

INTRODUCTION
The voltage-gated sodium channel Nav1.7 is a key determinant of nociceptor excitability and a major contributor to pain signalling in humans. Gain-of-function mutations of Scn9a, the gene that encodes Nav1.7, are associated with a spectrum of chronic pain conditions, including rare heritable syndromes such as inherited erythromelalgia (IEM) and paroxysmal extreme pain disorder (PEPD), as well as more common acquired pain disorders such as painful diabetic neuropathy and small fiber neuropathy (SFN) [1; 6]. Unlike diabetic polyneuropathy, which can involve all classes of fibers, SFN is characterized by damage to unmyelinated C-fibers and thinly myelinated Aδ- afferent fibers, as indicated by quantitative sensory testing (QST) and degeneration of intraepidermal nerve fibers (IENF) in the skin [3; 4]. Consistent with a selective involvement of small-diameter fibers, clinical presentation of SFN is characterized by a combination of pain, sensory deficits, and autonomic complaints [10]. SFN can be associated with multiple diseases, such as diabetes mellitus or autoimmune disorders, but is idiopathic in a substantial proportion (approximately 50%) of patients [15].
The mechanism underlying distal axon degeneration and of pain symptoms in SFN are not well understood. However, the identification of gain-of-function Nav1.7 variants in idiopathic SFN cases provides an avenue for investigating possible underlying molecular mechanisms [9]. SFN-associated Nav1.7 variants render dorsal root ganglion (DRG) neurons hyperexcitable, as demonstrated by abnormal spontaneous firing, lower action potential firing threshold and enhanced firing frequency [9]. Importantly, these mutations produce changes in the gating properties of Nav1.7 channel,s such as impaired slow-inactivation, depolarized slow- and/or fast-inactivation, or enhanced resurgent current [9]. Among the SFN Nav1.7 mutations, I228M is of particular interest because patients carrying this mutation experience a relatively early clinical onset (within first three decades) [8; 9]. A major unanswered question is whether the pain in these patients is secondary to the neuropathy or the consequence of nociceptor hyperexcitability. DRG neurons transfected with human Nav1.7 I228M channels exhibit a significant (~20%) reduction in neurite length [14] suggesting that the hyperexcitability may be linked to the neuropathy. Mechanistic investigations have demonstrated that reverse operation (Ca2+-importing) of the Na+/Ca2+ exchanger (NCX) [14] and damaged mitochondrial energy metabolism [12] may contribute to the impaired neurite integrity present in Nav1.7 I228M-transfected DRG neurons.
While in vitro studies have provided valuable insight into the underlying pathophysiology of the symptoms seen in SFN patients, models using over-expression of a mutant Nav1.7 1228M channel do not recapitulate the situation in patients. In this study we report the phenotypes of two Nav1.7 I228M knock-in (Nav1.7I228M) mouse lines generated independently in separate laboratories, by targeted homologous recombination or CRISPR editing. We investigate the physiological properties of their DRG neurons, assess morphological changes in axons and intra epidermal nerve fibers, and search for behavioural correlates of the pain symptoms found in patients.
METHODS
Generation of Nav1.7I228M knock-in mice
Nav1.7I228M knock-in mouse line by targeted homologous recombination
Scn9a p.I228M knock-in mice were generated by inGenious targeting Laboratory (Ronkonkoma, NY) using a targeted homologous recombination approach. Briefly, targeted iTL BF1 (C57BL/6 FLP) embryonic stem cells carrying the targeted I228M Scn9A mutant allele and the neomycin selection cassette were microinjected into Balb/c blastocysts. Resulting chimeras with a high percentage black coat color were mated to C57BL/6 WT mice to generate Germline Neo-Deleted mice. Final germline transmitted mice were confirmed for the deletion of the Neomycin cassette and the presence of the p.I228M allele by genotyping tail DNA from mice with black coat color, and sequencing the PCR product. A founder line was backcrossed to wild-type C57BL/6J for 6 generations before it was used for behavioural and electrophysiological assays.
Nav1.7I228M knock-in mouse line by CRISPR editing
Fertilized embryos from pregnant female C57/B6J mice were used to generate mice with the I228M mutation in Scn9a (Nav1.7). Fertilized embryos were removed and injected with Cas9 mRNA, synthetic CRISPR guide RNAs and a ssDNA donor sequence containing the base pair change to effect the desired mutation as well as 5 silent mutations. They were then placed into a donor mother and carried to term.
gRNA sequences: ATTCCAGGTAAGAAGTGATTGG and CACACCAATCACTTCTTACCTGG
I228M ssDNA donor sequence (103 bp): CAG CTC TTC GAA CTT TCA GAG TCT TGA GAG CTT TGA AAA CTA TTT CCG TTA TGC CGG GAA AGA AGT GAT TGG TGT GGA GCT TTA GAC TGC TCA ACT CCA GCT G
The resulting offspring were genotyped by PCR and Sanger sequencing. It was confirmed that two mice had an insertion of the desired ssDNA; one heterozygote and one homozygote founder. These mice were then each bred with wild-type mice to generate a colony.
PCR primers:
Forward: ACC TAG GCA ATG TTT CAG CTC TTC G
Reverse**: CTT CCT TCT CCA AGA CCC ATG CAA
**used for sequencing
Patch-clamp recordings
DRG neurons from 4-8 weeks Nav1.7 WT or Nav1.7 I228M knock-in mice by targeted homologous recombination (both sexes) were cultured using a protocol as described previously [5]. Whole-cell current–clamp recordings were obtained from small-diameter (<25 µm) DRG within 24 h after culture by using an EPC-9 amplifier and Pulse 8.5 (HEKA, Germany). Electrodes (1–3 MΩ) were filled with pipette solution (mM): 140 KCl, 0.5 EGTA, 5 HEPES, 3 Mg-ATP, pH 7.3 with KOH (adjusted to 315 mOsm with dextrose). The extracellular solution contained (mM): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, pH 7.3 with NaOH (adjusted to 320 mOsm with dextrose). Whole-cell configuration was obtained in voltage-clamp mode before proceeding to current-clamp. Cells with stable resting membrane potentials were used for data collection by PatchMaster (HEKA-Electronics) software.
Electrophysiological data were analyzed using Fitmaster (HEKA-Electronics) and Origin 8.5 (Microcal, Northampton, MA) and SPSS Statistics 24 (IBM), and were presented as means ± standard error. For multi-group statistical analysis, we used one-way ANOVA followed by Tukey post hoc test or Kruskal–Wallis followed by Dunn procedure depending on whether data showed a normal distribution. The 2-proportions z test was used to compare the proportion of spontaneously firing neurons.
Multi-electrode array (MEA)
Dissociated DRG neurons from 6-9-week old WT (n=3, 2 females, 1 male) and HomNav1.7I228M knock-in mice by targeted homologous recombination (n=4, 2 females, 2 males) were maintained at 37°C in a 5% CO2 incubator for 3 days before MEA recordings. Action potential firing was measured using a multi-well MEA system (Maestro, Axion Biosystems, Atlanta, GA, USA). A12-well recording plate with 768 electrodes was used. For each experiment, neurons from WT or HomNav1.7I228M mice were dissociated and plated at the same time in 6 wells each. The investigator was blinded to the identity of neurons. Precise temperature control of the MEA system was utilized to create temperature ramps and maintain temperatures at 30°, 33°, 37°, 40° and 43°C each for 200s. The size of the observed signal is impacted by many factors that include the current-density of the neuron as well as the geometry (distance) between the neuron and the electrode. We apply strict criteria for neuron plating to ensure comparable cell density per well, and data analysis with the signal to noise of spike detection is achieved by setting the threshold for a spike detection criterion of >6 standard deviations above background signals. Electrodes registering >5 recorded spikes over 200 s period were determined as active. Number of active electrodes and mean firing frequency were measured. Data was analysed using Axion Integrated Studio AxIS 2.1 (Axion Biosystems) and Neuro Explorer (NexTechnologies, Madison, AL, USA). Statistical significance was determined using Mann-Whitney U test. Data are expressed as mean ± S.E.M. A p-value of < 0.05 was considered significant.
Behavioural assays
Nav1.7 I228M knock-in mouse line by targeted homologous recombination
All animal procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC of the Veterans Administration Connecticut Healthcare System. Adult mice (8-12 week old) of both sexes were used in this study.
Hot plate:
Animals were placed on a metal plate (Hot Plate Analgesia Test Meter, IITC, USA) uniformly heated to a constant temperature of 52 ºC. The response latency for first licking or flinching of the hind paws was recorded. The mice were immediately removed from the hot plate upon showing nocifensive behaviors or if no response occurred within the 30s cut-off time.
Hargreaves test:
Animals were placed on a glass plate and plantar paw surface was exposed to radiant heat using a Hargreaves apparatus (IITC, Woodland Hills, CA) and the paw withdrawal latency was measured. The heat stimulation was repeated 3 times on both paws, with at least five minutes rest between consecutive tests. The cut-off value was set at 30 seconds to prevent tissue damage.
von Frey test:
Animals were placed on an elevated wire grid and the plantar surface of both paws were presented with a series of calibrated von Frey filaments (Stoeling, Wood Dale, IL). Each filament was applied for 5 seconds. A nocifensive response was recorded when flinching, withdrawal, paw licking, or toe spreading was observed. The 50% withdrawal threshold was determined using the “up-down” method which requires 6 stimuli straddling the threshold. At least five minutes of rest were allowed between each stimulus.
Thermal gradient test and preference test:
The thermal preference apparatus consisted of two heating/cooling plates (Bioseb, Chaville, France) placed side-by-side and enclosed in a plexiglass chamber. The temperatures of test and reference plates were set to 35 ºC vs 25 ºC, 15 ºC vs 25 ºC, or 38 ºC vs 33 ºC. Animal movements were recorded for 600s by an automated infrared camera tracking system. The time spent on the reference and test plates was calculated. The thermal gradient apparatus maintained a stable temperature gradient from about 6°C to 46°C using the two heating/cooling devices positioned at each end of a metal floor. The floor area was divided into 10 zones, each with a stable temperature measured using a thermometer (TM-3 three-scale temperature monitor, Warner Instruments). Tracking was performed using software provided by the manufacturer and the time spent in each zone over the 60 min test period determined.
Formalin test:
Mice were habituated to observation chambers for at least 30 min prior to experiments. For induction of short-term inflammation 10 μl of 5% formalin was injected subcutaneously (s.c.) into the plantar surface of the left hind paw. After injection, mice were immediately returned to the observation chamber and nociceptive behaviours (licking, flicking, lifting the injected paw) were recorded for 45 min. The duration of formalin-induced nociceptive behaviors in 5-min intervals was scored by an experimenter blind to treatment or genotype.
CFA injections:
Mice were anaesthetized by exposure to isoflurane (1-3 %), administered by a calibrated vaporizer. For induction of long-term inflammation, 10 µl of complete Freund’s adjuvant (CFA; Sigma, containing 1mg/ml of Mycobacteriom tuberculosis) were injected into the left hind paw using a 10 µl-Hamilton syringe (Hamilton company, Reno, Nevada).
Nav1.7 I228M knock-in mouse line by CRISPR editing
All mouse work at Boston Children Hospital was carried out in accordance with IACUC protocol 17-06-3494R. 8-12-week-old mice of both sexes were used in these studies.
von Frey:
These experiments were performed following standard protocol. Briefly, animals (n = 7 for WT, n = 5 for HetNav1.7I228M, n = 8 for HomNav1.7I228M) were placed on an elevated wire grid in a plastic enclosure and habituated twice for 2 hours preceding the day of measurement. On the day, they were habituated for 1 hour before probing the right hindpaw with von Frey filaments. All animals were tested with the 0.6g filament first, and then we proceeded using the up-down method.
Hot plate:
The hot plate was set at 50 C. Animals (n = 9 for WT, n = 9 for HetNav1.7I228M, n = 5 for HomNav1.7I228M) were placed on the plate and the latency to licking the hindpaw or jumping was recorded. After 60s the mice were removed from the plate if there was no response. Mice were tested only once to ensure they did not adapt to the assay.
Thermal gradient:
The thermal gradient assay was evaluated as previously described (PMID: 28481358). A continuous temperature gradient (5–50 ° C) was established along a metallic base plate (Bioseb, France) on which the mice (n = 7 for WT, n = 10 for HetNav1.7I228M, n = 4 for HomNav1.7I228M) walked freely while video-recorded from above (Noldus, VA, USA). Each evaluation lasted 1.5 h and two mice were recorded simultaneously in separate corridors. Each corridor was virtually divided into 20 zones of equal size (6 cm) with a different and stable temperature. After a period of exploration (60 min), the mouse showed a clear preference, indicating the most comfortable temperature range.
Immunohistochemistry (IHC) for intra-epidermal nerve fiber (IENF) density in skin
8-12-week-old WT and Nav1.7I228M knock-in mice by targeted homologous recombination (n=4 each genotype; both sexes) were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.) and transcardially perfused with 0.01M PBS (pH 7.4) followed by ice-cold 4% paraformaldehyde in 0.14 M Sorensen’s phosphate buffer (pH 7.4). Same regions of ventral hind paw glabrous skin were removed using 3 mm biopsy punch Foot pad skin tissues were removed, immersion-fixed in 4% paraformaldehyde (total fixation time 20 min) and cryo-protected with 30% (w/v) sucrose in PBS overnight at 4°C. Tissue sections were cut on a cryostat at 10 µm and mounted on slides (Fisher Scientific, Pittsburgh, PA). Sections were immediately processed for detection of target protein or stored at −20 °C for future use.
Sections were incubated in the following solutions: (1) blocking solution (PBS containing 4% normal donkey serum, 2% BSA, 0.1% Triton X-100, and 0.02% sodium azide) for 1 h at room temperature; (2) rabbit monoclonal anti-PGP 9.5 antibody (1:300, Abcam, Cat# ab108986, Batch# GR3231441-1) in blocking solution at 4°C overnight; (3) PBS, 3 × 10 min each; (4) Alexa Fluor 546-conjugated donkey anti-rabbit IgG (H+L) secondary antibody (1:1000, Invitrogen) in blocking solution for 1 h at room temperature; (5) PBS, 3 × 10 min. Tissue sections were mounted in antifade mounting medium with DAPI (H-1500, Vectashield) and were examined with a Nikon Eclipse E800 fluorescence microscope or a Nikon C1 confocal microscope (Nikon USA, Melville, NY).
Light microscopy analysis of axons in sciatic nerve
8-12-week-old WT or Nav1.7I228M knock-in mice by targeted homologous recombination (both sexes) were anesthetized with ketamine/xylazine (100/10 mg/kg, i.p.) and transcardially perfused with 0.01M PBS (pH 7.4) followed by ice-cold 4% paraformaldehyde in 0.14 M Sorensen’s phosphate buffer (pH 7.4). Sciatic nerves at mid-thigh level (distal to the trifurcation) were dissected and transferred to 2% paraformaldehyde plus 2% glutaraldehyde in 0.14 M Sorensen’s phosphate buffer (pH 7.4) at 4°C overnight. Samples were then post-fixed with 1% osmium (Polysciences, Warrington, PA, USA), dehydrated and blocked in 0.5 cm segments and embedded in Epox-812 (Ernest F. Fullam, Latham, NY, USA) using standard plastic embedding protocols. Semi-thin sections (1 µm) were collected from each tissue block and counterstained with methylene blue and azure II (0.5% each in 0.5% borax) for light microscopy. Sections were examined with a Nikon Ti Eclipse microscope and digital images were recorded with Nikon NIS-Elements imaging software.
Neurite growth assay
Nav1.7 I228M knock-in mouse line by targeted homologous recombination
DRG neurons were isolated from 6-8-week-old WT and Nav1.7 I228M mice (both sexes) and electroporated with the plasmid encoding red fluorescent protein (RFP). After the electroporation, cells were plated on the coverslips coated with laminin and cultured for 7 days. The resulting cultures were imaged and their total lengths per cell were assessed. Each experiment includes cultures from a pair of age/sex matched WT and Nav1.7 I228M mutant mice. DRG neurons from each mouse are plated onto 3 to 4 culture dishes. Each data point represents total neurite length per cell within one culture dish. Data are normalized to average WT values of the same experiment and presented as mean ± standard deviation.
Nav1.7 I228M knock-in mouse line by CRISPR editing
DRG culture:
DRGs were dissected from 8-12-week-old mice, and then dissociated and cultured according to standard protocol. Briefly, the samples were digested with collagenase/dispase and mechanically triturated in the presence of DNAse until a single-cell suspension was formed. Debris was removed using a BSA gradient. The neurons were then plated at a density of 1000 cells/well in a 96-well plate coated with PDL/laminin. Cells were grown in Neurobasal medium supplemented with PenStrep, B27, L-glutamine, NGF, GDNF, and AraC.
Neurite Outgrowth:
After 24 hours, the cells were fixed using 4% PFA and stained with Tuj1 and DAPI. The plates were then imaged on the ArrayScan system (Thermo Fischer) at 10x magnification. Cell bodies and neurites were automatically detected by the system and used to quantify neurite length per neuron within each well.
Spot culture:
Instead of plating in a 96-well plate, 30k DRG neurons were plated in a single spot (7 uL volume) in the center of a 35mm dish coated with PDL/laminin. After the cells had time to adhere (30 min), the rest of the media volume was added to the dish, with special care taken not to disturb the spot. The spots were then grown for up to 6 days and imaged on alternate days using brightfield microscopy. On the 6th day, the spots were fixed with 4% PFA and stained with Tuj1. They were imaged at 10x magnification, using an 8×8 tile, such that the 64 images captured the entirety of each spot.
Sholl analysis:
Sholl analysis was used to quantify neurite length. Briefly, the background of the image was removed, and then the image was thresholded such that neurites were easily visible. Then, using a macro in ImageJ, concentric circles were created around the center spot and intersections of neurites with these circles were quantified. The diameter of each ring was then plotted against the number of intersections and a proxy for the number of neurites of a given length.
RESULTS
Small-diameter DRG neurons from Nav1.7I228M mutant mice are hyperexcitable.
Knock-in mice, of both sexes, produced by targeted recombination were used for these studies. Using current-clamp recordings, we compared the excitability of small-diameter (<25 µm) DRG neurons from heterozygous (Het) and homozygous (Hom) Nav1.7I228M mutant mice with their wild-type (WT) littermate controls. A series of 200 ms depolarizing currents in 5 pA increments were used to determine the current threshold for the first all-or-none action potential. Figure 1A shows representative action potential traces from a small-diameter WT DRG neuron in response to graded membrane potential depolarizations, where an overshooting action potential was produced at a threshold of 125 pA. In contrast, representative action potentials recorded from HetNav1.7I228M (Figure 1B) and HomNav1.7I228M (Figure 1C) DRG neurons showed current thresholds of 60 pA and 55 pA, respectively. The average current threshold was significantly reduced (by half) in small-diameter DRG neurons from HetNav1.7I228M mice (62±9 pA, n=23, P<0.05) and HomNav1.7I228M mice (56±10 pA, n=23, P<0.01) compared with DRG neurons from WT mice (118±19 pA, n=28) (Figure 1D). Resting membrane potential (RMP) was also significantly depolarized in neurons from HetNav1.7I228M (−53.2±1.2 mV, n=23; P<0.05) and HomNav1.7I228M (−52.9±1.1 mV, n=23; P<0.05) mutant mice, compared to neurons from WT groups (−57.4±1.1 mV, n=28) (Fig. 1E). There was no statistically significant difference in input resistance (WT: 1.0±0.1 GΩ, n=28; HetNav1.7I228M: 1.2±0.1 GΩ, n=23; HomNav1.7I228M: 1.3±0.1 GΩ, n=23), action potential amplitude (WT: 120.1±1.7 mV, n=28; HetNav1.7I228M: 117.2±1.6 mV, n=23; HomNav1.7I228M: 117.7±1.6 mV, n=23) or action potential half width (WT: 4.27 ± 0.18 ms, n=28; HetNav1.7I228M: 4.37 ± 0.23 ms, n=23; HomNav1.7I228M: 4.43±0.17 mV, n=23) among the three groups.
Figure 1. Small-diameter DRG neurons from HetNav1.7I228M and HomNav1.7I228M mutant mice are hyper-excitable compared to WT control DRG neurons.
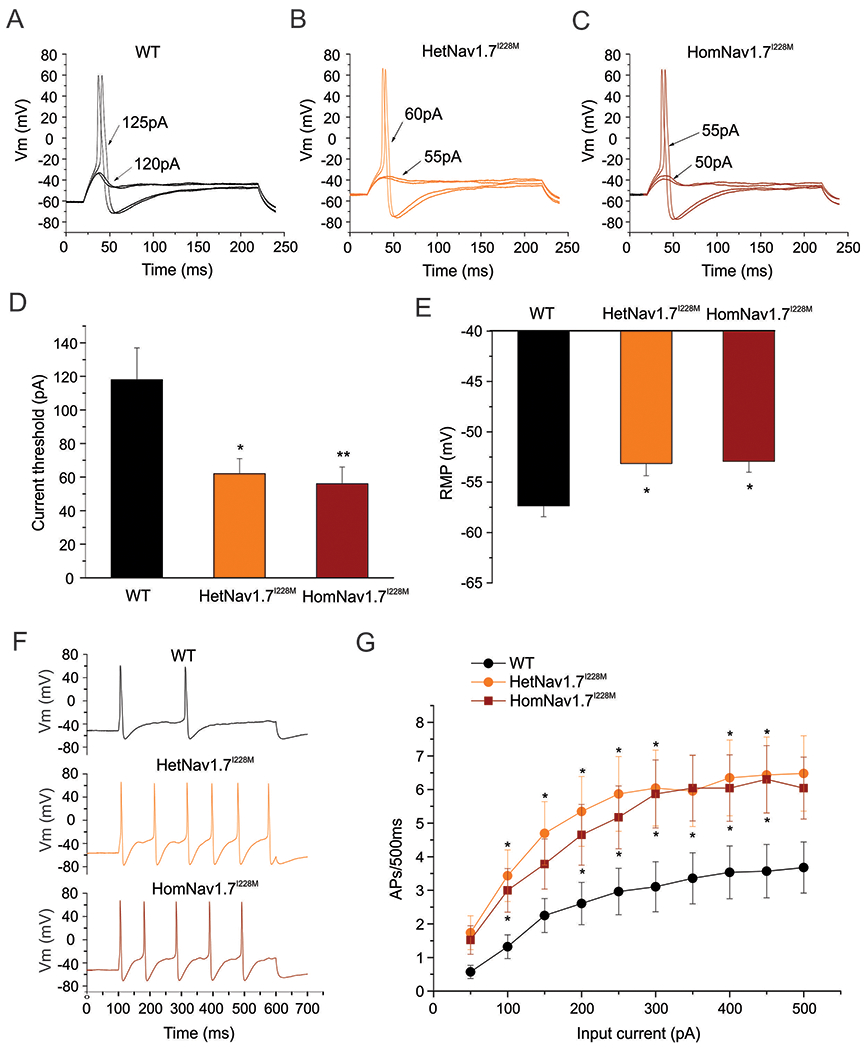
(A) Representative traces of a small-diameter DRG neuron from WT mouse, showing subthreshold responses to current injections up to 120 pA and subsequent action potentials evoked by current injections above 125pA (current threshold for this neuron). (B) Representative traces of a small-diameter DRG neuron from HetNav1.7I228M mouse, showing a lower current threshold (60 pA) for action potential generation. (C) Representative traces of a small-diameter DRG neuron from HomNav1.7I228M mouse, showing a lower current threshold (55 pA) for action potential generation. (D) Small-diameter DRG neurons from HetNav1.7I228M and HomNav1.7I228M mice display significantly reduced current threshold compared to WT control neurons (* p<0.05; ** p<0.01, one-way ANOVA followed by Tukey post hoc test). (E) Small-diameter DRG neurons from HetNav1.7I228M and HomNav1.7I228M mice display significantly depolarized RMP compared to WT control neurons (* p<0.05, one-way ANOVA followed by Tukey post hoc test). (F) Representative responses of small-diameter DRG neurons from WT, HetNav1.7I228M and HomNav1.7I228M mice, respectively, to 500 ms depolarizing current 3x the threshold for action potential generation. (G) Comparison of firing frequency of small-diameter DRG neurons from WT, HetNav1.7I228M and HomNav1.7I228M mutant mice, in response to graded 500 ms depolarising current stimuli from 50 to 500 pA in 50 pA increments. Nav1.7I228M mutant groups show significantly higher firing frequencies compared to WT group (* p<0.05, one-way ANOVA followed by Tukey post hoc test).
We next investigated the responses of small-diameter DRG neurons from WT, HetNav1.7I228M and HomNav1.7I228M mice to series of 500 ms sustained depolarizations. Representative action potential firing traces demonstrated that, compared to WT controls, small-diameter DRG neurons from HetNav1.7I228M and HomNav1.7I228M mice generated more action potentials in response to 3X threshold current injections (Figure 1F). As summarized in Figure 1G, compared to WT controls, small-diameter DRG neurons from HetNav1.7I228M and HomNav1.7I228M mice generated significantly more action potential firing in response to the current injections.
There was also an increased proportion of spontaneously firing neurons in both HetNav1.7I228M and HomNav1.7I228M mice (Figure 2). 34.3% (12 out of 35 neurons, p < 0.05) HetNav1.7I228M DRG neurons and 25.8% (8 out of 31 neurons) HomNav1.7I228M DRG neurons were spontaneously active, while only 4 out of 32 (12.5%) recorded DRG neurons from WT mice displayed spontaneous firing.
Figure 2. Spontaneously active DRG neurons.
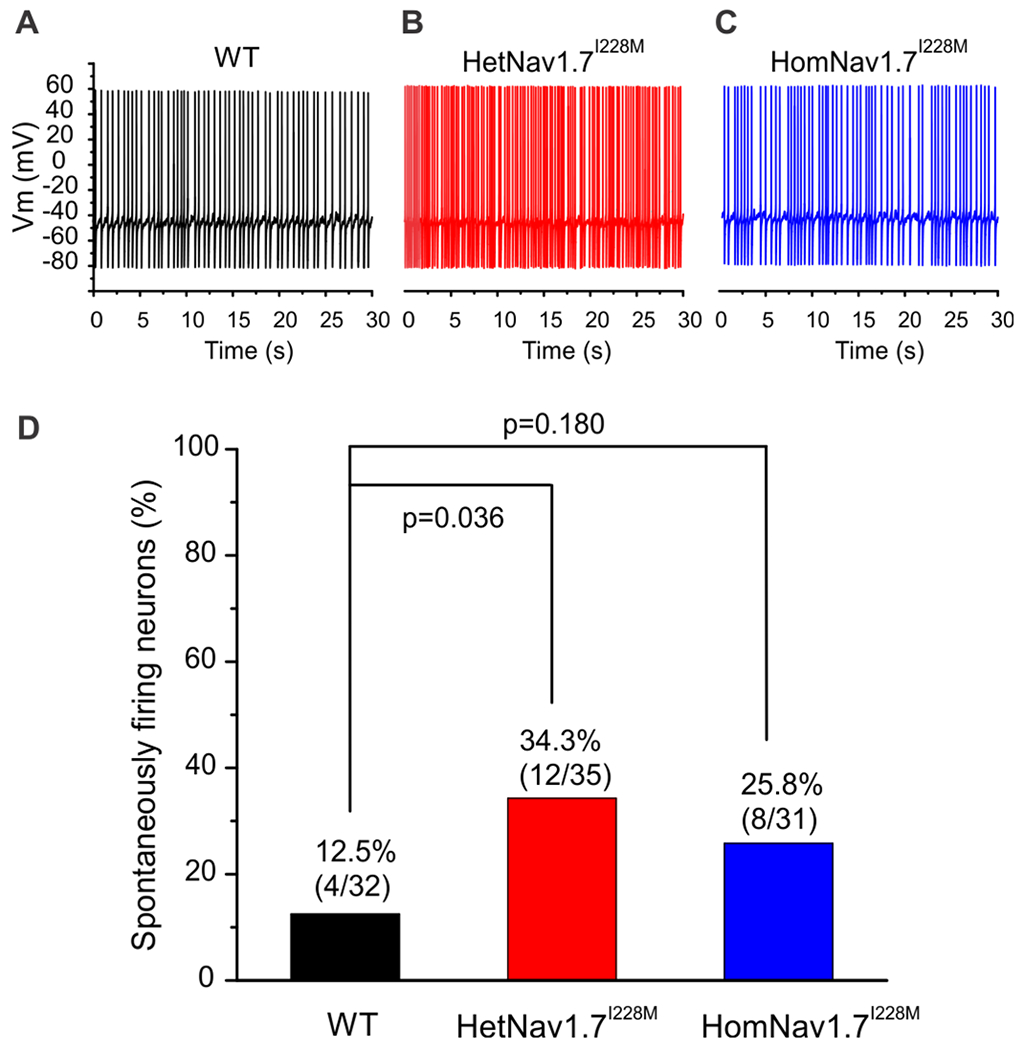
Representative recordings showing spontaneous firing (30 s) of DRG neuron from WT (A), HetNav1.7I228M (B) and HomNav1.7I228M (C) mice, respectively. Trace was recorded for 30 s without current injection. (D) Comparison of the proportion of spontaneously firing DRG neurons among WT, HetNav1.7I228M and HomNav1.7I228M groups. Compared with WT group, HetNav1.7I228M and HomNav1.7I228M groups displayed increased proportion of spontaneously firing neurons, but reached statistical significance only for the HetNav1.7I228M group (2-proportions z test was used to compare the proportion of spontaneously firing neurons).
DRG neurons from Nav1.7I228M mutant mice show enhanced responses to noxious heat in vitro
We recorded action potential firing of cultured DRG neurons from WT and HomNav1.7I228M mice (produced by targeted recombination) by multi-electrode array (MEA), an extracellular recording approach that permits study of the firing of many neurons without rupturing the membrane and dialyzing their content as in the current-clamp recordings. We examined activity at baseline temperature (30°C) to skin temperature (33°C), core body temperature (37°C), non-noxious warmth (40°C) and noxious heat (43°C). Representative raw traces of a 10s epoch (Figure 3A) show that DRG neurons from WT and HomNav1.7I228M mutant mice both produced clearly identifiable spikes, which exhibited a reproducible waveform in the MEA recordings in the absence of stimulation. Representative heat map plots of active electrode (colored circles) and fired action potentials (color scale) showed a difference in response to increasing temperatures between DRG neurons from WT and HomNav1.7I228M mutant mice (Figure 3B). Compared to WT controls, the HomNav1.7I228M group had significantly more active electrodes (WT: 1.9 ± 0.4; HomNav1.7I228M: 3.5 ± 0.7 active electrodes per well; p < 0.05) at the noxious temperature 43°C (Figure 3B), although the mean action potential firing frequency in those active electrodes appeared to be similar (Figure 3C). In addition, we observed that neurons from HomNav1.7I228M mice fired significantly more bursts (>5 spikes) than WT neurons (WT: 30.9 ± 18.0; HomNav1.7I228M: 120.9 ± 29.0 bursts per well; p<0.01) (Figure 3D).
Figure 3. DRG Neurons from HomNav1.7I228M mutant mice show enhanced response to heat in MEA recordings.
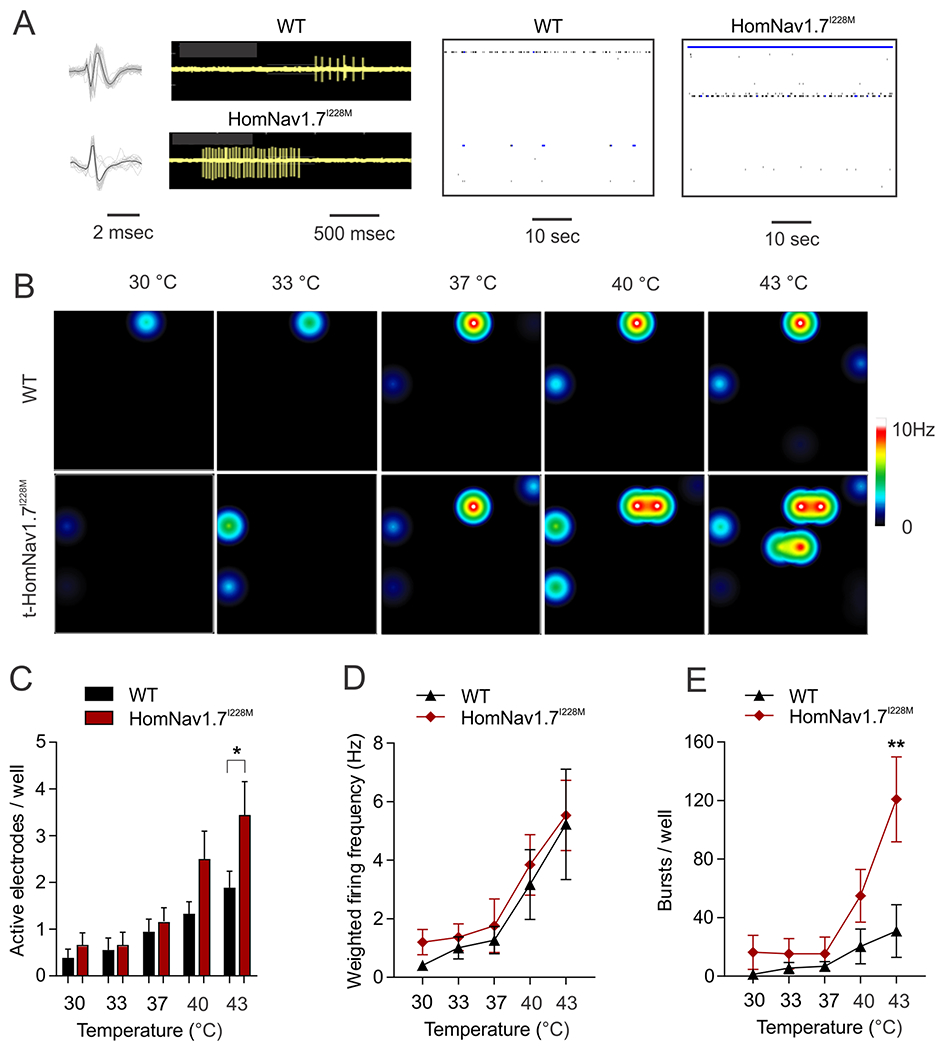
(A) Representative spike waveforms (left panels) recorded using MEA from DRG neurons expressing WT and HomNav1.7I228M mutant mice. Black line indicates the averaged waveform from 20 individual spikes. Grey lines indicate individual spikes. Raw traces from a single electrode recording DRG neurons from WT and HomNav1.7I228M mutant mice (Middle panels). DRG neurons from HomNav1.7I228M mutant mice produce more bursts compared with those from WT mice in the same recording period. Well-wide (64 electrodes) raster plot of MEA recordings of DRG neurons from WT and HomNav1.7I228M at 40°C. Each horizontal plot represents recording from one electrode. More electrodes recorded spikes from DRG neurons from HomNav1.7I228M than WT mice. The blue plots indicate burst firing as defined by default setting of the Axion analysis software. (B) Representative heat-map plots of MEA recordings of DRG neurons from WT and HomNav1.7I228M mutant mice at 30 °C, 33 °C, 37 °C, 40 °C, and 43 °C. Each colored circle represents an active electrode within an 8 × 8 electrode array. More active electrodes and higher firing frequency are recorded at higher temperatures. More active electrodes are seen in wells containing HomNav1.7I228M mutant DRG neurons. (C) Number of active electrodes in WT and HomNav1.7I228M groups at temperatures 30 °C, 33°C, 37°C, 40°C, and 43°C. HomNav1.7I228M group displays significantly more active electrodes compared to WT group at 43°C (* p<0.05, Mann-Whitney U test). (D) Weighted firing frequencies of DRG neurons from WT and HomNav1.7I228M mutant mice. No significant difference is seen between groups. (E) Number of bursts in DRG neurons from WT and HomNav1.7I228M mutant mice. HomNav1.7I228M group displays significantly more bursts compared to WT group at 43°C (** p<0.01, Mann-Whitney U test).
Basal and inflammatory pain behaviors are normal in Nav1.7I228M mutant mice
Given the robust hyperexcitability of cultured DRG neurons from Nav1.7 I228M mice and their enhanced response to noxious heat, we anticipated that pain behaviour in the mutant mice might be augmented and recapitulate aspects of the human clinical phenotype. However, surprisingly Nav1.7I228M mutant mice produced by targeted homologous recombination did not display any significant changes in thermal or mechanical pain thresholds compared to littermate controls. While HetNav1.7I228M and HomNav1.7I228M mutant mice showed a trend toward a reduction in thermal pain threshold in hot plate (52°C) test this did not reach statistical significance overall, nor in female or male sub-groups (Figure 4A). Withdrawal thresholds to thermal and mechanical stimuli assessed using the Hargreaves test and the von Frey test were also not significantly different between Nav1.7I228M mutant mice and wild-type controls (Figure 4B–C). In the thermal preference test, Nav1.7I228M mice displayed normal warmth seeking (35 ºC vs 25 ºC), cold avoidance (15 ºC vs 25 ºC), and heat avoidance behaviours (38 ºC vs 33 ºC) (Figure 4D). The thermal gradient test (Figure 4E) showed no difference between the genotypes. Behavioural examination in the CRISPR Nav1.7 I228M knock-in line showed similar findings. No differences in von Frey threshold (Figure 3F), 50 ºC hot plate response latency (Figure 4G), or preferred temperature based on the thermal gradient test (Figure 4H) were observed in these mice.
Figure 4. Nav1.7I228M mutant mice do not show thermal or mechanical hypersensitivity.
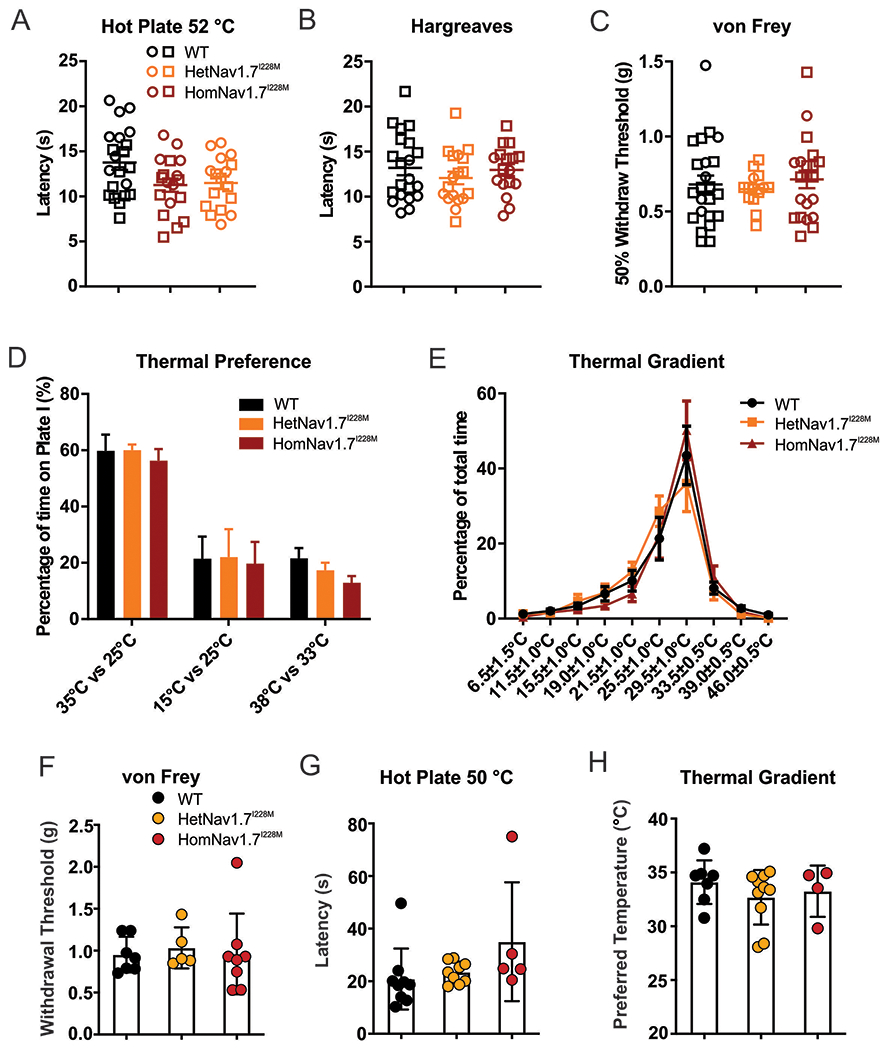
(A) Hot plate test at 52 °C. There is a trend toward lower thermal threshold in Nav1.7I228M mutant mice (HetNav1.7I228M, n=18; HomNav1.7I228M, n=17), but the difference compared to WT control littermates (n=21) does not reach statistical significance. Open circle and square represent female and male mice respectively. (B) Thermal threshold measured by Hargreaves test. No significant difference between WT (n=21), HetNav1.7I228M (n=18) and HomNav1.7I228M (n=17) groups. (C) Mechanical threshold measured by von Frey test. No significant difference between WT (n=23), HetNav1.7I228M (n=14) and HomNav1.7I228M (n=21) groups. (D) Thermal place preference test: the percentage of time spent at a test temperature versus control temperature (35 ºC vs 25 ºC, 15 ºC vs 25 ºC, and 38 ºC vs 33 ºC). The difference between groups does not reach significance (WT, n=8; HetNav1.7I228M, n=8; HomNav1.7I228M, n=8). (E) Thermal gradient test: the percentage of time spent at a range of temperature zones (6.5 ºC to 46 ºC). No significant difference is seen between WT (n=8), HetNav1.7I228M (n=8) and HomNav1.7I228M (n=8) groups. (F) von Frey test for mechanical sensitivity (Nav1.7I228M mutant mice by CRISPR gene editing). No significant difference is seen in withdrawal thresholds between WT (n=7), HetNav1.7I228M (n=5), and HomNav1.7I228M (n=8). (G) Hot plate test for thermal sensitivity at 50 °C (Nav1.7I228M mutant mice by CRISPR gene editing). No significant difference is seen between WT (n=9), HetNav1.7I228M (n=9), and HomNav1.7I228M (n=5). (H) Thermal gradient test for thermal sensitivity (Nav1.7I228M mutant mice by CRISPR gene editing). Freely moving animals placed on a gradient from 4 °C to 50 °C did not show a difference in preferred temperature between WT (n=7), HetNav1.7I228M (n=10), and HomNav1.7I228M (n=4).
We also examined the impact of HomNav1.7I228M on acute irritant-induced and inflammatory pain-related behavior induced by intra-plantar injection of formalin or complete Freund’s adjuvant (CFA) respectively. Behavioural responses (duration of paw licking) in early and late phases of formalin test in HomNav1.7I228M mutant mice produced by targeted homologous recombination were indistinguishable from those in control mice (Figure 5 A–B). CFA injection induced long-lasting (up to 2 weeks) mechanical and thermal hypersensitivity in both HomNav1.7I228M mutant mice and their wild-type control littermates. No statistical difference was observed between the groups (Figure 5 C–D)
Figure 5. Behaviour responses of HomNav1.7I228M mutant mice to formalin and CFA injections.
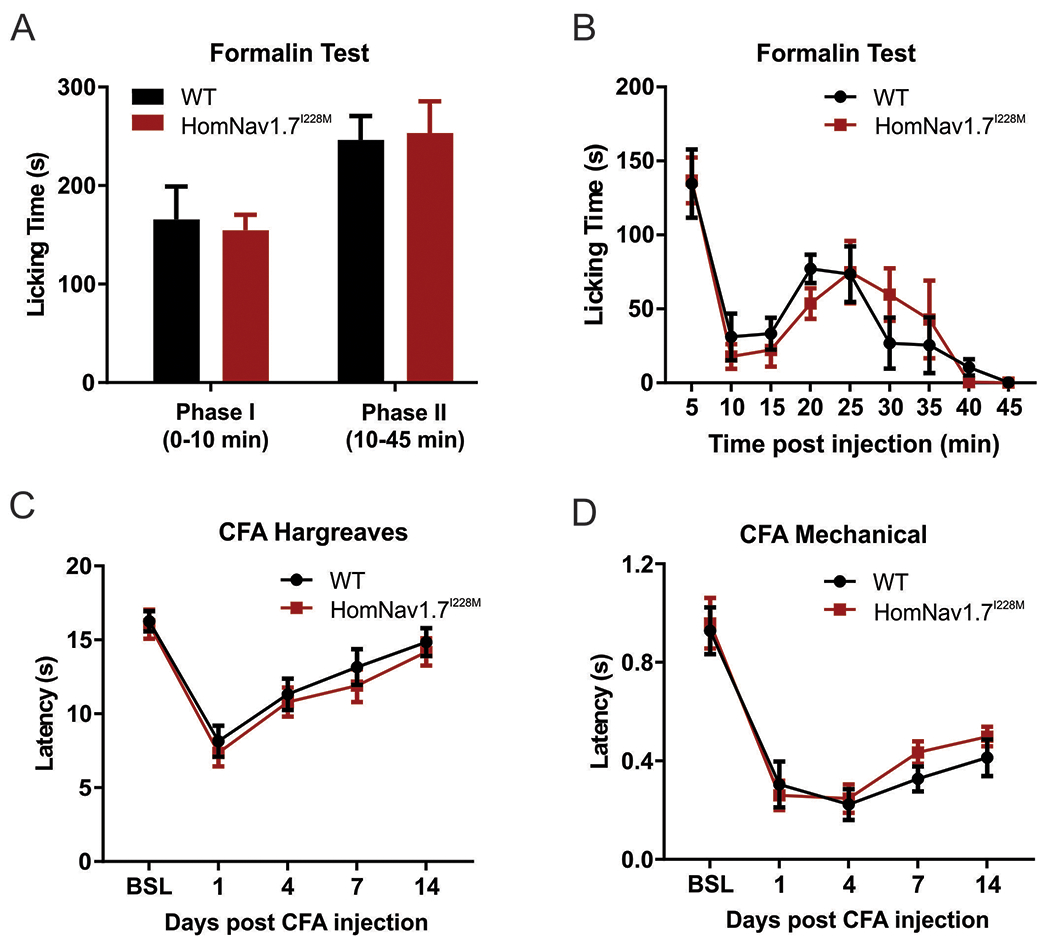
(A-B) Formalin test. HomNav1.7I228M mutant mice and their control littermates show similar licking durations in Phase I (WT, 165.6 ± 33.4 s, n=5; HomNav1.7I228M, 154.6 ± 15.8 s, n=5) and Phase II (WT, 246.4 ± 24.2 s, n=5; HomNav1.7I228M, 253.4 ± 32.2 s, n=5) of the formalin test. (C-D) Thermal and mechanical sensitivity measured by Hargreaves test and von Frey test following intra-plantar CFA injection. No significant difference is seen between WT (n=11) and HomNav1.7I228M (n=12) groups.
Nav1.7I228M channels do not cause IENF loss in mutant mice
As a variant associated with SFN, we tested whether Nav1.7I228M in mice had an impact on the integrity of distal nerve terminals. Using immunofluorescent staining, we evaluated the sensory innervation of the epidermis in skin biopsies taken from the same anatomical position in the ventral side of the glabrous foot pad from WT and Nav1.7I228M knock-in mice (targeted homologous recombination). As shown in Figure 6A, PGP9.5 staining of intra-epidermal nerve fiber (IENF) is similar between WT and HomNav1.7I228M mutant samples. Quantitative analysis did not reveal any statistical difference in IENF densities (WT, 19.6 ± 1.1 / mm, n=4; HomNav1.7I228M, 17.0 ± 0.5 / mm, n=4; p = 0.2, Mann-Whitney test) (Figure 6B). Semithin cross sections of plastic-embedded sciatic nerve samples demonstrated that the gross morphology of myelinated axons and integrity of myelin sheaths appeared normal and groups of nonmyelinated axons (arrows) could be identified in both WT and HomNav1.7I228M samples at the light microscopy level (100x) (Figure 6C). Analysis of size distribution of myelinated axons in tibial nerve showed that there was no significant difference between WT and HomNav1.7I228M groups (Figure 6D).
Figure 6. HomNav1.7I228M mutant mice have normal intra-epidermal nerve innervation and nerve fiber morphology.
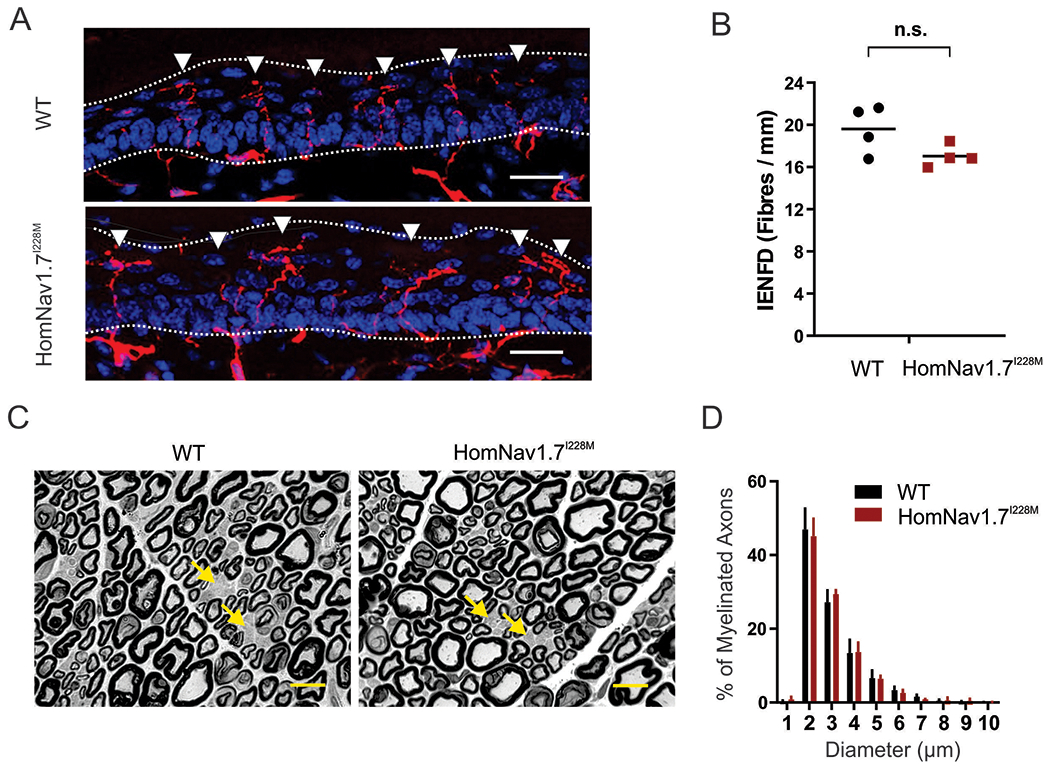
(A) confocal images of intra-epidermis nerve fibers (IENF) in hind paw skin samples taken from WT and HomNav1.7I228M mice. IENFs labelled with the pan-axonal marker PGP9.5 (red) extend from the subdermal plexus into the epidermis (nuclei labelled with DAPI, blue). The dashed line demarcates the division between the dermis and the epidermis layers. Scale bar = 20 μm. (B) Quantification of IENFs in hind paw skin samples taken from WT and HomNav1.7I228M mice. The analysis shows no statistically significant difference in the IENF density (unpaired student’s t-test; WT, 19.6 ± 1.1 / mm, n=4; HomNav1.7I228M, 17.0 ± 0.5 / mm, n=4). (C) Micrographs of semithin cross sections of tibial branch of the sciatic nerve from WT and HomNav1.7I228M mice. The morphology of the myelinated axons and unmyelinated axons (groups of unmyelinated axons in Remak bundles indicated by arrows) were all similar in WT and HomNav1.7I228M mice. Images were taken by a light microscope at 100x. Scale bar = 10 μm. (D) Size distribution of myelinated axons in the tibial branch of sciatic nerves from WT (n=3) and HomNav1.7I228M (n=3) mice. No significant differences exist between groups.
We next examined whether the presence of the gain-of-function I228M variant influenced the neurite integrity of RFP-labelled DRG neurons in in vitro cultures from WT (Figure 7A) and targeted recombination HomNav1.7I228M mice (Figure 7B). WT and HomNav1.7I228M DRG displayed similar neurite growth 7-day into culture (Figure 7 C–D). Quantification of mean neurite length per cell calculated from large-field images showed that WT and HomNav1.7I228M groups did not display any significant difference in the neurite lengths (WT 1.000 ± 0.1989, n = 19 cultures; HomNav1.7I228M 1.039 ± 0.4898, n = 19 cultures; p = 0.8338, nested t test) (Figure 7E).
Figure 7. DRG neurons from HomNav1.7I228M mutant mice display normal neurite length in vitro.
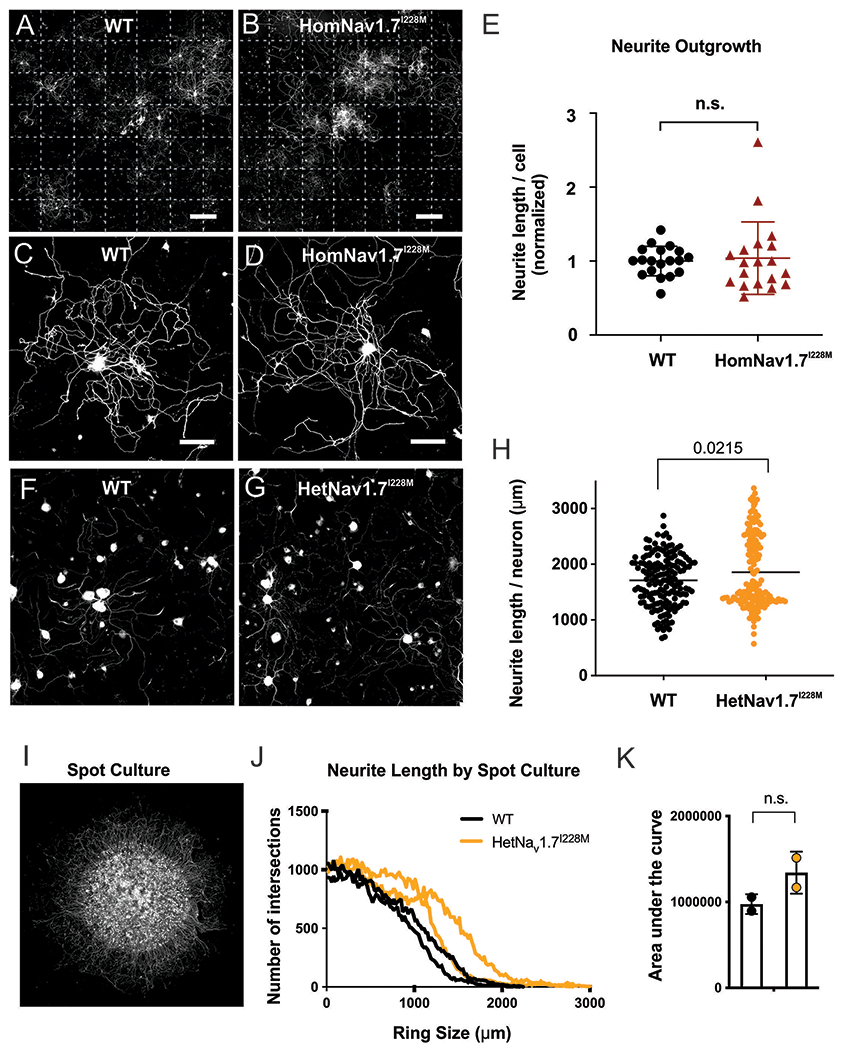
(A-B) Representative large-field montage images of 7-day DRG cultures from (A) WT mice and (B) HomNav1.7I228M mutant mice (6-8 weeks). Each large-field image consists of 7×7 field views. Dotted lines distinguish individual field-of-view. Scale bar, 1000 µm. (C-D) Representative images of individual (C) WT and (D) HomNav1.7I228M DRG neuron with neurite growth. Scale bar, 50 µm. (E) Quantification of total length per cell of WT and HomNav1.7I228M neurons. Average normalised neurite length per cell of WT (n = 19 culture dishes from 5 WT mice) and HomNav1.7I228M (n = 19 culture dishes from 5 Nav1.7 I228M mutant mice) are 1.000 ± 0.1989 and 1.039 ± 0.4898, respectively. There is no significant difference in neurite length (p = 0.8338, nested t test) between WT and HomNav1.7I228M groups. (F-G) Representative images of (F) WT and (G) HetNav1.7I228M dorsal root ganglion neurons (from HetNav1.7I228M mutant mice by CRISPR gene editing) cultured in a monolayer, then fixed and stained with Tuj1 after 24 hours. (H) Neurites of cultured HetNav1.7I228M neurons (n=156 wells in 96 well plate, from 4 HetNav1.7I228M mutant mice by CRISPR gene editing) do not show evidence of degeneration, and in fact display longer neurite lengths per neuron than WT (n=167 wells in a 96 well plate, 4 mice) (p = 0.0215, two-tailed t test). (I) Example image of dorsal root ganglion neurons cultured in a spot culture, where neurons are plated in a dense spot, allowing neurites to grow radially outward. After 6 days of growth, neurons were fixed and stained with Tuj1. (J) Neurite length calculated by Sholl analysis, whereby the intersections of neurites with concentric rings drawn around the spot are quantified as a proxy for neurite length. Intersections at higher diameters indicate longer neurite lengths (WT, n=2; HetNav1.7I228M mutant by CRISPR gene editing, n=2). (K) Area under the curve from (J). The area under the curve, representing total neurite length per spot is not significantly different between WT and HetNav1.7I228M (p = 0.195050, two-tailed t-test).
In the CRISPR Nav1.7I228M KI mouse line, 24-hour after monolayer culture, Tuj1-stained WT and HetNav1.7I228M mutant DRG neurons appeared identical (Fig 7 F–G). Quantification of neurite length revealed no decrease in the length of HetNav1.7I228M mutant neurons (Fig 7H). Neurite length was also assessed by spot culture where neurons were plated in a dense spot, allowing neurites to grow radially outward for 6 days (Fig 7I). Neurite length, quantified by Sholl analysis (the number of intersections of neurites with concentric rings drawn around the neuron), did not display a significant difference, supporting the conclusion that Nav1.7I228M mutant did not impair neurite length in vitro (Fig 7J–K).
DISCUSSION
We established two independent Nav1.7 I228M knock-in mouse lines, using both targeted homologous recombination and CRISPR editing. Using these mice, we provide the first detailed characterization of the excitability and axonal integrity of DRG neurons carrying the endogenous Nav1.7 gain-of-function variant, I288M. We also tested here whether three were signs of the positive aspects of pain that have been reported in SFN patients by inducing evoked responses to mechanical and thermal stimuli. We show that, while the presence of the I228M gain-of-function variant renders DRG neurons hyperexcitable, as expected, it does not alter thermal and mechanical sensitivity in these mice. In addition, at the age of the mice examined (up to 12 weeks when mice are considered adults (https://www.jax.org/news-and-insights/jax-blog/2017/november/when-are-mice-considered-old), the neurite/axonal integrity of cultured DRG neurons, the gross morphology of nerve fibers and IENFD in skin biopsies were also not altered. While the Nav1.7 I228M knock-in in vivo mouse models provide a useful model of hyperexcitability in DRG neurons, we do not see evidence that such a functional change alone induces the genetically driven pain and neuropathy phenotype seen in patients with SFN carrying this mutation. This raises several interesting questions about the relationship between hyperexcitability, neuropathy, and pain and the utility of mouse models as surrogates of certain human disorders.
Mismatch between hyper-excitability of DRG neurons and the lack of behavioural phenotype
Previous work has suggested that hyperexcitability of small DRG neurons is a major driver of neuropathic pain [1; 6]. However, because we found no indication of any alteration in pain-related behaviors in either mouse line, we suggest that in the case of the Nav1.7I228M mutation, and potentially other gain-of-function Nav1.7 mutations, the in vivo readout (behavior) might be modified or compensated for by additional factors that cannot be captured in the culture system.It is possible that in the case of this mutation the observed pain in patients is secondary to the neuropathy, which we do not see at the time-points measured in this study. This would be compatible with the decades it takes for pain to manifst in the patients. Further study into the relationship between the presence of a pain phenotype and the development of neuropathy in SFN in older mice will therefore be crucial. It is possible that in this mouse model and indeed in asymptomatic patients, developmental and compensatory changes within primary afferents or in spinal or supraspinal circuits mitigate the hyperexcitability effects of the mutation and, and in this way suppress pain hypersensitivity. In addition, the current standard methods used to detect pain-related behaviors in mice may also be a factor here [2]. Behavioral measures of pain in rodents are limited in scope, and standard stimulus-evoked-assays may be too insensitive to detect alterations in pain. The reflex-based behavioural measures used in this study assess withdrawal latencies and responses to thermal and mechanical stimuli, but this may not capture the persistent spontaneous pain that characterizes patients with neuropathy [3].
Lack of a neuropathy phenotype
While loss of IENF and presence of peripheral neuropathic pain as identified by quantitative sensory testing are used as the defining characteristics of SFN [4, 11] and are present in patients with SFN carrying the Nav1.7 I228M variant [8, 9], but overall diagnostic criteria continue for SFN continue to be reevaluated [3]. Over-expression of I228M in vitro impairs the integrity of sensory axons, possibly via mechanisms involving an increase in local intracellular Ca2+ in neurites [13; 14] and mitochondrial malfunction [12] and transient over-expression of I228M in zebrafish embryos causes decreased nerve density and increased temperature-induced behaviour [7]. Through to 12 weeks of age in the two independently generated mouse lines, the endogenous I228M variant did not cause any observable morphological changes in distal axon terminals in the skin. It is possible that degeneration seen in in vitro studies was partially due to the increased load of mutant channels that is caused by the overexpression of the mutant channel. Additionally, because degeneration in SFN is thought to occur in a dying-back fashion as a result of metabolic stress on long axons, the relative difference in axon length mice and humans is a potential explanation for the lack of epidermal denervation in these mice. While our current data does not preclude the possibility that a neuropathy phenotype may present beyond at later stage, it did allow us to characterize the behavior of the mice at a time-point when their DRG neurons were hyperexcitabile but their epidermal innervation remained intact. This provides insight into the question of whether the observed pain phenotype is due to hyperexcitability or to neuropathy, or a combination.
In three human patients with the Nav1.7I228M mutation onset of neuropathic pain symptoms occurred at ages 32, 36 and 46 [8]. Notably, two young adult sons of one patient harboured the I228M variant but did not have any SNF-like complaints at the time of study [8], which might be viewed as a parallel to our findings. Our behavioural assessments were conducted in adult mice (up to 12 weeks), and it is possible that neuropathic pain-associated behaviour like tactile allodynia might be observed at later time points, secondary to time-dependent development of neuropathy. This would leave unanswered, however, the discordance between DRG neuron hyperexcitability and the absence of demonstrable pain in the mouse lines we studied, and indeed presumably in asymptomatic patients. We suggest that while the Nav1.7 variants and the associated sensory neuron hyperexcitability may be important risk factors, but that a secondary insult, or aging may be required to cause intraepidermal nerve fiber degeneration, which may in turn trigger the neuropathic pain that is characteristic of SFN. I228M has been listed as a natural SNP in healthy control databases (103/60,146; gnomAD v2.1.1 Controls). A multi-hit model has been proposed to underlie the length-dependence and age-dependence of onset of peripheral neuropathies [13]. Other underlying factors, such as systemic diseases, modifier genes, or environmental factors, may be required, together with I228M, for the onset of clinical signs and symptoms of SFN, something not captured in the mouse models.
In conclusion, we have characterized two independent Nav1.7I228M knock-in in vivo models generated with targeted homologous recombination and CRISPR gene editing, respectively. These parallel mouse lines with the endogenous Nav1.7 I228M variant are the first mammalian models created for modelling Nav1.7-related SFN to date. Although the mutant mice recapitulated the DRG neuronal hyperexcitability that is expected in SFN patients with a gain of function mutation in Nav1.7, they do not recapitulate either the pain or neuropathy phenotypes seen in the patients. We believe that this suggests that the hyperexcitability induced by this mutation in mice is not sufficient by itself to induce behavioural changes indicative of pain, at least in the absence of a neuropathy phenotype in mice. We would stress that a limitation of our study is that our electrophysiological studies were confined to the soma of DRG neurons and recordings in vivo in both soma and nerve fibers will be needed to reach a more definitive answer on whether there is spontaneous firing of nociceptors. Nevertheless, our results suggest that DRG neuron hyperexcitability alone does not induce either SFN or a detectable pain phenotype in young adult mice. A parallel may exist in the clinical domain, where patients with Nav1.7 mutations that presumably produce hyperexcitability in early development only develop pain in adulthood, potentially secondary to development of neuropathy. The findings of the present study underscore the complexity of the relationship between hyperexcitability, neuropathy, and pain in Nav1.7 mutant generated SFN in an in vivo setting.
ACKNOWLEDGEMENTS
We thank Dr. Peng Zhao, Larry Macala and Pamela Zwinger for their technical support. This work was supported by a grant from the Nancy Taylor Foundation, and Center Grant B9253-C from the U.S. Department of Veterans Affairs Rehabilitation Research and Development Service (SDH and SGW). The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. This work was also supported by the NIH Award R35NS105076 (CJW) and a National Science Foundation fellowship (NKW).
Footnotes
COMPLETING INTERESTS
The author(s) declared no conflicting interests.
REFERENCE
- [1].Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol Rev 2019;99(2):1079–1151. [DOI] [PubMed] [Google Scholar]
- [2].Deuis JR, Dvorakova LS, Vetter I. Methods Used to Evaluate Pain Behaviors in Rodents. Front Mol Neurosci 2017;10:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Devigili G, Rinaldo S, Lombardi R, Cazzato D, Marchi M, Salvi E, Eleopra R, Lauria G. Diagnostic criteria for small fibre neuropathy in clinical practice and research. Brain 2019;142(12):3728–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Devigili G, Tugnoli V, Penza P, Camozzi F, Lombardi R, Melli G, Broglio L, Granieri E, Lauria G. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008;131(Pt 7):1912–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, Waxman SG. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc 2009;4(8):1118–1126. [DOI] [PubMed] [Google Scholar]
- [6].Dib-Hajj SD, Waxman SG. Sodium Channels in Human Pain Disorders: Genetics and Pharmacogenomics. Annu Rev Neurosci 2019;42:87–106. [DOI] [PubMed] [Google Scholar]
- [7].Eijkenboom I, Sopacua M, Otten ABC, Gerrits MM, Hoeijmakers JGJ, Waxman SG, Lombardi R, Lauria G, Merkies ISJ, Smeets HJM, Faber CG, Vanoevelen JM, Group PS. Expression of pathogenic SCN9A mutations in the zebrafish: A model to study small-fiber neuropathy. Exp Neurol 2019;311:257–264. [DOI] [PubMed] [Google Scholar]
- [8].Estacion M, Han C, Choi JS, Hoeijmakers JG, Lauria G, Drenth JP, Gerrits MM, Dib-Hajj SD, Faber CG, Merkies IS, Waxman SG. Intra- and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1.7. Mol Pain 2011;7(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Faber CG, Hoeijmakers JG, Ahn HS, Cheng X, Han C, Choi JS, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JP, Waxman SG, Merkies IS. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol 2012a;71(1):26–39. [DOI] [PubMed] [Google Scholar]
- [10].Hoeijmakers JG, Faber CG, Lauria G, Merkies IS, Waxman SG. Small-fibre neuropathies--advances in diagnosis, pathophysiology and management. Nat Rev Neurol 2012b;8(7):369–379. [DOI] [PubMed] [Google Scholar]
- [11].Hoitsma E, Reulen JP, de Baets M, Drent M, Spaans F, Faber CG. Small fiber neuropathy: a common and important clinical disorder. J Neurol Sci 2004;227(1):119–130. [DOI] [PubMed] [Google Scholar]
- [12].Lee SI, Hoeijmakers JGJ, Faber CG, Merkies ISJ, Lauria G, Waxman SG. The small fiber neuropathy NaV1.7 I228M mutation: impaired neurite integrity via bioenergetic and mitotoxic mechanisms, and protection by dexpramipexole. J Neurophysiol 2020;123(2):645–657. [DOI] [PubMed] [Google Scholar]
- [13].Persson AK, Hoeijmakers JG, Estacion M, Black JA, Waxman SG. Sodium Channels, Mitochondria, and Axonal Degeneration in Peripheral Neuropathy. Trends in molecular medicine 2016;22(5):377–390. [DOI] [PubMed] [Google Scholar]
- [14].Persson AK, Liu S, Faber CG, Merkies IS, Black JA, Waxman SG. Neuropathy-associated Na(V) 1.7 variant I228M impairs integrity of dorsal root ganglion neuron axons. Ann Neurol 2013;73(1):140–145. [DOI] [PubMed] [Google Scholar]
- [15].Themistocleous AC, Ramirez JD, Serra J, Bennett DL. The clinical approach to small fibre neuropathy and painful channelopathy. Pract Neurol 2014; 14(6):368–79. [DOI] [PMC free article] [PubMed] [Google Scholar]