

Summary
The mechanisms that drive systemic lupus erythematosus (SLE) patients to achieve remission are unknown; one possible explanation might be T cell exhaustion. The aim of the present study was to measure CD4+ and CD8+ T cell exhaustion in SLE patients in prolonged remission (PR‐SLE) and compared them with patients with active SLE (Act‐SLE) and healthy subjects. We included 15 PR‐SLE patients, 15 Act‐SLE and 29 healthy subjects. T cell exhaustion was determined by flow cytometry according to the expression of programmed cell death 1 (PD)‐1, T cell immunoglobulin and mucin 3 (Tim‐3), natural killer cell receptor (2B4), eomesodermin (EOMES) and T‐box transcription factor TBX21 (T‐bet) in CD4+ and CD8+ T cells. Dimensionality reduction using the T‐distributed stochastic neighbor‐embedding algorithm and clustering analysis was used for the identification of relevant populations. Percentages of CD3+, CD4+ and CD8+ T cells were similar among groups. We identified five subpopulations of CD8+ and seven of CD4+ cells. The CD4+T‐bet+CD45RO+ cells identified in the unsupervised analysis were significantly increased in PR‐SLE versus Act‐SLE [median = 0·20, interquartile range (IQR) = 1·74–30·50 versus 1·68, IQR = 0·4–2·83; P < 0·01]. CD4+EOMES+ cells were also increased in PR‐SLE versus Act‐SLE (5·24, IQR = 3·38–14·70 versus 1·39, IQR = 0·48–2·87; P < 0·001). CD8+EOMES+ cells were increased in PR‐SLE versus Act‐SLE (37·6, IQR = 24·9–53·2 versus 8·13, IQR = 2·33–20·5; P < 0·001). Exhausted and activated T cells presented an increased frequency of PD‐1, CD57 and EOMES in SLE patients versus healthy subjects. Some subpopulations of T cells expressing markers associated with exhaustion are increased in patients in remission, supporting T cell exhaustion as a tolerance mechanism in SLE. Exhaustion of specific populations of T cells might represent a potential therapeutic tool that will contribute to the goal of achieving sustained remission in these patients.
Keywords: clinical remission in SLE, clustering analysis, T cell exhaustion
T cells expressing markers associated with exhaustion are increased in patients in remission, supporting T cell exhaustion as a tolerance mechanism in SLE.

Introduction
Systemic lupus erythematosus (SLE) is an autoimmune, multi‐factorial disease with broad tissue and organ involvement. Its manifestations are usually heterogeneous, with a fluctuating and unpredictable course [1]. Despite the advances in the treatment and understanding of its pathogenesis, disease activity continues to be one of the major causes of morbidity and mortality. Progressive accumulation of irreversible damage is attributed to chronic inflammation, as well as to the prolonged use of steroids and immunosuppressants [2]. Many studies conducted in recent years have focused on the search for treatments and interventions that reduce or prevent relapses in patients with SLE, with the goal of reaching a sustained remission and limiting damage [3, 4]. Despite the information provided by these studies, very little is known about the clinical and serological features that characterize the group of patients with SLE who achieve sustained remission and the cellular, genetic or molecular factors involved in this task.
T cell exhaustion is a complex process that has been increasingly recognized in recent years in patients with chronic infections and cancer. T cell exhaustion is driven by persistent T cell receptor (TCR) stimulation in the absence of enough co‐stimulation and is characterized by limited proliferation, cytokine production and effector function [5]. There is no universally accepted set of markers that define the population of exhausted T cells; however, it is recognized that they express a variety of inhibitory receptors that reduce their response after activation, such as programmed cell death 1 (PD‐1), T cell immunoglobulin and mucin 3 (Tim‐3) or 2B4 and differentially express transcription factors such as eomesodermin (EOMES) and T‐box transcription factor TBX21 (T‐bet) [6, 7].
There are few reports concerning the role of exhausted T cells in autoimmunity, but they have linked their presence with an improved prognosis in several autoimmune diseases [8]. There is also evidence of an association of exhausted T cells and increased immune tolerance in transplantation [9]. Because of this potentially favorable effect of the exhaustion process in autoimmunity, we conducted this study with the aim of measuring CD4+ and CD8+ T cell exhaustion by quantifying surface inhibitory molecules and transcriptional factors by flow cytometry in SLE patients in prolonged remission and to compare them with active SLE patients and healthy subjects.
Material and methods
Patients and controls
This was a cross‐sectional study conducted at the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, a tertiary care center in Mexico City. We included 15 patients with SLE in long‐term remission without treatment (PR‐SLE). Prolonged remission was defined as a clinical SLE Disease Activity Index (SLEDAI‐2K) of 0 (regardless of serological activity, i.e. anti‐dsDNA antibodies and/or hypocomplementemia) and being free of glucocorticoids, immunosuppressive or anti‐malarial treatment for at least 10 years [10]. As controls, we included 15 patients with clinically active SLE (Act‐SLE) with a clinical SLEDAI‐2K ≥ 3 (excluding hypocomplementemia and high titers of anti‐dsDNA) and 29 healthy subjects (HS). All the healthy controls were confirmed negative for anti‐nuclear antibodies and with no personal or familial history of autoimmune diseases. All patients with SLE met the Systemic Lupus International Collaborating Clinics (SLICC) 2012 classification criteria [11].
Flow cytometry
One sample of peripheral blood was obtained from each patient according to standard procedures. Ethylenediamine tetraacetic acid (EDTA)‐treated blood samples were analyzed by eight‐color flow‐cytometry (Becton Dickinson Canto II Cytometer; Becton Dickinson, Franklin Lakes, NJ, USA) using several combinations of fluorescence‐labeled antibodies distributed in the following panels: panel 1, CD3 fluorescein isothiocyanate (FITC), CD4 allophycocyanin/cyanin 7 (APC/Cy7), CD25 Brilliant Violet 421 (BV421), CD127 APC and forkhead box protein 3 (FoxP3) phycoerythrin (PE); panel 2, CD3 peridinin chlorophyll (PerCP)/Cy5, CD4 APC/Cy7, CD8 Alexa Fluor 488, human leucocyte antigen (HLA)‐DR PE, CD244 (2B4) APC, CD279 (PD‐1) BV421, CD38 Brilliant violet 510 (BV510) and PE/Cy7 CD366 (Tim‐3); panel 3, CD3 PE/Cy7, CD4 PE/Cy5, CD8 BV510, CD27 APC/Cy7, CD57 APC, CD45RO FITC, T‐box transcription factor TBX21 (T‐bet) BV421 and eomesodermin (EOMES) PE; and panel 4, CD3 APC, CD4 BV510, CD8 PE, CD45RA FITC, CD197 APC/Cy7. All antibodies were purchased from Biolegend Inc. (San Diego, CA, USA) except EOMES PE (eBioscience, San Diego, CA, USA). Briefly, 250 µl of blood was incubated with fluorochrome‐conjugated antibodies for 30 min at room temperature prior to lysis with red blood cell lysis buffer (Biolegend, Inc., San Diego, CA, USA) and fixation with 3% formaldehyde in phosphate‐buffered saline (PBS). After surface staining, cells were fixed and permeabilized using the True‐Nuclear Transcription Factor Buffer Set (Biolegend, Inc.) for 60 min at room temperature. Incubation with antibodies for T‐bet, EOMES and FoxP3 was performed for 30 min at room temperature followed by washing and acquisition.
OneFlow™ Setup Beads (BD Biosciences) were used to adjust instrument settings, set fluorescence compensation and check instrument sensitivity. Fluorescence minus one controls were used to determine positive and negative staining boundaries for each fluorochrome. Two hundred and fifty thousand events were recorded for each sample and analyzed with FlowJo® software version 10.7.1 (FlowJo, LLC; TreeStar, Ashland, OR, USA). FlowAI plugin was used to perform an automatic or interactive quality control on the .fcs data [12]. Lymphocyte population was identified in a side‐scatter (SSC)‐A versus forward scatter (FSC)‐A plot, followed by doublets exclusion in a FSC‐height (FSC‐H) × FSC‐area (FSC‐A) scatter‐plot. Phenotypes used to define mayor and minor T cell populations are depicted in Table 1.
Table 1.
CD4+ and CD8+ memory subpopulations and Tregs
| Healthy subjects | Active SLE | PR‐SLE | P | ||
|---|---|---|---|---|---|
| n = 29 | n = 15 | n = 15 | |||
| CD3+ | % | 60·8 (56·2–69·2) | 68·0 (53·7–78·2) | 67·7 (61·4–74·4) | 0·23 |
| CD3+CD4+ | % | 35·4 (33·5–43·1) | 39·4 (25·8–46·7) | 39·0 (31·4–46·6) | 0·92 |
| Naive (CD45RA+CD197+) | % | 27·9 (19·4–33·1) | 23·2 (7·98–40·2) | 19·3 (11·3–32·5) | 0·28 |
| CM (CD45RA−CD197+) | % | 25·3 (16·1–34·6) | 26·5 (16·2–44·5) | 40·7 (22·1–45·3) | 0·04†† |
| EM (CD45RA−CD197−) | % | 40·9 (31·2–53·0) | 39·6 (36·2–62·7) | 39·9 (26·9–47·5) | 0·66 |
| TEMRA (CD45RA+CD197−) | % | 2·26 (1·40–4·26) | 2·63 (0·78–4·09) | 0·92 (0·66–2·14) | 0·11 † |
| CD25+ | % | 9·81 (5·68–14·8) | 11·2 (8·27–22·4) | 15·3 (9·13–32·0) | 0·09 † |
| CD127low | % | 5·19 (4·20–6·83) | 5·42 (4·96–6·85) | 5·74 (4·29–9·15) | 0·88 |
| CD25+CD127lowFoxP3+ | % | 1·16 (0·38–2·13) | 0·54 (0·23–1·52) | 1·13 (0·25–1·79) | 0·37 |
| CD3+CD8+ | % | 18·4 (14·8–21·9) | 18·9 (13·4–22·3) | 19·0 (12·5–25·2) | 0·95 |
| Naive (CD45RA+CD197+) | % | 21·5 (14·2–25·5) | 24·9 (15·3–51·7) | 14·7 (8·59–24·4) | 0·04 ‡ |
| CM (CD45RA−CD197+) | % | 3·13 (1·71–5·67) | 2·88 (1·42–6·45) | 8·73 (3·74–10·3) | 0·01 ‡ |
| EM (CD45RA−CD197−) | % | 41·4 (36·1–45·2) | 27·8 (18·0–38·8) | 38·0 (28·8–50·0) | 0·04 * |
| TEMRA (CD45RA+CD197−) | % | 33·8 (24·0–38·8) | 34·1 (19·0–45·0) | 35·2 (24·4–46·6) | 0·95 |
Results are shown as median with interquartile range and the Kruskal–Wallis test was used to perform comparisons between groups. The Mann–Whitney U‐test was performed to compare between two groups as a post‐hoc test and is represented by the following symbols. CM = central memory; EM = effector memory; TEMRA = effector memory T cells re‐expressing CD45RA. Treg = regulatory T cell; SLE = systemic lupus erythematosus; FoxP3 = forkhead box protein 3. Significant P values are in bold type.
Healthy subjects versus active (Act)‐SLE. * P < 0·05.
Healthy subjects versus prolonged remission (PR)‐SLE. † P < 0·05, †† P < 0·01.
Act‐SLE versus PR‐SLE. ‡ P < 0·05.
Dimensionality reduction and clustering analysis
After excluding death cells and doublets, the resulting flow cytometry data files were gated on the CD3+ population. Individual .fcs files from each subject from all groups (PR‐SLE, Act‐SLE and HS) were down‐sampled to equal cell numbers and concatenated in a single file. Barnes‐Hut implementation of t‐distributed stochastic neighbor‐embedding (tSNE) was performed in FlowJo version 10.7.1 with 1000 iterations, perplexity constant of 30 and learning rate of 45 767 [13, 14] to reduce dimensionality of the data followed by self‐organized maps (FlowSOM) [15] for unsupervised clustering of the data into eight subpopulations. tSNE plots were generated using the following T cell markers: CD3 PerCP/Cy5, CD4 APC/Cy7, CD8 Alexa Fluor 488, HLA‐DR PE, 2B4APC, PD‐1 BV421, CD38 BV510 and PE/Cy7 Tim‐3 (panel 2) and CD3 PE/Cy7, CD4 PE/Cy5, CD8 BV510, CD27 APC/Cy7, CD57 APC, CD45RO FITC, T‐bet BV421 and EOMES PE (panel 3). The resulting subpopulations were further analyzed for specific marker expression and frequency in each group. We also compared populations that have been reported in the literature and that were manually gated independently of the findings of the unsupervised clustering. These populations were: CD4/CD8 PD‐1+, CD4/CD8 T‐bet+, CD4/CD8 2B4, CD4/CD8 Tim‐3, CD4/CD8 EOMES, CD4/CD8 CD57, CD4/CD8 CD38+HLA‐DR+ (activated T cells) and CD4/CD8 CD38+HLA‐DR+PD‐1+ (exhausted T cells).
Statistical analysis
For descriptive statistics, continuous data are presented as median with interquartile ranges (IQR; 25–75). For comparisons of quantitative variables between two groups, the Mann–Whitney U‐test was used, while the Kruskal–Wallis test followed by Dunn’s test was used to compare three or more groups. Categorical values are presented as number and percentage. The χ2 test was used to compare categorical variables between groups. The statistical analysis was performed using spss software version 21.0 (IBM, Armonk, NY, USA). P‐value ≤ 0·05 was considered significant.
The study was approved by the Institutional Review Board of the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (Ref. 2232). All subjects gave their informed consent to participate according to the Declaration of Helsinki.
Results
Clinical and demographic characteristics
Of the 15 patients with PR‐SLE, 14 (93.3%) were women, with a median age of 49 years (48–60). All Act‐SLE patients were women with a median age of 33 years (30–46) and 28 (97%) of the healthy subjects were women, median age 43 years (32–52). Patients with Act‐SLE presented lymphopenia, with lower lymphocyte counts when compared with PR‐SLE and healthy controls [1·12 × 103/µl (0·74–1·71) versus 1·67 × 103 (1·23–2·76) versus 2·17 × 103 (1·61–2·41), P = 0·030]. The demographic and clinical characteristics at the time of recruitment are detailed in Table 2.
Table 2.
Demographic and laboratory characteristics of prolonged remission SLE patients, active SLE and healthy subjects
| Demographics | Healthy subjects | Active SLE | PR‐SLE | P |
|---|---|---|---|---|
| n = 29 | n = 15 | n = 15 | ||
| Sex | 0·61 | |||
| Female (%) | 28 (96·6) | 15 (100) | 14 (93·3) | |
| Age (years) | 43 (32–52) | 33 (30–46) | 49 (48–60) | < 0·01 |
| Body mass index | 24·74 (23·66–26·37) | 23·12 (20·57–25·43) | 24·45 (21·3–32·88) | 0·47 |
| Smoker (%) | 9 (31) | 6 (40) | 5 (33·3) | 0·70 |
| Laboratory characteristics | ||||
| Positive ANA (%) | 0 (0) | 14 (93·3) | 15 (100) | < 0·001 |
| ESR | 7·5 (4–10) | 5 (2–12) | 3 (2–5) | 0·15 |
| Positive ESR (%) | 0 (0) | 1 (6·7) | 1 (6·7) | 0·35 |
| Hemoglobin (g/dl) | 14·4 (14·1–15·2) | 13·5 (12·6–14·7) | 14·5 (12·6–15·7) | 0·10 |
| Hematocrit (%) | 44·9 (43·1–45·6) | 40·6 (37·4–44·2) | 42·1 (37·6–45·6) | 0·01 |
| Leukocytes (×103) | 6·3 (5·6–6·9) | 5·7 (4·1–9·2) | 5·3 (4·0–6·9) | 0·30 |
| Neutrophils (×103) | 3·84 (3·29–4·27) | 4·04 (2·94–6·10) | 3·36 (1·78–3·82) | 0·04 |
| Lymphocytes (×103) | 2,17 (1·61–2·41) | 1·12 (0·74–1·71) | 1·67 (1·23–2·76) | 0·03 |
| Platelets (K/µl) | 260·0 (235·0–289·0) | 263·0 (237·0–283·0) | 217·0 (150·0–256·0) | < 0·01 |
ANA = anti‐nuclear antibodies; ESR = erythrocyte sedimentation rate; n.a. = not applicable.
Results for quantitative variables are shown as median with interquartile range, and the Kruskal–Wallis test was used to perform comparisons between groups. For quantitative variables measured only in two groups, the Mann–Whitney U‐test was used to perform comparisons between both groups. The qualitative variables are presented in frequencies, using the χ2 test to compare between groups. The Mann–Whitney U‐test was performed to compare between two groups as a post‐hoc test, in which prolonged remission (PR)‐systemic lupus erythematosus (SLE) patients are older than active SLE patients (P = 0·001), and older than the healthy subjects (P = 0·017). Significant P values are in bold type.
No differences were found between in the SLICC 2012 classification criteria at diagnosis between both groups of patients. At inclusion, disease duration in PR‐SLE patients was 25·6 years (19·1–35·4) versus 9·25 (5·75–12·5) in Act‐SLE patients (P < 0·001). The median time in remission was 16·9 years (12·1–20·6). The PR‐SLE group had a SLEDAI‐2K score of 0 (0–2) versus 8 (6–16) in the Act‐SLE group (P = 0·001). The clinical manifestations, activity indices and accumulated damage and treatment of active patients are described in Table 3.
Table 3.
SLICC 2012 criteria, SLEDAI‐2K, accrual damage and treatment in active and prolonged remission SLE patients
| Active SLE | PR‐SLE | P | |
|---|---|---|---|
| n = 15 | n = 15 | ||
| Duration of SLE (years) | 9·25 (5·75–12·5) | 25·6 (19·1–35·4) | < 0·001 |
| Remission time (years) | n.a. | 16·9 (12·1–20·6) | n.a. |
| SLICC 2012 SLE classification criteria items | 7 (6–9) | 7 (6–8) | 0·50 |
| Acute cutaneous lupus | 12 (80) | 10 (66·7) | 0·63 |
| Chronic cutaneous lupus | 1 (6·7) | 0 (0) | 0·55 |
| Nasal or oral ulcers | 6 (40) | 9 (60) | 0·39 |
| Alopecia | 2 (13·3) | 4 (26·7) | 0·62 |
| Synovitis | 13 (86·7) | 13 (86·7) | 0·92 |
| Serositis | 2 (13·3) | 3 (20) | 0·82 |
| Renal | 6 (40) | 4 (26·7) | 0·50 |
| Neurological | 0 (0) | 3 (20) | 0·18 |
| Autoimmune hemolytic anemia | 1 (6·7) | 1 (6·7) | 0·96 |
| Leuko‐lymphopenia | 11 (73·3) | 12 (80) | 0·18 |
| Thrombocytopenia | 3 (20) | 3 (20) | 0·87 |
| Positive ANA | 14 (93·3) | 15 (100) | 0·55 |
| Positive anti‐dsDNA | 15 (100) | 11 (73·3) | 0·04 |
| Positive anti‐Smith | 4 (26·7) | 0 (0) | 0·01 |
| Anti‐phospholipid antibodies | 8 (53·3) | 6 (40) | 0·69 |
| Hypocomplementemia | 12 (80) | 9 (60) | 0·38 |
| Positive Coombs test | 4 (26·7) | 0 (0) | 0·06 |
| SLEDAI 2K | 8 (6–16) | 0 (0–2) | < 0·001 |
| Arthritis | 7 (46·6) | 0 (0) | 0·001 |
| Hematuria | 4 (26·6) | 0 (0) | 0·01 |
| Proteinuria | 8 (53·3) | 0 (0) | < 0·001 |
| Pyuria | 5 (33·3) | 0 (0) | < 0·01 |
| Low complement | 11 (73·3) | 7 (46·6) | 0·23 |
| Increased anti‐dsDNA titers | 12 (80·0) | 5 (33·3) | 0·02 |
| Fever | 1 (6·6) | 0 (0) | 0·45 |
| SLEDAI 2K serological items | |||
| C3 (mg/dl) | 81 (67–128) | 113 (79–128) | 0·24 |
| Low C3 (%) | 10 (66·7) | 4 (26·7) | 0·07 |
| C4 (mg/dl) | 11 (8–20) | 19 (12–30) | 0·10 |
| Low C4 (%) | 10 (66·7) | 7 (46·7) | 0·42 |
| Anti‐dsDNA (UI/ml) | 225·1 (20·2–586·9) | 7·2 (4·5–12·8) | < 0·01 |
| SLICC/ACR DI | 0 (0–1) | 1 (0–1) | 0·10 |
| Secondary APS | 2 (13·3) | 6 (40) | 0·36 |
| Treatment | 15 (100) | 0 (0) | n.a. |
| Prednisone | 7 (46·7) | n.a. | n.a. |
| Dose (mg) | 15 (6·25–27·5) | n.a. | n.a. |
| Immunosuppressor | 13 (86·6) | n.a. | n.a. |
| Azathioprine | 5 (33·3) | n.a. | n.a. |
| Methotrexate | 4 (26·7) | n.a. | n.a. |
| Mycophenolate mofetil | 4 (26·7) | n.a. | n.a. |
| Anti‐malarial | 10 (73·3) | n.a. | n.a. |
ANA = anti‐nuclear antibodies; n.a. = not applicable; SLE = systemic lupus erythematosus; PR = prolonged remission; SLEDAI = SLE Disease Activity Index; SLICC = Systemic Lupus International Collaborating Clinics; SLICC/ACR DI = Systemic Lupus International Collaborating Clinics/American College of Rheumatology Damage Index; APS = anti‐phospholipid syndrome. Results for quantitative variables are shown as median with interquartile range and the Kruskal–Wallis test was used to perform comparisons between groups. For quantitative variables measured only in two groups, the Mann–Whitney U‐test was used to perform comparisons between both groups. The qualitative variables are presented in frequencies using the c2 test to compare between groups.
Only the SLEDAI 2K items that were present in at least one study subject from either SLE group are shown.
Prednisone dose is shown from the seven active SLE patients who had it prescribed. Significant P values are in bold type.
Major lymphocyte populations
There were no differences in the relative percentage of CD3+, CD4+ or CD8+ T cells between groups. Among the subpopulations of CD4+ T cells, only the central memory compartment was significantly different between groups, being expanded in the PR group compared with the other two. More differences were observed in the subpopulations of CD8+ T cells. Similar to that observed within CD4+ T cells, patients in PR showed an increased central memory compartment and a concomitant reduction in the percentage of naive CD8+ cells when compared with the other groups. Meanwhile, patients with Act‐SLE presented a decreased percentage of CD8+ effector memory cells. The percentages of the subpopulations analyzed and the P‐values for the comparisons are described in Table 1.
T cell exhaustion markers and populations
As there is no clear consensus and little information about the specific marker combination that describes specific subpopulations of exhausted T cells in patients with SLE, we initially used an unsupervised approach to explore the behavior and clustering of surface markers and transcription factors that have been reported as relevant in exhausted T cells (see Methods for the complete list of markers included). As a first step, we generated tSNE maps using concatenated files with an equal number of CD3+ cells from all the patients included in each group. Figure 1a,b shows the tSNE plots for each group and the distribution of the included markers across the plots. In order to identify the clustering of these markers in subpopulations, we used a self‐organizing maps algorithm [15]. We could identify five subpopulations of CD8+, seven CD4+ cells and three CD3+CD4−CD8−. The heat‐maps and tables showing the relative expression of markers in each cluster across all the identified subpopulations are shown in Fig. 1c. Overlapping of these populations on the tSNE plots is shown in Fig. 1d. Histograms represent the relative expression of a CD3+CD8+ cell population that seems to be exclusive to the PR‐SLE group in both panels II and III (2·86% and 3·75%, respectively), and which expresses significantly transcription factors and surface markers associated with senescence and depletion.
Fig. 1.
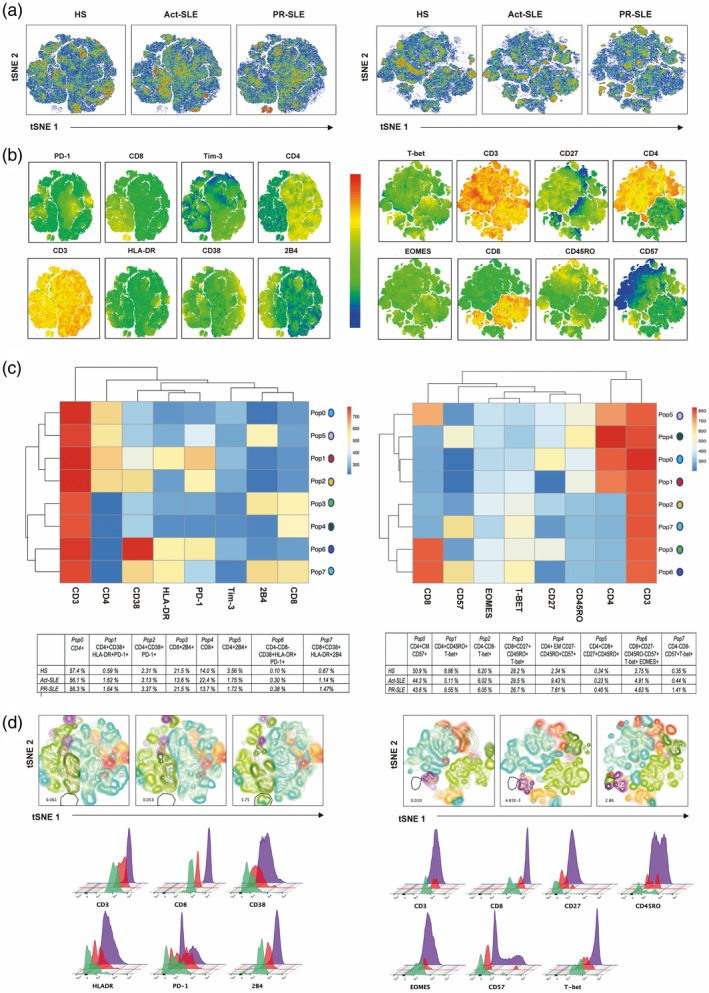
Dimensional reduction analysis and unsupervised clustering of subpopulations of CD3+ T cells using markers associated with cell exhaustion in panels 2 and 3 (described previously in methods). (a) T‐distributed stochastic neighbor‐embedding (tSNE) plots for each study group [healthy subjects (HS), active systemic erythematosus (Act‐SLE) and prolonged remission (PR)‐SLE]. Individual cell subsets of CD3+ cells from each subject in each group were randomly down‐sampled to an equal number of events and concatenated in a single file. The following T cell markers were used to generate tSNE plots: CD3 peridinin chlorophyll/cyanin 5 (PerCP/Cy5), CD4 allophycocyanin (APC)/Cy7, CD8 Alexa Fluor 488, human leukocyte antigen phycoerythrin (HLA‐DR PE), CD244 (2B4) APC, CD279 [programmed cell death 1 (PD‐1)] BV421, CD38 BV510 and PE/Cy7 CD366 [T cell immunoglobulin mucin‐3 (Tim‐3)] (panel 2) and CD3 PE/Cy7, CD4 PE/Cy5, CD8 BV510, CD27 APC/Cy7, CD57 APC, CD45RO fluorescein isothiocyanate (FITC), T‐box transcription factor TBX21 (T‐bet BV421) and eomesodermin (EOMES) PE (panel 3). (b) Distribution and relative expression of selected markers in T cells present in both panels are shown in tSNE plots. (c) Heat‐maps of selected markers using the self‐organizing maps (FlowSOM) algorithm according to the fluorescence intensity of the markers by flow cytometry. Eight clusters were identified and named Pop 0 to Pop 7). The heat‐map shows the relative fluorescence intensity and hierarchical clustering of each of the markers included in the subpopulations generated. Markers are presented as well as percentage relative to CD3+ cells are shown in tables below. (d) Distribution of clusters identified in (c) overlaid on the t‐SNE plots in each study group. In both panels, a CD8+ subpopulation that expresses exhaustion and senescence markers was identified exclusively in the PR‐SLE group. Overlaid histograms (HS: green; Act‐SLE: red; PR‐SLE: purple) below show relative frequencies of most abundant markers.
Populations differentially represented in the PR when compared with the Act‐SLE group
Within the CD4+ compartment, a population of T‐bet+CD45RO+ cells identified in the unsupervised analysis, which represents 10·20% (1·74–30·50) of the CD4+ cells in the PR group, was significantly increased compared with the Act‐SLE (1·68, IQR = 0·42–2·83) and HS (4.08, IQR = 1.64–5.48). The CD4+EOMES+ cells were also increased in the PR group compared with Act‐SLE and HS. The other population that was significantly different between PR and Act‐SLE was the CD4+Tbet+ cells, which were increased in the PR group and were similar to the HS.
Within the CD8+ T cells, the proportion of PD1+ cells was apparently increased only in the PR group, even though the difference was only significant when compared with the HS. The CD8+2B4+ cells, identified through the unsupervised clustering, were increased in the PR versus the Act‐SLE group. The most striking difference was observed in the EOMES+ cells, with a median of 37·6 (24·9–53·2) in the PR group versus 12·9 (6·78–24·8) in the HS and 8·13 (2·33–20·5) in the Act‐SLE groups.
Differences between SLE patients and healthy subjects
We observed a significant increase in the frequency of expression of cell activation and exhaustion markers in both the CD4+ and the CD8+ compartment between the SLE patients and the HS group. The T cell subpopulations in SLE patients presented an increase in the frequency of PD‐1, CD57, EOMES and exhausted and activated populations when compared with the HS group. Table 4 and Fig. 2 show all the populations that were identified as differentially expressed between groups.
Table 4.
T cell exhaustion in CD4+ and CD8+ lymphocytes
| Healthy subjects | Active SLE | PR‐SLE | P | ||
|---|---|---|---|---|---|
| n = 29 | n = 15 | n = 15 | |||
| CD4+ | |||||
| PD‐1+ | % | 31·40 (24·80–40·60) | 43·40 (32·10–52·30) | 42·90 (31·80–63·60) | < 0·01 * † |
| Tim‐3+ | % | 17·90 (11·60‐32·75) | 11·90 (7·99‐53·40) | 7·85 (3·18‐32·30) | 0·13 |
| CD38+HLA‐DR+ (activated) | % | 4·01 (2·56–5·72) | 6·86 (5·09–12·1) | 8·18 (4·66–10·5) | < 0·01***††† |
| POP 1 CD38 + HLA‐DR + PD‐1 + | % | 1·02 (0·62–1·92) | 3·48 (1·59–4·20) | 2·28 (1·47–3·96) | < 0·001***††† |
| POP 2 CD38 + PD‐1 + | % | 9·09 (5·63–13·25) | 15·20 (6·78–24·60) | 14·30 (8·57–24·10) | 0·01 † |
| POP 5 2B4 + | % | 2·79 (0·85–8·69) | 5·27 (2·47–11·10) | 5·05 (3·02–7·97) | 0·10 |
| CD57+ | % | 3·13 (1·79–6·25) | 4·19 (3·66–8·57) | 5·22 (2·12–10·4) | 0·24 † |
| EOMES+ | % | 1·59 (0·87–2·49) | 1·39 (0·48–2·87) | 5·24 (3·38–14·70) | < 0·001 † ††† |
| T‐bet+ | % | 4·93 (2·28–8·32) | 1·69 (1·30–4·02) | 5·05 (3·23–9·16) | 0·01 ‡ |
| POP 0 CM CD57 + | % | 3·90 (1·42–6·58) | 4·90 (1·45–39·60) | 12·00 (5·41–35·60) | 0·01 † |
| POP 1 CD45RO + T‐bet + | % | 4·08 (1·64–5·48) | 1·68 (0·42–2·83) | 10·20 (1·74–30·50) | < 0·01 † ‡‡ |
| POP 4 EM CD27‐CD45RO + | % | 2·36 (1·32–4·38) | 2·15 (0·45–12·50) | 3·16 (1·29–7·42) | 0·57 |
| CD8+ | |||||
| PD‐1+ | % | 27·9 (23·9–35·4) | 27·4 (13·6–42·9) | 38·6 (30·3–49·1) | 0·02 † |
| Tim‐3+ | % | 3·16 (1·77–7·34) | 2·58 (2·23‐3·95) | 3·09 (0·64‐6·22) | 0·84 |
| CD38+HLA‐DR+ (activated) | % | 6·28 (2·72–8·07) | 9·38 (6·30–17·3) | 12·5 (8·47–18·7) | < 0·01 † |
| CD38+PD‐1+ | % | 6·24 (4·38–9·23) | 12·0 (8·26–26·9) | 13·6 (10·7–27·7) | < 0·0001**††† |
| CD38+HLA‐DR+PD‐1+ (exhausted) | % | 1·24 (0·88–1·94) | 4·69 (1·70–10·2) | 4·38 (2·09–9·19) | < 0·001***††† |
| POP 3 2B4 + | % | 58·9 (49·3–68·3) | 46·7 (27·1–60·8) | 68·4 (49·7–79·0) | 0·02 ‡ |
| POP 7 CD38+HLA‐DR+2B4+ | % | 2·23 (1·17–3·06) | 7·11 (3·66–11·80) | 8·97 (4·00–14·80) | < 0·0001**††† |
| CD57+ | % | 28·0 (21·5–36·6) | 38·0 (31·4–45·4) | 37·6 (26·2–52·4) | 0·02 ‡ |
| EOMES+ | % | 12·9 (6·78–24·8) | 8·13 (2·33–20·5) | 37·6 (24·9–53·2) | < 0·001 ‡ ‡‡‡ |
| T‐bet+ | % | 32·8 (18·9–44·2) | 26·4 (10·7–44·2) | 47·8 (23·0–70·1) | 0·09 ‡ |
| POP 3 CD27+CD45RO+T‐bet+ | % | 2·06 (1·00–6·02) | 2·70 (0·76–5·91) | 5·75 (3·51–10·10) | 0·02 † |
| POP 5 DP CD4 + CD8 + CD27 + CD45RO + | % | 0·15 (0·10–0·30) | 0·20 (0·13–0·36) | 0·28 (0·23–0·47) | 0·15 |
| POP 6 CD27‐CD45RO‐CD57 + T‐bet + EOMES + | % | 0·81 (0·37–3·06) | 1·42 (0·33–6·27) | 4·54 (0·88–9·78) | 0·02 † |
| POP 7 DN CD4‐CD8‐CD57 + T‐bet + | % | 0·23 (0·11–0·49) | 0·32 (0·07–0·53) | 0·37 (0·21–0·68) | 0·35 |
Bold type represents populations identified by traditional manual gating. POP = populations that were inferred from the FlowSOM heat‐map are shown in italics. HLA = human leukocyte antigen; T‐bet = T‐box transcription factor TBX21; EOMES = eomesodermin; Tim‐3 = T cell immunoglobulin and mucin 3.
Results are shown as median with interquartile range and the Kruskal–Wallis test was used to perform comparisons between groups.
The Mann–Whitney U‐test was performed to compare between two groups as a post‐hoc test and is represented by the following symbols.
Healthy subjects versus active systemic lupus erythematosus (Act‐SLE). * P < 0·05, ** P < 0·01, *** P < 0·001.
Healthy subjects versus prolonged remission (PR)‐SLE. † P < 0·05, †† P < 0·01, ††† P < 0·001.
Act‐SLE versus PR‐SLE. ‡ P < 0·05, ‡‡ P< 0·01, ‡‡‡ P < 0·001.
Fig. 2.
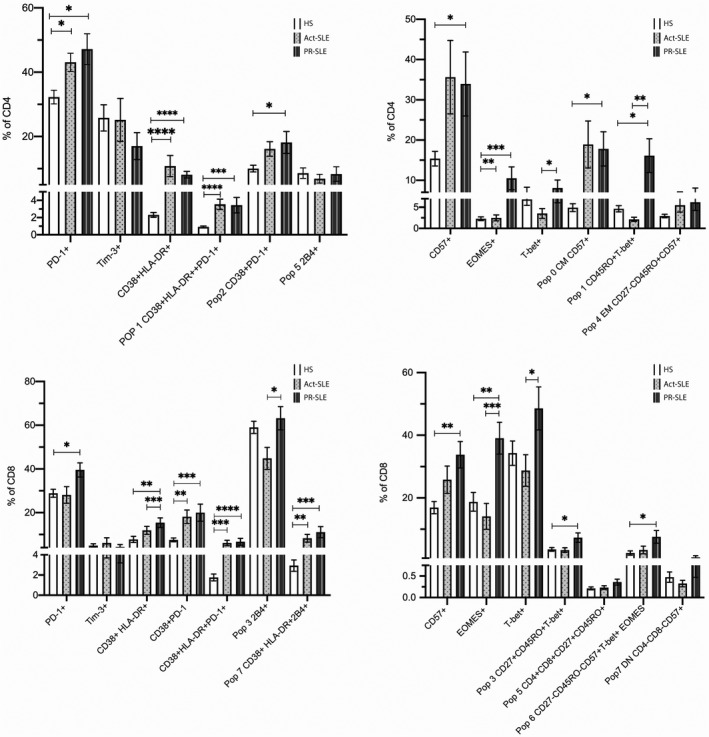
Bar graphs showing the frequencies (mean ± standard error of the mean (s.e.m.) of lymphocytes populations derived from traditional manual gating and self‐organizing maps (FlowSOM) clusters identified in the heatmap in panel (a). Mann–Whitney U‐test was performed to compare between two groups and differences are represented by the following symbols: * P < 0·05, ** P < 0·01, *** P < 0·001, **** P < 0·0001. HS (healthy subjects); Act‐SLE (active systemic lupus erythematosus); PR‐SLE (prolonged remission SLE).
Discussion
Remission in SLE implies the absence of major signs and symptoms, which translate to a lower probability of adverse outcomes when this state is sustained. In 2016, the conclusions from an international consensus of experts on the definitions of remission in SLE (DORIS) were published [16]. It was established that to catalog a patient in remission it is important to rely upon activity indices [such as SLEDAI‐2K, the British Isles Lupus Assessment Group (BILAG) or European Consensus Lupus Activity Measurements (ECLAM)]. The consensus also highlights the importance of differentiating patients in remission who are under treatment (low doses of glucocorticoids, maintenance immunosuppressants and/or biological maintenance) from those without treatment (or with anti‐malarials). All the patients classified and included as in remission in this study had a sustained clinical SLEDAI‐2K of 0 and were free of medication, including anti‐malarials for at least 10 years.
One of the strengths of this study is that the patients included were all totally free of treatment. This might be considered controversial, in view of the potential use of hydroxychloroquine. In the last two decades the notion has been consolidated that, unless there is a contraindication, all patients with SLE should receive this drug, mainly because it reduces the risk of relapse [17] and offers a beneficial effect on the lipid profile [18]. Most of our patients entered remission (and the decision of suspending the medication was taken) before the concept of relapse prevention with hydroxychloroquine was widely recognized [10]. The decision to restart a drug, when the patient has remained free of treatment and without clinical activity for too long, is not convincing for the patient, and there is no clear evidence supporting this conduct. The mechanism of action of anti‐malarials in SLE is complex. These drugs have a direct effect on lysosomal activity and signaling pathways. In antigen‐presenting cells, hydroxychloroquine interferes with antigen processing and presentation to T cells [19]. As mentioned, antigen presentation is a key step in the generation of exhausted T cells, and it is possible that anti‐malarials are able to modify this process. In this regard, it is an advantage that the population included in this study is free of these medications, as we were able to identify exhaustion without the confounder effect of therapy.
Patients with SLE, even after achieving long‐term remission, are not considered cured, and there is evidence confirming a subjacent autoimmune response: presence of autoantibodies or low complement [10] or relapses after long periods in remission (more than 10 years) [20]. How the altered immune response is kept under control in patients with lupus who achieve remission is not understood and is understudied. Our results support T cell exhaustion as a contributor in this process.
Using an unsupervised flow cytometric analysis, we found that multiple populations that express markers that have been associated with T cell exhaustion such as PD‐1, CD57 and EOMES are altered in patients with SLE when compared with HS. Remarkably, not all the populations with markers of exhaustion were different between SLE patients and HS, a finding consistent with previous reports suggesting that opposed to what occurs in models of chronic viral infection, patients with autoimmune disease have a selective up‐regulation of exhaustion associated receptors [8]. This phenomenon suggests that the signals that induce exhaustion in each condition are somehow context‐specific.
Exhausted T cells have a differential transcriptome and epigenome when compared with effector T cells [21, 22] and exhaustion is considered a stable differentiation fate of T cells, even though it can be reversed by therapeutic interventions. The presence of exhausted T cells in patients with an active autoimmune disease suggests that cells that are activated by an ongoing inflammatory response, or by the persistent presence of an autoantigen, cannot acquire co‐stimulatory signals above certain threshold, and are acquiring an exhausted phenotype [23]. The function of these cells is not completely known, but in patients with cancer exhausted cells in the tumor microenvironment contribute to the tumor immune evasion or induced tumor tolerance. This deleterious effect in cancer might have a positive implication in other diseases when tolerance has a positive connotation, as transplantation and autoimmunity [24].
In patients with autoimmune diseases, T cell exhaustion has been associated more with progression than with onset of the disease [8, 25]. In this regard, this is the first study, to our knowledge, that identifies specific subpopulations of exhausted T cells in patients with lupus in long‐term remission.
The mechanisms underlying the hyporesponsive state of exhausted cells are also subjects of study. We found an increased frequency of CD4 and CD8 EOMES+ cells in patients in remission. This transcription factor, a member of the T‐bet family, has also been described as altered in chronic viral infection [26] and cancer [27]. Remarkably, in patients with cancer, EOMES is necessary for the induction of cytotoxic anti‐tumor T cells; however, the deletion of one allele of this transcript factor reduced the number of exhausted T cells and improved tumor control [27]. This dual effect emphasizes the fine tuning involved in the generation of an exhausted versus effector response in T cells. A deeper understanding of the mechanisms that induce exhaustion of specific populations of T cells that are pathogenic in patients with SLE might represent a potential therapeutic tool that will contribute to the goal of achieving sustained remission.
There are some limitations in this study. The study is transversal; however, to identify and study longitudinally patients in long‐term remission free of medication before they achieve this status is probably not feasible. We believe that this group of patients provides an ideal group for this analysis, as there is no confounding effect caused by medications and there is no recent evidence of disease activity. We studied a relatively reduced number of markers associated with exhaustion; however, these were carefully selected and allowed us to identify populations that were differentially present in the groups of interest.
Our results confirm the presence of certain subpopulations of exhausted T cells in patients with JJ lupus that are unequivocally in remission. This suggests a potential role of T cell exhaustion as a mechanism of tolerance in patients with SLE. The populations identified in the present study warrant further investigation both as prognostic markers in longitudinal studies of these patients and in independent cohorts. The identification of the specific mechanisms behind the origin of these populations might provide new therapeutic options in SLE.
Disclosures
The authors declare no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Author contributions
G. L. and F. T.‐T. performed immunological experiments and analyzed results. Y. A.‐F. and L. L. conceived and supervised experiments, analyzed results and wrote the manuscript. H. F.‐L. supervised and analyzed clinical data and results. J. J.‐O. analyzed clinical data, analyzed results and wrote the manuscript.
Acknowledgements
This research was supported by a grant from the Consejo Nacional de Ciencia y Tecnología‐Fondo Sectorial de Investigación en Salud (CONACYT‐FOSIS), Grant No.289732, México. This report was the Master’s in Science degree thesis of Francisco Treviño‐Tello, Universidad Anáhuac del Norte, México.
Contributor Information
H. Fragoso‐Loyo, Email: jjakez_ocampo@yahoo.com, Email: hfragosol@gmail.com.
J. Jakez‐Ocampo, Email: jjakez_ocampo@yahoo.com, Email: hfragosol@gmail.com.
Data availability statement
The data that support the findings of this study are available from the corresponding author (J. J.‐O.) upon reasonable request.
References
- 1. Tsokos GC. Autoimmunity and organ damage in systemic lupus erythematosus. Nat Immunol 2020; 21:605–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sutton EJ, Davidson JE, Bruce IN. The systemic lupus international collaborating clinics (SLICC) damage index: a systematic literature review. Semin Arthritis Rheum 2013; 43:352–61. [DOI] [PubMed] [Google Scholar]
- 3. Urowitz MB, Feletar M, Bruce IN, Ibañez D, Gladman DD. Prolonged remission in systemic lupus erythematosus. J Rheumatol 2005; 32:1467–72. [PubMed] [Google Scholar]
- 4. Zen M, Iaccarino L, Gatto M et al. Prolonged remission in Caucasian patients with SLE: prevalence and outcomes. Ann Rheum Dis 2015; 74:2117–22. [DOI] [PubMed] [Google Scholar]
- 5. Wherry EJ. T cell exhaustion. Nat Immunol 2011; 12:492–9. [DOI] [PubMed] [Google Scholar]
- 6. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao Y, Shao Q, Peng G. Exhaustion and senescence: two crucial dysfunctional states of T cells in the tumor microenvironment. Cell Mol Immunol 2020; 17:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC. T‐cell exhaustion, co‐stimulation and clinical outcome in autoimmunity and infection. Nature 2015; 523:612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shahbazi M, Soltanzadeh‐Yamchi M, Mohammadnia‐Afrouzi M. T cell exhaustion implications during transplantation. Immunol Lett 2018; 202:52–8. [DOI] [PubMed] [Google Scholar]
- 10. Jakez‐Ocampo J, Rodriguez‐Armida M, Fragoso‐Loyo H, Lima G, Llorente L, Atisha‐Fregoso Y. Clinical characteristics of systemic lupus erythematosus patients in long‐term remission without treatment. Clin Rheumatol 2020; 39:3365–71. [DOI] [PubMed] [Google Scholar]
- 11. Petri M, Orbai A‐M, Alarcón GS et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012; 64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Monaco G, Chen H, Poidinger M, Chen J, de Magalhães JP, Larbi A. FlowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 2016; 32:2473–80. [DOI] [PubMed] [Google Scholar]
- 13. van der Maaten LJP, Hinton GE. Visualizing high‐dimensional data using t‐SNE. J Mach Learn Res 2008; 9:2579–605. [Google Scholar]
- 14. Belkina AC, Ciccolella CO, Anno R, Halpert R, Spidlen J, Snyder‐Cappione JE. Automated optimized parameters for t‐distributed stochastic neighbor embedding improve visualization and allow analysis of large datasets. Nat Commun 2019; 28:5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Gassen S, Callebaut B, Van Helden MJ et al. FlowSOM: using self‐organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015; 87:636–45. [DOI] [PubMed] [Google Scholar]
- 16. van Vollenhoven R, Voskuyl A, Bertsias G et al. A framework for remission in SLE: consensus findings from a large international task force on definitions of remission in SLE (DORIS). Ann Rheum Dis 2017; 76:554–61. [DOI] [PubMed] [Google Scholar]
- 17. Ponticelli C, Moroni G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin Drug Saf 2017; 16:411–9. [DOI] [PubMed] [Google Scholar]
- 18. Durcan L, Winegar DA, Connelly MA, Otvos JD, Magder LS, Petri M. Longitudinal evaluation of lipoprotein variables in systemic lupus erythematosus reveals adverse changes with disease activity and prednisone and more favorable profiles with hydroxychloroquine therapy. J Rheumatol 2016; 43:745–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schrezenmeier E, Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 2020; 16:155–66. [DOI] [PubMed] [Google Scholar]
- 20. Tselios K, Gladman DD, Touma Z, Su J, Anderson N, Urowitz MB. Clinical remission and low disease activity outcomes over 10 years in systemic lupus erythematosus. Arthritis Care Res 2019; 71:822–8. [DOI] [PubMed] [Google Scholar]
- 21. Pauken KE, Sammons MA, Odorizzi PM et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD‐1 blockade. Science 2016; 354:1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sen DR, Kaminski J, Barnitz RA et al. The epigenetic landscape of T cell exhaustion. Science 2016; 354:1165–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKinney EF, Smith KGC. Metabolic exhaustion in infection, cancer and autoimmunity. Nat Immunol 2018; 19:213–21. [DOI] [PubMed] [Google Scholar]
- 24. Pawelec G. Is There a positive side to T cell exhaustion? Front Immunol 2019; 10:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Long SA, Thorpe J, De Berg HA et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new‐onset type 1 diabetes. Sci Immunol 2016; 1:eaai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paley MA, Kroy DC, Odorizzi PM et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 2012; 338:1220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J, He Y, Hao J, Ni L, Dong C. High levels of eomes promote exhaustion of anti‐tumor CD8 (+) T cells. Front Immunol 2018; 9:2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author (J. J.‐O.) upon reasonable request.