

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is an inherited muscle disease caused by misexpression of the DUX4 gene in skeletal muscle. DUX4 is a transcription factor, which is normally expressed in the cleavage-stage embryo and regulates gene expression involved in early embryonic development. Recent studies revealed that DUX4 also activates the transcription of repetitive elements such as endogenous retroviruses (ERVs), mammalian apparent long terminal repeat (LTR)-retrotransposons and pericentromeric satellite repeats (Human Satellite II). DUX4-bound ERV sequences also create alternative promoters for genes or long non-coding RNAs, producing fusion transcripts. To further understand transcriptional regulation by DUX4, we performed nanopore long-read direct RNA sequencing (dRNA-seq) of human muscle cells induced by DUX4, because long reads show whole isoforms with greater confidence. We successfully detected differential expression of known DUX4-induced genes and discovered 61 differentially expressed repeat loci, which are near DUX4–ChIP peaks. We also identified 247 gene–ERV fusion transcripts, of which 216 were not reported previously. In addition, long-read dRNA-seq clearly shows that RNA splicing is a common event in DUX4-activated ERV transcripts. Long-read analysis showed non-LTR transposons including Alu elements are also transcribed from LTRs. Our findings revealed further complexity of DUX4-induced ERV transcripts. This catalogue of DUX4-activated repetitive elements may provide useful information to elucidate the pathology of FSHD. Also, our results indicate that nanopore dRNA-seq has complementary strengths to conventional short-read complementary DNA sequencing.
Introduction
The DUX4 gene, encoding a double homeobox transcription factor, was identified in the highly similar 3.3 kb subtelomeric macrosatellite D4Z4 repeats on chromosome 4q35 (1). Since the 4q35 region forms heterochromatin and no transcript was detected, DUX4 was initially thought to be a pseudogene. However, it was revealed that in facioscapulohumeral muscular dystrophy type 1 (FSHD1) patients (online mendelian inheritance in man (OMIM): 158900) with D4Z4 repeat contraction, DNA methylation of D4Z4 was reduced and consequently de-repression of DUX4 occurred (2,3). In FSHD type 2 (FSHD2) patients (OMIM: 158901) without D4Z4 contraction, a genetic mutation in SMCHD1, which is involved in chromatin modification, reduced D4Z4 methylation and caused DUX4 de-repression (4). Misexpression of DUX4 is thought to be the cause of FSHD because animal models expressing DUX4 demonstrate FSHD-like symptoms (5–8).
Two different DUX4 messenger RNA (mRNA) isoforms have been identified in human skeletal muscle: a full-length mRNA (DUX4-fl) and a spliced short isoform (DUX4-s) (9). Introduction of DUX4-fl in muscle cells activates a number of cleavage-stage genes and causes cell death (10). In contrast, DUX4-s does not promote transcription and has no cytotoxicity because DUX4-s lacks the C-terminal transactivation domain (11,12). Consistent with the results in muscle cells, DUX4-fl is transiently expressed in human preimplantation embryos at the four-cell stage and plays a physiological role in regulating gene expression specific to the cleavage stage as a master transcriptional regulator (13).
In addition to gene regulation, DUX4-fl strongly binds to the long terminal repeat (LTR) regions of endogenous retroviruses (ERVs) and mammalian apparent LTR-retrotransposons (MaLRs), and activates transcription of these repetitive elements (10,14). Since both ERVs and MaLRs have LTRs at their 5′ and 3′ termini, they are categorized as LTR retrotransposons (15). ERVs are thought to have originated from retroviruses, and therefore their genetic structures are similar to those of retroviruses, but MaLR internal sequences are relatively dissimilar, especially lacking the envelope genes. The LTRs, ranging in size from 300 to 1000 bp, have signals for RNA transcription initiation and 3′-end formation, and sometimes splice donor sites (16). Most ERV and MaLR elements in the human genome are no longer full length due to deletion, substitution or mutations over generations. In particular, many single LTRs (so-called ‘solo LTRs’) are present in the genome, presumably formed by recombination between the flanking LTRs causing loss of the internal region. Previous RNA-seq analyses detected upregulation of ERVs, MaLRs, fusion transcripts of these repetitive elements with a protein-coding gene and pericentromeric satellite repeats in DUX4-fl-expressing muscle cells (14). Among these repetitive elements, Human Satellite II (HSATII) has been shown to form double-stranded RNA (dsRNA) foci and partly play a role in DUX4-fl-induced cytotoxicity (17). In addition, human endogenous retrovirus L (HERVL) is activated by DUX4-fl in preimplantation embryos (13), suggesting the activation of repetitive elements by DUX4-fl may have important roles in FSHD pathology and developmental biology.
The findings to date on DUX4-fl-activated repetitive elements are based on data from short-read next-generation sequencers. However, there may be difficulty in analyzing transcripts comprehensively because there are numerous highly similar sequences in the human genome such as transposable elements (18). Therefore, an alternative approach to complement short-read sequencing may be required for further understanding of the repetitive element-derived transcripts that are activated by DUX4-fl. Long-read sequencing has advantages for analyzing repetitive elements because long enough reads may encompass whole repeat regions, allowing us to specify the genomic location of the repeat (19). In addition, long-read sequencing has better resolution for the transcriptome because it can clearly detect exon connectivity (20,21). Long-read full-length complementary DNA (cDNA) sequence has been shown to be useful for qualitative and quantitative transcriptome analysis (22,23). It was also shown that nanopore can sequence native RNA molecules; namely, direct RNA sequencing (dRNA-seq) (24). This approach has been also shown to be useful for transcriptome analysis, has more potential power to detect RNA modifications and avoids polymerase chain reaction (PCR) or reverse transcription bias (23,25,26).
In this study, we sequenced RNAs with polyA tail from DUX4-fl-expressing human muscle cells, using a nanopore direct RNA sequencer. We catalogued DUX4-fl-activated transcripts from repetitive elements with high confidence by statistically analyzing triplicated samples using DESeq2. Nanopore dRNA-seq confirmed the activation of cleavage-stage-specific genes in muscle cells. Furthermore, we found novel processed ERV transcripts and LTR fusion transcripts induced by DUX4-fl.
Results
Nanopore dRNA-seq
We sequenced polyA-enriched RNA extracted from DUX4-fl or DUX4-s overexpressing RD (rhabdomyosarcoma) cells using a single PromethION flowcell per sample in triplicates (DUX4-fl: fl-1, fl-2, fl-3; DUX4-s: s-1, s-2, s-3). Since DUX4-s does not have a transcriptional activation domain and its transcriptional activity is weak (5,11), we used DUX4-s overexpressing cells as a control. We obtained 5.9 million reads and 6.5 gigabases on average, which were mapped to the human reference genome GRCh38 using LAST (https://github.com/mcfrith/last-rna) (Supplementary Material, Table S1) according to the instructions (https://github.com/mcfrith/last-rna/blob/master/scripts.md). Median read length was 824 bases (658–922 bases) and all datasets show similar distribution of read length (Supplementary Material, Fig. S1). As with any nanopore sequencing, dRNA-seq is prone to errors. We calculated error probabilities using last-train (27) and aligned the reads to the reference genome using these probabilities (Supplementary Material, Fig. S2). Note that these probabilities contain both actual differences and nanopore errors. We could align ~75% of the reads on average to unambiguous sites in the reference genome, which amounts to 4.6 million reads on average (Supplementary Material, Table S1). The remaining reads either aligned ambiguously to two or more sites, or tended to be much shorter and had lower read quality (Supplementary Material, Table S1, Supplementary Material, Fig. S3).
Differentially expressed genes and repeats
Differentially expressed genes (DEGs) between cells with DUX4-fl and DUX4-s were estimated from nanopore count data, based on a negative binomial generalized linear model and Wald test using DESeq2 (28). dRNA-seq showed remarkable elevation of DUX4-fl-induced genes (10,13), including germline genes or early developmental genes (Fig. 1a and b, Supplementary Material, Table S2). This finding is supported by the correlation between mean read count per million from our triplicated nanopore data and transcripts per million from a public DUX4-fl-expressing myoblast dataset (14) (Fig. 1c). Gene ontology analysis shows significant enrichment of transcription regulation pathway and cell death pathway (Supplementary Material, Table S3), as reported previously (10).
Figure 1.
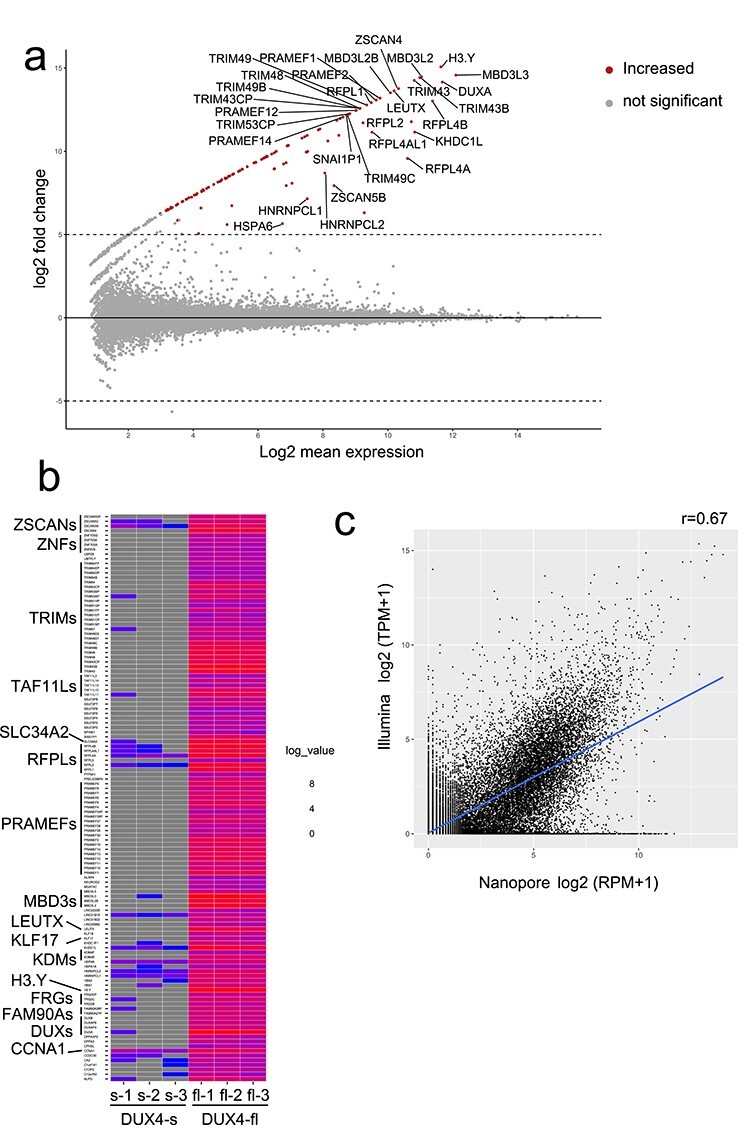
(a) MAplot for differentially expressed genes (DEGs) in triplicated nanopore dRNA-seq data from DUX4-fl and DUX4-s overexpressing RD cells. Only the top 30 genes are shown. The transcripts with adjusted P-value <0.001 and log2fold change ≥5 were determined as differentially expressed transcripts. (b) Heatmap of DEGs. Well-known DUX4 target genes and TAF11-like pseudogenes were significantly upregulated by DUX4-fl overexpression. (c) Correlation between Illumina short-read cDNA-seq and Nanopore long-read dRNA-seq expression in DUX4-fl-induced genes. Pearson correlation coefficients are shown.
Long reads can show exon connectivity clearly as well as isoform detection. For example, C12orf50 has several exons and some transcripts show different exon usage (Supplementary Material, Fig. S4a). Note that 5′-end of the dRNA-seq reads do not always agree with the transcription start site (TSS) because nanopore sequencing may end in the middle of the RNA. Nevertheless, we can observe that many reads originate from a novel ERV-derived TSS (arrow) (Supplementary Material, Fig. S4b). C12orf50 encodes an uncharacterized protein highly expressed in testis, which presumably has a role in embryogenesis along with other DUX4-induced genes. As another example, our data showed that DUX4-fl induced only the functional isoform of LEUTX (LEUTX.n), which has a complete homeodomain, but not the inactive isoform LEUTX.R, which has a partial homeodomain. LEUTX.n is a transcription factor specifically expressed in human preimplantation embryos that activates pluripotency-associated genes (Supplementary Material, Fig. S5) (29). Other isoform-specific transcripts were also detected by dRNA-seq (Supplementary Material, Fig. S6). These results show that our nanopore dRNA-seq robustly detects the DUX4-fl-inducible gene transcripts. In addition to previously reported DUX4-induced genes, our pipeline identified that expression of TAF11-like macrosatellite array pseudogenes was significantly induced by DUX4-fl (30).
Next, we counted transcripts from repetitive sequence by dRNA-seq, using two versions of RepeatMasker annotation: Repbase (version Open-3.0, downloaded from the UCSC (University of California, Santa Cruz) genome browser, https://genome.ucsc.edu) and Dfam (version 3.1, https://www.dfam.org). We then estimated differentially expressed repeats (DERs) using DESeq2 (Fig. 2a and b, Supplementary Material, Fig. S7a and b). Note that a DER represents transcripts that overlap one repeat locus. Most DERs (excluding tandem repeats) have overlapping Repbase and Dfam annotations, so we merged these annotations (Supplementary Material, Table S4). We checked overlap between Repbase and Dfam, and found 44 loci of 61 total DERs agreed (Fig. 2c). As tandem repeat annotation differs in the two databases (e.g. Repbase does not have HSATII annotation), we listed them separately (Supplementary Material, Table S5). LTR retrotransposons were significantly enriched as previously reported, as well as satellite repeats (Supplementary Material, Fig. S8, Supplementary Material, Tables S6 and S7) (10). Publicly available Illumina short-read data for repeats did not have good correlation with nanopore dRNA-seq, probably because short reads have difficulty in detecting the whole transcripts (Fig. 2d, Supplementary Material, Fig. S9). We also compared the detected DER loci with published DUX4–chromatin immunoprecipitation (ChIP) peaks (10). DUX4–ChIP peaks were near these DERs (Fig. 2e, Supplementary Material, Fig. S7c), suggesting DERs are actually activated by DUX4-fl.
Figure 2.
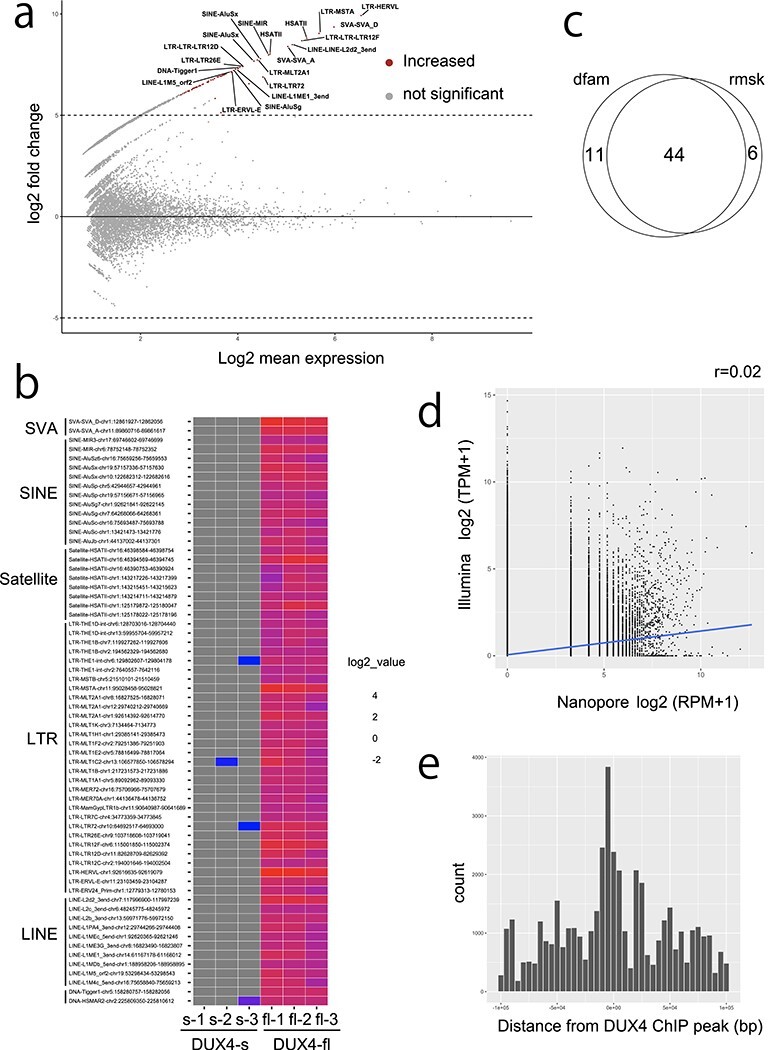
(a) MAplot for differentially expressed repeats (DERs) from Dfam repeats in triplicated dRNA-seq data from DUX4-fl and DUX4-s overexpressing RD cells. Only the top 20 repeats are shown. Transcripts from repetitive elements with adjusted P-value <0.001 and log2fold change ≥5 were determined as differentially expressed repeats. (b) Heatmap of DERs. Many of the upregulated DERs are ERV–MaLR, which confirms previous reports (14). (c) Venn diagram shows 44 repeats overlap in the Dfam and Repbase annotations. (d) Correlation is not observed (r = 0.02) between nanopore and Illumina read counts in Dfam repeats. Pearson correlation coefficients are shown. (e) DERs were located near the DUX4–ChIP peaks as previously reported (10).
To confirm the results of dRNA-seq, we selected four representative DERs induced by DUX4-fl [L1MD, L1MCa-antisense (AS), L1ME1 and MER72] and performed reverse transcription polymerase chain reaction (RT-PCR) analysis with DUX4-fl- or DUX4-s-transfected RD cells. The RT-PCR analysis showed that the four DERs were specifically induced by DUX4-fl (Supplementary Material, Fig. S10, primer sequences are in Supplementary Material, Table S8). We also examined expression of the four DERs in myocytes differentiated from FSHD1 and FSHD2 patient-derived induced pluripotent stem (iPS) cells as well as a healthy control (Fig. 3, Supplementary Material, Fig. S11). L1ME1 was specifically expressed in both FSHD1 and FSHD2 myocytes, and MER72 was specifically upregulated in both FSHD1 and FSHD2 myocytes (Fig. 3a, Supplementary Material, Fig. S12). Because one of the FSHD2 clones showed much higher expression of DUX4, comparison of the mean MER72 expression levels showed no statistically significant difference. However, the clone expressing DUX4 at a higher level showed higher MER72 expression, and expression levels of DUX4 were well correlated with the expression levels of L1ME1 and MER72 (Fig. 3b). When we compared the expression levels between FSHD1 myocytes and healthy control, we found a statistically significant difference (Student t-test, P < 0.01, data not shown). These results suggested that L1ME1 and MER72 are induced by DUX4 in FSHD patient-derived myocytes, where DUX4 is expressed at a physiological level. The expression of L1MD and L1MCa-AS was detected not only in FSHD myocytes, but also in the healthy control, although expression of these two DERs was undetected in the RD cells transfected with DUX4-s. This might be because of the differentiation state of the cell since some repetitive elements are activated in iPS-derived cells (31).
Figure 3.
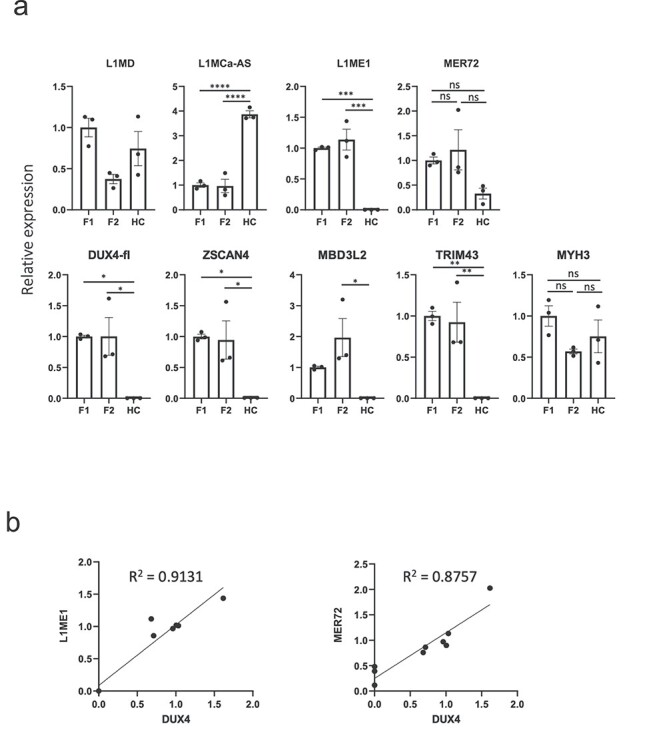
(a) RT-qPCR analysis of selected DERs and DUX4 target genes in myocytes differentiated from FSHD1 and FSHD2 patient-derived iPS cells. L1ME1 and MER72 were specifically upregulated in both FSHD1- and FSHD2-derived myocytes. The expression of MYH3 was measured as a muscle differentiation marker. Data are mean ± SEM. The differences are tested by one-way ANOVA followed by Tukey’s Multiple Comparison Test. P-values are as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (b) Expression level of DUX4 well correlated with the expression levels of L1ME1 and MER72. Pearson correlation coefficients are shown.
It was reported that DUX4-fl induces pericentric human satellite repeats HSATII bidirectionally and one strand is less expressed than the other (17). Our dRNA-seq showed that expression of satellite repeats from five loci was induced by DUX4-fl. Among them, we confirmed the expression of HSATII from chromosome 1 as previously reported (17). The satellite repeats from the five loci were expressed almost unidirectionally, but only a few opposite strand reads were detected (Supplementary Material, Fig. S13, Supplementary Material, Table S5), which corresponds to the previous finding that one strand is less expressed than the other (17). These results suggest that DUX4-fl may regulate transcription of satellite repeats on multiple chromosomes in a bidirectional but strand-biased manner. Interestingly, these transcripts from satellite repeats have polyA tails as long as those of mRNAs from protein-coding genes with the longest polyA (Supplementary Material, Fig. S14) (32).
Splicing is common in DUX4-induced ERVs
ERVs are known to be processed to produce mature transcripts (33). We found most of the DUX4-induced ERVs were spliced (Fig. 4a and b, Supplementary Material, Fig. S15) using the canonical splice donor and acceptor signals (Fig. 4c, Supplementary Material, Fig. S16), although the precise coordinates of the splice sites are uncertain because of error-prone nanopore sequencing (Supplementary Material, Fig. S2, Supplementary Material, Table S1).
Figure 4.
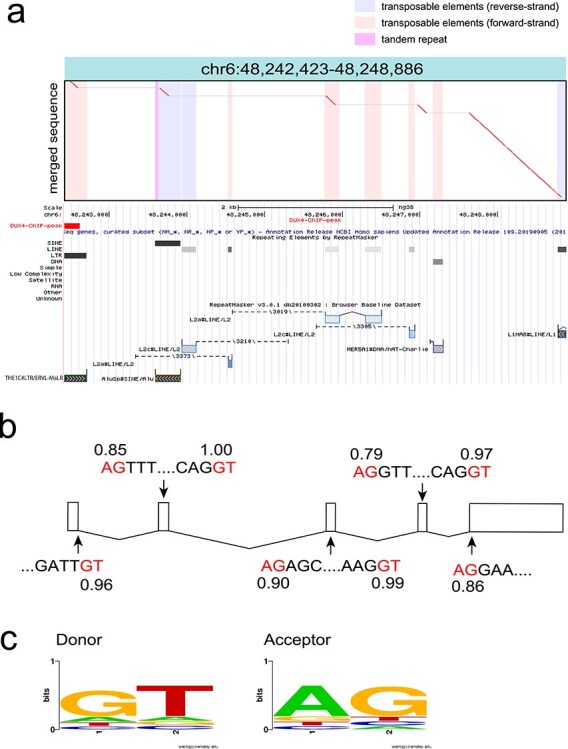
(a) An example dotplot of spliced repeat transcript. All detected transcripts from fl-1, fl-2 and fl-3 were merged using lamassemble, and then aligned to the reference genome. This transcript was transcribed from an ERVL–MaLR, and then four splicing events occurred. Merged sequence is on the vertical axis and reference genome is on the horizontal axis in the dotplot. Vertical colored striped represent repeats in the reference genome. Below the dotplot, RepeatMasker with Repbase annotation is shown from the UCSC genome browser. (b) Splicing occurred at canonical splice donor and acceptor signals (red letters). Splice site prediction was done using NNSPLICE (v.9.0) (https://www.fruitfly.org/seq_tools/splice.html). The splice donor and acceptor prediction scores are shown. All splicing sites were predicted from LAST alignment. (c) Donor and acceptor sequences are shown as a sequence logo. Sequence logo was created by WebLogo (https://weblogo.berkeley.edu/logo.cgi).
Furthermore, we found that non-LTR retrotransposons (e.g. Alu elements) are actually transcribed from nearby LTRs and processed (Supplementary Material, Fig. S17a). This AluSx on chromosome 10 has a polyadenylation signal at the 3′ end (Supplementary Material, Fig. S17b). Indeed, the measurement of polyA tail length using nanopore signals shows this AluSx has a polyA tail as long as mRNAs from protein-coding genes (Supplementary Material, Figs S14 and S17c). RNA folding prediction using RNAfold (34) shows that this AluSx could form dsRNA (Supplementary Material, Fig. S18).
Overall, long-read sequence revealed that 35 of 61 DERs were spliced (Supplementary Material, Table S4). We also found that 12 DERs were located downstream of DUX4-induced genes and six DERs were located within introns of DUX4-induced genes.
Fusion transcripts with LTR retrotransposons
The LTRs of DUX4-fl-induced ERVs were reported to act as promoters and to generate fusion transcripts of ERVs with nearby genes. We explored fusion transcripts including LTRs (called ‘LTR fusion transcripts’) using dRNA-seq. As one of the triplicated DUX4-fl replicates (fl-1) has less data than the other two replicates, we only listed LTR fusion transcripts that are present in both of the other replicates (fl-2 and fl-3) and never present in any of the DUX4-s overexpressing replicates. We listed 247 such LTR fusion transcripts with genes, pseudogenes or long non-coding RNAs (lncRNAs) (Supplementary Material, Fig. S19, Supplementary Material, Table S9), among which 216 were not reported previously (14). Some examples of DUX4-fl-induced LTR fusion transcripts are shown in Figure 5a and Supplementary Material, Figure S19. Among the 247 LTR fusion transcripts, 15 transcripts showed statistically significant increase of gene body expression (Fig. 5b). Although we confirmed 25 LTR fusion transcripts that were reported by Young et al. (14), we could not detect 86 LTR fusion transcripts. Most of these transcripts only have <100 raw Illumina reads, which may be hard to detect with smaller numbers of nanopore dRNA-seq reads (Supplementary Material, Fig. S20). Interestingly, the long-read sequencing detected splicing variants of some of the LTR fusion transcripts. For example, the previous study reported RBM26 as one LTR fusion transcript (Supplementary Material, Fig. S19g). However, our analysis revealed that two different splicing isoforms of the LTR–RBM26 fusion transcript are induced by DUX4-fl. This result suggests that long-read sequencing has an advantage for detecting whole isoforms of transcripts.
Figure 5.
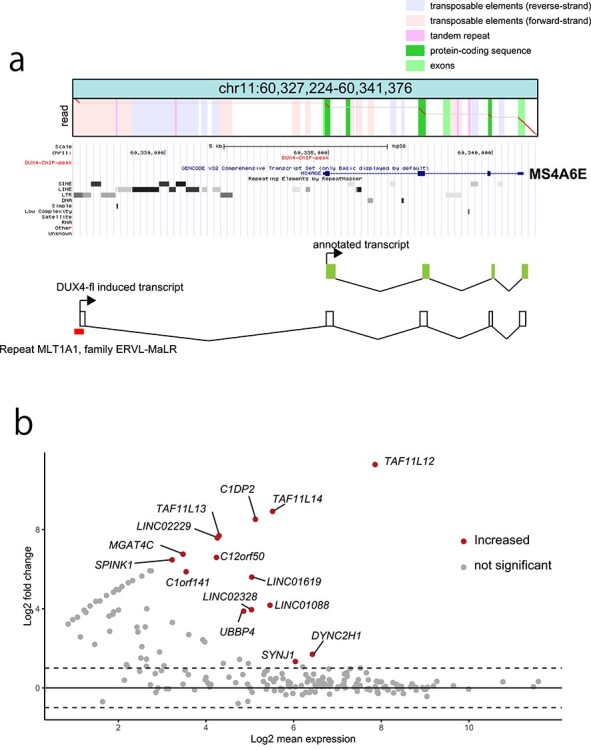
(a) An example fusion transcript read of an ERVL–MaLR with the MS4A6E gene, induced by DUX4-fl. Below the dotplot, RepeatMasker with Repbase annotation is shown from the UCSC genome browser. (b) MAplot of differentially expressed LTR fusion transcripts. Fifteen LTR fusion transcripts (genes) show significant upregulation in DUX4-fl-transfected cells compared with DUX4-s-transfected cells.
Discussion
Direct sequencing of RNA molecules is a highly anticipated technology because it avoids PCR or reverse transcription biases, and also has the possibility to provide RNA modification signatures. However, it is still not clear whether nanopore dRNA-seq is suited for much biology research because of the requirement for a large amount of RNA and low throughput (23). Our study showed that dRNA-seq using PromethION, a high-throughput nanopore sequencer, is applicable to detect differentially expressed transcripts including genes, lncRNA, satellite repeats and retrotransposons, and also provides example datasets to the research community.
The transcriptome of DUX4-fl-expressing myoblasts using an Illumina short-read sequencer was previously reported (10,14). We compared our results of nanopore long-read dRNA-seq to the previous findings. As we utilized public data of short-read sequencing, which has no biological replicates, fair comparison may be difficult. Nevertheless, we saw some consistency of gene expression between our long-read data and the previous short-read data. We observed activation of known DUX4-induced germline genes and cell cycle genes in our long-read dRNA-seq data, comparable with the previous reports by short-read sequencing. In addition, activation of ERVs and tandem repeats are also comparable with the previous results, which shows the feasibility of using long-read dRNA-seq to detect DUX4-activated repeats. We identified 79 DUX4-induced repetitive elements (including 18 tandem repeats, which may be overlapping) using two different repetitive sequence databases (Repbase and Dfam). Most of them were transcribed from DUX4-activated LTRs, as reported previously (14). In addition to the known findings, long-read dRNA-seq clearly shows that RNA splicing is a common event in DUX4-activated ERV transcripts.
ERVs have two LTR sequences that genuinely have promoter activity, and some of them are co-opted as promoters affecting the expression of nearby genes (35). It has been reported that binding of DUX4 to the LTRs activates genes or lncRNAs. Our dRNA-seq identified novel transcripts in addition to reported ones. DESeq2 analysis confirmed that some of these fusion genes are upregulated compared with DUX4-s, suggesting that these DUX4-fl-activated aberrant transcripts contribute to altered gene expression (Fig. 5b). We also identified several pseudogenes such as UBBP4 that were activated by DUX4. These pseudogenes may actually be genes, and may only be expressed in rare situations (e.g. cleavage-stage embryo or FSHD muscle).
It was reported that DUX4-fl also activates satellite repeats and causes toxic intranuclear dsRNA foci (17). Our dRNA-seq also detected transcripts from several satellite repeat loci. We did not observe obvious bidirectional transcription of satellite repeats: only a few antisense reads were observed, which may be concordant with a previous report (17). Nanopore sequencing error in reading the opposite strand seems an unlikely explanation, because several different tandem repeats showed a unidirectional pattern. Our results show that most of the DUX4-activated non-LTR retrotransposons (e.g. Alu elements) were transcribed from ERVs and underwent RNA processing, indicating the importance of analyzing full-length transcripts. Although our dRNA-seq analysis did not detect inverted tandem Alu elements that are often contained in human dsRNA, RNA folding prediction indicated that the AluSx induced by DUX4-fl may form dsRNA. Further study is needed to explore the roles of DUX4-induced Alu elements.
A large proportion of the annotated Alu elements in the human genome are embedded in the introns or 3′ untranslated regions of protein-coding genes and lncRNAs that are transcribed by RNA polymerase II (Pol II) (36,37). However, other ‘free Alu elements’ are transcribed from weak internal A and B boxes of the RNA polymerase III (Pol III) promoter, which are boosted by an upstream Pol III enhancer for efficient but very low transcription (36,37). These ‘free Alu elements’ usually do not have long polyA tails. We showed an interesting example where a DUX4-bound LTR transcribed an AluSx, which is not embedded in any gene. The dRNA-seq clearly shows the AluSx has a polyA tail, implying transcription by pol II and termination at a downstream polyadenylation signal. This showed that dRNA-seq has an advantage in providing native polyA length. In addition to the example of Alu elements, we showed the first evidence that DUX4-activated satellite repeats have long polyA tails. The exact mechanism by which satellite repeats are transcribed is unknown, but this novel finding indicates the usefulness of nanopore dRNA-seq for transcriptome analysis.
In conclusion, by nanopore long-read dRNA-seq with the PromethION, we obtained enough reads to detect DUX4-fl-induced genes and transposons with statistical significance. In addition, we could detect novel transcripts that were activated by DUX4-bound LTRs. Further improvements of the technologies (e.g. reducing the required input RNA amount or increased output) may expand the usefulness of this technology.
Materials and Methods
Cell culture, DUX4 transfection and extracting RNA
Human rhabdomyosarcoma-derived cell line, RD cells, were obtained from the American Type Culture Collection (ATCC) (CCL-136, University Boulevard Manassas, VA, USA). The RD cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (cat. D5796, Sigma-Aldrich, St Louis, MO, USA) supplemented with fetal bovine serum (cat. 10270-106, ThermoFisher Scientific, Grand Island, NY, USA) at 37°C in a humidified atmosphere of 5% CO2. The DUX4-fl and DUX4-s plasmids were prepared as described previously (5). The RD cells were plated on six-well plates at the density of ~30 × 104 cells per well. The next day, 2 μg of DUX4 expression constructs were transfected into RD cells with 6 μl of the X-treme GENE 9 DNA transfection reagent (cat. XTG9-RO, Roche Diagnostics, Indianapolis, IN, USA) diluted in 100 μl of Opti-MEM I (Gibco) following the manufacturer’s instructions. Twenty-four hour after transfection, total RNA was extracted with TRIzol reagent (cat. 15596018, Invitrogen) with DNase I treatment (cat.79254, QIAGEN). The total RNA solution from 12 wells was passed through one column of RNAeasy mini kit (cat.74104, QIAGEN) to obtain ~100 μg of purified total RNA. Then, polyA RNA was isolated from 60 to 90 μg of total RNA using μMACS mRNA Isolation Kits (Miltenyi Biotec Inc., CA, USA). PolyA tail-containing RNA (500 ng) was subjected to nanopore library preparation.
PromethION direct RNA nanopore sequencing
Library preparation was done using SQK-RNA002 Direct RNA Sequencing Kit according to the manufacturer’s protocol and sequenced by PromethION sequencer using one flowcell (FLO-PRO002) per sample (Oxford Nanopore Technologies, UK). Basecalling was done by MinKNOW v2.2 and Guppy v1.8.5 with default settings.
Read statistics were obtained using seqkit stat (https://bioinf.shenwei.me/seqkit/usage/). Exon bases before the alignment’s 5′-end were obtained using rna-alignment-stats (https://github.com/mcfrith/last-rna).
Mapping to the human reference genome
Nanopore direct RNA sequence reads (dRNA-seq.fastq) were mapped to the human reference genome GRCh38 according to the online instructions (https://github.com/mcfrith/last-rna/blob/master/last-long-reads.md).
Briefly, a reference database was generated using LAST v959 as follows:
lastdb -P8 -uNEAR -R01 GRCh38 GRCh38.fa.
Then, nanopore reads were aligned to GRCh38 with parameters for each dataset obtained using last-train:
last-train -Q0 GRCh38 dRNA-seq.fastq > train.out
parallel-fasta "lastal -p train.out -d90 -m20 -D10 mydb | last-split -d2 -g GRCh38" < dRNA-seq.fastq > aln.maf
Note the -d2 option is for cDNA of unknown/mixed strands. dRNA-seq reads are presumably of RNA forward strands; this option may be omitted.
Finding and counting genes and repeat overlapping reads
Aligned files (aln.maf) were subjected to read counting according to the online instructions (https://github.com/mcfrith/last-rna/blob/master/scripts.md).
Gene annotation (knownGene.txt) was obtained from the UCSC genome browser (http://hgdownload.soe.ucsc.edu/goldenPath/hg38/database/).
First, gene overlap regions were obtained as follows:
gene-overlap.sh genes file.maf > out
tail -n + 2 out | cut -f3 | cut -d, -f1 | uniq -c | awk '{print $2"\t"$1}' | sort > gene-count
For counting repeat regions, the above gene annotation was combined with Repbase (rmsk.txt) or Dfam annotation, and then only repeat regions were examined:
gene-overlap.sh genes+repeats file.maf > out
tail -n + 2 out | cut -f3 | uniq -c | grep 'rmsk,' | awk '{print $2"\t"$1}' | sort > repeat-count
Merging nanopore reads using lamassemble
Nanopore reads were merged into consensus sequence using lamassemble (38,39). We used fl-2 last-train output as a representative parameter set:
lamassemble train-out merged.fa > consensus.fa
The consensus sequence was realigned to GRCh38 and then dotplot pictures were drawn as previously described (38).
DEGs or DERs
We estimated DEGs or DERs using DESeq2 (28). For both genes and repeats, we determined the transcripts with adjusted P-value <0.001 and log2fold change ≥5 as differentially expressed transcripts.
Repeat enrichment analysis
We conducted an enrichment analysis of each repeat name (Dfam and Repbase), class and family (Repbase) of DERs by comparing the observed fraction of repeats with the fraction identified in the human genome (GRCh38). Based on those fractions and the number of repeats, Z-scores were calculated, and adjusted P-values were also obtained based on chi-squared tests with Bonferroni correction.
Finding fusion transcripts with LTRs
To find fusion transcripts including genes and LTRs, we used the gene fusion script from (https://github.com/mcfrith/last-rna/blob/master/scripts.md):
gene-fusions.sh genes.txt alignments.maf > fusions.txt
Transcripts fused with LTR annotations from RepeatMasker+Repbase that are upstream or in the gene bodies are counted. As the number of reads in one of the DUX4-fl replicates was about half of the other two replicates, we counted only the reads that were not expressed in any of three replicates with DUX4-s expression. Differentially expressed fusion transcripts were also determined by DESeq2.
Public Illumina cDNA-seq and ChIP-seq data
Publicly available Illumina short-read cDNA-seq data of DUX4-fl overexpressing human myoblast cells (MB-135, DUX4-fl or GFP was introduced by lentiviral infection. Accession numbers are SRR823337 and SRR8233378) were re-analyzed using STAR v2.7 (40). Ribosomal RNA sequence was removed using bowtie2 v2.2.4 (41) with iGenome indexes (https://jp.support.illumina.com/sequencing/sequencing_software/igenome.html). Only uniquely mapped reads (MAPQ 255) were counted.
Coordinates of DUX4 ChIP-seq peaks (10) were converted to GRCh38 using UCSC liftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver). We used the middle coordinate of the peak and that of the repeat to show the distance.
polyA tail analysis using nanopore sequencing
Nanopore fast5 files (signal data) from fl-3 that were mapped to satellite repeats, LTR transcribed AluSx and seven genes were collected, and their polyA lengths were estimated using tailfindr (https://github.com/adnaniazi/tailfindr) (42). For genes, we chose two housekeeping genes (ACTB and GAPDH) and five genes, which were previously shown to have longest (DDX17 and DDX5), median (SRP14) and shortest (OLA1 and RPS24) polyA length (26) in comparison.
FSHD patient-derived iPS cell analysis
iPS cell clones with Tetracycline (TET)-inducible myogenic potential were previously established from a patient with FSHD1, a patient with FSHD2 and a healthy control donor (43). Myocyte differentiation was conducted as previously described with minor modifications (43,44). Duration and concentration of doxycycline stimulation was optimized for each iPS cell clone, from 4 to 5 days and from 0.3 to 1.0 μg/ml, respectively, to achieve comparable myogenic differentiation. A total of 5 μM SB431542 (Nacalai) and 10 ng/ml human IGF-1 (Pepro Tech) were added after doxycycline stimulation for enhanced differentiation. Total RNA was extracted from differentiated myocytes using ReliaPrep RNA Miniprep (Promega) and cDNA was synthesized using SuperScript III reverse transcriptase (ThermoFisher Scientific) with an Oligo(dT)20 primer, and then subjected to reverse transcription quantitative polymerase chain reaction (RT-qPCR) as previously described (43). Primers used are shown in Supplementary Material, Table S8. Electrophoresis for RT-qPCR amplicons from each representative well were conducted with 2% agarose gel. To confirm myogenic differentiation, differentiated myocytes were fixed and immunostained against primary antibodies (mouse monoclonal IgG2 anti-myosin heavy chain antibody, eBioscience, 14-6503-82; rabbit monoclonal anti-MyoD antibody, Abcam, ab133627; mouse monoclonal IgG1 anti-myogenin antibody, Santa Cruz Biotechnology, sc-12 732) followed by secondary antibodies (anti-mouse IgG2b-Alexa Fluor 488, ThermoFisher Scientific, A21141; anti-rabbit IgG-Alexa Fluor 568, ThermoFisher Scientific, A11036; anti-mouse IgG1-Alexa Fluor 568, ThermoFisher Scientific, A21124), respectively.
Supplementary Material
Acknowledgements
The authors are grateful to Terumi Horiuchi, Kazumi Abe, Ayako Suzuki and Yutaka Suzuki for their support. Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics and partially performed on Matsumoto-lab computer at Yokohama City University.
Conflict of Interest statement: None declared.
Contributor Information
Satomi Mitsuhashi, Department of Genomic Function and Diversity, Tokyo Medical and Dental University, Tokyo 113-8510, Japan; Department of Human Genetics, Yokohama City University, Yokohama, Kanagawa 236-0004, Japan.
So Nakagawa, Micro/Nano Technology Center, Tokai University, Hiratsuka, Kanagawa 259-1292, Japan; Department of Molecular Life Science, Tokai University School of Medicine, Isehara, Kanagawa 259-1193, Japan.
Mitsuru Sasaki-Honda, Center for iPS Cell Research and Application (CiRA), Kyoto University, 53 Shogoin Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan.
Hidetoshi Sakurai, Center for iPS Cell Research and Application (CiRA), Kyoto University, 53 Shogoin Kawahara-cho, Sakyo-ku, Kyoto 606-8507, Japan.
Martin C Frith, Artificial Intelligence Research Center, National Institute of Advanced Industrial Science and Technology (AIST), Tokyo 135-0064, Japan; Graduate School of Frontier Sciences, University of Tokyo, Chiba 277-8561, Japan; Computational Bio Big-Data Open Innovation Laboratory (CBBD-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Tokyo 169-8555, Japan.
Hiroaki Mitsuhashi, Micro/Nano Technology Center, Tokai University, Hiratsuka, Kanagawa 259-1292, Japan; Department of Applied Biochemistry, School of Engineering, Tokai University, Hiratsuka, Kanagawa 259-1292, Japan.
Funding
JSPS KAKENHI (JP19K07977, 16H06279 (PAGS) to S.M.), (16H06429, 16 K21723, 17H05823, 20 K06775 to S.N.), (18 K07511 to H.M.) and (19 K16611, 20 J01478 to M.S.-H.). Partially supported by a grant from The Acceleration Program for Intractable Diseases Research utilizing Disease-specific iPS cells (#20bm0804005h0004), which were provided by the Japan Agency for Medical Research and Development (to H.S.).
Data Availability
Direct RNA-seq data is available under accession numbers DRA008398 (under project number PRJDB8318) from DDBJ DRA database (https://www.ddbj.nig.ac.jp/dra/index-e.html). Illumina human myoblast sequences were obtained under accession numbers SRR823337 and SRR823338.
Web Resources
UCSC genome browser: https://genome.ucsc.edu
LAST: https://gitlab.com/mcfrith/last
lamassemble: https://gitlab.com/mcfrith/lamassemble
STAR: https://github.com/alexdobin/STAR
Illumina iGenome: https://support.illumina.com/sequencing/sequencing_software/igenome.html
RNAfold: http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi
References
- 1. Gabriels, J., Beckers, M.C., Ding, H., De Vriese, A., Plaisance, S., van der Maarel, S.M., Padberg, G.W., Frants, R.R., Hewitt, J.E., Collen, D. and Belayew, A. (1999) Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene, 236, 25–32. [DOI] [PubMed] [Google Scholar]
- 2. Lemmers, R.J., van der Vliet, P.J., Klooster, R., Sacconi, S., Camano, P., Dauwerse, J.G., Snider, L., Straasheijm, K.R., van Ommen, G.J., Padberg, G.W. et al. (2010) A unifying genetic model for facioscapulohumeral muscular dystrophy. Science, 329, 1650–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Overveld, P.G., Lemmers, R.J., Sandkuijl, L.A., Enthoven, L., Winokur, S.T., Bakels, F., Padberg, G.W., van Ommen, G.J., Frants, R.R. and van der Maarel, S.M. (2003) Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat. Genet., 35, 315–317. [DOI] [PubMed] [Google Scholar]
- 4. Lemmers, R.J., Tawil, R., Petek, L.M., Balog, J., Block, G.J., Santen, G.W., Amell, A.M., van der Vliet, P.J., Almomani, R., Straasheijm, K.R. et al. (2012) Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet., 44, 1370–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitsuhashi, H., Mitsuhashi, S., Lynn-Jones, T., Kawahara, G. and Kunkel, L.M. (2013) Expression of DUX4 in zebrafish development recapitulates facioscapulohumeral muscular dystrophy. Hum. Mol. Genet., 22, 568–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pakula, A., Lek, A., Widrick, J., Mitsuhashi, H., Bugda Gwilt, K.M., Gupta, V.A., Rahimov, F., Criscione, J., Zhang, Y., Gibbs, D. et al. (2019) Transgenic zebrafish model of DUX4 misexpression reveals a developmental role in FSHD pathogenesis. Hum. Mol. Genet., 28, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bosnakovski, D., Chan, S.S.K., Recht, O.O., Hartweck, L.M., Gustafson, C.J., Athman, L.L., Lowe, D.A. and Kyba, M. (2017) Muscle pathology from stochastic low level DUX4 expression in an FSHD mouse model. Nat. Commun., 8, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones, T.I., Chew, G.L., Barraza-Flores, P., Schreier, S., Ramirez, M., Wuebbles, R.D., Burkin, D.J., Bradley, R.K. and Jones, P.L. (2020) Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle, 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Snider, L., Geng, L.N., Lemmers, R.J., Kyba, M., Ware, C.B., Nelson, A.M., Tawil, R., Filippova, G.N., van der Maarel, S.M., Tapscott, S.J. and Miller, D.G. (2010) Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet., 6, e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geng, L.N., Yao, Z., Snider, L., Fong, A.P., Cech, J.N., Young, J.M., van der Maarel, S.M., Ruzzo, W.L., Gentleman, R.C., Tawil, R. and Tapscott, S.J. (2012) DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell, 22, 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mitsuhashi, H., Ishimaru, S., Homma, S., Yu, B., Honma, Y., Beermann, M.L. and Miller, J.B. (2018) Functional domains of the FSHD-associated DUX4 protein. Biol Open, 7, bio033977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choi, S.H., Gearhart, M.D., Cui, Z., Bosnakovski, D., Kim, M., Schennum, N. and Kyba, M. (2016) DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res., 44, 5161–5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hendrickson, P.G., Dorais, J.A., Grow, E.J., Whiddon, J.L., Lim, J.W., Wike, C.L., Weaver, B.D., Pflueger, C., Emery, B.R., Wilcox, A.L. et al. (2017) Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet., 49, 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Young, J.M., Whiddon, J.L., Yao, Z., Kasinathan, B., Snider, L., Geng, L.N., Balog, J., Tawil, R., van der Maarel, S.M. and Tapscott, S.J. (2013) DUX4 binding to retroelements creates promoters that are active in FSHD muscle and testis. PLoS Genet., 9, e1003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mager, D.L. and Stoye, J.P. (2015) Mammalian endogenous retroviruses. Microbiol Spectr., 3, MDNA3-0009-2014. [DOI] [PubMed] [Google Scholar]
- 16. Peaston, A.E., Evsikov, A.V., Graber, J.H., de Vries, W.N., Holbrook, A.E., Solter, D. and Knowles, B.B. (2004) Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell, 7, 597–606. [DOI] [PubMed] [Google Scholar]
- 17. Shadle, S.C., Bennett, S.R., Wong, C.J., Karreman, N.A., Campbell, A.E., van der Maarel, S.M., Bass, B.L. and Tapscott, S.J. (2019) DUX4-induced bidirectional HSATII satellite repeat transcripts form intranuclear double stranded RNA foci in human cell models of FSHD. Hum. Mol. Genet., 28, 3997–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richardson, S.R., Doucet, A.J., Kopera, H.C., Moldovan, J.B., Garcia-Perez, J.L. and Moran, J.V. (2015) The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr., 3, MDNA3-0061-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mitsuhashi, S. and Matsumoto, N. (2020) Long-read sequencing for rare human genetic diseases. J. Hum. Genet., 65, 11–19. [DOI] [PubMed] [Google Scholar]
- 20. Bolisetty, M.T., Rajadinakaran, G. and Graveley, B.R. (2015) Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol., 16, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clark, M.B., Wrzesinski, T., Garcia, A.B., Hall, N.A.L., Kleinman, J.E., Hyde, T., Weinberger, D.R., Harrison, P.J., Haerty, W. and Tunbridge, E.M. (2020) Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol. Psychiatry, 25, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Byrne, A., Beaudin, A.E., Olsen, H.E., Jain, M., Cole, C., Palmer, T., DuBois, R.M., Forsberg, E.C., Akeson, M. and Vollmers, C. (2017) Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun., 8, 16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seki, M., Katsumata, E., Suzuki, A., Sereewattanawoot, S., Sakamoto, Y., Mizushima-Sugano, J., Sugano, S., Kohno, T., Frith, M.C., Tsuchihara, K. et al. (2019) Evaluation and application of RNA-Seq by MinION. DNA Res., 26, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garalde, D.R., Snell, E.A., Jachimowicz, D., Sipos, B., Lloyd, J.H., Bruce, M., Pantic, N., Admassu, T., James, P., Warland, A. et al. (2018) Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods, 15, 201–206. [DOI] [PubMed] [Google Scholar]
- 25. Soneson, C., Yao, Y., Bratus-Neuenschwander, A., Patrignani, A., Robinson, M.D. and Hussain, S. (2019) A comprehensive examination of nanopore native RNA sequencing for characterization of complex transcriptomes. Nat. Commun., 10, 3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Workman, R.E., Tang, A.D., Tang, P.S., Jain, M., Tyson, J.R., Razaghi, R., Zuzarte, P.C., Gilpatrick, T., Payne, A., Quick, J. et al. (2019) Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods, 16, 1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hamada, M., Ono, Y., Asai, K. and Frith, M.C. (2017) Training alignment parameters for arbitrary sequencers with LAST-TRAIN. Bioinformatics, 33, 926–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jouhilahti, E.M., Madissoon, E., Vesterlund, L., Tohonen, V., Krjutskov, K., Plaza Reyes, A., Petropoulos, S., Mansson, R., Linnarsson, S., Burglin, T. et al. (2016) The human PRD-like homeobox gene LEUTX has a central role in embryo genome activation. Development, 143, 3459–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tremblay, D.C., Alexander, G., Jr., Moseley, S. and Chadwick, B.P. (2010) Expression, tandem repeat copy number variation and stability of four macrosatellite arrays in the human genome. BMC Genomics, 11, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohnuki, M., Tanabe, K., Sutou, K., Teramoto, I., Sawamura, Y., Narita, M., Nakamura, M., Tokunaga, Y., Nakamura, M., Watanabe, A. et al. (2014) Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc. Natl. Acad. Sci. U. S. A., 111, 12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shafin, K., Pesout, T., Lorig-Roach, R., Haukness, M., Olsen, H.E., Bosworth, C., Armstrong, J., Tigyi, K., Maurer, N., Koren, S. et al. (2020) Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol., 38, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trejbalova, K., Blazkova, J., Matouskova, M., Kucerova, D., Pecnova, L., Vernerova, Z., Heracek, J., Hirsch, I. and Hejnar, J. (2011) Epigenetic regulation of transcription and splicing of syncytins, fusogenic glycoproteins of retroviral origin. Nucleic Acids Res., 39, 8728–8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lorenz, R., Bernhart, S.H., Honer Zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P.F. and Hofacker, I.L. (2011) ViennaRNA Package 2.0. Algorithms Mol. Biol., 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hurst, T.P. and Magiorkinis, G. (2017) Epigenetic control of human endogenous retrovirus expression: focus on regulation of long-terminal repeats (LTRs). Viruses, 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang, X.O., Gingeras, T.R. and Weng, Z. (2019) Genome-wide analysis of polymerase III-transcribed Alu elements suggests cell-type-specific enhancer function. Genome Res., 29, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hasler, J., Samuelsson, T. and Strub, K. (2007) Useful 'junk': Alu RNAs in the human transcriptome. Cell. Mol. Life Sci., 64, 1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frith, M.C.M.S. and Katoh, K. (2021) lamassemble: multiple alignment and consensus sequence of long reads. Methods Mol. Biol., 2231, 135–145. [DOI] [PubMed] [Google Scholar]
- 39. Mitsuhashi, S., Ohori, S., Katoh, K., Frith, M.C. and Matsumoto, N. (2020) A pipeline for complete characterization of complex germline rearrangements from long DNA reads. Genome Med., 12, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmead, B., Trapnell, C., Pop, M. and Salzberg, S.L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krause, M., Niazi, A.M., Labun, K., Torres Cleuren, Y.N., Muller, F.S. and Valen, E. (2019) tailfindr: alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA, 25, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sasaki-Honda, M., Jonouchi, T., Arai, M., Hotta, A., Mitsuhashi, S., Nishino, I., Matsuda, R. and Sakurai, H. (2018) A patient-derived iPSC model revealed oxidative stress increases facioscapulohumeral muscular dystrophy-causative DUX4. Hum. Mol. Genet., 27, 4024–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sasaki-Honda, M., Kagita, A., Jonouchi, T., Araki, T., Hotta, A. and Sakurai, H. (2020) Generation of a transgene-free iPSC line and genetically modified line from a facioscapulohumeral muscular dystrophy type 2 (FSHD2) patient with SMCHD1 p.Lys607Ter mutation. Stem Cell Res., 47, 101884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Direct RNA-seq data is available under accession numbers DRA008398 (under project number PRJDB8318) from DDBJ DRA database (https://www.ddbj.nig.ac.jp/dra/index-e.html). Illumina human myoblast sequences were obtained under accession numbers SRR823337 and SRR823338.