

Abstract
Aims
Murine models implicate phosphodiesterase 9A (PDE9A) as a nitric oxide‐independent regulator of cyclic guanosine monophosphate and promising novel therapeutic target in heart failure (HF) with preserved ejection fraction (HFpEF). This study describes PDE9A expression in endomyocardial biopsies (EMBs) and peripheral blood mononuclear cells (PBMNCs) from patients with different HF phenotypes.
Methods and results
Endomyocardial biopsies and PBMNCs were obtained from patients with HFpEF (n = 24), HF with reduced ejection fraction (n = 22), and inflammatory cardiomyopathy (n = 24) and patients without HF (n = 7). PDE9A expression was increased in EMBs and PBMNCs from patients with HFpEF as compared with other HF phenotypes or subjects without HF. Endomyocardial PDE9A expression in HFpEF correlated with the inflammatory cell count in EMBs, but not with cardiac fibrosis or left ventricular diastolic wall stress. PDE9A expression in PBMNCs was increased in HFpEF patients with higher high‐sensitivity C‐reactive protein levels and in response to pro‐inflammatory stimulation. As a validation cohort, 719 patients with HFpEF and 1106 subjects without HF were identified from the LIFE‐Heart study. PDE9A expression in PBMNCs was obtained from array data and displayed an age‐dependent distribution. PDE9A levels were elevated and conferred increased risk for HFpEF in middle‐aged subjects, but not in elderly HFpEF patients. Following age adjustment, lower PDE9A expression in PBMNCs was associated with worse survival in patients with HFpEF (log‐rank test P‐value <0.001).
Conclusion
Expression profiling indicates an up‐regulation of endomyocardial PDE9A in different HF phenotypes with the most robust increase in EMBs and PBMNCs from patients with HFpEF. An exclusive risk effect of PDE9A expression on HFpEF in middle‐aged patients and an unexpected association with survival calls for further studies to better characterize the role of PDE9A as a treatment target.
Keywords: Phosphodiesterase 9A, Heart failure, Heart failure with preserved ejection fraction, Fibrosis, Inflammation
Introduction
Medical management of patients with heart failure with preserved ejection fraction (HFpEF) remains a major challenge in clinical practice. Therapies targeting the renin–angiotensin–aldosterone system and beta‐blockers have been unable to demonstrate a definitive benefit in patients with HFpEF in large‐scale randomized trials. 1 , 2 Results of a recent meta‐analysis indicate that mineralocorticoid receptor antagonist treatment may reduce heart failure (HF) hospitalizations in HFpEF. 2 However, no effect of mineralocorticoid receptor antagonist treatment on mortality was observed in this analysis, emphasizing the need for novel therapeutic targets in patients with HFpEF.
Current pathophysiological concepts of HFpEF underline the importance of non‐cardiac co‐morbidities, ageing, and systemic inflammation as important drivers of cardiovascular perturbations in HFpEF. 3 Pro‐oxidative and pro‐inflammatory processes have been suggested to decrease myocardial levels of cyclic guanosine monophosphate (cGMP), a second messenger molecule that transduces nitric oxide (NO)‐coupled and natriuretic peptide‐coupled cardioprotective signalling. 3 , 4 Notably, blocking cGMP degradation by inhibition of phosphodiesterase 5 (PDE5) did not result in functional improvement in patients with HFpEF. 5 A recent study identified cGMP‐selective phosphodiesterase 9a (PDE9A) as a novel phosphodiesterase expressed in the mammalian heart, regulating GMP signalling independent of the NO pathway. 6 Genetic and pharmacological inhibition of PDE9A resulted in improved cardiac function in a murine model of left ventricular (LV) pressure overload. 6
We sought to investigate the relevance of these findings in humans by characterizing endomyocardial PDE9A expression in patients with different HF phenotypes and in a large gene expression analysis in peripheral blood mononuclear cells (PBMNCs) from patients with HFpEF and controls without HF enrolled in a large population‐based cohort study.
Methods
A detailed description of the methods is provided in the Supporting Information.
Patient cohorts
Right ventricular and LV endomyocardial biopsies (EMBs) were obtained from 22 patients with non‐ischaemic HF with reduced ejection fraction (HFrEF), 24 patients with inflammatory cardiomyopathy (iCMP), and 24 patients with HFpEF. The diagnosis of HFpEF was made according to current recommendations provided in the European Society of Cardiology Guidelines for the diagnosis and treatment of acute and chronic HF. 7 Differentiation of HFrEF and iCMP was based on histological findings, as described in detail in the Supporting Information. Commercially available total RNA purified from LV autopsy samples of seven subjects without known cardiovascular disease dying for non‐cardiac reason (mean age 38 years, 3 female), served as a control (BioChain, Newark, CA and Thermo Fisher Scientific, Waltham, MA).
To further characterize the association between PDE9A expression in PBMNCs, HFpEF risk, and clinical outcome in patients with HFpEF, gene array data were analysed from 719 patients with symptomatic HFpEF and 1106 control subjects without HF recruited in the prospective observational LIFE‐Heart study. Details about the LIFE‐Heart study are provided in the Supporting Information. The definition of HFpEF was based on current guidelines recommendations. 7 Control subjects were identified based on an LV ejection fraction (LVEF) ≥ 50% and N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP) level <125 ng/L.
Endomyocardial biopsy analysis
Histological and immunohistological analyses were performed to quantify myocardial inflammation and fibrosis, as outlined in the Supporting Information.
Quantification of phosphodiesterase 9A mRNA expression in endomyocardial biopsies
Total RNA was isolated from EMBs and processed for quantitative reverse transcription polymerase chain reaction (PCR). PDE9A expression was normalized to the respective TATA‐binding protein levels in EMBs.
Isolation and characterization of phosphodiesterase 9A expression in peripheral blood mononuclear cells
Peripheral blood mononuclear cells were isolated by Biocoll density gradient solution, and quantitative reverse transcription PCR for PDE9A expression was performed following RNA purification and cDNA synthesis. The effect of tumour necrosis factor‐α (TNF‐α) and interleukin‐1β (IL‐1β) on PDE9A expression in PBMNCs from healthy subjects and patients with different HF phenotypes was tested in separate experiments.
Calculation of left ventricular meridional diastolic wall stress
The calculation of end‐diastolic meridional wall stress was based on LV end‐diastolic pressure obtained during cardiac catheterization and LV end‐diastolic diameter as well as LV posterior wall thickness derived from echocardiography.
Gene expression preprocessing in the LIFE‐Heart study
Please refer to the Supporting Information for details about the processing of gene array expression data in patients with HFpEF and control subjects identified in the LIFE‐Heart study.
Results
Endomyocardial biopsy samples
Right ventricular and LV EMBs were obtained from 22 patients with non‐ischaemic HFrEF, 24 patients with iCMP, and 24 patients with HFpEF. The characteristics of the EMB cohort are displayed in Table 1 . Patients with HFpEF were older, more frequently female, and presented with a higher cardiovascular risk factor profile, including higher rates of arterial hypertension and dyslipidaemia (Table 1 ). NT‐proBNP levels were lower in patients with HFpEF as compared with patients with HFrEF and iCMP (Table 1 ). Compatible with the underlying aetiology of HF, patients with iCMP demonstrated an increased high‐sensitivity C‐reactive protein (hsCRP) level, a higher number of CD3‐positive T lymphocytes and CD68‐positive macrophages in EMBs, a higher percentage of virus‐positive EMBs, and enhanced endomyocardial major histocompatibility complex class II antigen expression (Table 1 ). The percentage of cardiac fibrosis, as assessed on histology, did not differ significantly between the three groups of patients (10.7 ± 6.2%, 13.9 ± 6.4%, and 11.4 ± 5.9% for patients with HFrEF, iCMP, and HFpEF, respectively).
Table 1.
Characteristics of the endomyocardial biopsy cohort (derivation cohort)
| HFrEF (n = 22) | iCMP (n = 24) | HFpEF (n = 24) | |
|---|---|---|---|
| Age (years) | 48.3 ± 15.1 | 45.0 ± 11.4 | 66.1 ± 8.6 a , b |
| Female, n (%) | 8 (36) | 10 (42) | 20 (83) a , b |
| BMI (kg/m2) | 26.8 ± 7.2 | 28.7 ± 6.2 | 29.8 ± 4.9 |
| Systolic BP (mmHg) | 119 ± 17 | 124 ± 18 | 148 ± 12 a |
| Diastolic BP (mmHg) | 78 ± 8 | 79 ± 6 | 81 ± 8 |
| Heart rate (b.p.m.) | 72 ± 9 | 76 ± 10 | 70 ± 11 |
| Creatinine (mg/dL) | 0.91 (0.75–1.02) | 0.86 (0.73–0.99) | 0.89 (0.75–0.97) |
| hsCRP, mg/L (%) | 2.0 (1.1–3.9) b | 7.6 (3.3–10.5) | 3.1 (1.9–6.2) a , b |
| NT‐proBNP (ng/mL) | 1831 (918–3547) b | 1051 (476–2089) | 342 (222–614) a , b |
| NYHA functional class | |||
| NYHA I, n (%) | 0 (0) | 6 (26) | 0 (0) |
| NYHA II, n (%) | 10 (45) | 9 (37) | 20 (83) |
| NYHA III, n (%) | 12 (55) | 9 (37) | 4 (17) |
| Cardiovascular risk factors, n (%) | 16 (73) | 13 (54) | 24 (100) b |
| Arterial hypertension, n (%) | 11 (50) | 10 (42) | 23 (96) a , b |
| Smoking, n (%) | 4 (18) | 6 (25) | 5 (21) |
| Diabetes mellitus, n (%) | 5 (23) | 4 (17) | 5 (21) |
| Dyslipidaemia, n (%) | 8 (36) | 7 (29) | 21 (88) b |
| Obesity, n (%) | 7 (32) | 8 (33) | 15 (63) |
| Atrial fibrillation, n (%) | 4 (18) | 4 (17) | 5 (21) |
| Chronic pulmonary disease, n (%) | 5 (23) | 3 (13) | 3 (13) |
| ACEI/ARB, n (%) | 17 (77) | 13 (54) | 18 (75) |
| Beta‐blocker, n (%) | 9 (41) | 6 (25) | 14 (58) |
| Aldosterone antagonist, n (%) | 5 (23) | 3 (13) | 5 (21) |
| Diuretic, n (%) | 14 (64) | 8 (33) | 11 (46) |
| Results of echocardiography | |||
| LV ejection fraction (%) | 30 (22–45) | 43 (27–54) | 61 (52–69) a |
| LV end‐diastolic diameter (mm) | 57 ± 8 | 52 ± 4 | 46 ± 4 |
| LV end‐systolic diameter (mm) | |||
| LA end‐systolic volume index (mL/m2) | 39.9 ± 14.5 | ||
| E/A ratio | |||
| E/E′ mean ratio | |||
| Results of LV endomyocardial biopsy | |||
| Number of CD3‐positive T lymphocytes | 2 (0–3) b | 13 (7–14) | 3 (1–4) b |
| Number of CD68‐positive macrophages | 10 (8–11) b | 30 (24–41) | 10 (7–12) b |
| Viral genome detection, n (%) | 6 (27) b | 13 (54) | 5 (21) b |
| Enhanced MHC class II antigen expression, n (%) | 4 (18) b | 24 (100) | 4 (17) b |
| LV fibrosis (%) | 10.7 ± 6.2 | 13.9 ± 6.4 | 11.4 ± 5.9 |
ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BMI, body mass index; BP, blood pressure; CD, cluster of differentiation; HFpEF, heart failure with preserved ejection fraction; HFrEF, heart failure with reduced ejection fraction; hsCRP, high‐sensitivity C‐reactive protein; iCMP, inflammatory cardiomyopathy; LA, left atrial; LV, left ventricular; MHC, major histocompatibility complex; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association.
P < 0.05 vs. HFrEF.
P < 0.05 vs. iCMP.
Myocardial phosphodiesterase 9A expression in patients with different heart failure phenotypes
As compared with commercially available autopsy samples from patients without known cardiovascular disease, LV endomyocardial PDE9A expression was increased in patients with all HF phenotypes (Figure 1 A ). Patients with HFpEF exerted higher LV endomyocardial PDE9A expression when compared with patients with non‐ischaemic HFrEF and iCMP (Figure 1 A ). Also, right ventricular endomyocardial expression of PDE9A was increased in patients with HFpEF as compared with patients with HFrEF and iCMP (Figure 1 B ).
Figure 1.
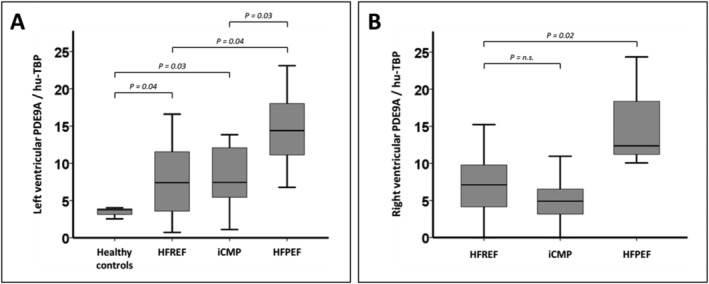
Quantification of endomyocardial phosphodiesterase 9A (PDE9A) expression in patients with different heart failure phenotypes. Endomyocardial biopsies were obtained from healthy subjects (n = 7) and patients with heart failure with reduced ejection fraction (HFrEF) (n = 22), inflammatory cardiomyopathy (iCMP) (n = 24), and heart failure with preserved ejection fraction (HFpEF) (n = 24). mRNA expression of PDE9A was quantified in left (A) and right (B) ventricular endomyocardial biopsies by reverse transcription PCR.
When LV endomyocardial PDE9A expression was compared with the percentage of cardiac fibrosis in patients with HFpEF, a modest and non‐significant inverse association was apparent (Figure 2 A ). LV endomyocardial PDE9A expression correlated with the LV inflammatory cell count in patients with HFpEF, with increased PDE9A levels being associated with a higher number of CD3‐positive and CD68‐positive cells on EMBs (Figure 2 B ). In contrast, LV endomyocardial PDE9A expression was not associated with the inflammatory cell count in EMBs from patients with HFrEF and iCMP (Supporting Information, Figure S1 A and S1 B). No relevant correlation between LV endomyocardial PDE9A levels and LV meridional diastolic wall stress was apparent in patients with HFpEF (Figure 2 C ).
Figure 2.
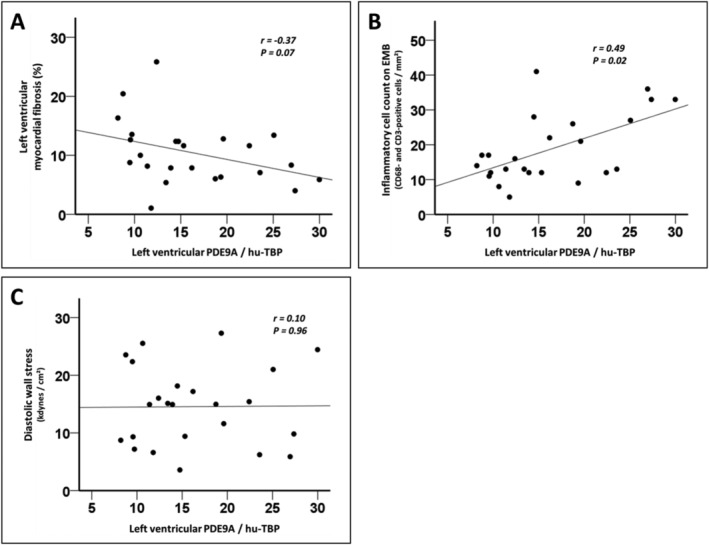
Association between left ventricular phosphodiesterase 9A (PDE9A) expression, cardiac fibrosis, myocardial inflammation, and diastolic wall stress. (A) In patients with heart failure with preserved ejection fraction (HFpEF), left ventricular PDE9A expression displayed a non‐significant inverse relationship with left ventricular endomyocardial fibrosis, as assessed by histology. (B) Left ventricular myocardial inflammation was analysed by immunohistochemistry in endomyocardial biopsies (EMBs) from patients with HFpEF, and the inflammatory cell count was derived from the total number of CD68‐positive macrophages and CD3‐positive T cells in EMBs. A significant positive correlation between left ventricular PDE9A levels and left ventricular myocardial inflammation was apparent in HFpEF. (C) Left ventricular meridional diastolic wall stress was calculated using haemodynamic and echocardiographic data from patients with HFpEF. No correlation was evident between left ventricular PDE9A levels and diastolic wall stress in patients with HFpEF.
Phosphodiesterase 9A expression in peripheral blood mononuclear cells from patients with different heart failure phenotypes
Given the association between LV endomyocardial PDE9A expression and histological markers of myocardial inflammation, we sought to investigate the expression of PDE9A in PBMNCs. PDE9A expression was increased in PBMNCs from patients with HFpEF as compared with PBMNCs from healthy control subjects and patients with HFrEF or iCMP (Figure 3 A ). A modest but significant correlation was evident when PDE9A expression in EMBs and PBMNCs from patients with HFpEF was compared (Supporting Information, Figure S2 ). Moreover, PDE9A expression in PBMNCs was higher in patients with HFpEF and hsCRP levels above the median than in patients with HFpEF and hsCRP levels below the median (Figure 3 B ). Stimulation of PBMNCs from HFpEF patients with TNF‐α or IL‐1β in vitro led to an increase in PDE9A expression in these cells (Figure 3 C ). This increase in PDE9A expression upon incubation with TNF‐α or IL‐1β was not evident in PBMNCs isolated from patients with HFrEF or iCMP and healthy subjects (Supporting Information, Figure S3 ).
Figure 3.
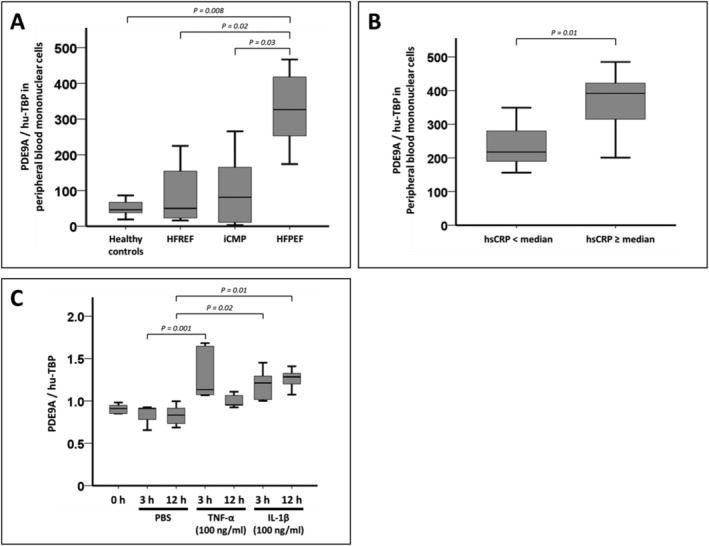
Phosphodiesterase 9A (PDE9A) expression in peripheral blood mononuclear cells (PBMNCs). (A) Quantification of PDE9A mRNA expression in PBMNCs from healthy subjects (n = 5) and patients with heart failure with reduced ejection fraction (HFrEF) (n = 22), inflammatory cardiomyopathy (iCMP) (n = 24), and heart failure with preserved ejection fraction (HFpEF) (n = 24) by reverse transcription PCR. (B) PDE9A expression in PBMNCs from patients with HFpEF (n = 24) stratified by median high‐sensitivity C‐reactive protein (hsCRP) level. (C) Effect of tumour necrosis factor‐α (TNF‐α) and interleukin‐1β (IL‐1β) on PDE9A expression in PBMNCs from patients with HFpEF (n = 5–9) after incubation for 3 or 12 h. PBS, phosphate‐buffered saline.
Patients with heart failure with preserved ejection fraction and control subjects from LIFE‐Heart
Baseline characteristics of patients with HFpEF and control subjects from LIFE‐Heart are shown in Table 2 . Consistent with data from the EMB cohort, patients with HFpEF in LIFE‐Heart were older, more likely to be female, and displayed a more pronounced cardiovascular risk factor profile. Levels of creatinine and hsCRP were higher in patients with HFpEF when compared with control subjects. Median NT‐proBNP level in patients with HFpEF was 283 ng/mL (inter‐quartile range 181–542 ng/mL). In addition, a higher percentage of patients with HFpEF had atrial fibrillation and coronary artery disease. Echocardiographic data suggested elevated LV wall thickness and LV filling pressures in patients with HFpEF, as indicated by increases in LV mass index, E/e′ mean ratio, and left atrial volume index. Right ventricular function, as assessed by tricuspid annular plane systolic excursion, was preserved in control subjects and patients with HFpEF.
Table 2.
Characteristics of patients with HFpEF and subjects without HF from the LIFE‐Heart study (validation cohort)
| Control (n = 1106) | HFpEF (n = 719) | P‐value | |
|---|---|---|---|
| Age (years) | 58.6 (52.2–66.9) | 68.4 (59.9–73.7) | <0.01 |
| Female, n (%) | 391 (35) | 325 (45) | <0.01 |
| BMI (kg/m2) | 29.0 (26.3–32.4) | 29.9 (27.3–33.7) | <0.01 |
| LV ejection fraction (%) | 63.0 (59.0–67.0) | 61.0 (56.0–66.0) | <0.01 |
| Systolic BP (mmHg) | 137 (125–149) | 144 (130–159) | <0.01 |
| Diastolic BP (mmHg) | 85 (78–91) | 84 (75–92) | 0.26 |
| Heart rate (b.p.m.) | 69 (62–80) | 66.0 (59–78) | <0.01 |
| Creatinine (mg/dL) | 0.85 (0.74–0.97) | 0.89 (0.75–1.03) | <0.01 |
| hsCRP, mg/L (%) | 1.7 (0.9–3.6) | 2.6 (1.3–5.3) | <0.01 |
| NT‐proBNP (ng/mL) | 57 (35–87) | 283 (181–542) | <0.01 |
| HbA1c (%) | 5.7 (5.4–6.1) | 5.8 (5.5–6.3) | <0.01 |
| NYHA functional class | |||
| NYHA I, n (%) | 709 (64) | 362 (50) | <0.01 |
| NYHA II, n (%) | 336 (31) | 272 (38) | <0.01 |
| NYHA III/IV, n (%) | 61 (5) | 85 (12) | <0.01 |
| Cardiovascular risk factors, n (%) | |||
| Arterial hypertension, n (%) | 843 (76) | 637 (89) | <0.01 |
| Smoking, n (%) | 219 (20) | 103 (14) | <0.01 |
| Diabetes mellitus, n (%) | 290 (26) | 236 (33) | 0.01 |
| Dyslipidaemia, n (%) (LAB/treatment) | 715 (65) | 478 (67) | 0.45 |
| Dyslipidaemia, n (%) (LAB) | 482 (44) | 248 (35) | <0.01 |
| Obesity, n (%) | 451 (41) | 354 (49) | <0.01 |
| Coronary artery disease, n (%) | 320 (30) | 287 (41) | <0.01 |
| Peripheral arterial disease, n (%) | 52 (5) | 47 (7) | 0.09 |
| Atrial fibrillation, n (%) | 18 (2) | 89 (12) | <0.01 |
| ACEI/ARB, n (%) | 738 (67) | 558 (78) | <0.01 |
| Beta‐blocker, n (%) | 564 (51) | 516 (72) | <0.01 |
| Aldosterone antagonist, n (%) | 15 (1) | 23 (3) | 0.01 |
| Diuretic, n (%) | 144 (13) | 216 (30) | <0.01 |
| LV mass index (g/m2) | 115 (94–138) | 128 (107–155) | <0.01 |
| E/e′ mean ratio (cm/s) | 8.0 (6.6–9.7) | 10.1 (7.9–13.0) | <0.01 |
| LA volume index (mL/m2) | 22.8 (18.8–28.1) | 27.1 (20.9–31.8) | 0.01 |
| TAPSE (mm) | 20.0 (19.0–23.0) | 20.0 (19.0–23.0) | 0.66 |
ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; BMI, body mass index; BP, blood pressure; HF, heart failure; HFpEF, heart failure with preserved ejection fraction; hsCRP, high‐sensitivity C‐reactive protein; LA, left atrial; LAB, based on laboratory lipid profiling; LV, left ventricular; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association; TAPSE, tricuspid annular plane systolic excursion.
Phosphodiesterase 9A expression and heart failure with preserved ejection fraction in LIFE‐Heart
Phosphodiesterase 9A expression in PBMNCs exhibited an age‐dependent association with HFpEF (P‐value for interaction of PDE9A expression with age = 0.003, Table 3 and Figure 4 ). With increasing age, PDE9A expression in PBMNCs decreased in control subjects without HF as well as in patients with HFpEF (Figure 4 ). We observed a strong risk effect in middle‐aged patients [age 50: odds ratio = 3.4 (95% confidence interval, CI 1.4–8.2) per PDE9A unit, P = 0.005] and a reduced or protective effect in elderly patients [age 80: odds ratio = 0.3 (95% CI 0.12–0.97) per PDE9A unit, P = 0.04, Table 3 ]. The same risk effect for PDE9A transcript levels was observed when only symptomatic patients with HFpEF in New York Heart Association (NYHA) Functional Classes II to IV were analysed (Supporting Information, Table S1 ).
Table 3.
Logistic regression analysis for the association of PDE9A transcript levels with HFpEF risk in HFpEF patients (NYHA Classes I to IV)
| Variables | OR | 95% CI | P‐value |
|---|---|---|---|
| Age (years) | 1.8 | 1.3–2.6 | 0.0008 |
| Sex (female/male) | 1.5 | 1.2–1.8 | 0.0004 |
| Diabetes (yes/no) | 1.1 | 0.8–1.3 | 0.653 |
| Smoking (yes/no) | 1.4 | 1.1–1.9 | 0.019 |
| High blood pressure (yes/no) | 1.5 | 1.2–1.8 | 0.0001 |
| Lymphocytes (increase of 10%) | 0.7 | 0.6–0.8 | <0.0001 |
| Monocytes (increase of 10%) | 1.1 | 0.7–1.7 | 0.790 |
| PDE9A expression (e.units, 50 years old) a | 3.4 | 1.4–8.2 | 0.005 |
| PDE9A expression (e.units, 80 years old) a | 0.3 | 0.12–0.97 | 0.04 |
| PDE9A expression–age interaction | 0.9 | 0.88–0.98 | 0.003 |
CI, confidence interval; e.units, expression units; HFpEF, heart failure with preserved ejection fraction; NYHA, New York Heart Association; OR, odds ratio; PDE9A, phosphodiesterase 9A.
The first entry in ‘(unit)’ represents the counted observation.
The main effect for PDE9A expression varies between different ages, which is well known when modelling interactions. Therefore, we present exemplarily results of the main effect for PDE9A for 50‐ and 80‐year‐old patients.
Figure 4.
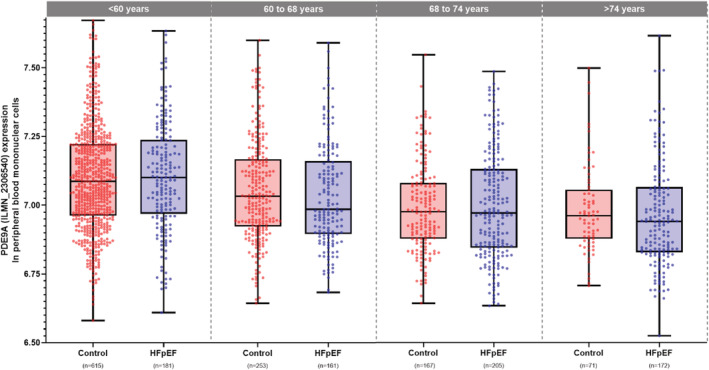
Age‐stratified expression of phosphodiesterase 9A (PDE9A) in peripheral blood mononuclear cells (PBMNCs) from subjects without heart failure (HF) and HF with preserved ejection fraction (HFpEF) in LIFE‐Heart. PDE9A (ILMN_2306540) expression data from PBMNCs were derived from HT‐12 v4 BeadChip analysis in subjects without HF (n = 1.106) and patients with HFpEF (n = 719). PDE9A (ILMN_2306540) expression reveals an age‐dependent association with HFpEF. Younger patients with HFpEF demonstrate higher PDE9A expression in PBMNCs when compared with patients without HF, whereas older patients with HFpEF have less PDE9A expression in PBMNCs (interaction P‐value 0.003; see also Table 3).
Phosphodiesterase 9A expression and survival in LIFE‐Heart
To investigate whether PDE9A has a prognostic impact in patients with HFpEF in LIFE‐Heart, we tested the relationship between PDE9A expression in PBMNCs and survival in these patients. During a median follow‐up of 2773 days (inter‐quartile range 2234–3366 days), 82 of 719 patients with HFpEF died. Patients with HFpEF whose PDE9A expression in PBMNCs was in the lower tertile had a significantly increased risk of mortality when compared with patients with PDE9A expression in the middle or upper tertile. This observation was evident in the overall cohort of asymptomatic and symptomatic patients with HFpEF (NYHA Classes I to IV, Figure 5 , global log‐rank test P‐value <0.001) and when symptomatic patients with HFpEF (NYHA Classes II to IV) were analysed separately (Supporting Information, Figure S4 , global log‐rank test P‐value <0.001). The protective effect of higher PDE9A expression was maintained even after adjustment for age in a multivariate model (PDE9A HRadj 0.15, 95% CI 0.04–0.54, per unit).
Figure 5.
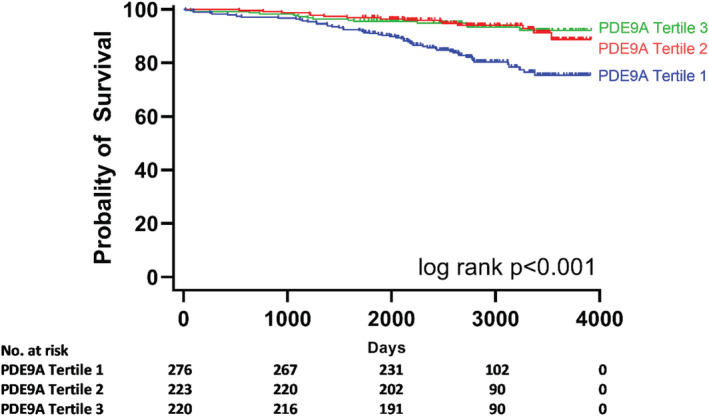
Phosphodiesterase 9A (PDE9A) expression in peripheral blood mononuclear cells (PBMNCs) and mortality in patients with heart failure with preserved ejection fraction (New York Heart Association Classes I to IV) in LIFE‐Heart. Kaplan–Meier curve of the probability of survival in patients with asymptomatic and symptomatic heart failure with preserved ejection fraction stratified to tertiles of PDE9A expression in PBMNCs. A strong difference in mortality was observed between patients with PDE9A levels in the lower tertile as compared with patients with PDE9A levels in the middle or upper tertile (global log‐rank test P‐value <0.001).
Discussion
After the description of PDE9A as a new potential therapeutic target in HFpEF, this is, to the best of our knowledge, the first study to assess the HF specific expression of PDE9A in humans, its association with HFpEF risk, and the prognostic implications of PDE9A levels in patients with HFpEF. The findings of the present study suggest that PDE9A expression is increased in patients with different HF phenotypes with the most pronounced elevation observed in patients with HFpEF. Endomyocardial PDE9A expression correlates with cellular myocardial inflammation in patients with HFpEF, and PDE9A expression in PBMNCs from patients with HFpEF is subject to pro‐inflammatory stimulation in vitro and increased in patients with higher hsCRP levels. Moreover, validation data in PBMNCs implied that expression level of PDE9A and its role as a risk marker for HFpEF vary with age. Whereas PDE9A expression is increased in middle‐aged patients with HFpEF as compared with subjects without HF and serves as a risk marker in this age group, the difference in PDE9A expression diminishes with advancing age and PDE9A is no longer a risk marker for HFpEF in elderly subjects. Notably, patients with HFpEF and lower expression of PDE9A in PBMNCs displayed increased mortality when compared with patients with higher PDE9A expression.
Recent interest has focused on pharmacological modulation of cardiac GMP/protein kinase G (PKG) signalling as a potential novel therapeutic approach in patients with HFpEF. cGMP in cardiomyocytes is produced by either NO‐mediated activation of soluble guanylate cyclase (sGC) or natriuretic peptide‐induced stimulation of receptor‐bound guanylate cyclase (i.e. GC‐A or GC‐B). 8 cGMP levels were profoundly reduced in EMBs from patients with HFpEF as compared with patients with HFrEF or aortic stenosis. 9 Blocking degradation of cGMP by PDE5 inhibition yielded conflicting results in clinical testing with regard to invasive haemodynamics and clinical parameters. 10 , 11 Overall results of the PDE5 Inhibition to Improve Clinical Status and Exercise Capacity in Heart Failure with Preserved Ejection Fraction (RELAX) trial failed to show a benefit for sildenafil in terms of exercise capacity or clinical status. 5
Several potential explanations have been put forward to explain the lack of benefit of sildenafil treatment in patients with HFpEF. One of them suggests that PDE5 primarily regulates NO‐generated cGMP, and therefore, PDE5 inhibition seems unlikely to yield benefit in patients with HF who are often characterized by a decrease in NO bioavailability. 8 Contrary to PDE5, PDE9A regulates natriuretic peptide rather than NO‐stimulated cGMP in cardiomyocytes 6 and, therefore, would be a more promising therapeutic target to raise cardiac cGMP levels in patients with HF. Lee et al. 6 suggested that genetic deletion as well as pharmacological inhibition of up‐regulated PDE9A improved LV dimensions and function in a pressure overload model in mice induced by transverse aortic constriction (TAC).
The present study provides a first systematic analysis of PDE9A levels in human HF. Our data imply that LV and right ventricular expression of PDE9A is not uniformly increased among HF phenotypes in humans but particularly elevated in patients with HFpEF. In contrast to observations by Lee et al. 6 and our group, a recent publication questioned the presence of PDE9A on a protein level in myocardium from patients with HFrEF sampled at the time of cardiac transplantation and cardiac muscle from a canine model of HFpEF. 12 However, the authors used lung samples from rats as a positive control, and hence, the specificity of this antibody for human samples needs further investigation. Notably, recent investigations also identified cardiac PDE9A expression in two rat HF models induced by abdominal aortic constriction or isoproterenol treatment, as well as in myocardium and thoracic aorta from rabbits fed with a high‐cholesterol diet. 13 , 14
Current data reinforce the concept that pathophysiology of HFpEF is fundamentally different to HFrEF and likely involves a multiple‐hit model. 15 Although primary unifying abnormalities of the disease still need to be identified, 15 a larger involvement of systemic inflammation in HFpEF than in HFrEF has been inferred from recent biomarker studies. 16 Besides increased markers of systemic inflammation, previous EMB findings also revealed signs of myocardial inflammation in patients with HFpEF as compared with patients with HFrEF or aortic stenosis. 17 Interestingly, PDE9A expression in the present study correlated with the amount of infiltrating CD68‐positive and CD3‐positive cells on EMBs in patients with HFpEF, but not in patients with HFrEF or iCMP. Also, PDE9A in PBMNCs was up‐regulated in response to TNF‐α and IL‐1β in PBMNCs from patients with HFpEF, whereas no such effect was observed in PBMNCs from other HF phenotypes, probably pointing towards an association with disease‐specific inflammatory pathways in HFpEF. Of note, in line with a recent study testing the effect of pharmacological PDE9A inhibition in a murine diastolic dysfunction model induced by TAC and deoxycorticosterone acetate administration, 18 PDE9A did not correlate with the percentage of myocardial fibrosis in the present study. These findings contrast with animal data from Lee et al. suggesting a decrease in interstitial myocardial fibrosis in PDE9A‐knockout mice after TAC. 6 Apart from the fact that the present data were obtained in humans, TAC represents an animal model of merely pressure overload‐induced LV hypertrophy and HF. Following an initial phase with concentric LV hypertrophy, diastolic dysfunction, and lung congestion, this model progresses to a ‘HFrEF‐like’ phenotype with LV chamber dilation and a decrease in systolic function later on. 19 Importantly, the TAC model lacks metabolic alterations, which are typically observed in patients with HFpEF and likely contribute to the complex pathophysiology of the disease. Furthermore, LV PDE9A expression was more pronounced in HFpEF than HFrEF and did not correlate with end‐diastolic wall stress in patients with HFpEF, supporting the concept that PDE9A is linked to inflammation rather than pure haemodynamic alterations.
Expression of PDE9A was increased in both EMBs and PBMNCs from patients with HFpEF. Previously, elevated PDE9A levels have been identified in circulating haematopoietic cells in patients with sickle cell disease, mainly in neutrophils. 20 Pharmacological inhibition of PDE9A reduced neutrophil surface expression of adhesion molecules, neutrophil adhesion to fibronectin in vitro, and leucocyte recruitment following TNF‐α‐induced acute vaso‐occlusion in a murine sickle cell disease model, pointing towards a pro‐inflammatory role of PDE9A in sickle cell disease. 21 The present findings suggest that PDE9A expression in isolated PBMNCs is subject to pro‐inflammatory stimulation and increased in patients with HFpEF as compared with other HF phenotypes or control subjects, particularly in those HFpEF patients with more pronounced systemic inflammation. However, considering the results of the present gene expression analysis in a larger cohort of HFpEF patients identified in the LIFE‐Heart cohort, the role of PDE9A expression in these cells in HFpEF is likely much more complex. First, PDE9A expression is highly age dependent and decreases in patients with HFpEF as well as subjects without HF with increasing age. Second, the age‐dependent decrease in PDE9A expression levels in PBMNCs is more profound in patients with HFpEF than in subjects without HF; that is, older patients with HFpEF have lower PDE9A expression in PBMNCs than patients without HF.
Our study was not designed to provide a detailed understanding of pathophysiological consequences of PDE9A expression in PBMNCs in HFpEF. Therefore, any conclusions on functional consequences of differential PDE9A expression in this analysis remain speculative. The expression pattern and prognostic implications of PDE9A observed in the present analysis rather argue for a compensatory mechanisms impacting on PDE9A levels; for example, PDE9A may be down‐regulated with increasing age as an adaptive mechanism associated with a decline in cGMP levels in the ageing patient with HFpEF. Such being the case, pharmacological inhibition of PDE9A would be most effective in younger patients with early stages of HFpEF. Yet aforementioned work in a diastolic dysfunction model in mice indicated that PDE9A inhibition may be a double‐edged sword. 18 Parallel to a reduction in ventricular diastolic chamber stiffness, chronic PDE9A inhibition led to impairment in LV contractility and ventricular–arterial coordination in these animals. 18 Hence, one may speculate that beneficial early effects of lower PDE9A levels on diastolic chamber stiffness are later offset by impairment in systolic LV function, for example, by mechanisms potentially mediated by negative inotropic effects of increased protein kinase G activity 8 , 22 or attenuation of cyclic adenosine monophosphate‐mediated signalling. 23 These effects may ultimately determine the prognostic impact of PDE9A.
Together, these data on PDE9A call for future studies to further examine potential long‐term consequences of modifying cGMP levels and PKG activity in patients with HFpEF. In this respect, recent experimental data in a murine HFpEF model induced by concomitant metabolic and hypertensive stress suggested an increase in inducible NO synthase activity and subsequent nitrosative stress, pointing towards excess NO production as a pathophysiological driver in HFpEF. 24 This is of particular importance given the lack of a clear short‐term benefit in recent trials testing pharmacological agents targeted at the NO/sGC/cGMP/PKG axis, such as inorganic nitrite 25 or the sGC stimulator vericiguat, 26 and the fact that the angiotensin receptor–neprilysin inhibitor sacubitril–valsartan failed to reduce HF hospitalizations and death from cardiovascular causes in PARAGON‐HF. 27
Limitations
A number of limitations need to be considered when interpreting the data of the present study. First, the present analysis does not provide mechanistic insights into the role of PDE9A in HFpEF, particularly for the observed association between PDE9A expression and markers of myocardial or systemic inflammation. Further research is mandatory to investigate whether this association is indeed of relevance for the pathogenesis of HFpEF. Second, the present results on PDE9A expression rely on mRNA detection and there is no confirmation on a protein or enzymatic activity level. This is primarily because EMB material was limited in the present cohort and precluded us from running western blot or activity assays. Of note, several attempts to reliably detect PDE9A on western blot analysis in a subgroup of patients with enough biopsy material failed owing to the limited specificity of commercially available antibodies. Third, as patients with HFpEF in the EMB were older, further EMB studies in larger cohorts of patients with different HF phenotypes are clearly required to definitely rule out an age‐dependent or disease stage‐specific bias in the comparison of endomyocardial PDE9A levels. Fourth, future studies are also needed to address the questions whether PDE9A expression in PBMNCs indeed serves as a readout for myocardial expression and whether findings obtained in PBMNCs can simply be transferred to the myocardium. Finally, patients in the EMB cohort underwent sampling for unexplained LV hypertrophy to rule out infiltrative cardiomyopathies and storage diseases. As such, we cannot rule out a potential selection bias in this cohort, and these patients likely do not represent the entire spectrum of HFpEF phenotypes. The same holds true for absolute differences in the distribution of HF medications among patients in the EMB cohort, although such differences did not reach statistical significance likely due to the limited number of patients.
Conclusion
The present data suggest an increase in endomyocardial PDE9A expression in patients with different HF phenotypes. When compared with patients with HFrEF and iCMP, the most robust increase in PDE9A expression is observed in EMBs and PBMNCs from patients with HFpEF. Specifically in these patients, PDE9A expression correlates with markers of myocardial and systemic inflammation. Validation of these findings in a large observational cohort implies that PDE9A expression in PBMNCs is age dependent with increased levels in middle‐aged subjects being associated with HFpEF risk, a finding that is no more evident in elderly patients. Even after age adjustment, lower PDE9A levels remain associated with higher mortality. While this translational study indicates a prognostic role of PDE9A in HFpEF and points towards an association with age and disease‐specific inflammation, more mechanistic and clinical data are needed to better define PDE9A as a potential treatment target.
Conflict of interest
None declared.
Funding
This study was funded by a research grant of the Heart Center Leipzig at University of Leipzig.
Supporting information
Table S1. Logistic regression analysis for the association of PDE9A transcript levels with HFpEF risk in symptomatic HFpEF patients (NYHA class II to IV).
Figure S1. Association between LV PDE9A expression and myocardial inflammation in patients with (A) HFrEF and (B) iCMP. PDE9A expression in LV EMB was quantified by RT‐PCR and the inflammatory cell count was calculated as the number of CD3‐ and CD68‐positive cells on immunohistological analysis of EMB.
Figure S2. Association between PDE9A expression in EMB and PBMNC. PDE9A expression in LV EMB and PBMNC was quantified by RT‐PCR.
Figure S3. Effect of TNF‐α and IL‐1β on PDE9A expression in PBMNC isolated from healthy subjects (n = 4) and patients with HFpEF (n = 4) or iCMP (n = 4) after incubation for 12 hours. PDE9A expression in LV EMB and PBMNC was quantified by RT‐PCR.
Figure S4. PDE9A expression in PBMNC and mortality in symptomatic patients with HFpEF (NYHA class II to IV). Kaplan–Meier curve of the probability of survival in patients with symptomatic HFpEF stratified to tertiles of PDE9A expression in PBMNC. A difference in mortality was observed between patients with PDE9A levels in the lower tertile as compared to patients with PDE9A levels in the middle or upper tertile irrespective of age.
Acknowledgements
The authors thank Frank Beutner, André Teren, and Martin Petzold for their excellent technical support in the study organization. Open access funding enabled and organized by Projekt DEAL.
Open access funding enabled and organized by Projekt DEAL.
Besler, C. , Rommel, K.‐P. , Kresoja, K.‐P. , Mörbitz, J. , Kirsten, H. , Scholz, M. , Klingel, K. , Thiery, J. , Burkhardt, R. , Büttner, P. , Adams, V. , Thiele, H. , and Lurz, P. (2021) Evaluation of phosphodiesterase 9A as a novel biomarker in heart failure with preserved ejection fraction. ESC Heart Failure, 8: 1861–1872. 10.1002/ehf2.13327
References
- 1. Parikh KS, Sharma K, Fiuzat M, Surks HK, George JT, Honarpour N, Depre C, Desvigne‐Nickens P, Nkulikiyinka R, Lewis GD, Gomberg‐Maitland M, O'Connor CM, Stockbridge N, Califf RM, Konstam MA, Januzzi JL Jr, Solomon SD, Borlaug BA, Shah SJ, Redfield MM, Felker GM. Heart failure with preserved ejection fraction expert panel report: current controversies and implications for clinical trials. JACC Heart Fail 2018; 6: 619–632. [DOI] [PubMed] [Google Scholar]
- 2. Martin N, Manoharan K, Thomas J, Davies C, Lumbers RT. Beta‐blockers and inhibitors of the renin‐angiotensin aldosterone system for chronic heart failure with preserved ejection fraction. Cochrane Database Syst Rev 2018; 6: CD012721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lam CSP, Voors AA, de Boer RA, Solomon SD, van Veldhuisen DJ. Heart failure with preserved ejection fraction: from mechanisms to therapies. Eur Heart J 2018; 39: 2780–2792. [DOI] [PubMed] [Google Scholar]
- 4. Kukreja RC, Salloum FN, Das A. Cyclic guanosine monophosphate signaling and phosphodiesterase‐5 inhibitors in cardioprotection. J Am Coll Cardiol 2012; 59: 1921–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O'Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 2013; 309: 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA. Phosphodiesterase 9A controls nitric‐oxide‐independent cGMP and hypertrophic heart disease. Nature 2015; 519: 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37: 2129–2200. [DOI] [PubMed] [Google Scholar]
- 8. Kim GE, Kass DA. Cardiac phosphodiesterases and their modulation for treating heart disease. Handb Exp Pharmacol 2017; 243: 249–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Heerebeek L, Hamdani N, Falcão‐Pires I, Leite‐Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012; 126: 830–839. [DOI] [PubMed] [Google Scholar]
- 10. Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase‐5 inhibition in a 1‐year study. Circulation 2011; 124: 164–174. [DOI] [PubMed] [Google Scholar]
- 11. Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, van Veldhuisen DJ, Voors AA. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J 2015; 36: 2565–2573. [DOI] [PubMed] [Google Scholar]
- 12. Li EA, Xi W, Han YS, Brozovich FV. Phosphodiesterase expression in the normal and failing heart. Arch Biochem Biophys 2018; 662: 160–168. [DOI] [PubMed] [Google Scholar]
- 13. Wang PX, Li ZM, Cai SD, Li JY, He P, Huang Y, Feng GS, Luo HB, Chen SR, Liu PQ. C33(S), a novel PDE9A inhibitor, protects against rat cardiac hypertrophy through upregulating cGMP signaling. Acta Pharmacol Sin 2017; 38: 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Priksz D, Bombicz M, Varga B, Kurucz A, Gesztelyi R, Balla J, Toth A, Papp Z, Szilvassy Z, Juhasz B. Upregulation of myocardial and vascular phosphodiesterase 9A in a model of atherosclerotic cardiovascular disease. Int J Mol Sci 2018; 19: pii: E2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Borlaug BA. Defining HFpEF: where do we draw the line? Eur Heart J 2016; 37: 463–465. [DOI] [PubMed] [Google Scholar]
- 16. Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, Metra M, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, Lang CC, Ng LL, Zannad F, Zwinderman AH, Hillege HL, van der Meer P, Voors AA. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol 2018; 72: 1081–1090. [DOI] [PubMed] [Google Scholar]
- 17. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, Leite‐Moreira AF, Musters R, Niessen HW, Linke WA, Paulus WJ, Hamdani N. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail 2016; 4: 312–324. [DOI] [PubMed] [Google Scholar]
- 18. Methawasin M, Strom J, Borkowski T, Hourani Z, Runyan R, Smith JE 3rd, Granzier H. Phosphodiesterase 9a inhibition in mouse models of diastolic dysfunction. Circ Heart Fail 2020; 13: e006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Valero‐Muñoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a “fishing expedition”. JACC Basic Transl Sci 2017; 2: 770–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Almeida CB, Traina F, Lanaro C, Canalli AA, Saad ST, Costa FF, Conran N. High expression of the cGMP‐specific phosphodiesterase, PDE9A, in sickle cell disease (SCD) and the effects of its inhibition in erythroid cells and SCD neutrophils. Br J Haematol 2008; 142: 836–844. [DOI] [PubMed] [Google Scholar]
- 21. Almeida CB, Scheiermann C, Jang JE, Prophete C, Costa FF, Conran N, Frenette PS. Hydroxyurea and a cGMP‐amplifying agent have immediate benefits on acute vaso‐occlusive events in sickle cell disease mice. Blood 2012; 120: 2879–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Layland J, Li JM, Shah AM. Role of cyclic GMP‐dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol 2002; 540: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stangherlin A, Gesellchen F, Zoccarato A, Terrin A, Fields LA, Berrera M, Surdo NC, Craig MA, Smith G, Hamilton G, Zaccolo M. cGMP signals modulate cAMP levels in a compartment‐specific manner to regulate catecholamine‐dependent signaling in cardiac myocytes. Circ Res 2011; 108: 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, Hill TM, Mammen PPA, Huang J, Lee DI, Hahn VS, Sharma K, Kass DA, Lavandero S, Gillette TG, Hill JA. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019; 568: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borlaug BA, Anstrom KJ, Lewis GD, Shah SJ, Levine JA, Koepp GA, Givertz MM, Felker GM, LeWinter MM, Mann DL, Margulies KB, Smith AL, Tang WHW, Whellan DJ, Chen HH, Davila‐Roman VG, McNulty S, Desvigne‐Nickens P, Hernandez AF, Braunwald E, Redfield MM. Effect of inorganic nitrite vs placebo on exercise capacity among patients with heart failure with preserved ejection fraction: the INDIE‐HFpEF randomized clinical trial. JAMA 2018; 320: 1764–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pieske B, Maggioni AP, Lam CSP, Pieske‐Kraigher E, Filippatos G, Butler J, Ponikowski P, Shah SJ, Solomon SD, Scalise AV, Mueller K, Roessig L, Gheorghiade M. Vericiguat in patients with worsening chronic heart failure and preserved ejection fraction: results of the SOluble guanylate Cyclase stimulatoR in heArT failurE patientS with PRESERVED EF (SOCRATES‐PRESERVED) study. Eur Heart J 2017; 38: 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, Martinez F, Packer M, Pfeffer MA, Pieske B, Redfield MM, Rouleau JL, van Veldhuisen DJ, Zannad F, Zile MR, Desai AS, Claggett B, Jhund PS, Boytsov SA, Comin‐Colet J, Cleland J, Düngen HD, Goncalvesova E, Katova T, Kerr Saraiva JF, Lelonek M, Merkely B, Senni M, Shah SJ, Zhou J, Rizkala AR, Gong J, Shi VC, Lefkowitz MP, PARAGON‐HF Investigators and Committees . Angiotensin–neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med 2019; 381: 1609–1620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Logistic regression analysis for the association of PDE9A transcript levels with HFpEF risk in symptomatic HFpEF patients (NYHA class II to IV).
Figure S1. Association between LV PDE9A expression and myocardial inflammation in patients with (A) HFrEF and (B) iCMP. PDE9A expression in LV EMB was quantified by RT‐PCR and the inflammatory cell count was calculated as the number of CD3‐ and CD68‐positive cells on immunohistological analysis of EMB.
Figure S2. Association between PDE9A expression in EMB and PBMNC. PDE9A expression in LV EMB and PBMNC was quantified by RT‐PCR.
Figure S3. Effect of TNF‐α and IL‐1β on PDE9A expression in PBMNC isolated from healthy subjects (n = 4) and patients with HFpEF (n = 4) or iCMP (n = 4) after incubation for 12 hours. PDE9A expression in LV EMB and PBMNC was quantified by RT‐PCR.
Figure S4. PDE9A expression in PBMNC and mortality in symptomatic patients with HFpEF (NYHA class II to IV). Kaplan–Meier curve of the probability of survival in patients with symptomatic HFpEF stratified to tertiles of PDE9A expression in PBMNC. A difference in mortality was observed between patients with PDE9A levels in the lower tertile as compared to patients with PDE9A levels in the middle or upper tertile irrespective of age.