

Abstract
The PHF6 mutation c.1024C > T; p.R342X, is a recurrent cause of Börjeson–Forssman–Lehmann Syndrome (BFLS), a neurodevelopmental disorder characterized by moderate–severe intellectual disability, truncal obesity, gynecomastia, hypogonadism, long tapering fingers and large ears (MIM#301900). Here, we generated transgenic mice with the identical substitution (R342X mice) using CRISPR technology. We show that the p.R342X mutation causes a reduction in PHF6 protein levels, in both human and mice, from nonsense-mediated decay and nonsense-associated alternative splicing, respectively. Magnetic resonance imaging studies indicated that R342X mice had a reduced brain volume on a mixed genetic background but developed hydrocephaly and a high incidence of postnatal death on a C57BL/6 background. Cortical development proceeded normally, while hippocampus and hypothalamus relative brain volumes were altered. A hypoplastic anterior pituitary was also observed that likely contributes to the small size of the R342X mice. Behavior testing demonstrated deficits in associative learning, spatial memory and an anxiolytic phenotype. Taken together, the R342X mice represent a good preclinical model of BFLS that will allow further dissection of PHF6 function and disease pathogenesis.
Introduction
Börjeson–Forssman–Lehman Syndrome (BFLS; MIM# 301900) is an X-linked disorder caused by mutations in the PHF6 (NM_001015877.2) gene (1). The characteristic clinical symptoms of BFLS are evident soon after birth and include moderate-to-severe mental retardation, large ears, small external genitalia and long tapering fingers. In addition, a range of endocrine problems that include gynecomastia and truncal obesity develop during adolescence, while a few individuals present with hypotonia and seizures (2–5). Males are predominantly affected with about 50% of obligate female carriers usually showing considerable X-chromosome inactivation skewing indicative of a selection against cells expressing a mutant PHF6 protein during early development (6). However, other female carriers do not present with X-inactivation skewing but have shown milder clinical features of BFLS (7,8). In addition, other females with de novo PHF6 mutations present with a distinct neurodevelopmental phenotype that overlaps with Coffin–Siris syndrome in younger patients and BFLS in older patients (6–10).
The PHF6 gene is composed of 11 exons and produces at least three alternatively spliced mRNA transcripts including one that retains intron 10 that alters the length of the 3′ UTR (1,11). The 365 amino acid PHF6 protein has two extended plant homedomain (PHD) zinc finger or ZaP domains and an acidic domain following the second PHD zinc finger (12). The PHF6 protein localizes to both the nucleoplasm and nucleolus where it has been shown to be involved in transcriptional regulation (1,13,14).
We and others have identified multiple PHF6 mutations, including missense, nonsense, duplication and small insertions/deletions that have been localized throughout the coding region (4,7,12,15). PHF6 mutations are not only associated with BFLS, but have also been implicated in malignant hematopoiesis, including acute lymphoblastic leukemia (T-ALL) and acute myeloid leukemia (AML) (16–18). Among PHF6 mutations, the nonsense mutation c.1024C > T (p.R342X), which predicts the truncation of the last 24 amino acids, is the most common recurrent mutation having been reported in several unrelated BFLS families and in numerous leukemia cohorts (1,3,6,16,19). Indeed, one report identified the R342X mutation in a BFLS case who also developed T-ALL at an early age (16). Despite the recurrence of this mutation, its specific consequence on PHF6 mRNA stability or the effect of the truncated PHF6 protein has not been determined.
Moreover, the precise function of PHF6 during brain development and disease pathogenesis also remains largely unclear. PHF6 protein has been demonstrated to interact with subunits of the nucleosome remodeling and deacetylation (NuRD) complex (14). While components of the NuRD complex are critical for corticogenesis and neurodevelopmental disease (20–22), the interplay between NuRD and PHF6 during corticogenesis and the specific targets remain to be defined. PHF6 also has a role in the regulation of rDNA expression either through a direct interaction with the upstream binding factor (UBF1), a nucleolar transcription factor, or indirectly via an upregulation of non-coding promoter-associated RNAs (13,23). In the hematopoietic system, PHF6 negatively regulates HSC self-renewal and is required for late stages of B- and T-cell development suggesting more of a role in cell-fate determination (17,24–27). Knockdown studies of PHF6 in the developing forebrain have indicated that PHF6 has a critical role in regulating the migration of upper layer neurons. One study identified an interaction between PHF6 and the polymerase II associated factor 1 (PAF1) transcriptional elongation complex (28). Acute knockdown of PHF6 or PAF1 resulted in migration defects that were attributed to reduced expression of the target gene Cspg5, a member of the neuregulin protein family involved in neuronal migration (28,29). The second study demonstrated that overexpression of miR-128 in neuroprogenitors prevented migration and dendritic arborization of upper layer neurons via direct targeting of PHF6, while restoration of PHF6 expression could rescue the defects (30).
Although acute knockdown experiments have indicated a key role for PHF6 in neuronal migration, mouse models have not shown similar results. Complete loss of PHF6 was lethal on a C57BL/6 background yet animals survived on a mixed background but had a postnatal growth defect that was attributed to altered neuroendocrine signaling (31,32). Migration defects were also not observed in the cortex of mice harboring the human patient mutation C99F, although these mice displayed some mild behavioral deficits and an increased susceptibility to seizures (31). In a separate study, conditional inactivation of PHF6 in the arcuate nucleus of the hypothalamus of adult mice increased the expression of activity-dependent immediate early genes (e.g. Egr1, cFos) that was linked to the hunger response (33). In this regard, PHF6 is thought to normally repress these genes under satiety conditions and is released from these promoters allowing their induction after fasting conditions. As such, the Phf6-null mice were resistant to body weight gain because they lacked the hunger-driven feeding motivation directed by this pathway. This discrepancy from the phenotype of BFLS patients who develop obesity in late childhood was presumed to derive from hormonal changes in the pituitary from germline PHF6 loss rather than the targeted loss of PHF6 specifically in the hypothalamus (33). Indeed, PHF6 deletion specifically in the nervous system altered the regulation of the growth hormone axis and anterior pituitary development which was attributed to the postnatal growth defect in these animals (32).
In this study, we generated mice with the commonly recurring R342X Phf6 mutation as a distinct model of the BFLS phenotype. The R342X mice displayed reduced body weight, longer tapering forepaw digits, and deficits in learning, memory and anxiety responses while lacking other features of the disease (e.g. microcephaly, obesity) that suggested this model should be considered as a mild representation of the BFLS phenotype. One key finding that was common in both mouse and human patients was that the R342X mutation enhances alternative splicing, increasing the level of intron 10 retention in human cells while inducing exon 10 skipping in mice. Taken together, the study has provided some additional insight into the pathogenesis of BFLS and specifically the effect of the R342X mutation.
Results
Generation of mice harboring the R342X mutation
The most recurrent mutation in the PHF6 gene is a nonsense mutation within exon 10 (c.1024C > T; p.R342X) that is located after the two PHD domains and truncates the last 24 amino acids of the protein (Fig. 1A; (1,4,12,16,19)). To investigate the pathophysiology of this mutation, we introduced the corresponding c.1024C > T mutation into exon 10 of the murine Phf6 gene using CRISPR-Cas9 technology. A highly specific spCas9 cleavage site located within 20 nucleotides of the target nucleotide was used to design the single guide RNA (Supplementary Material, Fig. S1A). The sgRNA was cloned into the pSpCas9n (BB)-2A-GFP plasmid (34) and then electroporated into R1 mESCs along with a 120-nucleotide single stranded oligomer (ssODN) repair template. The ssODN contained two silent nucleotide substitutions that created two restriction sites (a HindIII site for RT-PCR screening and a XhoI site that destroyed the Cas9 recognition sequence) in addition to the pathogenic mutation (Supplementary Material, Fig. S1A). Transfected cells were FACS sorted and 56 GFP-positive cells were expanded for screening. Of these, five contained the HindIII site suggesting homology-directed repair, but only one was perfectly recombined upon sequencing (Fig. 1B). Nuclease specificity was tested using the SURVEYOR assay (34), which confirmed that off-target sites with the greatest potential for cleavage remained intact within the clone (Supplementary Material, Fig. S1B). The targeted mESCs were next analyzed for Phf6 transcript and protein expression. RT-qPCR analysis demonstrated a significant reduction (2.10 ± 0.65 log2-fold change; n = 3; P < 0.01, Student’s t-test) in the R342X Phf6 transcript compared with control Phf6 transcript levels (Fig. 1C). Concomitantly, we did not detect any Phf6 protein by immunoblot (Fig. 1D). As such, these R342X targeted mESCs were used to generate chimeric mice and establish the R342X transgenic mouse line.
Figure 1 .
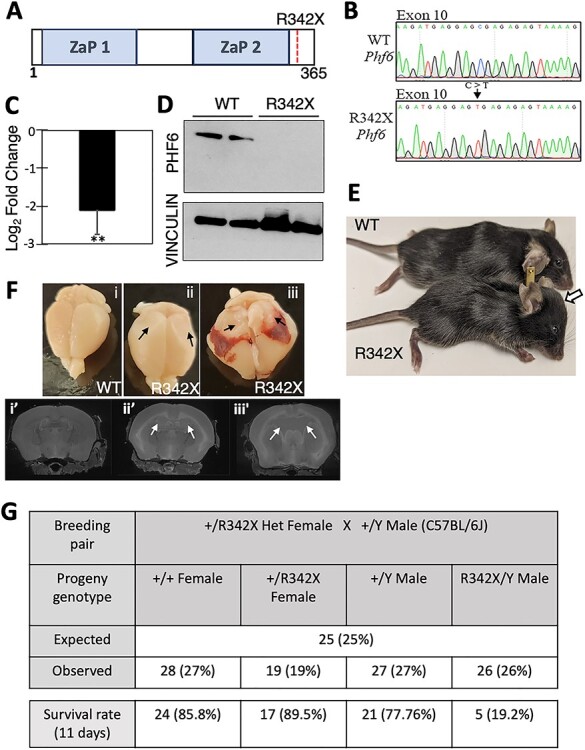
Generation of the R342X mice. (A) Schematic diagram of the PHF6 protein highlighting the location of the ZaP domains and the R342X nonsense mutation. (B) DNA sequence traces from the WT and R342X mESCs showing the engineered C to T transition at nucleotide 1024 within exon 10 of the Phf6 gene. (C) Plot of RT-qPCR log2-fold change of Phf6 from the R342X mESCs relative to WT mESCs. **, P < 0.01. (D) Phf6 immunoblot of extracts from WT or R342X mESCs. Vinculin was used as a loading control. (E) Photo of P40 WT and R342X mice on a C57BL/6 background. Arrow indicates dome-shaped head in mutant animal. (F) Dissected brains (top) or MRI scans (bottom) from WT (i) or R342X mice with mild (ii) or severe (iii) hydrocephaly at P100. Black arrows indicate collapsed cortical tissue due to loss of CSF from the enlarged ventricles which are indicated by white arrows in the bottom panel. (G) R342X male mice (C57BL/6) were born at normal Mendelian ratios but only had a 20% survival rate by P11.
The F0 generation of mice derived from the chimeric founders was viable and noticeably smaller in size. As we continued to backcross the animals onto the C57BL/6 J strain, we observed an increasing frequency of mutants with hydrocephaly in F3–F6 generations, as indicated by mice with dome-shaped heads (Fig. 1E). After dissection, the collapse of the cortex from CSF fluid release was evident (Fig. 1F, black arrows) and the enlarged ventricles observed by magnetic resonance imaging (MRI; Fig. 1F, white arrows) confirmed hydrocephaly, which was observed in 35% of the R342X animals (Supplementary Material, Fig. S1C). Consequently, these hemizygous male mice showed a high level of perinatal lethality (~80%) despite being born at normal Mendelian ratios (Fig. 1G) which precluded further analysis. However, when maintained on a mixed genetic background (C57BL/6 J:129/Sv or C57BL/6 J:FVB/N) or an FVB/N congenic strain the hemizygous male R342X mice survived normally and the hydrocephaly phenotype dissipated (Supplementary Material, Fig. S1D). All remaining experiments were performed on hemizygous males of either the mixed background lineage or the FVB/N congenic strain (as indicated).
R342X animals share some prominent clinical features with BFLS patients
Regardless of genetic background, the R342X mice were consistently smaller in size with a significant decrease in body weight at all timepoints examined; the size difference was maintained throughout their lifespan (Fig. 2A and B). BFLS patients are typically short in stature but they often present with truncal obesity (4,7) which we did not observe in the R342X mice. To confirm that the mice had no indication of truncal obesity, we examined three different subcutaneous white adipose depots, namely epididymal (WT: 0.029 ± 0.004, n = 5; R342X: 0.025 ± 0.008, n = 6; P = 0.7181, Student’s t-test), retroperitoneal (WT: 0.011 ± 0.001, n = 5; R342X: 0.010 ± 0.002, n = 6; P = 0.7468, Student’s t-test) and inguinal subcutaneous (WT: 0.018 ± 0.002, n = 5; R342X: 0.022 ± 0.004, n = 6; P = 0.4166, Student’s t-test) fat deposits but failed to observe any significant differences between WT and R342X mice when normalized to body weight (Fig. 2C–E).
Figure 2 .
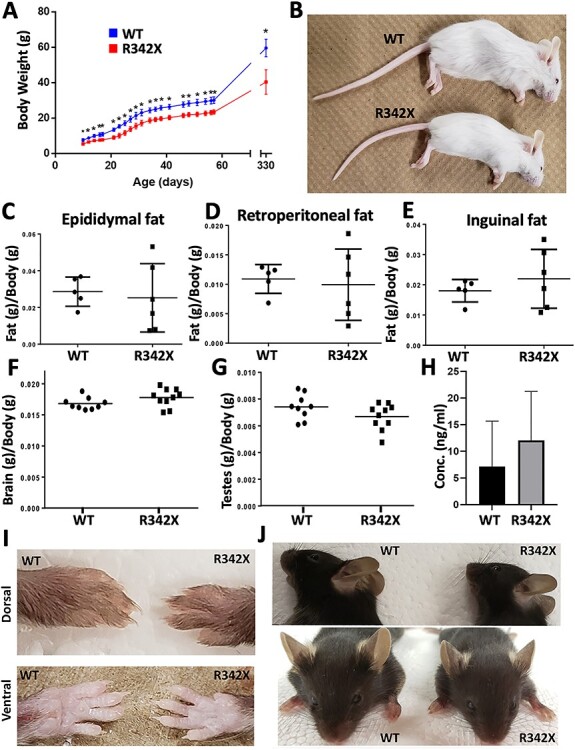
Analysis of R342X mice for clinical features of BFLS patients. (A) Weight measurements of a cohort of WT (Blue dots; n = 11) and R342X (Red dots; n = 12) mice were plotted beginning at P10 until P330. At all ages the R342X mice had a statistically significant (*, P < 0.05) reduction in body weight. (B) Image of WT and R342X mice (FVB/N) highlighting the difference in size at P25. (C-E) Plotted ratio of fat/body weight for WT and R342X mice from the epididymal (C), retroperitoneal (D) and inguinal (E) fat deposits. (F) Brain weight to body weight ratio, and (G) testes weight to body weight ratio show no difference between WT and R342X littermates. (H) Testosterone plasma concentration levels in WT and R342X mice. Images of forepaw digits (I) and ears (J) were assessed for morphological features of BFLS.
We next performed a general morphological analysis of the R342X animals to determine if any other prominent features of BFLS patients were present in the mice. Clinical reports of BFLS patients have indicated that brain size is variable ranging from a normal size to include individuals with both microcephalic and macrocephalic brains (4,5,7). The brains dissected from postnatal day 60 (P60) R342X and WT adult animals showed no significant difference in the brain weight/body weight ratio (WT: 0.017 ± 0.0003, n = 9; R342X: 0.018 ± 0.0004, n = 10; P = 0.1087, Student’s t-test; Fig. 2F). BFLS patients also have hypogonadism but the testes/body weight ratio of R342X to WT mice (P60) was not different (WT: 0.007 ± 0.0003, n = 9; R342X: 0.007 ± 0.0003, n = 10; P = 0.1154, Student’s t-test; Fig. 2G), nor was the level of circulating testosterone (WT: 7.15 ± 3.81 ng, n = 5 (each sample comprised plasma pooled from four mice); R342X: 12.06 ± 4.11 ng; n = 5 (as described for WT); P = 0.4060, Student’s t-test) in P40 plasma samples (Fig. 2H). We also investigated tissues where Phf6 expression was observed during mouse development (35), but found no significant difference in the organ/body weight ratios of the thymus, liver, lungs, spleen, kidneys or heart (Supplementary Material, Fig. S2A–F). Another prominent feature of BFLS patients are the presence of large ears and long tapering fingers (4,7). While we were unable to quantitatively analyze these features in detail, the forepaw digits of the R342X mice (P35) were relatively longer and more tapered than control animals (Fig. 2I). This was not observed in the hind limbs, nor did we observe broad and/or shortened digits that are common in the toes of human patients (Supplementary Material, Fig. S2G). No differences were apparent in the size of the ears (Fig. 2J).
We also examined the pituitary gland of the R342X mice since several clinical reports have documented reduced growth hormone (Gh) and/or altered levels of other pituitary hormones (8,36,37), while similar changes were observed in PHF6-null mice (32). Upon dissection of the pituitary from WT and R342X mice at P35, we observed that the mutant animals had a much smaller pituitary gland that was largely restricted to the anterior lobe (Fig. 3A). Sections immunostained for PHF6 showed a reduction in the anterior and intermediate lobes, while RT-qPCR of whole pituitary (P35) demonstrated decreased transcript levels of PHF6 (Fig. 3B and C). Similarly, RT-qPCR analysis demonstrated that the transcript levels for Gh and Prolactin (Prl) were significantly decreased (Fig. 3D and E), while genes coding for the follicle stimulating hormone (Fsh), Leuteinizing hormone (Lh), pro-opiomelanocortin (Pomc) and thyroid stimulating hormone (Tsh) showed no change in expression (Supplementary Material, Fig. S3A). Pituitary sections were immunostained for Gh and Prl to determine whether there were fewer cells producing the proteins or reduction in production per cell. Gh + or Prl + cells were quantified as a proportion of the total number of Hoescht positive nuclei demonstrating that there were fewer Prl-producing cells but no change in the number of Gh-producing cells in the mutants (Fig. 3F and G; Supplementary Material, Fig. S3B and C). Finally, we utilized ELISA assays to examine the plasma concentrations of Gh and Prl after fasting the mice for 24 h. Plasma levels of Prl showed a significant reduction in the R342X mice compared with WT littermates, while no change was observed in Gh levels. While the significance of reduced Prl levels remain to be determined, the R342X mice have pituitary defects that are consistent with alterations in pituitary hormone levels observed in some BFLS patients (3,7,38). Overall, we conclude from our phenotypic characterization that the R342X mice share some of the main physical characteristics of BFLS patients.
Figure 3 .
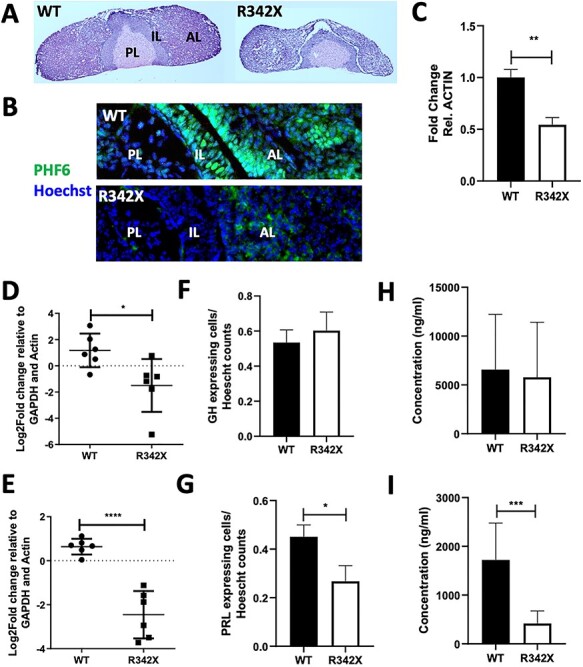
Pituitary defects observed in R342X mice. (A) H&E-stained sections of P35 pituitary gland isolated form WT and R342X mice. PL, posterior lobe, IL, intermediate lobe; AL, anterior lobe. (B) Immunostained pituitary sections stained for Phf6 (green) and counterstained with Hoechst. (C) RT-qPCR for Phf6 from RNA isolated from P35 pituitary samples of WT and R342X mice. Growth hormone (GH; D, F, H) and Prolactin (PRL; E, G, I) levels were quantified by RT-qPCR for transcript levels from whole pituitary extracts (D, E); for the proportion of immunopositive cells in the anterior pituitary (F, G); and for circulating protein levels in plasma by ELISA (H, I) from WT and R342X mice at P35. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
The R342X mice have variations in brain volume but normal cortical lamination
The Phf6 protein is highly expressed in the developing cerebral cortex (35), while acute Phf6 knockdown has indicated that it is required for proper cortical lamination (28,30). To assess cortical development, we first examined cortical sections from WT and R342X animals for alterations in cortical size and the expression of deep and upper cortical layer neuronal markers. Overall, the number of Hoescht-positive cells in the P0 cortex were similar (WT: 588.7 ± 12.99 cells; R342X: 637.7 ± 30.3 cells; n = 3; Unpaired t-test, P = 0.2114; Supplementary Material, Fig. S4A). Next, we used immunofluorescence (IF) to stain P0 sections for upper layer (SATB2) and deep layer (CTIP2, TBR1) neuronal markers for quantification (Fig. 4A). The upper layer marker SATB2 stains layers II–IV but also layer V at P0. We quantified the total number of SATB2+ cells as a proportion of Hoescht+ cells as well as the proportion specific to layers II–IV and layer V (Supplementary Material, Fig. S4B). All three analyses indicated that there was no difference in the total number of SATB2+ cells (WT: 33.64%; R342X: 32.88%; n = 3; Sidak’s multiple comparison test, P = 0.8794), or SATB2+ cell numbers within layers II–IV (WT: 12.78%; R342X: 11.59%; n = 3; Sidak’s multiple comparison test, P = 0.664), or in layer V (WT: 15.44%; R342X: 15.58%; n = 3; Sidak’s multiple comparison test, P = 0.9991). Similarly, the deep layer markers CTIP2 (layer V; WT: 5.49% ± 0.66; R342X: 5.08% ± 0.28; n = 3; Unpaired t-test, P = 0.5947) and TBR1 (layer VI; WT: 18.17% ± 0.84; R342X: 18.59% ± 0.74; n = 3; Unpaired t-test, P = 0.7276) also showed no proportional differences in cell number in the mutant mice (Fig. 4A; Supplementary Material, Fig. S4C and D). Previous studies have indicated that acute knockdown of Phf6 results in cortical migration defects at E18.5 (31). To examine whether there may be mild cortical migration defects that resolve spontaneously by P0, we stained E13.5 and E16.5 sections for CTIP2 and TBR1 but observed no differences further suggesting that cortical lamination proceeded normally in the R342X mice (Supplementary Material, Fig. S4E and F).
Figure 4 .
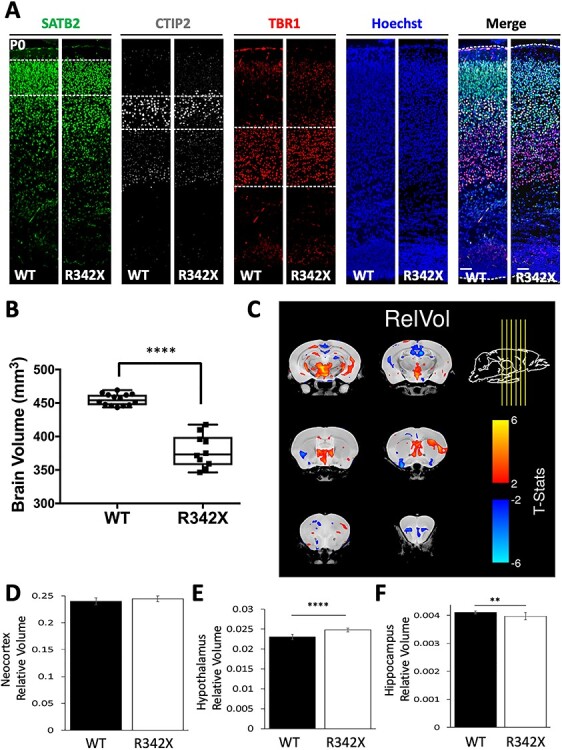
Analysis of corticogenesis and brain volume differences in R342X mice. (A) P0 sections from WT and R342X mice stained for layers II–IV (SATB2, green), layer V (CTIP2) and layer VI (TBR1) neuronal markers demonstrate no differences in cortical lamination. Cell counts are presented in Supplementary Material, Fig. S4. Hoechst served as a nuclear counterstain. (B) Plot of absolute brain volume differences obtained by MRI between WT and R342X animals (P100). (C) Coronal images from six different rostral–caudal locations (indicated by yellow arrows) showing the relative changes in brain volume identified in R342X mice compared with control animals. (D) The neocortex showed no difference in relative volume while statistically significant differences were observed in the hypothalamic (E) and hippocampal (F) brain regions. **, P < 0.01; ****, P < 0.0001.
Next, we performed MRI analyses to determine volumetric differences in whole brain and specific brain regions. The absolute brain volume was reduced in R342X mice at P100 compared with WT mice (WT: 455.02 ± 2.28 mm3, n = 14; R342X: 378.47 ± 7.73 mm3, n = 10; P < 0.0001, Student’s t-test; Fig. 4B). Given, that the R342X mice were smaller in size and that the brain weight/body weight ratio was not altered, the large difference in size can be mainly attributed to the reduced size of the animal. However, to assess differences in specific brain regions we compared relative volume changes by first normalizing the volume of each region to the absolute brain volume. In this way, we identified multiple regions with small yet significant differences in brain volume in the R342X mice, as indicated by red (increased volume) and blue (reduced volume) contour shading on 2D slices images of the forebrain (Fig. 4C). Importantly, this analysis confirmed that the neocortex of R342X mice when expressed as a proportion of overall brain volume (WT: 0.24 ± 0.002, n = 14; R342X: 0.244 ± 0.002, n = 10; P = 0.0641, Student’s t-test) was unchanged (Fig. 4D), while minor differences were observed in the hypothalamus (WT: 0.023 ± 0.0002, n = 14; R342X: 0.025 ± 0.0002, n = 10; P = 2.04 x 10−7, Student’s t-test) and the hippocampus (WT: 4.11 x 10−3 ± 1.3 x 10−5; R342X: 3.98 x 10−3 ± 4.10 x 10−5, n = 10; P = 0.002, Student’s t-test; Fig. 4E and F). Significant differences were also observed in the cerebellum, while a complete list of altered brain volumes is included in the supplemental information (Supplementary Material, Table S1). Taken together, we conclude that the R342X mutation has only minor effects on cortical development causing a slight reduction in the size of the hippocampus that may impair learning and memory.
The R342X mice have behavioral deficits
Many BFLS patients exhibit reduced cognitive function, display challenging behaviors (hyperactivity, ADHD, OCD and aggression) yet can also be overly friendly and score low on anxiety tests (5,7,38). To investigate whether the modest changes we observed in regional brain volumes altered anxiety or learning, we performed several behavioral tests on a cohort of WT and R342X mice at P60 maintained on a mixed genetic background. First, we challenged the mice in the elevated plus maze (EPM) test to assess an acute anxiety-like behavior. We observed that the R342X mice were far more active than control littermates, traveling 1.5 times the distance (WT: 2085 ± 136.8 cm, n = 17; R342X: 3216 ± 197.2 cm, n = 16; P < 0.0001) and making twice as many entries into both the open and closed arms (WT: 12.53 ± 1.12 open arm entries, 26.59 ± 2.40 closed entries, n = 17; R342X: 23.19 ± 2.17 open arm entries, 42.3 ± 2.20 closed entries, n = 16; P = 0.001 and P < 0.0001) as the WT mice (Supplementary Material, Fig. S5A–C). Despite the increased activity, the R342X mice spent significantly more time than WT mice in the open arms (WT: 68.19 +/− 5.85 s, n = 17; R342X: 122.6 +/− 17.72 s, n = 16, P = 0.0054) but an equivalent time in the closed arms (WT: 422.1 +/− 11.24 s, n = 17; R342X: 380.8 +/− 19.07 s, n = 16; P = 0.0675) suggesting a decreased anxiety-like behavior in the mutant animals (Fig. 5A; Supplementary Material, Fig. S5D). As a second test of anxiety, we performed the open field (OF) test. In this test, the R342X mice did not show any difference in the distance traveled during the test (WT: 14234 +/− 3077 cm, n = 18; R342X: 21023 +/− 4665 cm, n = 21; P = 0.2488; Supplementary Material, Fig. S5E) but did have a similar response to the EPM test in that they spent significantly more time in the center and less time in the corners than the WT cohort (WT: 7.44 +/− 1.12 s in center, 379.7 +/− 18.5 s in corner, n = 18; R342X: 14.90 +/− 2.72 s in center, 302.5 +/− 15.23 s in corner, n = 21; P = 0.0025 and P = 0.0250; Fig. 5B and C). Taken together both of these tests suggested that the R342X mice show reduced anxiety or an anxiolytic phenotype.
Figure 5 .
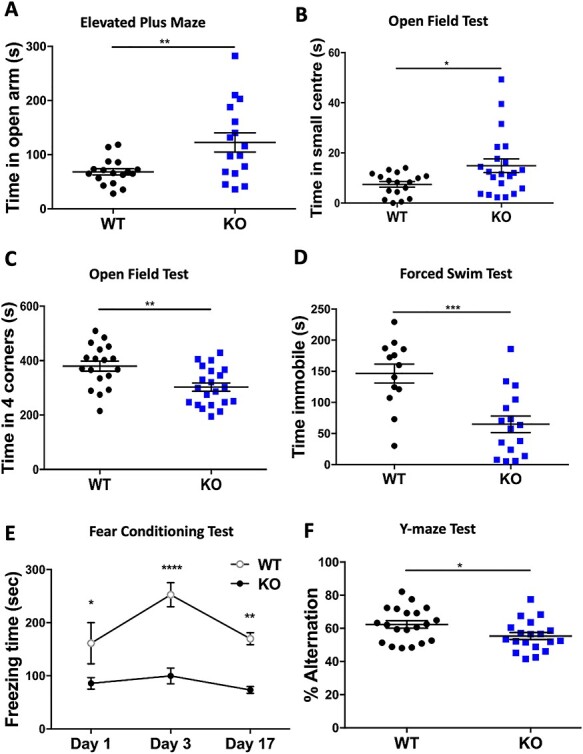
Behavioral testing of R342X mice. Cohorts of P60 control (WT, black dots) and R342X mice (KO, blue dots) were subjected to a battery of behavior tests. (A) Time spent in the open arms of the EPM. (B) OF test plot of time spent in the small center. (C) OF test plot of time spent in the four corners. (D) FST plot of the time spent immobile. (E) Plot of freezing time for mice in the contextual FC assay 1, 3 and 17 days after training. (F) Y-maze plot of the percentage of alternations between arms. Each dot (blue or black) represents data from a single animal. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Next, we subjected the mice to the forced swim test (FST). This test measures behavioral despair caused from an inescapable stress and it is used to assess a depression-like phenotype. The inescapable stress is the pool of water and the immobility of the animal represents the measure of despair. The R342X mice were far more active spending >2-fold less time immobile than their WT littermates (WT: 146.3 +/− 15.15 s, n = 13; R342X: 64.86 +/− 13.20 s, n = 16; P = 0.0004; Fig. 5D) suggesting that the R342X mutations are less prone to depressive-inducing stressors.
To assess learning and memory, we performed the contextual fear conditioning (FC) and Y-maze tests. In the contextual FC assay (39), the mice are exposed to a novel cue tone that is immediately followed by a foot-shock. The tone/shock training occurs twice on the first day and the freezing time is recorded during the 5-min session. The training period develops an association to the cue tone. The animals are then returned to the box 2 days later and the normal response is to show an increased level of freezing as the mice learn to associate the environment with the shock. The learned response typically dissipates with time, so testing is repeated a third time at day 17 to demonstrate a return to the freezing baseline. In this experiment, we observed that the R342X mice showed reduced freezing on all 3 days of testing (D1: WT = 161.4 s, R342X = 85.76 s, P = 0.0453; D3: WT = 252.7 s, R342X = 99.81 s, P < 0.0001; D17: WT = 169.9 s, R342X = 73.47 s, P = 0.0084; n = 5 both groups) suggesting that they have a severe impairment in associative learning, as they even had difficulty associating the cue tone with the shock (Fig. 5E). The Y-maze spontaneous alternation test measures the inclination of the animal to explore all three arms of the maze that each represent a new environment. A normal response is for the animal to alternate between different arms but to equally explore all arms over the length of the test. Animals with cognitive deficits will display a reduced tendency to enter a less recently visited arm. The R342X mice showed a mild but significant reduction in the percentage of alternation between arms compared with the control animals (WT: 62.36 +/− 2.27%, n = 20; R342X: 55.38 +/− 2.13%, n = 19, P = 0.0312) indicating they have a spatial recognition memory deficit (Fig. 5F). Overall, we conclude from these behavior tests that the R342X animals are showing reduced anxiety, associative learning and spatial memory deficits compared with their control littermates.
Phf6 protein levels are severely reduced in the cerebral cortex
Deletion of the last 24 amino acids of Phf6 had little effect on cortical lamination but did show behavioral deficits in all tests performed. To explore the molecular basis for these results, we next utilized RNAseq (n = 4/genotype; GSE156404) to analyze the expression of Phf6 and the cortical transcriptome at birth. RNAseq indicated that Phf6 expression was decreased almost 2-fold (log2 fold change = −0.932) in the R342X cortex compared with the control group (Supplementary Material, Table S2). RT-qPCR was used to validate the change in Phf6 expression and showed a similar 2-fold decrease in transcript levels (Fig. 6A). Apart from Phf6, use of the DESeq2 analysis package identified a total of 416 differentially expressed genes (DEGs) between WT and R342X samples but only 24 genes were significantly altered when a threshold (± 0.5 log2 fold change; s-value < 0.005) was applied (Fig. 6B). Moreover, most of the genes showed very modest expression differences with only four genes showing a greater than 2-fold difference. We performed a similar RNAseq experiment using RNA isolated from two BFLS lymphoblast cell lines with the R342X mutation and four control lines. In this experiment, we identified 609 DEGs (Supplementary Material, Table S2) which we then compared with the mouse DEG list using GeneOverlap. In this way, we identified 16 overlapping genes with dysregulated expression (Supplementary Material, Fig. S6C). Nonetheless, the significance of this data is unclear as 11 genes showed discordant changes in expression while only Phf6, Itgb3, Ccnd3, Lrig1 and Kif26a showed concordant expression changes between the two datasets. As a second test, we also performed an X–Y scatter plot to identify genes with correlated expression (Supplementary Material, Fig. S6C). While this analysis identified 342 genes with correlated expression patterns there was no statistical significance (Pearson’s r = −0.02324178, P = 0.6684) suggesting that PHF6 may have different roles in the two tissues.
Figure 6 .
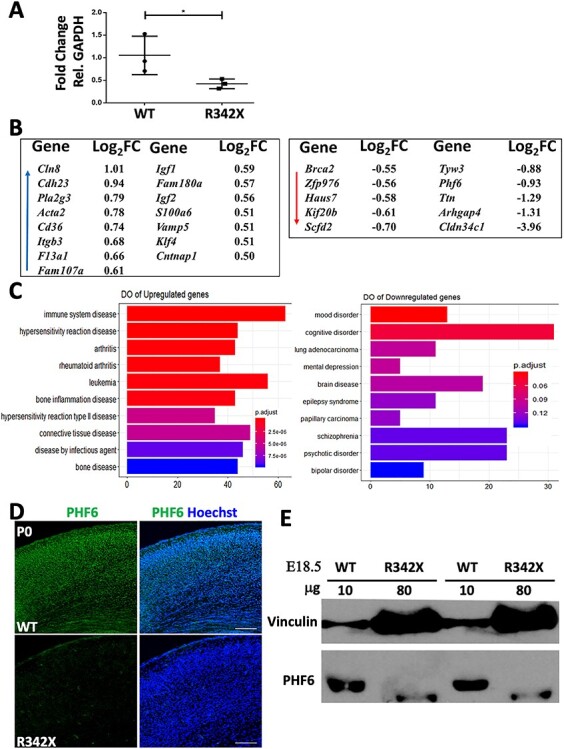
Analysis of PHF6 protein and gene expression differences in the P0 cortex. (A) RT-qPCR analysis shows that the R342X mice have reduced Phf6 transcript levels compared with WT littermates (n = 3). RNA levels were normalized to GAPDH expression. *, P < 0.05. (B) List of DEGs (± 0.5 log2 fold change; s-value < 0.005) identified in the R342X P0 cortex. Blue arrow indicates upregulated genes, while red arrow highlights downregulated genes. (C) DO plots of significant disease terms for upregulated (left) and downregulated gene lists (right). (D) Representative image of P0 coronal sections IF stained for PHF6 (green) from WT and R342X mice. Hoechst counterstained nuclei are blue. (E) Immunoblots of E18.5 cortical extracts for PHF6 from WT and R342X mice. Note the increased protein loading in the R342X lanes. Vinculin was used as a protein loading control.
To perform gene ontology (GO; Supplementary Material, Fig. S6) and disease ontology (DO; Fig. 6C) comparisons, we only used the larger mouse gene list. GO analysis indicated that upregulated genes were involved in biological processes related to the development of the vasculature, cardiovascular system and blood vessels. Downregulated genes were involved in chromatin organization, CNS development and organelle organization, which aligned with the known roles for PHF6. Similarly, DO analysis of upregulated genes suggested that Phf6 was involved in immune disorders and leukemia, while the downregulated genes indicated that Phf6 contributed to cognitive disorders, schizophrenia and psychotic disorders. We reasoned that the small DEG list might reflect that the C-terminally truncated PHF6 protein is stable and retains significant function during cortical development. Surprisingly, cortical sections of P0 mutant mice showed almost a complete loss of protein upon PHF6 immunostaining (Fig. 6D). Initial immunoblots also failed to detect the R342X truncated protein. However, when protein loading was increased 8-fold above the control samples, we observed a band corresponding in size to the expected truncated protein (Fig. 6E). Given the reduced intensity of the truncated protein, we estimate that the protein level is reduced by ~25-fold of normal levels in R342X mice.
The R342X mutation interferes with RNA processing and protein production
To understand if the R342X mutation had a similar effect on human PHF6 protein levels, we performed immunoblots on proteins extracted from lymphoblastoid cell lines (LCLs) from three BFLS patients containing the R342X mutation (R342X) and three unaffected individuals (WT). Similar to PHF6 protein levels in the R342X mouse, we observed a drastically reduced level of PHF6 protein in LCLs from patients compared with control samples (Fig. 7A). As a comparison, we also examined PHF6 expression in six LCLs from BFLS patients with different PHF6 mutations (C45Y; C99F; K234E; R257G; D333del; E337del). Protein levels were slightly decreased in three of the other patient samples (C45Y, C99F and K234E) with mutations within the PHD domains but protein levels were not affected for others including two different single amino acid deletions (D333del and E337del) that lie in close proximity to residue 342 (Fig. 7B).
Figure 7 .
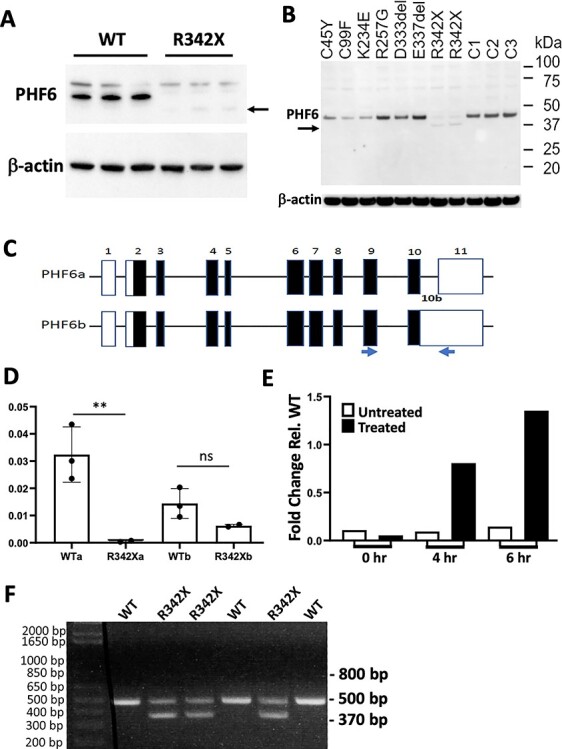
The R342X mutation results in reduced protein levels from NMD of the PHF6a transcript in human samples. (A) Immunoblots of LCLs derived from normal individuals (WT) or BFLS patients with the R342X mutation (R342X) stained with an antibody to PHF6 or β-actin as a loading control. Arrow indicates the position of the truncated PHF6 protein from patients with the R342X mutation. (B) Immunoblot showing comparative protein levels of PHF6 (~ 41 kDa) in BFLS patients and unaffected individuals. Protein expression in BFLS patients carrying the C45Y, C99F, K234E and R342X mutations is reduced while the R257G, D333del and E337del mutations are expressed at a similar level to the three unaffected individuals (C1, C2 and C3). Arrow denotes the band running at approximately 38.5 kDa in the patients with the R342X mutation, which is also present at reduced levels. Antibodies were the same as in (A). (C) Schematic diagram of the PHF6 gene structure comprising the PHF6a and PHF6b transcripts. Blue arrows denote primers used to amplify similar transcripts from the mouse forebrain. (D) A significant reduction in the PHF6a isoform compared with expression in the controls is detected in patients with the p.R342X mutation by RT-qPCR on cDNA derived from RNA extracted from LCL. Expression of the PHF6b isoform is not affected. **, P < 0.01; ns, not significant. (E) Expression of PHF6a isoform with the p.R342X mutation is restored following inhibition of the NMD pathway by treatment of LCL for 6 h with 100 μM cycloheximide. (F) RT-PCR analysis of cDNA derived from mouse forebrain (P0) samples. The primers shown in (C) were used to amplify the murine equivalent of the Phf6a (500 bp) and Phf6b (expected at 800 bp) transcripts. The Phf6b transcript was not observed but a smaller band (370 bp) corresponded to the ΔE10 transcript that splices out exon 10.
The human PHF6 gene contains 11 exons and two transcripts, one isoform that splices out intron 10 and a second isoform that retains it. The spliced and unspliced isoforms at this site have been previously labeled PHF6a and PHF6b, respectively (Fig. 7C; (1)). The normal stop codon is encoded by the final three nucleotides of exon 10 and thus alternative splicing does not alter the protein coding sequence. However, we reasoned that the novel premature termination codon generated by the c.1024 C > T mutation might promote nonsense-mediated decay (NMD) given its location 69 bp from the end of the exon. Furthermore, we predicted that this would have an effect on the PHF6a transcript but not the alternative PHF6b transcript. We designed PCR primers specific to either PHF6a or PHF6b and measured the amount of each transcript in affected individuals with the c.1024C > T mutation by real-time RT-PCR. A reduced level of expression of PHF6a but not PHF6b in affected individuals with the c.1024 C > T mutation was confirmed (Fig. 7D). Importantly, treatment of LCLs with the NMD inhibitor cycloheximide showed a time-dependent increase in the levels of the PHF6a transcript in both WT and R342X BFLS patients (Fig. 7E).
To confirm that a similar mechanism was occurring in the R342X mice, we first examined whether the mouse expressed both Phf6a and Phf6b transcripts. Primers were designed to amplify the Phf6 gene beginning in exon 9 and ending in exon 11 (Fig. 7C, blue arrows), which should generate a 830 bp fragment corresponding to the Phf6b transcript and a 500 bp band representing the Phf6a transcript. Following RT-PCR amplification from P0 cortical RNA samples, we observed the 500 bp fragment in both the WT and R342X samples but not the Phf6b specific 830 bp fragment (Fig. 7F), indicating that mice do not generate transcripts that retain intron 10 within the cortex. Surprisingly, we also observed a shorter truncated band of 375 bp that was present only in the mutant samples (Fig. 7F). Both Sanger sequencing of the purified fragment and analysis of the RNAseq data demonstrated that this product corresponds to an alternative spliceform that has skipped exon 10 and is present at equivalent levels to the normally spliced product (Figs 7F and 8A). Furthermore, we examined a range of tissues (thymus, lung, liver, spleen and kidney) and observed that exon 10 skipping was prevalent in all tissues examined (Supplementary Material, Fig. S7C).
Figure 8 .
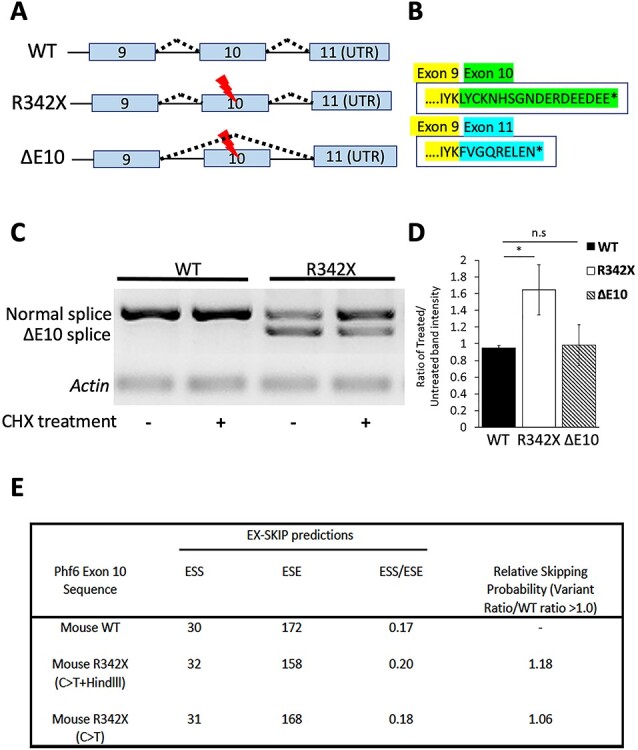
Characterization of the novel ΔE10 transcript. (A) Schematic diagram detailing the splicing events that generate the ΔE10 transcript. (B) Skipping of exon 10 generates a distinct PHF6 protein C-terminal isoform (blue amino acids) compared with the expected protein sequence of the R342X mutant protein (green). (C) Primary cortical neurons were treated for 6 h with 100 μM cycloheximide and then analyzed by RT-PCR for the normal and ΔE10 spliced transcripts. Cycloheximide treatment (CHX) increased the level of the normal transcript in the R342X samples. (D) Quantification of the change in transcript levels from cycloheximide treated and untreated samples, as shown in (C). R342X refers to the normally spliced transcript that incudes exon 10 but also has the R342X mutation. *, P < 0.05; ns, not significant. (E) The EX_SKIP tool was used to calculate the ESS and ESE within the WT exon 10 sequence, or exon that contains the C > T transition corresponding to the R342X mutation alone, or the sequence from the R342X mice which also contains an additional mutation that creates a HindII site that was used for genotyping. The ESS/ESE ratios are shown for the three transcripts and the relative exon skipping potential (variant ratio/WT ratio) is also shown. Values > 1.0 indicate an increased potential for alternative splicing.
This was an unexpected finding and suggested that two different truncated PHF6 proteins could be produced in the R342X mice, a 341 amino acid protein lacking the last 24 amino acids of PHF6 (R342X; 38.5 kDa) and a second protein we named ΔE10 that is 332 amino acids (37.4 kDa) in length due to a frameshift that creates a novel C-terminal end (Fig. 8A and B). As such, the R342X mice could produce two different PHF6 proteins differing in size by ~ 1.1 kDa. We tried to determine whether both proteins were produced by performing SDS-PAGE on a large apparatus to maximize migration distance for size extrapolation and to resolve any potential protein doublets. Following immunoblot we were unable to resolve two protein bands and while size extrapolation from the migration of molecular weight standards suggested that the R342X protein was produced (R2 = 0.9953, using the equation y = 0.0044X + 1.8687 the observed band was 39 kDa), we cannot completely exclude production of the ΔE10 protein (Supplementary Material, Fig. S7A).
Finally, we examined whether the R342X transcript was subject to degradation by NMD similar to our observations in the human samples. Primary cortical neuron cultures were isolated from WT and R342X mutant mice (E13.5) and treated with or without cycloheximide (CHX) for 6 h prior to transcript analysis by RT-PCR. As expected, there was no change in the level of the normally spliced product in the WT samples with or without CHX treatment (WT: 0.95 +/− 0.018, n = 3; Fig. 8C and D). Similarly, the level of the ΔE10 transcript did not significantly change (ΔE10: 0.983 +/− 0.142, n = 3, P = 0.8150). However, we observed an almost 2-fold increase in the level of the R342X transcript after CHX treatment (R342X: 1.645 +/− 0.174, n = 3, P = 0.01614) suggesting that this transcript is subject to NMD like the human PHF6a transcript containing the R342X mutation (Fig. 7C and D). We next repeated the experiment using caffeine treatment to block NMD. Caffeine seemed to have an even stronger effect as it not only increased the level of the R342X transcript but also showed a reduction in the level of the ΔE10 transcript (Supplementary Material, Fig. S7B). To gain an understanding for the presence of the ΔE10 transcript, we performed an in silico analysis to determine whether the R342X mutation increases the potential for exon 10 skipping. We used the EX-SKIP tool to calculate the number of exonic splicing enhancers (ESE) and silencers (ESS) within the WT exon 10 sequence. Sequence variants that alter the ESS/ESE ratio have been shown to alter exon splicing (40). We compared the WT exon 10 sequence with a sequence containing only the C1024T transition (C > T), and the sequence from the R342X mice that also harbors an additional nucleotide substitution that created a HindIII site for genotyping (C > T + HindIII). In this regard, both variant exon sequences increased the ESS/ESE ratio resulting in an increased propensity to induce exon skipping (Fig. 8E).
Discussion
In this study, we generated mice with a c.1024C > T substitution in the Phf6 gene to generate the p.R342X nonsense mutation that represents the most common recurring mutation identified in BFLS patients. The R342X mutation leads to a reduction in PHF6 protein levels in both mice and humans, which we demonstrate arises from NMD and nonsense-associated altered splicing. The absence of the C-terminal 24 amino acids and the severe reduction in protein levels did not significantly alter corticogenesis and had only minor effects on the relative volume of different brain regions when maintained on an FVB/N or a mixed genetic background. By contrast, mice on a C57BL/6 J background had a high incidence of hydrocephaly with a 20% survival rate through the early postnatal period that precluded further analysis. Behavior testing of the R342X mice (mixed background) demonstrated deficits in associative learning, spatial memory and indicated that they were also anxiolytic compared with their control littermates, both features of BFLS patients. The mice also displayed additional features of the disease including short stature, hypoplastic pituitary gland and long tapering digits while other clinical BFLS features (e.g. truncal obesity, gynecomastia and hypogonadism) were not readily observed in the mouse and may require additional stressors to tease out a phenotypic effect.
Mutations in many chromatin interacting proteins have been shown to participate in neurodevelopmental disorders, while mouse models have highlighted their roles in regulating neuroprogenitor homeostasis often resulting in reduced numbers that impact cortical lamination, learning and memory (20,41–47). Given that Phf6 is highly expressed in cortical progenitors, we anticipated that the R342X mice might similarly impact cortical development. Surprisingly, the cortex was proportional in size, contained no lamination defects or alterations in cell cycle kinetics, and few dysregulated genes in the P0 cortex. Similar findings were observed in mice with a C99F mutation in the Phf6 gene, although when we compared the list of DEGs there was no overlap with our study (31). The lack of any cortical defects contrast data from acute knockdown experiments of PHF6 or mIR-128 (an upstream regulator of PHF6) that impaired migration of the upper layer neurons. In related studies, knockdown of the NuRD complex constituents, CHD3 or CHD5, with which PHF6 interacts also impaired neuronal migration (14,20). One possible explanation is that the congenital reduction in PHF6 protein levels promotes compensatory mechanisms during corticogenesis that cannot be invoked during an acute knockdown experiment, or perhaps more simply, that even a low level of PHF6 protein is sufficient for proper cortical development. While we observed no differences in the production and lamination of the projection neurons in the cortex, it remains possible that interneuron number and/or migration into the cortex could be altered. Given that many ASD disorders are considered to represent an imbalance between excitatory and inhibitory neuronal function, the impact of PHF6 on interneuron production and migration should be examined in future studies.
Several groups have indicated that germline deletion of Phf6 resulted in perinatal lethality (24,31), yet it remains to be determined if this is related to a brain defect. The PHF6 phenotype is also subject to genetic background effects which could also influence cortical migration and/or brain development. Indeed, we observed clear phenotypic differences on distinct strains as hydrocephaly was only observed on the C57BL/6 background. It remains possible that the wide variation in brain size in BFLS patients may be linked to genetic background and/or specific modifier genes.
One of the most common features of BFLS are endocrine defects that are proposed to cause numerous defects in patients including short stature, truncal obesity, gynecomastia and hypogonadism (4). The R342X mice also display characteristics of abnormal neuroendocrine function. We demonstrated that the anterior pituitary was reduced in size with altered mRNA production of two pituitary hormones (growth hormone and prolactin), although it only affected the levels of circulating prolactin. A recent study reported that PHF6 KO mice had a postnatal growth defect and a similar reduction in pituitary gland size (32). They demonstrated reduced plasma levels of growth hormone and its downstream target insulin-like growth factor 1 (IGF-1), while the removal of SOCS2, a negative regulator of growth hormone signaling, rescued the growth defect (32). We propose that a similar mechanism is active in the R342X mice and suggest that our single measurement of growth hormone levels after fasting may not have been sensitive enough to detect alterations in circulating levels. Similarly, we did not measure IGF-1 levels. However, it is also likely that PHF6 affects neuroendocrine function in multiple ways. MRI analysis indicated that the hypothalamus was also altered in size in the R342X brain. The hypothalamus controls the secretion of hormones that stimulate the release of pituitary hormones and it is possible that these functions are perturbed. Moreover, the hypothalamus is involved in many essential bodily functions including appetite and weight control which could also contribute to the reduced body size. Indeed, it was recently shown that conditional deletion of Phf6 in the agouti-related peptide (AgRP) neurons of the hypothalamus resulted in an altered hunger response following fasting conditions (33). This study demonstrated that PHF6 dynamically regulates a subset of immediate-early genes (IEGs), whereby PHF6 is bound and represses these promoters in the satiety state but is released to activate the genes in the hunger state. In the mutant animals, the basal level of the IEGs was increased dramatically and could not be further activated by hunger thus decreasing feeding time and food intake. This mechanism seems counterintuitive from what might be expected in BFLS patients who develop obesity in adolescence but the authors argued that the pituitary, which retained PHF6 expression, may also participate in obesity development (33). Collectively, the R342X mice and two different Phf6 cKO animals all show alterations in the hypothalamus–pituitary axis that can contribute to the neuroendocrine defects that regulate postnatal growth. However, the mouse models do not appear to recapitulate some of the other endocrine defects including truncal obesity and hypogonadism and further studies are required to delineate the mechanisms underlying these phenotypic differences. In this regard, mice with different MeCP2-null alleles showed similar neuroanatomical changes but presented with strain dependent differences in body weight further supporting a role for the involvement of other tissues/factors in driving obesity (48).
One of the interesting findings from our analysis of the R342X mutation was its effect on protein levels and alternative splicing. In both mouse and human cells, we observed a massive reduction in PHF6 protein levels that indicated that the C-terminal truncation impaired protein stability. The mRNA levels were also reduced suggesting that NMD may be contributing to the low protein levels. NMD is a cellular mechanism designed to degrade mRNAs to control proper levels of gene expression and, in this instance, to remove mRNAs that potentially encode deleterious truncated proteins (49). Human cells produce two transcripts, PHF6a and PHF6b, the latter transcript failing to excise intron 10 to generate a larger 3′UTR sequence (the PHF6 termination codon resides within exon 10). The R342X mutation resulted in essentially undetectable levels of PHF6a transcript while PHF6b transcripts remained at normal levels. We reasoned that the premature termination codon (PTC) created by the mutation induced NMD of the PHF6a transcript but not the longer PHF6b transcript due to its proximity to the splice junction site. Indeed, blocking NMD by treating cells with cycloheximide restored the level of PHF6a transcripts. As such we looked for a similar splicing event in primary cortical neurons from the R342X mice but the equivalent Phf6b transcript is not made. However, we did observe a novel alternatively spliced transcript that skipped exon 10 (ΔE10). The ΔE10 transcript was not present in human LCLs from patients the R342X mutation and it represents a distinct mechanism in the R342X mice to evade PTCs referred to as nonsense-associated altered splicing (50). Similarly, cycloheximide was able to restore the level of the normally spliced transcript suggesting that in both species, the R342X mutation drives the cell to produce an isoform (ΔE10 or PHF6b) which is stable and does not undergo NMD. It is unclear whether the production of an alternative stable transcript is required so that a minimal threshold level of PHF6 protein can be produced for survival, or whether it is a bystander product of the mutation. In this regard, we used the EX-SKIP tool to examine the sequence of exon 10 to locate putative ESE and silencers (ESS) to calculate the ESS/ESE ratio (40,51). Nucleotide variants that disrupt or create novel ESS or ESE sequences can impact exon skipping (51). We determined that the C > T transition generating the PTC and the engineered HindIII site created to facilitate genotyping both altered the ESS/ESE ratio and are predicted to increase splicing suggesting that exon skipping might be an inappropriate effect of the position of the mutation. This also raises the possibility that a second truncated protein product is produced and while our preliminary studies indicated that the truncated Phf6 protein is the larger protein product truncated at R342X, further studies are required to confirm that both products are not present.
Overall, the R342X mice present with many clinical features of BFLS including short stature, a hypoplastic pituitary gland, impaired learning and memory skills, and an anxiolytic phenotype that indicate an important role for PHF6 in cognitive development and neuroendocrine function. As such, the R342X mice represent an exciting mouse model for further analysis into the pathophysiology of BFLS and the molecular pathways regulated by the PHF6 protein.
Materials and Methods
Cell culture
R1 (129/SV) mouse embryonic stem cells (mESCs) were cultured on gelatin coated plates (Sigma) and a monolayer of γ-irradiated mouse embryonic fibroblastic (MEF) cells in ES cell media (Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 15% ES grade FBS, 50 U/ml penicillin, 50 μg/mL streptomycin, 1% MEM non-essential amino acids (Gibco), 1.5 x 10−4 M beta-mercaptoethanol and 2% LIF). Epstein–Barr virus (EBV) immortalized B-cell lines (LCLs) from patients were cultured at 37°C with 5% CO2 in RPMI 1640 (Invitrogen) supplemented with 10% FCS, 2 mM L-glutamine, 0.017 mg/mL benzylpenicillin. Primary cortical neurons were established from E13.5 embryonic cortices as previously described (52). Cortices from one brain were equally divided into three wells of a six-well plate pre-coated with 0.1 mg/mL poly-D-lysine (PDL; Sigma). Cells were grown at 37°C with 5% CO2 in Neurobasal A media (Gibco) containing 1X B27 (Thermo Fisher Scientific), 1X Glutamax (Gibco) and 50 U/mL of penicillin/streptomycin (Gibco) that was refreshed every other day. For assessment of nonsense-mediated decay, 100 μM cycloheximide solution (Sigma) or a 10 mM caffeine solution (Sigma) was added directly to the primary cultures for 6 h.
Generation of R342X mice
Guide RNA sequences (Supplementary Material, Table S3) were designed to target exon 10 of the mouse Phf6 gene using the online CRISPR tool (crispr.genome-engineering.org) developed by Ran et al. (34). Guide sequences were annealed and cloned into the PX458 plasmid (pSpCas9(BB)-2A-GFP; Addgene #48138). A 120-nucleotide single stranded oligomer (Supplementary Material, Table S3) homologous to the targeted region and containing the C to T mutation (c.1024 C > T) and two restriction sites (HindIII for screening and XhoI that altered the Cas9 recognition sequence) was electroporated into the R1 mESCs with the targeting plasmid using the P3 Primary Cell 4D-Nucleofector X Kit manufacturer’s protocol (Lonza). Two days later, the mESCs were harvested, filtered through a cell strainer to generate a single cell suspension for FACS sorting (OHRI StemCore Flow Cytometry Facility). GFP fluorescent and 7-AAD negative cells were sorted into individual wells of gelatin and MEF coated 96 well plates for expansion and screening. Genomic DNA isolated from individual clones was used for PCR amplification and HindIII restriction analysis of the targeted region. Positive clones were confirmed by sequencing and subsequently screened for off-target indels using the SURVEYOR assay (Surveyor Mutation Detection Kit, IDT). The SURVEYOR assay was performed with PCR amplicons (Supplementary Material, Table S3) for the top four predicted off-target sites as described previously (34). Of 56 GFP-positive cells, five contained the HindIII restriction site and one remained positive after sequencing and SURVEYOR screening. This single clone was cultured and passaged for 7 days then harvested and provided to the University of Ottawa Transgenic Mouse Core Facility for blastocyst injections using a previously described protocol (53). Four chimeric mice harboring the Phf6 R342X mutation were obtained and two showed germline transmission when bred to C57BL/6 female mice.
Animal husbandry
R342X heterozygous agouti female progeny were bred with C57BL/6 J WT males that produced mixed background C57BL/6 J::129/Sv progeny. Mixed background R342X heterozygous females were subsequently backcrossed onto C57BL/6 J or FVB/N backgrounds for > eight generations. C57BL/6 J and FVB/N wild type mice were purchased from Charles River (Montreal, QC, Canada) and the Jackson Laboratory. The R342X mutant mice (Phf6R342X/Y; referred to as R342X mice) were generated by breeding Phf6+/R342X heterozygous females to Phf6+/Y male mice. All mice were housed and bred at the University of Ottawa animal facility under SPF (specific pathogen-free) conditions in a 12 h/12 h light:dark cycle with food and water ad libitum. All animal experiments were approved by the University of Ottawa’s Animal Care ethics committee, with the guidelines set out by the Canadian Council on Animal Care.
MRI analysis
Mice (P100) were anaesthetized and intracardially perfused at 3 mL/min (Ismatec REGLO ICC) with 10 mL of 0.1 M PBS with 1 μL/mL heparin (Sigma) and 2 mM ProHance (Bracco Diagnostics). Following the PBS flush, mice were perfused with 10 mL of 4% paraformaldehyde (PFA) with 2 mM ProHance. Mouse skulls were dissected, keeping the brain intact in the skull and removing the zygomatic bones. Skulls were then placed in scintillation vials (Sigma) of 4% PFA with 2 mM ProHance overnight at 4°C. The next day, the solution was replaced with autoclaved 0.1 M PBS with 2 mM ProHance and 0.02% sodium azide at 4°C for a month before MRI scanning. The parameters for the MRI scan were as follows: T2, 3D fast spin echo sequence, with cylindrical acquisition of k-space, a TR of 350 ms and TE of 12 ms per echo for six echoes. The field of view was 20 x 20 x 25 mm3 and the matrix size was 504 x 504 x 630 mm3 producing an image with 0.04 mm isotropic voxels (54). The total scanning time was ~ 14 h. The data are analyzed to create average representations of each genotype. Brain region volumes are compared in the context of predetermined segmentations of the brain (55–60). To perform statistical tests, linear models followed by false discovery rate correction for multiple comparisons were performed within the RMINC package for R.
Behavioral analysis
All behavioral tests were completed in the Behavior Core Facility at the University of Ottawa using standardized protocols. We have previously described the test conditions used for the EPM, FC, OF Test and the Y-maze (41,61). The FST was performed by placing each mouse each mouse in a transparent plexiglass cylinder (22 cm diameter; 37 cm height) filled with 23–25°C tap water up to 10 cm. A camera linked to the Ethovision 8 software recorded the movement of each mouse for 6 min. Mice were returned to their home cage after testing. For all behavior tests, the animals were habituated to the testing room for a minimum of 1 h before testing, and only male mice were assessed. A minimum of 15 animals of each genotype were used. The values are presented as the mean ± SEM and analyzed for statistical significance using either an unpaired t-test (EPM, OF, FST and Y-maze) or by two-way ANOVA (FC).
Tissue preparation and analysis
Hormone ELISAs were performed on mouse plasma. To reduce variability in circulating hormone levels in the plasma, mice were fasted for 6 h prior to blood collection. EDTA coated microvettes (Sarstedt) were used to collect trunk blood. The blood was supplemented with Halt Protease Inhibitor (Thermo Fisher Scientific) and DPPIV (1:100) (Millipore). Next, samples were centrifuged at 1000xg for 10 min at 4°C. The supernatant was diluted 1:50 in 1X autoclaved PBS and sent to Eve Technologies to be run on the Steroid-Thyroid Hormone 6-Plex Discovery Assay.
To calculate tissue weight ratios, the mice were first weighed then intracardially perfused with 0.1 M PBS prior to dissection and weighing of individual tissues. For brain analysis, mice older than P10 were flushed with 4% PFA (Sigma) while younger mice were decapitated first and then dissected brains were placed in 4% PFA overnight at 4°C for fixation. The following day brains were washed with autoclaved 0.1 M PBS and then submerged in 30% sucrose solution. Brains were left in the sucrose solution at 4°C until the sample sank to the bottom of the vial, then placed in a 1:1 solution of 30% sucrose:OCT (VWR) overnight at 4°C. Brains were flash frozen using an embedding mold (VWR) filled with 1:1 solution of 30% sucrose:OCT, floating on a dish placed on liquid nitrogen. Frozen brains were stored at −80°C.
Immunofluorescent histochemistry
Cryostat sections (12 μm) were mounted onto SuperFrost Slides (Fisher Scientific), washed three times in PBST (0.1 M PBS with 0.1% Triton X-100), and then heated in 10 mM sodium citrate buffer (pH 6.0) for antigen retrieval prior to blocking in 10% goat serum/PBST for 30 min at room temperature. Slides were incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: mouse anti-SATB2 (1:50; Abcam #51502); rat anti-CTIP2 (1:250, Abcam #18465); rabbit anti-TBR1 (1:100, Abcam #31940); and rabbit anti-PHF6 (1:50, Sigma#HPA001023). The following day, sections were washed five times in PBST and incubated (2 h, room temperature) with anti-mouse 488 Alexa Fluor or anti-rabbit 594 Alexa Fluor secondary antibodies (1:4000, Invitrogen #A21202 and A21206) against the IgG domains of the primary antibodies. All sections were counterstained with Hoescht 33 342 dye (ThermoFisher Scientific) and mounted on slides with DAKO Fluorescent mounting medium (Agilent Technologies). Tissue sections were examined and images acquired with a Zeiss Axiovert Observer Z1 epifluorescent/light microscope equipped with an AxioCam cooled-color camera (Zeiss). Images were exported to Adobe Photoshop CS5 (Adobe Systems Inc., CA, USA) and further processed for contrast when necessary.
Immunoblotting
mESCs were grown to ~ 80% confluency under feeder-free conditions (approximately 7 x 106 cells) and harvested with trypsin. Tissues were flash frozen in liquid nitrogen following dissections and homogenized prior to lysis. Samples were lysed and their protein was extracted for 2–4 hours at 4°C under gentle agitation using RIPA buffer (150 mM NaCl, 1% NP-40, 0.1% SDS, 50 mM Tris pH 8.0, 5 mM EDTA, 0.5% Na-deoxycholate) supplemented with Halt Protease Inhibitor (Thermo Fisher Scientific). Lysed samples were centrifuged at 12000xg for 30 min at 4°C to remove insoluble material and the supernatant containing soluble protein was retained. LCLs were lysed by incubation in RIPA buffer (65.3 mM Tris (pH 7.4), 150 mM NaCl, 1% Nonidet P40, 1 mM NaVO3, 1 mM NaF and 1 x protease inhibitor cocktail (Sigma, Australia)) on ice for 15 min followed by trituration through a 21G needle. Insoluble material was removed by centrifugation at 13 000xg for 15 min.
The concentrations of the protein extracts were quantified using the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad) and the extracts were denatured and reduced with NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) and separated by electrophoresis at 90–150 V using the Bio-Rad Mini-PROTEAN Tetra Cell. Electrophoresed proteins were wet-transferred from the polyacrylamide gel onto a PVDF membrane (Immobilon-P; Millipore, MA, USA) at 0.35 A for 90 min using the Bio-Rad Mini Trans-Blot cell. For larger gels and membranes, the PROTEAN II xi Cell (Bio-Rad) at 200 V and the Trans-Blot Cell (Bio-Rad) at 1.50 A for 2 h, were used. For hLCLs, protein was transferred to a BioTrace NT nitrocellulose membrane (PALL Life Sciences, USA). Membranes were blocked (1 h, RT) with 5% Skim milk in TBST and incubated (4°C, overnight) with the following primary antibodies: mouse anti-β-Actin (1:30 000; Sigma); mouse anti-Vinculin (1:2500; Sigma-Aldrich); rabbit anti-Vinculin (1:5000 WB mESCs, 1:500 WB cortical lysates; Abcam #129002); rabbit anti-PHF6 (1:500; Sigma-Aldrich; mESCs and mouse cortical extracts); and the previously described mouse monoclonal anti-PHF6 (clone 4B1B6; LCLs; (35)) overnight. Membranes were incubated (1 h, room temperature) with ImmunoPure® HRP-conjugated goat anti-rabbit or donkey anti-mouse secondary antibodies (1:50 000; Sigma #A4914 and A5906). Membranes were washed 5 x 5 min in TBST after antibody incubations, and signals were detected using the Pierce Supersignal West Fempto chemiluminescence substrate (Cat # 34095). For LCLs, the Clarity Western ECL substrate (Bio-Rad) was added to the membranes to detect the probed protein and the enhanced chemiluminescence method (GE Healthcare, Sweden) was used to develop the blots.
Reverse transcription and quantitative real-time PCR
Total RNA was extracted from cultured mESCs grown under feeder-free conditions, human LCLs, primary cortical neurons or flash frozen tissue using standard TRIzol Reagent extraction as described by the manufacturer’s protocol (Thermo Fisher Scientific). Glycogen (Thermo Fisher Scientific) was used as a carrier to improve yield. About 1 μg of total RNA was reverse-transcribed using the GoScript™ Reverse Transcriptase (Promega) when working with mESCs and RevertAid Reverse Transcriptase (Thermo Fisher Scientific) was used with tissue samples.
For mESCs, the cDNA was diluted 1:40 and qPCR was performed using 1X SYBR® Advantage® qPCR Premix (Clontech #639676) under the following conditions: one cycle at 95°C for 1 min, and then 40 consecutive cycles at 95°C for 10 s, 61°C for 10 s and 72°C for 20 s. All primers were analyzed by melt curve analysis after qPCR amplification. The ∆∆Ct method was used to compare fold-change. L32 and 18S mRNAs were used as normalizers in separate experiments. Triplicate or quadruplicate samples were performed per reaction and a minimum of three mice analyzed per genotype. Student’s t-test was used for statistical significance. For cortical tissue, total cDNA was diluted 1:10 and qPCR analysis performed using the SensiFAST SYBR Lo- ROX Kit (Bioline) with the annealing temperature set at 55°C. All qPCR primers are listed in Supplementary Material, Table S3.
RNA sequencing analysis
To perform RNA-seq, purified and DNA free RNA samples (50 ng/μL), were sent to the Centre d’expertise et de services Génome Québec. mRNA libraries were prepared using NEBNext dual adapters (NEBNext multiplex oligos for Illumina (Dual index primers set 1)). Samples were run on an Illumina NovaSeq 6000 S2. Paired-end sequencing was utilized and 50 million reads were targeted from each sample. Reads were aligned and quantified to the mouse genome (GENCODE vM19) with Salmon v0.12.0 using gcBias flag. Differential expression analysis was performed in R using the DESeq2 package (62) as follows: (i) Read transcript quantification data with tximport library and summarized to gene level quantification, (ii) Filtered to retain genes with at least five reads assigned in at least two samples, (iii) Constructed DESeqDataSet object with eight samples (four replicated of two conditions from two litters), using the model ~litter+condition (condition is cell type/treatment), (iv) Calculated fold changes using the lfcShrink function with the apeglm method. A similar strategy was used for the RNAseq analysis of human lymphoblast cell lines (four control samples, two BFLS patients with R342X mutations). The mouse RNAseq data have been deposited into the GEO database with the accession number GSE156404. The human RNAseq data are part of a larger Australian project but are available from the authors on request (mark.corbett@adelaide.edu.au). The human and mouse DEG lists were compared using GeneOverlap.
GO gene analysis was performed from gProfiler using the g:GOSt functional profiling tool. The query was ordered and an s-value threshold of <0.05 was used. Molecular function was used to categorize terms. All available data sources were used.
DO gene analysis was performed using the DOSE package on R. DOSE identifies the human transcripts which are associated with human disease. Human disease related transcripts orthologs were matched to the differentially expressed mouse transcripts from the RNA-seq DEG dataset. These targets were used to find the disease categories for our dataset.
PHF6 isoform analysis
PCR reactions were performed using primers located in exons 9 and 11 (Supplementary Material, Table S3) under the following conditions: one cycle at 94°C for 3 min, and then 40 consecutive cycles at 94°C for 45 s, 59°C for 30 s and 72°C for 40 s. PCR products were separated on a 1% agarose gel and imaged. To quantify bands from a gel, the agarose gel was imaged using a ChemiDoc-It Imager. Image J was used to analyze the intensity of the bands from the TIF file. The ‘label peaks function’ was used to measure the intensity of each band. Background was accounted for and the ‘Wand’ tool was used to measure the area under each peak. Phf6 isoform band intensities were normalized to the control gene band intensities. To determine the primary sequence of the amplified products, UV illuminated DNA bands were excised and extracted using the QIAquick Gel Extraction Kit (Qiagen) and sent to the StemCore Laboratory (OHRI) for Sanger sequencing on the Applied Biosystems 3730 DNA Analyzer after amplification with the Big Dye Terminator (BDT) v3.1 kit.
Statistics
To determine if pups were born in normal Mendelian ratios, Chi-square tests were used. A Student’s t-test was used to determine significant differences for experiments where indicated. *, **, *** and **** represent P-values of < 0.05, < 0.01, < 0.001 and < 0.0001, respectively. The t-tests were unpaired, parametric with equal SD. If the SD was not equal from the f-test, the t-test was performed with Welch’s correction. When evaluating the MRI data and RNA-seq data, an FDR approach was used.
Supplementary Material
Acknowledgements
The authors would like to thank Renee Carroll for technical assistance with the analysis of human lymphoblast samples. The RNA-sequencing was deposited to the GEO Database with the accession number GSE156404.
Conflict of Interest. The authors have no conflicts to declare.
Author Contributions
R.A. maintained the mouse colonies and performed the majority of the experiments. S.S. generated the CRISPR-Cas9 targeted ES cells; V.C. performed behavioral studies; J.H. analyzed cortical development; G.Z. provided bioinformatics support; L.R.Q. and J.P.L. performed MRI analysis; K.Y. and L.V. provided technical support; M.A.C. and J.G. designed and performed the human lymphoblast experiments; D.J.P. designed and supervised the project. D.J.P. and R.A. wrote the manuscript with input from all authors.
Funding Statement
The work was funded by an operating grant from the Canadian Institutes of Health Research (FRN159619) to D.J.P.
References
- 1. Lower, K.M., Turner, G., Kerr, B.A., Mathews, K.D., Shaw, M.A., Gedeon, A.K., Schelley, S., Hoyme, H.E., White, S.M., Delatycki, M.B.et al. (2002) Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat. Genet., 32, 661–665. [DOI] [PubMed] [Google Scholar]
- 2. Borjeson, M., Forssman, H. and Lehmann, O. (1962) An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med. Scand., 171, 13–21. [DOI] [PubMed] [Google Scholar]
- 3. Turner, G., Gedeon, A., Mulley, J., Sutherland, G., Rae, J., Power, K. and Arthur, I. (1989) Borjeson-Forssman-Lehmann syndrome: clinical manifestations and gene localization to Xq26-27. Am. J. Med. Genet., 34, 463–469. [DOI] [PubMed] [Google Scholar]
- 4. Turner, G., Lower, K.M., White, S.M., Delatycki, M., Lampe, A.K., Wright, M., Smith, J.C., Kerr, B., Schelley, S., Hoyme, H.E.et al. (2004) The clinical picture of the Borjeson-Forssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin. Genet., 65, 226–232. [DOI] [PubMed] [Google Scholar]
- 5. Visootsak, J., Rosner, B., Dykens, E., Schwartz, C., Hahn, K., White, S.M., Szeftel, R. and Graham, J.M. (2004) Clinical and behavioral features of patients with Borjeson-Forssman-Lehmann syndrome with mutations in PHF6. J. Pediatr., 145, 819–825. [DOI] [PubMed] [Google Scholar]
- 6. Crawford, J., Lower, K.M., Hennekam, R.C., Van Esch, H., Megarbane, A., Lynch, S.A., Turner, G. and Gecz, J. (2006) Mutation screening in Borjeson-Forssman-Lehmann syndrome: identification of a novel de novo PHF6 mutation in a female patient. J. Med. Genet., 43, 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carter, M.T., Picketts, D.J., Hunter, A.G. and Graham, G.E. (2009) Further clinical delineation of the Borjeson-Forssman-Lehmann syndrome in patients with PHF6 mutations. Am. J. Med. Genet. A, 149A, 246–250. [DOI] [PubMed] [Google Scholar]
- 8. Mangelsdorf, M., Chevrier, E., Mustonen, A. and Picketts, D.J. (2009) Borjeson-Forssman-Lehmann syndrome due to a novel plant homeodomain zinc finger mutation in the PHF6 gene. J. Child Neurol., 24, 610–614. [DOI] [PubMed] [Google Scholar]
- 9. Zweier, C., Kraus, C., Brueton, L., Cole, T., Degenhardt, F., Engels, H., Gillessen-Kaesbach, G., Graul-Neumann, L., Horn, D., Hoyer, J.et al. (2013) A new face of Borjeson-Forssman-Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype. J. Med. Genet., 50, 838–847. [DOI] [PubMed] [Google Scholar]
- 10. Zweier, C., Rittinger, O., Bader, I., Berland, S., Cole, T., Degenhardt, F., Di Donato, N., Graul-Neumann, L., Hoyer, J., Lynch, S.A.et al. (2014) Females with de novo aberrations in PHF6: clinical overlap of Borjeson-Forssman-Lehmann with Coffin-Siris syndrome. Am. J. Med. Genet. C Semin. Med. Genet., 166C, 290–301. [DOI] [PubMed] [Google Scholar]
- 11. Vallee, D., Chevrier, E., Graham, G.E., Lazzaro, M.A., Lavigne, P.A., Hunter, A.G. and Picketts, D.J. (2004) A novel PHF6 mutation results in enhanced exon skipping and mild Borjeson-Forssman-Lehmann syndrome. J. Med. Genet., 41, 778–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Todd, M.A., Ivanochko, D. and Picketts, D.J. (2015) PHF6 degrees of separation: the multifaceted roles of a chromatin adaptor protein. Genes (Basel), 6, 325–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Todd, M.A., Huh, M.S. and Picketts, D.J. (2016) The sub-nucleolar localization of PHF6 defines its role in rDNA transcription and early processing events. Eur. J. Hum. Genet., 24, 1453–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Todd, M.A. and Picketts, D.J. (2012) PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J. Proteome Res., 11, 4326–4337. [DOI] [PubMed] [Google Scholar]
- 15. Gecz, J., Turner, G., Nelson, J. and Partington, M. (2006) The Borjeson-Forssman-Lehman syndrome (BFLS, MIM #301900). Eur. J. Hum. Genet., 14, 1233–1237. [DOI] [PubMed] [Google Scholar]
- 16. Chao, M.M., Todd, M.A., Kontny, U., Neas, K., Sullivan, M.J., Hunter, A.G., Picketts, D.J. and Kratz, C.P. (2010) T-cell acute lymphoblastic leukemia in association with Borjeson-Forssman-Lehmann syndrome due to a mutation in PHF6. Pediatr. Blood Cancer, 55, 722–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Vlierberghe, P., Palomero, T., Khiabanian, H., Van der Meulen, J., Castillo, M., Van Roy, N., De Moerloose, B., Philippe, J., Gonzalez-Garcia, S., Toribio, M.L.et al. (2010) PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet., 42, 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Vlierberghe, P., Patel, J., Abdel-Wahab, O., Lobry, C., Hedvat, C.V., Balbin, M., Nicolas, C., Payer, A.R., Fernandez, H.F., Tallman, M.S.et al. (2011) PHF6 mutations in adult acute myeloid leukemia. Leukemia, 25, 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lower, K.M., Solders, G., Bondeson, M.L., Nelson, J., Brun, A., Crawford, J., Malm, G., Borjeson, M., Turner, G., Partington, M.et al. (2004) 1024C> T (R342X) is a recurrent PHF6 mutation also found in the original Borjeson-Forssman-Lehmann syndrome family. Eur. J. Hum. Genet., 12, 787–789. [DOI] [PubMed] [Google Scholar]
- 20. Nitarska, J., Smith, J.G., Sherlock, W.T., Hillege, M.M., Nott, A., Barshop, W.D., Vashisht, A.A., Wohlschlegel, J.A., Mitter, R. and Riccio, A. (2016) A functional switch of NuRD chromatin remodeling complex subunits regulates mouse cortical development. Cell Rep., 17, 1683–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weiss, K., Terhal, P.A., Cohen, L., Bruccoleri, M., Irving, M., Martinez, A.F., Rosenfeld, J.A., Machol, K., Yang, Y., Liu, P.et al. (2016) De novo mutations in CHD4, an ATP-dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am. J. Hum. Genet., 99, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Snijders Blok, L., Rousseau, J., Twist, J., Ehresmann, S., Takaku, M., Venselaar, H., Rodan, L.H., Nowak, C.B., Douglas, J., Swoboda, K.J.et al. (2018) CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun., 9, 4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang, J., Leung, J.W., Gong, Z., Feng, L., Shi, X. and Chen, J. (2013) PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. J. Biol. Chem., 288, 3174–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McRae, H.M., Garnham, A.L., Hu, Y., Witkowski, M.T., Corbett, M.A., Dixon, M.P., May, R.E., Sheikh, B.N., Chiang, W., Kueh, A.J.et al. (2019) PHF6 regulates hematopoietic stem and progenitor cells and its loss synergizes with expression of TLX3 to cause leukemia. Blood, 133, 1729–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyagi, S., Sroczynska, P., Kato, Y., Nakajima-Takagi, Y., Oshima, M., Rizq, O., Takayama, N., Saraya, A., Mizuno, S., Sugiyama, F.et al. (2019) The chromatin-binding protein Phf6 restricts the self-renewal of hematopoietic stem cells. Blood, 133, 2495–2506. [DOI] [PubMed] [Google Scholar]
- 26. Soto-Feliciano, Y.M., Bartlebaugh, J.M.E., Liu, Y., Sanchez-Rivera, F.J., Bhutkar, A., Weintraub, A.S., Buenrostro, J.D., Cheng, C.S., Regev, A., Jacks, T.E.et al. (2017) PHF6 regulates phenotypic plasticity through chromatin organization within lineage-specific genes. Genes Dev., 31, 973–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wendorff, A.A., Quinn, S.A., Rashkovan, M., Madubata, C.J., Ambesi-Impiombato, A., Litzow, M.R., Tallman, M.S., Paietta, E., Paganin, M., Basso, G.et al. (2019) Phf6 loss enhances HSC self-renewal driving tumor initiation and leukemia stem cell activity in T-ALL. Cancer Discov., 9, 436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang, C., Mejia, L.A., Huang, J., Valnegri, P., Bennett, E.J., Anckar, J., Jahani-Asl, A., Gallardo, G., Ikeuchi, Y., Yamada, T.et al. (2013) The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron, 78, 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rio, C., Rieff, H.I., Qi, P., Khurana, T.S. and Corfas, G. (1997) Neuregulin and erbB receptors play a critical role in neuronal migration. Neuron, 19, 39–50. [DOI] [PubMed] [Google Scholar]
- 30. Franzoni, E., Booker, S.A., Parthasarathy, S., Rehfeld, F., Grosser, S., Srivatsa, S., Fuchs, H.R., Tarabykin, V., Vida, I. and Wulczyn, F.G. (2015) miR-128 regulates neuronal migration, outgrowth and intrinsic excitability via the intellectual disability gene Phf6. elife, 4, e04263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng, C., Deng, P.Y., Ikeuchi, Y., Yuede, C., Li, D., Rensing, N., Huang, J., Baldridge, D., Maloney, S.E., Dougherty, J.D.et al. (2018) Characterization of a mouse model of Borjeson-Forssman-Lehmann syndrome. Cell Rep., 25, 1404, e1406–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McRae, H.M., Eccles, S., Whitehead, L., Alexander, W.S., Gecz, J., Thomas, T. and Voss, A.K. (2020) Downregulation of the GHRH/GH/IGF1 axis in a mouse model of Borjeson-Forssman-Lehman syndrome. Development, 147, dev187021. [DOI] [PubMed] [Google Scholar]
- 33. Gan, L., Sun, J., Yang, S., Zhang, X., Chen, W., Sun, Y., Wu, X., Cheng, C., Yuan, J., Li, A.et al. (2020) Chromatin-binding protein PHF6 regulates activity-dependent transcriptional networks to promote hunger response. Cell Rep., 30, 3717, e3716–3728. [DOI] [PubMed] [Google Scholar]
- 34. Ran, F.A., Hsu, P.D., Wright, J., Agarwala, V., Scott, D.A. and Zhang, F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc., 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voss, A.K., Gamble, R., Collin, C., Shoubridge, C., Corbett, M., Gecz, J. and Thomas, T. (2007) Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann syndrome of intellectual disability and obesity. Gene Expr. Patterns, 7, 858–871. [DOI] [PubMed] [Google Scholar]
- 36. Zhang, X., Fan, Y., Liu, X., Zhu, M.A., Sun, Y., Yan, H., He, Y., Ye, X., Gu, X. and Yu, Y. (2019) A novel nonsense mutation of PHF6 in a female with extended phenotypes of Borjeson-Forssman-Lehmann syndrome. J. Clin. Res. Pediatr. Endocrinol., 11, 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Birrell, G., Lampe, A., Richmond, S., Bruce, S.N., Gecz, J., Lower, K., Wright, M. and Cheetham, T.D. (2003) Borjeson-Forssman-Lehmann syndrome and multiple pituitary hormone deficiency. J. Pediatr. Endocrinol. Metab., 16, 1295–1300. [DOI] [PubMed] [Google Scholar]
- 38. de Winter, C.F., vanDijk, F., Stolker, J.J. and Hennekam, R.C. (2009) Behavioural phenotype in Borjeson-Forssman-Lehmann syndrome. J. Intellect. Disabil. Res., 53, 319–328. [DOI] [PubMed] [Google Scholar]
- 39. Maren, S. (2001) Neurobiology of Pavlovian fear conditioning. Annu. Rev. Neurosci., 24, 897–931. [DOI] [PubMed] [Google Scholar]
- 40. Raponi, M., Kralovicova, J., Copson, E., Divina, P., Eccles, D., Johnson, P., Baralle, D. and Vorechovsky, I. (2011) Prediction of single-nucleotide substitutions that result in exon skipping: identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat., 32, 436–444. [DOI] [PubMed] [Google Scholar]
- 41. Alvarez-Saavedra, M., Yan, K., De Repentigny, Y., Hashem, L.E., Chaudary, N., Sarwar, S., Yang, D., Ioshikhes, I., Kothary, R., Hirayama, T.et al. (2019) Snf2h drives chromatin Remodeling to prime upper layer cortical neuron development. Front. Mol. Neurosci., 12, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sokpor, G., Castro-Hernandez, R., Rosenbusch, J., Staiger, J.F. and Tuoc, T. (2018) ATP-dependent chromatin remodeling during cortical neurogenesis. Front. Neurosci., 12, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tuoc, T.C., Boretius, S., Sansom, S.N., Pitulescu, M.E., Frahm, J., Livesey, F.J. and Stoykova, A. (2013) Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell, 25, 256–269. [DOI] [PubMed] [Google Scholar]
- 44. Yip, D.J., Corcoran, C.P., Alvarez-Saavedra, M., DeMaria, A., Rennick, S., Mears, A.J., Rudnicki, M.A., Messier, C. and Picketts, D.J. (2012) Snf2l regulates Foxg1-dependent progenitor cell expansion in the developing brain. Dev. Cell, 22, 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feng, W., Khan, M.A., Bellvis, P., Zhu, Z., Bernhardt, O., Herold-Mende, C. and Liu, H.K. (2013) The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell, 13, 62–72. [DOI] [PubMed] [Google Scholar]
- 46. Shan, Y., Zhang, Y., Zhao, Y., Wang, T., Zhang, J., Yao, J., Ma, N., Liang, Z., Huang, W., Huang, K.et al. (2020) JMJD3 and UTX determine fidelity and lineage specification of human neural progenitor cells. Nat. Commun., 11, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berube, N.G., Mangelsdorf, M., Jagla, M., Vanderluit, J., Garrick, D., Gibbons, R.J., Higgs, D.R., Slack, R.S. and Picketts, D.J. (2005) The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J. Clin. Invest., 115, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Allemang-Grand, R., Ellegood, J., Spencer Noakes, L., Ruston, J., Justice, M., Nieman, B.J. and Lerch, J.P. (2017) Neuroanatomy in mouse models of Rett syndrome is related to the severity of Mecp2 mutation and behavioral phenotypes. Mol. Autism., 8, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Popp, M.W. and Maquat, L.E. (2013) Organizing principles of mammalian nonsense-mediated mRNA decay. Annu. Rev. Genet., 47, 139–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang, J., Chang, Y.F., Hamilton, J.I. and Wilkinson, M.F. (2002) Nonsense-associated altered splicing: a frame-dependent response distinct from nonsense-mediated decay. Mol. Cell, 10, 951–957. [DOI] [PubMed] [Google Scholar]
- 51. Grodecka, L., Buratti, E. and Freiberger, T. (2017) Mutations of pre-mRNA splicing regulatory elements: are predictions moving forward to clinical diagnostics? Int. J. Mol. Sci., 18, 1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Castro, M., Caprile, T., Astuya, A., Millan, C., Reinicke, K., Vera, J.C., Vasquez, O., Aguayo, L.G. and Nualart, F. (2001) High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem., 78, 815–823. [DOI] [PubMed] [Google Scholar]
- 53. Longenecker, G. and Kulkarni, A.B. (2009) Generation of gene knockout mice by ES cell microinjection. Curr. Protoc. Cell Biol., 44, 11–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Spencer Noakes, T.L., Henkelman, R.M. and Nieman, B.J. (2017) Partitioning k-space for cylindrical three-dimensional rapid acquisition with relaxation enhancement imaging in the mouse brain. NMR Biomed., 30. https://pubmed.ncbi.nlm.nih.gov/28902423/. [DOI] [PubMed] [Google Scholar]
- 55. Jung, H., Park, H., Choi, Y., Kang, H., Lee, E., Kweon, H., Roh, J.D., Ellegood, J., Choi, W., Kang, J.et al. (2018) Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci., 21, 1218–1228. [DOI] [PubMed] [Google Scholar]
- 56. Tamming, R.J., Dumeaux, V., Jiang, Y., Shafiq, S., Langlois, L., Ellegood, J., Qiu, L.R., Lerch, J.P. and Berube, N.G. (2020) Atrx deletion in neurons leads to sexually dimorphic dysregulation of miR-137 and spatial learning and memory deficits. Cell Rep., 31, 107838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Whittaker, D.E., Kasah, S., Donovan, A.P.A., Ellegood, J., Riegman, K.L.H., Volk, H.A., McGonnell, I., Lerch, J.P. and Basson, M.A. (2017) Distinct cerebellar foliation anomalies in a CHD7 haploinsufficient mouse model of CHARGE syndrome. Am. J. Med. Genet. C Semin. Med. Genet., 175. doi: 10.1002/ajmg.c.31595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Whittaker, D.E., Riegman, K.L., Kasah, S., Mohan, C., Yu, T., Pijuan-Sala, B., Hebaishi, H., Caruso, A., Marques, A.C., Michetti, C.et al. (2017) The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J. Clin. Invest., 127, 874–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gompers, A.L., Su-Feher, L., Ellegood, J., Copping, N.A., Riyadh, M.A., Stradleigh, T.W., Pride, M.C., Schaffler, M.D., Wade, A.A., Catta-Preta, R.et al. (2017) Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci., 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsai, P.T., Rudolph, S., Guo, C., Ellegood, J., Gibson, J.M., Schaeffer, S.M., Mogavero, J., Lerch, J.P., Regehr, W. and Sahin, M. (2018) Sensitive periods for cerebellar-mediated autistic-like behaviors. Cell Rep., 25, 357, e354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alvarez-Saavedra, M., De Repentigny, Y., Lagali, P.S., Raghu Ram, E.V., Yan, K., Hashem, E., Ivanochko, D., Huh, M.S., Yang, D., Mears, A.J.et al. (2014) Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat. Commun., 5, 4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Love, M.I., Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.