

Abstract
Bactobolin is a hybrid natural product with potent cytotoxic activity. Its production from Burkholderia thailandensis was reported as part of a collaboration between the Greenberg and Clardy laboratories in 2010. The collaboration sparked a series of studies leading to the discovery of new analogues and associated structure–activity relationships, the identification of the bactobolin biosynthetic gene cluster and assembly of its unusual amino acid building block, the molecular target of and resistance to the antibiotic, and finally an X-ray crystal structure of the ribosome– bactobolin complex. Herein, we review the collaborations that led to our current understanding of the chemistry and biology of bactobolin.
Graphical Abstract
INTRODUCTION
Collaborations among investigators in the Greenberg and Clardy laboratories began around 2008 but had their genesis in the 1980s, when the lives of Jon Clardy and Pete Greenberg first crossed paths at Cornell University. At the time, Clardy was on the faculty in Chemistry and Greenberg on the faculty in Microbiology. Although initially not friends or collaborators, these two scientists were vaguely aware of each other for several reasons both personal and professional. This early overlap at Cornell eventually led to later fruitful collaborations, and the particular collaboration on the antibiotic bactobolin is the topic of this review.
Early work by Greenberg at Cornell focused on bacterial luminescence-gene regulation by a phenomenon that has become known as quorum sensing (QS).1 QS involves self-produced acylated amino acid signals, notably the acylhomo-serine lactones (acyl-HSL), and detection of these molecules to regulate gene expression at high cell densities. Also at Cornell, Clardy used his skills as a natural product chemist to determine the structures of molecules produced by diverse microorganisms. Secreted small molecules formed the basis for scientific overlap between the two investigators, though this did not lead to a formal collaboration at the time. With fond memories of their time at Cornell both eventually moved on to other universities.
From the early 1990s on it became clear that acyl-HSL QS was common to a variety of different species of Proteobacteria and that different species used different acyl-HSLs to control a variety of different functions.1–4 Greenberg and others determined the structures of the signals produced by different bacteria. We now know they either are fatty acyl-HSLs with specificity residing in the fatty acyl tail or are aryl-HSLs. The signals are generated by members of the LuxI family of acyl-HSL synthases and detected by members of the LuxR family of transcription factors.5 Purification and identification of different acyl-HSLs became routine in the Greenberg laboratory. Among the genes shown to be controlled by QS were those for production of the antibiotics carbapenem by Serratia marcescens and violacein by Chromobacterium violacium.6,7 There is now evidence that many Proteobacteria control expression of biosynthetic gene clusters by acyl-HSL quorum sensing.8
Breck Duerkop and Josie Chanlder, then a graduate student and postdoctoral fellow in the Greenberg lab, respectively, became interested in the bacterial species Burkholderia thailandensis, a nonpathogenic close relative of the emerging pathogen Burkholderia pseudomallei.9 This bacterium has three acyl-HSL QS circuits, one of which is linked to a hybrid nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) gene cluster (Figure 1a). The Greenberg lab proceeded to show that this QS circuit was required for production of a potent, broad-spectrum antibiotic.10 Efforts to extract and concentrate the antibiotic were unsuccessful, and the chemistry required to identify this compound was beyond the capabilities of the Greenberg lab. Greenberg proposed to Chandler and Duerkop that he contact his colleague from their past lives at Cornell in hopes that Clardy would remember him and be willing to help in an effort to identify the polar antibiotic. Email excerpts that initiated the collaboration are included below:
“Jon,
I am writing you to see if I might be able to interest you in working with us on what we believe is a novel antibiotic. More on that below. I don’t know if you have any recollection of me. I was on the faculty in Microbiology at Cornell from 1977–1988. Eventually my family moved first to the University of Iowa Medical School and eventually to the University of Washington…
Pete Greenberg”
“Pete,
I do remember you… I’d be delighted to work on this problem, and there are several people in the lab who I imagine would like a crack at it.
Jon “
Figure 1.
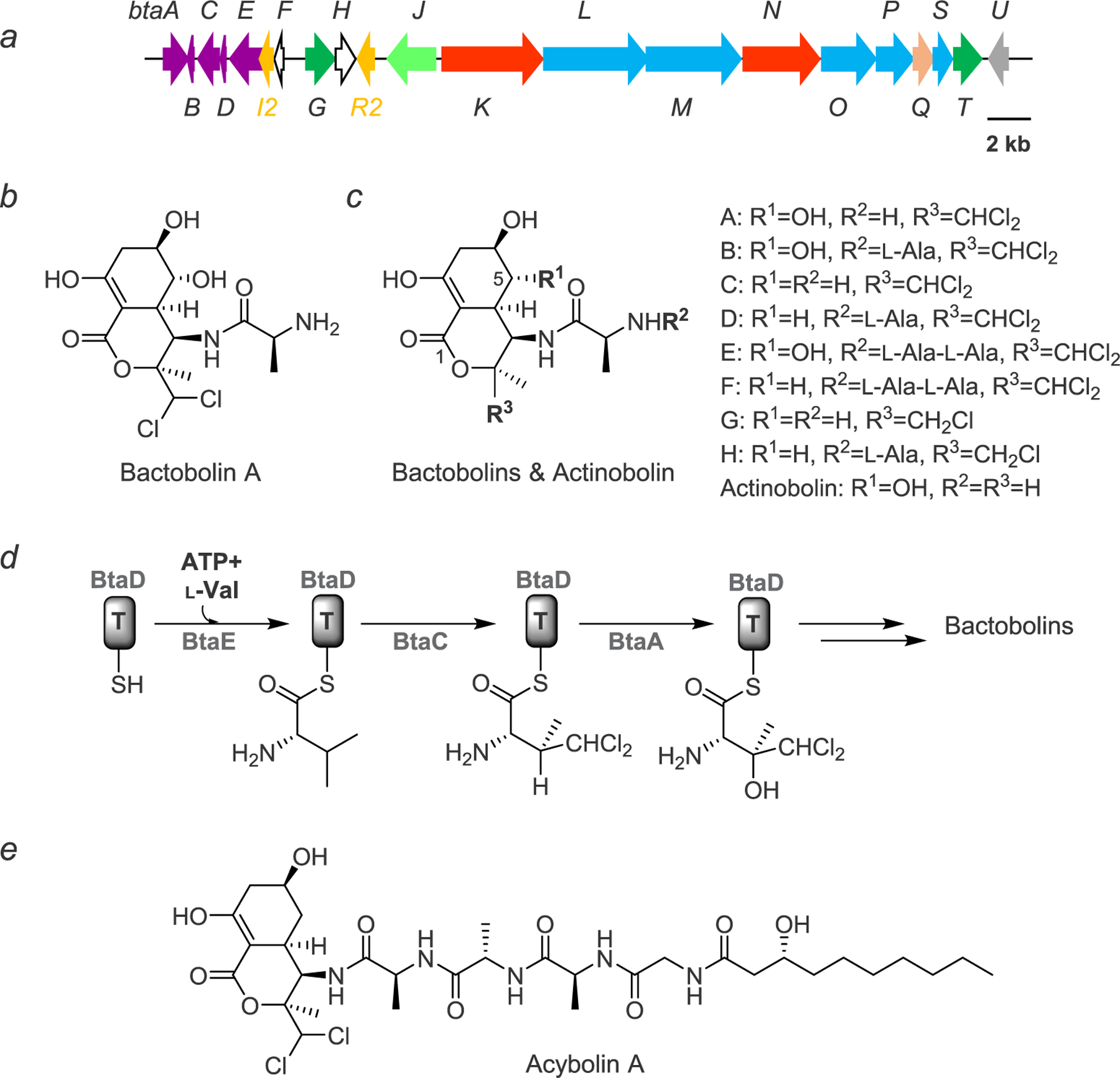
Bactobolin and the bta gene cluster. (a) The bta gene cluster. Genes are color-coded as follows: genes involved in the synthesis and incorporation of 3-hydroxy-4,4-dichloro-L-valine, purple; QS regulation, orange; genes of unknown function, white; transporters, dark green; oligopeptidase, light green; nonribosomal peptide synthetase, red; polyketide synthase, blue; acyltransferase, salmon; bactobolin hydroxylase, gray.(b) Structure of bactobolin A. (c) Structures of bactobolin variants B–H and actinobolin. (d) Biosynthesis of 3-hydroxy-4,4-dichloro-L-valine, as elucidated by mutational studies. Note that the timing of hydroxylation of valine has not yet been determined. (e) Structure of acybolin A with the bactobolin warhead.
Clardy recruited Mo Seyedsayamdost to tackle this problem, and in the Greenberg laboratory Josie Chandler generated B. thailandensis mutants involved in the regulation, synthesis, and export of bactobolin. This led to a series of studies to understand the bactobolin family of hybrid nonribosomal peptide–polyketide compounds, which are reviewed below, and to other fruitful collaborations between the two groups.
BURKHOLDERIA THAILANDENSIS STUDIES PRIOR TO COLLABORATION
The Greenberg lab’s interest in B. thailandensis began with QS. The QS systems of B. thailandensis and B. pseudomallei are nearly identical: each has three LuxR-I signal receptor and synthase pairs (QS-1, −2, and −3) and two unpaired LuxR receptors. Another closely related species, Burkholderia mallei, is a host-adapted pathogen with a reduced genome compared with the other two. B. mallei has QS-1 and QS-3 and the two unpaired receptors, but this species has lost the QS-2 system along with a flanking region of DNA, a biosynthetic gene cluster (BGC). The QS systems of these three species remained largely unstudied until around 2005. Several studies implicated QS-1 and, to a lesser extent, QS-3 of B. pseudomallei in animal infections.11–13 The QS systems of B. mallei were also initially shown to contribute to virulence.14 Although later results did not corroborate these initial findings,15 the conservation of QS-1 and QS-3 in B. mallei supported the idea that these two systems play a role during survival in the host. On the other hand, loss of QS-2 and flanking DNA from B. mallei suggested that this system and the surrounding BGC were important for surviving saprophytic soil environments.
At the same time that the QS systems of these three species were gaining interest, there was a parallel effort underway to characterize the secondary metabolomes of these bacteria, particularly B. thailandensis, because of the high number of unique natural product BGCs that remained largely uncharacterized.16–18 B. thailandensis, a nonpathogen with simple laboratory growth requirements and rapid growth rates, was considered an ideal system for study and a gold mine for new natural product discovery. However, a challenge at the time was the limited genetic tools for these species, which were needed to make gene deletions or for the induction of genes that are not normally expressed at high levels. To this end, Josie Chandler in the Greenberg lab developed a system for genetic engineering in the B. thailandensis–pseudomallei–mallei group for their QS research. The system made use of the few selective and counterselective markers that were available in these bacteria. Chandler used this methodology to generate a suite of B. thailandensis mutants with deletions in each of the QS genes.19 The suite of mutants would facilitate future studies of both the QS systems and new antibiotics, including bactobolin.
INITIAL DISCOVERY OF BACTOBOLIN
One of the first studies undertaken in B. thailandensis was focused on the QS-2 genes and the DNA flanking these genes. This region, a segment of about 120 kb of DNA, is missing from B. mallei. The region contains the btaR2 and btaI2 genes and 17 other genes predicted to be involved in a NRPS/PKS hybrid biosynthetic pathway (Figure 1a). The NRPS/PKS genes are on either side of btaR2 and btaI2. Breck Duerkop used a simple assay to test the hypothesis that the genes in this region are important for production of an antibiotic. Duerkop collected cell-free fluid from B. thailandensis cultures harboring mutations in the QS-2 genes or one of the antibiotic biosynthetic genes and tested the fluid for antibiotic activity. He did this by saturating paper diffusion disks with the fluid and placing the disks on an agar plate with a lawn of growing Bacillus subtilis. He found that the fluid from B. thailandensis inhibited growth of B. subtilis, as well as Staphylococcus aureus and Streptococcus pyogenes, but there was no growth inhibition with fluid from similarly grown QS-2 or antibiotic BGC mutants.10 He also used gene deletion mutants to show that btaR2 and btaI2 were required for transcription of the antibiotic biosynthetic genes. From these results he concluded that QS-2 regulates production of an antibiotic, and antibiotic production is induced by the QS-2 genes. Later, the Greenberg lab demonstrated that shifting B. thailandensis to lower temperatures (30 °C vs 37 °C) produced higher concentrations of the antibiotic.20
As outlined above, after failed attempts to purify the antibiotic by the Greenberg lab, the antibiotic was successfully purified by the Clardy lab, particularly his postdoc at the time, Mo Seyedsayamdost. The mutants and growth conditions established in the Greenberg lab helped identify the antibiotic, which was ultimately characterized as bactobolin.21
BACTOBOLIN ISOLATION AND CHARACTERIZATION
Metabolic profiling of wild-type B. thailandensis and the bactobolin biosynthetic mutants showed that the antibiotic was polar. Initial isolations utilized a five-column purification scheme, which was successful because of the robust production and stability of the antibiotic.21 The purification protocol was later simplified with increased understanding of the chromatographic and chemical properties of bactobolin.22 Activity-guided fractionation enabled purification of four variants. HR-MS analysis of the most abundant bactobolin variant, bactobolin A, was consistent with the presence of two chlorine substituents. Indeed, a singlet deshielded peak at 6.18 ppm, with a corresponding 74 ppm 13C shift, was symptomatic of a methine-carbon carrying two chlorine substituents. The structure of bactobolin A was ultimately solved by analysis of a full suite of 1D/2D NMR spectra and shown to be identical to that previously solved by Kondo and co-workers (Figure 1b).23 Detailed analysis of the NMR J-coupling patterns in conjunction with previous work on the related actinobolin (see below) established equatorial hydroxy groups at C-5 and C-6.21,24 Moreover, the NMR data allowed discrimination of axial–axial and axial–equatorial relationships between other protons in the bicycle and together with Marfey’s and Mosher’s ester analysis completed the full configurational assignment of bactobolin A. The data were consistent with a half-chair conformation within the carbocycle, a proposal later confirmed by the X-ray crystal structure of bactobolin A (see below). Three additional congeners were also structurally elucidated, with one of these, bactobolin D, representing a new analogue (Figure 1c).21
DIVERGENT BACTOBOLIN BIOSYNTHESIS
With the structure of bactobolin elucidated, its biosynthesis could be inferred using bioinformatic analysis of the bta gene cluster in combination with gene deletion studies.22 Specifically, synthesis of the unusual amino acid 3-hydroxy-4,4-dichloro-L-valine could be assigned to genes BtaE and BtaD as the valine-specific adenylation and peptidyl-carrier-protein (PCP) domains, respectively (Figure 1a). The bta cluster encodes three Fe/α-ketoglutarate-dependent enzymes that are important for the activity of bactobolin. By mutational analysis Carr et al. showed that BtaC installs the dichloro substituent, BtaA the hydroxy group onto valine, and BtaU the C-5 OH on the bactobolin carbocycle (Figure 1c,d).22 After loading of the valine derivative onto the assembly line, the remainder of the pathway utilizes a combination of NRPS/PKS with the incorporation of L-alanine at the A-domains and malonyl-CoA by the acyltransferase (AT) domains. Together, five NRPS/PKSs, BtaKNOML, generate the enol lactone within the fused bicycle to complete bactobolin A biosynthesis. Compared to variant A, bactobolins B and D contain an additional L-alanine residue, while bactobolins C and D lack the C-5 OH substituent.
Interestingly, Streptomyces griseoviridis synthesizes actinobolin, a relative of the bactobolin family of natural products. Actinobolins were discovered before the bactobolins and incorporate threonine in place of the valine derivative, therefore lacking the dichloro substituent (Figure 1c).24 This turns out to be significant, as side-by-side studies showed actionbolin to be 21-fold, 15-fold, 900-fold, and 1000-fold less active than bactobolin A in inhibition assays against E. coli, S. aureus MRSA, murine tumor models, and the ribosome, respectively.25 Inspection of the bactobolin BGC provides some insights into the origin of this substitution. The operon that introduces the dichloro moiety, btaA-F (Figure 1a,d), is located several kb removed from the NRPS/PKS genes that generate the bicyclic core.22 This locus may be a later evolutionary addition to the remainder of the gene cluster. The incorporation of 3-hydroxy-4,4-dichloro-L-valine into bactobolin in B. thailandensis and the Burkholderia lineage renders the antibiotic much more cytotoxic than actinobolin. In addition to the dichloro substituent, the C-5 OH, present in both bactobolin and actinobolin, is also important for the potency of the antibiotic, as bioactivity assays with both variants have demonstrated.21,22
The bta gene cluster turns out to have a highly divergent secondary metabolite output. Mutational analysis of the cluster revealed four additional variants that either contain a monochlorovaline derivative or incorporate a trialanyl moiety at the N-terminus of the antibiotic (Figure 1c).22 Moreover, the bta cluster responds to exogenous antibiotics, notably trimethoprim, by producing a diverse set of acylated derivatives with much longer peptide linkers, consisting of a combination of L-alanine and glycine residues, as well as a 3-hydroxydecanoyl acyl group, the same acyl group used by the QS-2 system, which caps the N-terminal end of the antibiotic.26,27 These molecules, the acybolins (Figure 1e), are also synthesized divergently, and the structures of nine variants were reported; thus at least 17 natural products can be attributed to the bta cluster.
The suite of bactobolins has facilitated more detailed structure–activity relationship studies.22 As expected from the actinobolin assays, the singly chlorinated congeners are less active than the corresponding doubly chlorinated ones. Moreover, variants carrying the C-5 OH are invariably more potent than those lacking the hydroxy group. Thus, the metalloenzymes BtaC and BtaU have an important impact on the bioactivity of bactobolins. The acybolins are less potent than bactobolin A, though they remain active against bactobolin-resistant B. subtilis,26 suggesting a disparate mechanism of action. The mode of action and site of bactobolin binding to its target have shed further light onto bactobolin SAR studies, as elaborated below.
ELUCIDATING THE BACTOBOLIN TARGET: A UNIQUE SITE ON THE RIBOSOME
Studies on bactobolin from the 1970s suggested that members of this family block translation. Further, the Clardy/Greenberg laboratories showed the activity of each of the bactobolin compounds against bacteria correlated with the activity against eukaryotic cells,20 indicating that the target site might be conserved across bacteria and eukaryotes. As an approach to identify the bactobolin target, the Greenberg lab isolated bactobolin-resistant mutants of B. subtilis and identified the mutations in these isolates using a whole-genome sequencing approach.20 The mutations mapped to rplB, which codes for the 50S ribosome-associated protein L2. Specifically, mutations at residue Glu236 to either alanine or glutamine, or insertion of glycine at position 235, lowered susceptibility to bactobolin. Ectopic expression of mutant L2 proteins in wild-type B. subtilis also conferred resistance, supporting the idea that the antibiotic target might be in or near the L2 protein.
L2 is quite conserved and also found in eukaryotes, where it is named L8e. L2/L8e is required for association of the 50S and 30S ribosomal subunits and is also involved in the activity of the peptidyl-transferase center (PTC) on the 50S subunit,28 which is critical for translation elongation. The B. subtilis mutations map to the C-terminus of L2,20 which interacts with the PTC.29 The location of these mutations suggested that bactobolin might specifically inhibit the peptidyl transfer step during ribosomal protein synthesis. Importantly, the activities of other antibiotics known to target the PTC or interfere with peptidyl transfer, such as chloramphenicol, clindamycin, and linezolid, are not affected by mutations in the L2 protein.20 These results suggested bactobolin might target a unique site on the ribosome.
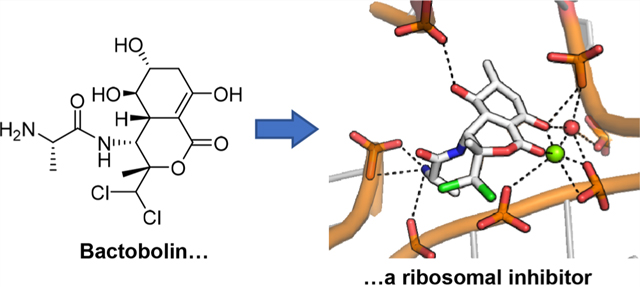
The potential novelty of bactobolin in its interaction with the ribosome piqued the interest of Dr. Venki Ramakrishnan, who shared the 2009 Nobel Prize in Chemistry for elucidating the structure of the ribosome. Ramakrishnan was brought into the collaboration through Greenberg, who knew Ramakrishnan through his sister Dr. Lalita Ramakrishnan, also a notable scientist and then a fellow faculty member in Greenberg’s department at the University of Washington. Ramakrishnan’s previous work relied on high-resolution (~3.4 Å) crystal structures of the Thermus thermophilus 70S ribosome,30 which revealed new insights into the structure and function of the ribosome during translation. To elucidate the precise binding site of bactobolin, Ramakrishnan’s postdoc Alexey Amunts soaked ribosome crystals with bactobolin A and determined the structure of the bactobolin-bound ribosome in the presence of tRNA and mRNA.31 The structure revealed that bactobolin forms direct interactions with the tRNA in the P-site of the ribosome, thereby causing structural distortions relative to the uninhibited ribosome. Specifically, the enol lactone moiety facilitates interactions of the antibiotic with the CCA terminus of P-site tRNA and with multiple sites on the 23S rRNA via binding to Mg2+ and an ordered water molecule (Figure 2a). The terminal amine and the C-5 OH, which is important for the potency of bactobolin A, make additional H-bonding interactions to different sites on the 23S subunit. Finally, the dichloro substituent contributes to additional interaction with the 23S rRNA. The conformational rearrangement of P-site tRNA caused by bactobolin binding suggests that ribosomal inhibition could be a consequence of hindered peptidyl transfer or translation termination.
Figure 2.

Structure of the ribosome–bactobolin A complex. (a) Magnified view of the binding pocket of bactobolin A, which is rendered in white and labeled. H-bonding interactions from the enol lactone, C-5 OH, and terminal amino group are shown with dotted lines. The enol lactone interacts with the 23S rRNA via a Mg2+ ion (green sphere) and an ordered water molecule (red sphere). Note that the C-6 OH group (star) was incorrectly defined as a methyl group in the deposited model. Glu236 on the L2 protein in B. subtilis (which corresponds to Glu237 in the T. thermophilus L2) is rendered in green. Some bases are omitted for clarity. (b) Interaction of L2-Glu237 with the ribosome and bactobolin. The Glu237 side chain interacts with a phosphate-oxygen on the 23S rRNA via a Mg2+ ion (green sphere) and with the 2′-OH group of the ribose of A2450, which is labeled and binds bactobolin A via its terminal amine.
The structure also provided an explanation as to why variants at Glu236 of the L2 protein led to lowered susceptibility to bactobolin. Glu236, which corresponds to Glu237 in the T. thermophilus L2 protein, contacts and binds A2450 in the 23S rRNA, which is also H-bonded to the terminal amino group of bactobolin A (Figure 2b).31 Substitutions at Glu236 likely disorder the bactobolin binding site, thereby lowering the affinity to the antibiotic. It is noteworthy that the bactobolin-resistant L2 mutants remain susceptible to blasticidin S,31 which causes a similar conformational rearrangement of P-site tRNA and has been proposed to inhibit the ribosome by impeding translation termination.32 Together, the structural studies revealed a novel binding and inhibition site on the ribosome and provided insights into the mode of action of the bactobolin antibiotics (Figure 2).
BACTOBOLIN: LOOKING FORWARD
The crystal structure of the bactobolin–ribosome complex brought our studies on bactobolin full circle, from its initial identification as a QS-regulated antibiotic, determination of the structures of numerous congeners, biosynthetic studies, detailed bioactivity profiles, discovery of its mode of action, and finally to pinpointing the target site on the ribosome. At the same time, our insights into bactobolin have opened new lines of inquiry. Specifically, given its potent bioactivity, can bactobolin serve as a broad-spectrum antibiotic lead? This is an especially pertinent question, given the pressing need for the introduction of new clinical antibiotics to combat multidrug-resistant pathogens.33,34 Clearly the inhibitory activity toward eukaryotic ribosomes needs to be suppressed while retaining potency against prokaryotic ribosomes. Total synthesis or semisynthetic modifications may be used to address this question.35 Moreover, what is the ecological importance of bactobolin? Why do B. thailandensis and B. pseudomallei produce this antibiotic in a QS-activated fashion, whereas B. mallei does not? Lastly, several interesting biosynthetic questions remain including the timing of valine hydroxylation, the mechanism of enol lactone formation, and control over the divergent synthesis of numerous variants from the same biosynthetic pathway. These and other questions are likely to be addressed in the future, thereby further deepening our understanding of this potent antibiotic.
CONCLUSIONS
The Greenberg-Clardy collaboration to study bactobolin began over a decade ago with an email from Pete asking Jon for help to purify an unknown antibiotic. Not only was the initial effort successful, but it paved the way for a series of publications involving more collaborative work, making the most of the two fields represented (chemistry and bacterial genetics). Collaborating in this way shed a whole new light on bactobolin, uncovering aspects of its biosynthesis,21,22,26 how it is regulated by QS,19,36,37 its target,20 and how it interacts with the ribosome.31 The project also led to a submitted patent on bactobolin and inspired new collaborations. Notably, Greenberg and Clardy have worked together on additional projects.38,39 Moreover, Chandler from the Greenberg lab and Seyedsaymdost from the Clardy lab are now PIs of their own research groups and continue to work together to characterize other antibiotics produced by B. thailandensis.40 The studies also indirectly inspired new investigations into the ecology of antibiotics produced by soil bacteria41–47 and the relationship between QS and antibiotic production.7,48
ACKNOWLEDGMENTS
We sincerely thank Dr. K. Davis for generating the structure images in Figure 2 and the National Institutes of Health (grants GM59026 to E.P.G., P20-GM103638 to J.R.C., and DP2-AI124786 to M.R.S.) for supporting the authors’ research on Burkholderia spp.
Footnotes
The authors declare no competing financial interest.
Contributor Information
E. Peter Greenberg, Department of Microbiology, School of Medicine, University of Washington, Seattle, Washington 98195, United States.
Josephine R. Chandler, Department of Molecular Biosciences, University of Kansas, Lawrence, Kansas 66045, United States
Mohammad R. Seyedsayamdost, Departments of Chemistry and Molecular Biology, Princeton University, Princeton, New Jersey 08544, United States.
REFERENCES
- (1).Fuqua WC; Winans SC; Greenberg EP J. Bacteriol 1994, 176, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Whitehead NA; Barnard AM; Slater H; Simpson NJ; Salmond GP FEMS Microbiol. Rev 2001, 25, 365–404. [DOI] [PubMed] [Google Scholar]
- (3).Abisado RG; Benomar S; Klaus JR; Dandekar AA; Chandler JR mBio 2018, 9, No. e01749–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Whiteley M; Diggle SP; Greenberg EP Nature 2017, 551, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fuqua WC; Greenberg EP Nat. Rev. Mol. Cell Biol 2002, 3, 685–695. [DOI] [PubMed] [Google Scholar]
- (6).Barnard AM; Bowden SD; Burr T; Coulthurst SJ; Monson RE; Salmond GP Philos. Trans. R. Soc., B 2007, 362, 1165–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).McClean KH; Winson MK; Fish L; Taylor A; Chhabra SR; Camara M; Daykin M; Lamb JH; Swift S; Bycroft BW; Stewart GS; Williams P Microbiology 1997, 143, 3703–3711. [DOI] [PubMed] [Google Scholar]
- (8).Brotherton CA; Medema MH; Greenberg EP mSystems 2018, 3, No. e00208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brett PJ; DeShazer D; Woods DE Int. J. Syst. Bacteriol 1998, 48, 317–20. [DOI] [PubMed] [Google Scholar]
- (10).Duerkop BA; Varga J; Chandler JR; Peterson SB; Herman JP; Churchill ME; Parsek MR; Nierman WC; Greenberg EP J. Bacteriol 2009, 191, 3909–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ulrich RL; Deshazer D; Brueggemann EE; Hines HB; Oyston PC; Jeddeloh JA J. Med. Microbiol 2004, 53, 1053–1064. [DOI] [PubMed] [Google Scholar]
- (12).Valade E; Thibault FM; Gauthier YP; Palencia M; Popoff MY; Vidal DR J. Bacteriol 2004, 186, 2288–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Song Y; Xie C; Ong YM; Gan YH; Chua KL J. Bacteriol 2005, 187, 785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ulrich RL; Deshazer D; Hines HB; Jeddeloh JA Infect. Immun 2004, 72, 6589–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Majerczyk C; Kinman L; Han T; Bunt R; Greenberg EP Infect. Immun 2013, 81, 1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liu X; Cheng YQ J. Ind. Microbiol. Biotechnol 2014, 41, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kunakom S; Eustáquio, A. S. J. Nat. Prod 2019, 82, 2018–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Baltz RH J. Ind. Microbiol. Biotechnol 2017, 44, 573–588. [DOI] [PubMed] [Google Scholar]
- (19).Chandler JR; Duerkop BA; Hinz A; West TE; Herman JP; Churchill ME; Skerrett SJ; Greenberg EP J. Bacteriol 2009, 191, 5901–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chandler JR; Truong TT; Silva PM; Seyedsayamdost MR; Carr G; Radey M; Jacobs MA; Sims EH; Clardy J; Greenberg EP mBio 2012, 3, No. e00499–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Seyedsayamdost MR; Chandler JR; Blodgett JA; Lima PS; Duerkop BA; Oinuma K; Greenberg EP; Clardy J Org. Lett 2010, 12, 716–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Carr G; Seyedsayamdost MR; Chandler JR; Greenberg EP; Clardy J Org. Lett 2011, 13, 3048–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kondo S; Horiuchi Y; Hamada M; Takeuchi T; Umezawa HJ Antibiot 1979, 32, 1069–1071. [DOI] [PubMed] [Google Scholar]
- (24).Antosz FJ; Nelson DB; Herald DL Jr.; Munk ME J. Am. Chem. Soc 1970, 92, 4933–4942. [DOI] [PubMed] [Google Scholar]
- (25).Hori M; Suzukake K; Ishikawa C; Asakura H; Umezawa HJ Antibiot 1981, 34, 465–468. [DOI] [PubMed] [Google Scholar]
- (26).Okada BK; Wu Y; Mao D; Bushin LB; Seyedsayamdost MR ACS Chem. Biol 2016, 11, 2124–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Seyedsayamdost MR Proc. Natl. Acad. Sci. U. S. A 2014, 111, 7266–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Diedrich G; Spahn CM; Stelzl U; Schafer MA; Wooten T; Bochkariov DE; Cooperman BS; Traut RR; Nierhaus KH EMBO J 2000, 19, 5241–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cooperman BS; Wooten T; Romero DP; Traut RR Biochem. Cell Biol 1995, 73, 1087–1094. [DOI] [PubMed] [Google Scholar]
- (30).Wimberly BT; Brodersen DE; Clemons WM Jr.; Morgan-Warren RJ; Carter AP; Vonrhein C; Hartsch T; Ramakrishnan V Nature 2000, 407, 327–339. [DOI] [PubMed] [Google Scholar]
- (31).Amunts A; Fiedorczuk K; Truong TT; Chandler J; Greenberg EP; Ramakrishnan VJ Mol. Biol 2015, 427, 753–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Svidritskiy E; Ling C; Ermolenko DN; Korostelev AA Proc. Natl. Acad. Sci. U. S. A 2013, 110, 12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nathan C Nature 2004, 431, 899–902. [DOI] [PubMed] [Google Scholar]
- (34).Clardy J; Fischbach MA; Walsh CT Nat. Biotechnol 2006, 24, 1541–1550. [DOI] [PubMed] [Google Scholar]
- (35).Adachi H; Nishimura Y; Kondo S; Takeuchi TJ Antibiot 1998, 51, 202–209. [DOI] [PubMed] [Google Scholar]
- (36).Majerczyk CD; Brittnacher MJ; Jacobs MA; Armour CD; Radey M; Schneider E; Phattarasokul S; Bunt R; Greenberg EP J. Bacteriol 2014, 196, 1412–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Majerczyk CD; Brittnacher MJ; Jacobs MA; Armour CD; Radey MC; Bunt R; Hayden HS; Bydalek R; Greenberg EP J. Bacteriol 2014, 196, 3862–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Puri AW; Mevers E; Ramadhar TR; Petras D; Liu D; Piel J; Dorrestein PC; Greenberg EP; Lidstrom ME; Clardy JJ Am. Chem. Soc 2018, 140, 2002–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Coutinho BG; Mevers E; Schaefer AL; Pelletier DA; Harwood CS; Clardy J; Greenberg EP Proc. Natl. Acad. Sci. U. S.A 2018, 115, 9785–9790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Klaus JR; Deay J; Neuenswander B; Hursh W; Gao Z; Bouddhara T; Williams TD; Douglas J; Monize K; Martins P; Majerczyk C; Seyedsayamdost MR; Peterson BR; Rivera M; Chandler JRJ Bacteriol. 2018, 200, No. e00008–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Evans KC; Benomar S; Camuy-Velez LA; Nasseri EB; Wang X; Neuenswander B; Chandler JR ISME J. 2018, 12, 1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Benomar S; Evans KC; Unckless RL; Chandler JR Appl. Environ. Microbiol 2019, 85, No. e00908–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Smalley NE; An D; Parsek MR; Chandler JR; Dandekar AA J. Bacteriol 2015, 197, 3154–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wong RR; Kong C; Lee SH; Nathan S Sci. Rep 2016, 6, 27475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Okada BK; Seyedsayamdost MR FEMS Microbiol. Rev 2017, 41, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wu Y; Seyedsayamdost MR Cell Chem. Biol 2017, 24, 1437–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wu Y; Seyedsayamdost MR Biochemistry 2018, 57, 4247–4251. [DOI] [PubMed] [Google Scholar]
- (48).Chandler JR; Heilmann S; Mittler JE; Greenberg EP ISME J. 2012, 6, 2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]