

Abstract
Mitochondrial respiratory chain disorders are empirically managed with variable antioxidant, cofactor and vitamin ‘cocktails’. However, clinical trial validated and approved compounds, or doses, do not exist for any single or combinatorial mitochondrial disease therapy. Here, we sought to pre-clinically evaluate whether rationally designed mitochondrial medicine combinatorial regimens might synergistically improve survival, health and physiology in translational animal models of respiratory chain complex I disease. Having previously demonstrated that gas-1(fc21) complex I subunit ndufs2−/− C. elegans have short lifespan that can be significantly rescued with 17 different metabolic modifiers, signaling modifiers or antioxidants, here we evaluated 11 random combinations of these three treatment classes on gas-1(fc21) lifespan. Synergistic rescue occurred only with glucose, nicotinic acid and N-acetylcysteine (Glu + NA + NAC), yielding improved mitochondrial membrane potential that reflects integrated respiratory chain function, without exacerbating oxidative stress, and while reducing mitochondrial stress (UPRmt) and improving intermediary metabolic disruptions at the levels of the transcriptome, steady-state metabolites and intermediary metabolic flux. Equimolar Glu + NA + NAC dosing in a zebrafish vertebrate model of rotenone-based complex I inhibition synergistically rescued larval activity, brain death, lactate, ATP and glutathione levels. Overall, these data provide objective preclinical evidence in two evolutionary-divergent animal models of mitochondrial complex I disease to demonstrate that combinatorial Glu + NA + NAC therapy significantly improved animal resiliency, even in the face of stressors that cause severe metabolic deficiency, thereby preventing acute neurologic and biochemical decompensation. Clinical trials are warranted to evaluate the efficacy of this lead combinatorial therapy regimen to improve resiliency and health outcomes in human subjects with mitochondrial disease.
Introduction
Mitochondrial disease is a remarkably heterogeneous yet collectively common group of energy deficiency disorders for which there exists no FDA-approved treatments or cures (1). Since the first genetic cause was identified three decades ago, an estimated 350 different gene disorders have been recognized to directly impair mitochondrial respiratory chain (RC) function (2). {McCormick, 2018 #2} Mitochondrial RC dysfunction may also secondarily result from a wide array of other genetic conditions, medications or environmental exposures, and is recognized to be a common pathophysiologic occurrence in metabolic syndrome, aging and neurodegenerative disorders such as Alzheimer and Parkinson’s diseases (3–7). Regardless of the cause, individuals with mitochondrial RC dysfunction are commonly managed on variable empiric combinations of antioxidants, cofactors and vitamin supplements, often referred to as ‘mitochondrial cocktails’ (8,9). However, there have been no single, gold-standard mitochondrial cocktail composition, validated dosing recommendations, nor clinical trials or objective pre-clinical evidence to support the safety or efficacy of administering cocktail-based combinatorial therapies for mitochondrial RC disease (10,11).
Mitochondrial RC disease clinical manifestations may result from diverse aspects of its downstream cellular pathophysiology. Impaired aerobic production by the RC of chemical energy in the form of adenosine triphosphate (ATP) is one major pathogenic factor. However, excessive oxidative stress that reflects the balance of oxidant production and scavenging also plays a contributing role. Indeed, the RC is a major site of oxidative stress generation at complexes I and III in the form of superoxide, which is near instantaneously converted by manganese superoxide dismutase (MnSOD) to yield hydrogen peroxide (12). Altered cellular reduction–oxidation (redox) balance also plays a key role in mediating the clinical pathophysiology of mitochondrial disease, most notably by failure of complex I (NADH dehydrogenase) to convert nicotinamide adenine dinucleotide that feeds electrons to the RC from its reduced (NADH) to oxidized (NAD+) form, a key redox couple whose balance regulates hundreds of other downstream cellular reactions (13). When considering these major RC products, it becomes clear that a multi-pronged therapeutic approach, or ‘cocktail’, may indeed be needed to replete diverse product deficiencies that result from primary RC disease. Such combinatorial therapies might specifically include antioxidants (to scavenge the increased oxidant burden (14,15)), signaling modifiers (to replete NAD+ or rebalance cellular capacity with demand (13,16)) and metabolic modifying compounds (providing alternative fuels to support, for example, ATP production by anaerobic glycolysis; or enzymatic cofactors that boost residual RC function). Thus, rationally designing ‘mitochondrial cocktail(s)’ might well play an important role in developing effective treatments for the diverse pathophysiologic sequelae that occur in mitochondrial disease, which on average causes 16 major symptoms per patient (17).
We sought to use preclinical models, both invertebrate (C. elegans, nematodes) and vertebrate (D. rerio, zebrafish) animals (18), to systematically evaluate whether rationally designed ‘mitochondrial cocktail(s)’ might yield objective synergistic effects to improve survival, organismal health and mitochondrial physiology in mitochondrial RC complex I disease (Fig. 1). Previously, we have screened 37 individual drug components of empirically postulated mitochondrial disease cocktail therapies at their maximal tolerated, non-lethal dose in a well-characterized and extensively validated C. elegans worm model of complex I disease, gas-1(fc21) (Supplementary Material, Fig. S1). These worms harbor a stable autosomal recessive p.R290K missense mutation in the orthologue to the nuclear gene encoding the NDUFS2 structural subunit of complex I, with significantly reduced lifespan and fertility, delayed development and substantially altered mitochondrial physiology and global intermediary metabolism (12,13,19–22). Overall, these RC disease mutants’ short lifespan were significantly rescued to variable extent by 17 drugs, which we categorized into three general treatment classes: antioxidants (14), signaling modifiers (13) and metabolic modifiers (21). Here, we evaluated whether randomly combining treatment leads from each of these three classes would synergistically improve gas-1(fc21) lifespan, as an integrated physiologic measure of their safety and therapeutic benefit (Supplementary Material, Fig. S2). Remarkably, synergy was found with only one of 11 combinatorial treatments tested, namely glucose, nicotinic acid and N-acetylcysteine (Glu + NA + NAC). Detailed mechanistic analyses in C. elegans of this synergistic ‘cocktail’ were performed to examine its effect on mitochondrial physiology (mitochondrial mass, membrane potential, oxidative stress and unfolded protein response (UPRmt)) and global metabolic effects on intermediary metabolism (transcriptome profiling by RNAseq, metabolite level and stable isotope flux profiling by HPLC and GC/MS analyses). Validation of the safety and beneficial effects of the Glu + NA + NAC synergistic combination we initially identified in the C. elegans invertebrate complex I disease worm model was performed at equivalent molar dosing in D. rerio zebrafish vertebrate animals with pharmacologic (rotenone) inhibition of mitochondrial complex I to assess its effects on animal swimming activity, prevention of brain death, and classical biomarkers of mitochondrial physiology (lactic acid, lactate:pyruvate and NADH/NAD+ ratios, ATP levels and glutathione levels).
Figure 1.
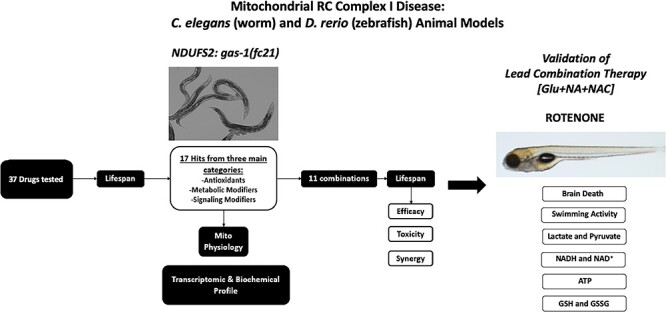
Schematic overview of experimental validation of combinatorial therapies in C. elegans and zebrafish models of respiratory chain complex I disease. Having previously identified 37 individual compounds in three major drug classes that significantly improved the short lifespan of complex I-deficient gas-1(fc21) mutant (that has a homozygous missense mutation in the nuclear-encoded complex I structural subunit NDUFS2 orthologue) C. elegans (worms) (Supplementary Material, Fig. S1), 11 combinations were randomly selected to evaluate the efficacy, toxicity and synergy of combining treatment classes on gas-1(fc21) worms’ lifespan as a primary outcome. Mechanistic analyses were performed at the level of mitochondrial physiology, transcriptome and metabolome analyses to interrogate the lead synergistic combination (glucose (Glu) + nicotinic acid (NAC) + N-acetylcysteine (NAC)). Validation of this lead combinatorial therapy was performed to assess brain death, swimming activity and effects on central biochemical readouts of mitochondrial dysfunction in a vertebrate zebrafish model of mitochondrial respiratory chain disease caused by pharmacologic (rotenone) inhibition of complex I function.
Results
17 individual treatments from three drug classes significantly improved the short lifespan of NDUFS2−/− mutant gas-1(fc21) worms
gas-1(fc21) worms have been extensively characterized by our research laboratory and others and shown to have consistent pathophysiology at multiple levels, including significantly reduced median lifespan at 20°C relative to N2 Bristol (wild-type, WT) worms (12,23,24). Over the past decade, we have manually evaluated lifespan effects in this model of 37 empirically recommended components of mitochondrial disease ‘cocktails’ (Supplementary Material, Fig. S1). Overall, 17 individual drug therapies were identified, many of which we previously reported in extensive detail (13–15,21), at doses that were non-toxic to wild-type animal development and significantly improved to variable extent the short median lifespan of gas-1(fc21) worms towards that of wild-type (N2 Bristol) worms (Supplementary Material, Fig. S2). These lead therapeutic candidates were organized into three general treatment categories, consisting of antioxidants, metabolic modifiers and signaling modifiers.
Only one combinatorial treatment regimen synergistically rescued lifespan of NDUFS2−/− mutant gas-1(fc21) worms
Treatment with 17 different drugs across three general treatment categories when initiated either at early development (L1 stage, where more pronounced effects were generally seen) or upon reaching adult life significantly, but often only partially, improved gas-1(fc21) lifespan. Therefore, we sought to determine if synergistic lifespan extension in these complex I disease animals might be achieved by combining drugs from each of the three treatment classes into rational therapeutic ‘cocktails’. We postulated this approach might collectively address the major downstream pathophysiology of RC disease, compare the relative efficacy of different combination regimens, and evaluate whether some drug combinations might unintentionally negate the component compounds’ individual beneficial effects (Fig. 1).
Eleven triplet drug combinations were randomly designed by our project bioinformatics scientist (Supplementary Material, Fig. S2), where each combination pooled three of the 17 lead compounds selected, with inclusion of one from each of the three major drug categories (Supplementary Material, Figs. S2 and S3). Initial lifespan screening of these 11 combinatorial regimens from early larval (L1 stage) exposure led to the identification of only two combinatorial regimens that significantly increased the lifespan of short-lived gas-1(fc21) worms (Fig. 2A and B and Supplementary Material, Fig. S4). These regimens were Glu + NA + NAC (glucose + nicotinic acid + N-acetylcysteine, 55% improvement in median lifespan with P < 0.0001 relative to buffer-only gas-1(fc21)) and Resv+FA + Cyst (resveratrol + folinic acid + cysteamine, and 33% improvement in median lifespan with P < 0.0001 relative to buffer-only gas-1(fc21)). Interestingly, the Glu + NA + NAC combinatorial-treated gas-1(fc21) worms’ median lifespan was significantly longer even than wild-type (N2 Bristol) worms (P = 0.011) (Fig. 2A and Supplementary Material, Fig. S4).
Figure 2.
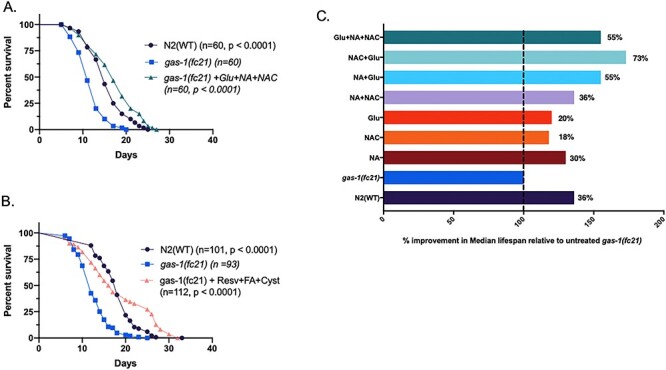
Analysis of combinatorial treatments on C. elegans gas-1(fc21) complex I mutant worms’ lifespan. Lifespan analyses were performed at 20°C in gas-1(fc21) worms treated from the L1 early larval development stage with one of 11 randomly selected triple therapy combinations of three treatment classes that had previously been found to each significantly increase to variable degrees these complex I mutant worms’ lifespan (Supplementary Material, Figs. S2 and S3). All drug-treated lifespan analyses were performed relative to buffer-only treated gas-1(fc21) and N2 (WT) worms. (A-B) Lifespan results are shown for the 2 of 11 tested combinatorial therapy regimens that synergistically rescued gas-1(fc21) lifespan, even beyond that of WT worms. Key indicates number of worms studied across two biological replicate experiments per condition (n). Statistical analysis was performed by log-rank (Mantel-Cox) test comparing N2 (buffer only) or gas-1(fc21) worms treated with the indicated combination relative to buffer-only treated gas-1(fc21) worms. Subsequent analyses indicated that apparent efficacy of combination #6 (resveratrol + folinic acid + cysteamine bitartrate) was driven by resveratrol effect, without further synergy from additive therapies tested. (C) Glu + NA + NAC in triple or pairwise combinations showed synergistic rescue effect of gas-1(fc21) worms’ median lifespan relative to each individual component(s). Results convey median lifespan normalized to buffer-only exposed gas-1(fc21) worms. Detailed results are shown in Supplementary Material, Fig. S4.
To determine if the observed lifespan extension resulted from true triple compound synergy rather than the lead action of a dominant component in each treatment combination, we performed manual lifespan analysis of treatment from early larval (L1) stage with all possible single, double and triple permutations of each of the two lead combinatorial regimens (Fig. 2C and Supplementary Material, Fig. S4). Results of these assays demonstrated that triple compound synergy occurred only with Glu + NA + NAC cocktail combination therapy (Fig. 2). By contrast, resveratrol alone drove the lifespan extension effect apparent with the Resv+FA + Cyst cocktail, without further lifespan benefit of any double or triple combinations (Supplementary Material, Fig. S4). However, two-way combinations of either Glu + NAC or Glu + NA both synergistically improved lifespan relative to their individual components in gas-1(fc21) worms (P < 0.0001), and Glu + NAC treated gas-1(fc21) worms also lived significantly longer than wild-type (N2 Bristol) worms (P < 0.002) (Supplementary Material, Fig. S4). Overall, gas-1(fc21) worms treated with the Glu + NA pairwise combination showed lifespan extension similar to that observed with the full triple combination regimen, and Glu + NAC treated gas-1(fc21) worms showed the longest median survival that surpassed even the triple combination regimen.
Glu + NA + NAC combinatorial treatments differentially modulated the abnormal mitochondrial physiology of complex I-deficient NDUFS2−/− mutant gas-1(fc21) worms
As mitochondrial complex I is the largest and rate-limiting RC enzyme complex, complex I deficiency is well recognized to disrupt broader mitochondrial physiology. Previous studies have shown that gas-1(fc21) worms have significantly reduced mitochondrial membrane potential and mitochondrial mass, as well as increased mitochondrial oxidant burden, which all may be improved to variable degrees by supplementation with pharmacological agents (12,21,23,24). In our previously published studies, NA (1 mM) significantly reduced the increased mitochondrial oxidant burden, NAC (2.5 mM) significantly rescued the decreased mitochondrial membrane potential, and glucose (10 mM) increased the mitochondrial membrane potential and mitochondrial mass in gas-1(fc21) worms (13,14,21). Here, we sought to determine if these disrupted aspects of mitochondrial physiology in gas-1(fc21) worms were synergistically rescued by Glu + NA + NAC combinatorial treatment, when given at the same concentrations we had found to synergistically rescue the primary outcome measure of animal lifespan. Interestingly, Glu + NA + NAC combined treatment from the early larval (L1) stage through 24 h of adult life significantly improved gas-1(fc21) membrane potential (21.5% increased mean TMRE fluorescence relative to buffer-only gas-1(fc21), P < 0.001), but neither rescued nor exacerbated their reduced mitochondrial mass or increased mitochondrial matrix oxidant burden (Fig. 3A). Upon mitochondrial physiology evaluation of the pairwise treatment combination effects, only Glu + NAC significantly improved membrane potential (30% increased mean TMRE fluorescence relative to buffer-only gas-1(fc21)), P < 0.001) with synergistic effect seen relative to individual treatments with either glucose or NAC alone (Fig. 3A). Interestingly, mitochondrial matrix oxidant burden showed further significant increase with Glu + NAC treatment (16.5% increased mean MitoSOX fluorescence relative to buffer-only gas-1(fc21), P < 0.001), by a similar magnitude to that observed with glucose treatment alone (19.7% increased mean MitoSOX fluorescence relative to buffer-only gas-1(fc21), P < 0.001). Glu + NA significantly improved the complex I disease worms’ low mitochondrial mass (25% increased MitoTracker Green mean fluorescence relative to buffer-only gas-1(fc21), P < 0.001). No significant changes in any mitochondrial parameter were observed with NA + NAC treatment. Taken together, Glu + NA + NAC triple combination therapy significantly improved mitochondrial membrane potential that is a readout of integrated RC capacity and mitochondrial health, without exacerbating mitochondrial oxidant stress (as occurred with Glu given alone or in combination with NAC) or modulating mitochondrial mass (as occurred with Glu given alone or in combination with NA).
Figure 3.
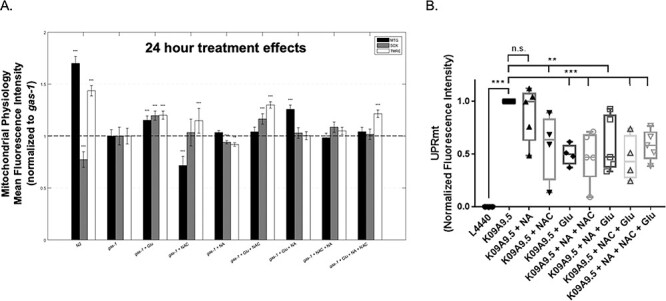
Glu + NA + NAC combinatorial treatment effects on C. elegans gas-1(fc21) worms’ mitochondrial physiology. (A) In vivo fluorescence analyses of mitochondrial content, mitochondrial oxidant burden and mitochondrial membrane potential were performed by terminal pharyngeal bulb (PB) relative fluorescence microscopy quantitation of MitoTracker Green FM (MTG), MitoSOX (SOX) and TMRE, respectively. gas-1(fc21) mutant synchronized young adult worms were treated for 24 h with Glu + NA + NAC combination therapy, or its individual and pairwise components, were compared with buffer-only treated gas-1(fc21) and N2 (WT) worms, and normalized to buffer-only gas-1(fc21) worms. Significant differences in mean fluorescence intensity between strains under different experimental conditions were assessed by mixed-effect ANOVA to account for potential batch effect due to samples being experimentally prepared, processed and analyzed on different days by including a batch random effect in the model. Statistical significance threshold was set at P < 0.05, and statistical analyses were performed in SAS 9.3. P-value conveys the significance of the difference between untreated N2 and untreated gas-1(fc21) (strain effect) or the difference between gas-1(fc21) plus drug(s) and untreated gas-1(fc21) (treatment effect). For each parameter, each drug treatment assay was repeated in 3 to 10 independent trials, with n = 50 worms per trial. Bars and error bars convey mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus concurrent gas-1(fc21) buffer control. (B) Feeding RNA interference (RNAi) knockdown of gas-1 (K09A9.5) in an hsp-6p::gfp labeled wild-type reporter worm strain resulted in significantly elevated UPRmt in first day young adult stage worms relative to empty vector (L4440) RNAi control, which was significantly reduced by treatment from early development (L1 stage) with glucose (Glu) or N-acetylcysteine (NAC) and any pairwise or triple combinatorial regimens in which they were present. Nicotinic acid (NA) alone did not reduce UPRmt in this model. hsp-6p::gfp C. elegans was used as a positive control. All tests were carried out using three biological replicate independent trials, with approximately 300 worms per condition in each replicate. Boxes depict 10th and 90th percentile for the normalized fluorescence intensity. Whiskers depict minimum and maximum values for each condition. Statistical analysis was performed by unpaired t-test (**, P < 0.01; ***, P < 0.001) for each comparison, as indicated.
Glu + NA + NAC combinatorial treatments reduced UPRmt induction in NDUFS2 (gas-1) RNAi knockdown C. elegans
The C. elegans mitochondrial unfolded protein response (UPRmt) pathway is similar to that of mammals, with induction of nuclear-encoded mitochondrial chaperones in response to either misfolded proteins within mitochondria or to stoichiometric imbalance of mitochondrial respiratory complexes (25–29). Specifically, UPRmt induction in C. elegans leads to transcriptional upregulation of mitochondrial chaperone proteins hsp-6 and hsp-60, as has been shown by GFP reporter studies following knockdown of electron transport chain components or when mitochondrial stress is present by the L3/L4 larval stage transition (25). To quantify UPRmt response to NDUFS2-based complex I inhibition in worms, we used feeding RNA interference (RNAi) to knockdown its C. elegans orthologue, K09A9.5 (gas-1), in the hsp-6p::gfp reporter strain. A strong UPRmt response was induced in this model, as quantified at the whole animal level by live animal flow cytometry (Union Biometrica BioSorter®) of GFP fluorescence intensity. Glu + NA + NAC combination therapy individual components, except NA, significantly reduced the mean UPRmt induction when given from the L1 larval stage individually by 40–50% (Fig. 3B, Glu P < 0.001 and NAC P < 0.01). Synergistic reduction of UPRmt induction by approximately 60% was observed with all three pairwise combinations. Interestingly, the Glu + NAC combinations that showed the largest reduction in UPRmt also yielded the greatest lifespan extension effect in gas-1(fc21) worms (Fig. 3B). The Glu + NA + NAC triple combination also showed significantly reduced UPRmt induction relative to buffer-only K09A9.5 RNAi controls, but without further synergy as compared to the pairwise combinations. Collectively, these data showed that Glu + NA + NAC combinatorial therapy reduced mitochondrial stress at the integrated level of the UPRmt as quantified in complex I disease living worms.
Glu + NA + NAC combination therapies significantly normalized global gene and pathway expression profiles in complex-I-deficient NDUFS2−/− mutant gas-1(fc21) worms
Extensive studies previously reported by our laboratory and others have clearly demonstrated that global transcriptome adaptations consistently occur in gas-1(fc21) relative to wild-type (N2 Bristol) adult worms (16,20,30). To evaluate whether Glu + NA + NAC therapy would normalize this transcriptional response, RNAseq-based pathway expression fold changes (increased and decreased) were calculated between gas-1(fc21) and wild-type synchronized adult worm populations in buffer-only basal conditions, with correlation coefficients used to assess whether single or combinatorial drug treatments normalized expression alterations of gas-1(fc21) mutants either completely or partially back toward that of wild-type worms (Fig. 4). Indeed, many transcriptionally dysregulated pathways in gas-1(fc21) reflect a coordinated response to reverse cellular energy deficiency that occurs from CI dysfunction, including basic intermediary metabolism pathways involved in oxidative phosphorylation, glycolysis, fatty acid metabolism, alpha ketoglutarate metabolism, alanine, aspartate and glutamate metabolism, and pentose and glucuronate interconversions. While the individual component therapies each showed some restoration of globally disrupted pathway expression profile of gas-1(fc21) worms (particularly NA and Glu), all pairwise combined treatments of NAC + NA, NAC + Glu or NA + Glu significantly normalized expression of both downregulated and upregulated biochemical pathways to a greater degree than did any of the individual treatments alone (Fig. 4). Similarly, gene-level expression heat map analysis showed that decreased expression of 198 genes in untreated gas-1(fc21) compared to N2 (WT) worms were normalized to a greater extent by pairwise or triple combination therapy than any individual treatment, with most significant rescue by either NAC + NA and NAC + Glu treatments (Supplementary Material, Fig. S5A). Pairwise and triple drug combination drug treatments also significantly normalized multiple aspects of the integrated nutrient sensing and signaling network (NSSN) (16), including those related to WNT, TGF-beta, mTOR, Notch, FOXO and MAPK pathway signaling, as well as the key cellular processes (translation and protein processing by endoplasmic reticulum, autophagy and endocytosis) and biochemical pathways (oxidative phosphorylation, glycolysis/gluconeogenesis, fatty acid metabolism, multiple aspects of amino acid metabolism, calcium signaling and cell defenses (P450 and glutathione metabolism) that these signaling pathways regulate (Fig. 4 and Supplementary Material, Fig. S5B). No difference was seen in the pentose phosphate pathway (Supplementary Material, Fig. S5B) that has been recently suggested to be one mechanism by which complex I-deficient cells in culture protect against oxidative stress and inflammation (31). Overall, Glu + NA + NAC short-term (24 h) combinatorial therapy significantly reversed the expression of global metabolic pathways that are dysregulated in gas-1(fc21) adult worms, including central NSSN and mito-regulatory pathways, highlighting their central contributions to the pathogenesis of mitochondrial complex I disease and their role as therapeutic targets.
Figure 4.
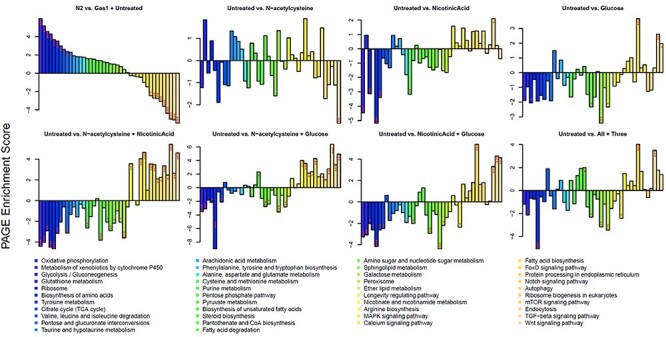
Overall transcriptome dysregulation at the level of KEGG biochemical pathways was normalized by Glu + NA + NAC pairwise combinatorial therapies in C. elegans gas-1(fc21) worms. Parametric analyses of gene set enrichment (PAGE) scores are shown for selected KEGG pathways that showed significant dysregulation in gas-1(fc21) worms compared to N2 (WT) at baseline, and/or showed significant modulation in gas-1(fc21) worms when treated with Glu + NA + NAC triple combination therapy, or its individual or pairwise components, for 24 h at the young adult stage. For each comparison, the increased or decreased expression is shown for the second group named in the title relative to the first group. The greatest overall normalization of dysregulated pathway-level expression in gas-1(fc21) worms occurred with the 3 pairwise drug comparisons studied, * P < 0.05, **, P < 0.01, and ***, P < 0.001 for each two-way comparison, as shown. Additional transcriptome results are shown in Supplementary Material, Fig. S5.
Glu + NA + NAC combinatorial treatment improved metabolic profiles of complex I-deficient NDUFS2−/− mutant gas-1(fc21) worms
Targeted profiling by HPLC analysis of intermediary metabolites in gas-1(fc21) synchronized adult worms showed their characteristic pattern of steady-state amino acid alterations, similarly as we previously reported (20,22). This pattern included trends toward elevated branched-chain amino acids (leucine, isoleucine, valine), alanine (the most predominant amino acid in C. elegans), glycine, threonine, lysine and ornithine levels, along with modest reduction in glutamate (Fig. 5 and Supplementary Material, Fig. S6). Glu or NAC alone did not significantly alter this amino acid profile, potentially due to the limited sample size of biological replicates with variable analyte levels between replicates, similarly as we previously observed in gas-1(fc21) (13). However, NA individual treatment significantly reduced levels of glycine and aspartate (P < 0.05) relative to buffer-only gas-1(fc21) (P < 0.05) (Supplementary Material, Fig. S6). Significant but modest increases in gas-1(fc21) were seen upon treatment with NAC+NA in glutamine, serine and citrulline levels (P < 0.05) and upon treatment with NAC + Glu in glutamate levels (P < 0.05) (Supplementary Material, Fig. S6). While no amino acid steady state levels were significantly altered with triple combination Glu + NA + NAC therapy, a trend was seen toward normalized concentration of several amino acids including alanine, citrulline, ornithine and glycine.
Figure 5.
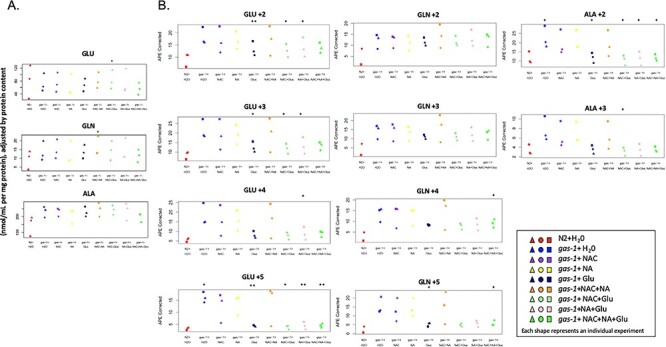
Combinatorial Glu + NA + NAC therapy partially restored altered intermediary metabolism of gas-1(fc21) mutant C. elegans. Whole worm concentrations and stable isotopic enrichment in free amino and organic acids of gas-1(fc21) worms were evaluated in synchronous young adult populations treated for 24 h with Glu + NA + NAC (glucose, nicotinic acid, and n-acetylcysteine) triple combination therapy or its single components and their pairwise combinations. (A) Whole worm free amino acid concentrations were measured by HPLC and normalized to overall protein concentration (nmol/mL per mg protein) for glutamic acid (GLU), glutamine (GLN) and alanine (ALA). Symbol color denotes experimental condition as defined in key, with separate symbols shown to indicate results of three independent replicate experiments performed. Random effects ANOVA statistical analysis was used to account for experimental batch effects. * P < 0.05 when compared to buffer (water) only exposed gas-1(fc21) worms. Results for additional HPLC amino acid levels are shown in Supplementary Material, Fig. S6. (B) Absolute isotopic enrichment of each amino acid metabolite in all molecular species for glutamate (GLU +2, +3, +4 and + 5), glutamine (GLN +2, +3, +4 and + 5), and alanine (ALA+2 and + 3) (indicating the number of carbon atoms on which 13C enrichment was present from U-13C6-glucose fed to the worms) was quantified by GC/MS in synchronous adult worm populations following U-13C6-glucose feeding to N2 (WT) or gas-1(fc21) worms for 24 h on the first day of adulthood. gas-1(fc21) worms were maintained in buffer (water)-only conditions or with Glu + NA + NAC as triple, pairwise, or single component therapies for 24 h, and compared to untreated N2 (WT) worms. Three biological replicate experiments were performed * P < 0.05 and ** P < 0.01 when compared to buffer (water) only exposed gas-1(fc21) worms. Results for additional GC/MS analytes are shown in Supplementary Material, Figs. S7–S8.
Metabolic flux through glycolysis, pyruvate metabolism and the tricarboxylic acid cycle (TCA) is altered in gas-1(fc21) worms, as seen by GC–MS analysis of isotopic enrichment of amino and organic acids from 13C6-glucose universally labeled adult worms (22). Here, we observed gas-1(fc21) worms showed a consistent trend, which reached statistical significance in some molecular species, for increased isotopic enrichment in glutamate and glutamine (indicating impaired flux from labeled glucose to alpha ketoglutarate in the TCA) and alanine (a metabolite in equilibrium with pyruvate, which indicates impaired pyruvate dehydrogenase flux) (Fig. 5A). Similar trends were seen toward increase in the +1, +2 and + 3 molecular species of the amino acids aspartate (a metabolite in equilibrium with malate, a TCA metabolite), serine (indicative of altered one carbon metabolism) and glycine (Supplementary Material, Fig. S7). Isotopic enrichment was also increased in organic acids that serve as TCA cycle metabolites (citrate, succinate, malate) and lactate (often increased in complex I RC disease as a result of increased NADH:NAD+ ratio that drives the lactate dehydrogenase (LDH) equilibrium reaction from pyruvate to lactate) (Supplementary Material, Fig. S8).
Glu + NA + NAC therapy components in multiple instances partially normalized the intermediary metabolic flux alterations that occur in gas-1(fc21) toward that of wild-type (N2) worms. While NA treatment alone did not normalize isotopic enrichment of any molecular species, NAC significantly reduced enrichment in +1 lactate species, as well as +4 malate species (Supplementary Material, Fig. S8). Glu significantly reduced enrichment in +1 succinate, with a trend toward decrease in the other molecular species of succinate (Supplementary Material, Fig. S8) as well as all molecular species of serine and + 1 glycine (Supplementary Material, Fig. S7). Glu treatment alone, and when used in pairwise combination with NA or NAC, also significantly normalized enrichment for nearly all molecular species of glutamate and the +2 (and in some cases +3) species of alanine (Fig. 5B). Finally, Glu + NA + NAC triple combination therapy did not exacerbate any metabolic flux abnormalities of gas-1(fc21) worms, but instead significantly decreased enrichment toward that of wild-type worms (P < 0.05) in +2 alanine, +5 glutamate, +4 and + 5 glutamine (Fig. 5B), as well as +2 serine (Supplementary Material, Fig. S7). Thus, the components of Glu + NA + NAC therapy alone (particularly glucose) and when given in pairwise or triple combination improved intermediary metabolic pathway flux through glycolysis, pyruvate metabolism and the TCA cycle in the complex I disease mutant worms.
Vertebrate animal validation of Glu + NA + NAC combinatorial treatment beneficial effects in D. rerio zebrafish rotenone model of mitochondrial complex I inhibition
We have previously demonstrated that mitochondrial complex I dysfunction can be modeled in zebrafish larvae by exposure to the potent complex I pharmacologic inhibitor, rotenone, that induces a ‘gray brain’ phenotype indicative of brain death along with abnormal neuromuscular responses, reduced swimming activity and, ultimately, animal death (32). We previously found that individual NAC therapy partially prevents brain death caused by rotenone exposure (14). Here, we evaluated whether Glu + NA + NAC combinational pre-treatment would synergistically improve prevention of the gray brain phenotype caused by pharmacologic (rotenone) inhibition of complex I in wild-type (AB) zebrafish larvae (Fig. 6A panels). Indeed, Glu + NA + NAC pre-treatment of AB larvae from 5 days post-fertilization (dpf) yielded significantly protection from developing the gray brain phenotype when larvae were subsequently exposed to 150 nM rotenone in ethanol buffer for 5 h on 7 dpf, as compared to 0.075% ethanol buffer-only control (86% reduction in gray brain phenotype, P < 0.0001) (Fig. 6A).
Figure 6.
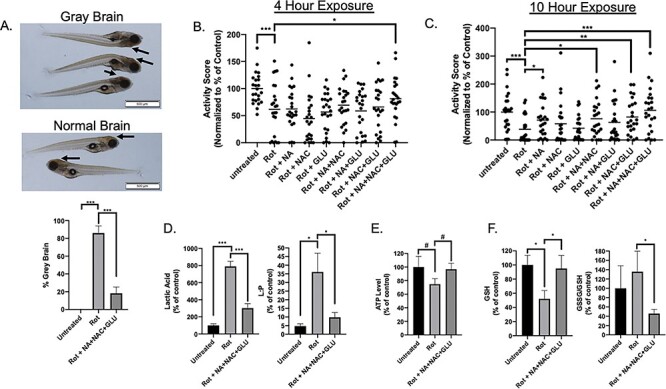
Combination Glu + NA + NAC treatment protected zebrafish larvae from rotenone-induced brain death, reduced swimming activity, and key biochemical hallmarks of mitochondrial complex I dysfunction. (A) Brain death (gray brain, indicated by arrows) was induced with 150 nM rotenone exposure in wild-type (AB) fish for approximately 5 h on 7 days post-fertilization (dpf). Pre-treating wild-type (AB) zebrafish beginning at 5 dpf with combinational GLU + NA + NAC therapy significantly prevented the brain death (golden color of brain, indicated by arrows) upon co-exposure to 150 nM rotenone at 7 dpf and combinational treatment for 5 h (P < 0.001). Three independent biological replicates were performed per condition, with n ≥ 15 animals in each condition per replicate. (B) Zebrafish larval swimming activity was quantified by exposure for 10 h to repetitive light cycles of 60% light for 20 min followed by 0% light for 20 min during rotenone co-exposure at 7 dpf. Independent experimental data was collected by ZebraBox (Viewpoint Life Sciences) analysis, with activity scores in the first 5 min of each dark period averaged across three independent biological replicate experiments with data normalized as percent of wild-type buffer-only control for each independent biological replicate (see Supplementary Material, Fig. S9). Effects of pre-treatment with Glu + NA + NAC triple combination therapy, along with each of the single and double permutations were evaluated, with activity score shown after rotenone (150 nM) co-exposure for 4 h, to assess biochemical mechanism before the 5-h peak when maximal brain death was observed. Data are represented in a scatter plot, with n = 8 animals per condition studied in each replicate. (C) Zebrafish larval swimming activity was quantified following pre-treatment with Glu + NA + NAC triple combination therapy, along with each of the single and double permutations, with activity score shown after 10 h co-exposure to rotenone (150 nM). n = 8 zebrafish larvae per condition in each replicate. (D) Zebrafish lactate and lactate:pyruvate ratios. Lactate levels were quantified in wild-type (AB) zebrafish (20 fish per replicate for each condition) in buffer or after rotenone exposure for 4 h either with or without Glu + NA + NAC pre-treatment from 5 dpf. Significant differences were seen between all treatment group (***, P < 0.001). Specifically, lactic acid mean levels were 279 pmol/larvae in buffer-only control larvae (n = 6 biological replicates), 2202 pmol/larvae in 150 nM rotenone ×4 h exposed larvae (n = 4 biological replicates), and 841 pmol/larvae in Glu + NA + NAC pre-treated rotenone ×4 h exposed larvae (n = 6 biological replicates). Pyruvate levels were not significantly changed between groups (Supplementary Material, Fig. S9). However, lactate-to-pyruvate (L:P) ratio was significantly increased in rotenone-exposed larvae and partially normalized with Glu + NA + NAC pre-treatment (*, P < 0.05). (E) ATP levels. Wild-type (AB) zebrafish (20 fish per replicate for each condition) pre-treated from 5 dpf with GLU + NA + NAC combinational therapy (n = 6 biological replicates) showed a trend toward improved ATP concentration upon evaluation after 4h rotenone exposure at 7 dpf as compared to rotenone (150 nM) exposed larvae for 4 h at 7 dpf without pre-treatment (n = 4 biological replicates). Specifically, ATP mean concentration in untreated AB zebrafish was 346 pmol per larvae, in rotenone-exposed AB zebrafish was 259 pmol per larvae, and in Glu + NA + NAC pre-treated zebrafish before rotenone exposure was 335 pmol per larvae. #, P ≤ 0.1. (F) Glutathione levels. Wild-type (AB) zebrafish (20 fish per replicate for each condition) exposed to 150 nM rotenone for 4 h (n = 7 biological replicates) had lower GSH mean concentration (85 pmol per larvae) than buffer-only control larvae (161 pmol per larvae (n = 6 biological replicates). Pre-treatment from 5 dpf with GLU + NA + NAC combinational therapy (n = 7 biological replicates) normalized GSH level (153 pmol per larvae), similar to that seen in buffer-control wild-type larvae. Glu + NA + NAC pre-treatment also significantly lowered the GSSG/GSH ratio relative to rotenone-only exposure without pre-treatment. Statistical significance between experimental conditions was assessed by Student’s t-test in Graphpad Prism 7.04, #, P ≤ 0.1, *, P < 0.05, **, P < 0.01, and ***, P < 0.001. Bars indicate mean and standard error of mean.
Glu + NA + NAC triple combination pre-treatment from 5 dpf also significantly rescued larval zebrafish neuromuscular function when subsequently co-exposed to acute complex I inhibition with rotenone and combinational therapy, as assessed at 7 dpf by quantifying their swimming activity in repetitive dark–light cycles (Fig. 6B and C). Specifically, Glu + NA + NAC combinational treatment significantly rescued the maximum startle response of the zebrafish that occurs during the first 5 min of each dark cycle (Supplementary Material, Fig. S9A) after being co-exposed to rotenone for 4 h (Fig. 6B, P < 0.05) or 10 h (Fig. 6C, P <0.001). Indeed, neuromuscular activity was better preserved by pre-treatment with Glu + NA + NAC combination therapy as compared to any of its individual or pairwise component therapies. Upon statistical analysis using rigorous multiple t tests with false discovery rate (FDR) correction for multiple testing, statistically significant differences were seen only with Glu + NA + NAC triple combination therapy pre-treatment relative to rotenone-only exposure (**, q-value = 0.0012) and rotenone exposure relative to untreated buffer-only control (***, q-value = 0.00093).
Given improved brain morphological appearance and swimming activity upon rotenone-induced acute complex I inhibition in the zebrafish larvae pre-treated with Glu + NA + NAC, we sought to evaluate whether this therapy normalized biochemical hallmarks of mitochondrial respiratory chain disruption, which typically include alterations in lactate, pyruvate (33), lactate to pyruvate (L:P) ratio (34), reduced glutathione (GSH) (35), oxidized glutathione (GSSG) or GSSG to GSH ratio (36) and adenosine triphosphate (ATP) levels (37). We found that an identical rotenone regimen to that which induced severe neurologic dysfunction in zebrafish larvae also significantly increased their lactate levels by more than 7.5-fold compared to untreated AB zebrafish (Fig. 6D). Impressively, Glu + NA + NAC combinational therapy pre-treatment prior to rotenone exposure led to dramatic rescue of lactate induction by more than 50 % (P < 0.001, with only a 3-fold increase in lactic acid level seen in Glu + NA + NAC pre-treated larvae upon rotenone exposure relative to buffer-only treated AB zebrafish (P < 0.001) (Fig. 6D). Similarly as expected, given pyruvate levels are typically unchanged in RC complex I disease (33), pyruvate levels in zebrafish larvae did not increase with rotenone exposure (Supplementary Material, Fig. S9B). The L:P ratio is classically considered to be among the most reliable ways to differentiate mitochondrial disease where it is elevated above 20, from pyruvate metabolism disorders where it is classically below 20 (34,38). Indeed, the L:P ratio was significantly increased upon rotenone exposure at 35 (P < 0.05), but significantly reduced (10, P < 0.05 relative to rotenone-only exposed worms) when larvae were pre-treated with Glu + NA + NAC therapy. The L:P ratio reflects cytosolic NADH:NAD+ balance, as pyruvate plus NADH are enzymatically converted to lactate via the lactate dehydrogenase equilibrium reaction. Thus, the increased L:P ratio seen upon acute complex I inhibition in the zebrafish larvae directly reflects the increased NADH:NAD+ ratio that is driven by impaired complex I (a.k.a., NADH dehydrogenase) activity (39). NADH and NAD+ levels are challenging to measure directly in whole animal samples, given their subcellular compartmentalization into nuclear, cytosolic and mitochondrial pools. Interestingly, HPLC analysis of whole animal NAD+ levels did show a significantly decrease by approximately 40% upon rotenone exposure (P < 0.05) relative to buffer control (Supplementary Material, Fig. S9C), consistent with the known NAD+ deficiency that occurs in cells with complex I disease (16). However, no significant differences were detected in whole animal NADH levels or the NADH/NAD+ ratio upon rotenone exposure (Supplementary Material, Fig. S9C and S9E), which we suspect relates to the loss of discrimination of the subcellular pools by the whole animal HPLC analysis approach.
Wild-type (AB) zebrafish larvae showed a trend toward a 25% reduction in whole fish ATP level when exposed at 7 dpf to rotenone (with dead animals excluded prior to selection for biochemical analysis), which appeared to largely be prevented when larvae were pre-treated from 5 dpf with Glu + NA + NAC combinatorial therapy (Fig. 6E). While Glu + NA + NAC therapy is not directed or anticipated to restore complex I activity, we postulate this finding relates to improved cytosolic redox balance and provision of nutrients (glucose) to increase anaerobic ATP production via cytosolic glycolysis.
Oxidative stress is widely recognized to be increased in mitochondrial RC complex I disease, due in part to increased oxidant production with insufficient oxidant scavenging capacity (14,40). Here, glutathione redox components and ratio were measured to assess whether this central endogenous regulator of oxidative stress was altered by rotenone and improved with Glu + NA + NAC therapy. Indeed, acute rotenone exposure significantly decreased levels of reduced glutathione (GSH, active form) by 48% (P < 0.05) as compared to untreated wild-type (AB) zebrafish (Fig. 6F). Glu + NA + NAC combinational therapy pre-treatment prior to rotenone exposure fully normalized GSH levels (P < 0.05) back to that seen in wild-type controls (Fig. 6F). The predominant form of glutathione in cells is predominantly GSH (~95%), with much lower concentrations of its oxidized form, GSSG. While no significant different was apparent in whole animal GSSG concentration (Supplementary Material, Fig. S9F), the ratio of GSH:GSSH that is broadly recognized as an important indicator of cellular health (36) trended toward increase in the rotenone-treated animals and was significantly reduced with Glu + NA + NAC therapy (Fig. 6F).
Overall, these data provide clear validation of Glu + NA + NAC triple combination treatment in an evolutionarily distinct animal model of complex I deficiency. Indeed, studies of Glu + NA + NAC therapy in the rotenone-exposed wild-type AB zebrafish larvae animal model demonstrate it acts synergistically to provide in vivo resiliency upon acute RC complex I inhibition. Specific beneficial effects of this combinatorial therapy included synergistic prevention of brain death and impaired swimming capacity, and improved biochemical hallmarks of mitochondrial complex I dysfunction, including reduced lactate and L:P ratio, preserved ATP levels and improved GSH concentration and GSSG/GSH ratio upon acute complex I (rotenone) inhibition.
DISCUSSION
We utilized two evolutionarily distinct species, namely C. elegans (worms, invertebrates) and D. rerio (zebrafish, vertebrates), as a translational research platform in which to study well-established animal models of mitochondrial RC complex I dysfunction, with a goal to objectively evaluate the therapeutic potential, safety and synergy of rationally designed combinatorial therapy regimens or colloquially named ‘mitochondrial cocktails’. Treatment regimens were selected from individual drugs across three general treatment groups that we had previously shown to rescue lifespan and other aspects of mitochondrial pathophysiology in complex I disease worms. While our previous work showed that antioxidants, metabolic modifiers and signaling modifiers (13–15,21) variably improved the short lifespan of complex I disease autosomal recessive NDUFS2−/− genetic mutant gas-1(fc21) worms, 10 of 11 triplet drug combinations tested from these groups failed to synergistically improve these animals’ lifespan relative to their individual components. Interestingly, while no lifespan extension was observed for 10 of these random combinations, neither were they found to be toxic at the level of overall animal survival; this is similar to the outcome commonly reported by human mitochondrial disease patients and clinicians when they empirically create unique ‘cocktails’ of drugs, vitamins or supplements in similar treatment classes. Overall, these data suggest that empirically combining therapies is not universally beneficial, and may in some cases modify physiology sufficiently to negate beneficial (i.e. lifespan-extending) effects otherwise achieved with individual components.
Only one combinatorial treatment regimen, Glu + NA + NAC, consistently yielded a synergistic lifespan benefit, beyond that of any individual component, in complex I disease gas-1(fc21) mutant worms. Indeed, detailed lifespan analysis of all individual, two-way and triplet permutations of this treatment regimen revealed some degree of synergy with all multi-drug combinations. Interestingly, combining glucose with either of the other therapies (e.g. Glu + NA and Glu + NAC) maximally extended gas-1(fc21) lifespan even beyond that of healthy wild-type (N2) worms.
Mechanistic dissection of the Glu + NA + NAC combination revealed its components partially rescued different aspects of the disrupted mitochondrial physiology that occur in complex I disease worms. Glu + NAC synergistically improved mitochondrial membrane potential that is studied as a means to indicate integrated mitochondrial respiratory chain capacity, but also significantly increased mitochondrial matrix oxidant burden to a similar degree as occurred with glucose alone. Similarly, we have previously reported that another single agent, resveratrol (sirtuin agonist), significantly improved gas-1(fc21) lifespan despite causing a substantial increase in mitochondrial matrix oxidant burden (13). These findings once again raise the question as to whether reduction of mitochondrial oxidant burden is a relevant therapeutic goal and required to improve overall health in primary mitochondrial disease (14). Interestingly, only Glu + NA significantly improved the reduced mitochondrial mass that occurs in gas-1(f21) worms, as plausibly may relate to an NAD+-dependent effect on mitochondrial biogenesis. Collectively, Glu + NA + NAC combination therapy after 24 h yielded maximal synergistic effect to improve gas-1(fc21) mitochondrial membrane potential, without changing their overall mitochondrial mass or oxidant burden. Significant rescue of mitochondrial proteostatic stress, as quantified at the level of UPRmt induction following feeding RNAi knockdown of gas-1 (K09A9.5) in the hsp-6p::gfp reporter line, occurred with all Glu + NA + NAC combinations that contained both Glu and/or NAC. These data are consistent with the observed improvement with this regimen in mitochondrial membrane potential, as reduced membrane potential is a known cause of UPRmt induction in C. elegans. Overall, these data suggest that while Glu + NA + NAC combination therapy may well negate some of its individual component's effects on mitochondrial physiology, given together this regimen improves overall animal survival with an associated improvement of integrated mitochondrial respiratory chain function.
Glu + NA + NAC combination therapy further improved downstream cellular alterations that are recognized to contribute to the broader clinical pathophysiology of primary mitochondrial RC disease. Consistent with previous studies by our research laboratory and others that have highlighted the therapeutic potential of targeting NSSN pathways and/or central signaling nodes (13,16), transcriptome profiling by RNAseq showed that treating gas-1(fc21) worms with Glu + NA + NAC combination or its component regimens that included glucose maximally normalized expression of dysregulated signaling pathways including mTOR, FOXO, MAPK, as well as WNT, TGF-beta and Notch, that modulate mitochondrial activities and animal fitness (8,41–46). Transcriptome profiling also revealed that Glu + NA + NAC combinations normalized globally dysregulated biochemical pathways, a finding we validated by direct quantitation of intermediary metabolism at the level of steady state analyte and stable isotopic flux analyses. Glucose-containing combinations, in particular, normalized metabolic flux through glycolysis, pyruvate metabolism and the TCA cycle, a finding consistent with normalized expression of the upregulated glycolysis pathway that was seen in complex I disease mutant worms on transcriptome profiling. This finding is further consistent with an increased reliance on anaerobic glycolysis to generate energy in cells with impaired aerobic mitochondrial energy capacity and highlights the key role of carbohydrate-based nutrition in primary mitochondrial disease to therapeutically exploit their increased glycolytic capacity. Indeed, glucose itself seems to be a particularly important driver of combinatorial treatment efficacy in the gas-1(fc21) worms, providing strong rationale for improving strategies to optimize nutrition in individuals with primary mitochondrial disease.
Dysfunction of mitochondrial complex I (NADH dehydrogenase) is well recognized to increase both NADH:NAD+ redox balance and oxidative stress, pathophysiologic targets that are improved with NA and NAC therapies, respectively (13,14,16). Thus, these central regulators of the global cellular pathophysiology that occur in mitochondrial respiratory chain disease highlight key therapeutic opportunities to meaningful improve overall health. Indeed, a recent study of niacin in a small cohort of mitochondrial myopathy patients with mitochondrial DNA deletions showed that niacin therapy improved blood and tissue NAD+ levels, as well as myopathy (47). Similarly, glutathione deficiency is commonly detected in blood from mitochondrial disease patients and may be directly improved with NAC treatment (9,40). Here, we showed that Glu + NA + NAC combination therapy in mitochondrial disease models had therapeutic benefit, as did some two-way combinations, on diverse phenotypes and biochemical biomarkers above any one of these therapies alone. We interpret these data to suggest that this combination (1) provides an alternative nutrition source that can be used to generate sufficient cellular energy by utilizing the mitochondrial disease cell’s increased glycolytic capacity to anaerobically generate ATP (here represented by glucose (Glu) at physiologic concentrations) (48); (2) normalizes disrupted NAD+ metabolism by providing nicotinic acid (NA) as an NAD+ agonist (47); and (3) reduces total cellular oxidative stress by providing N-acetylcysteine (NAC) to restore total cellular glutathione-based oxidant scavenging capacity (40). Figure 7 schematically depicts this overall therapeutic rationale for utilizing Glu + NA + NAC combination therapy to replete key metabolic products (ATP, oxidant:antioxidant balance and NAD+) that consistently become deficient in primary mitochondrial RC complex I disease.
Figure 7.
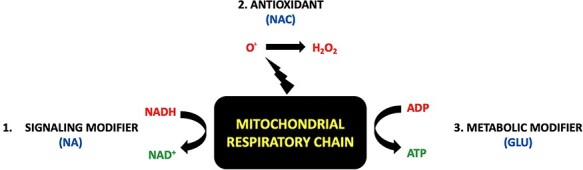
Schematic rationale supporting use of Glu+NA+NAC multi-drug combinatorial treatment regimen to replace major metabolite deficiencies in primary mitochondrial respiratory chain complex I disease. Understanding treatment approaches for the biochemical sequelae of respiratory chain (RC) disease may be simplified by reducing the RC to a ‘bioenergetic factory’ (black box). While the RC is the major site at which chemical energy is produced in the form of adenosine triphosphate (ATP), the RC also generates other several other essential ‘products’ needed to support normal cellular function. These include (A) nicotinamide adenine dinucleotide (NAD+) as generated in mitochondria by conversion of NADH-reducing equivalents generated through intermediary metabolism that are donated at CI (official enzyme name of CI is ‘NADH dehydrogenase’), and (B) oxidants, which are generated in mitochondria both with the mitochondrial matrix (via CI) and in the intermembrane space (via CIII). O., superoxide. H2O2, hydrogen peroxide. This simplified RC product view highlights major categories of therapies for RC dysfunction (red font), including (1) NAD+ supplementation (e.g. NA, nicotinic acid) (13), (2) antioxidants (e.g., NAC, to scavenge oxidants within mitochondria or throughout the cell) (14), and (3) optimizing production of ATP via anaerobic glycolysis (e.g. glucose or diet changes, to optimize cellular nutrition and therapeutically exploit upregulated glycolytic capacity). As shown by experimental modeling in C. elegans and zebrafish animal models of complex I disease, Glu + NA + NAC combinatorial therapies to correct all of these metabolite deficiencies that occur downstream of complex I dysfunction synergistically improves animal survival and health outcomes at diverse levels including mitochondrial physiology, intermediary metabolism, cellular signaling, stress resistance and overall animal activity.
Glu + NA + NAC synergistic efficacy at both organ and whole animal levels was clearly validated in a vertebrate animal, D. rerio, in which the potent pharmacologic inhibitor of RC complex I, rotenone, induces severe neurologic dysfunction and animal death. Remarkably, Glu + NA + NAC pre-treatment prior to induction of acute RC failure significantly mitigated the incidence of acute brain death, synergistically improved neuromuscular function as assessed by maximal swimming capacity, and restored lactic acid, L:P ratio, ATP levels, GSH concentration and GSSG/GSH ratio toward normal in rotenone-exposed zebrafish larvae. Thus, Glu + NA + NAC combination therapy objectively improves animal resiliency in the face of stressors that cause severe metabolic deficiency to prevent acute neurologic and biochemical decompensation. The value of improving resiliency in this setting has high potential relevance to the pressing need to prevent decompensation in the analogous occurrence of ‘metabolic strokes’ that are often induced by acute stressors, such as infection or fasting, in human mitochondrial disease patients. These animal model data further suggest that combinatorial Glu + NA + NAC treatment may directly improve health outcomes consistently prioritized by primary mitochondrial disease patients, including improvements of activity and exercise capacity (17). In summary, these translational data objectively demonstrate a synergistic benefit of combinatorial Glu + NA + NAC therapy on lifespan, healthspan and relevant biochemical biomarkers in two pre-clinical animal models of both chronic genetic-based as well as acute stressor-based mitochondrial RC complex I disease. Further evaluation of Glu + NA + NAC combination therapy is warranted in a randomized, double-blinded, controlled clinical treatment trial in human mitochondrial disease patients to evaluate its clinical utility to prevent acute neurologic decompensation, preserve activity and exercise capacity, and improve overall quality of life.
Materials and Methods
Lifespan analysis of combinatorial treatment effects in C. elegans
Animals were maintained at 20°C throughout the experiment. Synchronized nematode cultures were initiated by bleaching young adults to obtain eggs. Collected eggs were allowed to hatch overnight on 10 cm un-spread (i.e. without E. coli) nematode growth media (NGM) agar plates, after which L1-arrested larvae were transferred to 10 cm NGM plates spread with OP50 E. coli. Upon reaching the first day of egg laying, synchronous young adults were moved to fresh 3.5 cm NGM plates seeded with OP50 E. coli (lifespan experiment ‘Day 0’) and evaluated by classical microscopic lifespan methods. About, 40 to 60 nematodes were studied per strain, divided on two 3.5 cm NGM plates. Mortality was confirmed by manually stimulating nematodes lightly with a platinum wire; nematodes that did not move after stimulation were scored as dead and removed from the plate. Worms that died of protruding/bursting vulva, bagging or crawling off the agar were censored. For higher throughput lifespan screening to prevent offspring from nematodes under study from also reaching adulthood, fluorodeoxyuridine (FUdR) was added to OP50-spread NGM plates to a final concentration of 100 μg/ml (49). Median lifespan was determined for each strain relative to concurrently studied wild-type worms. All treatment effects were compared to buffer-only controls. Treatment compounds were obtained from Sigma, unless otherwise specified.
Relative quantitation of mitochondrial matrix superoxide burden, mitochondrial membrane potential and mitochondria content by fluorescence microscopy in young adult C. elegans
Mitochondrial oxidant burden (MitoSOX Red), membrane potential (tetramethylrhodamine ethyl ester, TMRE) and mitochondrial content (MitoTracker Green FM, MTG) were performed at 20°C using in vivo terminal pharyngeal bulb relative fluorescence microscopic quantitation, as previously described (15). Briefly, synchronous populations of Day 0 young adults were moved to 35 mm NGM plates spread with OP50 E. coli, a desired drug treatment 2.5 mM N-acetylcysteine, nicotinic acid, glucose and the double or triple combinations of these drugs or buffer control (S-basal/water for all other drugs) was performed on NGM plates. Simultaneously with the drug treatments, worms were treated with either 10 μM MitoSOX Red (matrix oxidant burden), 100 nM TMRE (mitochondrial membrane potential) or 2 μM MitoTracker Green FM (mitochondria content) for 24 h. The next day, worms were transferred with a pick onto 35 mm agar plates spread with OP50 E. coli without dye for 1 h to allow clearing of residual dye from the gut. Worms were then paralyzed in situ with 5 mg/ml levamisole. Photographs were taken in a darkened room at 160× magnification with a Cool Snap cf2 camera (Nikon, Melville, NY). A CY3 fluorescence cube set (MZFLIII, Leica, Bannockburn, IL) was used for MitoSOX and TMRE. A GFP2 filter set (Leica) was used for MitoTracker Green FM. Respective exposure times were 2 s, 320 ms and 300 ms for each of MitoSOX, TMRE and MitoTracker Green FM. The resulting images were background subtracted, and the nematode terminal pharyngeal bulb was manually circled to obtain mean intensity of the region by using Fiji Is Just ImageJ (50). Fluorescence data for each strain were normalized to its same day control to account for day-to-day variation. A minimum of three independent experiments of approximately 50 animals per replicate were studied per strain per dye. The significance of the difference in the mean fluorescence intensity between strains under different experimental conditions was assessed using mixed-effects ANOVA, which takes into account potential batch effect due to samples being experimentally prepared, processed and analyzed on different days by including a batch-specific random effect in the model. Post hoc two-sample comparisons were performed as necessary with false discovery rate (FDR) adjusted Q value provided to correct for multiple testing using Benjamini and Hochberg approach (51). A statistical significance threshold was set at P < 0.05. All statistical analyses were performed in SAS 9.3 (Cary, NC: SAS Institute Inc.).
Quantification of UPRmt induction using RNAi in C. elegans by large particle flow cytometry
K09A9.5 (gas-1) RNAi bacteria was inoculated into LBAmp media and seeded onto NGM plates with 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and 100 μg/mL ampicillin. Hsp-6p::GFP reporter worms were hypochlorite treated, and eggs were transferred to NGM plates seeded with RNAi bacteria. GFP reporter worms were grown to the young adult stage (after 3 days), were washed from the RNAi plates, and mean green fluorescence for each worm was obtained using the Union Biometrica BioSorter®. Fluorescence of each worm was normalized to its size, approximated by time of flight (TOF)*extinction outputs from the BioSorter. Fluorescence of all worms for a given replicate were averaged, and are represented relative to untreated empty vector L4440 control (0) and untreated K09A9.5 (1). All replicates are shown relative to their corresponding same-day controls. The significance between each condition was assessed by Student’s t-test in Graphpad Prism 7.04 (San Diego, CA: GraphPad Software Inc.).
Whole worm amino acid profiling and stable-isotopic intermediary metabolic flux analysis in C. elegans
Whole worm free-amino acid profiling and metabolic flux analysis were performed as previously described. Briefly, synchronous populations of 1000 to 1500 worms were grown to adulthood on NGM plates (22). About, 10 mM universally labeled 13C6-glucose and appropriate drug in desired concentration were added to plates before first day adult worms were transferred to fresh plates. Following 24 h of incubation with drug, adult worms were washed clear of bacteria five times with S. basal. Worm number was estimated by counting. Three biological triplicate experiments were performed per condition. Metabolic reactions were stopped by the addition of 4% perchloric acid (PCA) containing 20 nmol internal standard ( -aminocaproic acid, 16.7 μM). Samples were ground using a plastic homogenizer and motorized drill until visual inspection confirmed worm disruption. Precipitated protein was removed, re-dissolved in one normal NaOH and protein concentration was determined by DC Protein Assay (Bio-Rad). About, 50 μL of neutralized samples were separated for HPLC analysis in the CHOP Metabolomics Core Facility. From the remaining neutralized samples, amino acids and organic acids were extracted using ion exchange resin (Bio-Rad) in AG50 and AG1 columns, respectively, to measure relative enrichment in amino acids and organic acids by mass spectrometry, as previously described (22). MS analyses were performed in the CHOP Clinical Biochemical Laboratory. Stable isotopic enrichment was calculated for each species as previously described, according to the following formula:
-aminocaproic acid, 16.7 μM). Samples were ground using a plastic homogenizer and motorized drill until visual inspection confirmed worm disruption. Precipitated protein was removed, re-dissolved in one normal NaOH and protein concentration was determined by DC Protein Assay (Bio-Rad). About, 50 μL of neutralized samples were separated for HPLC analysis in the CHOP Metabolomics Core Facility. From the remaining neutralized samples, amino acids and organic acids were extracted using ion exchange resin (Bio-Rad) in AG50 and AG1 columns, respectively, to measure relative enrichment in amino acids and organic acids by mass spectrometry, as previously described (22). MS analyses were performed in the CHOP Clinical Biochemical Laboratory. Stable isotopic enrichment was calculated for each species as previously described, according to the following formula:
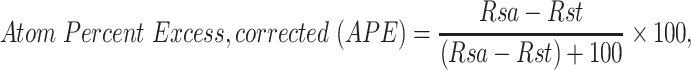 |
(1) |
where 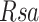 for ratio of the sample and
for ratio of the sample and 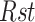 for ratio of the standard (13,52). Statistical comparison between groups was performed using mixed-effects ANOVA (JMP version 10, SAS Institute, Cary NC).
for ratio of the standard (13,52). Statistical comparison between groups was performed using mixed-effects ANOVA (JMP version 10, SAS Institute, Cary NC).
Transcriptome profiling of drug treatment effect by RNAseq analysis
Sample preparation for gene expression profiling by RNAseq technique was performed, as previously described (14,30). Briefly, wild-type (N2 Bristol) and mitochondrial RC complex-I-deficient gas-1(fc21) animals were maintained at 20°C by established protocol (15,30). Synchronous young adult populations of approximately 1000 to 2000 nematodes were obtained and treated on the first day of egg laying for 24 h on NGM plates spread with the desired drug or drug combination, per established protocol. After drug treatment, total RNA was isolated and prepared for transcriptome profiling, as previously described (14,15). Briefly, total RNA was isolated using the Trizol method and RNA concentration was measured using the NanoDrop-1000. RNA quality was determined by using Agilent Bioanalyzer in the Nucleic Acid and Protein Core Facility at The Children’s Hospital of Philadelphia (CHOP) Research Institute, where RIN number between 8 and 10 as required for further sample analysis. Library preparation was performed using the Illumina Truseq Stranded Total RNA Sample Preparation Kit (San Diego, CA), with indexing to enable eight samples run per lane. Samples were submitted to the BGI@CHOP Sequencing Core Facility at CHOP for next generation sequencing (RNAseq) analysis on Illumina HiSeq 2000 instruments. Samples were run in High Throughput Mode of 100 base pair paired end reads with eight samples per lane to generate an estimated 20 million reads per sample. Library quality was assessed by Bio A analysis to check concentration, library size and contamination, as we well as by gel analysis to assess degradation. Quantitative PCR was then performed to determine optimal sample concentrations using the Applied Biosystems Step One Plus Real Time PCR machine to enable proper sample pooling. Sequencing reads saved in FASTQ files were aligned to obtain gene-level expression data for bioinformatics analysis. Data quality issues, such as total throughput, confounding factors, and outlier samples, were fully evaluated to ensure validity of analysis results. Gene and KEGG Pathway-level analyses of combinatorial treatment effects in mutant relative to wild-type worms were performed, as previously described (14,15,30). PAGE was used for gene set enrichment analysis (53). A total of 8691 gene sets were tested. Enrichment scores were summarized and compared between the eight comparisons. All RNAseq data were submitted in the gene expression omnibus public database (GEO ID #GSE134535).
Zebrafish brain death and neuromuscular activity assessment in rotenone-induced RC complex I dysfunction
All protocols and methods were performed in accordance with CHOP IACUC number 18–001154 regulations for care and use of D. rerio at the Children’s Hospital of Philadelphia Research Institute. Embryos and larvae were maintained at 28°C. Adult zebrafish (AB strains) were set in pairwise in undivided mating tanks as described (15) in order to collect and sort embryos. For brain death experiments, embryos were placed in E3 with phenylthiourea (PTU) at 0.03 μg per liter to prevent larval pigment formation. For Zebrabox (ViewPoint Life Sciences) neuromuscular activity assessment, embryos were not exposed to PTU so that the system could observe the highest contrast of the zebrafish embryos. Unless otherwise specified, all reagents were obtained from Sigma-Aldrich (St Louis, MO, USA). AB strain zebrafish larvae were pre-treated with either 1 mM nicotinic acid (NA), 2.5 mM N-acetylcysteine (NAC), 10 mM glucose (Glu) or combinations of these treatments starting at 5 days post-fertilization (dpf). Rotenone stock solutions (1 mM) were prepared in ethanol by sonication. Drug stocks of NA (100 mM) pH 7.0, NAC (250 mM) pH 7.0 and glucose (1 M) were prepared in E3 with Tris (10 mM) pH 7.2 and 0.1% DMSO. Zebrafish were co-exposed to combinational treatments and 150 nM of rotenone on 7 dpf, and larvae were score for toxic effects. Brain death was scored by presence or absence of gray brain phenotype after approximately 5 h of co-exposure of rotenone, as previously described by (32). Neuromuscular assessment was scored after approximately 4 and 10 h of co-exposure with 150 nM rotenone. Larvae were transferred on 7 dpf to 96-well 650 μL square well plates, with one larva per well (catalogue number 7701–1651), with a final volume 200 μL. The combinational treatments were evaluated using the automated imaging system ZebraBox, data were collected using the ZebraLab software (ViewPoint Life Sciences). Larvae were acclimatized to 60% light for 20 min to achieve a stable level of activity (54). Larvae were then exposed to 20 min of dark (0% light) to produce a startle response, in which locomotion increases to a maximum movement for the first 5 min and then decreases gradually to a stable level. This light cycling of 20 min of on and off was repeated to observe the rescue of the compound. The maximum movement of the larvae for the first 5 min of the dark cycles was analyzed in Graphpad Prism 7.04. Sample comparisons were performed with a FDR adjusted Q value provided to correct for multiple testing using Benjamini and Hochberg approach.
Zebrafish larval treatment and sample preparation for biochemical analyses
Zebrafish embryos were exposed to drugs dissolved in E3 buffer with Tris (10 mM) pH 7.2 and 0.1% DMSO at 7 dpf for 4 h (with rotenone only or rotenone plus combination drug solutions containing 1 mM nicotinic acid (NA), 2.5 mM N-acetylcysteine (NAC) and/or 10 mM glucose (Glu). About, 20 embryos were collected per tube and washed two times with E3 buffer. After buffer removal, embryos were immediately frozen in liquid nitrogen, then stored at −80°C until ready for analysis.
For ATP, lactate, pyruvate or NAD+ assays, frozen embryos were homogenized in 100 μL of ice-cold 0.5 molar perchloric acid (PCA) by a combination of grinding, 1 s sonication and freeze/thaw cycles in liquid nitrogen and water. After centrifuging at 16000 xg for 15 min at 4°C, the supernatant was collected and neutralized by ice-cold 1 molar potassium carbonate. Pellets were dissolved in one molar sodium hydroxide (NaOH) and kept at −80°C until protein estimation.
For NADH assays, frozen embryos were homogenized in 100 μL of argon-bubbled ice-cold acetonitrile/50 mM ammonium acetate (1:1 v/v) containing 50 mM NaOH by grinding on ice for 2 min. The suspension was vigorously vortexed followed by freeze/thaw cycles in liquid nitrogen and water, and then centrifuged at 16000 × g for 15 min to remove insoluble material. The supernatant was transferred to a spin column (50 kDa MWCO) and subsequently centrifuged at 16000 × g for 90 min to remove macromolecules in the sample. The eluate was transferred to a new tube and further dried up under pure argon gas stream for 60 min and stored at −80°C until HPLC analysis. Dried up samples were reconstituted with 20 mM Tris–HCl buffer right before HPLC analysis.
For GSH and GSSG assays, frozen embryos were homogenized in 150 μL of ice-cold water by a combination of grinding, 1 s sonication and freeze/thaw cycles in liquid nitrogen and water. After centrifuging at 20000 xg for 15 min at 4°C, the supernatant was collected and divided into three tubes at 40, 80 and 10 μL volumes for GSH, GSSG and protein analyses, respectively. For GSH analysis, 120 μL of ice-cold 1:1 mixture of methanol and ethanol solution was added to 40 μL of the supernatant for deproteinization. Samples were vigorously vortexed and kept on ice for 15 min. After centrifuging at 20000 xg for 15 min at 4°C, the supernatant was collected and stored at −80°C until HPLC analysis. For GSSG analysis, N-ethylmaleimide (NEM), a thiol modifier, was added to 80 μL of the supernatant at a final concentration of 2 mM. Samples were vigorously vortexed for 30 s and kept on ice for 30 min. Then, in order to remove GS-NEM (modified GSH with NEM) and excess NEM, ice-cold dichloromethane (DCM) was added at 1:1 ratio followed by vortexing for 15 s. After centrifuging at 1000 xg for 10 min, the upper (aqueous) layer was collected and followed by deproteinization (as described above for GSH sample treatment). The resultant supernatant was further dried under pure argon gas stream for 30 min and stored at −80°C until HPLC analysis. Dried samples were reconstituted with dH2O, and freshly prepared sodium borohydride was added at a final concentration of 10 mM to reduce GSSG to GSH right before HPLC analysis, typically 3 min before each injection.
ATP, NAD+ and NADH measurements by HPLC coupled with photodiode array detection
Separation of ATP, NAD+ and NADH was carried out on an YMC-Pack ODS-A column (5 μm, 4.6 × 250 mm) preceded by a guard column at 5°C. Flow rate was set at 0.4 mL/min. For ATP analysis, the mobile phase was initially 100% of mobile phase A1 (0.1 M sodium phosphate buffer, pH 6.0). The methanol was linearly increased with mobile phase B (0.1 M sodium phosphate buffer, pH 6.0, containing 25% methanol) increasing to 20% over 10 min. The column was washed by increasing mobile phase B to 100% for 5 min. For NAD+ and NADH analyses, the mobile phase was initially 100% of mobile phase A2 (0.1 M sodium phosphate buffer, pH 6.0, containing 3.75% methanol). The methanol was linearly increased with mobile phase B (0.1 M sodium phosphate buffer, pH 6.0, containing 25% methanol) increasing to 50% over 15 min. The column was washed after each separation by increasing mobile phase B to 100% for 5 min. UV absorbance was monitored at 260 and 340 nm with Shimadzu SPD-M20A. Pertinent peak areas were integrated by the LabSolution software from Shimadzu, and quantified using standard curves.
GSH and GSSG measurements by HPLC coupled with electrochemical detection
Separation of GSH was carried out on an YMC-Pack ODS-A column (5 μm, 4.6 × 250 mm) preceded by a guard column at 25°C with flow rate of 0.4 mL/min. An HPLC system consisting of an LC-20 AD pump (Shimadzu, Kyoto, Japan) and an electrochemical detection-300 electrochemical detector (Eicom, Kyoto, Japan) equipped with a WE-AU gold electrode (Eicom) and a 25 μm GS-50 gasket (Eicom) was used. The mobile phase consisted of 100 mM sodium phosphate buffer (pH 2.5), 75 mg/L sodium octanesulfonate and 5 mg/L EDTA. The voltage of the gold electrode was set at +400 mV against the Ag/AgCl reference electrode. The chromatograms were analyzed by PowerChrom v2 (eDAQ) and the amounts of GSH and GSSG content in homogenates of zebrafish larvae were calculated using standard curves with freshly prepared GSH.
Lactate and pyruvate analyses
Lactate and Pyruvate assays were done by spectrophotometric methods and performed at 37°C in 170 μL final volume using a Tecan Infinite 200 PRO plate reader. For the L-lactate assay, we have developed a highly sensitive lactate oxidase (LOX)-based colorimetric assay for lactate using a unique dye, (carboxymethylaminocarbonyl)-4,4′-bis (dimethylamino) diphenylamine sodium salt (DA-64), which has been shown to be a highly sensitive indicator for the detection of hydrogen peroxide (55). LOX catalyzes the following reaction: L-lactate + O2—> pyruvate + hydrogen peroxide. Five μL of neutralized PCA extract were added to 155 μL of lactate assay reaction mixture (0.2 mM DA-64 and 5 U/mL horseradish peroxidase (HRP) in 100 mM HEPES pH 7.4), mixed thoroughly and then incubated at 37°C for 3 min. After, 10 μL of LOX (freshly prepared at 2 U/mL) was added to each well and the absorbance was measured at 727 nm for 15 min at every 20 s intervals. Lactate concentrations in samples were calculated using standard curves with sodium L-lactate. For the Pyruvate assay, we have newly developed a pyruvate oxidase (POX)-based colorimetric determination of pyruvate that uses DA-64. POX uses oxygen and phosphate to catalyze oxidative decarboxylation of pyruvate to acetylphosphate and hydrogen peroxide. In this reaction, co-factors flavin adenine dinucleotide (FAD), thiamine pyrophosphate (TPP) and magnesium (Mg2+) are required. Pyruvate + phosphate + oxygen + water ---- > acetylphosphate + carbon dioxide + water. Twenty-five or fifty μL of neutralized PCA extracted were added to 110–135 μL of pyruvate assay reaction mixture (100 μM FAD, 2 mM TPP, 10 mM MgCl2, 0.2 mM DA-64 and 5 U/mL horseradish peroxidase (HRP) in 100 mM KPB pH 6.5), mixed thoroughly and then incubated at 37°C for 3 min. After 10 μL of POX (freshly prepared at 2 U/mL) was added to each well, the absorbance was measured at 727 nm for 15 min at every 20 s intervals. Pyruvate concentrations in samples were calculated using standard curves with sodium pyruvate.
Author Contributions
M.J.F. conceived of and designed the study. J.O. and S.G. performed lifespan analysis in C. elegans. J.O., E.P. and M.B. performed biochemical analyte and flux profiling studies. Y.J.K., S.G. and C.K. performed fluorescence analyses of mitochondrial physiology in C. elegans. ENO devised and performed ATP, lactate, pyruvate, glutathione and NADH/NAD+ HPLC analyses. E.P. performed RNAseq library sample preparation in C. elegans, with bioinformatics analysis by Z.Z. N.D.M., E.N.O. and C.S. performed zebrafish studies. R.X. assisted with all statistical analyses. S.G., N.D.M., C.K., E.O. and M.J.F. wrote the manuscript. All authors approved of the final version.
Supplementary Material
Acknowledgements
We are grateful to Evgueni Daikhin, MD, PhD, and Ilana Nissim for their assistance with sample analysis in the Children’s Hospital of Philadelphia Metabolomics Core Facility, Shana McCormack, MD, for her assistance with metabolomics data analysis, and Shiven Sharma for experimental assistance with zebrafish husbandry and treatment exposures.
Conflict of Interest statement. MJF and ENO are co-inventors on International Patent Application No. PCT/US19/39631 based on U.S. Serial No. 62/690718 filed on 6/27/2018 and U.S. Serial No. 62/830850 filed on 4/8/2019 Entitled, ‘Compositions and Methods for Treatment of Mitochondrial Respiratory Chain Dysfunction and Other Mitochondrial Disorders and Methods for Identifying Efficacious Agents for the Alleviating Symptoms of the Same,’ filed in the Name of The Children’s Hospital of Philadelphia on 6/27/2019. M.J.F. is a co-founder of MitoCUREia, Inc., scientific advisory board member with equity interest in RiboNova, Inc., and scientific board member as paid consultant with Khondrion and with Larimar Therapeutics. M.J.F. has previously been or is currently engaged with several companies involved in mitochondrial disease therapeutic preclinical and/or clinical stage development as a paid consultant (Astellas [formerly Mitobridge] Pharma Inc., Casma Therapeutics, Cyclerion Therapeutics, Epirium Bio, Imel Therapeutics, Minovia Therapeutics, NeuroVive Pharmaceutical AB, Reneo Therapeutics, Stealth BioTherapeutics, Zogenix, Inc.) and/or a sponsored research collaborator (AADI Bioscience, Cardero Therapeutics, Cyclerion Therapeutics, Epirium Bio, Imel Therapeutics, Minovia Therapeutics Inc., Mission Therapeutics, NeuroVive Pharmaceutical AB, Raptor Therapeutics, REATA Inc., RiboNova Inc., Standigm Inc., and Stealth BioTherapeutics). MJF also receives royalties from Elsevier and educational honorarium from PlatformQ.
Contributor Information
Sujay Guha, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Neal D Mathew, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Chigoziri Konkwo, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Julian Ostrovsky, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Young Joon Kwon, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Erzsebet Polyak, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Christoph Seiler, Aquatics Core Facility, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Michael Bennett, Department of Pathology and Laboratory Medicine, The Children’s Hospital of Philadelphia and University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Rui Xiao, Department of Biostatistics, Epidemiology and Informatics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Zhe Zhang, Center for Biomedical Informatics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Eiko Nakamaru-Ogiso, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA.
Marni J Falk, Mitochondrial Medicine Frontier Program, Division of Human Genetics, Department of Pediatrics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA; Department of Pediatrics, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA.
Funding
The National Institutes of Health (R01-HD065858, R01-GM120762, R35-GM134863, and T32-NS007413). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Muraresku, C.C., McCormick, E.M. and Falk, M.J. (2018) Mitochondrial disease: advances in clinical diagnosis, management, therapeutic development, and preventative strategies. Curr Genet Med Rep, 6, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McCormick, E.M., Zolkipli-Cunningham, Z. and Falk, M.J. (2018) Mitochondrial disease genetics update: recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr., 30, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Park, J.S., Davis, R.L. and Sue, C.M. (2018) Mitochondrial dysfunction in Parkinson's disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep., 18, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moon, H.E. and Paek, S.H. (2015) Mitochondrial dysfunction in Parkinson's disease. Exp Neurobiol, 24, 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng, Y. and Bai, F. (2018) The association of tau with mitochondrial dysfunction in Alzheimer's disease. Front. Neurosci., 12, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Onyango, I.G., Dennis, J. and Khan, S.M. (2016) Mitochondrial dysfunction in Alzheimer's disease and the rationale for bioenergetics based therapies. Aging Dis., 7, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Swerdlow, R.H. (2018) Mitochondria and mitochondrial cascades in Alzheimer's disease. J. Alzheimers Dis., 62, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parikh, S., Saneto, R., Falk, M.J., Anselm, I., Cohen, B.H., Haas, R. and Medicine Society, T.M. (2009) A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol., 11, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barcelos, I., Shadiack, E., Ganetzky, R.D. and Falk, M.J. (2020) Mitochondrial medicine therapies: rationale, evidence, and dosing guidelines. Curr Opin Pediatr, Curr Opin Pediatr, 32, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parikh, S., Goldstein, A., Koenig, M.K., Scaglia, F., Enns, G.M., Saneto, R. and Mitochondrial Medicine Society Clinical Directors Working, G. and Clinical Director's Work, G (2013) Practice patterns of mitochondrial disease physicians in North America. Part 2: treatment, care and management. Mitochondrion, 13, 681–687. [DOI] [PubMed] [Google Scholar]
- 11. Parikh, S., Goldstein, A., Karaa, A., Koenig, M.K., Anselm, I., Brunel-Guitton, C., Christodoulou, J., Cohen, B.H., Dimmock, D., Enns, G.M. et al. (2017) Patient care standards for primary mitochondrial disease: a consensus statement from the mitochondrial medicine society. Genet. Med., 19, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dingley, S., Polyak, E., Lightfoot, R., Ostrovsky, J., Rao, M., Greco, T., Ischiropoulos, H. and Falk, M.J. (2010) Mitochondrial respiratory chain dysfunction variably increases oxidant stress in Caenorhabditis elegans. Mitochondrion, 10, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McCormack, S., Polyak, E., Ostrovsky, J., Dingley, S.D., Rao, M., Kwon, Y.J., Xiao, R., Zhang, Z., Nakamaru-Ogiso, E. and Falk, M.J. (2015) Pharmacologic targeting of sirtuin and PPAR signaling improves longevity and mitochondrial physiology in respiratory chain complex I mutant Caenorhabditis elegans. Mitochondrion, 22, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Polyak, E., Ostrovsky, J., Peng, M., Dingley, S.D., Tsukikawa, M., Kwon, Y.J., McCormack, S.E., Bennett, M., Xiao, R., Seiler, C. et al. (2018) N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab., 123, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guha, S., Konkwo, C., Lavorato, M., Mathew, N.D., Peng, M., Ostrovsky, J., Kwon, Y.J., Polyak, E., Lightfoot, R., Seiler, C. et al. (2019) Pre-clinical evaluation of cysteamine bitartrate as a therapeutic agent for mitochondrial respiratory chain disease. Hum. Mol. Genet., 28, 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang, Z., Tsukikawa, M., Peng, M., Polyak, E., Nakamaru-Ogiso, E., Ostrovsky, J., McCormack, S., Place, E., Clarke, C., Reiner, G. et al. (2013) Primary respiratory chain disease causes tissue-specific dysregulation of the global transcriptome and nutrient-sensing signaling network. PLoS One, 8, e69282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zolkipli-Cunningham, Z., Xiao, R., Stoddart, A., McCormick, E.M., Holberts, A., Burrill, N., McCormack, S., Williams, L., Wang, X., Thompson, J.L.P. et al. (2018) Mitochondrial disease patient motivations and barriers to participate in clinical trials. PLoS One, 13, e0197513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falk, M.J. (2020) The pursuit of precision mitochondrial medicine: harnessing preclinical cellular and animal models to optimize mitochondrial disease therapeutic discovery. J. Inherit. Metab. Dis., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kayser, E.B., Morgan, P.G. and Sedensky, M.M. (1999) GAS-1: a mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans. Anesthesiology, 90, 545–554. [DOI] [PubMed] [Google Scholar]
- 20. Falk, M.J., Zhang, Z., Rosenjack, J.R., Nissim, I., Daikhin, E., Nissim, I., Sedensky, M.M., Yudkoff, M. and Morgan, P.G. (2008) Metabolic pathway profiling of mitochondrial respiratory chain mutants in C. elegans. Mol. Genet. Metab., 93, 388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kwon, Y.J., Guha, S., Tuluc, F. and Falk, M.J. (2018) High-throughput bio sorter quantification of relative mitochondrial content and membrane potential in living Caenorhabditis elegans. Mitochondrion, 40, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vergano, S.S., Rao, M., McCormack, S., Ostrovsky, J., Clarke, C., Preston, J., Bennett, M.J., Yudkoff, M., Xiao, R. and Falk, M.J. (2014) In vivo metabolic flux profiling with stable isotopes discriminates sites and quantifies effects of mitochondrial dysfunction in C. elegans. Mol. Genet. Metab., 111, 331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kayser, E.B., Morgan, P.G., Hoppel, C.L. and Sedensky, M.M. (2001) Mitochondrial expression and function of GAS-1 in Caenorhabditis elegans. J. Biol. Chem., 276, 20551–20558. [DOI] [PubMed] [Google Scholar]
- 24. Kayser, E.B., Sedensky, M.M. and Morgan, P.G. (2004) The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegans. Mech. Ageing Dev., 125, 455–464. [DOI] [PubMed] [Google Scholar]
- 25. Durieux, J., Wolff, S. and Dillin, A. (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell, 144, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bennett, C.F., Vander Wende, H., Simko, M., Klum, S., Barfield, S., Choi, H., Pineda, V.V. and Kaeberlein, M. (2014) Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat. Commun., 5, 3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoneda, T., Benedetti, C., Urano, F., Clark, S.G., Harding, H.P. and Ron, D. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci., 117, 4055–4066. [DOI] [PubMed] [Google Scholar]
- 28. Benedetti, C., Haynes, C.M., Yang, Y., Harding, H.P. and Ron, D. (2006) Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics, 174, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haynes, C.M., Petrova, K., Benedetti, C., Yang, Y. and Ron, D. (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell, 13, 467–480. [DOI] [PubMed] [Google Scholar]
- 30. Polyak, E., Zhang, Z. and Falk, M.J. (2012) Molecular profiling of mitochondrial dysfunction in Caenorhabditis elegans. Methods Mol. Biol., 837, 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balsa, E., Perry, E.A., Bennett, C.F., Jedrychowski, M., Gygi, S.P., Doench, J.G. and Puigserver, P. (2020) Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat. Commun., 11, 2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Byrnes, J., Ganetzky, R., Lightfoot, R., Tzeng, M., Nakamaru-Ogiso, E., Seiler, C. and Falk, M.J. (2018) Pharmacologic modeling of primary mitochondrial respiratory chain dysfunction in zebrafish. Neurochem. Int., 117, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harper, L., Balasubramanian, D., Ohneck, E.A., Sause, W.E., Chapman, J., Mejia-Sosa, B., Lhakhang, T., Heguy, A., Tsirigos, A., Ueberheide, B. et al. (2018) Staphylococcus aureus responds to the central metabolite pyruvate to regulate virulence. MBio, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parikh, S., Goldstein, A., Koenig, M.K., Scaglia, F., Enns, G.M., Saneto, R., Anselm, I., Cohen, B.H., Falk, M.J., Greene, C. et al. (2015) Diagnosis and management of mitochondrial disease: a consensus statement from the mitochondrial Medicine Society. Genet. Med., 17, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Atkuri, K.R., Cowan, T.M., Kwan, T., Ng, A., Herzenberg, L.A., Herzenberg, L.A. and Enns, G.M. (2009) Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc. Natl. Acad. Sci. U. S. A., 106, 3941–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ballatori, N., Krance, S.M., Notenboom, S., Shi, S., Tieu, K. and Hammond, C.L. (2009) Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem., 390, 191–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu, H.Q., Zhu, X.Z. and Weng, E.Q. (2005) Intracellular dopamine oxidation mediates rotenone-induced apoptosis in PC12 cells. Acta Pharmacol. Sin., 26, 17–26. [DOI] [PubMed] [Google Scholar]
- 38. Debray, F.G., Mitchell, G.A., Allard, P., Robinson, B.H., Hanley, J.A. and Lambert, M. (2007) Diagnostic accuracy of blood lactate-to-pyruvate molar ratio in the differential diagnosis of congenital lactic acidosis. Clin. Chem., 53, 916–921. [DOI] [PubMed] [Google Scholar]
- 39. Williamson, D.H., Lund, P. and Krebs, H.A. (1967) The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J., 103, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Enns, G.M. and Cowan, T.M. (2017) Glutathione as a redox biomarker in mitochondrial disease-implications for therapy. J. Clin. Med., 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andreux, P.A., Houtkooper, R.H. and Auwerx, J. (2013) Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov., 12, 465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang, Z. and Falk, M.J. (2014) Integrated transcriptome analysis across mitochondrial disease etiologies and tissues improves understanding of common cellular adaptations to respiratory chain dysfunction. Int. J. Biochem. Cell Biol., 50, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morita, M., Gravel, S.P., Hulea, L., Larsson, O., Pollak, M., St-Pierre, J. and Topisirovic, I. (2015) mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle, 14, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ramanathan, A. and Schreiber, S.L. (2009) Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. U.S.A., 106, 22229–22232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Javadov, S., Jang, S. and Agostini, B. (2014) Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol. Ther., 144, 202–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheng, Z., Tseng, Y. and White, M.F. (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab., 21, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pirinen, E., Auranen, M., Khan, N.A., Brilhante, V., Urho, N., Pessia, A., Hakkarainen, A., Kuula, J., Heinonen, U., Schmidt, M.S. et al. (2020) Niacin cures systemic NAD(+) deficiency and improves muscle performance in adult-onset mitochondrial myopathy. Cell Metab., 31, 1078, e1075–1090. [DOI] [PubMed] [Google Scholar]
- 48. Falk, M.J. (2018) Nutritional Inadequacies in Mitochondrial-Associated Metabolic Disorders. The National Academies Press The National Academies Press, Washington, DC, pp. 24–30. [Google Scholar]
- 49. Hamilton, B., Dong, Y., Shindo, M., Liu, W., Odell, I., Ruvkun, G. and Lee, S.S. (2005) A systematic RNAi screen for longevity genes in C. elegans. Genes Dev., 19, 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Benjamini, Y. and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Methodol., 57, 289–300. [Google Scholar]
- 52. Falk, M.J., Rao, M., Ostrovsky, J., Daikhin, E., Nissim, I. and Yudkoff, M. (2011) Stable isotopic profiling of intermediary metabolic flux in developing and adult stage Caenorhabditis elegans. J. Vis. Exp., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim, S.Y. and Volsky, D.J. (2005) PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics, 6, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Colwill, R.M. and Creton, R. (2011) Imaging escape and avoidance behavior in zebrafish larvae. Rev. Neurosci., 22, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Takagi, K., Tatsumi, Y., Kitaichi, K., Iwase, M., Shibata, E., Nakao, M., Matsumoto, T., Takagi, K. and Hasegawa, T. (2004) A sensitive colorimetric assay for polyamines in erythrocytes using oat seedling polyamine oxidase. Clin. Chim. Acta, 340, 219–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.