

Summary
Extrachromosomal, circular DNA (ecDNA) is emerging as a prevalent, yet less characterized oncogenic alteration in cancer genomes. We leverage ChIA-PET and ChIA-Drop chromatin interaction assays to characterize genome-wide ecDNA-mediated chromatin contacts that impact transcriptional programs in cancers. EcDNAs in glioblastoma patient-derived neurosphere and prostate cancer cell cultures are marked by widespread intra-ecDNA and genome-wide chromosomal interactions. EcDNA-chromatin contact foci are characterized by broad and high-level H3K27ac signals converging predominantly on chromosomal genes of increased expression levels. Prostate cancer cells harboring synthetic ecDNA circles comprised of characterized enhancers result in the genome-wide activation of chromosomal gene transcription. Deciphering the chromosomal targets of ecDNAs at single-molecule resolution reveals an association with actively expressed oncogenes spatially clustered within ecDNA-directed interaction networks. Our results suggest that ecDNA can function as mobile transcriptional enhancers to promote tumor progression and manifest a potential synthetic aneuploidy mechanism of transcription control in cancer.
Keywords: ecDNA, Mobile enhancers, ChIA-PET, Chromatin interactions, ChIA-Drop
Graphical Abstract
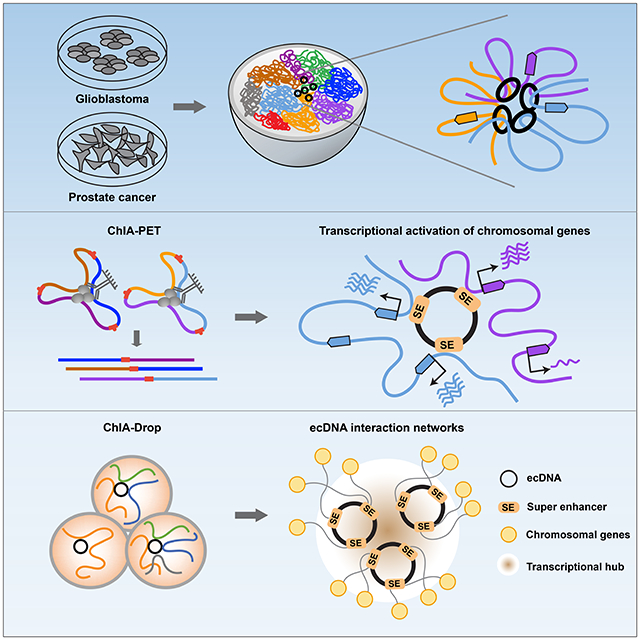
eTOC Blurb
Zhu et al. report the chromatin connectivity networks of circular and extrachromosomal DNA elements (ecDNA) in cancer, revealing that ecDNAs can function as mobile super-enhancers which drives genome-wide transcriptional amplification, including of oncogenes. These findings support an expanded role for ecDNA in trans-regulating chromosomal genes in promoting tumor growth.
Introduction
Extrachromosomal, circular DNA (ecDNA) are extrachromosomal circular chromatin elements that frequently carry oncogenes (Cox et al., 1965; Spriggs et al., 1962; Turner et al., 2017; Verhaak et al., 2019; Wu et al., 2019). First described as ‘double-minutes’ in the karyotypes of cancer cells by microscopic imaging (Cox et al., 1965), ecDNAs exist as extrachromosomal, histone-packaged chromatin bodies and are thought to be a mode of gene amplification associated with in vitro drug resistance (Alt et al., 1978; deCarvalho et al., 2018; Nathanson et al., 2014; Xu et al., 2019). More recently, ecDNAs are found to be common in primary cancers (Kim et al., 2020; Turner et al., 2017) and to constitute a bona fide mechanism and adaptive reservoir for oncogene amplification (Kohl et al., 1983). EcDNA can rapidly accumulate in cancer cells through uneven segregation (Verhaak et al., 2019), which offers a competitive advantage in response to selective pressures in the tumor microenvironment and in response to cytotoxic therapeutic agents (deCarvalho et al., 2018; Xue et al., 2017). Rapid fluctuation in ecDNA levels as a result of disjointed inheritance patterns (deCarvalho et al., 2018) likely contributes to the mechanism of tumor evolution. While the presence of ecDNAs and their structure information are extensively characterized (deCarvalho et al., 2018; Sanborn et al., 2013; Turner et al., 2017; Xu et al., 2019), the mechanism(s) by which ecDNA are deployed to modulate tumor growth and to contribute to cancer drug resistance is not yet well understood. Their open and accessible chromatin features, together with co-amplified enhancers demonstrated by recent studies (Koche et al., 2020; Morton et al., 2019; Wu et al., 2019), implicate a regulatory function beyond serving as vehicles for oncogene amplifications.
Inside the nucleus, chromosomes are extensively folded into chromatin loops which occupy distinct chromatin territories (Cremer and Cremer, 2010). Such highly organized 3-dimensional (3D) chromatin conformation provides a topological basis for many genome functions, including transcription, by bringing distal regulatory elements and their targeted genes into close spatial proximity (Sexton and Cavalli, 2015). The spatiotemporal organization of these chromatin interactions is critical to maintain normal cell state and function (Bertolini et al., 2019; Ngan et al., 2020). Our existing knowledge of 3D chromatin organization has been largely restricted to the twenty-three pairs of chromosomes. Little is known about the chromatin organization of extrachromosomal DNA elements and its impact on genome-wide expression regulation. Previous analysis of a set of unique glioblastoma (GBM)-derived neurosphere cultures using whole genome sequencing (WGS), computational and cytogenetic image approaches detected multiple ecDNAs harboring oncogenes including EGFR, MYC and CDK4 (deCarvalho et al., 2018). ecDNAs are also frequently observed in many cancer cell models including prostate cancer cell line PC3 (Wu et al., 2019). Here, we applied the ChIA-PET and ChIA-Drop technologies (Tang et al., 2015; Zheng et al., 2019) to characterize both general spatial chromatin organization and RNA polymerase II (RNAPII) mediated long-range chromatin interactions on the same ecDNAs. We demonstrate that known ecDNAs are readily recognizable through a pattern of dense intra- and inter-molecular genome-wide chromatin contacts. Importantly, in deciphering the RNAPII-mediated ecDNA connectomes and their chromosomal partners, we discovered an association between ecDNA and actively expressed chromosomal genes. Their contact regions share the key characteristics of super-enhancers known to drive high-level transcription of oncogenes in many tumor cells (Loven et al., 2013). Our data suggests that ecDNA, beyond manifestation of oncogene amplification, function as mobile transcription-amplifying elements in human cancers.
Results
EcDNAs display widespread, genome-wide chromatin interactions
We reasoned that, in contrast to the subnuclear compartments occupied by large chromosomes, the small and circular nature of an extrachromosomal chromatin body enables its mobility within the nucleus and potentially establish chromosomal interactions. To explore ecDNA-associated chromatin conformation, we performed a ChIP-free ChIA-PET analysis, similar to Hi-C procedure (Dixon et al., 2012), on three GBM patient-derived neurosphere cell lines (Figure 1A). Two of the three neurosphere lines were ecDNA (+) (HF-2354, HF-2927) and one line was ecDNA (−) (HF-3035) (deCarvalho et al., 2018). We previously reported that HF-2927 harbored a chr7p11/EGFR containing ecDNA (deCarvalho et al., 2018), referred to as ecEGFR, whereas HF-2354 contained a chr8q24/MYC ecDNA, referred to as ecMYC. Genomic regions amplified on these ecDNAs and their defined copy numbers in their respective cell lines were summarized in Table S1. Hi-C revealed general chromatin contacts within spatial topologically chromatin associated domains (TADs) in these cell lines (Figure S1A). Moreover, from the chromatin contact maps, chromosomal structural variants frequently observed in GBM, such as deletions of PTEN and CDKN2A & CDKN2B on chromosomes 10q23 and 9p21, were readily visualized as an elimination of chromatin interactions (Figure S1B). We also detected a 600 Kb deletion of the chrX:31.4-32 Mb common fragile site (Ma et al., 2012) involving the DMD gene in HF-2927, a 15 Mb extensive rearrangement of chr3:168-183 Mb and a double translocation event of 3.5 and 11.5 Mb between chr3 and chr6 in HF-2354 genomes (Figure S1C).
Figure 1. EcDNAs mediate widespread contacts with chromosomes.

(A) Hi-C and ChIA-PET were used to characterize the general ecDNA-associated chromatin conformation and their specific chromatin contacts associated with RNAPII. In RNAPII ChIA-PET assays, RNAPII ChIP was performed to capture all RNAPII-bound chromatin interactions, while in Hi-C, no ChIP was performed. The chromatin fragments were ligated by proximity ligation followed by sequencing analysis. Pair-end reads were aligned to the genome to characterize the interactions between ecDNA regions and their chromosomal targets.
(B) Distributions of copy-number adjusted nTIFs (adjnTIF) across 23 chromosomes at 50-Kb resolution in HF-2927 and HF-2354 as well as ecDNA (−) HF-3035 lines. In their respective cell lines, the distributions of adjnTIFs of chromosomes 7 and 8 (shown in the zoomed-in adjnTIF plots) reveal the location of the expected ecDNAs to be encompassing respectively EGFR and MYC. adjnTIFs in the known ecDNA regions are marked in brown stars. Genome-wide 2D chromatin contact heatmaps show distinct pairs of lines at regions on chromosomes 7p11 and 8q24 indicating intensive contacts with the entire genomes.
(C) Violin plots display the adjnTIFs between the known ecDNA regions and chromosomal background in HF-2927 and HF-2354 lines. Statistical analyses by one-sided Wilcoxon Rank-Sum Test. For boxplots, center line, median; boxes, first and third quartiles; whiskers, 1.5 × the interquartile range (IQR); points, outliers.
As shown on the chromatin contact heatmaps, both ecDNA loci exhibited extensive contacts across the entire genome (Figure 1B), suggesting a widespread ecDNA connectivity from the extrachromosomal genetic elements. To quantify the degree of chromatin contacts between chromosomal regions, we developed a metric that faithfully describes the average genome-wide trans-chromosomal interaction frequencies (nTIF) normalized across all 23 chromosomes at 50-Kb resolution. We observed a significant enrichment (one-sided Wilcoxon Rank-Sum Test with P values < 5 × 10−9 in both lines) of the nTIFs specifically in the ecDNA amplified regions (Table S2). To remove the effects resulted from the high number of copies and verify that the elevated nTIFs were specific to the circular and extrachromosomal conformation nature of the ecDNAs, we used an established linear regression model (Seaman et al., 2017; Wu and Michor, 2016) to adjust for the impact of copy number (CN) on nTIFs (adjusted nTIFs as adjnTIFs). We found that the adjnTIFs mediated by ecMYC and ecEGFR remained significantly higher after CN normalization (one-sided Wilcoxon Rank-Sum Test with P values < 5 × 10−9 in both lines) (Figure 1B-C). The adjnTIFs contributed by each ecDNA copy (adjnTIFs/CN) were also significantly higher (median adjnTIFs/CN were ~ 0.3-1.1) than per copy of each chromosome (median adjnTIFs/CN were ~ 0.2) in both ecDNA (+) lines (one-sided Wilcoxon Rank-Sum Test with P values 1 × 10−3 to 6 × 10−32) (Figure S1D). Therefore, we conclude that ecDNAs exhibit extensive chromatin connectivity and the elevated contact frequency of ecDNA across the genome is driven by its autonomous capacity.
Based on the high level of RNA expressed from genes amplified within the ecDNAs (Figure S2A), we reasoned that ecDNA is highly associated with the RNAPII complex within the active chromatin domains and establishes specific chromosomal interactions which may exert unique regulatory activities that contribute to gene transcription regulation. Therefore, we applied the ChIA-PET assays to pull down RNAPII-associated chromatin and characterized ecDNA-associated chromatin interactomes, including both intra-ecDNA and ecDNA-chromosome interactions (Figure 1A, Figure S2B). In addition to the above three lines, we also included HF-3016 and HF-3177, two neurosphere lines derived from a primary and a recurrent GBM from the same patient (deCarvalho et al., 2018). In both lines, three genes were found to be amplified extrachromosomally (Figure S2C), of which chr7p11/EGFR and ch12q14.1/CDK4 were demonstrated to be co-amplified on ecDNA while chr8q24/MYC was also found on ecDNA (referred to as respectively ecEGFR, ecCDK4 and ecMYC). We detected sharp elevation in ecMYC, ecEGFR and ecCDK4 adjnTIF levels as well as cross-interactions between the three loci in HF-3016 and HF-3177 (Figure 2A-B), suggesting that the dominant ecDNA in these lines carries all three oncogenes or that they have a close inter-molecular proximity. Similar to the adjnTIFs observed in the Hi-C data, we observed significantly higher adjnTIFs in the ecDNA regions (median adjnTIFs were ~ 4-24) than the chromosomes (median adjnTIFs were ~ 0.2-0.4) in HF-3016, HF-3177 as well as the HF-2927 and HF-2354 ecDNA (+) lines (one-sided Wilcoxon Rank-Sum Test with P values 5 × 10−9 to 9 × 10−76) but not in the HF-3035 ecDNA (−) cells (median adjnTIF was 0.2) (Figure 2C). Comparing to the chromosomal amplified regions (CNs ranged from 5 to 15), which are expected to be constrained within the chromosomal territories, these extrachromosomally amplified regions also exhibited significantly higher adjnTIFs (median adjnTIFs between 4 – 24 vs 0.7 – 1.8, one-sided Wilcoxon Rank-Sum Test with P values < 8 × 10−8 in all four lines). Table S2 summarizes CN-adjusted ecDNA-chromosomal interaction frequencies and statistical analyses. The enrichment in the chromosomal interactions from ecDNA elements associated with RNAPII binding suggests that ecDNA molecules have an important transcriptionally regulatory function.
Figure 2. EcDNA signatures can be distinguished by the high chromosomal interaction frequency (adjnTIF) across 23 chromosomes.

(A) Distribution of genome-wide adjnTIFs at 50-Kb resolution in the ecDNA (+) HF-3016 and HF-3177 cell lines in comparison with the ecDNA (−) HF-3035 line. Elevated adjnTIFs are observed on the chromosomes 7, 8 and 12 regions known to be amplified on ecDNAs. Distributions of adjnTIFs along the chromosomes 7, 8 and 12 are shown and regions with elevated adjnTIF values are well-matched with known ecEGFR, ecMYC and ecCDK4 regions.
(B) Circos plots of the interactions mediated by ecDNA regions across all 23 chromosomes in HF-3016 and HF-3177 ecDNA (+) cell lines. Extensive connections between ecMYC, ecEGFR and ecCDK4 regions are shown.
(C) Box plot displays the adjnTIFs between the known ecDNA regions and chromosomal background in four ecDNA (+) cell lines. Statistical analyses by one-sided Wilcoxon Rank-Sum Test. For boxplots, center line, median; boxes, first and third quartiles; whiskers, 1.5 × the interquartile range (IQR); points, outliers.
EcDNA-chromosomal contacts converge on non-coding regions with high H3K27ac signals
To address how RNAPII-mediated ecDNA chromatin interactions associated with transcriptional regulation, we performed RNAPII binding and H3K27ac modification ChIP-seq profiling to mark active promoters and transcriptional enhancers in these cells. To correct the bias resulted from CN variation, all ChIP-seq data was generated through an unique molecular identifier (UMI) approach (Kivioja et al., 2011). Furthermore, sequencing data from input (no-ChIP) libraries was used to normalize the copy number variation in each of the four ecDNA (+) cells. The sequencing summary and resulted peaks were summarized in Table S1 and Figure S2B. From the RNAPII tethered and CN-normalized chromatin contacts mapped within the known ecDNA regions, we observed that ecDNA regions exhibit 5-17 fold enrichment in CN-normalized cis-interaction frequencies compared to the cis-interaction frequencies of their corresponding native chromosomal regions in ecDNA (−) cells (Figure 3A, Table S2). These high frequency intra-ecDNA interactions including distinct pairs of loops and foci of intense contacts were observed in the 530 Kb ecEGFR region in HF-2927, and between the two segments of the ecMYC region in HF-2354 (Figure 3B) as well as in the ecMYC, ecEGFR and ecCDK4 from the paired primary HF-3016 and recurrent HF-3177 lines (Figure S3A). We confirmed that such high intra-ecDNA interaction frequencies were not caused by potential tandem duplications in the native chromosomal loci. Based on the optical mapping of ultra-long DNA molecules through BioNano Saphyr platform (Mak et al., 2016), these loci showed no evidence of local amplification (Data not shown). Thus, such intense cis-chromatin interaction patterns are collectively derived from contacts between the native chromosomal loci, different ecDNA molecules and folding within individual ecDNAs. While we cannot unambiguously differentiate their origins due to their near-identical sequences, we expect that most of the interactions observed were derived from the ecDNA molecules because their copy numbers far exceed the copies of chromosomal alleles and likely reflected the clustering of ecDNA molecules mediated by RNAPII binding. Vast majority (92% and 89%) of the intra-ecDNA loops detected in HF-3016 and HF-3177 were unique, presumably due to that the structures subjected to extrachromosomal amplification were highly variable (Figure S2C).
Figure 3. EcDNAs are bound by RNAPII and mediate extensive intra-ecDNA and chromosomal interactions.

(A) Copy-number normalized 2D contact heatmaps from ecDNA (+) HF-2927 (left), HF-2354 (right) and ecDNA (−) HF-3035 cell lines within ecEGFR and ecMYC regions. RNAPII bound cis-interactions and fold enrichment of RNAPII binding within the ecEGFR region (chr7: 54,860,254-55,535,856) in HF-2927 and the corresponding chromosomal EGFR locus in HF-3035 as well as the two segments of ecMYC regions (chr8: 128,032,011-128,806,493 and chr8: 129,573,241-130,968,628) in HF-2354 and the corresponding chromosomal MYC locus in HF-3035 are shown.
(B) Circos plots of ecDNA regions defined in HF-2927 and HF-2354 ecDNA (+) cell lines. From inner to outer circles: intra-ecDNA interaction loops, blue: distribution of intra-ecDNA interaction frequency, green: distribution of ecDNA-chromosomal interaction frequency, cyan: H3K27ac fold enrichment, red: RNAPII binding enrichment. The signal tracks are at 1-Kb resolution. Examples of the high concordance regions between H3K27ac signals and interaction frequency are highlighted in grey.
(C) Enrichment or depletion of promoter, intergenic and intragenic regions associated with ecDNA interaction anchors compared to genomic background on ecDNA regions. The interaction anchors from ecDNAs mediated cis (I) and trans (II) as well as their chromosomal contacts (III) in HF-2927 and HF-2354 are shown.
In total, there were 220, 271, 587 and 455 RNAPII-mediated chromatin interactions between ecDNAs and their chromosomal partners defined in HF-2354, HF-2927, HF-3016 and HF-3177, respectively (Figure S2B). The contact sites on ecDNAs and their chromosomal targets were mostly enriched at promoters (defined as TSS ± 2.5 Kb) (Figure 3C, Figure S3B). The affinity between ecDNAs and chromosomal promoters was also independently confirmed by Hi-C which detected significant contacts between ecDNAs and the chromosomal gene promoters (binomial test P value < 3 × 10−30). We next examined the potential trans-regulation of the three oncogenes amplified on ecDNA by evaluating their chromosomal interaction regions. From the promoters amplified on the ecDNAs, we detected 9, 20, 68 and 67 noncoding interacting regions on chromosomes in HF-2354, HF-2927, HF-3016 and HF-3177 cell lines, respectively. They exhibited high levels of H3K27ac enrichment and overlaps (56-95%) with H3K27ac peaks in their respective cell lines (Figure 4A), suggesting that the transcription of the oncogenes on ecDNAs is further enhanced by engaging chromosomal enhancers through chromatin contacts. Similarly, we also observed co-occurrence of high-frequency contact foci and H3K27ac peaks within ecDNAs (Figure 3B, Figure S3A). Within the 530 Kb ecEGFR in HF-2927, H3K27ac peaks aligned consistently with high-interaction frequency regions (Figure 4B), suggesting enhancer signals accumulated on ecDNA chromatin contact sites. In comparison to H3K27ac peaks of the chromosomal EGFR region in HF-3035 ecDNA (−) cells, the H3K27ac peaks on ecDNAs exhibited higher enrichment and broader spans. H3K27ac immunostaining on metaphase HF-2927 cells also confirmed overlapping H3K27ac and DAPI ecDNA signals, further validating the association between enhancer function and ecDNA (Figure S3C). These findings confirm previous report of an enhancer function for ecDNA sequences (Morton et al., 2019; Wu et al., 2019) and extend those by providing their regulatory genes targeted by the active ecDNA-chromosome interactions.
Figure 4. EcDNAs are enriched with super-enhancer signature.

(A) H3K27ac modification enrichment density within ± 3 Kb of the chromosomal non-coding regions interacting with ecDNA promoters across four ecDNA (+) cell lines. Densities from regions detected in each corresponding cell line are highlighted.
(B) Concordance between the chromatin interaction frequency and H3K27ac signal density across the ecEGFR region (chr7: 54,929,292-55,441,765) in HF-2927. Lower panel: Zoom-in view of the two super-enhancer regions on ecEGFR region, H3K27ac signal density and peaks in HF-2927 (blue) and HF-3035 (red) are shown. Peak sizes are labeled.
(C-D) Box plots show the fold enrichment (C) and span size (D) distributions of H3K27ac peaks within foci of high interaction frequency on ecDNAs (Group A, n = 7, 4, 30 and 12), their corresponding chromosomal partners (Group B, n = 770, 1,502, 2,374 and 1,522) and genome-wide remaining chromosomal peaks (Group C, n = 35,755, 32,867, 33,580 and 50,181) from each of the four ecDNA (+) lines. In ecDNA (−) HF-3035 cells, Group A (n = 45) refers as the H3K27ac peaks found in the native chromosomal regions corresponding to ecDNA foci of high interaction frequencies. Group C (n = 45,723) represents the remaining genome-wide H3K27ac peaks. Y-axis are in log10 scales. *: P value < 0.005 compared to Group C (One-sided Wilcoxon Rank-Sum Test).
(E) Proportion of trans-interacting PET counts associated with ecDNA super enhancers (SEs) (n = 7, 3, 27, 22) and typical enhancers (TEs) (n = 14, 7, 47, 46).
(F) Percentage of spans occupied by SEs on ecDNA and ecDNA SE-associated interactions vs. total spans and numbers of interactions. Fold enrichment of SE-mediated interactions are labeled. All H3K27ac signals in A-D were copy-number normalized. For all boxplots, center line, median; boxes, first and third quartiles; whiskers, 1.5 × the interquartile range (IQR); points, outliers.
EcDNAs are enriched for enhancer signatures and associated with transcriptionally active chromosomal genes
We next investigated the chromosomal contact regions on the ecDNA molecules. We found that the majority of the contact sites on the ecDNAs were converged onto only a few distinct (7-26) loci and they shared high overlaps with H3K27ac peaks in each of the four ecDNA (+) lines (Figure S3D). These few regions, while only accounted for 1-2.4% of the ecDNA sizes, mediated 17-59% of total chromosomal interactions. Such distinct contact pattern indicates the highly selective nature of ecDNA-chromosomal interactions. To quantify the H3K27ac signal associated with ecDNA-mediated chromatin interactions, we compared fold enrichment and span size from the H3K27ac peaks detected in these high frequency interaction foci on the ecDNAs (Group A), their corresponding interacting chromosomal partners (Group B), and the genome-wide chromosomal H3K27ac peaks which have no contact with ecDNA (Group C) in each of the four ecDNA (+) cell lines (Table S3). As expected, Group A showed significantly higher enrichment compared to Group C (median value 11-31 vs. 6-7, P values 1 × 10−2 to 5 × 10−6, one-sided Wilcoxon Rank-Sum Test) (Figure 4C). Group B, reflecting chromosomal ecDNA anchors, also showed a significant increase in H3K27ac signal compared to Group C (median: 12-20, P values 7 × 10−111 to 5× 10−264, one-sided Wilcoxon Rank-Sum Test), suggesting that ecDNA-chromosome connectivity converges on transcriptionally active regions. The enhancement of H3K27ac signal was ecDNA-specific as Group A and Group B showed higher fold enrichment compared to the ecMYC, ecEGFR and ecCDK4 equivalent regions in the ecDNA (−) HF-3035 neurospheres (median value: 7). EcDNA H3K27ac peaks also showed significantly larger spans than the chromosomal H3K27 peaks with no ecDNA contact (median spans of 2.3-4.1 Kb in Group A vs. 1 Kb in Group C, P values 2 × 10−3 to 5 × 10−8, one-sided Wilcoxon Rank-Sum Test) (Figure 4D). Among the three ecDNA (+) cell lines carrying either ecMYC or ecEGFR, the H3K27ac peaks detected on ecMYC and ecEGFR are mostly distinct (Figure S3E), potentially due to the underlying variation of ecDNA physical structures. Sequence analysis of H3K27ac peaks on ecDNAs (Group A) relative to chromosomal H3K27ac peak regions shows an enrichment of transcription-factor binding motifs (q < 0.001, enrichment > 1.5) known to promote glioma growth and Wnt signaling pathway, including TCF12, ZNF264 and LEF1 (Table S3).
Enhancers with super-high intensity and large domains of H3K27ac signals have been referred to as super-enhancers (SEs) (Hnisz et al., 2013), which are known to drive high transcriptional activity of oncogenes in cancers (Loven et al., 2013). To evaluate whether these high-frequency chromosomal contact loci on the ecDNA are SE-like, we applied the ROSE (the Ranking Of Super Enhancer) algorithm (Loven et al., 2013; Whyte et al., 2013) to define typical enhancers (TEs) and super enhancers (SEs) on linear chromosomes and ecDNAs. We found H3K27ac signals were significantly enriched (3-18 fold, P = 0.014, one-sided Wilcoxon Rank-Sum Test) in SE regions on ecDNA compared to chromosomes. In contrary, none of the H3K27ac peaks on the ecDNA-corresponding chromosomal regions in the ecDNA (−) line exhibited high H3K27ac signals or were classified as SE (Table S3). SEs are not only preferentially enriched on ecDNAs; they also function as the foci for ecDNA-mediated chromosomal interactions. SEs on ecDNAs were associated with high percentages of interacting PETs (Figure 4E). Following a copy-number adjustment, we observed significant enrichment in interactions originating from ecDNA SEs (1.9 to 3.7-fold, P values 7 × 10−4 to 1 × 10−6) compared to the ratio of interactions originating from chromosomal SEs in their respective ecDNA (+) lines (Table S3). In total, there were 7, 3, 27 and 22 SEs defined on ecDNAs in HF-2354, HF-2927, HF-3016 and HF-3177 cell lines, respectively and 33-100% of these high-frequency interaction SE foci were found in non-coding regions. While only occupying for 2%-18% of the total sizes of ecDNAs, these SE loci accounted for 31%-73% of the total ecDNA-mediated interactions (Figure 4F). For example, the two SEs defined on ecEGFR, both resided in the non-coding regions, mediated 62% of the total chromosomal interactions in HF-2927.
Interestingly, the ecDNA-associated SEs shared little overlap between the different ecDNA (+) cell lines. The lack of significant overlaps is potentially due to the highly variable ecDNA structures (Figure S2C, Figure S4A). When we specifically examined the SEs detected in the ecMYC regions commonly amplified in HF-2354, HF-3016 and HF-3177 cell lines, high concordance was detected. Among total 14 SEs detected, 8 SEs appeared to be shared in 2 or more cell lines. These common SEs connected extensively with the ecDNA-targeted genes of active transcription. For example, proto-oncogene c-FOS was found to be connected to the SEs commonly found in HF-2354 and HF-3016. Collectively, the convergence of SE signals on the ecDNA regions and their juxtaposition to chromosomal promoters corroborates a function of ecDNA as a genome-wide transcriptional amplifier. We recently reported a pan-cancer analysis of ecDNA frequency, identifying at 579 Circular and extrachromosomal amplicons across 3,200 tumors (Kim et al., 2020; Turner et al., 2017). We annotated amplicons for the presence of protein-coding genes, including oncogenes, and enhancer elements, and found that 22 of 579 amplicons contain one or more enhancers but no protein-coding genes, suggesting selection of enhancer elements on extrachromosomal DNA elements. In contrast, we detected 13 enhancer-only amplicons out of 1,327 amplicons classified as Breakage-Fusion-Bridge or Heavily-rearranged, which was significantly less (P = 0.0001, Fisher’s Exact Test). While the frequency of enhancer-only amplifications was higher among amplicons classified as Linear (465 of 3,895), so was the frequency of amplicons containing no enhancers or protein-coding genes (98, versus 3 across all other classes), suggesting that this class of amplifications undergoes less selection (Figure S4B).
Since super-enhancers are known to facilitate high target gene transcription (Loven et al., 2013), their prevalence on ecDNAs could mediate transcriptional activation of chromosomal target genes. We next determined whether the increase in SE signals on ecDNA results in active transcription, through the analysis of RNA expression from the same four lines. From 220, 271, 587 and 455 RNAPII-mediated ecDNA-associated chromatin interactions, we found 214, 294, 592 and 399 chromosomal genes whose promoters (located within Group B described in Figure 4C-D) made contacts with ecDNAs in HF-2354, HF-2927, HF-3016 and HF-3177 ecDNA (+) cell lines, respectively. These genes showed significantly higher levels of expression (FPKM median value 12.9-20.4) compared to genes whose promoters were bound by RNAPII but with either no trans-chromosomal contact (FPKM median value 0.4-2.7, P value 4 × 10−23 to 7 × 10−48, one-sided Wilcoxon Rank-Sum Test) (Figure 5A) or with trans-chromosomal contact but not with ecDNA (FPKM median value 9.5-11.3, P value 6 × 10−3 to 2 × 10−7, one-sided Wilcoxon Rank-Sum Test) (Figure S5A-B). Table S3 summarizes all the statistical analyses of the RNA expression among different groups of genes. Furthermore, the expression level of ecDNA-connecting genes positively correlated with ecDNA contact frequency as measured by the number of independent interactions (Figure 5B). 214, 294, 592 and 399 genes were in contact with ecDNAs in their respectively ecDNA (+) cell lines and their function is commonly enriched in multiple biological processes involved in cellular metabolic (GO:0008152) and biosynthetic processes (GO:0009058) (FDR < 0.05, Panther online GO analysis) (Mi et al., 2019). We also found significant enrichment in representation of a set of 729 chromosomal oncogenes (Forbes et al., 2015; Repana et al., 2019) (n = 71, P < 0.05, one-sided Wilcoxon Rank-Sum Test) among the list of 1,412 ecDNA-contact genes. Consistent with the effects of ecDNA on transcription activation, ecDNA-targeted oncogenes showed five to sixteen-fold FPKM increases compared to median transcription levels across all genes in each of the four ecDNA (+) lines (P values 4 × 10−5 to 9 × 10−9, one-sided Wilcoxon Rank-Sum Test) (Figure S5C).
Figure 5. EcDNA-mediated chromatin interactions associated with genes of active transcription.

(A) The distributions of RNA expression (FPKM) of chromosomal genes trans-interacting with ecDNA (n = 214, 294, 592 and 399, respectively) and genes with no trans-chromosomal interactions (n = 618, 430, 755 and 533, respectively) in each of the four ecDNA (+) cell lines. Statistical analyses by one-sided Wilcoxon Rank-Sum Test. For boxplots, center line, median; boxes, first and third quartiles; whiskers, 1.5 × the interquartile range (IQR); points, outliers.
(B) The distributions of gene expression (FPKM) of chromosomal genes with increasing degree (0-5) of ecDNA contact frequency. For each ecDNA (+) line, 95% confidence interval of the fitted values are shaded. Smoothened FPKM is represented as the solid fitted line.
(C) Synthetic ecDNA enhancer assay. H3K27ac regions from ecDNAs were amplified, circularized and transfected into ecDNA (−) cells. RNA expression was measured by RNA-seq in triplicates.
(D) Volcano plot of differentially expressed genes between En-circles and Ctrl-circles (FPKM > 1). Significantly dysregulated genes (∣log2(Fold change)∣ > 1, q < 0.05) are marked in red. The horizonal line in the −Log10(q value) was resulted from the identical q values from the differentially expressed genes of different fold changes.
(E) The normalized read coverages of the differentially expressed MMP13, ATF3, and TNFAIP3 genes in En-circles and Ctrl-circles transfected cells are shown in triplicates.
See also Figure S5, Figure S6, Figure S7, Table S3 and Table S4.
Multiple lines of evidence corroborate the specificity of interactions inferred by ChIA-PET, the high H3K27ac signals and gene expression activities detected in the ecDNA-associated contact loci. From fluorescence in-situ hybridization (FISH), we validated the interactions between ecEGFR and its chromosomal targets (JUND, HSF1, ZC3H4 and KLF6) in HF-2927 (Figure S5D-E). Secondly, comparisons of adjnTIF among regions sub-sampled on ecDNAs and chromosomes with matched H3K27ac fold enrichment reaffirmed the higher interaction frequencies mediated from ecDNAs in their respective cell lines (Figure S6A). Thirdly, among genes with equivalent RNAPII binding enrichment, genes connecting with ecDNAs displayed higher expression levels than those without ecDNA interactions (Figure S6B). Lastly, chromosomal genes found with the highest expression levels in each cell line displayed 5-6 fold lower ecDNA contact frequencies, represented by the total sums of PET counts, than ecDNA-specific targets (Figure S6C). Taken together, these series of comparisons provided further support that the ecDNA connectivity captured by the ChIA-PET is highly specific and function to connect SEs to their targeted genes in trans.
Perturbation of ecDNA identity and level resulted in the dysregulation of chromosomal gene transcription.
To validate the regulatory role of ecDNAs on chromosomal gene expression, we designed a synthetic ecDNA assay by constructing artificial circular DNAs carrying genomic regions enriched for H3K27ac modification identified from the known ecDNA regions and transfecting them into ecDNA-negative cells to evaluate their genome-wide RNA expression (Figure 5C). We adopted a pair of human prostate cancer (PC3) cell lines for their known high transfection efficiency with confirmed ecDNA-positive (ecMYC) (Wu et al., 2019) and ecDNA-negative status (Seim et al., 2017) (Figure S7A-D). We conducted an extensive, multi-omics analysis to characterize the ecMYC associated-chromatin interactions and H3K27ac enhancer peaks as well as transcriptome profiling in PC3 cells (Figure S7E-G). The results have reaffirmed the observations we made in GBM lines. Twelve of these enhancer segments were selected for PCR amplification and individually circularized as putative enhancer circles (En-circles). Regions of similar size absence of H3K27ac signatures were used as control circles (Ctrl-circles). En-circles and Ctrl-circles were separately transfected at equivalent amounts into PC3 ecDNA (−) cells and their genome-wide gene expression after transfection were compared by RNA-seq analysis. 55 out of 57 significantly differentially expressed genes were found to be upregulated (Fold change > 2, q < 0.05) while only two genes were downregulated (Figure 5D, Table S4), supporting that the circular DNA molecules harboring enhancer sequences can activate chromosomal gene expression in cancer cells. Examples of three genes found among the most significantly induced genes including transcription factor ATF3, matrix metallopeptidase MMP13 and TNF alpha induced protein TNFAIP3 were shown (Figure 5E). Collectively, by manipulating the nature of ecDNAs, synthetic ecDNA assays support a mobile enhancer role of ecDNA in transcriptional activation of chromosomal genes, beyond serving as the reservoirs of oncogene amplification.
ChIA-Drop precisely deciphers the complexity of ecDNA-chromosome contacts at single-molecule resolution
The enrichment of SEs on the ecDNAs and majority of the ecDNA-chromosome contacting loci were converged on selective SE regions suggest that ecDNAs may act as foci of multivalent, aggregated chromatin hubs. To characterize the co-aggregation of the RNAPII mediated ecDNA chromatin interactomes, we adopted the ChIA-Drop assay (Chromatin interaction analysis by droplet sequencing) (Zheng et al., 2019) to decipher the multiplicity of chromatin interactions at single-molecule resolution. In ChIA-Drop, each chromatin complex is partitioned into individual droplets for droplet-specific barcoding, such that all of the chromatin fragments within one complex will have the same barcodes and can be distinguished. Sequencing reads from each GEM (gel beads-in-emulsion, defined as individual chromatin complexes with fragments of the same barcodes) are grouped to infer multivalent, combinatorial chromatin interactions between multiple loci connected simultaneously within a single chromatin complex. From PC3 ecDNA (+) cells, we produced ChIA-PET libraries in two replicates and generated over 2.5 million GEMs from over 250 million barcoded sequences in each replicate. Mover than 1.5 million distinct GEMs composed of interacting fragments with ≥ 2 read support. Among them, ~180K GEMs associated with regions from ecMYC (Figure 6A, Table S1). To verify that ChIA-Drop successfully captures the expected chromatin contacts, particularly within ecDNAs, we examined the chromatin interaction signals aggregated from the individual ecDNA-associated chromatin complexes and found that they displayed high concordance with the interaction signals derived from bulk ChIA-PET data, as observed from their aggregated contact heatmaps in the ecMYC region (Figure S8A). The replicate datasets showed a high correlation (Pearson correlation coefficient 0.92) and the combined data shared significant overlaps (P = 1 × 10−12, Binomial test) among individual interactions between ecMYC-interacting genes detected by ChIA-PET and ChIA-Drop. For example, the eight loops within the ecMYC circles detected by ChIA-PET can be found in three separate ecDNA-associated chromatin complexes in three different combinations (Figure S8B). Approximate 17,000 RNAPII-bound chromatin complexes comprising ecMYC-interacting loci with ≥ 2 reads support were defined in each replicate (Figure 6A). Of which, ~ 9,000 complexes (54%) harbored ≥ 2 chromosomal promoters (Figure 6B) and their contact loci were converged on the SEs on ecMYC, consistent with the patterns found by ChIA-PET (Figure 6C).
Figure 6. EcDNAs associated multiplex promoter interactions are converged on SEs.

(A) ChIA-Drop analysis. RNAPII-bound, ecMYC-associated complexes were defined from two independent replicates. Complexes harnessing multiple chromosomal interaction sites with read support ≥ 2 were selected for downstream analysis.
(B) Proportions of ecMYC-associated chromatin complexes harnessing different numbers of chromosomal promoters.
(C) Proportions of ecMYC-associated chromatin complexes harnessing one or more ecDNA SEs.
(D) Two examples of ecMYC-associated complexes and their chromosomal targets. Reads of the identical barcodes were shown in a 10-kb window for each fragment with their corresponding RNAPII and H3K27ac fold enrichment. The annotated gene promoters were labeled.
In total, we detected 1,415 distinct ecMYC interacting genes appeared in ≥ 3 individual ecMYC SE complexes from both replicates (Table S5). Their functions were enriched in the TNFα, MYC and p53 pathways (FDR 7 × 10−11 to 1 × 10−25) from Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). 84 of them were oncogenes found in NCG 6 (Repana et al., 2019) and COSMIC v90 (Forbes et al., 2015). Among the 55 upregulated genes in PC3 cells harboring synthetic enhancer circles (En-circles), 25 genes whose promoters have shown ecMYC contacts and they mediated a total of 186 and 174 ecDNA chromatin contacts in each of the ChIA-Drop replicates, which were significantly higher (P value = 2 × 10−308, one-sided Wilcoxon Test) than random sets (randomization 10,000x) of equivalent numbers of genes selected for comparable transcription abundance (Figure S8C), underscoring the functional relevance of the synthetic ecDNA enhancer assays and further affirming the direct targets of ecMYC. Specifically, ATF3-ecMYC interactions were detected in 17 unique chromatin complexes. The contacts from 12 of the 17 chromatin complexes were mediated from SE sites on ecMYC. Other genes found in the ATF3-ecMYC complexes are MAPS1S and IER5 (Figure 6D). Such co-aggregation of oncogenes by ecDNA-SE presumably serves a structure-based mechanism to achieve coordinated transcriptional co-activation to promote tumorigenesis.
Discussion
In summary, we report here the use of the chromatin interaction assays to characterize the ecDNA transcription interactomes and regulation in glioblastoma and prostate cancer cells. Through a multi-omics integrative analysis of genome-wide ecDNA connectivity foci, ecDNA associated chromosomal genes, SE characterization, RNA expression and the functional interrogation of ecDNA identity, we demonstrate that ecDNAs, residing in the active chromatin compartments, can act as mobile super-enhancer elements that make contacts with multiple chromosomal genes co-localized within the same chromatin domains and regulate their transcription (Figure 7). The multi-copy and mobile nature of ecDNAs, combined with the dynamics of ecDNA sequences and abundance among different cancers and across tumor progression (deCarvalho et al., 2018; Kim et al., 2020; Turner et al., 2017; Xu et al., 2019), raise an exciting possibility that ecDNA can act as a trans-activator, akin to a transcription factor complex, that can transverse the nucleus to increase transcription from their cognate promoters. This model offers a versatile mode of transcription regulation that could enable rapid expansion of clonal diversity and drive intratumor heterogeneity. We propose the possibility that ecDNAs can actively recruit a high numbers of RNA polymerases and transcriptional factor complexes to form molecular condensates as a separate phase in the nuclei. Additional functional characterization of such “ecDNA associated phase-separation” may be important to understand how ecDNAs function to promote oncogenesis.
Figure 7. Model illustrating how ecDNAs contribute transcription and tumorigenesis.

The transcriptionally active, extra-chromosomal chromatin particles make contacts with specific chromosomal genes through RNAPII-mediated chromatin interactions. The contacts converged on super enhancers on ecDNAs and activate expression of genes relevant to oncogenesis pathways.
While introducing the concept of mobile enhancer model, we recognize that this current study has not fully established the causality between the ecDNA-chromosomal interactions and transcriptional activation. This is limited by the technical challenges to precisely and efficiently manipulate ecDNA copy numbers, regulatory sequences and genomic contexts in the intact nuclei. Therefore, a stochastic collision model could be an alternative, non-exclusive mechanism that ecDNAs adopt to enhance transcription and promote oncogenesis. Additional studies are needed to further explore mechanisms of ecDNA targeting to realize the functional significance of this exciting concept. It is worth pointing out the lack of significant overlaps of these enhancers among different ecDNA (+) cell lines, which is potentially due to the highly variable ecDNA structures (Wu et al., 2019). Analysis of ecDNAs of different regulatory sequences in the absence of oncogene amplification in both primary tumors and multiple different cancer models will be of great importance and clinical value.
In addition to offering the detailed characterization of ecDNA targeted chromatin interactomes in cancers, the use of the ChIA-PET approach provides an effective means to precisely map the amplified genomic domains within ecDNAs based on their widespread and prevalent chromatin contacts with the chromosomes. Existing methods adopted to characterize ecDNA are either through imaging-based analysis (Turner et al., 2017) or structural analysis of regions with copy number gain (Deshpande et al., 2019). Comparing with these methods, direct measuring the inter-chromosomal contact frequencies through chromatin interaction assays like ChIA-PET or Hi-C offers an unbiased approach to uncover ecDNA signatures of different sizes, copy numbers or sequence context. Furthermore, the contact frequency and pattern between different regions of ecDNA molecules can provide insight into their physical structure and continuity, much like the ability of 3D chromatin conformation to aid the characterization of genome structural variation and assembly (Dixon et al., 2018; Spielmann et al., 2018). With further protocol optimization to reduce required input cell numbers, combined with fluorescence-activated cell sorting to enrich for transfected tumor cells, direct profiling ecDNA-associated chromatin interactomes and their target genes in primary tumor specimens could provide new clinical utility.
Taken together, our results propose a transcription mechanism deployed by extra-chromosomal chromatin particles that could profoundly expand the current view of how eukaryotic genes are regulated. Given the prevalence of ecDNA in cancers and the unique genomic dynamics of this extrachromosomal structure, we anticipate new approaches in targeting ecDNA and their activated chromosomal genes in the future.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Chia-Lin Wei (chia-lin.wei@jax.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data described in this study are being deposited in NCBI's Gene Expression Omnibus GSE124769. Whole-genome sequencing data for PC3 ecDNA (+) and ecDNA (−) cell lines were obtained from SRA SRR4009277 and SRR5263237, respectively. All software tools used in this study are listed in the STAR Methods description.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Glioblastoma neurosphere cell lines were generated from the resected brain tumor specimens collected at Henry Ford Hospital (Detroit, MI). Sex details of the cell lines used are as below: HF2354: Male, HF2927: Female, HF3016 and HF3177: Male, HF3035: Female. Specimens were obtained with written informed consent from patients with a protocol approved by the Henry Ford Hospital Institutional Review Board. Briefly, tumor specimens were dissociated and cultured as neurospheres in DMEM/F12 medium (11330-032, Gibco) supplemented with N-2 supplement (17502-048, Gibco), bovine serum albumin (BSA) and growth factors (EGF and FGF-basic). Cells with passages between 15 and 26 were used for experiments and were authenticated by short tandem repeat (STR) profiling at Michigan State University. The PC3 ecDNA (+) cancer cell line was a gift from the Dr. Paul Mischel lab/Ludwig Institute for Cancer Research, University of California at San Diego (Turner et al., 2017) and the PC3 ecDNA (−) line (Seim et al., 2017) was obtained from ATCC (CRL1435, ATCC). PC3 cells were cultured following the manuals from ATCC in F-12K medium (ATCC, 30-2004) supplemented with 10% FBS. PC3 cells were derived from a male patient. All cell lines were cultured at 37°C with 5% CO2 and tested for mycoplasma using MycoAlert Mycoplasma Detection Kit (Lonza).
METHOD DETAILS
ChIA-PET experiments
Ten million cells were dual crosslinked with 1.5 mM EGS (21565, Thermo Fisher) for 45 min followed by 1% formaldehyde (F8775, MilliporeSigma) for 20 min at room temperature (RT) and then quenched with 0.125 M Glycine (G8898, MilliporeSigma) for 5 min. The crosslinked cells were washed twice with 1× PBS and lysed in 100 μL of 0.55% SDS at room temperature, 62°C and 37°C sequentially for 10 min each, followed by 37°C for 30 min with addition of 25 μL 25% Triton-X 100 to quench the SDS. To fragmentize the chromatin, nuclei were digested with 50 μL AluI (R0137L, NEB) at 37°C for 12 hours and the digested nuclei were resuspended in 500 μL of dA-tailing solution containing 50 μL 10× CutSmart buffer, 10 μL BSA (B9000S, NEB), 10 μL of 10 mM dATP (N0440S, NEB), 10 μL Klenow (3’- 5’ exo-) (M0202L, NEB), and 420 μL H2O for 1 hour at RT and then subjected to proximity ligation by adding 200 μL 5× ligation buffer (B6058S, NEB), 6 μL biotinylated bridge linker (200 ng/μL), 10 μL T4 DNA ligase (M0202L, NEB) and 284 μL H2O and incubated at 16°C overnight. The ligated chromatins were then sheared by sonication and immunoprecipitated with anti-RNAPII antibody (920102, Biolegend). The immunoprecipitated DNA was tagmented, selected with streptoavidin-beads followed by library preparation and sequencing as described (Tang et al., 2015). The ChIP-free ChIA-PET (Hi-C) experiments were performed with the same procedures in the absence of the ChIP step.
ChIA-PET interaction analysis
We processed ChIA-PET data with ChIA-PET Utilities, a scalable re-implementation of ChIA-PET Tools (Li et al., 2010) (see code availability). In brief, the sequencing adaptors were removed from the pair-end reads, the bridge linker sequences were identified and the tags flanking the linkers were extracted. Tags identified (≥ 16 bp) were mapped to hg19 using BWA alignment and mem (Li and Durbin, 2009) according to their tag length. The uniquely mapped, non-redundant pair-end tags (PETs) were classified as either inter-chromosomal (left tags and right tags aligned to the different chromosomes), intra-chromosomal (left tags and right tags aligned to the same chromosomes with genomic span > 8 Kb) and self-ligation (left tags and right tags aligned to the same chromosomes with genomic span ≤ 8 Kb) PETs. PETs aligned to chr M, chr Y and problematic genomic regions (defined as Blacklist and Greylist) were excluded. The blacklist was established by the public ENCODE community (Amemiya et al., 2019) (accession code ENCFF001TDO) while the Greylist represents the genomic artifact regions introduced specifically by the procedures in ChIA-PET experiments, including the preferential tagmentation by transposases at specific sequence contexts. They were defined based on their highly enriched interaction signals outside of the ecDNA amplicons that are commonly found in multiple ChIP-free ChIA-PET datasets made by transposase procedure. Peaks were called using MACS (version 2.1.0.20151222) with q < 0.001. The fold-enrichment of each peak was transformed into z-scores and peaks with z-score > 3 (~0.4% of the top-ranking peaks) were selected as the most enriched regions.
Interacting PETs (iPETs), the uniquely mapped, non-redundant pair-end tags (PETs) from both the inter- (left tags and right tags from different chromosomes) and intra-chromosomal (left tags and right tags with genomic span > 8 Kb) PETs, were extended by 500 bp which was the average length of the sheared chromatin fragments. Multiple iPETs overlapping at both ends were then clustered as iPET-2, 3 …(clusters with 2, 3 … iPETs) to represent their interaction strength. To define significant trans-chromosomal interactions, we adopted the previously established statistical model (Duan et al., 2010; Kaufmann et al., 2015; Kruse et al., 2013) to assess their significance. This model assumes the probability (m) of observing any particular inter-chromosomal interaction is uniform, that is m=1/M, where M is the total number of all possible inter-chromosomal pairs in a representation. To ensure we have sufficient read coverage to calculate the average interaction probabilities, we merged the overlapping PET anchors as node. M was computed as a product of the number of nodes between any two interacting chromosomes. We used the binomial distribution to estimate the P values that reflect contact significance for each pair of the chromosomal interactions. The P value of observing a particular interaction pair (PET) at least k times is expressed as: p-value = , where n is the total number of observed PET counts between two interacting chromosomes where the PET anchors stem. The P value of each interaction was converted into a q value (defined as the false discovery rate threshold at which the interaction was deemed significant) using Benjamini-Hochberg procedure. To remove the bias on n resulting from the chromosome size, the q values were further normalized (Kaufmann et al., 2015) , where chrA and chrB are the chromosomes that node A and B involved in an trans-interaction cluster are on, scaled against the longest (chr1) and second longest (chr2) chromosome. Based on the >2 order of magnitude difference between the qnorms of IPET-2+ and iPET-1 interactions (Figure S9), and with iPET-1 interactions being considered mostly transient and noisy, iPET ≥ 2 trans-interactions with both interaction anchors supported by RNAPII binding were used for downstream analyses.
For ecDNA-mediated chromosomal interactions, we required that each interaction be trans in nature, which means two anchors are from different chromosomes, and that only one of the two anchors overlapped an ecDNA region. Note that this preclude ecMYC-ecEGFR, ecMYC-ecCDK4 and ecEGFR-ecCDK4 trans-interactions from consideration. They were defined as intra-ecDNA interactions.
We used ChiaSigScaled, a scalable re-implementation of ChiaSig (Paulsen et al., 2014) to perform the statistical assessments of cis-interactions. Cis-interaction clusters with member size 3 and above (iPET 3+), q < 0.05 and RNAPII binding at both anchors are reported. All interactions within the individual ecMYC, ecEGFR and ecCDK4 regions are defined as the intra-ecDNA interactions. The anchors regions from each of the interactions were further annotated based on their overlaps with GENCODE gene models (Release 19, excludes all pseudogene and all RNAs except miRNA) with priority given to promoter (P) region (defined as ± 2.5 Kb of TSS) followed by gene-coding region (G). Anchors that do not overlap with any intragenic region are classified as intergenic (I). The oncogenes from the union list of NCG 6 (Repana et al., 2019) and COSMIC v90 (Forbes et al., 2015) were used to annotate the ecDNA-targeted genes.
Copy number analysis
Genome-wide DNA copy number of five GBM cell lines and two PC3 cell lines was determined from whole-genome sequencing (WGS) data (deCarvalho et al., 2018; Miller et al., 2011; Seim et al., 2017; Turner et al., 2017). WGS reads were aligned to the human genome (hg19) and duplicates were removed with GATK MarkDuplicates tool. We then used readDepth method (Miller et al., 2011) to compute the average estimated copy numbers (CNs) from the deduplicated reads with default parameters (https://github.com/chrisamiller/readdepth). Following the author’s guidance, we used the first read (R1) when the WGS data was in paired-end mode. The computed absolute copy numbers were filtered to remove overlaps with the Blacklist, Greylist and UCSC hg19 gap regions with “bedtools subtract” command.
Copy number-adjusted trans-chromosomal interaction analysis
We define a metric known as normalized trans-chromosomal interaction frequency (nTIF) to represent a genomic locus’s interaction activity, such that it is comparable within sample and across samples. We first segmented the hg19 genome at 50-Kb interval, giving a total of 60,739 non-overlapping bins. Next, the genome-wide interaction frequency (IF) matrix at 50-Kb resolution was derived from the ChIA-PET data. The trans-chromosomal element, TIF(i,j), is the number of PETs whose left tags and right tags fall into the segmented genomic bin i and j where both bins are from different chromosomes. The total TIF for bin i is where k is the number of bins (60,739). The corresponding number of possible trans-chromosomal interaction bins is TIFBinCount(i) = ∣{j: j ∈ [1, k], (i, j) ∈ trans }∣. For comparability, a useful baseline is the number of TIF expected based on uniform distribution, defined by . Finally, the bin i interaction activity fold enrichment is nTIF(i) = totalTIF(i)/TIFBinCount(i)/aveTIF.
To determine the Pearson’s correlation between nTIF and copy number (CN), we assigned the CN analysis result to each 50-Kb genomics bin based on the genomics coordinate. If a 50-Kb bin overlap with multiple CN segments, the average CNs were assigned. Regions overlapped with the ecDNA regions, Blacklist, Greylist, UCSC hg19 gaps, with uncalled CN or CNs less than 0.5 were excluded. Next, a linear regression model was derived to determine the expected nTIF as a function of CN. We then adjusted for the effect of CNs on the nTIFs by subtracting the CN-fitted nTIFs from the observed nTIFs. The results, defined as adjusted nTIFs (adjnTIFs), are considered as the surplus nTIF over the expected CN nTIF. The distribution of genome-wide adjnTIFs at 50-Kb resolution between ecDNA regions and the chromosomes were then compared. Wilcoxon Rank-Sum hypothesis test was used to determine the significance of the adjnTIFs from ecDNAs > adjnTIFs from the chromosomes. To determine the contribution of the adjnTIFs from each copy of the ecDNAs (adjnTIF/CN), we divided the adjnTIFs by their corresponding copy numbers. The same analysis was also performed on the Hi-C data.
Copy number normalization of cis-interaction frequency in the ecDNA regions
RNAPII ChIA-PET raw data of HF-2354, HF-2927, HF-3035, and HF-3177 were first downsampled to the same sequencing depth as HF-3016 ChIA-PET, the one with the least amount of sequencing reads (~ 172 million read pairs). Then the read-pairs were processed by ChIA-PET Utilities as described in Methods to produce the uniquely mapped PETs. To normalize with CN, the uniquely mapped PETs in the ecDNA regions were further downsampled by randomly removing PETs with a probability of (1-2/<ecDNA copy number>). The remaining uniquely mapped PETs were then de-duplicated and clustered into interactions as described in ChIA-PET Interaction analysis. The reported RNAPII bound significant cis-interactions (FDR < 0.05) were used for analysis.
AmpliconArchitect analysis
AmpliconArchitect (Deshpande et al., 2019) was run on PC3 ecDNA (+) and ecDNA (−) whole genome sequencing data with default parameters as described in the documentation (https://github.com/virajbdeshpande/AmpliconArchitect). Only aligned reads in regions with copy number greater than six above the sample ploidy were used as seeds.
ChIP-seq library construction and data analysis
For each ChIP-seq experiment, two million cells were crosslinked and lysed as described in the ChIA-PET protocol. After lysis, the nuclei pellets were sonicated and immunoprecipitated with anti-H3K27ac (39133, Active Motif) or anti-RNAPII (920102, Biolegend) antibody. 4 ng DNA from each of the immunoprecipitated chromatin and matched input DNA were subjected to end-repair, A-tailing and adaptor ligation (IDT xGen Dual Index UMI Adapters) using KAPA Hyper Prep Kit (KK8505, KAPA Biosystems). Adaptor-ligated DNA fragments were PCR amplified with KAPA Library Amplification ReadyMix (KK2612, Kapa Biosystems) and sequenced on Illumina platform. The raw reads were quality trimmed using Trim Galore version 0.4.3 (options: --stringency 3 -q 30 -e .20 --length 15) and mapped to the hg19 genome using BWA 0.7.12 (command: mem) (Li and Durbin, 2009). Reads with mapping quality greater than 30 were retained and the reads sharing the same mapping coordinate and UMI barcodes were deduplicated. We applied MACS2.1.0.20151222 (macs2 callpeak options: --nomodel --extsize 250 -B --SPMR -g hs --keep-dup all) (Liu, 2014) to call peaks from the high quality, non-redundant reads and peaks defined with FDR < 0.05 were used in all subsequent analyses. MACS2.0 was run with the flag --SPMR so that the generated signal/coverage density tracks for both ChIP and input were normalized to read per million. (SPMR = Signal Per Million Reads.). The fold change data tracks were then computed by running MACS2 with the command “bdgcmp”.
H3K27ac fold enrichment and super-enhancer analysis
To produce the heatmap in Figure 4A, the non-promoter, chromosomal sites contacting with promoters on ecDNAs were merged to form non-overlapping regions in each of the four ecDNA (+) cells. They were individually extended ± 3Kb from the centers and their average H3K27ac fold-enrichment were plotted. Each region is shown as a single row, which was ordered according to the cell line where interacting regions originated from and ranked based on their weighted average of signals from highest to lowest. For ranking purpose, the weights for averaging signals were favored towards the middle. To generate the heatmaps in Figure S3E, H3K27ac peaks in ecEGFR and ecMYC were collected, respectively. The peaks were extended ± 3Kb from the centers and their average H3K27ac fold-enrichment were plotted as described above.
To construct groups of H3K27ac peaks depicted in Figure 4C & D, we first defined the high interaction foci on ecDNAs. EcDNAs were divided into 2.5-Kb bins. In each bin, sum of interacting PET counts from RNAPII bound trans-interactions were calculated. Consecutive bins with sum of interacting PET > mean + 2 × standard deviation were merged to define a set of loci in each cell line and referred as ecDNA high interaction frequency foci. Next, we grouped H3K27ac peaks in ecDNA (+) lines into three types: Group A (H3K27ac peaks that were located within ecDNA high interaction frequency foci), Group B (H3K27ac peaks that overlap with chromosomal anchors from Group A and Group C (the remaining H3K27ac peaks that were not found in Groups A and B). In ecDNA (−) cell line, H3K27ac peaks found in the corresponding regions ecDNA high interaction frequency regions from all other ecDNA+ lines were included as Group A. Group C included all remaining H3K27ac peaks that were not found in Group A. Super-enhancers were defined from the genome wide H3K27ac peaks (P < 10−9) by the Ranking of Super Enhancer (ROSE) algorithm set at the default parameters (Loven et al., 2013; Whyte et al., 2013).
Optical mapping by Bionano Syphyr
High molecular weight DNA was extracted from HF-2927 and HF-3016 cells using Bionano DNA Isolation Kit (80004, Bionano Genomics), labeled by Direct Label and Stain (DLS) Labeling Kit (80005, Bionano Genomics) and loaded into SaphyrChips (Bionano Genomics). Optical mapping data was collected for 96 hours and the data was processed with Bionano Solve analysis software (Bionano Genomics). Only single molecules with a minimum length of 150 Kb and 9 labels per molecule were used.
Immunofluorescence and FISH analysis
Metaphase cells were dropped to the slides preincubated in KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 0.1% (v/v) Triton X-100) for 10 min at RT. Slides were blocked in 1% (w/v) BSA/KCM buffer for 30 min at RT, followed by incubation with primary H3K27ac antibody (39133, Active Motif) (1:500 dilution) in 2% BSA overnight at 4°C. After washing twice in KCM buffer for 10 min at RT, slides were incubated with goat anti-rabbit Alexa Fluor 488 secondary antibody (A32731, Invitrogen) (1:1000 dilution) in 2% BSA for 30 min at RT. After washing with KCM buffer twice, slides were crosslinked with in 4% (v/v) formaldehyde/KCM for 15 min, mounted with the coverslip in 50 μL Prolong Gold Antifade (P36931, Invitrogen) and sealed with clear nail polish. For FISH analysis, cells were fixed with methanol: acetic acid (3:1) solution and dropped onto the slides. FISH probes were ordered from Empire Genomics, denatured at 73°C for 5 min and kept at 37°C for 10–30 min until 10 μl of probe was applied to each sample slide. Slides were coverslipped and allowed to hybridize overnight at 37°C. The post-hybridization washes were carried out using 0.4× SSC at 73 °C followed by 2× SSC/0.2% Tween-20 at room temperature. Slides were air dried and counterstained with VECTASHIELD Antifade mounting medium with DAPI (Vector Laboratories). The slides were scanned under Leica STED 3X/DLS Confocal. Colocalization was analyzed with ImageJ (Schneider et al., 2012).
Synthetic circular enhancer assay
Primers for the H3K27ac peak regions on ecMYC detected in PC3 ecDNA (+) cells and negative control regions were designed and used for PCR amplification (Table S4). ~ 5 Kb PCR products were purified, digested with restriction enzymes at the sites built-in in the primer design and then subjected to circularization at the concentration of 0.1 ng/ul. The circularization was confirmed by inverse qPCR. For each PCR product, 0.5 ng of linear and circularized DNA were subjected to qPCR using PCR primers across the circularized junctions and their Ct values were compared. qPCR was performed as per manufacturer’s protocol (KM4620, KAPA Biosystems) in technical triplicates. All linear DNA, except one, showed non-detectable Ct after 40 cycles of PCR while the Ct values for all circularized DNA were < 24. The circularized enhancer elements (En-circles) and negative controls (Ctrl-circles) were purified and pooled separately at equal molar ratio. 500 ng of En-circles and Ctrl-circles were transfected separately into ecDNA-negative PC3 cells in triplicate using Lipofectamine 3000 (Invitrogen) as per manufacturer’s protocol. Briefly, ecDNA-negative PC3 cells were plated to reach 70-80% confluence. DNA-lipid complexes were allowed to form at room temperature for 15 minutes and mixtures were added to cells. Cells were incubated at standard conditions and harvested 24 hours post-transfection for RNAseq analysis.
ChIA-Drop experiments and data analysis
RNAPII ChIA-Drop was performed as described (Zheng et al., 2019). In brief, two million PC3 ecDNA (+) cells were crosslinked, permeabilized and digested with HindIII enzyme (R0104M, NEB). The digested nuclei were collected for chromatin sonication followed by immunoprecipitation with anti-RNAPII antibody (920102, Biolegend). RNAPII-enriched chromatin was collected with EB Buffer and cleared through the Amicon Ultra-0.5 100 K column (Z740183-96EA, MilliporeSigma) following the manufacture’s instruction to remove small fragments. The RNAPII-associated chromatin complexes were adjusted to a concentration of 0.5 ng/μl and proceeded to ChIA-Drop library construction using Chromium Genome v2 Library Kit & Gel Bead Kit (PN-120258, 10X Genomics) following the manufacturer’s protocol. The final libraries were sequenced on Illumina platform with 150 bp pair-end sequencing. We use the ChIA-DropBox (CDB), a data-processing and visualization pipeline for multiplex chromatin interaction analysis (Tian et al., 2019), to process the raw reads into chromatin complex interaction data. Briefly, CDB aligns the reads to the reference genome (hg19) using 10X Genomics Long Ranger pipeline (v2.1.5) and identify their corresponding droplet-specific barcode. The uniquely mapped reads with MAPQ ≥ 30 and read length ≥ 50 bp are extended by 500 bps from its 3’ end, and those with the same droplet-specific barcode within 3kb distance were merged. The collection of all merged reads with the same droplet-specific barcode represents the DNA fragments interacting simultaneously at a single-chromatin complex. 2D interaction heatmap of this data is generated by CDB via Juicer (v1.22.01) and JuiceBox (v1.11.08). Multiplex interactions are visualized with ChIA-view (v1.0).
For analysis presented in Figure S8C, 31 genes with RNAPII binding peaks at promoters were selected from the 55 up-regulated genes in the synthetic circular enhancer assay as a test gene set. Similarly, the remaining genome wide genes where their TSS were enriched with RNAPII were pooled as the background. For each of the tested gene, a random gene was picked from the background pool with the following criteria: FPKM range is 90% to 110% if tested gene FPKM > 1, or +/− 0.1 if tested gene FPKM was between 0.1 and 1, or 0 < FPKM < 0.1 if tested gene FPKM is above 0 to 0.1 (all the 31 tested gene had FPKM > 0). This process yielded a set of 31 background genes representing a random set. The total number of unique RNAPII bound ecMYC-associated chromatin complexes associated with the 31-gene set was evaluated. The distribution of unique complex counts from background sets after 10,000 random selection was drawn and compared with that from the test set.
Transcription factor motif enrichment analysis
We applied Homer2 (v4.11.1) (Heinz et al., 2010) to search known motifs specifically enriched within ecDNA H3K27ac peaks against chromosomal H3K27ac peaks as normalized background sequences. The enriched motifs were selected based on the following criteria: q value < 0.001, enrichment > 1.5, motifs present in > 25% of the target sequences.
RNA-seq library construction and data analysis
Total RNA was isolated using AllPrep DNA/RNA Mini Kit (80204, QIAGEN). Strand-specific RNA libraries were generated from 300 ng of total RNA using KAPA Stranded mRNA Sequencing Kit (KK8502, KAPA Biosystems) following the manufacturer’s instruction. Libraries were sequenced on the Illumina platforms with either 75 or 150 bp paired-end sequencing. The raw sequencing reads were trimmed using Trim Galore version 0.4.3 (options: --stringency 3 -q 20 -e .20 --length 15 --paired) and aligned to the hg19 genome using hisat 2.1.0 (options: --dta-cufflinks) (Kim et al., 2019). The transcripts were assembled using Cufflinks 2.2.1 (Trapnell et al., 2010) and the differential gene expression analysis was performed using Cuffdiff (options: --library-type fr-firststrand) (Trapnell et al., 2013). All RNA-seq experiments from GBM neurospheres were performed in biological replicates and triplicates in PC3 cells.
Quantification and statistical analysis
Statistical analyses were performed using R statistical software. The Wilcoxon Rank-Sum Test, Binomial Test, Fisher’s Exact Test, and t-test were used in this study. Details of statistical tests used are specified in Results and figure legends.
Supplementary Material
Table S1. Defined ecDNA regions, ecDNA copy number and sequencing summary, Related to Figure 1.
Table S2. Statistics summary of copy number adjusted trans and cis interaction frequency analysis, Related to Figures 1-3.
Table S3. H3K27ac enrichment, super-enhancer (SE) enrichment, and RNA expression associated with ecDNA, ecDNA connected anchors and genes, Related to Figures 4-5.
Table S4. Primers used and gene significantly differentially expressed in synthetic circular enhancer assay, Related to Figure 5.
Table S5. EcMYC associated complexes and genes detected by ChIA-Drop in PC3 ecDNA (+) line, Related to Figure 6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| RNA Polymerase II | BioLegend | Cat#: 664912; RRID: AB_2650945 |
| H3K27ac | Active Motif | Cat#: 39134; RRID: AB_2722569 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Invitrogen | Cat#: A32731; RRID: AB_2633280 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fetal Bovine Serum | Gibco | Cat#: 10082147 |
| F-12K Medium | ATCC | Cat#: 30-2004 |
| DMEM/F-12, HEPES | Gibco | Cat#: 11330032 |
| N-2 Supplement (100X) | Gibco | Cat#: 17502048 |
| Animal-free recombinant human EGF | Peprotech | AF-100-15 |
| Recombinant human FGF-basic | Peprotech | 100-18B |
| EGS (ethylene glycol bis(succinimidyl succinate)) | Thermo Fisher | Cat#: 21565 |
| Formaldehyde | MilliporeSigma | Cat#: F8775 |
| Glycine | MilliporeSigma | Cat#: G8898 |
| AluI | NEB | Cat#: R0137L |
| BSA | NEB | Cat#: B9000S |
| dATP | NEB | Cat#: N0440S |
| Klenow (3’- 5’ exo-) | NEB | Cat#: M0202L |
| 5× ligation buffer | NEB | Cat#: B6058S |
| T4 DNA ligase | NEB | Cat#: M0202L |
| ProLong™ Gold Antifade Mountant with DAPI | Invitrogen | Cat#: P36931 |
| KCl | Invitrogen | Cat#: AM9640G |
| NaCl | Invitrogen | Cat#: AM9760G |
| Tris-HCl | Invitrogen | Cat#: 15568025 |
| EDTA | Invitrogen | Cat#: AM9260G |
| Triton X-100 | MilliporeSigma | Cat#: T8787-50ML |
| Methanol | MilliporeSigma | Cat#: 34860-100ML-R |
| Acetic acid | Thermo Fisher | Cat#: A38-500 |
| HindIII | NEB | Cat#: R0104M |
| Critical Commercial Assays | ||
| KAPA Hyper Prep Kit | KAPA Biosystems | Cat#: KK8505 |
| KAPA Library Amplification Kit | KAPA Biosystems | Cat#: KK2612 |
| IDT xGen Dual Index UMI Adapters | IDT | N/A |
| Lipofectamine™ 3000 Transfection Reagent | Invitrogen | Cat#: L3000015 |
| Amicon Ultra-0.5 100 K column | MilliporeSigma | Cat#: Z740183-96EA |
| Chromium Genome v2 Library Kit & Gel Bead Kit | 10x Genomics | Cat#: PN-120258 |
| KAPA SYBR FAST qPCR Master Mix (2X) ROX Low | KAPA Biosystems | Cat#: KK4620 |
| Bionano DNA Isolation Kit | Bionano Genomics | Cat#: 80004 |
| Direct Label and Stain (DLS) Labeling Kit | Bionano Genomics | Cat#: 80005 |
| KAPA Stranded mRNA Sequencing Kit | KAPA Biosystems | Cat#: KK8502 |
| AllPrep DNA/RNA Mini Kit | QIAGEN | Cat#: 80204 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE124769 |
| WGS for PC3 ecDNA(+) | Wu et al., 2019 | SRA: SRR4009277 |
| WGS for PC3 ecDNA(−) | Seim et al., 2017 | SRA: SRR5263237 |
| WGS for GBM cell lines | deCarvalho et al., 2018 | European Genome-phenome Archive (EGA): EGAS00001001878 |
| Experimental Models: Cell Lines | ||
| HF-2354 | Ana C. deCarvalho, Henry Ford Hospital | N/A |
| HF-2927 | Ana C. deCarvalho, Henry Ford Hospital | N/A |
| HF-3016 | Ana C. deCarvalho, Henry Ford Hospital | N/A |
| HF-3035 | Ana C. deCarvalho, Henry Ford Hospital | N/A |
| HF-3177 | Ana C. deCarvalho, Henry Ford Hospital | N/A |
| PC3 ecDNA(+) | Provided by Dr. Paul Mischel lab/Ludwig Institute for Cancer Research, University of California at San Diego | RRID: CVCL_0035 |
| PC3 ecDNA(−) | ATCC | Cat#: CRL-1435; RRID: CVCL_0035 |
| Oligonucleotides | ||
| See Table S4 | This paper | N/A |
| Software and Algorithms | ||
| ChIA-PET Tools | Github | https://github.com/cheehongsg/CPU |
| ChiaSigScaled | Github | https://github.com/cheehongsg/ChiaSigScaled |
| cnadjTIF | Github | https://github.com/WeAllOneCode/cnTIF |
| ChIA-DropBox | Github | https://github.com/TheJacksonLaboratory/ChIA-DropBox |
| ChIA-View | Github | https://github.com/TheJacksonLaboratory/ChIA-view |
| Long Ranger | 10x Genomics | https://support.10xgenomics.com/genome-exome/software/pipelines/latest/installation |
| Juicer | Github | https://github.com/aidenlab/juicer |
| Homer2 (v4.11.1) | Heinz et al., 2010 | http://homer.ucsd.edu/homer/motif/ |
| Cufflinks 2.2.1 | Trapnell et al., 2010; Trapnell et al., 2013 | https://github.com/cole-trapnell-lab/cufflinks |
| Trim Galore version 0.4.3 | Github | https://github.com/ime-tools/trimgalore |
| hisat 2.1.0 | Kim et al., 2019 | https://github.com/DaehwanKimLab/hisat2 |
| Bionano Solve analysis software | Bionano Genomics | https://bionanogenomics.com/support/software-downloads/ |
| ROSE | Loven et al., 2013; Whyte et al., 2013 | http://younglab.wi.mit.edu/super_enhancer_code.html |
| MACS2.1.0.20151222 | Liu, 2014 | https://pypi.org/project/MACS2/ |
| BWA 0.7.12 | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| AmpliconArchitect | Deshpande et al., 2019 | https://github.com/virajbdeshpande/AmpliconArchitect |
| readDepth | Miller et al., 2011 | https://github.com/chrisamiller/readdepth |
| Fiji (ImageJ) | Open source | https://fiji.sc/ |
Highlights.
EcDNAs show intense chromatin connectivity and are in contact with chromosomal DNA.
Chromosomal ecDNA contacts are associated with transcriptional activity.
Oncogenes are co-localized within the multi-valent, aggregated ecDNA chromatin hubs.
EcDNA functions as mobile regulatory elements leading to synthetic aneuploidy.
Acknowledgments
The authors thank Drs. Edison Liu and Rachel Goldfeder for their feedback and comments on the manuscript, Dr. Qianru Yu for her support in imaging analysis. Research reported in this publication was partially supported by the 4DN (U54 DK107967) and ENCODE (UM1 HG009409) consortia. C.-L.W., R.G.W.V., C.Y.N. and the research here are supported by NCI under Award Number P30CA034196. Y.Z., G. L., and C-L.W. are supported by grants from the US National Institutes of Health R01 GM127531 and R33 CA236681. R.G.W.V. and A.G. are supported by grants from the US National Institutes of Health R01 CA190121, R01 CA237208, R21 NS114873, the Musella Foundation and the B*CURED Foundation, the Brain Tumour Charity, and the Department of Defense W81XWH1910246. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of Interests
R.G.W.V. is a co-founder of Boundless Bio, Inc (BB), and a member of its scientific advisory board. BB was not involved in the research presented here. C-L.W., C-H.W., H.T., and R.G.W.V. are co-inventors on a patent application (WO2020223309A1 "Extrachromosomal DNA Identification and Methods of Use") submitted by The Jackson Laboratory. The other authors have declared no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- Alt FW, Kellems RE, Bertino JR, and Schimke RT (1978). Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J Biol Chem 253, 1357–1370. [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini JA, Favaro R, Zhu Y, Pagin M, Ngan CY, Wong CH, Tjong H, Vermunt MW, Martynoga B, Barone C, et al. (2019). Mapping the Global Chromatin Connectivity Network for Sox2 Function in Neural Stem Cell Maintenance. Cell Stem Cell 24, 462–476 e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D, Yuncken C, and Spriggs AI (1965). Minute Chromatin Bodies in Malignant Tumours of Childhood. Lancet 1, 55–58. [DOI] [PubMed] [Google Scholar]
- Cremer T, and Cremer M (2010). Chromosome territories. Cold Spring Harb Perspect Biol 2, a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCarvalho AC, Kim H, Poisson LM, Winn ME, Mueller C, Cherba D, Koeman J, Seth S, Protopopov A, Felicella M, et al. (2018). Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat Genet 50, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande V, Luebeck J, Nguyen ND, Bakhtiari M, Turner KM, Schwab R, Carter H, Mischel PS, and Bafna V (2019). Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 10, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, et al. (2018). Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 50, 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, and Noble WS (2010). A three-dimensional model of the yeast genome. Nature 465, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et al. (2015). COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 43, D805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann S, Fuchs C, Gonik M, Khrameeva EE, Mironov AA, and Frishman D (2015). Inter-chromosomal contact networks provide insights into Mammalian chromatin organization. PLoS One 10, e0126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Nguyen NP, Turner K, Wu S, Gujar AD, Luebeck J, Liu J, Deshpande V, Rajkumar U, Namburi S, et al. (2020). Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat Genet 52, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivioja T, Vaharautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, and Taipale J (2011). Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9, 72–74. [DOI] [PubMed] [Google Scholar]
- Koche RP, Rodriguez-Fos E, Helmsauer K, Burkert M, MacArthur IC, Maag J, Chamorro R, Munoz-Perez N, Puiggros M, Dorado Garcia H, et al. (2020). Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat Genet 52, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl NE, Kanda N, Schreck RR, Bruns G, Latt SA, Gilbert F, and Alt FW (1983). Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell 35, 359–367. [DOI] [PubMed] [Google Scholar]
- Kruse K, Sewitz S, and Babu MM (2013). A complex network framework for unbiased statistical analyses of DNA-DNA contact maps. Nucleic Acids Res 41, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, et al. (2010). ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol 11, R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T (2014). Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 1150, 81–95. [DOI] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K, Qiu L, Mrasek K, Zhang J, Liehr T, Quintana LG, and Li Z (2012). Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int J Mol Sci 13, 11974–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AC, Lai YY, Lam ET, Kwok TP, Leung AK, Poon A, Mostovoy Y, Hastie AR, Stedman W, Anantharaman T, et al. (2016). Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 202, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47, D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CA, Hampton O, Coarfa C, and Milosavljevic A (2011). ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One 6, e16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AR, Dogan-Artun N, Faber ZJ, MacLeod G, Bartels CF, Piazza MS, Allan KC, Mack SC, Wang X, Gimple RC, et al. (2019). Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 179, 1330–1341 e1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, et al. (2014). Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 343, 72–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngan CY, Wong CH, Tjong H, Wang W, Goldfeder RL, Choi C, He H, Gong L, Lin J, Urban B, et al. (2020). Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat Genet 52, 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen J, Rodland EA, Holden L, Holden M, and Hovig E (2014). A statistical model of ChIA-PET data for accurate detection of chromatin 3D interactions. Nucleic Acids Res 42, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repana D, Nulsen J, Dressler L, Bortolomeazzi M, Venkata SK, Tourna A, Yakovleva A, Palmieri T, and Ciccarelli FD (2019). The Network of Cancer Genes (NCG): a comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol 20, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanborn JZ, Salama SR, Grifford M, Brennan CW, Mikkelsen T, Jhanwar S, Katzman S, Chin L, and Haussler D (2013). Double minute chromosomes in glioblastoma multiforme are revealed by precise reconstruction of oncogenic amplicons. Cancer Res 73, 6036–6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman L, Chen H, Brown M, Wangsa D, Patterson G, Camps J, Omenn GS, Ried T, and Rajapakse I (2017). Nucleome Analysis Reveals Structure-Function Relationships for Colon Cancer. Mol Cancer Res 15, 821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seim I, Jeffery PL, Thomas PB, Nelson CC, and Chopin LK (2017). Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3 (Bethesda) 7, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, and Cavalli G (2015). The role of chromosome domains in shaping the functional genome. Cell 160, 1049–1059. [DOI] [PubMed] [Google Scholar]
- Spielmann M, Lupianez DG, and Mundlos S (2018). Structural variation in the 3D genome. Nat Rev Genet 19, 453–467. [DOI] [PubMed] [Google Scholar]
- Spriggs AI, Boddington MM, and Clarke CM (1962). Chromosomes of human cancer cells. Br Med J 2, 1431–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. (2015). CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163, 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian SZ, Capurso D, Kim M, Lee B, Zheng M, and Ruan Y (2019). ChIA-DropBox: a novel analysis and visualization pipeline for multiplex chromatin interactions. bioRxiv 10.1101/613034. [DOI] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RGW, Bafna V, and Mischel PS (2019). Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat Rev Cancer 19, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, and Michor F (2016). A computational strategy to adjust for copy number in tumor Hi-C data. Bioinformatics 32, 3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Ding L, Chang TC, Shao Y, Chiang J, Mulder H, Wang S, Shaw TI, Wen J, Hover L, et al. (2019). Structure and evolution of double minutes in diagnosis and relapse brain tumors. Acta Neuropathol 137, 123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, Chaudhary N, Riely GJ, Li BT, Scott K, et al. (2017). An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med 23, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Tian SZ, Capurso D, Kim M, Maurya R, Lee B, Piecuch E, Gong L, Zhu JJ, Li Z, et al. (2019). Multiplex chromatin interactions with single-molecule precision. Nature 566, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Defined ecDNA regions, ecDNA copy number and sequencing summary, Related to Figure 1.
Table S2. Statistics summary of copy number adjusted trans and cis interaction frequency analysis, Related to Figures 1-3.
Table S3. H3K27ac enrichment, super-enhancer (SE) enrichment, and RNA expression associated with ecDNA, ecDNA connected anchors and genes, Related to Figures 4-5.
Table S4. Primers used and gene significantly differentially expressed in synthetic circular enhancer assay, Related to Figure 5.
Table S5. EcMYC associated complexes and genes detected by ChIA-Drop in PC3 ecDNA (+) line, Related to Figure 6.
Data Availability Statement
All data described in this study are being deposited in NCBI's Gene Expression Omnibus GSE124769. Whole-genome sequencing data for PC3 ecDNA (+) and ecDNA (−) cell lines were obtained from SRA SRR4009277 and SRR5263237, respectively. All software tools used in this study are listed in the STAR Methods description.