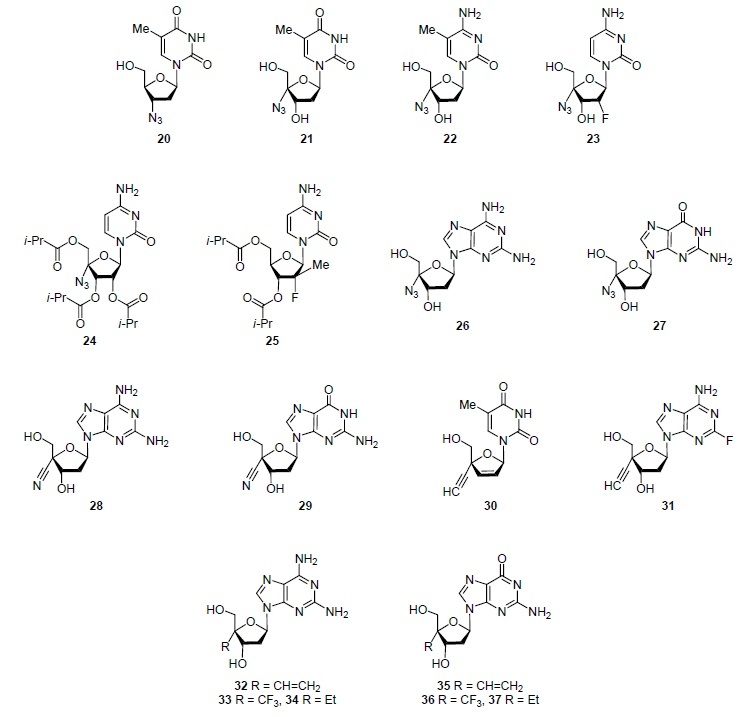
Nucleoside analogs with a variable substituent at the C-4 'atom.
It is interesting to trace how the antiviral activity of zidovudine (20) changed upon modification (Table 1). Figure 3 shows analogs of nucleosides 21–24, 26, 27 in which, in contrast to zidovudine (20), the azido group is located at the C-4' atom. These compounds became known for their high activity against hepatitis B (compounds 21–23, 26, 27)13 or hepatitis C (compounds 24, 25) viruses.3,14 It can be assumed that it is the introduction of the azido group at the С-4' position of 2'-deoxycytidine (6), 2'-deoxyadenosine (7), and 2'-deoxyguanosine (8) (Fig. 1) that provided the emergence of activity against the hepatitis B virus. Note that the different nature of the heterocyclic fragment in compounds 21 (thymine), 22 (5-methylcytosine), 23 (cytosine), 26 (2-aminoadenine), and 27 (guanine) does not affect the purposefulness of their action (Table 1). Purposefulness was achieved by acylation of hydroxy groups at the C-2', C-3', and C-5' atoms. Thus, balapiravir (24) and mericitabine (25) (Fig. 3), in contrast to compounds 21–23, 26, 27, inhibit the replication of the hepatitis C virus,15,16 belonging to the class of RNA viruses (Table 1). In 2018, it was found that 4'-substituted analogs of 2'-deoxyadenosine (7) and 2'-deoxyguanosine (8), which are highly active against the hepatitis B virus, a DNA virus, are highly active against HIV (related to RNA viruses) as well.17 This property is possessed by both 4'-azidosubstituted analogs of nucleosides 26 and 27 and analogs of nucleosides 28 and 29 (Fig. 3), which contain not the azido but the cyano group at the C-4' position.17 Substitution of the azido group and cyano groups at the C-4' position by the ethynyl group does not affect the antiviral activity. Thus, thymidine (2) analog festinavir (30)16 and adenosine (4) analog islatravir (31)14,18 inhibit HIV reverse transcriptase (Table 1). Moreover, replication of hepatitis B virus and HIV is inhibited by 2-amino-2'-deoxyadenosine analogs 32–34 and 2'-deoxyguanosine analogs 35–37 containing at the position C-4' the vinyl (compounds 32, 35), trifluoromethyl (compounds 33, 36), and even ethyl (compounds 34, 37) groups.17
2.2. Variation of substituents at the С-1' and С-2' atoms
Gemcitabine (38) (2'-deoxy-2',2'-difluorocytidine) is the best known among the nucleoside analogs with substituents at C-1' or C-2' carbon atoms (Fig. 4). In 1996, gemcitabine (38) was approved by the FDA for the treatment of various types of cancer (cancer of the pancreas, ovaries, lungs, breast, and bladder).1 However, in recent years, this compound has been found to exhibit potent antiviral activity against HIV, hepatitis C virus, influenza A virus, and a number of enteroviruses19 (Table 1). A significant limitation of the use of gemcitabine (38) in vivo is its rapid metabolism by cytidine deaminase. Therefore, its derivative 39 is currently being studied in which the amino group is protected from deamination by the valproic acid functionality.19 It was noted above that 6-azauridine (12) (Fig. 2) possesses a pronounced antiviral activity against dengue, Zika, yellow fever, and West Nile fever flaviviruses,3 being an inhibitor of the RNA-dependent RNA polymerase of these viruses. Adenosine analogs 40–43 with an ethynyl group at the C-2' atom or cyano group at the C-1' atom (Fig. 4) demonstrated a clear dependence of the antiviral activity both on the structure of the heterocyclic fragment of the molecule and on the nature of the substituents at the C-1' and C-2' atoms. In contrast to 6-azauridine (12), 2'-ethynyladenosine (40) is active only against the dengue flavivirus.3 The absence of the N-7 atom endows compound 41 with activity against the Zika flavivirus3 (Table 1). A radical change in the structure, in comparison with N-nucleosides 40, 41, of C-nucleoside 42 (the absence of nitrogen atoms in positions 7 and 9 of the adenine fragment, the appearance of a nitrogen atom in position 4 and a cyano substituent at the C-1' atom) leads to a significant change of biological activity – it is active against hepatitis C, Ebola, SARS-CoV viruses20,21 (Table 1). At the same time, a seemingly insignificant change in the structure, the replacement of the hydroxy group at the C-2' atom by the fluorine atom in compound 42 (structure 43), led to a loss of antiviral activity against hepatitis virus, flaviviruses, and Ebola virus.21
Figure 4.
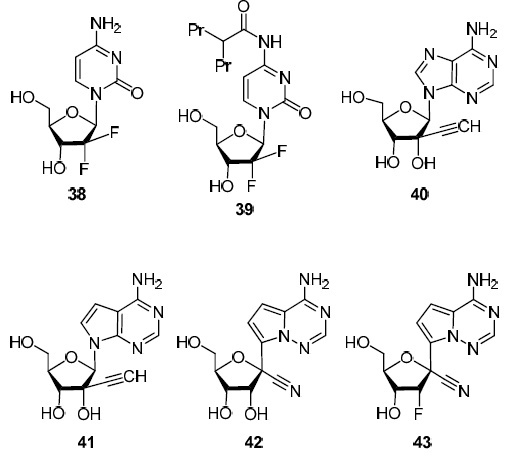
Nucleoside analogs with variable substituents at the C-1' and C-2' atoms.
2.3. 2',3'-Dideoxynucleosides
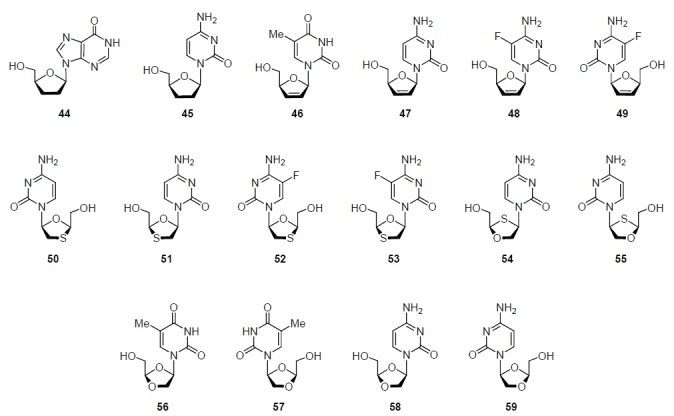
It is to this group of nucleoside analogs that the most well-known drugs for the treatment of HIV infection belong. Following zidovudine (20), the other five nucleoside analogs that inhibit HIV reverse transcriptase and are approved by the FDA for the treatment of HIV infection – didanosine (44), stavudine (46), lamivudine (50), emtricitabine (52) (Fig. 5), and abacavir (62) (Fig. 6) – became known as nucleoside reverse transcriptase inhibitors (NRTIs). HIV is an RNA virus, but since DNA is formed during its replication, it is classified in a special group of RNA viruses called retroviruses. Therefore, the listed NRTIs are often referred to as antiretroviral drugs.
Figure 5.
2',3'-Dideoxynucleosides.
Figure 6.
Carbocyclic nucleoside analogs.
Didanosine (2',3'-dideoxyinosine) (44) (Fig. 5), synthesized in 1964 and approved by the FDA in 1991, was the second NRTI after zidovudine (20) for the treatment of HIV infection.2,4 Zalcitabine (2',3'-dideoxycytidine) (45), synthesized in the late 1960s and launched on the pharmaceutical market in 1992, became the third nucleoside analog approved by the FDA for the treatment of HIV infection (Table 1). However, limited efficacy, unfavorable pharmacokinetics, and side effects led to this compound being withdrawn from the pharmaceutical market in June 2006, becoming the first antiretroviral drug to be phased out.2,4 Stavudine (3'-deoxy-2',3'-didehydrothymidine) (46) (Fig. 5) was synthesized in the late 1960s, but its ability to inhibit HIV replication was discovered only in 1982 during the AIDS epidemic that began in New York. Stavudine (46), approved for use in 1994, became the fourth drug among nucleoside analogs for the treatment of HIV infection.2,4
Cytidine analog of stavudine (46), 3'-dideoxy-2',3'-didehydrocytidine (47), has approximately the same activity against HIV (EC50 0.009 and 0.005 μM, respectively22). The introduction of a fluorine atom into the C-5 position of the cytosine ring decreases the activity, and compound 48 inhibits HIV replication at EC50 of 0.046 μM.23 Judging from the literature data,23,24 elvucitabine (49) (Fig. 5), which is undergoing clinical trials,16 has practically the same activity as its enantiomer 48 (Table 1). This is surprising, since elvucitabine (49) belongs to L-nucleosides, an unusual group of nucleoside analogs.
It is strictly regulated in nature that amino acids have the L-configuration, whereas sugars and, therefore, nucleosides and nucleotides have the D-configuration. Hence, it can be assumed that the active centers of transmembrane proteins and intracellular enzymes are primarily kinases that are complementary specifically to D-nucleosides and their analogs. Consequently, any properties of L-nucleosides and their analogs that affect cellular metabolism should be less pronounced than those of their D-enantiomers or be absent altogether.
However, when comparing the antiviral activity against HIV and hepatitis B virus of enantiomeric analogs of nucleosides 50 and 51 (Fig. 5, Table 1), it can be seen that in reality the opposite is true: the activity of L-enantiomer 50 is, respectively, 100 and 50 times higher than the activity of D-enantiomer 51.25 L-Nucleoside 50, approved by the FDA in 1996 and named lamivudine, became chronologically the fifth drug among nucleoside analogs for the treatment of HIV infection.1,2 Fluorination at the C-5 position of the cytosine ring of lamivudine (50) and its D-enantiomer 51 practically did not change the antiviral activity (Table 1). L-Nucleoside 52, named emtricitabine, like L-nucleoside 50, exceeds its D-enantiomer 53 in antiviral activity against HIV and hepatitis B virus by about 100 times25 (Table 1). Emtricitabine (52) was among the seven nucleoside reverse transcriptase inhibitors approved by the FDA for the treatment of HIV infection.4
Nucleoside analogs 50–53 are derivatives of zalcitabine (45) in which the C-3' atom is replaced by a sulfur atom, that is, the 2',3'-dideoxy-D-ribofuranosyl ring is replaced by the 1,3-oxathiolane ring (Fig. 5). The analogs of nucleosides 54 and 55, the D- and L-enantiomers, respectively, are their isosteres in which the oxygen and sulfur atoms are interchanged (Fig. 5). While the antiviral activity of L-enantiomers of nucleoside analogs 50–53 exceeded that of their D-enantiomers (Table 1), enantiomers 54 and 55 had practically the same ability to inhibit HIV replication (IC50 0.2–0.3 μM).26,27 The most famous of them – apricitabine (54) – is still in clinical trials due to commercial and legal hurdles.16
Replacement of the sulfur atom by an oxygen atom in compounds 50–55 leads to another group of 2',3'-dideoxynucleosides, dioxolane derivatives 56–59 (Fig. 5). They also have high antiviral activity against HIV and hepatitis B virus.28 However, unlike compounds 50–53, (+)-L-nucleoside 57 is less active than its (–)-D-enantiomer 56 (Table 1).
Substitution of thymine for cytosine in compounds 56 and 57, i.e., the transition to 1-(1,3-dioxolan-4-yl)cytosines 58 and 59 leads to an increase in the antiviral activity against HIV by 20 and 2000 times, respectively28 (Table 1). In addition, the substitution of cytosine for thymine led to an inversion of activity in favor of the L-enantiomer. Thus, L-nucleoside 59 inhibits in vitro the replication of HIV and hepatitis B virus, respectively, 8 and 20 times better than D-nucleoside 58 (Table 1). Despite the fact that statistically the antiviral activity of L-nucleosides is better than that of D-nucleosides, special examination of the dependence of the antiviral activity of nucleoside analogs on their D- or L-configuration is required.
2.4. Carbocyclic nucleoside analogs
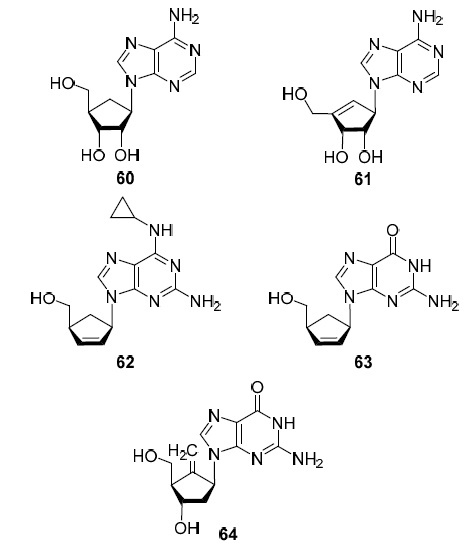
Aristeromycin (60) synthesized in 19662 (Fig. 6) is the first representative of carbocyclic nucleoside analogs in which the D-ribofuranosyl moiety is replaced by a cyclopentane or cyclopentene ring. Interestingly, two years later it was isolated from Streptomyces citricolor.2 Later, in 1981, neplanocin A (61)2 (Fig. 6), another carbocyclic analog of adenosine was found in nature which differs from aristeromycin (60) by the presence of the С(4')=С(6') double bond. These two carbocyclic analogs of adenosine and their various synthetic derivatives exhibit a broad spectrum of antiviral activity against RNA and DNA viruses: HIV, hepatitis C, hepatitis B, and enteroviral vesicular stomatitis viruses29,30 (Table 1).
In abacavir (62), a carbocyclic analog of 2-amino-10-cyclopropyladenosine, and carbovir (63), a carbocyclic analog of guanosine, the double bond of the cyclopentene ring is located between the C2' and C3' atoms (Fig. 6). Both nucleoside analogs exhibited high antiviral activity against HIV; however, abacavir (62) was approved by the FDA for the treatment of HIV infection in 1998, and carbovir (63), due to its cytotoxicity, low solubility, and poor oral bioavailability had limited clinical use.2,4 It is interesting to note that abacavir (62) in vivo is first phosphorylated by intracellular kinases converting to the 5'-triphosphate form which then undergoes deamination converting to the 5'-triphosphate form of carbovir (63).2 Thus, it turns out that abacavir (62) is a prodrug of carbovir (63).
One of the most interesting carbocyclic nucleosides from the structural point of view is entecavir (64). This is an analog of 2'-deoxyguanosine (8) in which an exocyclic double bond is introduced in place of the ribofuranosyl oxygen atom (Fig. 6). In 2005, entecavir (64) was approved for the treatment of both hepatitis B and HIV infection2,31 (Table 1). It is assumed that it is the exocyclic double bond that provides the high activity of this compound against hepatitis B, since it is the key fragment of the molecule that ensures its effective binding to the hydrophobic pocket of the active domain of the DNA-dependent DNA polymerase of the hepatitis B virus.2
3.5. 5'-PHOSPHORYLATED NUCLEOSIDE ANALOGS
In 1986–1990, it was shown in a series of articles that zidovudine (20) and stavudine (46) in uninfected and HIV-infected cells are metabolized by various cellular kinases (phosphotransferases) to the corresponding 5'-mono-, 5'-di-, and 5'-triphosphate derivatives.32 Later, it was suggested that all nucleoside analogs are susceptible to such metabolism and viral replication is inhibited not by themselves but by their 5'-triphosphate derivatives.1,3,33 These data focused the attention of chemists on the synthesis of 5'-triphosphate analogs of nucleosides, primarily those that have been approved as drugs for the treatment of HIV infection and hepatitis. Summarizing the data from more than 100 studies devoted to the synthesis of 5'-triphosphate derivatives of nucleoside analogs which were compiled in the latest review on this topic,34 three most rational strategies can be identified.
The first strategy is shown in Scheme 1. The reaction of nucleoside analog 65 with protected 2',3'-hydroxy groups (if any) with salicyl chlorophosphite (2-chloro-1,3,2- benzodioxaphosphorin-4-one) (66) in pyridine or PCl3 in CH2Cl2 followed by hydrolysis to give 5'-phosphite 67 whose protecting groups (PG) are then removed. Then, 5'-phosphite 68 is activated with trimethylchlorosilane in pyridine, oxidized with iodine to a 5'-monophosphate derivative, and its reaction with tris(tetra-n-butylammonium) pyrophosphate gives 5'-triphosphate derivative 69 of the starting nucleoside analog 65 (Scheme 1).34 A crucial disadvantage of this strategy is the need to protect the hydroxy groups of the D-ribofuranosyl or cyclopentane ring from highly reactive salicyl phosphite (66) which adds two extra steps (introduction of protection and its removal). But for 2',3'-dideoxy analogs of nucleosides, this approach is quite acceptable.
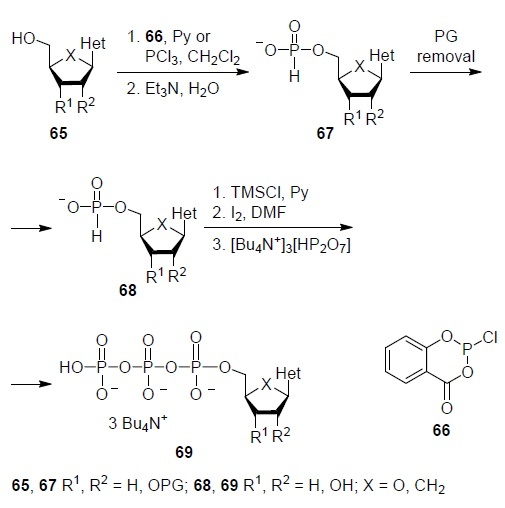
Scheme 1
The strategy presented in Scheme 2 does not have this drawback. Its difference is that not salicyl chlorophosphite (66) is involved in the reaction with the analog of nucleosides 70 but the product of its reaction with an excess of tris(tetra-n-butylammonium) pyrophosphate, compound 71. Due to its significant steric bulk, phosphorylation proceeds regioselectively at the C-5' position which makes it possible to involve in the reaction analogs of nucleosides 70 with unprotected hydroxy groups.34 The resulting intermediate 72 containing threeand pentavalent phosphorus atoms is then oxidized with a solution of iodine in aqueous pyridine and hydrolyzed to afford the target 5'-triphosphate derivative 69 of the original nucleoside analog 70.34
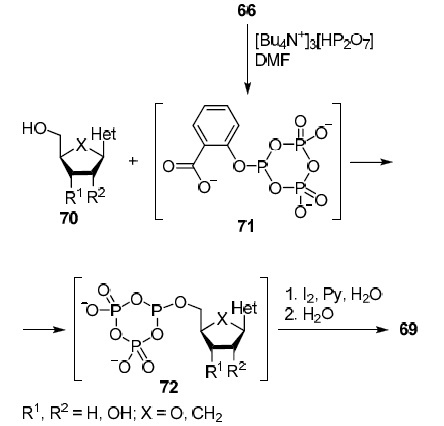
Scheme 2
The third strategy is shown in Scheme 3. This the most famous three-step one-pot synthesis of 5'-triphosphate derivatives of nucleoside analogs was developed by Ludwig in 1981.34 The reaction of the unprotected nucleoside analog 70 with phosphorus oxychloride in trimethyl phosphate gives 5'-dichlorophosphate 73 which is immediately involved in the reaction with a fivefold excess of tris(tetra-n-butylammonium) pyrophosphate. The resulting intermediate 72 is hydrolyzed with triethylammonium bicarbonate (TEAB) to give the target 5'-triphosphate 69.34
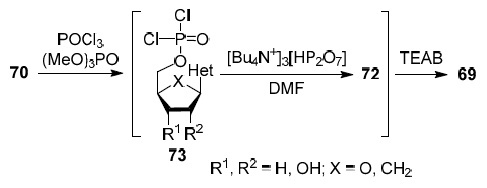
Scheme 3
Surprisingly, despite of the many publications on the synthesis of 5'-triphosphate derivatives of nucleoside analogs, we found in the literature in the last decade only a few studies on their antiviral activity35,36 and the ability to inhibit viral polymerases thus interrupting the synthesis of viral DNA.37,38 However, Krayevsky's group, known for their work on the synthesis of thymidine (2) derivatives,39,40 already in 1989 synthesized 5'-phosphite derivative of 3'-azido-3'-deoxythymidine 74 (Scheme 4) called phosphazide, which demonstrated antiHIV activity and low toxicity on MT-4 cell line.41,42 Over the next 10 years, phosphazide (74) underwent a full range of preclinical and clinical trials and in 1999 was registered in the Russian Federation as an antiviral agent used in combination antiretroviral therapy against HIV infection called nikavir.40,42 Thus, phosphazide (74) became the first and, according to our information, the only 5'-phosphite analog of nucleosides among antiretroviral drugs and a prodrug form of zidovudine (20).
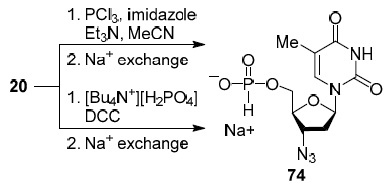
Scheme 4
The synthesis of phosphazide (74) (Scheme 4) differed from the synthesis of 5'-phosphite 67 presented in Scheme 1. Instead of the highly reactive salicyl chlorophosphite (66), the less reactive tris(1,3-imidazolyl) phosphine41 or monon- butylammonium phosphate in the presence of 1,3-dicyclohexylcarbodiimide (DCC) was used (Scheme 4).43 This approach, in principle, makes it possible to apply this strategy for the synthesis of 5'-phosphite derivatives of nucleoside analogs with unprotected hydroxy groups at the C-2' and C-3' atoms.
Curiously, the success of Krayevsky's group, which for the first time actually showed that synthetic 5'-phosphorylated derivatives of nucleoside analogs can become antiretroviral drugs, did not cause a response among scientists searching for antiviral nucleoside analogs. The probable reason for such a long lack of interest among biologists in synthetic nucleotides was the statement expressed in the early 1960s that negatively charged 5'-triphosphate analogs of nucleosides cannot penetrate into cells through lipid-rich cell membranes.44 This statement turned out to be erroneous, since during 2000–2010 integral proteins were identified in human cells with the help of which not only nucleoside analogs but also various organic anions penetrate through membranes.16,33 Thus, the ability of 5'-triphosphate nucleoside analogs to penetrate into the cell was rehabilitated.
Nevertheless, for 40 years, in parallel with the synthesis of nucleoside analogs containing a 5'-triphosphate fragment, the synthesis of their derivatives in which in order to facilitate the penetration of this hydrophilic anionic fragment through the lipid cell membrane it is “masked” with various protective hydrophobic groups has been developing. It was suggested that inside the cell these groups are removed by hydrolases and the formed active 5'-triphosphate forms interrupt the synthesis of viral RNA or DNA.16,19,44,45 Since this approach began to be applied primarily to nucleoside analogs approved as drugs, their derivatives with protected 5'-phosphate groups came to be called prodrugs. Analysis of the literature makes it possible to distinguish three currently used strategies for the synthesis of prodrugs.
The first strategy is the synthesis of 5'-triphosphate derivatives of nucleoside analogs containing two terminal hydrophobic fragments readily hydrolyzed by cell hydrolases (Scheme 5). At the first stage, the carbocyclic analog of nucleosides 46 or 63 is converted into the corresponding 5'-monophosphate 75 or 76 via reaction with phosphorus oxychloride by the Sowa and Ouchi method.36,45 Scheme 5 shows chemical transformations using the antiviral drugs stavudine (46) and carbovir (63) as examples, but it can be any 2',3'-dideoxynucleoside, including with a carbocyclic structure.
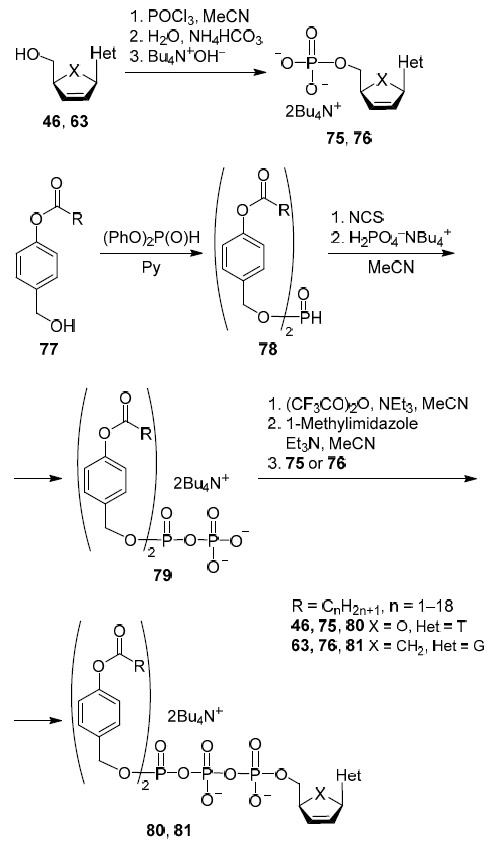
Scheme 5
At the second stage, diaryl phosphite 78 is obtained by the reaction of 4-acyloxybenzyl alcohols 77 with diphenyl phosphite which after activation with N-chlorosuccinimide is converted into pyrophosphate 79 by the reaction with tetra-n-butylammonium monophosphate. At the final stage, the reaction of pyrophosphate 79, previously converted into the amidophosphate form, with 5'-monophosphates 75, 76 produces the target 5'-triphosphates 80 and 81 with two terminal hydrophobic fragments. According to Meier et al.,45 antiviral activity against HIV of stavudine prodrug 80 exceeded the activity of stavudine itself (46) by 10 times45 (Table 1) while the activity of carbovir prodrug 81 exceeded the activity of carbovir (63) only three times36 (Table 1).
The second strategy, presented in Scheme 6, was named cyclosaligenin scheme after the name of the key reagent, salicylic alcohol (saligenin) (84)46 which is involved in the reaction with 5'-dichlorophosphates 83 obtained from 2',3'-dideoxy- and 2',3'-dideoxy-2',3'-didehydronucleosides 82 leading to the production of prodrug 5'-cyclosaligenyl nucleosides 85.46,47 It is assumed that cellular enzymes hydrolyze prodrugs 85 to 5'-monophosphate derivatives of nucleoside analogs 82 and salicylic alcohol (84) which is nontoxic and does not exhibit antiviral activity.46
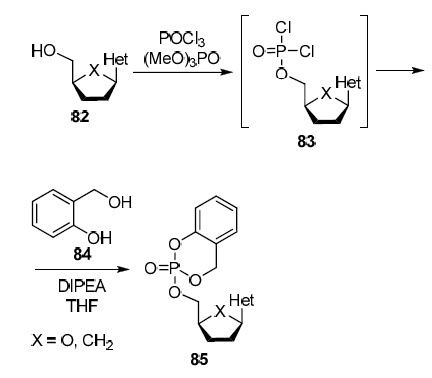
Scheme 6
It is interesting to compare the antiviral activity of some nucleoside analogs, namely, stavudine (46), 2',3'-dideoxyadenosine (87), 2',3'-dideoxy-2',3'-didehydroadenosine (89), abacavir (62), carbovir (63), and brivudine (11) with their prodrug forms, the corresponding 5'-cyclosaligenyl nucleosides 86, 88, 90–93 (Fig. 7). It was found that the antiviral activity against HIV of stavudine (46) and its prodrug 86 is practically the same47 (Table 1). Functionalization of 2',3'-dideoxyadenosine (87) with the 5'-cyclosaligenyl fragment led to an increase of the anti- HIV activity of its derivative 88 by a factor of 100,48,49 and the antiHIV activity of prodrug 90 exceeded the activity of the starting 2',3'-dideoxy-2',3'-didehydroadenosine (89) 400-fold.49 The effectiveness of 5'-cyclosaligenyl derivative 91 of abacavir (62) improved 4-fold,50 and the functionalization of carbovir (63) with the 5'-cyclosaligenyl fragment increased the antiHIV activity of its derivative 92 30 times50 (Table 1). Functionalization of brivudine (11) with a cyclosaligenyl fragment led to the appearance for its derivative 93 of pronounced activity against the herpes virus46 (Table 1).
Figure 7.
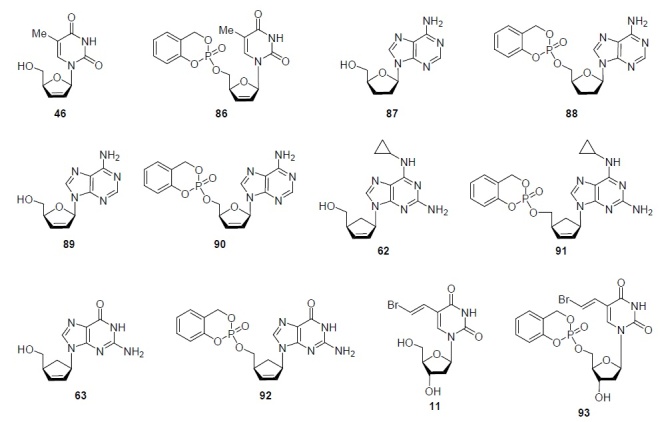
Some antiviral nucleoside analogs and their prodrug 5'-cyclosaligenyl derivatives.
The third strategy is the synthesis of prodrug forms of nucleoside analogs in the form of their 5'-amidophosphate derivatives. Scheme 7 shows the synthesis of 5'-arylamidophosphate derivatives of nucleoside analogs.51,52 The reaction of 5'-phosphite of any 2',3'-dideoxynucleoside 67 with arylamine in the presence of diphenyl chlorophosphate gives almost quantitative yield of 5'-amidophosphites 94 which are oxidized in situ with iodine solution in aqueous pyridine. The target 5'-amidophosphates 95 are isolated in good yields by flash chromatography.
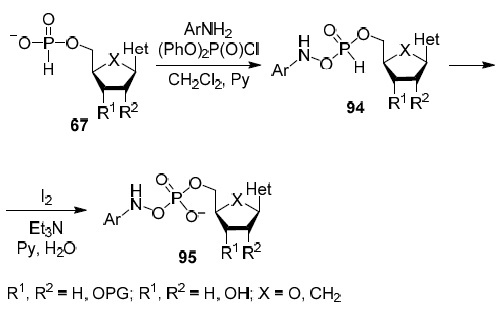
Scheme 7
The synthesis of 5'-arylamidophosphates containing an amino acid moiety proposed by McGuigan in 1993 to obtain a prodrug of zidovudine (20) is much more popular.53 He used methyl ester of L-alanine hydrochloride 98 (Scheme 8, Alk = Me);53,54 however, this three-step approach can in principle be used for any phenol, any ester of any amino acid, and any nucleoside analog. In the first step, aryl dichlorophosphate 97 is obtained by the reaction of phenol 96 with phosphorus oxychloride (Scheme 8). In the second step, the condensation of aryl dichlorophosphate 97 with an L-amino acid ester hydrochloride (alanine in Scheme 8) in the presence of Et3N at –78°C gives aryl chloroamidophosphate 99, the reaction of which with a nucleoside analog 65 in the presence of N-methylimidazole leads after the removal of protective groups (if any) to the target 5'-amidophosphate in the form of a mixture of diastereomers. This strategy is used at present for the synthesis of amidophosphate prodrugs of various nucleoside analogs 100.55,56

Scheme 8
The amidophosphate strategy for converting nucleoside analogs into their prodrug forms (Scheme 8) proved to be very effective for obtaining highly active antiviral agents. According to Kandil et al.,57 amidophosphate derivatives of stavudine 101 (Fig. 8) outperformed stavudine (46) in activity against HIV by a factor of 10.57 If zidovudine (20) and alovudine (3'-deoxy-3'-fluorothymidine) (104) are known as inhibitors of HIV reverse transcriptase,2,4,58 then their amidophosphate derivatives 102, 105, as well as 5'-triphosphate derivatives 103 and 106 are inhibitors of the RNA-dependent RNA polymerase of SARS-CoV-2.59
Figure 8.
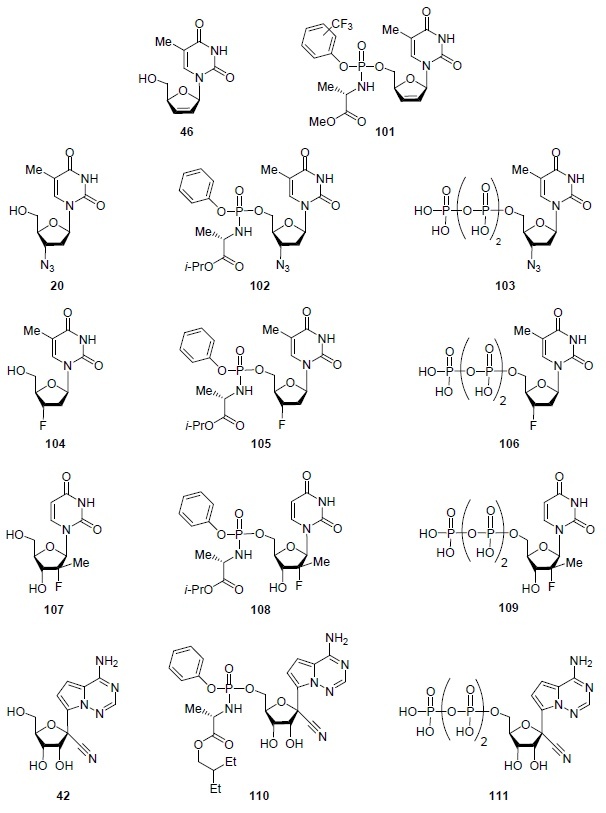
Amidophosphate and 5'-triphosphate derivatives of some antiviral nucleoside analogs.
Uridine analog 107 is not active against herpes simplex virus C,55 but its prodrug amidophosphate 108 (sofosbuvir) is approved for the treatment of hepatitis C and is also effective against a number of other RNA viruses (HIV, dengue, Zika, West Nile).19 Intracellular kinases metabolize sofosbuvir (108) to its 5'-triphosphate derivative 10960 which inhibits the RNA-dependent RNA polymerase of SARS-CoV-2.59
The adenosine analog C-nucleoside 42 (Fig. 8) exhibits pronounced activity against a number of filoviruses, pneumoviruses, paramyxoviruses (Table 1) and is currently undergoing clinical trials as a drug for the treatment of Ebola hemorrhagic fever.61 In addition, C-nucleoside 42 is effective in treating cats infected with feline coronavirus (FCoV) and those with infectious feline peritonitis.62
The prodrug of C-nucleoside 42, amidophosphate 110, called remdesivir, also selectively inhibits Ebola virus replication (Table 1) and is undergoing clinical trials as a drug for the treatment of Ebola virus infection.63 Most importantly, however, remdesivir (110) effectively suppress the replication of SARS-CoV and MERS-CoV in primary cultures of the human respiratory tract epithelium (Table 1), being metabolized in the cell to a 5'-triphosphate derivative 111.61,62 Remdesivir (110) is currently undergoing clinical trials in several countries as a therapeutic agent for COVID-19 infections and was approved by the FDA for emergency use in 2020.64
The reason for the activity of amidophosphate and 5'-triphosphate analogs of nucleosides shown in Figure 8 regarding the SARS-CoV-2 RNA-dependent RNA polymerase is that coronaviruses are RNA viruses that share properties with other known RNA viruses such as hepatitis C, influenza, hemorrhagic fever, and Coxsackie viruses.60 The active site of RNA-dependent RNA polymerase responsible for viral RNA replication is approximately the same for all RNA viruses, including SARS-CoV and MERS-CoV, and is not very selective for the structure of substrates64,65 which makes it an attractive drug target. Therefore, all nucleoside analogs with the ability to inhibit the replication of RNA viruses are a promising scaffold for creating drugs to combat COVID-19.
More than 50 years of the synthesis of cyclic nucleoside analogs and the study of their biological activity have shown that representatives of this class of nitrogen-containing organic compounds are among the best antiviral drugs for the treatment of socially significant diseases such as HIV infection and hepatitis. It should be noted that there has been a qualitative leap in the methodology of searching for antiviral nucleoside analogs in the last decade. If in the 1960-1990s the modification of native nucleosides was carried out almost randomly, then now, when not only the structure of the genomes of HIV, hepatitis viruses, influenza, and 2019-nCoV coronavirus but also the crystal structure of most of the proteins and enzymes encoded by them has been established, and such a powerful tool like molecular docking is in the hands of chemists, the search for new effective antiviral nucleoside analogs has become more meaningful and substantive. Despite the achieved successes, the intensity of research in this area of medicinal chemistry is not diminishing because biologists and chemists have yet to reveal the effect on the antiviral activity of the correlation of the structure of nucleoside analogs not only with the structure of active domains of viral proteins, but also with the structure of integral proteins that ensure their penetration into the cell, as well as the kinases of both normal and infected cells which are responsible for their metabolism. And, of course, the most important task of overcoming the strong variability of viral genomes remains.
The authors would like to thank the Russian Science Foundation for financial support (grant 19-13-00003).
Footnotes
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2021, 57(4), 326–341
References
- 1.Pastuch-Gawołek G, Gillner D, Król E, Walczak K, Wandzik I. Eur. J. Pharmacol. 2019;865:172747. doi: 10.1016/j.ejphar.2019.172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seley-Radtke KL, Yates MK. Antiviral Res. 2018;154:66. doi: 10.1016/j.antiviral.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eyer L, Nencka R, De Clercq E, Seley-Radtke K, Růžek D. Antiviral Chem. Chemother. 2018;26:1. doi: 10.1177/2040206618761299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Clercq E, Li G. Clin. Microbiol. Rev. 2016;29:695. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnard D. Curr. Opin. Invest. Drugs. 2002;3:1585. [PubMed] [Google Scholar]
- 6.De Clercq E, Cools M, Balzarini J, Snoeck R, Andrei G, Hosoya M, Shigeta S, Ueda T, Minakawa N, Matsuda A. Antimicrob. Agents Chemother. 1991;35:679. doi: 10.1128/AAC.35.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krajczyk F, Kulinska K, Kulinski T, Hurst BL, Day CW, Smee DF, Ostrowski T, Januszczyk P, Zeidler J. Antiviral Chem. Chemother. 2014;23:161. doi: 10.3851/IMP2564. [DOI] [PubMed] [Google Scholar]
- 8.Elayadi, H.; Mesnaoui, M.; Korba, B. E.; Smietana, M. Vasseur, J. J.; Secrist, J. A.; Lazrek, H. B. ARKIVOC2012, (viii), 76.
- 9.St. Amant, A. H.; Bean, L. A.; Guthrie, J. P.; Hudson, R. H. E. Org. Biomol. Chem.2012, 10, 6521. [DOI] [PubMed]
- 10.Parmenopoulou V, Chatzileontiadou DSM, Manta S, Bougiatioti S, Maragozidis P, Gkaragkouni D-N, Kaffesaki E, Kantsadi AL, Skamnaki VT, Zographos SE, Zounpoulakis P, Balatsos NAA, Komiotis D, Leonidas DD. Bioorg. Med. Chem. 2012;20:7184. doi: 10.1016/j.bmc.2012.09.067. [DOI] [PubMed] [Google Scholar]
- 11.Chatzileontiadou DSM, Tsika AC, Diamantopoulou Z, Delbé J, Badet J, Courty J, Skamnaki VT, Parmenopoulou V, Komiotis D, Hayes JM, Spyroulias GA, Leonidas DD. ChemMedChem. 2018;13:259. doi: 10.1002/cmdc.201700688. [DOI] [PubMed] [Google Scholar]
- 12.Andreeva OV, Garifullin BF, Zarubaev VV, Slita AV, Yesaulkova IL, Saifina LF, Shulaeva MM, Belenok MG, Semenov VE, Kataev VE. Mol. Diversity. 2020;25:473. doi: 10.1007/s11030-020-10141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Onitsuka, K.; Tokuda, R.; Kuwata-Higashi, N.; Kumamoto, H.; Aoki, M.; Amano, M.; Kohgo, S.; Das, D.; Haraguchi, K.; Mitsuya, H.; Imoto, S. Nucleosides, Nucleotides Nucleic Acids2020, 39, 518. [DOI] [PMC free article] [PubMed]
- 14.Kasthuri M, Li C, Verma K, Russell OO, Dickson L, McCormick L, Bassit L, Amblard F, Schinazi RF. Molecules. 2020;25:1258. doi: 10.3390/molecules25061258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coats SJ, Garnier-Amblard EC, Amblard F, Ehteshami M, Amiralaei S, Zhang H, Zhou L, Boucle SRL, Lu X, Bondada L, Shelton JR, Li H, Liu P, Li C, Cho JH, Chavre SN, Zhou S, Mathew J, Schinazi RF. Antiviral Res. 2014;102:119. doi: 10.1016/j.antiviral.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jordheim LP, Durantel D, Zoulim F, Dumontet C. Nat. Rev. Drug Discovery. 2013;12:447. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 17.Kohgo S, Imoto S, Tokuda R, Takamatsu Y, Higashi-Kuwata N, Aoki M, Amano M, Kansui H, Onitsuka K, Maeda K, Mitsuya H. ChemistrySelect. 2018;3:3313. doi: 10.1002/slct.201800527. [DOI] [Google Scholar]
- 18.Kamata M, Takeuchi T, Hayashi E, Nishioka K, Oshima M, Iwamoto M, Nishiuchi K, Kamo S, Tomoshige S, Watashi K, Kamisuki S, Ohrui H, Sugawara F, Kuramochi K. Biosci. Biotechnol. Biochem. 2020;84:217. doi: 10.1080/09168451.2019.1673696. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Yogarajah T, Lee RCH, Kaur P, Inoue M, Tan YW, Chu JJH. Sci. Rep. 2020;10:8159. doi: 10.1038/s41598-020-65152-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho A, Saunders OL, Butler T, Zhang L, Xu J, Vela JE, Feng JY, Ray AS, Kim CU. Bioorg. Med. Chem. Lett. 2012;22:2705. doi: 10.1016/j.bmcl.2012.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siegel D, Hui HC, Doerffler E, Clarke MO, Chun K, Zhang L, Neville S, Carra E, Lew W, Ross B, Wang Q, Wolfe L, Jordan R, Soloveva V, Knox J, Perry J, Perron M, Stray KM, Barauskas O, Feng JY, Xu Y, Lee G, Rheingold AL, Ray AS, Bannister R, Strickley R, Swaminathan S, Lee WA, Bavari S, Cihlar T, Lo MK, Warren TK, Mackman RL. J. Med. Chem. 2017;60:1648. doi: 10.1021/acs.jmedchem.6b01594. [DOI] [PubMed] [Google Scholar]
- 22.Lin T-S, Schinazi RF, Prusoff WH. Biochem. Pharm. 1987;36:2713. doi: 10.1016/0006-2952(87)90253-X. [DOI] [PubMed] [Google Scholar]
- 23.Chen S-H. Curr. Med. Chem. 2002;9:899. doi: 10.2174/0929867024606696. [DOI] [PubMed] [Google Scholar]
- 24.Bryant ML, Bridges EG, Placidi L, Faraj A, Loi A-G, Pierra P, Dukhan D, Gosselin G, Imbach J-L, Hernandez B, Juodawlkis A, Tennant B, Korba B, Cote P, Marion P, Cretton-Scott E, Schinazi RF, Sommadossi J-P. Antimicrob. Agents Chemother. 2001;54:229. doi: 10.1128/AAC.45.1.229-235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang P, Hong JH, Cooperwood JS, Chu CK. Antiviral Res. 1998;40:19. doi: 10.1016/S0166-3542(98)00041-2. [DOI] [PubMed] [Google Scholar]
- 26.de Muys J-M, Gourdeau H, Nguyen-Ba N, Taylor DL, Ahmed PS, Mansour T, Locas C, Richard N, Wainberg A, Rando RF. Antimicrob. Agents Chemother. 1999;43:1835. doi: 10.1128/AAC.43.8.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mansour TS, Jin H, Wang W, Hooker EU, Ashman C, Cammack N, Salomon H, Belmonte AR, Wainberg MA. J. Med. Chem. 1995;38:1. doi: 10.1021/jm00001a001. [DOI] [PubMed] [Google Scholar]
- 28.Kim HO, Shanmuganatban K, Alves AJ, Jeong LS, Beach JW, Schinazi RF, Chang CN, Cheng Y-C, Chu CK. Tetrahedron Lett. 1992;33:6899. doi: 10.1016/S0040-4039(00)60890-0. [DOI] [Google Scholar]
- 29.Rawala RK, Bariwal J, Singh V. Curr. Top. Med. Chem. 2016;16:3258. doi: 10.2174/1568026616666160506145300. [DOI] [PubMed] [Google Scholar]
- 30.De Clercq E. Antimicrob. Agents Chemother. 1985;28:84. doi: 10.1128/AAC.28.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherer de Fraga R, Van Vaisberg V, Alfaia Mendes LC, Carrilho FJ, Ono SKJ. Gastroenterology. 2020;55:496. doi: 10.1007/s00535-020-01680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balzarini J. Pharm. World Sci. 1994;16:113. doi: 10.1007/BF01880662. [DOI] [PubMed] [Google Scholar]
- 33.Cano-Soldado P, Pastor-Anglada M. Med. Res. Rev. 2012;32:428. doi: 10.1002/med.20221. [DOI] [PubMed] [Google Scholar]
- 34.Roy R, Depaix A, Périgaud C, Peyrottes S. Chem Rev. 2016;116:7854. doi: 10.1021/acs.chemrev.6b00174. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Mao S, Hager MW, Becnel KD, Schinazi RF, Liotta DC. Bioorg. Med. Chem. Lett. 2007;17:3398. doi: 10.1016/j.bmcl.2007.03.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weising S, Sterrenberg V, Schols D, Meier C. ChemMedChem. 2018;13:1771. doi: 10.1002/cmdc.201800361. [DOI] [PubMed] [Google Scholar]
- 37.Shi J, Zhou L, Zhang H, McBrayer TR, Detorio MA, Johns M, Bassit L, Powdrill MH, Whitaker T, Coats SJ, Götte M, Schinazi RF. Bioorg. Med. Chem. Lett. 2011;21:7094. doi: 10.1016/j.bmcl.2011.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toti KS, Derudas M, Pertusati F, Sinnaeve D, Van den Broeck F, Margamuljana L, Martins JC, Herdewijn P, Balzarini J, McGuigan C, Van Calenbergh S. J. Org. Chem. 2014;79:5097. doi: 10.1021/jo500659e. [DOI] [PubMed] [Google Scholar]
- 39.Krayevsky, A. A.; Watanabe, K. A. Modified Nucleosides as Anti-Aids Drugs: Current Status and Perspectives; Bioinform: Moscow, 1993.
- 40.Malin, A. A.; Ostrovskii, V. A. Russ. J. Org. Chem. 2001, 37, 759. [Zh. Org. Khim.2001, 37, 801.]
- 41.Tarusova, N. B.; Khorlin, A. A.; Kraevsky, A. A.; Korneeva, M. N.; Nosik, D. N.; Kruglov, I. V.; Galedov, G. A.; Bibilashvili, R. Sh. Mol. Biol. (Moscow)1989, 23, 1716. [Mol. Biol.1989, 23, 1716.] [PubMed]
- 42.Khandazhinskaya A, Matyugina E, Shirokova E. Expert Opin. Drug Metab. Toxicol. 2010;6:701. doi: 10.1517/17425251003713501. [DOI] [PubMed] [Google Scholar]
- 43.Kraevsky, A. A.; Tarusova, N. B.; Zhu, Q.-Y.; Vidal, P.; Chou, T.-C.; Baron, P.; Polsky, B.; Jiang, X.-J.; Matulič- Adamič, J.; Rosenberg, I.; Watanabe K. A. Nucleosides Nucleotides1992, 11, 177.
- 44.Wagner, C. R.; Iyer, V. V.; McIntee, E. J. Med. Res. Rev. 2000, 20, 417. [DOI] [PubMed]
- 45.Jia X, Schols D, Meier C. J. Med. Chem. 2020;63:6003. doi: 10.1021/acs.jmedchem.0c00271. [DOI] [PubMed] [Google Scholar]
- 46.Meier C. Mini Rev. Med. Chem. 2002;2:219. doi: 10.2174/1389557023406205. [DOI] [PubMed] [Google Scholar]
- 47.Meier C, Balzarini J. Antiviral Res. 2006;71:282. doi: 10.1016/j.antiviral.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 48.Meier C, Knispel T, Marquez VE, Siddiqui MA, De Clercq E, Balzarini J. J. Med. Chem. 1999;42:1615. doi: 10.1021/jm981097r. [DOI] [PubMed] [Google Scholar]
- 49.Meier C, Knispel T, De Clercq E, Balzarini J. J. Med. Chem. 1992;42:1604. doi: 10.1021/jm981096z. [DOI] [PubMed] [Google Scholar]
- 50.Balzarini J, Haller-Meier F, De Clercq E, Meier C. Antiviral Chem. Chemother. 2001;12:301. doi: 10.1177/095632020101200505. [DOI] [PubMed] [Google Scholar]
- 51.Romanowska J, Sobkowski M, Szymańska-Michalak A, Kołodziej K, Dąbrowska D, Lipniacki A, Piasek A, Pietrusiewicz ZM, Figlerowicz M, Guranowski A, Boryski J, Stawiński J, Kraszewski A. J. Med. Chem. 2011;54:6482. doi: 10.1021/jm2001103. [DOI] [PubMed] [Google Scholar]
- 52.Kolodziej K, Romanowska J, Stawinski J, Boryski J, Dabrowska A, Lipniacki A, Piasek A, Kraszewski A, Sobkowski M. Eur. J. Med. Chem. 2015;100:77. doi: 10.1016/j.ejmech.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 53.McGuigan C, Pathirana RN, Balzarini J, De Clercq E. J. Med. Chem. 1993;36:1048. doi: 10.1021/jm00060a013. [DOI] [PubMed] [Google Scholar]
- 54.McGuigan C, Cahard D, Sheeka HM, De Clercq E, Balzarini J. J. Med. Chem. 1996;39:1748. doi: 10.1021/jm950605j. [DOI] [PubMed] [Google Scholar]
- 55.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang H-R, Bansal S, Espiritu C, Keilman M, Lam AM, Micolochick Steuer HM, Niu C, Otto MJ, Furman PA. J. Med. Chem. 2010;53:7202. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 56.Singh, U. S.; Chu, C. R. Nucleosides, Nucleotides Nucleic Acids2020, 39, 52. [DOI] [PubMed]
- 57.Kandil S, Pannecouque C, Chapman FM, Westwell AD, McGuigan C. Bioorg. Med. Chem. Lett. 2019;29:126721. doi: 10.1016/j.bmcl.2019.126721. [DOI] [PubMed] [Google Scholar]
- 58.Parang K, El-Sayed NS, Kazeminy AJ, Tiwari RK. Molecules. 2020;25:2343. doi: 10.3390/molecules25102343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chien M, Anderson TK, Jockusch D, Tao C, Li X, Kumar S, Russo JJ, Kirchdoerfer RN, Ju J. J. Proteome Res. 2020;19:4690. doi: 10.1021/acs.jproteome.0c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murakami E, Tolstykh T, Bao H, Niu C, Micolochick Steuer HM, Bao D, Chang W, Espiritu C, Bansal S, Lam AM, Otto MJ, Sofia MJ, Furman FA. J. Biol. Chem. 2010;285:34337. doi: 10.1074/jbc.M110.161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pruijssers AJ, Denison MR. Curr. Opin. Virol. 2019;35:57. doi: 10.1016/j.coviro.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan VC, Muller FL. ACS Med. Chem. Lett. 2020;11:1361. doi: 10.1021/acsmedchemlett.0c00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cardile AP, Warren TK, Martins KA, Reisler RB, Bavari S. Annu. Rev. Pharmacol. Toxicol. 2017;57:329. doi: 10.1146/annurev-pharmtox-010716-105055. [DOI] [PubMed] [Google Scholar]
- 64.Jockusch S, Tao C, Li X, Anderson TK, Chien M, Kumar S, Russo JJ, Kirchdoerfer RN, Ju J. Antiviral Res. 2020;180:104857. doi: 10.1016/j.antiviral.2020.104857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jácome, R.; Campillo-Balderas, J. A.; Ponce de León, S.; Becerra, A.; Lazcano, A. Sci. Rep.2020, 10, 9294. [DOI] [PMC free article] [PubMed]
- 66.de Clercq E, Descamps J, Schmidt CL, Mertes MP. Biochem. Pharm. 1979;28:3249. doi: 10.1016/0006-2952(79)90117-5. [DOI] [PubMed] [Google Scholar]
- 67.Aoki, F. Y. In Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases; Bennett, J. E.; Blaser, M. J.; Dolin, R., Eds.; Elsevier: Amsterdam, 2000, 4th ed., Vol. 1, p. 546.
- 68.De Clercq E, Descamps J, De Somer P, Barr PJ, Jones AS, Walker RT. Proc. Natl. Acad. Sci. U. S. A. 1979;76:2947. doi: 10.1073/pnas.76.6.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dapp, M. J.; Clouser, C. L.; Patterson, S.; Mansky, L. M. J. Virology2009, 83, 11950. [DOI] [PMC free article] [PubMed]
- 70.Clouser CL, Holtz CM, Mullett M, Crankshaw DL, Briggs JE, O'Sullivan MG, Patterson SE, Mansky LM. Antimicrob. Agents Chemother. 2012;56:1942. doi: 10.1128/AAC.06161-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Markland W, Mcquaid TJ, Jain J, Kwong AD. Antimicrob. Agents Chemother. 2000;44:859. doi: 10.1128/AAC.44.4.859-866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sidwell RW, Bailey KW, Wong M-H, Barnard DL, Smee DF. Antiviral Res. 2005;68:10. doi: 10.1016/j.antiviral.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 73.Sidwell RW, Barnard DL. Antiviral Res. 2006;71:379. doi: 10.1016/j.antiviral.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 74.Chung D-H, Kumarapperuma SC, Sun Y, Li Q, Chu Y-K, Arterburn JB, Parker WB, Smith J, Spik K, Ramanathan HN, Schmaljohn CS, Jonsson CB. Antiviral Res. 2008;79:19. doi: 10.1016/j.antiviral.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McDowell M, Gonzales SR, Kumarapperuma SC, Jeselnik M, Arterburn JB, Hanley KA. Antiviral Res. 2010;87:78. doi: 10.1016/j.antiviral.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takamatsu Y, Tanaka Y, Kohgo S, Murakami S, Singh K, Das D, Venzon DJ, Amano M, Higashi-Kuwata N, Aoki M, Delino NS, Hayashi S, Takahashi S, Sukenaga Y, Haraguchi K, Sarafianos SG, Maeda K, Mitsuya H. Hepatology. 2015;62:1024. doi: 10.1002/hep.27962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gentile I, Coppola N, Buonomo AR, Zappulo E, Borgia G. Expert Opinion Invest. Drugs. 2014;23:1211. doi: 10.1517/13543784.2014.921680. [DOI] [PubMed] [Google Scholar]
- 78.Nitanda T, Wang X, Kumamoto H, Haraguchi K, Tanaka H, Cheng Y-C, Baba M. Antimicrob. Agents Chemother. 2005;49:3355. doi: 10.1128/AAC.49.8.3355-3360.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Markowitz, M.; Sarafianos, S .G. Curr. Opin. HIV AIDS2018, 13, 294. [DOI] [PMC free article] [PubMed]
- 80.Beran, R. K. F.; Sharma, R.; Corsa, A. C.; Tian, Y.; Golde, J.; Lundgaard, G.; Delaney, W. E., IV; Zhong, W.; Greenstein, A. E. PLoS One2012, 7, e30286. [DOI] [PMC free article] [PubMed]
- 81.Daluge, S. M.; Good, S. S.; Faletto, M. B.; Miller, W. H.; St. Clair, M. H.; Boone, L. R.; Tisdale, M.; Parry, N. R.; Reardon, J. E.; Dornsife, R. E.; Averett, D. R.; Krenitsky, T. A. Antimicrob. Agents Chemother. 1997, 41, 1082. [DOI] [PMC free article] [PubMed]
- 82.Samunuri R, Toyama M, Pallaka RS, Neeladri S, Jha AK, Baba M, Bal C. RSC Med. Chem. 2020;11:597. doi: 10.1039/D0MD00059K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu H-T, Colby-Germinario SP, Hassounah SA, Fogarty C, Osman N, Palanisamy N, Han Y, Oliveira M, Quan Y, Wainberg MA. Sci. Rep. 2017;7:6345. doi: 10.1038/s41598-017-06612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bullard-Feibelman, K. M.; Govero, J.; Zhu, Z.; Salazar, V.; Veselinovic, M.; Diamond, M. S.; Geiss, B. J. Antiviral Res.2017, 137, 134. [DOI] [PMC free article] [PubMed]
- 85.Lo MK, Jordan R, Arvey A, Sudhamsu J, Shrivastava-Ranjan P, Hotard AL, Flint M, McMullan LK, Siegel D, Clarke MO, Mackman RL, Hui HC, Perron M, Ray AS, Cihlar T, Nichol ST, Spiropoulou CF. Sci. Rep. 2017;7:43395. doi: 10.1038/srep43395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cho A, Zhang L, Xu J, Lee R, Butler T, Metobo S, Aktoudianakis V, Lew W, Ye H, Clarke M, Doerffler E, Byun D, Wang T, Babusis D, Carey AC, German P, Sauer D, Zhong W, Rossi S, Fenaux M, McHutchison JG, Perry J, Feng J, Ray AS, Kim CU. J. Med. Chem. 2014;57:1812. doi: 10.1021/jm400201a. [DOI] [PubMed] [Google Scholar]