

Abstract
Pain management devoid of serious opioid adverse effects is still far from reach despite vigorous research and development efforts. Alternatives to classical opioids have been sought after for years and mounting reports of individuals finding pain relief with kratom have recently intensified research on this natural product. Although the composition of kratom is complex, the pharmacological characterization of its most abundant alkaloids has drawn attention to three molecules in particular, owing to their demonstrated antinociceptive activity and limited side effects in vivo. These three molecules are: mitragynine (MG), its oxidized active metabolite 7-hydroxymitragynine (7OH), and the indole-to-spiropseudoindoxy rearrangement product of MG known as mitragynine pseudoindoxyl (MP). Although these three alkaloids have been shown to preferentially activate the G protein signaling pathway by binding and allosterically modulating the μ-opioid receptor (MOP), a molecular level understanding of this process is lacking and yet important in order to design improved therapeutics. The molecular dynamics (MD) study and experimental validation reported here provide an atomic-level description of how MG, 7OH, and MP bind and allosterically modulate the MOP, which can eventually guide structure-based drug design of improved therapeutics.
Graphical Abstract
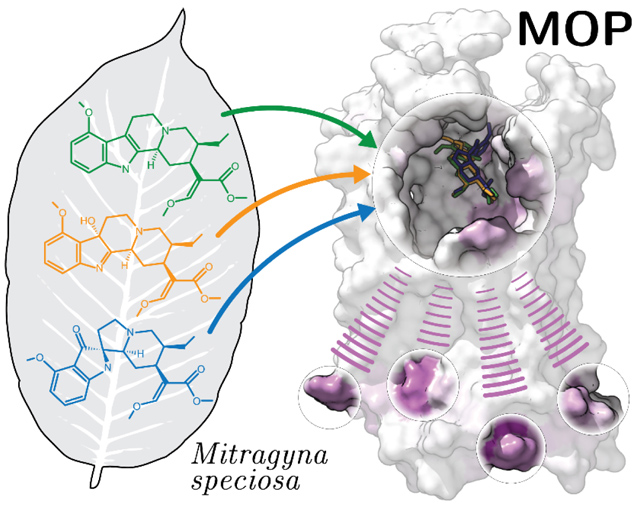
INTRODUCTION
Kratom, also known as Mitragyna speciosa (Korth.), is a tropical evergreen tree indigenous to Southeast Asia that shares close phylogeny with the Rubiaceae family of flowering plants, which includes coffee plants.1 Kratom has been traditionally used in Thailand and Malaysia for centuries.2, 3 Its leaves have been prevalently chewed or brewed for their stimulant effects to stave off the fatigue of rural living (when consumed at low concentrations), but also for their analgesic and euphoric effects (when consumed at high concentrations).4, 5
Outlawed in Thailand and Malaysia, kratom leaves continue to be consumed illegally in those countries.6 In the West, kratom’s use has been steadily gaining popularity as a safer alternative to classical opioids.7, 8 Sold as a dietary or herbal supplement, kratom is classified as an uncontrolled substance in the United States, and it is reportedly utilized for self-management of chronic pain, but also for a variety of other conditions, including anxiety, depression, alcohol use disorder (AUD),9 and opioid use disorder (OUD).7, 8 Although kratom is considered to be less dangerous than typical opioids since it does not cause respiratory depression,10 there have been conflicting reports of its toxicity, as well as its potential for dependence and addiction in humans.11–16 Not only have these observations made kratom’s role as an effective therapeutic difficult to ascertain, but its potentially harmful impact has started to alarm regulatory agencies, which have recently considered kratom’s removal from the market.17
The presence of wildly varying combinations of ~40 different alkaloids in individual kratom leaves18–20 is likely the reason for the variety of different effects reported for this natural product. The most abundant alkaloid in kratom leaves (approximately two-thirds of total alkaloid content) is mitragynine (MG), alongside MG stereoisomers speciociliatine, speciogynine, and mitraciliatine, as well as the related compound paynantheine (see chemical structures in Figure S1 of Supporting Information), the latter making up to 10% of total alkaloid content.2, 3, 21 Other MG congeners, which constitute less than 1% of the total kratom alkaloid content, include the oxidized MG analog 7-hydroxymitragynine (7OH). Additional scaffolds (see Figure S1) are either pyran-fused MG congeners with an additional ring in the core structure (e.g., ajmalicine), or indole-to-oxindole rearrangement products of MG (e.g., corynoxine) or indole-to-spiropseudoindoxyl rearrangement product of MG (e.g., mitragynine pseudoindoxyl; MP) with or without an additional pyran-fused ring.
Although structurally different from classical opioids, MG, 7OH, MP, and several other MG congeners were shown to exert opioid activities by Takayama et al.22, 23 more than twenty years ago (also see ref24 for a recent review). Since then, several receptor-level functional characterizations, including one we co-authored,25 showed that, in general, MG congeners preferentially bind to the human μ-opioid receptor (MOP), followed by κ-opioid receptor (KOP), and δ-opioid receptor (DOP). A more recent report also showed that MG has higher affinity for opioid receptors (Ki = 161–198 nM) compared to α-adrenergic receptors (e.g., Ki = 1340–9290 nM).21 Notably, MG (Ki = 230 ± 47, 231 ± 21, 1011 ± 49 nM at MOP, KOP, and DOP, respectively) was found to have lower affinity towards opioid receptors in Chinese Hamster Ovary (CHO) cells compared to 7OH (Ki = 37 ± 4, 132 ± 7, 91 ± 8 nM at MOP, KOP, and DOP, respectively) and MP (Ki = 0.8 ± 0.2, 24 ± 0.9, 3.0 ± 1.3 nM at MOP, KOP, and DOP, respectively).26 Our co-authored functional studies in Human Embryonic Kidney 293 (HEK 293) cells showed that MG and 7OH acted as partial agonists at human MOP and competitive antagonists at human KOP and human DOP though with very low potency at the latter.25 Notably, both MG and 7OH were found to preferentially recruit G protein over β-arrestin2, leading us to speculate that the functional selectivity of kratom alkaloids might be responsible for their beneficial effects and reduced side effects compared to classical opioids.25, 27 G protein bias was also confirmed in CHO cells for MG, 7OH, and MP, with the latter showing higher G-protein efficacy at MOP.26
The agonistic activity of MG, 7OH, and MP was confirmed in animal studies, particularly in mice and rats.21, 26 Notably, MG showed the weakest analgesic effect, which was significantly lower than morphine, in both the radiant heat tail flick assay in mice and hot plate assay in rats. In contrast, 7OH was shown to be significantly more potent than morphine in both assays, but less potent than fentanyl.21, 26 Although MP was only 1.5-fold more potent than morphine in the radiant heat tail flick assay in mice, reports of reduced side effects such as respiratory depression, physical dependence, and constipation26 make this molecule and other MG congeners worthy of further study from a drug discovery perspective. Notably, since 7OH and MP form as metabolites of MG in vivo, a metabolism-dependent mechanism must be considered to properly evaluate the analgesic effects of MG congeners, given that their effective concentration would depend on the route of administration.28, 29
Although the recent pharmacological characterization of kratom alkaloids has greatly advanced our understanding of how this traditional medicine might work, little is known about how, at a molecular level, these atypical opioids with demonstrated antinociceptive profile in vivo bind and activate the MOP. Among their notable structural features, the molecules are based on an indole scaffold and lack the phenol group seen in endogenous opioids and morphine derivatives. Structural information thus far has been limited to docking studies of a few MG congeners to a rigid MOP structure (e.g., see ref25). Herein, we provide a molecular dynamics (MD)-based description of how MG, 7OH, and MP bind and allosterically modulate the MOP, substantiated by cyclic adenosine monophosphate (cAMP) inhibition experiments on wild-type and mutant receptors.
METHODS
System Setup and Molecular Docking.
Initial binding poses of MG, 7OH, and MP at the orthosteric binding site of the MOP were obtained by flexible ligand-rigid protein docking, using three-dimensional conformations of the three ligands retrieved from the ZINC database (ZINC13473305, ZINC13473311, ZINC13473306 codes for MG, 7OH, and MP, respectively), and the crystal structure of active murine MOP bound to the morphinan agonist BU72 (PDB code 5C1M).30 Prior to docking, the N-terminal fragment of the protein (residues Gly52 to Pro63), as well as the crystal water molecules, were removed. Missing protein side chains (Lys269, Glu2706.25, and Arg2736.28) and hydrogen atoms were added with protonation assigned by the default protocol of the Protein Prep Wizard in the Schrödinger suite 2019–2,31 which includes optimization of hydrogen bonding networks and energy minimization using the OPLS3e force-field.32 Residue numbers, above and throughout this manuscript, refer to the mouse MOP sequence with dot-separated superscript numbers following the Ballesteros-Weinstein generic numbering scheme33 when located in transmembrane (TM) helices and the Isberg’s numbering scheme34 when in loops. According to these schemes, the first number either refers to the helix (e.g., 2 refers to TM2) or the loop (e.g., “23” refers to the loop between TM2 and TM3) to which that residue belongs, whereas the second number refers to the residue position relative to the most conserved residue, defined as number 50.
Ligands were docked at MOP using the DOCK6.9 program suite.35 Briefly, docking spheres within 10 Å of the crystal ligand BU72 were generated with sphgen in the DOCK6.9 program suite35 and used for anchor orientation. Docking grids that store pre-computed van der Waals and electrostatic energetic terms representing the rigid receptor were generated with grid using a 0.3 Å grid spacing inside a cubic box with an 8 Å margin along each dimension surrounding these docking spheres.
To ensure identification of ligand poses with the charged amino group directly interacting with Asp1473.32, a key residue in the orthosteric binding site, the “pruning_max_orients” number was set to 3000 and the “max_orientations” quantity was set to 2000 for each ligand-receptor docking system. Up to 100 top-scored conformations of each ligand according to DOCK6.9 grid energy score were retained and subsequently clustered with a root mean square deviation (RMSD) threshold of 2 Å. The clusters were ranked according to the best score they contained, and the pose with the best score from each cluster was chosen as its representative. For each ligand, the three top-scored representatives were then subject to metadynamics rescoring.
Metadynamics Rescoring of Docking Poses.
Metadynamics rescoring simulations were run separately on each of the 3 aforementioned top-scored cluster representatives identified for each ligand, thus simulating a total of 9 different ligand-MOP complexes. Following a method originally introduced by Clark et al.,36 which was incorporated and further validated by Fusani et al.,37 as a Binding Pose Metadynamics module into the Schrödinger suite 2019–2,31 we carried out 10 independent metadynamics simulations for each selected ligand pose. Ligands were parameterized using the default protocol of LigPrep in the Schrödinger suite 2019–2,31 which assigns OPLS3e partial charges.32 Ligand’s dihedral parameters that were not included in the standard OPLS3e distribution were generated using the Force Field Builder in the Schrödinger suite. The RMSD of the ligand heavy atoms relative to the starting pose, after alignment of the residues within 3 Å from the ligand, was used as collective variable. Ten well-tempered metadynamics38 simulations were run with Desmond31 for 10 ns each with a bias factor of 7.74, Gaussian hills with initial height 0.05 kcal/mol and width 0.02 Å, and a deposition rate of 1 ps. Binding pose stability was assessed based on a score that reflects the average RMSD corrected by a term indicating hydrogen bond persistence. Specifically, for each trajectory, we calculated the composite score (CompScore) defined as:
where F(RMSD) is the free energy of the system as a function of the collective variable RMSD, kB is the Boltzmann’s constant, and T is the temperature; 〈H〉 is the fraction of intermolecular hydrogen bonds interactions detected in the input structure, and maintained over the last 2 ns of each independent simulation. CompScore values were averaged over the 10 metadynamics simulations and are reported here as average values alongside 25%−75% confidence intervals.
Molecular Dynamics Refinement of Ligand-Bound Complexes.
The ligand-MOP complexes containing the top metadynamics-rescored poses of MG, 7OH, and MP were refined using unbiased MD simulations carried out with Desmond. All three ligand-receptor complexes were embedded in a 1-palmitoyl-2-oleoyl phosphatidyl choline (POPC) bilayer, and solvated in an orthorhombic box with a 10 Å buffer in each dimension consisting of simple point charge (SPC) water molecules and 0.15 M NaCl, and neutralized with chloride ions using System Builder in the Schrödinger suite 2019–2.32 MD simulations were run using default simulation parameters, including timesteps of 2 fs for bonded forces and short-range non-bonded forces, and 6 fs for long-range non-bonded forces using the RESPA integrator.39 Following the standard membrane relaxation protocol in Desmond, 72 ns constant-pressure, constant-temperature (NPT) equilibration runs were carried out in 19 steps, the first 18 of which employed gradually relaxing positional restraints on the heavy atoms of lipids, protein sidechains, protein backbone, ligand ring atoms, and lastly, the remaining ligand atoms. The last step consisted of a 3 ns unrestrained NPT equilibration run. During equilibration, system temperature and pressure were maintained at 300 K and 1 bar, respectively, using the Nose-Hoover thermostat40 and a semi-isotropic MTK barostat. Short-range Coulomb interactions were cut off at 9 Å. Four independent replicas of 250 ns production runs were then performed for each ligand-receptor complex, with structural data collected every 0.5 ns for a total of 2000 frames for each complex.
Trajectory Clustering.
To identify and visually inspect highly populated conformations of each ligand in the orthosteric binding site of MOP, clustering was performed for each ligand using Schrödinger’s trj_cluster.py script, which employs the affinity propagation clustering algorithm described by Fray and Dueck.41 Specifically for each ligand, 500 snapshots of the trajectory with a stride of 2 ns were fitted to the same frame using the protein heavy atoms within 8 Å of the ligand prior to clustering to provide an overlay of the orthosteric binding site but prevent overfitting of the ligand. Pairwise RMSD values of the same selected group of atoms were then used as input for the affinity propagation clustering algorithm that calculates the clusters and a representative member of each cluster.
Structural Interaction Fingerprints (SIFT) Analyses.
Following data collection, SIFT analyses were performed using an in-house python script. The interactions between ligands and receptor residues were calculated as a 9-bit representation based on the following nine types of interactions: apolar interactions (carbon-carbon atoms in contact), face-to-face (Aro_F2F) and edge-to-face (Aro_E2F) aromatic interactions, hydrogen-bond interactions with the protein as hydrogen-bond donor (Hbond_proD) or hydrogen-bond acceptor (Hbond_proA), electrostatic interactions with positively (Elec_ProP) or negatively charged (Elec_ProN) residues, 1-water mediated H-bond (Hbond_1wat), and 2-water mediated H-bond (Hbond_2wat). A distance cutoff of 4.5 Å was used to define apolar interactions, while a cutoff of 4 Å was used to describe aromatic and electrostatic interactions. Interactions were calculated for both backbone and sidechain atoms. The probability of each interaction was estimated using a two-state Markov model, sampling the transition matrix posterior distribution using standard Dirichlet priors for the transition probabilities as described by Noé et al.42
Transfer Entropy Analysis.
The transfer entropy between two structural descriptors X and Y is defined as their conditional mutual information
This metric quantifies the directed (time-asymmetric) communication between pairs of structural descriptors, which yields molecular determinants for allosteric communication within proteins.43 Similar concepts have been shown to effectively capture allosteric communication during GPCR activation.44–48 Here, we employed the MDEntropy package49 to compute all pairwise transfer entropy values between 2,115 residue-residue contact pairs and 284 ligand-residue contacts, yielding a transfer entropy matrix of 2,399 structural descriptors built using the MD trajectory for each ligand-bound MOP system. The residue-residue contacts were defined as residue pairs with any pair of receptor heavy atoms within a 7 Å cutoff in the active MOP crystal structure. A directed graph was built for each resulting transfer entropy matrix derived from a ligand-bound MOP system using the NetworkX python library,50 with each node representing a contact (residue-residue or ligand-residue), and the weight of the directed edge between two nodes defined as the transfer entropy between them.
Contacts formed by residues located at least 8 positions apart on the intracellular side of the receptor in the 5C1M crystal structure (residues 93–105, 164–184, 255–279, and 339–347) were considered as possible target contacts of the MOP allosteric modulation induced by the three different simulated ligands. The information flow from the ligand orthosteric binding site to the intracellular region of MOP was described by the set of paths Γ connecting the set of ligand-residue contacts and the set of target residue-residue contacts in the intracellular region, where the “length” of each edge is the negative logarithm of the transfer entropy value of that edge. The importance of each node in allosteric communication was measured by their contribution in terms of accumulated normalized flux within the shortest 5,000 paths in Γ. Specifically, the contribution of a given contact qi was calculated as:
where qi is one of the contact descriptors, Γi is the set of paths containing qi and connecting the ligand-residue nodes to target residue-residue nodes in the MOP intracellular region, γ is one path in this set, ej is an edge between two structural descriptors in path γ, and wj is the transfer value of that edge. The contact-based normalized flux was then converted to residue-based normalized flux by summing all normalized flux values that involved a given residue. Residues that contributed the most to allosteric communication (> 2% flux) are listed in Table S1.
Gαi-mediated cAMP inhibition assays.
To measure Gαi-mediated cAMP inhibition, HEK 293T (ATCC CRL-11268) cells were co-transfected with human opioid receptor OPRM_HUMAN (hMOP or mutants) along with a luciferase-based cAMP biosensor and the assay was performed as reported previously.51 Briefly, transfected cells were plated into 96-well white clear bottom cell culture plates with Dulbecco’s Modified Eagle Medium (DMEM) + 1% dialyzed Fetal Bovine Serum (FBS) and incubated at 37 °C with 5% CO2 overnight. The next day, drug solutions were prepared in fresh drug buffer [20 mM HEPES, 1X Hanks’ Balanced Salt solution (HBSS), pH 7.4] at 3X drug concentration. Plates were decanted and received 60 μL per well of drug buffer (20 mM HEPES, 1X HBSS) followed by an addition of 30 μL of drug solution (3 wells per condition) for 15 min in the dark at room temperature. To stimulate endogenous cAMP production, 30 μL luciferin (4 mM final concentration) supplemented with forskolin (30 μM final concentration) were added per well. Cells were again incubated in the dark at room temperature for 15 min, and luminescence intensity was quantified using a luminescence counter. Results (relative luminescence units) were plotted as a function of drug concentration, normalized to % DAMGO stimulation, and analyzed using “log(agonist) vs. response” in GraphPad Prism 8.0.
RESULTS AND DISCUSSION
Preferred Binding Mode of MG, 7OH, and MP to MOP from Metadynamics Rescoring.
Initial binding poses of MG, 7OH, and MP at the orthosteric site of the rigid crystal structure of MOP (pdb:5C1M) were obtained with DOCK6.9. RMSD clustering of the 100 top-scoring, docked conformations obtained for each of the three ligands (see Methods for details) yielded 38, 34, and 73 unique pose clusters for MG, 7OH, and MP, respectively. Since the DOCK6.9 scores of the 3 top-ranked cluster representatives for each ligand were similar (Figure S2), we applied metadynamics rescoring for final pose selection. Table S2 shows the metadynamics CompScore calculated by the Schrödinger’s Binding Pose Metadynamics module (see Methods section for definitions and calculation details) and used to assess the stability of each pose in the binding pocket. MG pose #2, 7OH pose #1, and MP pose #1 (Figures S2B, S2D, and S2G, respectively), shown overlaid in the MOP binding pocket in Figure 1A, were selected as preferred binding poses for each ligand based on their lowest CompScore value (Table S2 and Figure 1B). The time evolution of the average RMSD of the heavy atoms of each of the three simulated ligands from their initial docking conformation across the 10 independent metadynamics simulations used for CompScore ranking is reported in Figure S3 for each pose.
Figure 1.
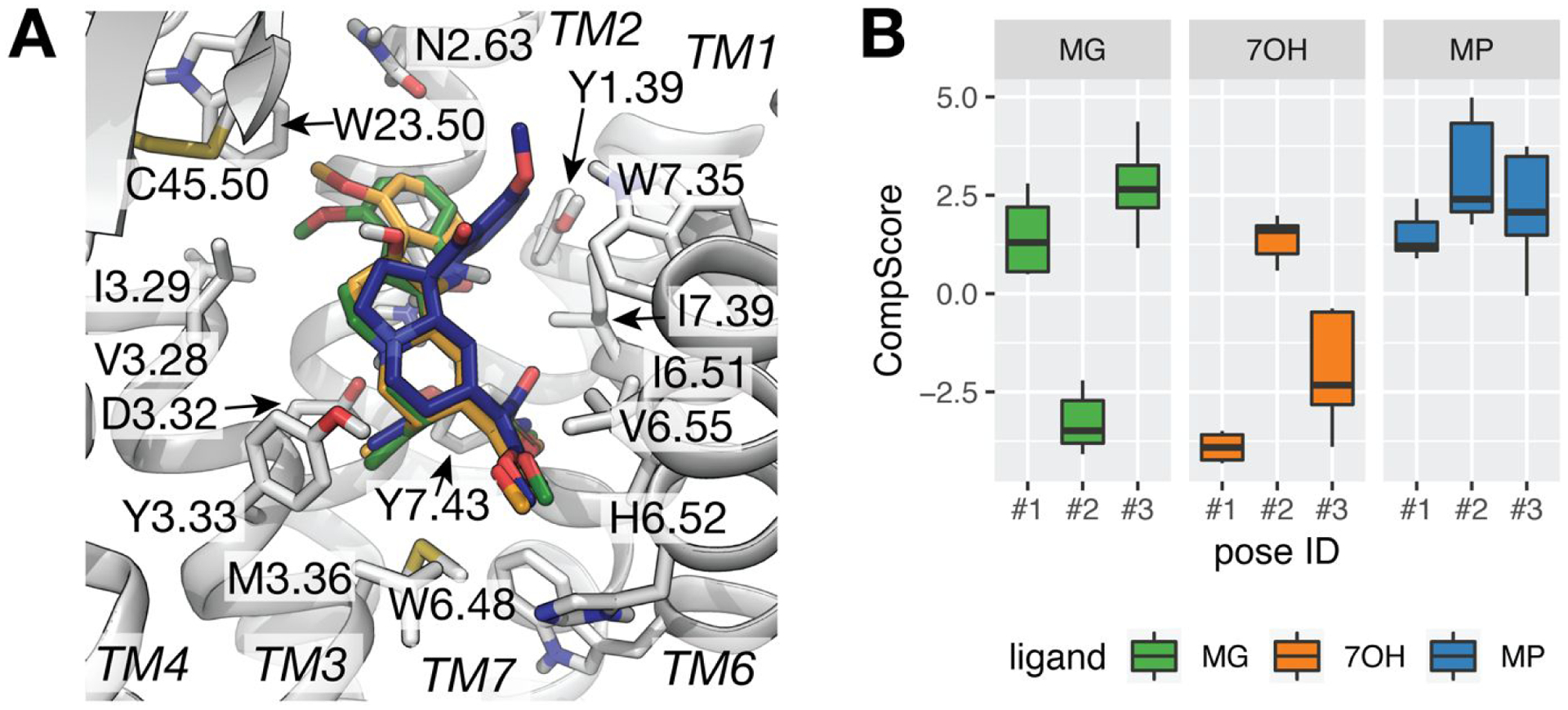
Preferred docking poses of MG, 7OH, and MP at MOP as predicted by metadynamics rescoring. (A) Docked poses with top-ranking CompScore (i.e., pose #2 of MG, in green sticks, and poses #1 for 7OH and MP in orange and blue sticks, respectively) within the MOP receptor depicted in light grey cartoon representation. Residues with heavy atoms within 4 Å of any of the three ligands are shown as sticks. TM5 is not shown for clarity. (B) Box plots of the CompScore values calculated for the selected top three cluster representatives of each ligand over 10 independent metadynamics runs. Error bars denote the 25th and 75th percentiles.
As shown in Figure 1A, the three preferred docking poses of MG, 7OH, and MP are oriented similarly in the MOP binding pocket, forming the salt bridge between their charged amino groups and Asp1473.32 (see Figure S2) that is known to be a critical interaction across classical opioid agonists and antagonists.30, 52 Another similarity among these best-scored MG, 7OH, and MP binding poses is the orientation of the ethyl group at position C19 (cyan color in Figure S1), which forms apolar interactions with surrounding residues within 4 Å from the ligand’s heavy atoms (Met1513.36, Tyr1483.33, and Tyr3267.43). Notably, these are the same residues that interact with the N-methyl group of the co-crystallized ligand BU72 in the active MOP crystal structure 5C1M. Among the noticeable differences in the preferred MG, 7OH, and MP docking poses is the direction of the methyl ester group within the β-methoxyacrylate moiety (purple color in Figure S1), which points towards TM6-TM7 in the MP pose or TM5-TM6 in the MG and 7OH poses (Figure 1A). Specifically, the carbonyl oxygen of the methyl ester group within the β-methoxyacrylate moiety of MP is located between Ile2966.51 and Ile3227.39 in the MP predicted binding pose and towards His2976.52 in the MG and 7OH binding poses. Another difference relates to the position of the methoxyindole group (red color in Figure S1). The preferred docking poses of MG and 7OH show this group leaning toward a hydrophobic pocket formed by residues of TM2 (Asn1272.63) and TM3 (Ile1443.29), and pointing towards Trp13323.50. In contrast, the indole-to-spiropseudoindoxyl substitution of MP forces this part of the molecule to interact with Trp3187.35 in the preferred docking pose for this compound.
MD-based Analysis of the Preferred Binding Poses of MG, 7OH, and MP.
Complexes of the aforementioned metadynamics-rescored top binding poses of MG, 7OH, and MP with MOP were embedded in a POPC bilayer and simulated in four independent standard MD replicas of 250 ns each, totaling 3 μs simulations for the three ligands.
During simulation, the MOP TM bundle and ligand conformations fluctuate only slightly as assessed by the time evolution of the RMSD of the receptor TM alpha carbons from the MOP crystal structure and the RMSD of the ligand heavy atoms from their initial docked pose (Figures S4 and S5, respectively). In particular, during 4 different MD simulations of the MG-bound MOP system, the ligand heavy atoms and receptor alpha carbons maintain aggregated RMSD averages from the docked conformation of 1.71 Å (1.24, 1.95 Å) and 1.52 Å (1.33, 1.68 Å), respectively, with values in parenthesis indicating the 25% and 75% quantiles of the distribution. Similarly, the ligand heavy atoms and receptor alpha carbons of the simulated 7OH-bound MOP system exhibit average RMSD values of 1.63 Å (1.23, 2.00 Å) and 1.50 Å (1.24, 1.74 Å), respectively, while corresponding values for the simulated MP-bound MOP system are 2.38 Å (2.05, 2.69 Å) and 1.49 Å (1.30, 1.67 Å), respectively.
The differences noted in the preferred docking poses of 7OH and MG versus that of MP were maintained during simulations despite slightly different dynamic behavior among the three ligands. While the binding poses of MG and 7OH did not change much during simulation (Figure S6A and 6B, respectively) and maintained their β-methoxyacrylate moiety orientation (see Figure S7), MP moved slightly closer to TM2 and TM3 during simulations, apparently due to a rotation of the Gln1242.60 side chain, which incidentally allowed the MP’s indole ring to get closer to Tyr3267.43. Notably, this slight change in the MP binding pose did not affect the orientation of both the β-methoxyacrylate moiety and the ethyl group at position C19 (see Figure S6C).
Structural interaction fingerprint analysis was used to measure the stability of interactions formed by MG, 7OH, and MP with MOP during simulation. The nature of these interactions (see definitions in the Methods section) and their probabilities of being formed during the four independent MD simulations carried out for the three ligand-MOP systems are shown in Figure 2, while Figure 3 provides a structural visualization of the most probable interactions (probability > 75%) formed by representative conformations of MG, 7OH, and MP from the most populated MD simulation clusters.
Figure 2.
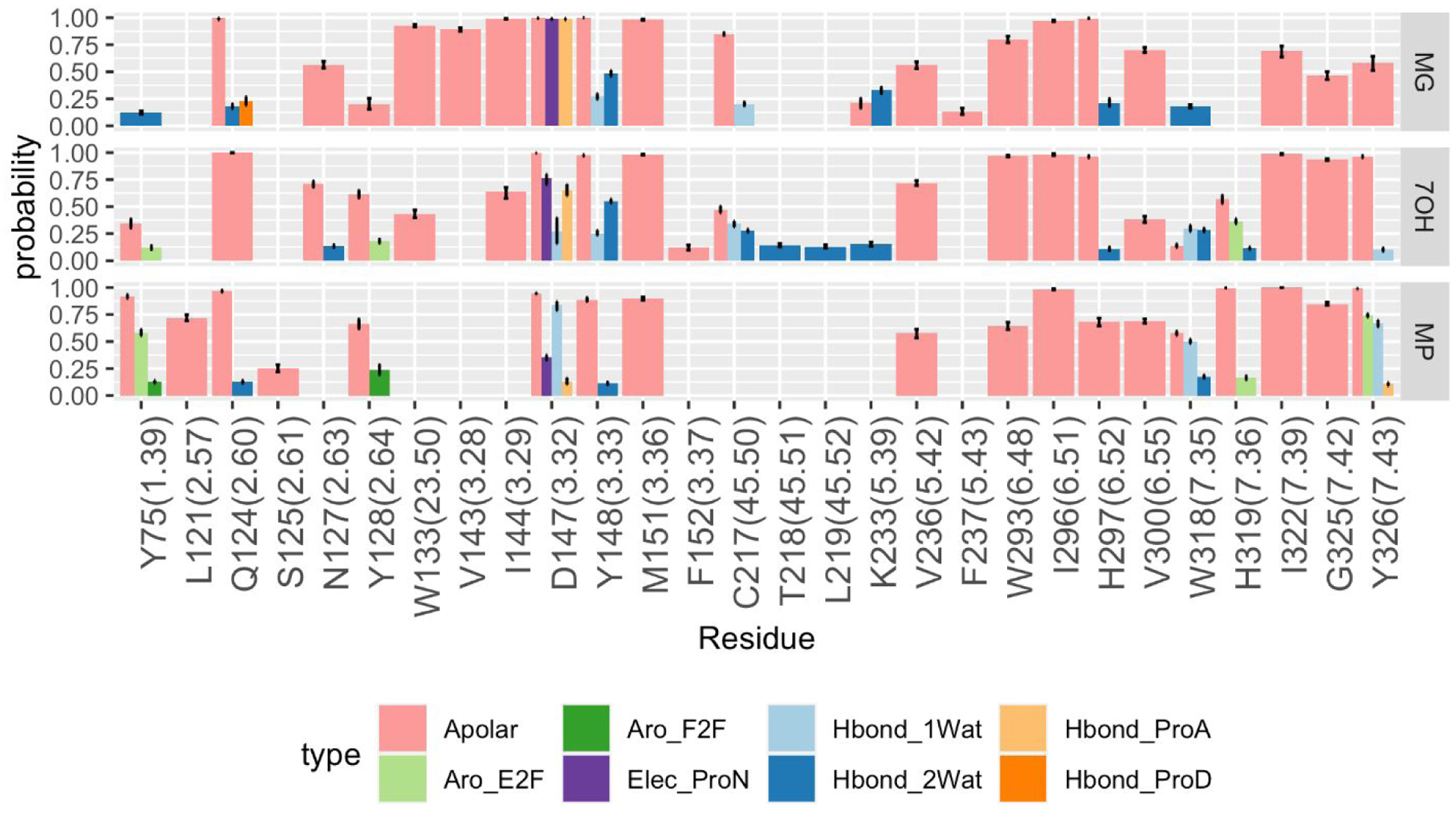
Probabilities of ligand-receptor interactions formed during MD simulations of MG-MOP, 7OH-MOP, and MP-MOP. The eight interaction types formed by the ligands with protein backbone and side chains are: carbon-carbon atomic interactions (Apolar, pink), face-to-face aromatic (Aro_F2F, light green), edge-to-face aromatic (Aro_E2F, dark green), hydrogen bond with the protein as hydrogen bond donor (Hbond_ProD, dark orange), hydrogen bond with the protein as hydrogen bond acceptor (Hbond_ProA, light orange), electrostatic interaction with the protein negatively charged (Elec_ProN, purple), one-water mediated and two-water mediated hydrogen bond interactions (Hbond_1Wat and Hbond_2Wat, light and dark blue, respectively). Only interactions with an average probability above 10% are displayed.
Figure 3.
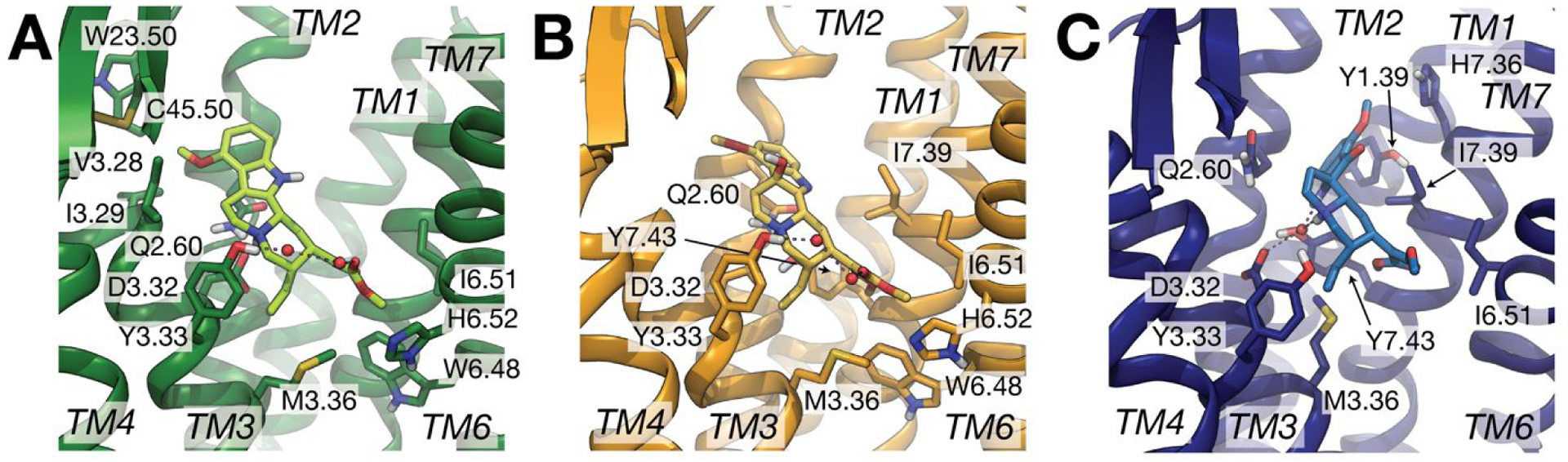
Representative conformations of the largest MD simulation clusters obtained for (A) MG-MOP (green), (B) 7OH-MOP (orange), and (C) MP-MOP (blue) from the combined 1 μs trajectory for each system. Only residues involved in highly probable interactions (>75%) with the ligand are displayed as sticks. Red spheres indicate water molecules involved in high probability (>50%) interactions with both the ligand and the protein. Specifically, 2-water-mediated interactions connecting Tyr3.33 to the carbonyl group of the β-methoxyacrylate moiety of MG and 7OH are indicated with dotted lines in panels (A) and (B), respectively, while 1-water-mediated interactions connecting Asp3.32 or Tyr7.43 to MP are indicated in panel (C).
The analysis points to several ligand-receptor interactions that remain stable with high probability (>75%) during simulations of all three MG-MOP, 7OH-MOP, and MP-MOP systems suggesting common molecular recognition of the three kratom alkaloids by the receptor. Specifically, these highly probable and common interactions are with Gln1242.60, Asp1473.32, Tyr1483.33, Met1513.36, and Ile2966.51, with Trp2936.48 and His2976.52 just below the 75% threshold for MP and Ile3227.39, Gly3257.42, and Tyr3267.43 just below the 75% threshold for MG. It is worth noting that - with the exception of Gln1242.60, Asp1473.32, and Tyr1483.33, which are interacting with the indole group, the charged amino group, and the quinolizidine group, respectively - these residues are all interacting with the β-methoxyacrylate moiety of the three ligands. As the β-methoxyacrylate moiety does not change orientation during the simulated time (Figure S7), and we cannot exclude the possibility that this is due to the sampling limitations of standard MD simulations, we cannot draw any specific conclusion on the preferred orientation of the β-methoxyacrylate moiety in the three ligands. Our simulations suggest that the MP methyl ester group within the β-methoxyacrylate moiety forms stable apolar interactions with Trp2936.48, Ile2966.51, Ile3227.39, and Tyr3267.43, whereas the MG and 7OH methyl esters form apolar interactions with Met1513.36, Trp2936.48, Ile2966.51, and His2976.52, with the latter residue also involved in interaction through a 2-water-mediated H-bond network, albeit with very low probability.
Notably, the simulated ligands display different propensities for water-mediated interactions, with 7OH forming the largest number (11 residues above 10% probability), followed by MG (7 residues), and then MP (5 residues). The hydroxyl group of 7OH at position C7 appears to be involved in unique water-mediated hydrogen bonds, specifically with Thr21845.51 and Leu21945.52 on the EL2, as well as with Asn1272.63 and His3197.36 (Figure 2), although with relatively low probability (12–14%). The different orientation of the indole group between 7OH and MG we had previously drawn attention to25 enables more stable interactions of 7OH with Tyr1282.64 and His3197.36 compared to MG (Figures 2 and 3).
The difference in C9 methoxy group orientation in MP compared to MG and 7OH may provide an explanation for MP’s increased affinity for MOP. Specifically, interactions with Tyr751.39 and His3197.36 occur with a probability higher than 75% in the MP-MOP system, but with a much lower probability in 7OH-MOP or even below 10% in the MG-MOP system. In contrast, the MG and 7OH methoxy groups at the C9 position are oriented towards a hydrophobic pocket formed by residues with which MP did not form appreciable contact (e.g., Trp13323.50, Ile1443.29, and Cys21745.50; see Figure 2).
Experimental Validation of the Predicted Differential Involvement of Certain Residues in Ligand-Induced MOP Activation.
The calculated probabilities of ligand-receptor interactions formed during MD simulations of MG-MOP, 7OH-MOP, and MP-MOP (Figure 2) drew attention to specific residues on the receptor whose mutations are expected to differentially affect the ability of the three ligands to activate the receptor. Among them are Asn1272.63 and His3197.36, where mutation of the former is hypothesized to affect the ability of MG and 7OH, but not MP, to activate the MOP, while mutation of the latter may primarily affect MP and 7OH, based on the data shown in Figure 2. The corresponding Asn1272.63 residue in hMOP (Asn129) was mutated to lysine to match the corresponding DOP residue and significantly alter the interaction with the ligands. As predicted, the potency of MP was not altered at the Asn129K mutant (0.9 nM) compared to the wild-type receptor (EC50 =0.55 nM) while the MG and 7OH potencies were reduced 23 fold (237 nM to 5524 nM) and 5 fold (19.5 nM to 96.5 nM), respectively (Figure 4A–B).
Figure 4. Asn129K mutation decreases MG and 7OH potency but not MP potency whereas H321F decreases both 7OH and MP potency and efficacy.
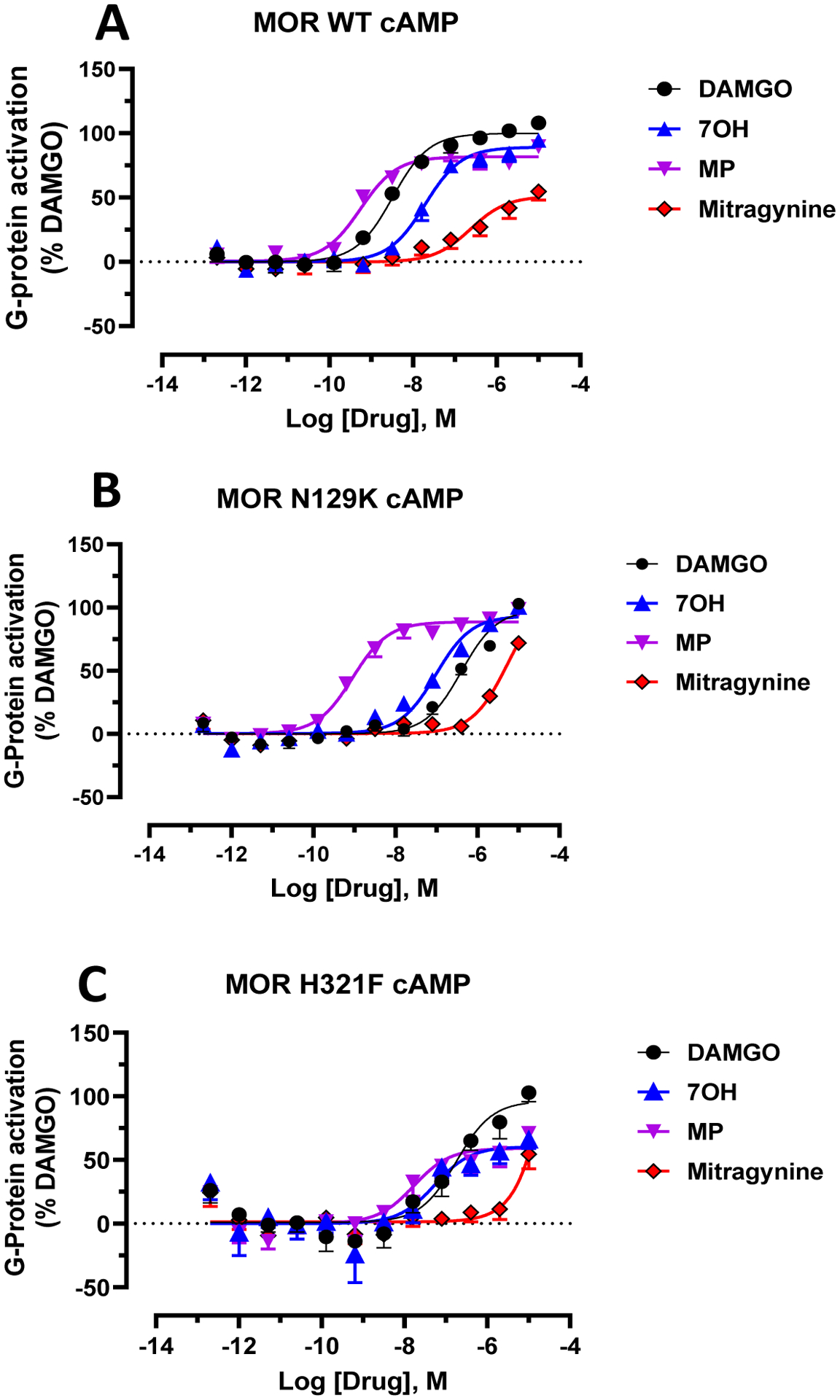
A) At wild type (WT) hMOP, DAMGO, MG, 7OH and MP showed dose dependent G-protein activation in the cAMP assay. Gi: DAMGO, EC50 = 3.23(8.49 ± 0.087) nM; MG, EC50 = 237 (6.62±0.21) nM), Emax = 51±5.4; 7OH, EC50 = 19.5 (7.71 ± 0.1) nM, Emax = 89±3.8 and MP, EC50 = 0.55 (9.28±0.11) nM, Emax = 81±3.6. B) At the hMOP Asn129K mutant both MG and 7OH potency were reduced along with DAMGO. Gi: DAMGO, EC50 = 404(6.39 ± 0.1) nM; MG, EC50 = 5524 (5.26 ± 0.084) nM), Emax = 72±3; 7OH, EC50 = 96.5 (7.02 ± 0.05) nM, Emax = 92 ± 3.2 and MP, EC50 = 0.90 (9.04 ± 0.084) nM, Emax = 88 ± 2.97. C) At the hMOP H321F mutant both 7OH and MP potency were reduced along with DAMGO. Gi: DAMGO, EC50 = 17.9 (6.75 ± 0.18) nM; MG, EC50 = nd, Emax = 54.6±8.19; 7OH, EC50 = 53.7 (7.27 ± 0.34) nM, Emax = 60 ± 9 and MP, EC50 = 16.9 (7.77 ± 0.27) nM, Emax = 58.9 ± 6.7. Data from cAMP assays at hMOP were normalized to Emax of DAMGO. The dose response curves were fit using a three-parameter logistic equation in GraphPad Prism and the data are presented as mean EC50 (pEC50 ± SEM) for assays run in triplicate.
The corresponding His3197.36 residue in hMOP (His321) was mutated to phenylalanine to maintain an aromatic interaction while eliminating the possibility of an additional polar interaction. At this mutant receptor the potency of MP was reduced 32 fold (EC50=16.9 nM) whereas the potency of 7OH was reduced 2.5 fold (EC50=34.5 nM). The greater reduction of MP potency compared to 7OH at this mutant is in line with our modeling, which suggests that the probability of MP side-chains interacting with His is higher than that of 7OH (99.6% versus 56.6%; see Figure 2). We are unable to comment on the effect of the His321F mutation on MG because of the ligand’s poor potency and the shallow slope seen in the cAMP assay, which does not allow an accurate measurement of EC50. Of note, we see a >25% decrease in efficacy for both 7OH and MP (Figure 4C).
Residues Contributing to Coupling between Ligand-Binding Pocket and Receptor Intracellular Region in the Three Ligand-Bound MOP Complexes.
To understand how MG, 7OH, and MP compare in terms of their induced allosteric modulation of MOP, we assessed their effect on the communication between the ligand-binding pocket and the intracellular region of the receptor using transfer entropy analysis and derived normalized flux contributions for each receptor residue in the three ligand-bound MOP systems.
Table S1 lists the MOP residues that are most significantly contributing (> 2% flux) to the transfer entropy between the orthosteric ligand binding site and the intracellular region of the receptor for the three simulated ligand-MOP systems. Twenty-one of them are common between all three systems, suggesting they may contribute to a common allosteric mechanism leading to the G protein-biased agonism demonstrated experimentally for MG, 7OH, and MP. These residues are: Lys10012.51, Thr1012.37, Thr1032.39, Asn1042.40, Ile1052.41, Arg1653.50, Cys1703.55, Pro1814.39, Met2555.61, Ile2565.62, Leu2575.63, Arg2585.64, Leu2595.65, Lys2716.26, Asn2746.29, Leu2756.30, Arg2776.32, Leu3397.56, Asp3408.47, Glu3418.48, Asn3428.49. Among them, Thr1032.39, Ile1052.41, Cys1703.55, Pro1814.39, Lys2716.26, Leu2756.30, Glu3418.48, and Asn3428.49 and may be worthy of experimental testing since they contribute more than 10% to the transfer entropy between the binding pocket and intracellular region of the receptor in at least one of the three simulated ligand-MOP systems. Notably, two other residues contributing more than 10% to the transfer entropy are unique to 7OH (Thr21845.51) and MP (Trp2936.48). The latter observation is interesting in view of the fact that the tryptophan at position 6.48 is the rotamer toggle switch that has been noted in the literature for contributions to GPCR activation and whose involvement here might suggest a specific role in the observed higher efficacy of MP compared to the partial agonism of MG and 7OH, the latter in less amplified systems.25, 26 While a small number of transfer entropy-contributing residues are common between MG and 7OH (Asp1643.49, Pro17234.50, Lys2605.66), a much larger number of residues are found that are common between 7OH and MP (Thr9712.48, Met9912.50, Thr1202.56, Leu1292.65, Tyr1493.34, Asn1503.35, Phe1523.37, Ser2615.67, Val2625.68, Arg263IL3, Asp2726.27, and Phe3438.50). The latter are particularly interesting and worthy of experimental testing owing to the observed higher potency of MP and 7OH compared to MG. Figure 5 shows the receptor surface colored according to the residue contribution to the information flux between the ligand-binding pocket and the intracellular region of the receptor in the simulated MG-MOP, 7OH-MOP, and MP-MOP systems. The figure clearly illustrates the many similarities in the distribution of the residues that most contribute to the ligand-induced allostery, which suggests a common mechanism of G protein biased agonism at MOP for the three compounds.
Figure 5.
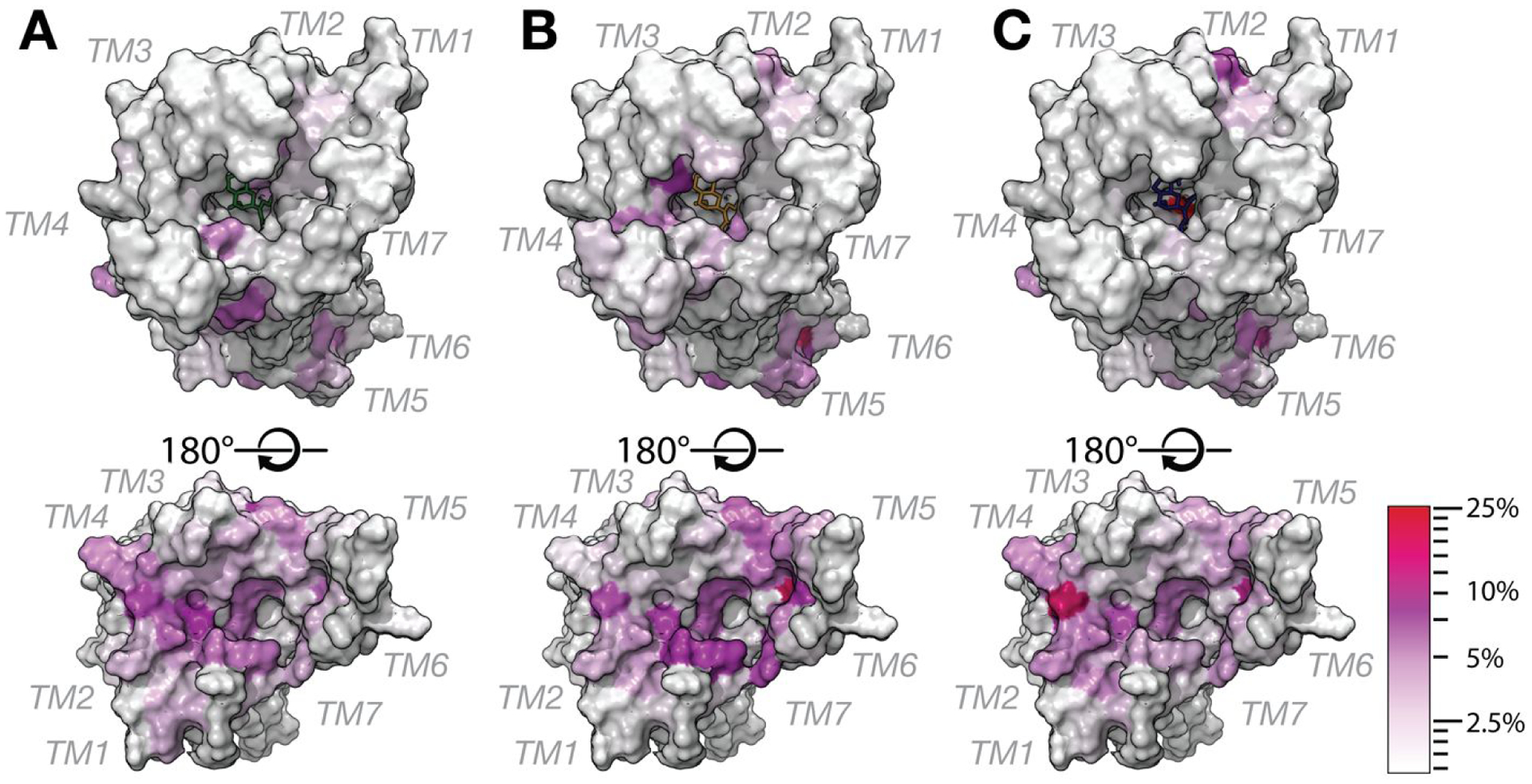
Structural location of receptor residues that are most significantly contributing to the transfer entropy between the ligand-binding pocket and the intracellular region of the receptor in the simulated (A) MG-MOP, (B) 7OH-MOP, and (C) MP-MOP systems. For simplicity, residues are depicted on the 5C1M crystal structure shown in horizontal view with extracellular and intracellular sides of the receptor on the top and bottom panels, respectively. The residue color indicates the residue contribution to the overall flux, from low (white) to high (red) values of the transfer entropy.
Notably, the intracellular side of TM6, the helix known to undergo the largest conformational change upon activation, is not the only region involved in the transfer of information from the ligand binding pocket to the G protein-binding region of the receptor, but several high contributions are observed at the intracellular side of TM4 and TM2, with residues such as Ile1052.41 and Pro1814.39 hypothesized to play a major role in G protein-biased signaling.
CONCLUSIONS
The MD study and experimental validation reported herein shed light not only on the preferred binding mode of three kratom alkaloids that have been recently reported to exert antinociception in vivo, specifically MG, 7OH, and MP, but also on their induced allosteric modulation of the MOP resulting in G protein-biased signaling. Metadynamics rescoring carried out to discriminate among similarly scored docking poses of these three molecules reveals similar binding modes in the MOP orthosteric binding pocket, but also important differences that are mostly due to intrinsic conformational changes induced by the oxidization or indole-to-spiropseudoindoxyl rearrangement of the MG chemotype, and possibly responsible for the different potency of MP, MG, and 7OH. These similarities and differences are largely maintained during MD simulations, and experimental validation of the differential impact of Asn1272.63 and His3197.36 mutations on the ability of MG, 7OH, and MP to activate MOP instills confidence in the dynamic modeling. The several common residues that are highly contributing to the transfer entropy between the orthosteric ligand binding site and the intracellular region of the receptor among the three simulated ligand-MOP systems point to a common allosteric mechanism that may form the basis for the experimentally observed G protein-biased agonism by the three kratom active alkaloids. Of note, this analysis generates a number of mechanistic hypotheses that are worthy of experimental testing to achieve an even better molecular-level understanding of how kratom active alkaloids bind and activate MOP, which is useful information to design improved therapeutics.
Supplementary Material
ACKNOWLEDGEMENTS
Computations were run on resources available through the Office of Research Infrastructure of the National Institutes of Health under award numbers S10OD018522 and S10OD026880, as well as the Extreme Science and Engineering Discovery Environment under MCB080077, which is supported by National Science Foundation grant number ACI-1548562.
FUNDING
This work was supported by National Institutes of Health grants DA034049, DA045884, and DA046487.
Footnotes
SUPPORTING INFORMATION
MOP residues that are most significantly contributing to the transfer entropy; Binding Pose Metadynamics CompScore of the 3 top cluster representatives of MG, 7OH, and MP from docking at MOP; Structures of kratom alkaloids cited in this paper; Binding poses and DOCK6.9 scores for MG, 7OH, and MP; Time evolution of the RMSD of MG, 7OH, and MP heavy atoms from their initial docking conformations; Time evolution of the RMSD of the alpha-carbons of the MG-bound, 7OH-bound, and MP-bound MOP transmembrane region from the corresponding region in the 5C1M crystal structure; Time evolution of the RMSD of the heavy atoms of MG, 7OH, and MP from those in the docked conformations; Representative binding conformations of the largest MD simulation clusters obtained for MG-MOP, 7OH-MOP, and MP-MOP overlapped onto their respective starting, docking conformations; Time evolution of the C19-C14-C15-C16 dihedral angle of the β-methoxyacrylate moiety of MG, 7OH, and MP.
REFERENCES
- [1].Davis A (2006) Rubiaceae of Thailand-A pictorial guide to indigenous and cultivated genera, Oxford University Press. [Google Scholar]
- [2].Eastlack SC, Cornett EM, and Kaye AD (2020) Kratom—Pharmacology, clinical implications, and outlook: a comprehensive review, Pain and Therapy 9, 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ramanathan S, and McCurdy CR (2020) Kratom (Mitragyna speciosa): worldwide issues, Current Opinion in Psychiatry 33, 312–318. [DOI] [PubMed] [Google Scholar]
- [4].Singh D, Narayanan S, Vicknasingam B, Corazza O, Santacroce R, and Roman-Urrestarazu A (2017) Changing trends in the use of kratom (Mitragyna speciosa) in Southeast Asia, Human Psychopharmacology: Clinical and Experimental 32, e2582. [DOI] [PubMed] [Google Scholar]
- [5].Singh D, Narayanan S, Müller CP, Swogger MT, Chear NJY, Dzulkapli EB, Yusoff NSM, Ramachandram DS, León F, and McCurdy CR (2019) Motives for using Kratom (Mitragyna speciosa Korth.) among regular users in Malaysia, Journal of ethnopharmacology 233, 34–40. [DOI] [PubMed] [Google Scholar]
- [6].Likhitsathian S, Jiraporncharoen W, Aramrattana A, Angkurawaranon C, Srisurapanont M, Thaikla K, Assanangkornchai S, Kanato M, Perngparn U, and Jarubenja R (2018) Polydrug use among kratom users: Findings from the 2011 Thailand National Household Survey, Journal of Substance Use 23, 384–389. [Google Scholar]
- [7].Coe MA, Pillitteri JL, Sembower MA, Gerlach KK, and Henningfield JE (2019) Kratom as a substitute for opioids: Results from an online survey, Drug Alcohol Depend 202, 24–32. [DOI] [PubMed] [Google Scholar]
- [8].Grundmann O (2017) Patterns of kratom use and health impact in the US—results from an online survey, Drug and alcohol dependence 176, 63–70. [DOI] [PubMed] [Google Scholar]
- [9].Gutridge AM, Robins MT, Cassell RJ, Uprety R, Mores KL, Ko MJ, Pasternak GW, Majumdar S, and van Rijn RM (2020) G protein-biased kratom-alkaloids and synthetic carfentanil-amide opioids as potential treatments for alcohol use disorder, Br J Pharmacol 177, 1497–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson LL, Harris HM, Eans SO, Brice-Tutt AC, Cirino TJ, Stacy HM, Simons CA, Leon F, Sharma A, Boyer EW, Avery BA, McLaughlin JP, and McCurdy CR (2020) Lyophilized Kratom Tea as a Therapeutic Option for Opioid Dependence, Drug Alcohol Depend 216, 108310. [DOI] [PubMed] [Google Scholar]
- [11].Post S, Spiller HA, Chounthirath T, and Smith GA (2019) Kratom exposures reported to United States poison control centers: 2011–2017, Clinical toxicology 57, 847–854. [DOI] [PubMed] [Google Scholar]
- [12].Nelsen JL, Lapoint J, Hodgman MJ, and Aldous KM (2010) Seizure and coma following Kratom (Mitragynina speciosa Korth) exposure, Journal of Medical Toxicology 6, 424–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kronstrand R, Roman M, Thelander G, and Eriksson A (2011) Unintentional fatal intoxications with mitragynine and O-desmethyltramadol from the herbal blend Krypton, Journal of analytical toxicology 35, 242–247. [DOI] [PubMed] [Google Scholar]
- [14].Henningfield JE, Grundmann O, Babin JK, Fant RV, Wang DW, and Cone EJ (2019) Risk of death associated with kratom use compared to opioids, Preventive medicine 128, 105851. [DOI] [PubMed] [Google Scholar]
- [15].Eggleston W, Stoppacher R, Suen K, Marraffa JM, and Nelson LS (2019) Kratom use and toxicities in the United States, Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 39, 775–777. [DOI] [PubMed] [Google Scholar]
- [16].Kuehn B (2019) Kratom-Related Deaths, JAMA 321, 1966. [DOI] [PubMed] [Google Scholar]
- [17].Administration, U. S. F. a. D. (2018) Statement from FDA Commissioner Scott Gottlieb, M.D., on the agency’s scientific evidence on the presence of opioid compounds in kratom, underscoring its potential for abuse
- [18].Adkins JE, Boyer EW, and McCurdy CR (2011) Mitragyna speciosa, a psychoactive tree from Southeast Asia with opioid activity, Curr Top Med Chem 11, 1165–1175. [DOI] [PubMed] [Google Scholar]
- [19].Cinosi E, Martinotti G, Simonato P, Singh D, Demetrovics Z, Roman-Urrestarazu A, Bersani FS, Vicknasingam B, Piazzon G, Li JH, Yu WJ, Kapitany-Foveny M, Farkas J, Di Giannantonio M, and Corazza O (2015) Following “the Roots” of Kratom (Mitragyna speciosa): The Evolution of an Enhancer from a Traditional Use to Increase Work and Productivity in Southeast Asia to a Recreational Psychoactive Drug in Western Countries, Biomed Res Int 2015, 968786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leon F, Habib E, Adkins JE, Furr EB, McCurdy CR, and Cutler SJ (2009) Phytochemical characterization of the leaves of Mitragyna speciosa grown in U.S.A, Nat Prod Commun 4, 907–910. [PMC free article] [PubMed] [Google Scholar]
- [21].Obeng S, Kamble SH, Reeves ME, Restrepo LF, Patel A, Behnke M, Chear NJ, Ramanathan S, Sharma A, Leon F, Hiranita T, Avery BA, McMahon LR, and McCurdy CR (2020) Investigation of the Adrenergic and Opioid Binding Affinities, Metabolic Stability, Plasma Protein Binding Properties, and Functional Effects of Selected Indole-Based Kratom Alkaloids, J Med Chem 63, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Takayama H (2004) Chemistry and pharmacology of analgesic indole alkaloids from the rubiaceous plant, Mitragyna speciosa, Chem Pharm Bull (Tokyo) 52, 916–928. [DOI] [PubMed] [Google Scholar]
- [23].Takayama H, Ishikawa H, Kurihara M, Kitajima M, Aimi N, Ponglux D, Koyama F, Matsumoto K, Moriyama T, Yamamoto LT, Watanabe K, Murayama T, and Horie S (2002) Studies on the synthesis and opioid agonistic activities of mitragynine-related indole alkaloids: discovery of opioid agonists structurally different from other opioid ligands, J Med Chem 45, 1949–1956. [DOI] [PubMed] [Google Scholar]
- [24].Chakraborty S, and Majumdar S (2020) Natural Products for the Treatment of Pain: Chemistry and Pharmacology of Salvinorin A, Mitragynine, and Collybolide, Biochemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kruegel AC, Gassaway MM, Kapoor A, Váradi A, Majumdar S, Filizola M, Javitch JA, and Sames D (2016) Synthetic and receptor signaling explorations of the mitragyna alkaloids: mitragynine as an atypical molecular framework for opioid receptor modulators, Journal of the American Chemical Society 138, 6754–6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Váradi A, Marrone GF, Palmer TC, Narayan A, Szabó MR, Le Rouzic V, Grinnell SG, Subrath JJ, Warner E, Kalra S, Hunkele A, Pagirsky J, Eans SOE, Medina JM, Xu J, Pan Y-X, Borics A, Pasternak GW, McLaughlin JP, and Majumdar S (2016) Mitragynine/corynantheidine pseudoindoxyls as opioid analgesics with mu agonism and delta antagonism, which do not recruit β-arrestin-2, Journal of medicinal chemistry 59, 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Majumdar S, and Devi LA (2018) Strategy for making safer opioids bolstered, Nature 553, 286–288. [DOI] [PubMed] [Google Scholar]
- [28].Kamble SH, Sharma A, King TI, Leon F, McCurdy CR, and Avery BA (2019) Metabolite profiling and identification of enzymes responsible for the metabolism of mitragynine, the major alkaloid of Mitragyna speciosa (kratom), Xenobiotica 49, 1279–1288. [DOI] [PubMed] [Google Scholar]
- [29].Kruegel AC, Uprety R, Grinnell SG, Langreck C, Pekarskaya EA, Le Rouzic V, Ansonoff M, Gassaway MM, Pintar JE, and Pasternak GW (2019) 7-Hydroxymitragynine is an active metabolite of mitragynine and a key mediator of its analgesic effects, ACS central science 5, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, and Kobilka BK (2015) Structural insights into micro-opioid receptor activation, Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schrödinger L, New York, NY. (2019) Small-Molecule Drug Discovery Suite 2019–2, 2018-3 ed., Schrödinger, LLC. [Google Scholar]
- [32].Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C, Dahlgren MK, Mondal S, Chen W, Wang L, Abel R, Friesner RA, and Harder ED (2019) OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules, J Chem Theory Comput 15, 1863–1874. [DOI] [PubMed] [Google Scholar]
- [33].Ballesteros JA, and Weinstein H (1995) [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors, In Methods in Neurosciences (Sealfon SC, Ed.), pp 366–428, Academic Press. [Google Scholar]
- [34].Isberg V, de Graaf C, Bortolato A, Cherezov V, Katritch V, Marshall FH, Mordalski S, Pin JP, Stevens RC, Vriend G, and Gloriam DE (2015) Generic GPCR residue numbers - aligning topology maps while minding the gaps, Trends Pharmacol Sci 36, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, and Rizzo RC (2015) DOCK 6: Impact of new features and current docking performance, J Comput Chem 36, 1132–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Clark AJ, Tiwary P, Borrelli K, Feng S, Miller EB, Abel R, Friesner RA, and Berne BJ (2016) Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations, J Chem Theory Comput 12, 2990–2998. [DOI] [PubMed] [Google Scholar]
- [37].Fusani L, Palmer DS, Somers DO, and Wall ID (2020) Exploring Ligand Stability in Protein Crystal Structures Using Binding Pose Metadynamics, J Chem Inf Model 60, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Barducci A, Bussi G, and Parrinello M (2008) Well-tempered metadynamics: a smoothly converging and tunable free-energy method, Phys Rev Lett 100, 020603. [DOI] [PubMed] [Google Scholar]
- [39].Tuckerman M, Berne BJ, and Martyna GJ (1992) Reversible multiple time scale molecular dynamics, The Journal of Chemical Physics 97, 1990–2001. [Google Scholar]
- [40].Nosé S (1984) A unified formulation of the constant temperature molecular dynamics methods, The Journal of Chemical Physics 81, 511–519. [Google Scholar]
- [41].Frey BJ, and Dueck D (2007) Clustering by passing messages between data points, Science 315, 972–976. [DOI] [PubMed] [Google Scholar]
- [42].Noe F, Schutte C, Vanden-Eijnden E, Reich L, and Weikl TR (2009) Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations, Proc Natl Acad Sci U S A 106, 19011–19016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hacisuleyman A, and Erman B (2017) Entropy Transfer between Residue Pairs and Allostery in Proteins: Quantifying Allosteric Communication in Ubiquitin, PLoS Comput Biol 13, e1005319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bhattacharya S, Salomon-Ferrer R, Lee S, and Vaidehi N (2016) Conserved Mechanism of Conformational Stability and Dynamics in G-Protein-Coupled Receptors, J Chem Theory Comput 12, 5575–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bhattacharya S, and Vaidehi N (2014) Differences in allosteric communication pipelines in the inactive and active states of a GPCR, Biophys J 107, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nivedha AK, Tautermann CS, Bhattacharya S, Lee S, Casarosa P, Kollak I, Kiechle T, and Vaidehi N (2018) Identifying Functional Hotspot Residues for Biased Ligand Design in G-Protein-Coupled Receptors, Mol Pharmacol 93, 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vaidehi N, and Bhattacharya S (2016) Allosteric communication pipelines in G-protein-coupled receptors, Curr Opin Pharmacol 30, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kapoor A, Provasi D, and Filizola M (2020) Atomic-Level Characterization of the Distinct Methadone-Induced Conformational Sampling and Activation Kinetics of the μ-Opioid Receptor by Molecular Simulations, Biophysical Journal 118, 92a. [Google Scholar]
- [49].Hernández C (2015) mdentropy: v0.2, Version 0.2 ed
- [50].Hagberg AA, Schult DA, and Swart P (2008) Exploring network structure, dynamics, and function using NetworkX, In Proc. SciPy (Varoquaux G, V. T, Millman J, Ed.), pp 11–16. [Google Scholar]
- [51].Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee MY, Pardon E, Steyaert J, Huang XP, Strachan RT, Tribo AR, Pasternak GW, Carroll FI, Stevens RC, Cherezov V, Katritch V, Wacker D, and Roth BL (2018) Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor, Cell 172, 55–67 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, and Granier S (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist, Nature 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.