

Abstract
Vitamin E deficiencies can impact normal growth and development in humans and animals, and assessment of circulating levels of vitamin E and its metabolites may be an important endpoint for evaluation. Development of a sensitive method to detect and quantify low concentrations of vitamin E and metabolites in biological specimens allows for a proper diagnosis for patients and animals that are deficient. We developed a method to simultaneously extract, detect, and quantify the vitamin E compounds alpha-tocopherol (α-TP), gamma-tocopherol (γ-TP), alpha-tocotrienol (α-TT), and gamma-tocotrienol (γ-TT), and the corresponding metabolites formed after β-oxidation of α-TP and γ-TP, alpha-carboxymethylbutyl hydroxychroman (α-CMBHC) and alpha- or gamma-carboxyethyl hydroxychroman (α- or γ-CEHC), respectively, from equine plasma and serum. Quantification was achieved through liquid chromatography–tandem mass spectrometry. We applied a 96-well high-throughput format using a Phenomenex Phree plate to analyze plasma and serum. Compounds were separated by using a Waters ACQUITY UPLC BEH C18 column with a reverse-phase gradient. The limits of detection for the metabolites and vitamin E compounds were 8–330 pg/mL. To validate the method, intra-day and inter-day accuracy and precision were evaluated along with limits of detection and quantification. The method was then applied to determine concentrations of these analytes in plasma and serum of horses. Alpha-TP levels were 3–6 µg/mL of matrix; the metabolites were found at much lower levels, 0.2–1.0 ng/mL of matrix.
Keywords: α/γ-tocopherol, alpha-carboxymethylbutyl hydroxychroman, gamma-carboxyethyl hydroxychroman, liquid chromatography–mass spectrometry, vitamin E
Introduction
Vitamin E is a well-known antioxidant that protects against oxidative stress, specifically the propagation of lipid peroxidation.3 Vitamin E is a collective term used to describe a group of 8 potent and lipophilic vitamin E isoforms; alpha-, beta-, gamma-, and delta-tocopherol (α-TP, β-TP, γ-TP, δ-TP, respectively) as well as alpha-, beta-, gamma-, and delta-tocotrienol (α-TT, β-TT, γ-TT, δ-TT, respectively), which have a large number of beneficial attributes.1,7,13,26,30–32 Following ingestion, vitamin E isoforms are metabolized via P450-mediated ω-oxidation to longer chain metabolites, such as carboxymethylbutyl hydroxychroman (CMBHC), followed by β-oxidation to shorter chain metabolites, such as carboxyethyl hydroxychromans (CEHCs). In addition to the TP and TT isoforms being beneficial, several metabolites including γ- and α-CEHC demonstrate similar properties.11,13,38 The precursor metabolite to α-CEHC, alpha-5-(6-hydroxy-2,5,7,8-tetramethyl-chroman-2-yl)-2-methyl-pentanoic acid (α-CMBHC), is a minor vitamin E metabolite formed from α-TP and has been investigated as a biomarker of excess/adequate vitamin E.28 By monitoring vitamin E and vitamin E metabolite levels in biological matrices, it may be possible to determine the level at which vitamin E can protect tissues against oxidative injury.41
Vitamin E deficiencies and resulting neurologic diseases affect both humans and animals.1,2,7,13,26,30,31 In particular, horses receive their dietary intake of vitamin E from grazing pasture, and genetically susceptible foals who do not have access to enough fresh pasture during the first year of their lives may develop equine neuroaxonal dystrophy (eNAD). eNAD is a disease characterized by axonal degeneration within the brainstem and spinal cord, leading to general proprioceptive ataxia.6,7 In addition to the lack of vitamin E in foals, adult horses that have been vitamin E deficient for 18 mo can develop lower motor neuron weaknesses, termed equine motor neuron disease (EMND).5
Vitamin E and its metabolites have previously been analyzed using high-performance liquid chromatography–ultraviolet visible spectroscopy (HPLC-UV-VIS),8,29 gas chromatography–mass spectrometry (GC-MS),21 and liquid chromatography–mass spectrometry (LC-MS).16,25 As the underlying etiologies for both eNAD and EMND remain unknown, quantification of vitamin E metabolites in diseased horses may provide further insight into the pathophysiology of these conditions. We aimed to develop and validate a sensitive method to measure vitamin E and the vitamin E metabolites (Fig. 1) at low concentrations using liquid chromatography–tandem MS (LC-MS/MS), allowing for the establishment of reference intervals (RIs) in healthy horses maintained on irrigated grass pasture. Additionally, we sought to determine if there were significant differences in vitamin E and vitamin E metabolites in matched serum versus plasma samples.
Figure 1.
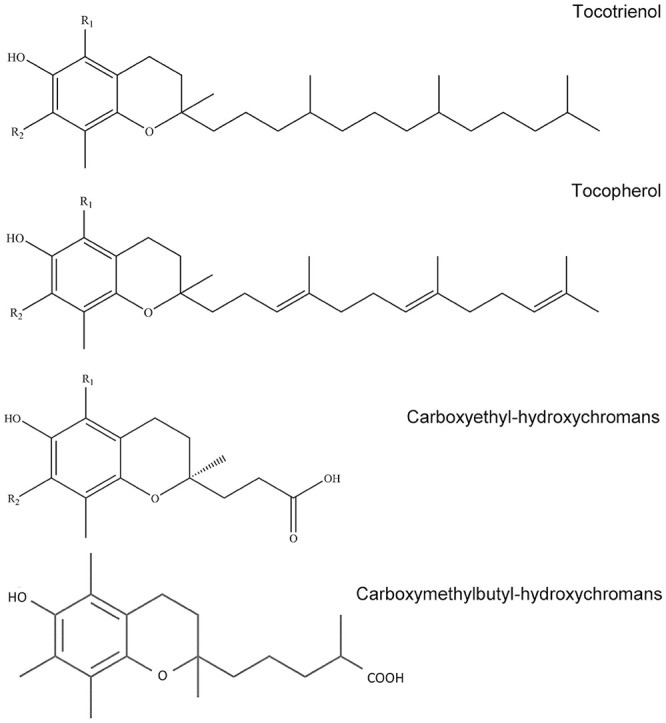
Chemical structures of targeted analytes. R1 = methyl and R2 = methyl for α-TP, α-TT, and α-CEHC. R1 = H and R2 = methyl for γ-TP, γ-TT, and γ-CEHC. The structure for α-CMBHC is shown without substitutions.
Materials and methods
Chemicals and materials
DL-α-TP (> 99%), γ-TP (> 96.3%), α-TT (> 97%), γ-TT (> 97%), L-ascorbic acid (pharmaceutical secondary standard, > 99%), and chlorpropamide were purchased from Sigma-Aldrich. The vitamin E metabolites (α-CEHC > 98%, γ-CEHC > 98%, α-CMBHC > 98%) and (12-((cyclohexylamino)carbonyl)amino)-dodecanoic acid (CUDA) were purchased from Cayman Chemical. α-TP-D6 (5-methyl-D3, 7-methyl-D3, > 98%) was purchased from Cambridge Isotope Laboratories. The antioxidant butylated hydroxytoluene (BHT) was purchased from MP Biomedicals. Glacial acetic acid (> 99.7%, analytical grade), methanol (analytical grade), and acetonitrile (ACN; analytical grade) were purchased from Fisher Scientific. Isopropyl alcohol (IPA; analytical grade) was purchased from Millipore Sigma. The 96-well Phree plates and deep-well plates (96/2,000 μL) were purchased from Phenomenex. The vacuum manifold for plate processing was purchased from Waters Corp. Ultrapure water (18.2 mΩ) was obtained using a Milli-Q system (Millipore).
Preparation of stock standard solutions and antioxidant solutions
Stock standard solutions of target analytes and internal standards were prepared in methanol at various concentrations. Working solutions containing all 7 target analytes were prepared at 12.5, 1,250, and 4,000 ng/mL. Additionally, methanolic solutions of α-TP-D6 (1,000 ng/mL), chlorpropamide (50 ng/mL), BHT (10 mg/mL), and CUDA (2 ng/mL) were prepared. Ascorbic acid was prepared fresh daily at 10 mg/mL in water.
Biological sample collection
Matched serum and plasma samples were collected from 16 healthy Thoroughbred horses (2–24 y old, 9 mares and 7 geldings) to establish baseline vitamin E and metabolite levels. These horses were maintained on irrigated pasture for 24 h/d for ≥3 mo prior to sampling, with no supplemental feed provided. Light-protected plasma and serum samples were obtained with signed owner consent under a protocol approved by the University of California’s Institutional Animal Care and Use Committee. Plasma samples were collected in lavender-top tubes containing K2EDTA as the anticoagulant. Serum samples were collected in either empty red-top tubes or serum separator tubes without anticoagulant. Hemolysis was assessed at collection, and no hemolyzed samples were included in the analysis. All samples were centrifuged at 4°C within 4 h of collection and frozen at −80°C until analyzed. Charcoal-stripped negative control serum was prepared in-house using dextran-coated activated charcoal.17
Sample extraction from plasma and serum samples
To extract vitamin E and vitamin E metabolites, frozen plasma and serum samples were placed in a refrigerator at 4°C until thawed. Once thawed, samples were inverted and mixed to ensure homogeneity. Six hundred µL of ACN was added to each well of a Phree plate. Ten µL of ascorbic acid (10 mg/mL) and BHT (10 mg/mL) as antioxidants, along with 10 µL of α-TP-D6 (1 μg/mL in methanol) and chlorpropamide (50 ng/mL in methanol) as internal standards, were added to each tube. Select wells were then spiked with 10 μL of working solutions containing all 7 analytes at various concentrations for calibrators and quality control (QC) samples, or 10 μL of methanol for samples. Then, 150 μL of plasma/serum sample or water for controls and calibration curve was added to each well and mixed by pipette. A gentle vacuum of 5–10 mm Hg was then applied to collect the flow-through containing the metabolites. After the wells appeared to be dry, 800 μL of an ACN/IPA mix (9:1) was added to the wells to extract the tocopherol and tocotrienol compounds, and eluent was collected. The contents were transferred to a 2-mL microcentrifuge tube and evaporated to dryness (45°C) in vacuo (VacuFuge; Eppendorf). Each residue was then reconstituted in 50 μL of methanol containing CUDA (2 ng/mL) and vortexed, sonicated (2 min), vortexed, and centrifuged (8 min) at 14,000 × g. Approximately 40 μL of supernatant was transferred into HPLC vials for analysis.
Instrumental analysis
The analysis of vitamin E and metabolites was accomplished using a LC-MS/MS system, which consisted of an Advance HPLC system coupled with an EVOQ Elite triple-quadrupole mass spectrometer (Bruker). The chromatography system was fitted with a 130 Å, 1.7 μm, 2.1 mm × 100 mm ACQUITY BEH C18 column with a corresponding Van Guard pre-column (Waters). Mobile phases consisted of ultrapure water (18.2 mΩ) with 0.1% acetic acid (channel A) and methanol with 0.1% acetic acid (channel B) delivered over a reverse-phase gradient. Initially, 5% B was increased to 45% B over 2 min, which was then increased to 98% B over 6 min, held for 8 min, and returned to 5% B immediately after 16 min, and then held for 6 min for re-equilibration to initial conditions. The column was maintained at 50°C, and the flow rate was set at 250 μL/min. Analytes were monitored by multiple reaction monitoring (MRM) in positive mode electrospray ionization for the tocopherols and tocotrienols, and negative mode electrospray ionization for the metabolites (Table 1, Figs. 2, 3). Nitrogen gas was used as the nebulizer gas, and argon was used as the collision gas. Other source parameters were as follows: nebulizer gas flow 45 arbitrary units (a.u.), probe gas flow 35 a.u, temperature of the heated probe 300°C, cone gas flow 15 a.u., cone temperature 300°C, and the capillary voltage 4,000 V. The injection volume per sample was 5 μL. MSWS 8.1 software (Bruker) was used for all data analysis and processing.
Table 1.
Tandem mass spectrometry parameters and multiple reaction monitoring (MRM) transitions in positive and negative electrospray ionization mode.
| Compound | Retention time (min) | MRM transition (m/z) | Collision energy (eV) | Ionization mode |
|---|---|---|---|---|
| Chlorpropamide | 5.6 | 275.7 → 191.0, 127.1, 80.2 | 12, 22, 35 | Negative |
| γ-CEHC | 5.9 | 263.3 → 136.1, 149.1 | 23, 17 | Negative |
| α-CEHC | 6.2 | 277.3 → 233.1, 163.1, 150.1 | 10, 19, 15 | Negative |
| α-CMBHC | 7.5 | 319.4 → 150.1, 163.1, 275.1 | 15, 20, 14 | Negative |
| CUDA | 7.8 | 339.0 → 214.1, 240.1 | 22, 14 | Negative |
| γ-TT | 10.9 | 411.0 → 151.0, 191.0, 205.1 | 18, 13, 11 | Positive |
| α-TP | 12.6 | 431.7 → 165.0, 137.1, 111.2 | 18, 36, 14 | Positive |
| α-TT | 11.2 | 425.7 → 165.0, 205.1, 137.1 | 16, 12, 33 | Positive |
| γ-TP | 12.2 | 417.7 → 151.0, 123.1, 97.2 | 15, 37, 15 | Positive |
| α-TP-D6 | 12.6 | 437.4 → 171.1, 143.1, 97.2 | 18, 37, 17 | Positive |
Product ions used for quantitation are shown in bold.
Figure 2.

Representative LC-MS/MS chromatograms for chlorpropamide, γ-CEHC, α-CEHC, and α-CMBHC in a 250 ng/mL quality control sample.
Figure 3.

Representative LC-MS/MS chromatograms for CUDA, γ-TT, α-TT, γ-TP, α-TP, and α-TP-D6 in a 250-ng/mL quality control sample.
Method validation
To determine the performance of the method, intra- and inter-day accuracy and precision were evaluated. An external calibration curve in water was utilized because a truly blank matrix (serum or plasma) was unavailable given the endogenous nature of the targeted analytes. Nine calibration standards of 0.1, 0.5, 1, 5, 10, 50, 100, 500, and 1,000 ng/mL for all 7 analytes were used, plus additions of 200 ng/mL of internal standard α-TP-D6, 10 ng/mL of internal standard chlorpropamide, and 2 ng/mL of the instrument internal standard CUDA. An additional 3 calibration standards were prepared at 2,000 ng/mL, 5,000 ng/mL, and 10,000 ng/mL, only containing α-TP and the internal standards to account for high levels of α-TP expected in serum and plasma of healthy horses. Three QC samples at low, medium, and high levels were prepared at final concentrations of 2.5, 250, and 800 ng/mL in water, respectively (n = 5/level). The recovery of the methodology was assessed by comparing the response of neat standards to extracted samples in water. The matrix effects of the method were assessed by calculating the ratio of peak area response from a charcoal-stripped serum sample to an extracted standard. In addition to the positive control samples, a water negative control was run each day to monitor contamination or significant carry-over.
Data analysis
Statistical analyses were performed with Prism v.7.0 (GraphPad). Normality was evaluated using a Shapiro–Wilk test and evaluation of residuals using a Q-Q plot. Correlation between matched plasma and serum samples was assessed using a Pearson coefficient for normally distributed data, and a Spearman coefficient for non-normally distributed data. An unpaired t-test with a Welch correction was used to assess sex differences for all analytes in both plasma and serum samples. Statistical significance was set at p ≤ 0.05.
Results
The spray voltage (in both positive and negative mode), cone temperature, cone gas flow, heated probe temperature, probe gas flow, and nebulizer gas flow were optimized following either the infusion of standards (10 ng/µL) or injection of an extracted sample to obtain the greatest instrument sensitivity. Source and MRM scan parameters were evaluated for vitamin E and vitamin E metabolites in both positive and negative mode using 3 different acidic modifiers (0.1% formic acid, 0.1% acetic acid, or 1.0% acetic acid) in MeOH/H2O 50:50. Based on signal intensities, the metabolites ionized with greater efficiency with 0.1% acetic acid as the modifier. Decreasing the spray voltage below 4,000 V resulted in decreased peak areas for all analytes. The peak areas were not affected when switching between cone temperatures of 200, 250, and 300°C. Heated probe temperature was tested at 250, 300, and 350°C, with 300°C having the largest response for most of the compounds. Cone gas flow was evaluated at 15 and 20 a.u. with a nebulizer gas flow at 40 and 50 a.u., respectively, with the lower flows producing better results.
Calibration curves were generated by calculating relative responses (analyte peak area/internal standard peak area) for each point on the standard curve. The calibration was fitted with a linear regression for vitamin E compounds and a quadratic regression for the metabolites (Table 2). Method accuracy, precision, recovery, and matrix effects were evaluated during method validation. The limits of detection and limits of quantification were determined by diluting a standard containing all 7 analytes in methanol until signal-to-noise ratios of 3:1 and 10:1 were obtained, respectively (Table 2). The linear/quadratic regression for all compounds was 0.1–1,000 ng/mL (α-TP regression extended to 10,000 ng/mL) with coefficients of determination (R2) > 0.99 for each day.
Table 2.
Limits of detection (LOD), limits of quantification (LOQ), upper limits of quantification (ULOQ), percent recovery, matrix effect ratio, type of regression used for the calibration curve, and representative regression equations of targeted compounds.
| Compound | LOD (ng/mL) | LOQ (ng/mL) | ULOQ (ng/mL) | % Recovery | Matrix effect ratio | Calibration curve regression utilized | Representative regression equation |
|---|---|---|---|---|---|---|---|
| α-TP | 0.008 | 0.025 | 10,000 | 44 | 0.42 | Linear | 0.0038x + 0.0084 |
| γ-TP | 0.333 | 1.11 | 1,000 | 41 | 0.22 | Linear | 1.37e−4x + 2.87e−5 |
| α-TT | 0.029 | 0.095 | 1,000 | 44 | 0.36 | Linear | 0.0047x + 4.4e−5 |
| γ-TT | 0.125 | 0.417 | 1,000 | 42 | 0.35 | Linear | 0.002x − 4.21e−4 |
| α-CMBHC | 0.010 | 0.032 | 1,000 | 66 | 0.42 | Quadratic | −2.1e−5x2 + 0.069x + 0.270 |
| α-CEHC | 0.022 | 0.074 | 1,000 | 93 | 0.54 | Quadratic | −1.2e−5x2 + 0.054x + 0.130 |
| γ-CEHC | 0.026 | 0.087 | 1,000 | 96 | 0.53 | Quadratic | −3.9e−6x2 + 0.016x + 0.056 |
The accuracy and precision of all analytes were evaluated by determining intra-day and inter-day statistics (Table 3) using QC samples spiked with low, medium, and high concentrations. Intra-day accuracies were 73.9–121%, depending on the target analyte. Assay precision was evaluated, and the coefficient of variation (% CV) was calculated for each concentration level. For intra-day precision, % CVs were 0.96–24.4%. The inter-day accuracy was based on the average obtained over 3 d, at each concentration level. Inter-day accuracies were 85.8–117%, 75.3–117%, and 80.9–111%, for the low, medium, and high concentration levels, respectively. The inter-day precision was determined by the average obtained in days 1–3. The inter-day precisions were 5.29–15.8%, 5.07–19.6%, and 4.55–16.5%, for the low, medium, and high concentration levels, respectively. The matrix effects (matrix effect ratio) and recovery were evaluated by comparing peak area responses from neat standard to either a charcoal-stripped serum sample or extracted standard at equivalent concentrations (Table 2).
Table 3.
Intra-batch accuracy and precision, and inter-batch accuracy and precision using liquid chromatography–mass spectrometry.
| Analyte | Nominal concentration (ng/mL) | Day 1 |
Day 2 |
Day 3 |
Inter-day |
||||
|---|---|---|---|---|---|---|---|---|---|
| Accuracy* (%) | Precision† (%) | Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | Accuracy‡ (%) | Precision§ (%) | ||
| α-TP | 2.50 | 114 | 6.58 | 119 | 2.50 | 109 | 5.00 | 114 | 5.83 |
| 250 | 99.1 | 5.23 | 115 | 3.63 | 101 | 6.14 | 105 | 7.96 | |
| 800 | 103 | 4.95 | 108 | 2.23 | 102 | 4.44 | 104 | 4.55 | |
| γ-TP | 2.50 | 102 | 17.5 | 81.4 | 9.63 | 103 | 5.67 | 95.6 | 15.8 |
| 250 | 113 | 7.52 | 76.9 | 4.53 | 82.6 | 6.99 | 93.7 | 19.6 | |
| 800 | 101 | 8.57 | 79.3 | 21.5 | 84.0 | 7.80 | 88.2 | 16.5 | |
| α-TT | 2.50 | 84.3 | 5.10 | 80.4 | 9.97 | 92.6 | 7.95 | 85.8 | 9.53 |
| 250 | 94.6 | 7.98 | 109 | 4.39 | 109 | 7.12 | 104 | 9.11 | |
| 800 | 91.9 | 14.9 | 107 | 2.03 | 109 | 1.58 | 103 | 10.6 | |
| γ-TT | 2.50 | 115 | 8.06 | 93.1 | 18.7 | 106 | 5.21 | 104 | 13.7 |
| 250 | 75.7 | 6.63 | 76.5 | 22.1 | 73.9 | 11.5 | 75.3 | 14.0 | |
| 800 | 78.0 | 15.3 | 87.7 | 11.4 | 77.2 | 3.29 | 80.9 | 12.0 | |
| α-CMBHC | 2.50 | 99.5 | 7.11 | 116 | 2.86 | 98.8 | 24.4 | 105 | 8.88 |
| 250 | 98.3 | 5.60 | 116 | 0.96 | 98.9 | 11.8 | 105 | 10.6 | |
| 800 | 101 | 12.5 | 109 | 3.14 | 107 | 2.87 | 106 | 7.57 | |
| α-CEHC | 2.50 | 114 | 5.65 | 118 | 2.88 | 110 | 4.12 | 114 | 10.2 |
| 250 | 108 | 5.46 | 118 | 2.33 | 116 | 4.18 | 114 | 5.48 | |
| 800 | 101 | 11.5 | 110 | 1.96 | 112 | 2.81 | 108 | 7.46 | |
| γ-CEHC | 2.50 | 114 | 2.87 | 124 | 3.27 | 115 | 5.48 | 118 | 5.29 |
| 250 | 117 | 1.88 | 121 | 3.55 | 113 | 6.80 | 117 | 5.07 | |
| 800 | 109 | 6.02 | 108 | 2.47 | 118 | 6.09 | 111 | 6.20 | |
Intra-day accuracy was measured as the amount of target analytes added to a blank matrix, carried throughout the extraction procedure, and then analyzed. The % accuracy was based on the mean of 5 samples at low, medium, and high concentration levels.
Inter-day precision was measured as the amount of target analytes added to a blank matrix, carried throughout the extraction procedure, and then analyzed. The inter-day % precision is determined by the average % recoveries obtained in days 1–3.
Intra-day accuracy was measured by the amounts obtained from the coefficients of variation (% CV). The % accuracy is based on the averages of the % CVs of 5 samples at low, medium, and high concentration levels.
§ Inter-day precision was measured by the amounts obtained from the coefficients of variation (% CV). The inter-day precision % is determined by the average % precisions obtained in days 1–3.
Serum and plasma α-TP, γ-TP, α-CMBHC, α-CEHC, and γ-CEHC concentrations were significantly positively correlated (Fig. 4A, 4B, 4D, 4E, respectively). Serum α-TP, γ-TP, and γ-CEHC concentrations were higher than matched plasma samples, whereas plasma α-CEHC was higher than matched serum samples. Serum and plasma values of α-TT were not significantly correlated (Fig. 4C), with higher values in plasma versus serum. Serum and plasma γ-TT concentrations from healthy horses maintained on pasture were below the reporting limit for most data points and therefore excluded from further analysis.
Figure 4.

Correlations of matched plasma and serum: A. α-TP, B. γ-TP, C. α-TT, D. α-CMBHC, E. α-CEHC, and F. γ-CEHC. Significant positive correlations were identified for all analytes except α-TT.
Discussion
We developed and ultimately applied our LC-MS/MS method to analyze plasma and serum from 16 healthy Thoroughbred horses maintained on pasture to determine endogenous levels of vitamin E and vitamin E metabolites. Our data generated are similar to the findings of a previous study,7 in which α-TP levels in healthy Thoroughbred horses were 3–5 μg/mL in plasma or serum. Our data support this claim; our α-TP serum and plasma concentrations were 4.42 ± 1.02 μg/mL and 3.72 ± 0.68 μg/mL, respectively. Results were higher than previously reported α-TP plasma (2.8 ± 0.9 μg/mL) levels of healthy Thoroughbreds, Quarter Horses, and other breeds maintained on hay and alfalfa pellets.33 This is likely the result of different diets, with our Thoroughbred population maintained on irrigated pasture 24 h/d. With regards to sex, α-TP was the only analyte that showed significant differences, with mares demonstrating higher concentrations of α-TP in both plasma and serum (p = 0.007 and 0.01, respectively).
Various analytical approaches have been utilized to detect vitamin E and its metabolites9,10,12,14,16,19–25,27,29,37,38,40 and are summarized in in-depth reviews.36,37 Given its high endogenous concentration in serum or plasma, α-TP measurement does not require a highly sensitive methodology, but measurement of other vitamin E isoforms and metabolites requires highly sensitive approaches with circulating levels commonly observed at either low ng/mL or sub ng/mL concentrations.12,19,27 The use of a triple-quadrupole mass spectrometer allows for improvements in detection levels compared to single-quadrupole instruments.14 Most notably, our approach allows very low level detection of both α- and γ-CEHC at over an order of magnitude lower concentrations compared to other studies. Others have found similar accuracy and precision values in plasma and liver homogenates using GC-MS and LC-MS approaches.19,25 Although our results showed slightly greater variance in precision, and lower recoveries for γ-TP and γ-TT, we have demonstrated simultaneous detection of vitamin E and vitamin E metabolites using a 22-min method, minimizing sample preparation and run times compared to previous investigations.19,25 Some studies have compared the differences in detecting vitamin E through LC-MS/MS analysis to GC-MS analysis after derivatization to trimethylsilyl derivatives.16,21,25 Derivatizing vitamin E samples for GC-MS analysis not only adds a time-consuming and costly step, but also adds the risk of hydrolysis.16
The use of internal standards during bioanalytical sample preparation and analysis is a commonly utilized approach to account for losses observed during processing or to correct for matrix effects that are observed during the electrospray ionization process.34 Given the potential for degradation or oxidation of analytes during sample preparation and to compensate for varied instrument responses, we used 2 internal standards. Alpha-TP-D6 was used to correct for vitamin E responses, and chlorpropamide was used to correct the responses for the metabolites. Alpha-TP-D6 was initially used as an internal standard for all 7 target analytes. Because of the differences in chemical structure and ionizing modes between the metabolites and vitamin E compounds, α-TP-D6 did not properly correct for the metabolites detected in negative mode with observed concentrations ~120–230% of predicted values, indicating that the metabolites required a different internal standard. Initially, Trolox was evaluated as an internal standard based on structure similarities to the metabolites and data from previous studies,19,22 but resulted in poor recoveries (0.2–22.8%). Chlorpropamide was ultimately evaluated and selected as an internal standard for the metabolites.15 Our data suggest that the internal standards were helpful in correcting for both recovery and ion suppression or enhancement, although the use of stable isotope–labeled internal standards for all of the targeted analytes that are not commercially available would further strengthen the methodology.
The analysis of vitamin E and its metabolites is challenging for a variety of reasons, and our study has some limitations that should be noted and can be addressed in future work. First, our approach uses only one stable isotope–labeled internal standard (α-TP-D6) given the lack of commercial stable isotope–labeled reference standards for the remaining compounds, which increases the likelihood of uncorrected biases being introduced during sample processing.34 Second, the endogenous nature of the targeted compounds included in our analysis makes obtaining a “blank” matrix that does not contain the compounds of interest challenging. There are several potential bioanalytical approaches to address these challenges, such as background subtraction, method of standard addition, or use of a surrogate matrix.35,39 Thus, similar to the work of others, we used a surrogate matrix (water) to prepare calibration curves.12,14,20 Although this approach is not ideal given the use of non–matrix-matched calibrators potentially resulting in unaccounted-for biases in results, it was suitable as part of our preliminary evaluation of vitamin E and its metabolites in serum or plasma. One alternative surrogate matrix is charcoal-stripped serum/plasma samples, although, based on our data (not shown), charcoal stripping was more effective at reducing the background levels of the metabolites, it was less effective for less polar compounds, such as α-TP, and was not utilized. Further studies would be strengthened using the background subtraction approach in the analysis of vitamin E and its metabolites.10 Third, instability during sample processing is also of concern when working with vitamin E isoforms, and thus our approach utilized a combination of BHT and ascorbic acid to attempt to mitigate degradation during sample preparation.14,36,37 Likewise, the stability of vitamin E and its metabolites in biological fluids after collection and storage is of further interest and, although not addressed specifically in our study, is an area for further investigation. Others have shown that α-TP concentrations are stable when stored refrigerated or frozen for 1–2 wk, although extended storage did result in decreased levels after 6 mo of storage below −10°C.4,12,23 Additionally, given that several of the metabolites analyzed in our study are excreted as phase II metabolites (glucuronide, sulfate, amino acid conjugates) in urine, our approach focusing on non-conjugated compounds may not assess the full spectrum of vitamin E metabolites present in circulation.36,37 Last, it should be noted that others have shown that hemolysis of samples can have dramatic impacts on α-TP concentrations and it is likely that other similar compounds will be impacted in the same fashion, and thus care should be taken when collecting and analyzing samples.4,12,18
Acknowledgments
We thank Anthony Valenzuela for his assistance with analytical instrumentation, Sunjay Sethi for his assistance with statistical analysis, and Heidi Kucera for laboratory assistance.
Footnotes
Declaration of conflicting interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: This work was supported by the National Institutes of Health (NIH) award to Carrie J. Finno (K01OD015134, L40 TR001136). This project was supported by the Center for Equine Health with funds provided by the State of California pari-mutuel fund and contributions by private donors.
ORCID iD: Benjamin C. Moeller  https://orcid.org/0000-0003-2945-3620
https://orcid.org/0000-0003-2945-3620
Contributor Information
Hadi Habib, Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA.
Carrie J. Finno, Department of Population Health and Reproduction, School of Veterinary Medicine, University of California, Davis, CA
Ingrid Gennity, Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA.
Gianna Favro, Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA.
Erin Hales, Department of Population Health and Reproduction, School of Veterinary Medicine, University of California, Davis, CA.
Birgit Puschner, Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, CA.
Benjamin C. Moeller, K. L. Maddy Equine Analytical Chemistry Laboratory, California Animal Health and Food Safety Laboratory, School of Veterinary Medicine, University of California, Davis, CA.
References
- 1. Brigelius-Flohe R, et al. Vitamin E: function and metabolism. FASEB J 1999;13:1145–1155. [PubMed] [Google Scholar]
- 2. Burns EN, Finno CJ. Equine degenerative myeloencephalopathy: prevalence, impact, and management. Vet Med (Auckl) 2018;9:63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burton G, et al. Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function. Acc Chem Res 1986;19:194–201. [Google Scholar]
- 4. Craig AM, et al. Variability of alpha-tocopherol values associated with procurement, storage, and freezing of equine serum and plasma samples. Am J Vet Res 1992;53:2228–2234. [PubMed] [Google Scholar]
- 5. De la Rua-Domenech R, et al. Association between plasma vitamin E concentration and the risk of equine motor neuron disease. Vet J 1997;154:203–213. [DOI] [PubMed] [Google Scholar]
- 6. Finno CJ. Equine neuroaxonal dystrophy. In: Sprayberry KA, Robinson NE, eds. Robinson’s Current Therapy in Equine Medicine. 7th ed. Elsevier, 2015:384–386. [Google Scholar]
- 7. Finno CJ, Valberg SJ. A comparative review of vitamin E and associated equine disorders. J Vet Intern Med 2012;26:1251–1266. [DOI] [PubMed] [Google Scholar]
- 8. Gámiz-Gracia L, et al. Fully automated determination of vitamins A and E in milk-based products using a robotic station-HPLC arrangement. Analyst 1999;124:801–804. [DOI] [PubMed] [Google Scholar]
- 9. Giusepponi D, et al. LC-MS/MS assay for the simultaneous determination of tocopherols, polyunsaturated fatty acids and their metabolites in human plasma and serum. Free Radic Biol Med 2019;144:134–143. [DOI] [PubMed] [Google Scholar]
- 10. Giusepponi D, et al. Determination of tocopherols and their metabolites by liquid-chromatography coupled with tandem mass spectrometry in human plasma and serum. Talanta 2017;170:552–561. [DOI] [PubMed] [Google Scholar]
- 11. Grammas P, et al. Anti-inflammatory effects of tocopherol metabolites. Biochem Biophys Res Commun 2004;319:1047–1052. [DOI] [PubMed] [Google Scholar]
- 12. Hooser SB, et al. Effects of storage conditions and hemolysis on vitamin E concentrations in porcine serum and liver. J Vet Diagn Invest 2000;12:365–368. [DOI] [PubMed] [Google Scholar]
- 13. Jiang Q, et al. γ-Tocopherol, the major form of vitamin E in the US diet, deserves more attention. Am J Clin Nutr 2001;74:714–722. [DOI] [PubMed] [Google Scholar]
- 14. Jiang Q, et al. Analysis of vitamin E metabolites including carboxychromanols and sulfated derivatives using LC/MS/MS. J Lipid Res 2015;56:2217–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson CH, et al. Novel metabolites and roles for α-tocopherol in humans and mice discovered by mass spectrometry-based metabolomics. Am J Clin Nutr 2012;96:818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lauridsen C, et al. Quantitative analysis by liquid chromatography–tandem mass spectrometry of deuterium-labeled and unlabeled vitamin E in biological samples. Anal Biochem 2001;289:89–95. [DOI] [PubMed] [Google Scholar]
- 17. Leake RE, Green B. Steroid Hormones: A Practical Approach (The Practical Approach Series, 30). IRL Press, 1987. [Google Scholar]
- 18. Lear AS, et al. Evaluation of sample handling effects on serum vitamin E and cholesterol concentrations in alpacas. Vet Med Int 2014;2014:537213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leonard SW, et al. Quantitation of rat liver vitamin E metabolites by LC-MS during high-dose vitamin E administration. J Lipid Res 2005;46:1068–1075. [DOI] [PubMed] [Google Scholar]
- 20. Leonard SW, Traber MG. Measurement of the vitamin E metabolites, carboxyethyl hydroxychromans (CEHCS), in biological samples. Curr Protoc Toxicol 2006;29:7.8.1–7.8.12. [DOI] [PubMed] [Google Scholar]
- 21. Liebler DC, et al. Gas chromatography-mass spectrometry analysis of vitamin E and its oxidation products. Anal Biochem 1996;236:27–34. [DOI] [PubMed] [Google Scholar]
- 22. Lodge JK, et al. A rapid method for the extraction and determination of vitamin E metabolites in human urine. J Lipid Res 2000;41:148–154. [PubMed] [Google Scholar]
- 23. Mitsioulis A, Judson GJ. Stability of vitamin E in blood and plasma from cattle, sheep, and pigs. J Vet Diagn Invest 2000;12:364–365. [DOI] [PubMed] [Google Scholar]
- 24. Morton LW, et al. Evidence for the nitration of γ-tocopherol in vivo: 5-nitro-γ-tocopherol is elevated in the plasma of subjects with coronary heart disease. Biochem J 2002;364:625–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mottier P, et al. Comparison of gas chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry methods to quantify α-tocopherol and α-tocopherolquinone levels in human plasma. Anal Biochem 2002;301:128–135. [DOI] [PubMed] [Google Scholar]
- 26. Mustacich DJ, et al. Vitamin E. In: Litwack G, ed. Vitamins and Hormones. Vol. 76. Academic Press, 2007:1–21. [DOI] [PubMed] [Google Scholar]
- 27. Nierenberg DW, Lester DC. Determination of vitamins A and E in serum and plasma using a simplified clarification method and high-performance liquid chromatography. J Chromatogr 1985;345:275–284. [DOI] [PubMed] [Google Scholar]
- 28. Pope SA, et al. Synthesis and analysis of conjugates of the major vitamin E metabolite, α-CEHC. Free Radic Biol Med 2002;33:807–817. [DOI] [PubMed] [Google Scholar]
- 29. Romeu-Nadal M, et al. Determination of γ-and α-tocopherols in human milk by a direct high-performance liquid chromatographic method with UV–VIS detection and comparison with evaporative light scattering detection. J Chromatogr A 2006;1114:132–137. [DOI] [PubMed] [Google Scholar]
- 30. Saito H, et al. γ-Tocotrienol, a vitamin E homolog, is a natriuretic hormone precursor. J Lipid Res 2003;44:1530–1535. [DOI] [PubMed] [Google Scholar]
- 31. Sen CK, et al. Tocotrienols: vitamin E beyond tocopherols. Life Sci 2006;78:2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soll J, Schultz G. Comparison of geranylgeranyl and phytyl substituted methylquinols in the tocopherol synthesis of spinach chloroplasts. Biochem Biophys Res Commun 1979;91:715–720. [DOI] [PubMed] [Google Scholar]
- 33. Steiss JE, et al. Alpha tocopherol concentrations in clinically normal adult horses. Equine Vet J 1994;26:417–419. [DOI] [PubMed] [Google Scholar]
- 34. Tan A, et al. Use of internal standards in LC-MS bioanalysis. In: Wenkui L, et al., eds. Handbook of LC-MS Bioanalysis. Wiley, 2013:217–227. [Google Scholar]
- 35. Thakare R, et al. Quantitative analysis of endogenous compounds. J Pharm Biomed Anal 2016;128:426–437. [DOI] [PubMed] [Google Scholar]
- 36. Torquato P, et al. Analysis of vitamin E metabolites. In: Niki E, ed. Vitamin E: Chemistry and Nutritional Benefits: Best Practices, Experimental Protocols, and Regulations (Food Chemistry, Function and Analysis Series, 11). The Royal Society of Chemistry, 2019:208–227. [Google Scholar]
- 37. Torquato P, et al. Analytical strategies to assess the functional metabolome of vitamin E. J Pharm Biomed Anal 2016;124:399–412. [DOI] [PubMed] [Google Scholar]
- 38. Traber MG, et al. Synthetic as compared with natural vitamin E is preferentially excreted as α-CEHC in human urine: studies using deuterated α-tocopheryl acetates. FEBS Lett 1998;437:145–148. [DOI] [PubMed] [Google Scholar]
- 39. van de Merbel NC. Quantitative determination of endogenous compounds in biological samples using chromatographic techniques. Trends Anal Chem 2008;27:924–933. [Google Scholar]
- 40. Vaule H, et al. Vitamin E delivery to human skin: studies using deuterated α-tocopherol measured by APCI LC-MS. Free Radic Biol Med 2004;36:456–463. [DOI] [PubMed] [Google Scholar]
- 41. Wu JH, Croft KD. Vitamin E metabolism. Mol Aspects Med 2007;28:437–452. [DOI] [PubMed] [Google Scholar]