

Abstract
Most of the fascinating phenomena studied in cell biology emerge from interactions among highly organized multimolecular structures embedded into complex and frequently dynamic cellular morphologies. For the exploration of such systems, computer simulation has proved to be an invaluable tool, and many researchers in this field have developed sophisticated computational models for application to specific cell biological questions. However, it is often difficult to reconcile conflicting computational results that use different approaches to describe the same phenomenon. To address this issue systematically, we have defined a series of computational test cases ranging from very simple to moderately complex, varying key features of dimensionality, reaction type, reaction speed, crowding, and cell size. We then quantified how explicit spatial and/or stochastic implementations alter outcomes, even when all methods use the same reaction network, rates, and concentrations. For simple cases, we generally find minor differences in solutions of the same problem. However, we observe increasing discordance as the effects of localization, dimensionality reduction, and irreversible enzymatic reactions are combined. We discuss the strengths and limitations of commonly used computational approaches for exploring cell biological questions and provide a framework for decision making by researchers developing new models. As computational power and speed continue to increase at a remarkable rate, the dream of a fully comprehensive computational model of a living cell may be drawing closer to reality, but our analysis demonstrates that it will be crucial to evaluate the accuracy of such models critically and systematically.
INTRODUCTION
Twenty-first century cell biology has been transformed by rapid development of new technologies that have delivered our field into an era where scientists can now easily generate vast amounts of quantitative data, providing a broad and comprehensive view of problems that were previously accessible only by brief, laboriously achieved glances through tiny chinks. Northern blots have been replaced by RNA sequencing, Western blots are being replaced by proteomics, and the central cell biological tool of imaging has been revolutionized by a wealth of dramatic improvements in labeling methods, optical design, and digital imaging. While currently available data are just a tiny fraction of the amount that will eventually be needed to model entire cells, sufficient data are available for a number of cell biological processes to ask whether and how we can use it to understand the biological mechanisms underlying those processes.
Mathematical modeling can help (Cohen, 2004; Gunawardena, 2014). By generating a model of a process that we want to study, where we are able to define and control all the inputs and parameters, we can directly determine whether a mechanism we have hypothesized is actually sufficient to explain the phenomenon we have observed. While mathematical modeling of cell biological processes can take many forms, here we will focus exclusively on computational simulation. One of our goals is to discuss the current state of the art in computational cell biology and remaining open challenges, particularly regarding the spatial organization of signaling processes, the inclusion of stochastic effects, and the multiscale nature of many cell biological processes.
Computers are much better than humans at keeping track of large, complex systems with many interacting parts and also at mercilessly following predetermined rules. One subfield of cell biology that has been able to make great use of these abilities is the study of signal transduction, where typically many different molecular species interact with one another in branching and reticular networks. Biochemical and genetic experiments have been able to map out and characterize many individual pairwise interactions in multicomponent signaling pathways. However, the resulting “wiring diagrams” have provided little direct insight into how these networks of molecular interactions could give rise to the diverse and fascinating outputs of these systems, which might be able to generate oscillations, high-pass or low-pass filtering of receptor-mediated input signals, conversion of analog signals into switch-like binary outputs, and so forth. Computational simulations have been instrumental in bringing order and insight into this tangled web so that now it is possible to recognize recurrent motifs in the design of signal transduction systems and, sometimes, accurately predict cellular responses to external stimuli (Eungdamrong and Iyengar, 2004; Kestler et al., 2008; Janes and Lauffenburger, 2013; Cao et al., 2016).
Within this context, a very fertile ecosystem of computational tools for modeling cellular biochemistry has flourished (Bartocci and Lio, 2016).There are now also approaches that enable scientists who are not computational experts themselves to translate their hypotheses about cellular signaling mechanisms into formal models in compact and intuitive ways; these include using textual rules (Faeder, 2011; Maus et al., 2011; Tiger et al., 2012; Harris et al., 2016; Boutillier et al., 2018) or iconographic symbols (Zhang et al., 2013; Schaff et al., 2016; Sekar et al., 2017) to specify molecular interactions. Based on those specifications, the tools generate the resulting computational representations of the signaling networks and allow modelers to easily modify their assumptions to explore the consequences of such simulated manipulations on cellular behavior (Lopez et al., 2013). To allow for tool-independent formulation and sharing of computational models, the widely used “systems biology markup language” SBML was developed. (Hucka et al., 2003) SBML is continuously evolving and is supported by a large number of software systems for simulation and data analysis (Keating et al., 2020; SBML.org). While there are many variations, generally simulations of this kind are able to keep track of concentrations and interactions of many individual molecular species as they change over time, often the most interesting dimension for the study of signal transduction.
However, many cell biological processes, including some kinds of signaling, cannot be analyzed without the notion of space. Cell–cell communication is frequently based on the exchange of soluble messenger molecules, such as hormones, cytokines or chemokines, that diffuse through extracellular space before being captured by specific receptors at particular locations on cellular membranes. Direct cell–cell contacts also typically involve only a few of the receptors on a cell and generate localized signals that activate cascades of protein interactions and modifications to propagate from the membrane into the cytoplasm. Spatial simulation of cell biological phenomena is not new; in 1952, Hodgkin and Huxley simulated the propagation of an action potential down a neuronal axon (Hodgkin and Huxley, 1952; Hellander et al., 2015), and in the same year, Alan Turing used computational simulation to demonstrate how chemical systems featuring both diffusion and reaction could generate regular spatial patterns from an initial uniform state (Turing, 1952). More recently, many researchers have developed computational simulations that employ state-of-the-art knowledge about the properties and interactions of individual molecular components to attempt spatially resolved reproduction of complex cell biological phenomena. Reaction–diffusion models of intracellular biochemistry have been used to explore a variety of cellular symmetry-breaking processes, including the establishment of cell polarity (Jilkine and Edelstein-Keshet, 2011). Although the exact mechanistic details vary, spatial simulation approaches have yielded insights for symmetry-breaking systems as diverse as yeast bud site selection (Wedlich-Soldner et al., 2003), asymmetric cell division in early Caenorhabditis elegans embryos (Dawes and Munro, 2011) and neutrophil chemotaxis (Onsum and Rao, 2007). Establishment of spatial gradients that determine cell fate has been explored in cells ranging from giant syncytial Drosophila embryos (Gregor et al., 2007) to tiny individual bacteria (Chen et al., 2011). Simulations that explicitly consider spatial effects as one cell communicates with its neighbors have been used to understand the formation of regular stripes in Drosophila embryos (von Dassow et al., 2000) and bizarre noncell-autonomous effects in the patterning of wing bristles in adult flies (Amonlirdviman et al., 2005).
In many of the modeling efforts that we have mentioned so far, the computational model and/or simulation was formulated explicitly for the problem at hand. While this approach has enabled important scientific insights, we believe that spatial simulation for cell biological processes can become a much more widely used tool in the cell biologist’s toolbox if there was more general access to user-friendly implementations of general spatial modeling frameworks that do not require extensive computational expertise. Consider, for example, the wide variety of user-friendly open-source software packages now available for analysis of sequencing data (Rice et al., 2000; Trapnell et al., 2012). One particularly important benefit of more standardized approaches to spatial simulations in cell biology is that standardization may help to resolve whether conflicting conclusions arise because of fundamental scientific differences in model assumptions or because of details of numerical implementation.
Another aspect gaining importance as we zoom in closer on the building blocks of cellular structures is the fact that, at the molecular level, cellular biochemistry is governed by stochastic processes such as thermal Brownian motion and the collisions and interactions among individual particles (Schnoerr et al., 2017). Only in the limit of high concentrations and homogenous spatial distributions can the behavior of the molecular components of cellular signaling pathways be described in terms of deterministic reaction rate equations. Many subcellular mechanisms operate far from this limit either due to highly nonhomogenous clustering of receptors and of the signaling components they recruit, such as studied in the MAPK signaling pathway (Takahashi et al., 2010), or due to locally low copy numbers, for instance of multimolecular complexes regulating transcription in the nucleus (Cho et al., 2018). To accurately capture the stochastic characteristics of such processes, computational models have to simulate the motion and interactions of individual molecules. A full simulation of Brownian dynamics (BD), following each single “Brownian hop” of all molecules of a cellular region would, however, in most cases be too computationally expensive and time consuming. Moreover, by choosing a particular time step for recreating Brownian hops on the computer, we would impose this timescale on our simulations, missing events, such as molecular encounters, that may occur ‘in between’ our time steps. Many approaches have been developed to deal with this problem and we will discuss several of them below. A common theme among all of them is, however, saving computational cost through temporal and spatial coarse graining without sacrificing too much accuracy in the simulation results. This challenge unites researchers looking at spatially resolved modeling (without a focus on stochastic effects) and those who try to capture the manifestations of stochastic fluctuations in cellular systems.
The question of how the field could best go about building and sharing broadly applicable computational tools for spatial and stochastic modeling of cell biological processes was the focus of our working group, organized by J.R.F. and R.F.M., which met with the support of the National Institute for Mathematical and Biological Synthesis (NIMBioS). We chose to focus specifically on the biochemical scale of molecular interactions. We did not attempt to include the enormous field of molecular dynamics (MD) simulations that explores forces and movements of individual atoms within proteins or other macromolecules. The reason is that, because of their intensive computational demands, MD simulations are currently limited to exploration of very small biological systems (a few macromolecules) over very short periods of time (typically in the microsecond range or below), too small and too fast to be incorporated into cell-scale computational simulations. Conversely, we also limited ourselves to considering simulations of interactions within systems of molecular complexes with specific stoichiometry rather than extending our analysis to mesoscopic-scale models that abstract the behaviors of complex molecular systems into continuum physical descriptions, such as those describing cytoskeletal filaments as elastic beams (Nedelec and Foethke, 2007; Odell and Foe, 2008) or the plasma membrane as a flexible thin film (Fowler et al., 2016). While these kinds of models are enormously useful in cell biology, they rely on fundamental simplifying assumptions. In contrast, we are specifically interested in exploring whether detailed simulations of the behaviors and interactions among biomolecular complexes can succeed in predicting certain kinds of mesoscopic phenomena and hence may help determining under which conditions the simplifying assumptions are justified. As we will discuss in our conclusion, it will be an exciting future direction for the field of biological simulation when all these three levels of spatially resolved simulations can be seamlessly interconnected.
In this article, we will first briefly survey several existing approaches for spatial and stochastic cell simulations and then apply them to a series of “unit tests” and benchmark problems. The problems we chose are categorized to cover a variety of different aspects of spatially resolved and/or stochastic simulations of cellular behavior. Conceptionally, they are relatively simple and are meant to capture specific challenges related to essential aspects of biological processes. They must be accurately modeled by a simulation tool to ensure that that tool’s results are reproducible for the problem category covered by the unit tests or benchmark problems. With these examples in hand, we then summarize how features of a particular cell biological problem should guide selection of the appropriate modeling approach. One should note that, in practice, finding or developing the appropriate approach for a given cell biological phenomenon involves far more decisions than “just” whether the model should capture spatial or stochastic aspects (or both). A similarly important and related challenge is to find the right balance between biological realism and detail on the one hand and computational manageability and parameter estimation on the other hand. However, a systematic exploration of methods for identifying the right degree of model granularity would go beyond the scope of an article exploring the roles of space and stochasticity. Here, we will, therefore, limit our discussion of topics such as model reduction and parameter estimation to pointing out where we think these aspects become particularly important. Finally, we will present the results of our wide-ranging and, sometimes, highly opinionated discussions on future directions and challenges for the field. We all share an ambitious vision of the future power of spatial cell simulations both for exploring hypotheses about mechanisms and for coming to grips with the massive amounts of quantitative data now available to cell biologists. Our overall goal here is to map out where the field currently stands and propose a trajectory for the future.
MODELING APPROACHES
Overview of approaches for spatial and stochastic modeling and simulation of molecular reactions underlying cell biological phenomena
Computational models that simulate the biochemistry underlying cell biological processes need to be able to describe molecular players and their reactions. However, depending on the particular question at hand, taking into account spatial aspects and stochastic effects (Figure 1) may or may not be essential, as we show in Results. The addition of spatial resolution is computationally demanding, and stochastic simulations are usually more costly than their deterministic counterparts. To attempt a whole-cell simulation, for example, one must choose whether more components and a more complex reaction network are necessary, generally requiring the sacrifice of spatial resolution (Tomita et al., 1999; Sanghvi et al., 2013), or if spatial resolution is necessary, then the reaction network must be simplified (Ghaemi et al., 2020). If spatial resolution is a priority, must species be resolved as individual particles, capturing fluctuations in copy numbers but at considerable extra expense (Supplemental Table S3), or is an efficient deterministic approach sufficient?
FIGURE 1:

Overview of nonspatial and spatial simulation approaches for describing the time dependence of interacting and reacting species. Different colors represent different species. Spatial models are illustrated at one point in time. Left: deterministic approaches to modeling biological systems solve (a) ordinary or (c) partial differential equations (ODE/PDE) using standard numerical methods, benefiting from extensive method development across science, math, and engineering fields. For example, PDEs can be numerically solved on a mesh as shown. Right: stochastic simulation approaches sample from a time- (and space-) dependent probability distribution that typically models unimolecular and bimolecular reactions, as well as diffusion for spatial methods. (d) Reaction–diffusion Master Equation (RDME) methods are the extension of the (b) chemical master equation (CME) methods on to a spatial lattice, where integer copy numbers of species are tracked and can diffuse between lattice subvolumes. (e) Single-particle methods propagate individual particles undergoing diffusion in continuous space, where bimolecular reactions can occur only on collision or colocalization in space. We provide a guide to corresponding software tools of each approach in Supplemental Table S1.
Whether to include spatial or stochastic resolution will also affect the kind of data and (a priori known) parameters that are required and the results that can be expected. Differential equation-based models can incorporate phenomenological elements such as Hill-type functions bridging different model elements whose interdependencies are either not well understood or whose details are considered less important for the overall quality of a modeling effort. Moreover, more abstract models sometimes permit identifying components (species or mechanisms) whose kinetics contribute little to the behavior of a model or that can be lumped with other species to simplify its computational representation (Rao et al., 2014). This can be particularly useful for very large systems with well-defined constraints, such as metabolic network models (Masid et al., 2020). With decreasing complexity of a model, it also becomes easier to perform robust parameter estimations and to determine how well the model is justified based on the available data (Raue et al., 2009). However, model abstractions are also at risk of losing the ability to describe the behavior of biological entities (for instance molecular signaling components) considered relevant by experimental biologists and, ultimately, the choice between model simplicity and biological realism can be difficult (Meier-Schellersheim et al., 2019).
Nonspatial modeling approaches
In many situations, we can describe a biochemical system adequately in terms of the overall concentrations of interacting molecule types and complexes (collectively called “species”), while neglecting the spatial variations in these concentrations. Reaction rate equations (see Box 1) describe how the species concentrations evolve in time. The terms in these equations arise from the rates of the reactions that can occur in the system, which are often described by the Law of Mass Action. The rate of reaction between two interacting species can be given by the product of their concentrations and a rate constant, for example, kon for the ligand-receptor binding described in Box 1. The bimolecular rate constants that appear in these equations are sometimes referred to as “macroscopic” rate constants because they describe the average rate of reaction assuming homogenous distribution of the reacting species. In contrast, “microscopic” rate constants, which govern reaction kinetics at the scale of interacting particles, may take into account more details about the way the molecules approach each other, as discussed below.
BOX 1: reaction rate equations
Technically speaking, reaction rate equations are ordinary differential equations (ODEs). Here, the “ordinary” refers to the fact that they involve only time (as opposed to, for instance, time and space). To describe the time evolution of multiple interacting molecule types, one uses coupled differential equations that express how the components’ concentration changes are linked (or coupled). For applications, see, for example, Aldridge et al. (2006) and Tyson et al. (2003). From a numerical/mathematical point of view, ODEs describing biochemical reactions are typically simple, and many tools exist that can solve them to obtain the temporal evolution of the concentrations in ODE models.
Consider a simple model of a receptor binding to a ligand. We call R the concentration of the receptor, L that of the ligand, and RL that of the complex formed by the binding of the two. The rate equations giving the time derivatives of RL, R and L for this reaction could be written as
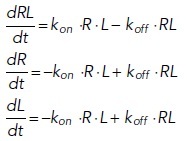 |
Here, kon and koff are the association and dissociation constants, respectively. The time course of RL would look similar to the red curve in Figure 1a, whereas time courses of R and L would be similar to the blue curve. These equations can be solved analytically, but the additional complexity of most biologically relevant models generates equations that require numerical solution by computer.
The chemical master equation (CME; McQuarrie, 1967; Gillespie, 1992; Ge and Qian, 2013) considers the discrete and stochastic nature of the biochemical system, which can cause differences from deterministic rate equations (Samoilov and Arkin, 2006). The CME describes how the probability of the system being in a specific state evolves over time by using reaction probabilities (likelihood of occurrence per unit time) rather than the equivalent reaction rates. Just like reaction rate equations, the CME assumes well-stirred (homogeneous) systems. In most practical modeling applications, the CME cannot be solved analytically (i.e., with a closed-form expression), but simulations of the CME are conceptually straightforward and widely used (see Box 2).
Both the reaction rate approach and the CME approach simply cannot capture effects of inhomogeneous distributions of molecules in space, such as receptors clustered on membranes, or intrinsic time delays due to diffusion to localized targets such as membrane-bound receptors. For more realistic simulations of cellular process, we must turn to different computational approaches that explicitly include space.
Spatial modeling approaches
The molecular components of living systems are not distributed homogeneously and the high spatial resolution of today’s fluorescence microscopy is continuously giving us more examples of biological phenomena where the spatial arrangement of the underlying biochemical processes is fundamentally important. To model such phenomena, we have to switch from nonspatial to spatial simulations. However, this switch is frequently not easy due to a growth in the number of model and system features that must be specified (Figure 2a). The most important difference between nonspatial and spatial simulations is that the latter take into account the translocation of the interacting molecules. In the simplest case, this means that, in addition to reactions, the diffusion of molecular species in space must be simulated. Along with diffusion, the system geometry must be specified and what happens at the “walls.” This is significantly more challenging to implement when the spatial system containing the interacting molecules is not simply a square box with rigid walls. Realistic spatially resolved models often aim to capture aspects such as particular cell morphologies; examples include synaptic structures with narrow regions connected to larger cell bodies (Rangamani et al., 2016; Cugno et al., 2019) or geometries that are flat and almost two-dimensional, like lamellipodial extensions in migrating eukaryotic cells (Nickaeen et al., 2017) (Figure 2b). Geometries for cell simulations can be designed by hand, derived from microscope images (Schaff et al., 2000), or generated from machine-learned cell models (Majarian et al., 2019). Currently, there are only a few simulation tools that can model cellular biochemistry within dynamic morphologies (Angermann et al., 2012; Tanaka et al., 2015) and the computational treatment of reaction–diffusion processes within domains that exhibit moving boundaries is still a very active field of research (Wolgemuth and Zajac, 2010; Novak and Slepchenko, 2014). We note that models allowing moving boundaries (which usually represent the membrane) do not necessarily capture the biophysics of membrane dynamics (Figure 2d). They can be decoupled methodologically, although fundamentally, they are not.
FIGURE 2:

Demands on spatial approaches and their extensibility. (a) For all spatial models, in addition to treating fundamental reaction types in all dimensions (3D, 2D,1D) and between all dimensions (for instance molecular exchange between bulk [3D] and surfaces [2D]), they must specify an equation of motion, typically diffusion, and boundary conditions on the system geometry. (b) More advanced treatments of curved and complex boundaries require additional care for treating reactions and diffusion. (c) Single-particle spatial methods can be classified based on the model they use and further whether they support large time steps. In both models, the ability to take large time steps generally requires reaction probabilities (preact) that are determined by finding analytical solutions for the fraction of diffusive trajectories that are reactive (not all collisions lead to reactions when the microscopic rate ka<∞). In the volume-reactive methods here, short-time step approximations are used, except MCell, which is described in the text. (d) Single-particle methods have the capacity to build in higher-resolution features, although these will alter their equation of motion, requiring new definitions of preact. We note that PDE-based models can also expand beyond purely diffusive dynamics. In Supplemental Table S2, we summarize features available in commonly used software tools.
Spatial models have the capacity to build in many additional features such as mechanics, electrostatics, or coarse molecular structure that may be particularly important for useful simulations of cell biological processes (Figure 2d). However, modeling efforts that take these additional features into account to render their simulations of cellular behavior more realistic are exceedingly rare. One reason for this is that they introduce further challenges to both the numerical implementation and the mathematical descriptions of the physical model. Another reason for shying away from this level of detail is that, frequently, it would be difficult or (currently) impossible to measure sufficient parameters to be able to estimate the remaining, unknown, parameters through computational “fitting.” Limiting themselves to smaller regions of cellular membranes, several studies have integrated (simple) biochemical processes with membrane biophysics to explore how actin dynamics drive membrane protrusions (Mogilner and Rubinstein, 2005; Atilgan et al., 2006). There is reason for hope that such efforts may, at some point, be scaled up to comprise larger membrane domains or even entire cells, given the ever increasing resolution and quantitative information contents in imaging-based measurements of the underlying molecular diffusion processes (Saha et al., 2016; Swaminathan et al., 2017).
Reaction–diffusion equations represent the most straightforward extension of reaction rate equations for the inclusion of spatial aspects. Instead of depending just on time as a variable, the behavior of molecular species now additionally depends on spatial coordinates. Equations describing these reactions must therefore be formulated as partial differential equations (PDEs) rather than as ODEs (Figure 1c). As is frequently the case for mathematical models of biological phenomena, only very simple situations can be described through equations that can be explicitly solved in such a way that the solution describes the behavior of the modeled system as a continuous function of space and time (Lipkow and Odde, 2008). In most cases, one has to explore reaction–diffusion equations through numerical simulations that divide space into subvolumes (frequently called ‘voxels’) and calculate how diffusion leads to exchange among the subvolumes. Reaction–diffusion equations have been widely used to model spatially resolved biomolecular dynamics and interactions of cell biological systems (Loew and Schaff, 2001). The spatial dynamics can be extended beyond pure diffusion (e.g., to include advection), and reactions can be defined phenomenologically (Hill-type or Michaelis–Menten). Like reaction rate equations, deterministic PDE reaction–diffusion equations do not capture stochastic fluctuations in species numbers and thus cannot, for example, capture pattern formation driven by a system’s sensitivity to low copy numbers (Howard and Rutenberg, 2003).
One way that stochastic reaction–diffusion equations can be formulated is the spatial extension of the CME, known as the reaction–diffusion master equation (RDME) (Figure 1d). In the RDME, instead of only defining how a system switches from one set of numbers of molecules in particular states to another (for instance when a complex in the system decays into two molecules), the RDME includes “hops” from one location to another. Importantly, just like the CME, the RDME describes discrete changes. That means, it requires a spatial discretization into subvolumes within each of which well-mixed conditions are assumed to prevail. Diffusion events of molecules are tracked only when they occur between adjacent subvolumes, not within an individual subvolume (Fange et al., 2010). Similar to the nonspatial case, it is usually not possible to solve the RDME analytically, and instead it is standard practice to compute solutions by simulating a particular stochastic time evolution of the system with, for instance, the SSA (see Box 2) that adds diffusional hops to the list of events that can occur, as is done with Lattice Microbes (Roberts et al., 2013), StochSS (Drawert et al., 2016), and STEPS (Wils and De Schutter, 2009; Chen and De Schutter, 2017). However, care must be taken to choose the right degree of spatial resolution that strikes the appropriate balance between capturing spatial details and avoiding subvolume sizes that are so small that discretization dilutes the molecules to a point where they essentially do not “see” their potential reaction partners anymore because the molecules are spread out over distinct subvolumes (see Box 3). A recent review (Smith and Grima, 2019) discusses the relationship between the RDME model and the particle-based models of reaction–diffusion.
BOX 2: the Chemical Master Equation (CME)
The CME is a set of coupled linear ODEs that describe the time dependence of the probability to occupy each of a set of discrete states, with each state defined by the copy numbers of molecular species. The CME may be written as
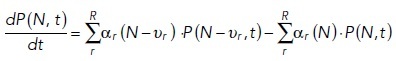 |
where the composition vector N is composed of copy numbers of each molecular species, and thus one has a set of equations, one for each possible instantiation of N. Here, the sum runs over all possible reactions R. The υr is the stoichiometric vector of reaction r that describes how this reaction changes the number of molecules in the composition vector N and αr (N) is the probability per unit time that reaction r occurs, given that the system is in the state described by N. The CME is important from a conceptual point of view as it represents a framework to describe probabilistic transitions and thus captures the stochasticity underlying all molecular interactions (Grima and Schnell, 2008; Schnoerr et al., 2017). The computational cost of solving the CME equations scales exponentially with the number of chemical species, and, although clever approaches have extended the size of systems for which the CME can be solved (Munsky and Khammash, 2006), high computational cost still limits biological applications. An intuitively simple way to calculate a solution of the CME would be to set up a simulation where time ticks forward in small, discrete intervals (time steps). However, the fixed time step in this integration scheme has finite error that is only eliminated in the limit Δt → 0, since reactions may occur even during shorter time steps than the one chosen to propagate the system in time.
One popular and precise method used to generate trajectories through the state space sampled by the CME without the need to choose a discrete time step is the stochastic simulation algorithm (SSA) (Gillespie, 1976; see Gillespie et al., 2013, for a detailed review). In an SSA simulation, the time interval until the next reaction occurs is itself sampled, as is the type of reaction that will occur (Gillespie, 1976). Molecular species that can react quickly and have many possible interaction partners will be selected frequently, while rarer molecules associated with slower reactions will be selected rarely. Since the simulations proceed with one reaction at a time, the computational cost depends strongly on the number of particles and reaction rates in the system. In contrast, the effort required to integrate (or solve) reaction rate equations depends mostly on how many molecule types are involved and whether their interactions occur on different or similar timescales. Various approaches have been developed to increase the efficiency of both exact (Gupta and Mendes, 2018) and approximate (Schnoerr et al., 2017) stochastic simulations of the CME. In addition, efficient methods have been developed to compute distributions and moments directly from the CME itself (Hasenauer et al., 2014; Hellander et al., 2017). A stochastic simulation of the example system from Box 1 would look similar to Figure 1b. Note, however, that such a trajectory represents only one possible time course compatible with the underlying CME. This means that many stochastic simulation trajectories must be collected to determine probability distributions and moments of the CME.
BOX 3: spatial discretization of reaction–diffusion equations
To numerically solve reaction–diffusion processes modeled as PDEs, it can be challenging to choose the appropriate spatial discretization of the modeled biological geometry. A discretization that is too coarse will suppress many spatial details and will represent a poor approximation of the underlying biology. However, keeping track of the contents of many very small voxels will not only be very expensive computationally but may also lead to situations where the assumptions of mass-action kinetics no longer strictly hold since the fraction of the molecules in the system that populate a single voxel becomes so small that the very concept of an average concentration becomes problematic. Furthermore, the accuracy and efficiency of PDE solvers is not just sensitive to the resolution of the spatial discretization (sometimes called lattice or mesh) but also to the discretization scheme as manifested, for instance, in the shape of the voxels. The practice of designing adaptive meshes, that is, combining voxels of different shape and size in one simulation to capture small-scale spatial details where needed while keeping the total number of voxels as low as possible, is a field of active research. The structure of the mesh also has to be adjusted to the numerical method chosen to solve the reaction–diffusion PDEs. Finite volume methods directly simulate diffusional exchange between voxels. In contrast, finite element algorithms optimize the coefficients of interpolation functions at the nodes of the mesh to achieve good approximations of concentration profile resulting from the combination of reactions and diffusion. See, for example, Richmond et al. (2005). We note that mesh-free approaches to solving PDEs provide an alternate to spatial discretization methods.
Similar to PDEs, the accuracy and cost of the RDME is sensitive to the spatial mesh; this problem is inherent to all spatially discretized simulations. Computational costs grow rapidly as the mesh resolution increases. Importantly, the accuracy of an RDME model does not always increase with a finer mesh. A very small mesh size violates the assumption that species are dilute and their own molecular volume is small relative to the voxel (Erban and Chapman, 2009; Isaacson, 2009; Wolf et al., 2010; Isaacson and Zhang, 2018). For specific nonfundamental reaction types, RDME has an additional limitation in that it does not always converge to the CME solutions in the limit of fast diffusion, as expected (Smith and Grima, 2016). Hence, the RDME may be viewed as a nonconvergent approximation of more microscopic spatially continuous models, such as the Smoluchowski model discussed below. We note that a variety of lattice methods have been designed to overcome the small voxel size issue (Chew et al., 2018) and to address convergence to a more microscopic model (Isaacson, 2013; Isaacson and Zhang, 2018).
Particle-based spatial simulation methods take into account the stochastic motion and interactions of individual molecules in continuous time and space and are thus capable of modeling biochemical processes that involve low copy numbers and strongly heterogeneous molecular spatial distributions (Figure 1e). These methods have the highest resolution (Figure 2c), but they come with a high computational cost. Importantly, simulations that treat each particle as an individual also offer the possibility of building in more detailed molecular features (Figure 2d). Typically, particle-based approaches resolve a bimolecular reaction A+B→C of a pair of molecules as two physically distinct stochastic processes. First, the molecules’ diffusive (Brownian) motion leads to their encounter. Then, the molecules either form a bond with a reaction probability determined by the reaction rate constant and their current separation or else they diffuse away from each other. Because the macroscopic kinetics of association must depend on both diffusion and reaction rate constants, this generally results in a distinction between a microscopic and macroscopic rate (see Box 4). Numerical approaches to single-particle reaction–diffusion calculations for biological systems can perhaps be best categorized into classes based on whether they model reactions to occur on collisions (von Smoluchowski, 1917; Collins and Kimball, 1949), or whether they model reactions to occur within a reactive volume (Doi, 1976; Erban and Chapman, 2009). Within both of these classes, different algorithms introduce approximations that affect accuracy in recovering the underlying physical model, flexibility, and connection to experimental (macroscopic) rates (Figure 2c).
BOX 4: microscopic versus macroscopic rates, and the sensitivity of strong binding to diffusion
For all nonspatial models, as well as PDE and RDME models, bimolecular association reactions are parameterized by the macroscopic rate constants, kon, corresponding to the rates one would measure from a binding experiment in bulk solution. This is because in all these spatial models, species that are localized in a small volume are assumed well mixed, thus obeying the same mass-action kinetics used in nonspatial models. In the single-particle methods, however, molecular interaction kinetics is split into two steps, as described in the text. This results in a purely diffusive contribution to the bimolecular encounter and then what is effectively an energetic contribution defined by a microscopic on-rate. In the Smoluchowski model, the encounter occurs on collision at a specific binding radius σ with microscopic rate ka. In 3D, this results in the long-known relationship:
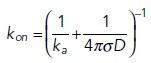 |
A |
where D is the sum of both species’ diffusion coefficients. With this relationship we can directly assess the impact of diffusion on controlling macroscopic kinetics. As shown in the image on the left below, for large macroscopic rates, the macroscopic kinetics of the A+B→∅ reaction is noticeably dependent on diffusion. For smaller rates, as shown in the Figure on the right, the effect of diffusion is negligible, despite DA = DB dropping from 100 to 1 µm2/s. Here, A0 = B0 = 1000 particles (here corresponding to 62 μM). Hence, large ka, or strong binding, is diffusion-limited, and small ka is rate-limited.

In two and one dimensions (e.g., on surfaces and filaments), the relation between microscopic and macroscopic parameters is more complicated, due to the properties of diffusion in lower dimensions smaller than three. No single relationship exists between microscopic and macroscopic rates (see, e.g., Yogurtcu and Johnson, 2015a), but meaningful theoretical relationships can be defined if the system size is considered (Szabo et al., 1980), where in 2D we further correct for system density using (Yogurtcu and Johnson, 2015a):
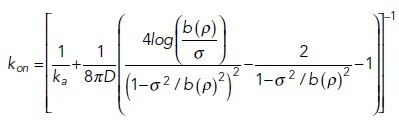 |
B |
Where 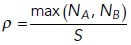 and
and 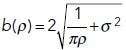
NA and NB are the copy numbers of reactants A,B and the system size is S.
For our test cases below, we thus always derive the microscopic rates ka to reproduce the macroscopic rates using Eq. A or Eq. B. This is because it is already clear from nonspatial models that changes to the macroscopic rates will necessarily alter the reaction kinetics. Since we are not focused on probing the influence of kinetic parameters on molecular behavior, but rather the role of explicit spatial representations in controlling species distributions and encounter times, we preserve all kon values. However, it is worth noting that for a reaction pair with a large ka value, if diffusion slows throughout the simulation due to, for instance, formation of large complexes, then the macroscopic kinetics will also slow down, an effect that is naturally captured in GF-based methods.
The definition of the reaction probability is the primary challenge and distinguishing feature of different single-particle algorithms. For the first class of collision-based numerical methods, the reaction probabilities are derived or matched to the Smoluchowski model of diffusion-influenced reactions, which naturally captures excluded volume (Figure 2c). We note that excluded volume is a critical feature for dense systems and for single-particle simulations of clustering or assembly where molecular structure/volume impacts interactions between species. For either class of models, the Green’s function (GF) approaches provide the most accurate solution by predicting the encounter probability for pairs of particles based on the particles’ initial positions. GF methods allow for much larger time steps, although they can only calculate the encounter probability for two particles at a time, meaning that the simulated system has to be segmented into two-particle subsystems (see Box 5). In practice, this turns out to be feasible for many interesting biological problems. Frameworks that have been developed to take advantage of this GF approach include FPR (Johnson and Hummer, 2014), NERDSS (Varga et al., 2020), GFRD (van Zon and ten Wolde, 2005), and eGFRD (Sokolowski et al., 2019). The SpatioCyte methods are performed on a lattice and recover the correct kinetics (beyond short times) and equilibrium of the Smoluchowski model (Chew et al., 2018, 2019). Smoldyn is also derived to use large time steps (albeit without excluded volume), and it is simpler to implement than GF approaches (Andrews and Bray, 2004a; Andrews et al., 2010; Andrews, 2017). However, the reaction parameters are coupled to the time step size, rather than representing independent model features (e.g., binding radii and microscopic rates), resulting in a time dependence (and 2D equilibrium) that is not as rigorously correct.
BOX 5. single-particle reaction–diffusion simulations
Single-particle methods simulate the stochastic behavior of individual and (in spite of the name) pairs of interacting molecules. Any biochemical network whose description does not include ad-hoc phenomenological processes (such as, for example, Hill coefficients describing nonlinear dose–response characteristics) can be described as composed of uni- and bimolecular reactions. As unimolecular reactions are only time-dependent, they are typically modeled as Poisson processes. For bimolecular reactions, the distance between a pair of particles influences the probability that they will diffuse to either collision or their reactive volume and react with one another in a time step. The time evolution of the molecules’ positions is described by a stochastic differential equation, the overdamped Langevin equation (Van Kampen, 2007). Its numerical implementation, known as BD (Ermak and Mccammon, 1978; Northrup et al., 1984), requires tiny time steps to accurately resolve molecular encounters, which renders the BD scheme highly inefficient. It is, however, possible to derive reaction probabilities for pairs of molecules that are nonetheless propagated using BD updates but use larger steps. GFRD is the only method that does not use BD updates for reactive pairs. To calculate the distance-dependent reaction probabilities for pairs of molecules, the most accurate approach is to use the GF defined below. This can enhance the efficiency of BD simulations by resolving bimolecular reactions within one large time step, Δt, without approximation.
The GF p(r, Δt|r0) can be obtained as the solution of the diffusion equation that describes a pair of molecules A, B that diffuse with diffusion constants DA,DB, respectively, and may undergo a reaction A + B→ as follows
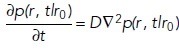 |
where D = DA +DB and r, r0 refer to the distance between A and B after and before the time step, respectively. In accordance with the two-step picture described in the main text, reactions are incorporated by imposing boundary conditions that specify the physics at or within an encounter distance r = σ. In the collision-based Smoluchowski model, r is always ≥ σ, and the Collins-Kimball Boundary Conditions (Collins and Kimball, 1949) in 3D is written as:
where ka refers to the intrinsic reaction constant. In the volume reactive, or Doi model, reactions occur whenever r ≤ σ, with intrinsic rate λ, creating a reactive sink between the two particles (Doi, 1976). It is worth noting that finding the appropriate GF to capture the desired properties of the molecular interactions can be a challenge. For the case of reversible diffusion-influenced bimolecular reactions of an isolated molecule pair in 2D, the GF was derived only in 2012 (Prüstel and Meier-Schellersheim, 2012).
For the second class of volume-reactive numerical methods, the reaction probabilities are derived based on a distance cutoff between particles (often called Doi model), which thus naturally lacks excluded volume (Figure 2c). There are no GF-based algorithms for this model. Erban and Chapman derived reaction probabilities and corresponding microscopic rates for this model in the limit of small time steps (Erban and Chapman, 2009), which is the basis for the implementations ReaDDy (Schoneberg and Noe, 2013), and SpringSaLaD (Michalski and Loew, 2016). Both these implementations introduce methods to capture excluded volume via, for example, repulsive short-range forces, which ultimately require adjustments to properly recover reversible reactions, as done in ReaDDY 2 (Hoffmann et al., 2019). With these methods, the ability to reproduce the underlying model will depend on using small time steps and assessment of the extent to which the modeled forces introduce error into the kinetics of many-body systems. Last, the widely used implementation MCell (Kerr et al., 2008) is not based on either the Smoluchowski or the Doi model, and instead derives reaction probabilities that quantify collisions within a volume, where this instantaneous volume depends on time step and diffusion constants. MCell lacks excluded volume, but it can take large time steps and recover the proper equilibrium in reversible reactions.
The future extensibility of all methods hinges on the feasibility of finding mathematical expressions for the crucial reaction probabilities that incorporate additional features and details, such as curved surfaces, intramolecular constraints, and external and internal deterministic forces (Figure 2, b and d). Reaction probabilities for multisite molecules have been derived, for example, by assuming rigid molecules and simplifying the dependence on orientation (Johnson, 2018). The addition of any interaction potentials (and therefore forces) between particles can significantly alter reaction kinetics (Zhou, 1990), and quantifying reaction probabilities has required either substantial computational overhead (Johnson and Hummer, 2014) or steady-state assumptions in dilute systems (Dibak et al., 2019). Careful validation of these additional features is critical for producing models that can be quantitatively reproduced across multiple simulation platforms.
RESULTS
We present a series of test problems relevant to spatial modeling of cellular and subcellular processes. This list is not meant to be exhaustive, but rather to permit a manageably sized survey of the kinds of problems that different simulation programs may be challenged to solve within a larger biological study. Our selection thus includes very simple problems that can be solved by many simulation tools as well as complex problems that can be solved (at present) by only a few. By framing the problems explicitly, we facilitate a direct comparison among different simulation packages both with respect to accuracy of execution and how they encode these particular scenarios. While all the models presented have been simulated previously, they have not been subject to the quantitative comparative analysis performed here across multiple model and method types. This comparison provides us with distinctive insight into the sensitivity of quantitative and qualitative behavior that emerges with specific biologically relevant features, which we summarize at the end of Results.
For these test cases, we contrast results from stochastic, deterministic, spatial, and nonspatial modeling approaches. We use Virtual Cell software for all nonspatial simulations and spatial deterministic simulations. For spatial stochastic simulations, we use the single-particle softwares NERDSS, Smoldyn, MCell, and eGFRD. We provide executable model inputs and numerical outputs for each model in our publicly accessible repository (https://github.com/spatialnimbios/testcases/). We summarize a broader range of actively developed tools and their features in Supplemental Table S2, as distinct software tools have introduced selected complex features of RD systems (also discussed previously; Takahashi et al., 2005; Schoneberg et al., 2014), and this is an additional consideration for users when selecting a tool for their biological problem.
Category 1: “unit test” cases
This category of problems represents fundamental building blocks for which there is a known correct answer (at least at steady state). We emphasize that because these reactions form the basis of much more complex models and geometries, it is essential that they be carefully tested for accuracy with regard to reaction kinetics and, in the case of bimolecular reactions, reversibility. For all of these, we initialize simulations with well-mixed components, and thus one may expect that any modeling approach would give the same outcome. However, due to differences in both approaches and algorithmic choices, we find that differences do in fact emerge in specific parameter regimes, particularly at short times before the system approaches steady state (Figure 3).
FIGURE 3:
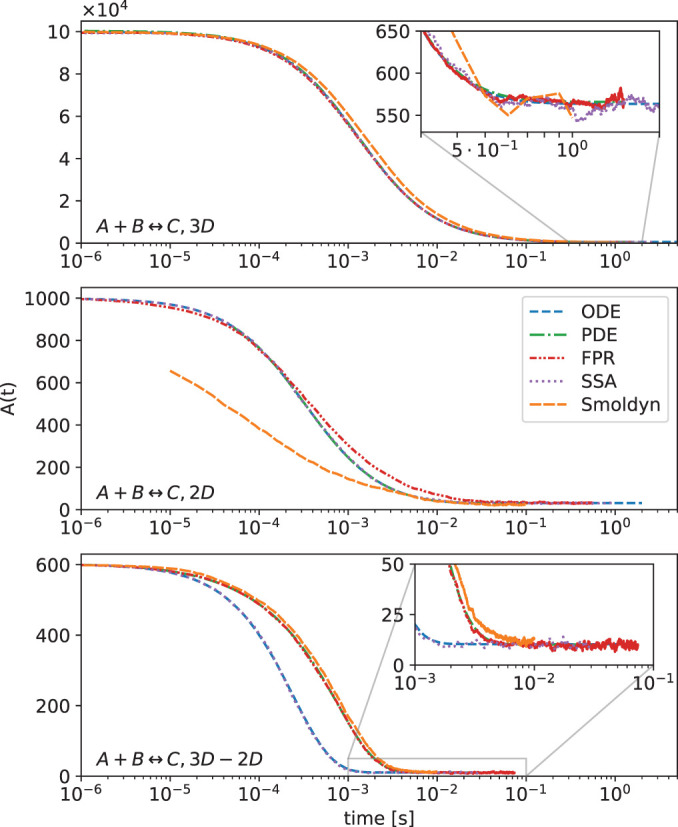
Reversible bimolecular reactions provide fundamental building blocks for any complex biological systems and thus warrant careful testing. In all dimensions, we use well-mixed initial conditions in closed systems, such that the reactions will reach a well-defined thermodynamic equilibrium. We test them using the nonspatial ODE and SSA methods, which match each other for all systems. The analytical solution to the rate equation is essentially exactly captured by the ODE. We compare the spatial PDE solved using VCell with single-particle methods solved via the FPR algorithm (NERDSS) and Smoldyn. For the 3D→2D reaction, we see the first difference between the ODE and PDE solution, because in the spatial models the reactants are well mixed in distinct locales (volume vs. surface), whereas in the nonspatial models, they are all well mixed in one volume. For 2D, single-particle methods will differ from other approaches, with NERDSS essentially capturing the theoretical diffusion-influenced reaction dynamics in 2D (Yogurtcu and Johnson, 2015a).
1A: bimolecular association in 3D, 2D, and from 3D to 2D.
Although seemingly simple, bimolecular association events require both a diffusional encounter and a reactive event; thus, the rate-constants and the kinetics are dependent on the dimensionality of the systems, and even for well-mixed systems, spatial details can cause deviations from nonspatial models. For reversible bimolecular association of well-mixed reactants in a closed system, the equilibrium state is analytically solveable and the kinetics of nonspatial rate equations also have analytical solutions. The effects of diffusion (Agmon and Szabo, 1990; Zhou and Szabo, 1996; Gopich et al., 2001; Gopich and Szabo, 2002a, b), electrostatics (Zhou, 1993; Schreiber et al., 2009), orientation (Shoup et al., 1981; Zhou, 1990), and dimensionality (Szabo et al., 1980; Torney and McConnell, 1983; Prüstel and Meier-Schellersheim, 2012; Yogurtcu and Johnson, 2015) on reaction kinetics have also received considerable theoretical study, providing a rich basis for understanding spatial effects. One may note that when bimolecular association is reversible, recovering the proper equilibrium is a simple test that can nonetheless be challenging for single-particle methods. Reaction probabilities and the placement of reactants on un/binding events must be derived to ensure equilibrium is reached (Box 5).
In 3D, all models and tools produce nearly identical results, even for this strongly diffusion-influenced reaction (kon = 1.48 107 M–1s–1). This is as expected (Figure 3, top panel). Although at short times the kinetics is slightly faster for Smoluchowski-type simulations (NERDSS), the kinetics rather rapidly converges to the macroscopic rate equations (Johnson and Hummer, 2014). Differences between single-particle and nonspatial methods can also emerge for reversible reactions as they approach equilibrium (Mattis and Glasser, 1998; Tauber et al., 2005), but these can only be effectively observed with high numerical precision and statistics—usually they are dwarfed by the copy number fluctuations.
Unlike in 3D, macroscopic rate equations in 2D only approximate the dynamics captured in Smoluchowski-type approaches at all times (Fange et al., 2010; Hellander et al., 2012; Yogurtcu and Johnson, 2015) (see Box 4). All macroscopic rate-based methods produce the same kinetics as each other (Figure 3, middle panel). Here we see distinctions between the spatial PDE and the spatial single-particle methods. Although species diffuse in the PDE, because they are present at all positions in space (due to uniform initial conditions), association is not dependent on their spatial distribution. For single-particle methods, there is always a distribution of starting separations between species that leads to some very fast reactions initially and at long times produces slower reactive collisions as particles that started off close to each other have already been consumed in the reaction. The Smoldyn method uses the steady-state solution to the Smoluchowski model to derive reaction parameters (Andrews and Bray, 2004b), but in 2D there is no steady state, and thus the reaction parameters are approximate. Because of this, Smoldyn can generate inaccurate kinetics in certain parameter regimes, with deviations being typically small in 3D but significant in 2D.
For binding between 3D particles and 2D particles (relevant for biological cases where soluble cytoplasmic proteins bind to membrane proteins or lipids), all models produce the same equilibrium, but the spatial models have slower kinetics delayed by diffusion to the surface (Figure 3, bottom panel). The extent of divergence between the nonspatial and the spatial models is driven by three factors, the “height” the solution volume stretches from the membrane surface, the diffusion coefficient of the 3D particles, and the speed of the binding. Here we simulated a fast, strongly diffusion-influenced reaction (8.4 × 107 M–1s–1), meaning nearly every collision results in a reaction. For a simulation box with h = 0.2 μm, one can estimate an average time to diffuse to a surface particle would be ∼60 μs (D3D = 30 μm2/s), which is relatively fast. However, with the numerous 2D particles mixed in the solution volume for a nonspatial simulation, the time to diffuse to a “surface” particle drops to ∼6 μs. Thus we find a mean relaxation time of ∼200 μs without space versus ∼700 μs with space (Figure 3, bottom panel). By dropping the reaction rate to more moderate protein–protein interaction levels, the spatial and nonspatial results begin to converge. Smoldyn shows excellent agreement for larger steps, here 10–6 s, although the kinetics shift slower for shorter steps. We note that when particles can only collide with one another from one side (because one is embedded in a surface, for example), this reduces the binding by a half, and solvers should explicitly account for this so that user-defined rates produce the equilibrium expected from a nonspatial model.
Last, transitions to the surface can be modeled using adsorption, which uses an effectively 1D rate. This is more efficient but importantly, it need not reach the same equilibrium as explicit particle simulations, because the occupancy of surface binding sites is not accounted for. Modeling explicit particles thus gives more control over the surface properties, and algorithms for binding to surfaces while accounting for site occupancy using implicit sites rival adsorption models in speed (Fu et al., 2019). Not all tools allow for all types of surface binding; hence, it is important to recognize these distinctions between adsorption versus single-site binding.
1B: crowding.
Inside of living cells, the extremely high density of macromolecules (with typical spacing on the order of nanometers) can alter the speed of molecular diffusion (Ando and Skolnick, 2010) and kinetics of intermolecular reactions, either increasing or decreasing biochemical reaction rates as compared with rates in dilute solution, depending on the size and mobility of the crowders (Zimmerman and Minton, 1993; Schreiber et al., 2009). As crowders become larger and less mobile (e.g., vesicles), they act more like barriers to encounters, slowing down rates of association (Minton, 2006; Zhou et al., 2008). However, as we see here in our test-case, when crowders are of comparable sizes and similar mobility to the reactant species, they drive up rates of association. This rate increase is due to a reduction in the total volume available to the reactants, effectively concentrating them without providing substantial barriers to encounters (Minton, 2006; Zhou et al., 2008). To quantify how increasing concentrations of crowding agents alter bimolecular association rates, here we simulated the bimolecular reaction A+B→B+C in the presence of additional, inert, C crowders, where all species are mobile, the same size, and exclude volume (Figure 4a). The analytical solution for no crowding/no excluded volume, A (t) = A0 exp (–kmacro Btott), provides a convenient baseline and fit function for interpreting deviations due to crowding/excluded volume (Figure 4b).
FIGURE 4:

The effects of volume exclusion, or crowding, can be tested by a simple model of bimolecular association in the presence of inert particles. (a) For the simple model B+A→B+C, the total population of catalytic B molecules (blue) remains fixed. A molecules (red) are converted into C molecules (green) when they collide with B molecules. C molecules do not react but do exclude volume and so can act as physical crowders. A molecules (red) are depleted over time. (b) The kinetics of A depletion depend strongly on the initial concentration of inert crowders C. For each simulation, the rate of depletion of A is fit to a single-exponential based on the solution to the rate equation (see Materials and Methods). The best fit rate is plotted here as a function of the volume fraction occupied by all A, B, and C particles. (c) For low crowding fraction where the initial concentration of C molecules C0 = 0, the kinetics is well described by a single exponential with a rate close to the nonspatial solution of 63.5 nm3/µs (65.4 FPR, 64.9 GFRD). (d) For high crowding with a large initial C0, the simulated kinetics is not as well described by a single exponential, exhibiting slower decay as the density of A approaches zero. Snapshots display the actual volume of the particles (r = 0.5 nm) given the full volume (23.2 nm box length), with 100 initial A particles (13.3 mM), 100 initial B particles (13.3 mM), and variable C. The walls use periodic boundary conditions. Simulations were performed with the NERDSS and eGFRD software using the FPR and GFRD algorithms, respectively.
For the single-particle algorithms that capture excluded volume (GFRD, van Zon and ten Wolde, 2005; and FPR, Johnson and Hummer, 2014), two main results emerge. First, the overall kinetics of association increase with increasing crowding fraction, up from kon = 63.5 nm3/µs at zero crowding/no excluded volume (3.8 × 107 M–1s–1) to ∼85–100 nm3/µs with 25% crowding fraction. This result is consistent across both algorithms, indicating that for GF-based solutions to the Smoluchowski model applied to a strongly diffusion-influenced reaction, small mobile crowders will enhance reaction rates for concentrated reactants (here A0 = 13.3 mM). This same trend was observed for simulations at lower reactant concentrations but comparable rate constants (Kim and Yethiraj, 2009). In contrast, simulations that immobilized the crowders caused them to act as a rigid barrier, leading to a reduction in reaction rates despite using the same reactant concentrations studied here (Andrews, 2020). Second, for high crowding regimes, we find that the kinetics is not described by a single rate constant (Figure 4d), whereas at low crowding, the results fit extremely well to the nonspatial analytical solution, with a new rate-constant (Figure 4c). This is perhaps not surprising; for diffusion-influenced reactions, short-timescale kinetics is dominated by reactants that are already close together where we expect crowding agents to promote their repeated collisions. Then, as the reactant populations decrease either to zero or toward an equilibrium state, the kinetics slows relative to nonspatial predictions, although this shift may be hard to detect (Mattis and Glasser, 1998; Tauber et al., 2005; Johnson and Hummer, 2014; Yogurtcu and Johnson, 2015a). Our results suggest that crowding agents exacerbate this slow search for the final reactants, similar to what happens in 2D, making the deviations from a single-rate constant kinetics easier to detect.
Although we show here that mobile crowders impact observed reaction rates, the changes are often quite modest. For the strongly diffusion-influenced reaction simulated here, we measure clear increases in rates, but for more rate-limited reactions (kon = 6 × 105 M–1s–1), the effect of crowders on the reaction rate is minimal (data not shown). Finally, FPR and GFRD are not in perfect agreement in terms of the quantitative size of the change in kinetics, although qualitatively they both predict a higher rate. At these high densities, GFRD converts to a brute-force BD algorithm, rather than its exact event-driven method, because the overhead cost of propagating GFRD is more expensive in dense systems. Because FPR combines the GF approach with simple Brownian updates and reweighting corrections, it performs well in dense systems, but still must also take extremely small time steps (10–10 to 10–12 s) to prevent particle overlap. It is not clear which method is more correct, as neither will preserve exactly two-body problems at each step. Further, we note that over the span of a picosecond time step (10–12 s), particle dynamics is not truly diffusive but is inertial. Capturing these dynamics would require a different model (e.g., generalized Langevin Dynamics; Van Kampen, 2007) that tracked both positions and velocities of particles and would be a valuable comparison in future work.
Category 2: “intermediate” cases
This category includes slightly more realistic biological cases with interesting emergent properties. These are particularly useful for illustrating the fundamental conceptual differences among the different modeling approaches.
2A: exploiting membrane localization to stabilize protein–protein interactions.
In a variety of biological processes, including clathrin-mediated endocytosis and initiation of signal transduction, multivalent proteins localize to membranes and assemble into larger, multiprotein complexes. Reducing dimensionality from a 3D search to a 2D search can accelerate kinetics of receptor binding (Adam and Delbruck, 1968; Berg and Purcell, 1977; Axelrod and Wang, 1994) and stabilize interactions for macromolecules restricted to the surface (Minton, 1995; Kholodenko et al., 2000; Abel et al., 2012). For soluble cytoplasmic protein binding partners, however, localization to the 2D membrane can further drive dramatic increases in protein complex stability, as was recently quantified using a simple model of two proteins that bind to one another and to a specific membrane lipid (Yogurtcu and Johnson, 2018) (Figure 5, a and b). The origin of the increased stability is largely a concentration effect, where the proteins collide with one another more frequently on the surface than in solution. While a change also must occur in 3D versus 2D equilibrium constants (Wu et al., 2011), the magnitude is usually much smaller than the change in 3D versus 2D search space (V/A > K3D/K2D) (Yogurtcu and Johnson, 2018).
FIGURE 5:
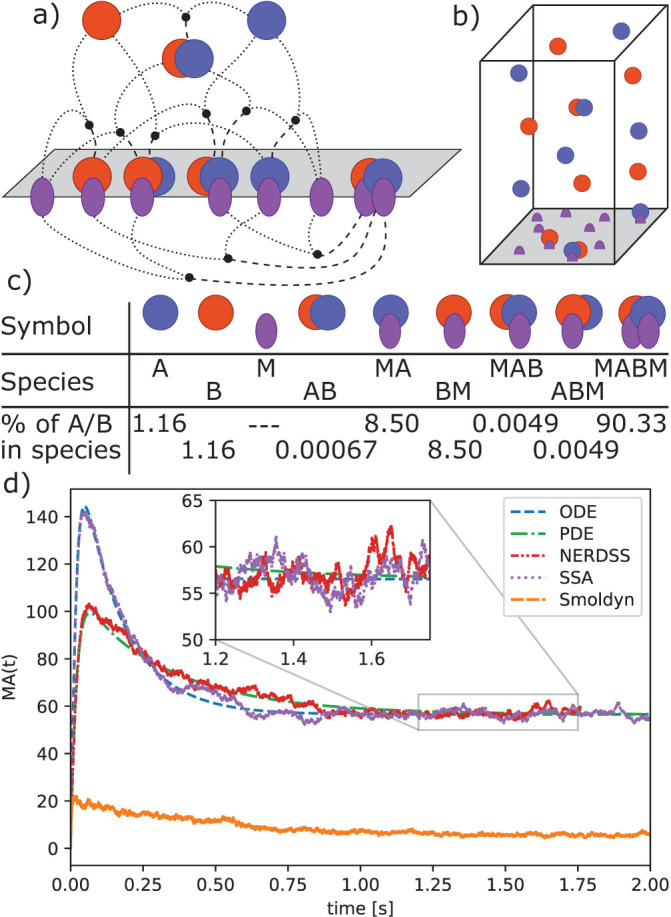
Membrane localization can tune speed and stability of protein complex formation. (a) A pair of protein binding partners A and B that can also localize to the membrane surface through binding a lipid M can exploit the 2D search space to promote complex formation. There are three binding interactions (below the plane) that involve 2D interactions between protein A and B on the surface, or between a protein and the lipid M. (b) The species can partition reversibly between solution and the membrane, forming nine distinct species, which are listed in c. (c) Model species and fraction of A or B proteins in each at equilibrium. (d) Time-dependent formation of a single protein–lipid complex MA. (The time course for BM is identical due to the choice of model parameters.) The initial rise in MA concentration is followed by a drop as the MA and BM complexes combine to form MABM, which dominates at equilibrium. The spatial simulations (PDE and NERDSS) exhibit slower MA formation kinetics and a lower peak concentration than the well-mixed simulations (ODE and SSA) due to slower recruitment of proteins to the surface, which is limited by diffusion. Relaxation of MA to equilibrium is also slower for the same reason. For smaller simulation volumes, where diffusion to the surface is fast, these differences disappear. Smoldyn does not reproduce the proper equilibrium due to inaccuracies of binding in 2D.
With the ability to localize to the membrane and reduce their search space, over 90% of proteins end up bound to one another (Figure 5c), which contrasts markedly with the purely 3D solution binding result calculated at equilibrium where only 4.6% would end up bound (for KD = 20 μM and [A]0 = [B]0 = 1 μM). Although all simulation methods are expected to produce the same equilibrium, and in this example stochastic effects are minimal, we find here that spatial effects arise because diffusion slows the localization of A and B molecules to the membrane (Figure 5d). The height of the box in the spatial simulations is 5 μm, and so despite efficient solution diffusion (50 μm2/s), the proteins are recruited more slowly to the membrane in spatial simulations than in the well-mixed simulations (compare red and green curves in Figure 5d with purple and blue curves). The protein–lipid complexes plotted in Figure 5d peak before dropping, as they bind to one another to equilibrate, and the peak is lower when they do not localize rapidly to the membrane. Errors in the results calculated using Smoldyn arise due to approximate treatment of purely 2D interactions, which lead to quantitative deviations from expected equilibria and kinetics for these reactions. This current limitation is being actively addressed in Smoldyn software (S. Andrews, personal communication).
Overall, these types of reactions form a critical component of more complex models of membrane-mediated assembly. Due to the quantitative differences we observe here in time dependence of species numbers, if the model is coupled to reactions that drive it out of an equilibrium steady state, this could then drive qualitative changes in the biological outcomes (as in case 2B below).
2B: increasing stochastic fluctuations in a system with multiple steady states.
Positive feedback in combination with other interactions can give rise to systems with multiple, distinct steady states. Such systems can exhibit large differences in their dynamics depending on whether they are simulated deterministically or stochastically. A simple model that illustrates such effects is the autophosphorylating kinase model first introduced by Lisman (Lisman, 1985) and studied more recently by Agarwal et al. (Agarwal et al., 2012), who analyzed a stochastic version of the model. Figure 6a shows a diagram of the model, which consists of a kinase that can activate itself through phosphorylation (reactions 2 and 3) and a phosphatase that can bind and dephosphorylate the active kinase (reactions 4 and 5). Because the production rate of active kinase (Ap) increases with the amount of Ap starting at low concentrations (Figure 6b, red curve), the system exhibits positive feedback. The rate balance plot shown in Figure 6b illustrates a case where the production and degradation rates of Ap as a function of Ap intersect multiple times to give rise to multiple steady states. This model has three steady states, also called “fixed points,” two of which are stable (Figure 6b, filled circles). For a stable fixed point, the balance of production and decay returns the system back to the fixed point following any slight changes in Ap away from the steady state value, whereas at an unstable fixed point, the balance of rates carries the system away from the fixed point as a result of any tiny fluctuation.
FIGURE 6:
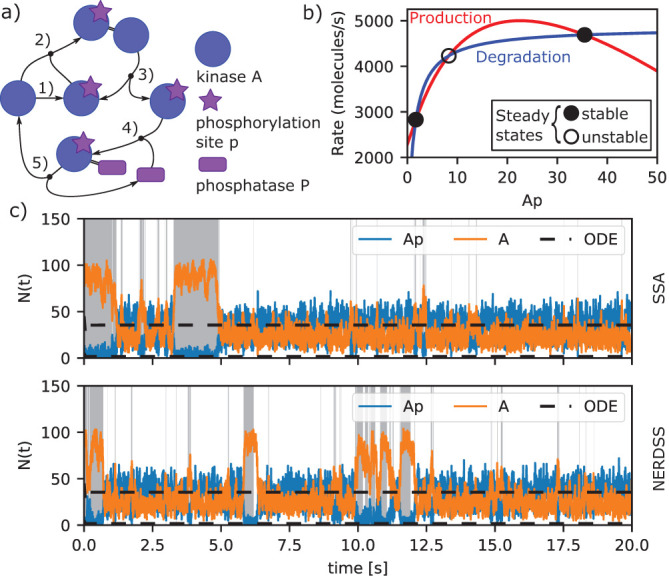
Positive feedback induces stochastic switching in a kinase autophosphorylation circuit. (a) Model reactions. The kinase A (blue circle) becomes active on phosphorylation (purple start; reaction 1) and serves as its own substrate (reactions 2 and 3). A phosphatase P (purple rectangle) binds and dephosphorylates the phosphorylated kinase (reactions 4 and 5). The activation reactions 2 and 3 form a positive feedback for kinase activation. A low rate of spontaneous activation of the kinase (reaction 1) is also included to prevent the system from being trapped in a state with no active kinase. (b) Rate balance plot identifying steady state concentrations of phosphorylated kinase (Ap). The red and blue lines show the rates of production (sum of rates of reactions 1 and 3) and degradation (rate of reaction 4) of Ap for the parameter values and initial concentration simulated here (Materials and Methods). Intersections of these curves indicate points at which production and degradation rates are equal and hence give rise to a steady state of the system. The two intersection points shown with filled circles indicate the stable steady states of the system, which occur at Ap concentrations of 1.7 and 35.5 molecules, respectively. A third steady state (open circle) indicates an unstable steady state, which occurs at a value of 8.3 Ap molecules. (c) Model trajectories computed with deterministic and stochastic methods. Deterministic trajectories starting from different initial conditions may relax to either of the two stable steady states (black dashed lines). Stochastic trajectories exhibit fluctuations about each of the steady states and occasionally switch between states with low and high kinase activation as shown by the blue trajectory. The top panel shows the result of a nonspatial stochastic simulation (SSA) and the bottom panel shows the result of a spatial stochastic simulation using the same paramenters (NERDSS). Regions of the trajectories shaded gray indicate where the system is in the state with low kinase activation with correspondingly high levels of inactive kinase (orange lines). In the regions with no shading the system is in a state with high kinase activation, as indicated by the active kinase level (blue lines) generally being above the inactive kinase level. The spatial stochastic simulations performed with NERDSS exhibit small differences from the nonspatial simulations carried out using SSA when performed in 3D with all species having a diffusion constant of 100 μm2 /s, and the same macroscopic rate is targeted (see Materials and Methods for full details on parameters used in simulation).
Deterministic simulations of the system, whether spatial or not, converge rapidly to one of the stable steady states depending on the initial level of Ap (Figure 6c, black dashed lines). Simulations starting from a state with low initial Ap will reach the steady state with lower Ap and vice versa for high Ap. Stochastic trajectories, on the other hand, may initially stay in the vicinity of the closer steady state, but fluctuations due to noise occasionally induce switches between states. In both trajectories shown in Figure 6c, the system starts in the lower steady state (as indicated by gray shading) but after a minute or so switches to the higher steady state where it continues to display fluctuations, some of which lead to short-lived excursions back to the lower state. For the set of rate parameters we used here, the system spends more total time in the higher state than the lower state (88% vs. 12%). In addition, the length of each segment in the higher state, called the residence time, is longer on average (2.6 s vs. 0.34 s) (see Materials and Methods and Supplemental Figure S1).
Despite the dramatic differences between the deterministic and the stochastic simulations for this model, we find the addition of space has relatively modest effects, with spatial models still producing bistable switching as diffusion slows. Figure 6c shows a nonspatial SSA simulation in the top panel (SSA) and an explicitly spatial simulation in the lower panel (NERDSS). For the geometries and diffusion constant values chosen here, these two simulations yield small differences between the probabilities and the residence times for each state (Supplemental Table S4). When the diffusion constant is reduced by a factor of 10, the probabilities of the lower state drop significantly although not dramatically, and the residence times of both states drop (Supplemental Figure S2 and Supplemental Table S4). The shorter residence times in spatial simulations relative to the nonspatial likely result from larger fluctuations in copy numbers due to transient spatial inhomogeneities, making both steady-states slightly less stable. Reduction of the diffusion coefficient by another factor of 10 makes simulation incompatible with the macroscopic reaction rates specified in the model. In other words, the reaction rate is limited by diffusion (see Box 4). We found that the effect of varying diffusion constant was quantitatively similar in the same model, but with all rates reduced by a factor of 10, reducing the fastest rate from 8 × 108 to 8 × 107 M–1s–1 (Supplemental Table S5). A study on a similar model of bistability found that slow diffusion limited the parameter regimes where one observed bistable switching due to fluctuations in local concentration, but also by changing the effective rates (Endres, 2015). Here we kept the macroscopic rates the same as the nonspatial model even as diffusion constants slow, and our results therefore indicate that timescales are sensitive to spatial inhomogeneities that persist longer with slower diffusion.
Category 3: “application” cases
Proper implementation of the test cases in this category requires the building blocks described above. These well-defined problems yield rich, biologically interpretable outputs that are sensitive to parameter choices.
3A: stochastic effects in gene expression.
Proteins that are important for controlling circadian clocks exhibit regular oscillations in expression levels. These oscillations are quite robust even in the presence of stochastic copy-number fluctuations. In fact, stochastic fluctuations can support oscillations in regimes that a purely deterministic model cannot (Supplemental Figure S3). Using a simple model of circadian oscillations (Vilar et al., 2002) (Figure 7a), we simulated the behavior of an activator protein A and repressor protein R that are produced from mRNA transcribed from a single copy of a gene (one for each protein). Coupling of A and R expression is driven by positive feedback of the activator A, which binds to each gene’s promoters to enhance transcription. Protein R also binds to A to effectively degrade it, and all proteins and mRNA are also degraded spontaneously at a constant rate. If the spontaneous degradation rate of protein R is slow, the oscillations will quench in the deterministic model, but persist in the stochastic solutions, as demonstrated in the original work and reproduced in Supplemental Figure S3.
FIGURE 7:
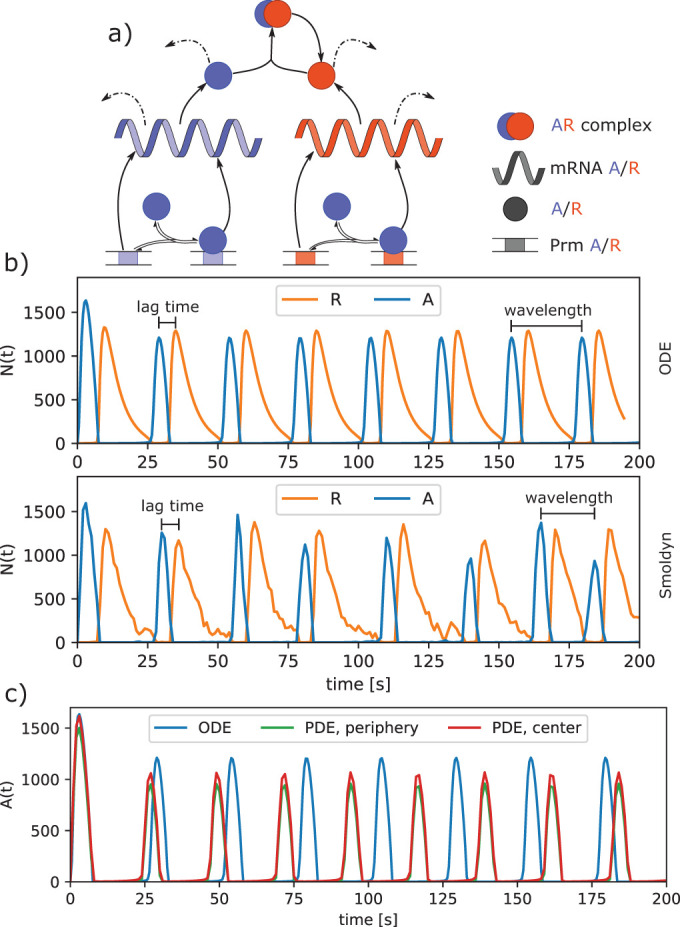
Circadian clock model shows robustness of oscillations to stochastic fluctuations. (a) Model of two proteins, A and R, translated from corresponding mRNA and transcribed from the corresponding genes (Prm A/R). Protein A is an activator that binds to the promoter of both genes and increases their transcription. Protein R acts as a repressor that binds to A and catalyzes its degradation. All proteins and mRNA also undergo spontaneous degradation with a specific lifetime (dashed arrows). (b) Growth of A copy numbers (dashed) peaks and then decreases as it is depleted by repressor R. Initially most R molecules are present in the AR complex, but the number of unbound R molecules increases as A is degraded. Next, the free R peaks and then begins to decrease because not enough A is present to promote its transcription at a rate that is greater than its rate of spontaneous degradation. The cycle then restarts, with the ODE solution producing highly regular oscillations of 25.2 s between A peaks and a 6 s lag between A and R peaks. The stochastic single-particle simulator Smoldyn produces noisier oscillations, but the average behavior is very comparable to the deterministic ODE model, with peak oscillations at 25.9 s and a 6.1 s lag between A and R peaks. (c) After localization of the promoters to the cell center and slowing of DA from 10 to 2 μm2/s, the A molecules become more concentrated in the cell center (red vs. green), which leads to noticeably faster oscillations relative to the nonspatial solution (blue).
We find that with the addition of space to the model, with all species diffusing at D = 10 μm2/s, the oscillation times show no real significant differences in single-particle or deterministic solutions relative to the nonspatial model (Supplemental Table S6). To quantitatively compare the kinetics across all models, we cannot use simple steady-state values due to the oscillations. These time-dependent oscillations are nearly perfectly regular in the deterministic models, but are quite imperfect (although recurring) in all stochastic and single-particle methods (Figure 7b). We therefore measure the average time interval between peaks in the expression of A and the lag time between the appearance of a peak in A expression followed by a peak in R (see Materials and Methods). The similar results across all methods show how with relatively small spatial dimensions (sphere of R = 1 μm), purely 3D reactions, and all species diffusing at the same D = 10 μm2/s, no spatial dependence was distinguishable.
The lack of any significant spatial dependence is somewhat surprising because we pushed the reaction rates into the strongly diffusion-influenced regime. The model was actually formulated to describe the slow oscillations of gene expression regulation, with all rates reported in units of hr–1, rendering diffusion times irrelevant. Here, we chose to accelerate the rates by a factor of 3600 (from hr–1 to s–1) due to the computational expense of simulating spatial models with explicit diffusion. This result overall indicates that the reactants still mixed faster than any spatial inhomogeneities could emerge, preventing deviations between spatial and nonspatial simulations.
We were finally able to measure a significant dependence on diffusion when we simulated the same set of reactions but we localized and immobilized the promoters for A and R to a small nucleus in the center of the volume, with unrestricted diffusion in and out of this nucleus (Figure 7c). For the same diffusion constants of D = 10 μm2/s for all other species, this immobilization of promoters was not enough to have an effect, and oscillation times remained not significantly different (Supplemental Figure S4 and Supplemental Table S7). However, when the diffusion constant of the activator A was slowed to 2 μm2/s, we observed a persistently higher concentration of A and its mRNA near the cell center relative to the cell periphery (Figure 7 and Supplemental Figure S4). This localization of A near the gene promoters had the effect of shortening the oscillation period in the PDE from 25 to 22.5 s (Supplemental Table S7). This change was independent of the diffusion constant of R, which does not bind the genes. Further, this same trend was observed in the single-particle simulations, which also produced faster oscillations as DA slowed to 2 μm2/s (Supplemental Table S7). Overall, we thus found that while slowing the search time for the Activator for both genes does modulate the oscillation times, the oscillations are quite robust and strongly dictated by total copy numbers and the unimolecular decay reactions, which are inherently independent of diffusion and spatial dimensions. This fairly complicated scenario is one where intuitively we had expected to see strong spatial effects, so it is rather interesting that the actual spatial effects are minor in this case. Certainly this outcome might vary depending on the parameter regimes selected.
3B: spatial and temporal oscillations in MinCDE.
When coupled to reactions, diffusion that is sufficiently slow or occurs over large enough length scales can establish spatial gradients of concentrations and subsequent oscillations in space and time. By construction, spatial oscillations are lost in nonspatial models, and no temporal oscillations occur either (Supplemental Figure S5). In a simplified model of bacterial cell division, the MinD and MinE proteins (Huang et al., 2003) spatially control the location of cell division by creating an oscillating spatial gradient of both proteins in the cytosol and on the membrane (Figure 8). The site of cell division is determined by assembly of a ring constructed from the bacterial tubulin homolog FtsZ. Because the Min proteins inhibit the assembly of the FtsZ ring, the spatial oscillation of the Min proteins from one end to the other in the cylindrical bacterial cell results in the localization of the FtsZ ring to a site very close to the geometrical center (Raskin and de Boer, 1999). This model involves 3D and 3D→2D bimolecular reactions and unimolecular reactions with no 2D reactions. Instead of forming explicit polymers, the membrane-bound proteins act to locally increase protein density through recruitment.
FIGURE 8:

Spatial oscillations in protein abundance for the MinCDE cell division model. (a) The MinD protein can exist in two states, ADP or ATP bound (named MinD-ADP and MinD-ATP). Only MinD-ATP localizes to the membrane in a 3D→2D reaction (producing MinD-ATP2D). Once on the membrane MinD-ATP2D can recruit additional cytoplasmic MinD-ATP (3D→2D) or MinE (3D→2D). The MinD-ATP.MinE2D complex on the membrane recruits MinD-ATP from solution as well (3D→2D), or it dissociates to return MinE to solution and MinD-ATP2D to MinD-ADP. None of these six reactions is thus reversible. (b) Simulations in a cylindrical cell with L = 4 µm and R = 0.5 µm, with 2.1143 μM MinD-ATP and 0.74 μM MinE initially well mixed. (c, d) The kymographs show how the copy numbers of MinD-ATP2D on the membrane in molecules/µm² oscillate in space and time. (c) Single-particle simulation Smoldyn. (d) PDE with uniform initial concentrations has faint symmetric oscillations visible up to ∼300 s before symmetry breaks. (e) Time dependence of MinD-ATP2D at a point on the left (blue) and right (orange) end of the cell. The oscillations are perfectly symmetric until ∼300 s in the top panel. However, if the error tolerance is tightened on the numerical integration, the symmetric oscillations persist longer out to ∼550 s in the bottom panel, illustrating the dependence on numerical precision.
We are able to produce very similar spatial and temporal oscillations of the MinD-ATP protein on the membrane in both a stochastic single-particle model (Smoldyn; Andrews, 2017) and a deterministic PDE solution (using Vcell; Moraru et al., 2008; Schaff et al., 2016). The major distinction between the two models is that the deterministic PDE is able to support symmetric or striped oscillations in the protein, as would be generated by two traveling waves in opposite directions. However, we find that these dynamics are not stable even in the deterministic solution—the accumulation of numerical precision error eventually breaks the symmetry and transitions to pole-to-pole oscillations. We show that by tightening the error tolerance on the numerical integration (by a factor of 10) or by increasing the PDE mesh resolution (by 2), one can delay the onset of this transition, clearly demonstrating the dependence on numerical precision (Figure 8e and Supplemental Figure S6). The stochastic single-particle method is therefore not able to support the symmetric oscillations due to copy number fluctuations and always produces pole-to-pole oscillations (Figure 8b). This shows quite clearly that the pole-to-pole oscillations are more robust to stochastic fluctuations in copy numbers for this geometry, and in the wild-type biological systems, these are the types of oscillations always observed for cells of this length (Raskin and de Boer, 1999).
A challenging aspect of model comparison for any simulations tracking the spatial and time-dependent concentration of species is deciding how best to quantify the results. Analysis is often defined in a problem-specific way. For the MinCDE model, depending on the cell geometry and model parameters, it may not even reach a steady state over long timescales, but can continue to oscillate and change oscillation amplitude or frequency. For symmetric geometries (like cylinders), we reduced the analysis to a function of time by tracking the MinD-ATP2D molecules at a specific point on the membrane (Supplemental Figure S7). Then, similar to the analysis for Figure 6, we calculated the average period between oscillations (Materials and Methods), finding values of 43.0 ± 0.7 s for Smoldyn and 41.3 ± 0.3 s for the PDE.
The oscillations in this model are fairly sensitive to initial concentrations of species (Supplemental Figure S6) and their relative stoichiometry. If the initial concentrations were cut in half, we found the oscillations disappeared in both models. The MinCDE model has been extensively studied, with the major determining features of oscillations being initial concentrations and cell geometry (see Halatek and Frey, 2012 and reference therein). The oscillations are relatively robust even to small changes in the reaction network, as long as membrane recruitment (3D→2D) and ATP hydrolysis are included (Halatek and Frey, 2012). We note that for Smoldyn, the oscillations were sensitive to the diffusion coefficients of the membrane-bound particles, set here to D = 0.05 μm2/s. If proteins did not diffuse on the membrane, the oscillations disappeared in Smoldyn, whereas they were retained in the PDE.
Summary of test case outcomes
By simulating the same biological cases using a range of spatial, nonspatial, stochastic, and deterministic methods, we have shown here how specific biological features and in some cases algorithm selection (e.g., classes of single-particle approaches) can alter quantitative and and even qualitative outcomes. As summarized in Figure 9, quantitative differences can be small, medium, or large based on system geometry and parameter regimes. Our results with fast versus slow diffusion are consistent with a recent review on spatial simulations that contain a more detailed analysis of the RDME method (Smith and Grima, 2019). We find that qualitative differences can be minor, but can also be major, producing entirely new behavior patterns that emerge. For example, major impacts could be observed in application scenario A2, which combines an elongated geometry with a comparatively slow diffusion (in comparison to the reaction rate), and includes both 3D and 2D dynamics. In general, we find that effects of introducing stochasticity to a system may be orthogonal to its spatial properties, as wrongly applying a deterministic method is likely to dominate all other choices. This can be most clearly seen for the results as shown in I2. Overall, we found that, while stochasticity played a minor role in models with reversible interactions (U1, I1), it was capable of driving major changes in models with irreversible interactions and feedback (I2, A1). Quantitatively, crowding might have a large impact, in particular if combined with slow diffusion and high reaction rates. Also, the impact on dynamics that involve reactions in 2D, be this 3D→2D or 2D dynamics, is significant and cannot be ignored in many cases. The suitability of specific simulation methods and their limitations must also be taken into account when any investigator is making choices about which methods to employ for a particular biological problem (see Supplemental Table S2). For example, if crowding and sterics need to be considered, particle-based approaches, in particular those with excluded volume, should be applied. However, in the case of very dense crowding, these approaches do not scale well because of their computational cost.
FIGURE 9:

Summary of the impact of spatial modeling and simulation approaches on quantitative (small, medium, large) and qualitative (major/minor) biochemical behavior. For quantitative behavior, our tree structure separates parameter and geometry regimes to specifically identify the scale of change observed and expected. For qualitative behavior, we note that, especially for reaction-network elements and stochastic effects, observing major changes also depends on additional parameter specifications, but in a less predictable way than the spatial effects (e.g., relative sizes of distinct rates or copy numbers). Minor changes are the default, as we observe for models with purely reversible reactions (rxns). The test cases range from very simple problems (U1: bimolecular association in 3D, 2D, and from 3D to 2D, U2 crowding) via intermediate tests (I1: exploiting membrane localization to stabilize protein–protein interactions; I2: increasing stochastic fluctuations in a system with multiple steady states) to applications that combine different spatial features (A1: stochastic effects in gene expression; A2: spatial and temporal oscillations in MinCDE). Various tools have been applied in the test cases: U1: ODE, PDE, SSA/Gillespie, particle-based (NERDSS, Smoldyn); U2: particle-based (NERDSS, eGFRD); I1: ODE, PDE, SSA/Gillespie, particle-based (NERDSS, Smoldyn); I2: ODE, SSA/Gillespie, particle-based (NERDSS); A1: ODE, PDE, SSA/Gillespie, particle-based (NERDSS, MCell, Smoldyn); A2: PDE, particle-based (Smoldyn). A detailed description of the theoretical basis and the features offered by these tools/methods can be found in Supplemental Table S2.
The spatial dimension in our test cases primarily drives quantitative changes in outcomes of varying magnitude. However, once multiple features are combined, as in the A2 MinCDE model, major qualitative differences emerge. We expect that our results will extrapolate to informing model selection in a broader range of biological processes. For example, the effect of clustered molecules acts to spatially localize interactions, which similar to the membrane localization (I1) can drive dramatic increases in complex stability and changes to kinetics. Compartmentalized or highly elongated and narrow geometries also can act to locally alter species concentrations, not unlike the role of dimensionality in driving quantitative changes (U1, I1) or qualitative changes (A2). Last, in all the models studied here, all species were initially well mixed in their system volumes, albeit in U1 (3D→2D) and I1 there was both a membrane and a solution volume. By introducing an explicit spatial concentration gradient via localized sources or sinks of molecules, spatial effects are inevitable and can only be captured with explicit spatial representations.
DISCUSSION
Since the use of GFP as a genetically encoded fluorescent tag came into widespread use over a quarter-century ago (Chalfie et al., 1994), methods for live-cell light microscopy have advanced spectacularly, and the spatial resolution and temporal resolution now possible for imaging experiments in living cells is truly awe-inspiring (Chen et al., 2014; Guo et al., 2018; Liu et al., 2018). At the same time, a great deal of technical development has gone into the generation of live-cell fluorescent reporters for biological readouts including signaling events, enzymatic activity, and force generation (Greenwald et al., 2018; Yasunaga et al., 2019). Many of the cell biological phenomena illuminated by direct observation of dynamic molecular events in living cells reveal a spatial component that could not be fully appreciated during the era when the most sensitive assays for molecular events were biochemical in nature (Machacek et al., 2009; Tay et al., 2010).
At this point, the quality of time-resolved live cell imaging data for cell biological events, and its quantitative accuracy, is far ahead of our ability to model these events computationally. As a community we could avoid doing the work to develop detailed, spatially resolved models when the data quality was relatively poor, relying on images of fixed cells at poor spatial resolution, but now that the data are excellent there is no excuse. Quantitative data demand quantitative models.
Accurate, spatially resolved 3D modeling of subcellular processes is inherently challenging, particularly as compared with the larger field of time-resolved cellular modeling where space is not taken into account. In exploring the currently available tools for 3D modeling, and dreaming about what the future might hold, we have identified several sticking points that will require focused community effort to overcome.
Spatially accurate modeling is intrinsically computationally expensive
Most interesting cell biological events unfold over minutes, hours, or days, requiring that relevant computational models be able to simulate comparable lengths of real time. For spatially resolved models, keeping track of the positions as well as the numbers and identities of relevant molecular species requires substantially greater computational overhead than simpler models that include only numbers (or concentrations) changing over time. In practice, this difficulty often limits spatially resolved models to keeping track of only a few molecular species.
An interesting analogy can be drawn to the field of MD simulations, which aim to model relative movements of atoms within proteins (Dror et al., 2012), while models of the scales that we are discussing typically aim to simulate the relative movements and transformations of molecules within cells. The equations of motion for atoms within protein molecules are relatively well understood, but accurately calculating their trajectories over even the millisecond timescales relevant for protein folding or conformational changes requires massive computational power (Shaw et al., 2010). Generally, increases in complexity and timescale for simulations of atoms in proteins have benefited from two types of efforts, improvements in hardware including purpose-built computers and distributed computing architectures for parallelization (Snow et al., 2002), accompanied by improvements in statistical sampling approaches to capture rare but significant events (Zuckerman, 2011). Even so, most MD simulations still cannot simulate processes central to the lives of proteins such as enzymatic catalysis, because they essentially treat atoms as hard balls subject to defined forces, but ignore the quantum mechanics that would be required to describe chemical transformations. Some hybrid methods have shown promise by including quantum-mechanical detail only in the enzyme’s active site (Karelina and Kulik, 2017), but naturally this improvement in chemical detail comes at substantial computational cost.
Similarly, for spatially resolved models at the cell biological scale, there is room for substantial improvement in efficient design of hardware and computational parallelization, development of appropriate statistical sampling methods, and hybrid methods that could smoothly integrate full detail in some particular locations or at some particular times with computationally efficient approximations elsewhere (Hellander et al., 2017). GPU architectures offer many advantages for efficient parallelization and have been effectively used by several groups to push cell biological models to much larger scales (Hallock et al., 2014; Ghaemi et al., 2020), though these approaches have not yet been widely adopted. There are also some kinds of modeling problems that intrinsically cannot be parallelized, where each step in time depends on the state of the system as a whole (Enculescu et al., 2010).
Several features of cell-scale modeling render this a massively more difficult computational challenge as compared with MD. One, of course, is the scale; a protein may include thousands or tens of thousands of atoms, while a typical cell will include 2–4 million proteins per cubic micrometer of volume (Milo, 2013), or over 1010 in an average mammalian cell, not to mention lipids, nucleic acids, carbohydrates, small metabolites and ions that may all be relevant to propagation of cellular signals. At present, all spatially resolved models for cell biology therefore necessarily include only a very small sampling of the molecular species that may influence the process. Furthermore, most parameters as fundamental as molecular concentrations and reaction rate constants are not known, or known only approximately (Gutenkunst et al., 2007; Schillings et al., 2015). For that reason, cell biological models must typically also perform a broad sampling of “parameter space.” There are increasing efforts to accelerate and optimize this parameter search (Mitra et al., 2018; Shockley et al., 2018; Mitra et al., 2019), but the size of parameter space nonetheless limits the depth to which any particular instantiation of a spatially resolved model can be explored.
There is no common framework for sharing and reproducing spatially resolved models
Over the past decade, there has been a humbling realization that a large fraction of published results in experimental biology, including many that have led directly to significant efforts toward drug development, cannot be reproduced (Prinz et al., 2011; Begley and Ellis, 2012). This has led to widespread reform in both the performance and the publication of experimental research, notably with the US National Institutes of Health (NIH) now requiring explicit training in experimental rigor and reproducibility and altering the structure of research grant applications to address these issues directly (Health, 2018). In order for the modeling enterprise to advance, it will be necessary for our community to establish standards for sharing and reproducing spatially resolved computational models as well.
Sharing and reproducing models are only possible if every quantitative parameter is strictly defined in a meaningful way. This will require a common “language” that is flexible enough to describe a wide variety of complex cell biological problems, and it is particularly challenging to define parameters that can be implemented in an equivalent way for both PDE-based and particle-based simulations of the same process. Even within the confines of our small working group, where we had all agreed to work together closely and were in constant communication throughout the process, we frequently found it challenging to run the “same” model on different platforms because of inconsistent parameter definitions. We also found that published models often lack critical parameters even if they appear at first glance to be completely described.
For time-resolved cell biological models that do not include an explicit spatial component, the SBML represents a large-scale community effort to improve standardization of model representation. An extension to SBML explicitly designed for spatially resolved modeling, SBML-spatial, is nearing completion (SBML.org) and is supported to varying degrees by some modeling systems, although the specification has not yet been formally released and is not widely adopted. Some leaders in the field have begun to organize resources to aid in efforts toward verifying simulation results (https://reproduciblebiomodels.org). Several related fields that require highly complex computational models, such as finite element modeling in biomechanics, have also begun to define community standards for reporting and verifying model parameters and results (Erdemir et al., 2012).
Minimal steps toward ensuring reproducibility in cell biological spatial modeling must include a general expectation that all code will be readily accessible (via GitHub or equivalent) and directly runnable by outside users, not dependent on local files or libraries. All code for spatial modeling should include explicitly defined unit tests, such as those we have included here. Furthermore, all parameters used in any model must include meaningful units. This is not a trivial consideration; for example, if the problem being studied involves a transition between a soluble form and a membrane-associated form of a particular molecular species, we have found that rate constants and their units are usually not carefully documented in most published models, and in some cases it is not even clear that different researchers agree on what the “right” units are for some typical problems.
It is difficult to compare outcomes of different types of models and to compare model results to experimental data
A typical goal for cell biological modeling is to determine whether a particular underlying mechanistic hypothesis is consistent with experimental observation. For processes with a spatially dependent stochastic component, which includes many cell biological problems of interest, each computational run (and indeed each independent experiment) will give a slightly different result. To compare experimental results with simulations, and to compare outcomes of different simulations with one another, the simplest approach is to use some kind of summary statistics that described probabilities of various outcomes. However, it is not often trivial to determine which summary statistics are most appropriate. For example, one influential early model illustrating the importance of stochasticity in genetic regulation leading to cell fate determination was an exploration of the events underlying the switch for bacteriophage lambda from a lysogenic to a lytic state, published in 1998 (Arkin et al., 1998). However, more than a decade passed before a useful method for calculating summary statistics on these extremely rare switching events was developed (Morelli et al., 2009). Ironically, it may in many cases be easier to repeat an experiment for thousands or tens of thousands of cells, for example using automated videomicroscopy (Cai et al., 2018) than to run a computationally intensive spatial simulation the same number of times. In general, the field would benefit from greater attention to scalability in model construction.
Some statistical strategies that were originally developed in other fields have been creatively adapted for applications in spatially defined stochastic simulations. These include extensions to the singular spectrum analysis methods that are widely used for analysis of time-series data (Shlemov et al., 2015) and the weighted ensemble sampling method that is useful when rare events are of particular interest (Donovan et al., 2016).
Visualization of model outcomes should also be standardized. This is much more conceptually challenging for spatially resolved models than for models where the only output is a change in concentration of molecular species over time. Here recent developments in the field of visual analytics, which combines statistical analysis with visualization and user interactions to cope with large complex data sets, may offer some promise (Matković et al., 2018). Visual analytics take the human into the loop for exploring data and the space of simulation experiments. Other approaches aim at testing requirements and confirming behavioral expectations automatically. One example of a promising avenue for automatic analysis would be spatio-temporal model checking approaches, which rely on explicitly stating spatio-temporal properties to be checked on the produced trajectories of the spatial simulation (Bartocci et al., 2015).
It may not be possible to address distinct model features required for different kinds of cell biological problems within a single modeling framework
In our comparative work, we emphasized simple biochemical interactions because we were able to implement these examples in comparable ways across multiple different computational platforms. We chose to use only very simple geometries for our example cases, but some of the stochastic simulation platforms we considered are capable of incorporating complex cell geometries, and complex geometries are also standard in most PDE solvers. However, none of the packages we used here are designed to incorporate correct subcellular physics for cytoskeletal mechanics, force generation, fluid flow, or electrostatics. Clearly these physical elements are important in accurate simulations of cellular behaviors. It has been widely recognized for over a century that cellular organization is tightly coupled to cellular structure (Abbot, 1916).
In our opinion, the highest priorities for integration of correct subcellular physics with stochastic molecule-based methods of the kind described here are accurate mechanical models for cytoskeletal filaments, molecular motors, and membranes. Interactions of the cytoskeleton and its associated motors with membranes determine overall cell shape and organelle localization, and these also underlie interactions of cells with one another and with their extracellular matrix. Actions of cytoskeletal filaments and motors directly deform membranes to produce invaginations or protrusions, and membrane surfaces influence cytoskeletal filament growth both because of their mechanical resistance (Keren et al., 2008) and because of their ability to accumulate and spatially organize key regulators (Mullins et al., 2018). Simulation frameworks that could properly deal with these kinds of mechanical and biochemical interactions should be adaptable for a wide variety of related problems, for example, exploring the assembly of viral “replication factories” on intracellular membranes of host cells, a process which requires large-scale oligomerization of viral polymerase (Spagnolo et al., 2010).
While we recognize that all models are necessarily approximations, we believe that some approximations are better than others. For membrane mechanics, the Helfrich model (Helfrich, 1973) is the most widely used framework for calculation of energies associated with membrane bending and deformation (Guckenberger and Gekle, 2017). While the theory itself is simple and elegant, calculation of energies for highly complex geometries in a continuum mechanics model can become complicated. A few interesting approaches have been used to enable cellular geometries to adapt in response to biochemical reactions (Angermann et al., 2012; Tanaka et al., 2015), but so far none of them have attempted to incorporate correct Helfrich-derived bending energies. PDE-based models for membrane–protein interactions can better approximate the correct underlying physics, but have so far been limited to fairly simple geometries (Rangamani et al., 2014; Wu et al., 2018). Ultimately, the membrane mechanics must be coupled to the proteins and protein assemblies that provide most of the force necessary to induce membrane bending and remodeling.
Capturing the dynamics of protein assemblies, both as highly ordered filaments or lattices (e.g., microtubules or clathrin-coated vesicles) and as disordered condensates, will be essential for bridging biochemical interactions with mechanics. Recent advances in extending single-particle methods to include rigid (Varga et al., 2020) or polymer-like structures (Michalski and Loew, 2016; Hoffmann et al., 2019) have enabled simulations of highly ordered clathrin lattices and viral shells (Varga et al., 2020), or disordered condensate-like assemblies (Chattaraj et al., 2019), respectively. For these simulations, excluded volume is a critical feature to reproduce realistic molecular features and prevent unphysical overlap between multicomponent assemblies. For cytoskeletal mechanics, in many cases a simple approximation of cytoskeletal filaments as elastic rods and motor proteins as discrete localized generators of quantized directional force can give impressively realistic simulation outcomes (Nedelec and Foethke, 2007). This approach has been widely used for studies of dynamic microtubule and actin structures in eukaryotic cells (Karsenti et al., 2006; Odell and Foe, 2008; Rubinstein et al., 2009). It has recently been demonstrated that mechanically accurate modeling of cytoskeletal filament dynamics can be fruitfully combined with a continuum model for membrane bending in order to simulate endocytosis (Akamatsu et al., 2020).
More generally, it is clear that much more work needs to be done in the development of multiscale simulations and hybrid computational approaches (Groen et al., 2019). For example, in some intermediate regimes, approximate subvolume-based simulations may be able to bridge the gap between well-mixed and particle-based simulations. It will require careful work to understand what approximations are generally useful when jumping across scales in hybrid models and what approximations give rise to propagating errors.
Overall, the members of our working group have thoroughly enjoyed this opportunity to critically evaluate the state of tools that are currently available for simulation of cell biological processes. Although we have of course identified important gaps, we are generally optimistic about recent developments and look forward to incorporation of more sophisticated computational methods to improve the efficiency and accuracy of cell biological modeling as these methods are developed. Spatially accurate cellular simulation is a field still in its infancy, but its future is bright.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Simulation solvers
ODE, PDE, and SSA simulations were all run using Virtual Cell, version 7 (Moraru et al., 2008). Unless otherwise noted, the PDE solver used a fully implicit finite volume, regular grid (variable time step), max step of 0.1 s. Absolute error tol: 10–9, relative error: 10–7. The mesh used the default values, which typically resulted in mesh side lengths of ∼0.02 μm. We note that for the PDE, the mesh, volume, or concentrations sometimes had to be adjusted slightly to ensure the copy numbers were the same for accurate comparisons. The ODE used the Combined Stiff Solver (IDA/CVODE), abs error tol: 10–9, relative error tol: 10–9. SSA simulations used the Gibson–Bruck method, an exact SSA, implemented in Virtual cell, or for the autophosphorylation model, RuleBender (Smith et al., 2012).
All single-particle solvers propagated BD, with no additional forces added. Smoldyn simulations were run either through Virtual Cell, version 7, or using the stand-alone Smoldyn software, version 2.60/2.61 (Andrews, 2017). FPR or NERDSS simulations were run using the NERDSS software, version 1 (Varga et al., 2020), which uses the FPR algorithms (Johnson and Hummer, 2014; Yogurtcu and Johnson, 2015; Johnson, 2018) to solve the single-particle reaction–diffusion model. GFRD simulations were run using eGFRD software (Sokolowski et al., 2019) from August 3, 2019. MCell simulations were run using MCell version 3.3 (Kerr et al., 2008).
Models and simulation parameters
Unless otherwise noted, species in spatial simulations were initialized as uniformly distributed.
1. Units tests.
1a. 3D Reversible binding A+B C. Model parameters: V = 3.1934 μm3 cube, [A]0 = [B]0 = 52 μM, or 100,000 copies, [C]0 = 0. kon = 1.476 × 107 M–1s–1, koff = 0.02451 s–1. Microscopic rates, ka = 1000 nm3μs–1, kb = 1s–1. KD = 0.00166 μM. For FPR/NERDSS, σ = 1 nm. DA = DB = DC = 1 μm2/s. [A]eq = 0.293 μM, or 563.5 copies, reached after ∼1 s. Sim parameters: single-particle methods ∆t = 10–7-10–6 s. FPR/NERDSS Ntraj = 18, SSA Ntraj = 10, and Smoldyn Ntraj = 1.
1b. 2D Reversible binding A+B ⇌ C. Model parameters: A = 1 μm2, flat surface. [A]0 = [B]0 = 1000 μm–2, or 1000 copies, [C]0 = 0. kon = 3.07 μm2s–1, koff = 3.07 s–1. Microscopic rates, ka = 10 μm2s–1, kb = 10 s–1. KD = 0.00166 μM. For FPR/NERDSS, σ = 1 nm. DA = DB = 1 μm2/s, DC = 0.5 μm2/s. [A]eq = 31.1267 μm–2, reached after ∼0.1 s. Sim parameters: single-particle methods ∆t = 10–7s. FPR/NERDSS Ntraj = 5, SSA Ntraj = 5, Smoldyn Ntraj = 2.
1c. 3D→2D Reversible binding A+B ⇌ C. Model parameters: V = 1 μm3 cube, flat membrane surface of 2.2 μm × 2.2 μm. [A]0 = 1 μM, or 602 copies, [B]0 = 6045 μm–2, or 29,258 copies, [C]0 = 0. kon = 8.44 × 107 M–1s–1, koff = 70.085 s–1. Microscopic rates, ka3D = 500 nm3μs–1, kb = 250 s–1. KD = 0.83 μM. σ = 1 nm. DA = 30 μm2/s, DB = 1 μm2/s, DC = 0.97 μm2/s. [A]eq = 10.3 copies, or 0.017 μM, reached after ∼0.02 s. Sim parameters: single-particle methods ∆t = 10–7s. FPR/NERDSS Ntraj = 10, SSA Ntraj = 5, and Smoldyn Ntraj = 3. Smoldyn ∆t = 10–6 s, with the longer steps converging to the PDE solution.
1d. Crowding A+B→B+C. Model parameters: V = 12,500 nm3 cube (side length of 23.21 nm), periodic boundary conditions enforced. [B] = 100 copies (13.3 mM). [A]0 = 100 copies. [C]0 varied across 6 systems: [0, 994, 2187, 3381, 4575, 5768], corresponding to total volume fractions occupied of [0.00838, 0.05, 0.1, 0.15, 0.2, 0.25]. Reactions: 1. A+B→B+C ka = 85 nm3/μs, 2. A+A ka = 0, 3. A+C ka = 0, 4. B+B ka = 0, 5. B+C ka = 0, 6. C+C ka = 0. With no volume exclusion, Rxn 1 has kon = 63.5 nm3/μs = 3.82 × 107 M–1s–1. σ = 1 nm, and thus each particle is effectively modeled as a volume-excluding sphere of diameter = 1 nm. DA = DA = Dc = 10 μm2/s.
Sim parameters: for FPR/NERDSS(Varga et al., 2020), a maximal ∆t for each system was limited by the density, values for each crowding fraction: [10–11 s, 10–11 s, 10–11 s, 5 × 10–12 s, 5 × 10–12 s, 10–12 s]. Ntraj = 40–80 for each crowding fraction. Another Ntraj = 40–80 were run at another time step (5 times faster) to verify quantitatively similar results. For eGFRD (Sokolowski et al., 2019), Ntraj = 10 for each crowding fraction. Algorithmic adjustments to standard FPR: first, when an A+B→B+C reaction occurred, the products were kept at their same coordinates as the reactants. Second, at each time step, particle positions are updated, and they must avoid overlapping (r ≥ σ) all other particles, considering all particles within collision distance (r < Rmax) (Johnson and Hummer, 2014). Due to the high crowding, we used a more stringent criteria to update particle positions. We defined clusters that contained all the particles with a capacity to possibly overlap all their partners. The positions of all particles in this cluster were simultaneously updated to avoid any overlap. For large clusters, to reduce the number of rejected updates, the time step was reduced by a factor of 10 and then updated 10 times to the full ∆t. Third, initial configurations for high crowded simulations were generated on a grid. This crystalline configuration was then “melted” by simply setting the rate of reaction 1 to zero to prevent binding events. After 0.1 s, the now disordered but well-mixed configuration was used to start simulations.
2. Intermediate tests.
2a. Membrane localization model. Model parameters: V = 1.1045 μm3, A = 0.2209 μm2, rectangular cube, 0.47 μm × 0.47 μm surface, height = 5 μm. [A]0 = [B]0 = 1 μM or 665 copies, [M]0 = 5.6474 μM = 17,000/μm2, or 3755 copies. [AM]0 = [BM]0 = [AB]0 = [ABM]0 = . [MAB]0 = [MABM]0 = 0. Reactions: 1. A+B ⇌ AB kon = 0.05 μM–1s–1. 2. A+M ⇌ AM kon = 2 μM–1s–1. 3. B+M ⇌ BM kon = 2 μM–1s–1. 4. A+BM ⇌ ABM kon = 0.05 μM–1s–1. 5. B+MA ⇌ MAB kon = 0.05 μM–1s–1. 6. AB+M ⇌ ABM kon = 2 μM–1s–1. 7. AB+M ⇌ MAB kon = 2 μM–1s–1. 8. MA+BM ⇌ MABM 0.0415 μm2s–1. 9. MAB+M ⇌ MABM 1.6693 μm2s–1. 10. M+ABM ⇌ MABM 1.6693 μm2s–1. For all reactions, koff = 1 s–1. At equilibrium, 90.3% of proteins were in complex with one another on the membrane (MABM), or 600 copies, and 1.16% were remaining in solution, or 7.7 copies. σ = 1 nm for all rxns, DA = DB = 50 μm2/s. DAB = 25 μm2/s. DAM = DBM = 0.495 μm2/s. DABM = DMAB = 0.49 μm2/s. DMABM = 0.248 μm2/s DM = 0.5 μm2/s. All membrane bound had Dz = 0.
Sim parameters: single-particle methods ∆t = 10–7s. FPR/NERDSS Ntraj = 10. SSA Ntraj = 10 Smoldyn Ntraj = 10. We note that the model includes 10 coupled pairwise reactions (three are 3D, four are 3D→2D, and three are 2D), although only 6 equilibrium constants are unique. This is because there are only 3 reactants and multiple thermodynamic cycles that can result in the final assemblies (see, e.g., MABM in Figure 5a). To preserve a thermodynamic equilibrium at steady state, not all rates can be defined independently.
2b. Autophosphorylation: model parameters A: V = 0.003 μm3. [A]0 = 103 copies, [Ap]0 = 5 copies, [P] 0 = 9 copies. [A_Ap]0 = 0, [Ap_P]0 = 0. 1. A→Ap at 21.2 s–1. 2. A+Ap→A_Ap at 107M–1s–1, ka = 16.7 nm3μs–1, σ = 1 nm. 3.A_Ap→Ap+Ap at 200 s–1. 4. Ap+P→Ap_P at 8*108 M–1s–1, ka = 2820 nm3μs–1, σ = 1 nm and 5. Ap_P→A+P at 539 s–1. DA = DP = 100 μm2/s. A second spatial stochastic simulation had DA = DP = 10 μm2/s, which required new bimolecular parameters for rxn 2: ka = 17.8 nm3μs–1, σ = 1 nm, rxn 4: ka = 11191 nm3μs–1, σ = 6 nm. Note: the binding radius had to be increased to recover the large macroscopic binding rates for rxn 4. A third spatial stochastic simulation had DA = DP = 20 μm2/s, which required new bimolecular parameters for rxn 2: ka = 17.2 nm3μs–1, σ = 1 nm, rxn 4: ka = 3919 nm3μs–1, σ = 4 nm.
Sim parameters: single particle methods: ∆t = 2*10–8s, Ntraj = 2. For D = 10, ∆t = 1*10–7s, Ntraj = 2. For D = 20, ∆t = 1*10–7s, Ntraj = 2. All simulations were ≥200 s to obtain statistics on switching times.
B version of model with all rates reduced by 10: 1. 2.12 s–1. 2. 106 M–1s–1, ka = 1.67 nm3μs–1, σ = 1 nm. 3. 20 s–1. 4. 8*107 M–1s–1, ka = 282.0 nm3μs–1, σ = 1 nm and 5. 53.9 s–1. DA = DP = 10 μm2/s. A second spatial stochastic simulation had DA = DP = 100 μm2/s, which required for rxn 4: ka = 140.31 nm3μs–1, σ = 1 nm. A third spatial stochastic simulation had DA = DP = 1 μm2/s, which required for rxn 2: ka = 1.78 nm3μs–1, σ = 1 nm. rxn 4: ka = 1119.14 nm3μs–1, σ = 6 nm.
Sim parameters: ∆t = 2*10–7s, Ntraj = 2. For D = 100, ∆t = 2*10–8s, Ntraj = 2. For D = 1, ∆t = 1*10–6 s, Ntraj = 2. All statistics were collected for at least 2000 s, except the fast diffusion constant. With only 100 s for D = 100, we observed transitions but insufficient statistics to determine trends relative to the other models.
3. Application tests.
3a. Circadian clock oscillator model. Model parameters A: 4.189 μm3, cube, or a sphere with R = 1 μm. [prmA]0 = [prmR]0 = 1 copy, or 0.000397 μM. [A]0 = [R]0 = [C]0 = [mRNAA]0 = [mRNAR]0 = [prmA.A]0 = [prmR.A]0 = 0. Reactions: 1. A + R → C kon = 1204 µM–1s–1, ka = 356,263 nm3μs–1, s = 8 nm. 2. prmA → prmA + mRNAA k2 = 50 s–1. 3. prmA.A → prmA.A + mRNAA k3 = 500 s–1. 4. prmR → prmR + mRNAR k4 = 0.01 s–1. 5. prmR.A → prmR.A + mRNAR k5 = 50 s–1. 6. prmA + A ↔ prmA.A k6f = 602 µM–1s–1, k6b = 50 s–1, ka = 4889 nm3μs–1, kb = 244.5 s–1 s = 5 nm. 7. prmR + A↔ prmR.A k7f = 602 µM–1s–1, k7b = 100 s–1, ka = 4889 nm3μs–1, kb = 489 s–1 σ = 5 nm. 8. mRNAA → mRNAA + A k8 = 50 s–1. 9. mRNA_R → mRNA_R + R k9 = 5 s–1. 10. mRNA_A → NULL k10 = 10 s–1. 11. mRNA_R → NULL k11 = 0.5 s–1. 12. A → NULL k12 = 1 s–1. 13. R→ NULL k13 = 0.2 s–1. 14. C→ R k14 = 1 s–1. D = 10 μm2/s (for all 9 species).
B version of the model was run with one modification from the original A model: 13. R→ NULL k13 = 0.05 s–1, eliminating oscillations in the deterministic model Supplemental Figure S3.
C version of this model was created with modifications from original A model: a) promoters were spatially localized to a sphere at the center of the volume of R = 0.1 μm. b) DprmA = DprmR = DprmA.A = DprmA.R = 0. c) [prmA]0 = [prmR]0 = 1 copy, or here 0.3964 μM. No barrier existed for proteins to access this ‘nucleus’ in the cell center. Full details are in the Supplemental Supporting Methods.
Sim parameters: NERDSS ∆t = 0.5, 2, and 10 μs, and Smoldyn ∆t = 100 μs. SSA Ntraj = 10. Smoldyn Ntraj = 2, NERDSS Ntraj = 2 (600 s each); see Supplemental Tables S6 and S7. Time steps were limited by the fast unbinding reaction of the single promoter, Rxn 7, rather than the particle density. With only a single event possible due to N = 1, a larger time step results in slightly fewer events occuring relative to the theoretical value.
3b. Model: MinCDE cell division model. Model parameters: cylindrical volume, h = 4 μm, R = 0.5 μm. Five species: [MinD-ATP]0 = 2.1143 μM, or 4000 copies. [MinE]0 = 0.74 μM, or 1400 copies. [MinD-ADP]0 = [MinD-ATP2D]0 = [MinD-ATP.MinE2D]0 = 0, where 2D indicates membrane bound. Six irreversible reactions. 1. MinD-ATP→MinD-ATP2D, k1 = 0.025 µm.s–1. 2. MinD-ATP + MinD-ATP2D→2MinD-ATP2D, k2 = 0.903 s–1.µM–1. 3. MinD-ATP + MinD-ATP.MinE2D→MinD-ATP2D + MinD-ATP.MinE2D, k3 = 0.903 s–1.µM–1. 4. MinE+MinD-ATP2D→MinD-ATP.MinE2D, k4 = 56.0 s–1.µM–1. 5. MinD-ATP.MinE2D→MinD-ADP + MinE, k5 = 0.7 s–1. 6. MinD-ADP→MinD-ATP, k6 = 1 s–1. D = 2.5 μm2/s for MinD-ATP, MinD-ADP, and MinE. D = 0 μm2/s for MinD-ATP2D and MinD-ATP.MinE2D in the PDE. Initial species all well-mixed in PDE. In Smoldyn, MinD-ATP copies split between solution and membrane. Necessary modification for Smoldyn to produce oscillations: D = 0.05 μm2/s for MinD-ATP2D and MinD-ATP.MinE2D.
Sim parameters: Smoldyn: Ntraj = 4, ∆t = 2 ms.
Analysis methods
Equilibria and error: for all stochastic methods, we calculated averages and variances of copy numbers in time over N trajectories. We used the standard error of the mean at time points: SEM(t) = 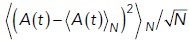 . For equilibrium values, we used differences from the mean to determine when equilibrium was reached. We note that for all models, we selected parameters such that the equilibrium was not too close to zero, which is important for assessing clear deviations from the proper equilibrium values.
. For equilibrium values, we used differences from the mean to determine when equilibrium was reached. We note that for all models, we selected parameters such that the equilibrium was not too close to zero, which is important for assessing clear deviations from the proper equilibrium values.
Clock oscillator timescales: the average time separation between A peaks, between R peaks, and the lag time between A and R peaks was calculated in two ways. First, by finding the peak maxima and simply calculating the separation between neighboring peaks, this was then averaged over all peaks. Second, to calculate time period of peaks, we used a discrete FFT that was zero-padded out to 5000 s to increase the resolution of the frequencies sampled. The frequency with the largest coefficient, fmax, was used to define the period of the oscillation (1/fmax). The cross-correlation between the A and R time-series was used to identify the lag-time, again keeping the time with the largest correlation coefficient. Both methods produced similar results, and were performed using MATLAB functions. We report in the Supplemental tables the periods determined from the find peaks method, with SEMs reporting the calculated 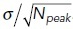 .
.
Fit to kinetics of crowding simulations: to define the kfit values for the crowding simulations as a function of packing fraction, we fit the copies of A versus time A(t) = A0 exp (–kfit Btott), where Btot is 0.008/nm3, and we performed nonlinear fitting to the exponential rather than the logarithm of A(t). We fit each individual trajectory and report the mean of these values 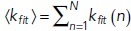 , where N is total trajectories. The SEM is calculated based on the variance around this mean value. Fits to the averaged trajectory gave similar values, and fits using the ln(A(t)) produced the same trends, but with slightly lower kfit values. The same approach was used for both FPR and GFRD trajectories.
, where N is total trajectories. The SEM is calculated based on the variance around this mean value. Fits to the averaged trajectory gave similar values, and fits using the ln(A(t)) produced the same trends, but with slightly lower kfit values. The same approach was used for both FPR and GFRD trajectories.
MinCDE timescales: for the MinCDE models, we calculated the time-scales of oscillations by choosing fixed points in space at either end of the cylinder (Supplemental Figure S7). Once the oscillations were pole-to-pole, we quantified the period of oscillations in time using the same approach as applied to the clock oscillator model above by finding the peak maxima and taking the average of time separations between adjacent peaks. This produced periods of 43.0 ± 0.7 s for Smoldyn and 41.3 ± 0.3 s for the PDE.
Autophosphorylation state assignments: to assign stochastic trajectories to states, 1 = low phosphorylation, 2 = high phosphorylation, we report results based on thresholding the values of A(t) and Ap(t). We created a transition region, where points could be in either 1 or 2 depending on their previous state, to minimize rapid recrossings between states (see Supplemental Figure S1). If [A(t) > 62 + Ap(t)], assign to state 1, else if [A(t) < 35 + Ap(t)] assign state 2, else [if(State(t-1) = i, State(t) = i], where i = [1,2]. We then counted the number of times in each state, length of intervals spent in each state, and transitions between points separated by a time δt to construct 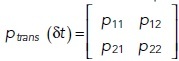 . We calculated
. We calculated 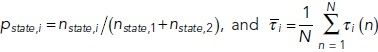 , and
, and 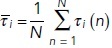 , where N is the number of intervals spent in state i, each of duration τi. Distributions of these residence times are shown in Supplemental Figure S1. To calculate errors on the probability of each state, each trajectory was split into 10 chunks (length at least the larger residence time), and these 10 state probabilities were used to calculate a mean and SD/SEM.
, where N is the number of intervals spent in state i, each of duration τi. Distributions of these residence times are shown in Supplemental Figure S1. To calculate errors on the probability of each state, each trajectory was split into 10 chunks (length at least the larger residence time), and these 10 state probabilities were used to calculate a mean and SD/SEM.
Supplementary Material
Acknowledgments
This work was funded by NIMBioS. We thank Carlos Lopez for stimulating discussions. M.E.J. also gratefully acknowledges computing resources from Johns Hopkins University’s MARCC supercomputer and XSEDE through XRAC MCB150059 and support from an NIH MIRA R35GM133644. M.M.S. and T.P. acknowledge support by the intramural research program of the National Institute of Allergy and Infectious Diseases, NIH. P.H. and A.M.U. acknowledge support from the DFG (German Research Foundation) Collaborative Research Center 1270/1 - 299150580 ELAINE.
Abbreviations used:
- BD
Brownian dynamics
- CME
chemical master equation
- GF
Green’s function
- MD
molecular dynamics
- ODE
ordinary differential equation
- PDE
partial differential equation
- RDME
reaction diffusion master equation
- SBML
systems biology markup language
- SSA
stochastic simulation algorithm.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-08-0530) on November 25, 2020.
REFERENCES
- Abbot ES (1916). The causal relations between structure and function in biology. Am J Psychol 27, 245–250. [Google Scholar]
- Abel SM, Roose JP, Groves JT, Weiss A, Chakraborty AK (2012). The membrane environment can promote or suppress bistability in cell signaling networks. J Phys Chem B 116, 3630–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam G, Delbruck M (1968). Reduction of dimensionality in biological diffusion processes. In: Structural Chemistry and Molecular Biology, San Francisco: Freeman, 198–215. [Google Scholar]
- Agarwal A, Adams R, Castellani GC, Shouval HZ (2012). On the precision of quasi steady state assumptions in stochastic dynamics. J Chem Phys 137, 044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agmon N, Szabo A (1990). Theory of reversible diffusion-influenced reactions. J Chem Phys 92, 5270–5284. [Google Scholar]
- Akamatsu M, Vasan R, Serwas D, Ferrin MA, Rangamani P, Drubin DG (2020). Principles of self-organization and load adaptation by the actin cytoskeleton during clathrin-mediated endocytosis. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge BB, Burke JM, Lauffenburger DA, Sorger PK (2006). Physicochemical modelling of cell signalling pathways. Nat Cell Biol 8, 1195–1203. [DOI] [PubMed] [Google Scholar]
- Amonlirdviman K, Khare NA, Tree DR, Chen WS, Axelrod JD, Tomlin CJ (2005). Mathematical modeling of planar cell polarity to understand domineering nonautonomy. Science 307, 423–426. [DOI] [PubMed] [Google Scholar]
- Ando T, Skolnick J (2010). Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc Natl Acad Sci USA 107, 18457–18462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SS (2017). Smoldyn: particle-based simulation with rule-based modeling, improved molecular interaction and a library interface. Bioinformatics 33, 710–717. [DOI] [PubMed] [Google Scholar]
- Andrews SS (2020). Effects of surfaces and macromolecular crowding on bimolecular reaction rates. Phys Biol 17, 045001. [DOI] [PubMed] [Google Scholar]
- Andrews SS, Addy NJ, Brent R, Arkin AP (2010). Detailed simulations of cell biology with Smoldyn 2.1. PLoS Comput Biol 6, e1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SS, Bray D (2004a). Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys Biol 1, 137–151. [DOI] [PubMed] [Google Scholar]
- Andrews SS, Bray D (2004b). Stochastic simulation of chemical reactions with spatial resolution and single molecule detail. Phys Biol 1, 137–151. [DOI] [PubMed] [Google Scholar]
- Angermann BR, Klauschen F, Garcia AD, Prustel T, Zhang F, Germain RN, Meier-Schellersheim M (2012). Computational modeling of cellular signaling processes embedded into dynamic spatial contexts. Nature Methods 9, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin A, Ross J, McAdams HH (1998). Stochastic kinetic analysis of developmental pathway bifurcation in Phage λ-infected Escherichia coli cells. Genetics 149, 1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilgan E, Wirtz D, Sun SX (2006). Mechanics and dynamics of actin-driven thin membrane protrusions. Biophys J 90, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod D, Wang MD (1994). Reduction-of-dimensionality kinetics at reaction-limited cell surface receptors. Biophys J 66, 588–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartocci E, Bortolussi L, Milios D, Nenzi L, Sanguinetti G (2015). Studying emergent behaviours in morphogenesis using signal spatio-temporal logic. In: Hybrid Systems Biology. HSB 2015. Lecture Notes in Computer Science, tructural Chemistry and Molecular Biology, ed. Abate A, Šafránek D, Springer. [Google Scholar]
- Bartocci E, Lio P (2016). Computational modeling, formal analysis, tools for systems biology. PLoS Comput Biol 12, e1004591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley CG, Ellis LM (2012). Raise standards for preclinical cancer research. Nature 483, 531–533. [DOI] [PubMed] [Google Scholar]
- Berg HC, Purcell EM (1977). Physics of chemoreception. Biophys J 20, 193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutillier P, Maasha M, Li X, Medina-Abarca HF, Krivine J, Feret J, Cristescu I, Forbes AG, Fontana W (2018). The Kappa platform for rule-based modeling. Bioinformatics 34, i583–i592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Hossain MJ, Hériché J-K, Politi AZ, Walther N, Koch B, Wachsmuth M, Nijmeijer B, Kueblbeck M, Martinic-Kavur M, et al. (2018). Experimental and computational framework for a dynamic protein atlas of human cell division. Nature 561, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Terebus A, Liang J (2016). State space truncation with quantified errors for accurate solutions to discrete chemical master equation. Bull Math Biol 78, 617–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC (1994). Green fluorescent protein as a marker for gene expression. Science 263, 802–805. [DOI] [PubMed] [Google Scholar]
- Chattaraj A, Youngstrom M, Loew LM (2019). The interplay of structural and cellular biophysics controls clustering of multivalent molecules. Biophys J 116, 560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, De Schutter E (2017). Parallel STEPS: large scale stochastic spatial reaction-diffusion simulation with high performance computers. Front Neuroinform 11, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B-C, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, et al. (2014). Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 346, 1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YE, Tropini C, Jonas K, Tsokos CG, Huang KC, Laub MT (2011). Spatial gradient of protein phosphorylation underlies replicative asymmetry in a bacterium. Proc Natl Acad Sci USA 108, 1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew WX, Kaizu K, Watabe M, Muniandy SV, Takahashi K, Arjunan SNV (2018). Reaction-diffusion kinetics on lattice at the microscopic scale. Phys Rev E 98, 032418. [DOI] [PubMed] [Google Scholar]
- Chew WX, Kaizu K, Watabe M, Muniandy SV, Takahashi K, Arjunan SNV (2019). Surface reaction-diffusion kinetics on lattice at the microscopic scale. Phys Rev E 99, 042411. [DOI] [PubMed] [Google Scholar]
- Cho W-K, Spille J-H, Hecht M, Lee C, Li C, Grube V, Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JE (2004). Mathematics is biology’s next microscope, only better; biology is mathematics’ next physics, only better. PLoS Biol 2, e439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FC, Kimball GE (1949). Diffusion-controlled reaction rates. J Colloid Sci 4, 425–437. [Google Scholar]
- Cugno A, Bartol TM, Sejnowski TJ, Iyengar R, Rangamani P (2019). Geometric principles of second messenger dynamics in dendritic spines. Sci Rep 9, 11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes AT, Munro EM (2011). PAR-3 oligomerization may provide an actin-independent mechanism to maintain distinct par protein domains in the early Caenorhabditis elegans embryo. Biophys J 101, 1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibak M, Frohner C, Noe F, Hofling F (2019). Diffusion-influenced reaction rates in the presence of pair interactions. J Chem Phys 151. [DOI] [PubMed] [Google Scholar]
- Doi M (1976). Stochastic theory of diffusion-controlled reaction. J Phys a-Math Gen 9, 1479–1495. [Google Scholar]
- Donovan RM, Tapia J-J, Sullivan DP, Faeder JR, Murphy RF, Dittrich M, Zuckerman DM (2016). Unbiased rare event sampling in spatial stochastic systems biology models using a weighted ensemble of trajectories. PLoS Comput Biol 12, e1004611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawert B, Hellander A, Bales B, Banerjee D, Bellesia G, Daigle BJ, Douglas G, Gu MY, Gupta A, Hellander S, et al. (2016). Stochastic simulation service: bridging the gap between the computational expert and the biologist. PLoS Comput Biol 12, 1005220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE (2012). Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys 41, 429–452. [DOI] [PubMed] [Google Scholar]
- Enculescu M, Sabouri-Ghomi M, Danuser G, Falcke M (2010). Modeling of protrusion phenotypes driven by the actin-membrane interaction. Biophys J 98, 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres RG (2015). Bistability: requirements on cell-volume, protein diffusion, thermodynamics. PLoS One 10, e0121681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erban R, Chapman SJ (2009). Stochastic modelling of reaction-diffusion processes: algorithms for bimolecular reactions. Phys Biol 6, 046001. [DOI] [PubMed] [Google Scholar]
- Erdemir A, Guess TM, Halloran J, Tadepalli SC, Morrison TM (2012). Considerations for reporting finite element analysis studies in biomechanics. J Biomech 45, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermak DL, Mccammon JA (1978). Brownian dynamics with hydrodynamic interactions. J Chem Phys 69, 1352–1360. [Google Scholar]
- Eungdamrong NJ, Iyengar R (2004). Computational approaches for modeling regulatory cellular networks. Trends Cell Biol 14, 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faeder JR (2011). Toward a comprehensive language for biological systems. BMC Biol 9, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fange D, Berg OG, Sjoberg P, Elf J (2010). Stochastic reaction-diffusion kinetics in the microscopic limit. Proc Natl Acad Sci USA 107, 19820–19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler PW, Hélie J, Duncan A, Chavent M, Koldsø H, Sansom MSP (2016). Membrane stiffness is modified by integral membrane proteins. Soft Matter 12, 7792–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Yogurtcu ON, Kothari R, Thorkelsdottir G, Sodt AJ, Johnson ME (2019). An implicit lipid model for efficient reaction-diffusion simulations of protein binding to surfaces of arbitrary topology. J Chem Phys 151, 124115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Qian H (2013). Chemical Master Equation, New York: Springer. [Google Scholar]
- Ghaemi Z, Peterson JR, Gruebele M, Luthey-Schulten Z (2020). An in-silico human cell model reveals the influence of spatial organization on RNA splicing. PLoS Comput Biol 16, e1007717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie DT (1976). General method for numerically simulating stochastic time evolution of coupled chemical-reactions. J Comput Phys 22, 403–434. [Google Scholar]
- Gillespie DT (1992). A rigorous derivation of the chemical master equation. Physica A 188, 404–425. [Google Scholar]
- Gillespie DT, Hellander A, Petzold LR (2013). Perspective: Stochastic algorithms for chemical kinetics. J Chem Phys 138, 170901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopich I, Szabo A (2002a). Asymptotic relaxation of reversible bimolecular chemical reactions. Chemical Physics 284, 91–102. [Google Scholar]
- Gopich I, Szabo A (2002b). Kinetics of reversible diffusion influenced reactions: The self-consistent relaxation time approximation. J Chem Phys 117, 507–517. [Google Scholar]
- Gopich IV, Ovchinnikov AA, Szabo A (2001). Long-time tails in the kinetics of reversible bimolecular reactions. Phys Rev Lett 86, 922–925. [DOI] [PubMed] [Google Scholar]
- Greenwald EC, Mehta S, Zhang J (2018). Genetically encoded fluorescent biosensors illuminate the spatiotemporal regulation of signaling networks. Chem Rev 118, 11707–11794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregor T, Wieschaus EF, McGregor AP, Bialek W, Tank DW (2007). Stability and nuclear dynamics of the bicoid morphogen gradient. Cell 130, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grima R, Schnell S (2008). Modelling reaction kinetics inside cells. Essays Biochem 45, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen D, Knap J, Neumann P, Suleimenova D, Veen L, Leiter K (2019). Mastering the scales: a survey on the benefits of multiscale computing software. Philos Trans A Math Phys Eng Sci 377, 20180147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guckenberger A, Gekle S (2017). Theory and algorithms to compute Helfrich bending forces: a review. J Phys Condens Matter 29, 203001. [DOI] [PubMed] [Google Scholar]
- Gunawardena J (2014). Models in biology: ‘accurate descriptions of our pathetic thinking’. BMC Biol 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li D, Zhang S, Yang Y, Liu J-J, Wang X, Liu C, Milkie DE, Moore RP, Tulu US, et al. (2018). Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175, 1430–1442.e1417. [DOI] [PubMed] [Google Scholar]
- Gupta A, Mendes P (2018). An Overview of network-based and -free approaches for stochastic simulation of biochemical systems. Computation (Basel) 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP (2007). Universally sloppy parameter sensitivities in systems biology models. PLoS Comput Biol 3, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halatek J, Frey E (2012). Highly canalized MinD transfer and MinE sequestration explain the origin of robust MinCDE-protein dynamics. Cell Rep 1, 741–752. [DOI] [PubMed] [Google Scholar]
- Hallock MJ, Stone JE, Roberts E, Fry C, Luthey-Schulten Z (2014). Simulation of reaction diffusion processes over biologically relevant size and time scales using multi-GPU workstations. Parallel Comput 40, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LA, Hogg JS, Tapia JJ, Sekar JA, Gupta S, Korsunsky I, Arora A, Barua D, Sheehan RP, Faeder JR (2016). BioNetGen 2.2: advances in rule-based modeling. Bioinformatics 32, 3366–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenauer J, Wolf V, Kazeroonian A, Theis FJ (2014). Method of conditional moments (MCM) for the Chemical Master Equation. J Math Biol 69, 687–735. [DOI] [PubMed] [Google Scholar]
- Health UNIO (2018). Enhancing reproducibility through rigor and transparency. Available at: https://grants.nih.gov/policy/reproducibility/index.htm (accessed 1 August 2020).
- Helfrich W (1973). Elastic Properties of Lipid Bilayers - Theory and Possible Experiments. Z Naturforsch C J Biosci 28, 693–703. [DOI] [PubMed] [Google Scholar]
- Hellander S, Hellander A, Petzold L (2012). Reaction-diffusion master equation in the microscopic limit. Phys Rev E 85, 042901. [DOI] [PubMed] [Google Scholar]
- Hellander S, Hellander A, Petzold L (2015). Reaction rates for mesoscopic reaction-diffusion kinetics. Phys Rev E Stat Nonlin Soft Matter Phys 91, 023312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellander S, Hellander A, Petzold L (2017). Mesoscopic-microscopic spatial stochastic simulation with automatic system partitioning. J Chem Phys 147, 234101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Frohner C, Noe F (2019). ReaDDy 2: Fast and flexible software framework for interacting-particle reaction dynamics. PLoS Comput Biol 15, e1006830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard M, Rutenberg AD (2003). Pattern formation inside bacteria: fluctuations due to the low copy number of proteins. Phys Rev Lett 90, 128102. [DOI] [PubMed] [Google Scholar]
- Huang KC, Meir Y, Wingreen NS (2003). Dynamic structures in Escherichia coli: spontaneous formation of MinE rings and MinD polar zones. Proc Natl Acad Sci USA 100, 12724–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, Arkin AP, Bornstein BJ, Bray D, Cornish-Bowden A, et al. (2003). The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531. [DOI] [PubMed] [Google Scholar]
- Isaacson SA (2009). The reaction-diffusion master equation as an asymptotic approximation of diffusion to a small target. SIAM J Appl Math 70, 77–111. [Google Scholar]
- Isaacson SA (2013). A convergent reaction-diffusion master equation. J Chem Phys 139. [DOI] [PubMed] [Google Scholar]
- Isaacson SA, Zhang Y (2018). An unstructured mesh convergent reaction-diffusion master equation for reversible reactions. J Comput Phys 374, 954–983. [Google Scholar]
- Janes KA, Lauffenburger DA (2013). Models of signalling networks—what cell biologists can gain from them and give to them. J Cell Sci 126, 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilkine A, Edelstein-Keshet L (2011). A comparison of mathematical models for polarization of single eukaryotic cells in response to guided cues. PLoS Comput Biol 7, e1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME (2018). Modeling the self-assembly of protein complexes through a rigid-body rotational reaction-diffusion algorithm. J Phys Chem B 122, 11771–11783. [DOI] [PubMed] [Google Scholar]
- Johnson ME, Hummer G (2014). Free-propagator reweighting integrator for single-particle dynamics in reaction-diffusion models of heterogeneous protein-protein interaction systems. Physical Review X 4, 031037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelina M, Kulik HJ (2017). Systematic quantum mechanical region determination in QM/MM simulation. J Chem Theory Comput 13, 563–576. [DOI] [PubMed] [Google Scholar]
- Karsenti E, Nédélec F, Surrey T (2006). Modelling microtubule patterns. Nat Cell Biol 8, 1204–1211. [DOI] [PubMed] [Google Scholar]
- Keating SM, Waltemath D, Konig M, Zhang FK, Drager A, Chaouiya C, Bergmann FT, Finney A, Gillespie CS, Helikar T, et al. (2020). SBML Level 3: an extensible format for the exchange and reuse of biological models. Mol Syst Biol 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren K, Pincus Z, Allen GM, Barnhart EL, Marriott G, Mogilner A, Theriot JA (2008). Mechanism of shape determination in motile cells. Nature 453, 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr RA, Bartol TM, Kaminsky B, Dittrich M, Chang JCJ, Baden SB, Sejnowski TJ, Stiles JR (2008). Fast Monte Carlo simulation methods for biological reaction-diffusion systems in solution and on surfaces. SIAM J Sci Comput 30, 3126–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kestler HA, Wawra C, Kracher B, Kuhl M (2008). Network modeling of signal transduction: establishing the global view. Bioessays 30, 1110–1125. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN, Hoek JB, Westerhoff HV (2000). Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol 10, 173–178. [DOI] [PubMed] [Google Scholar]
- Kim JS, Yethiraj A (2009). Effect of macromolecular crowding on reaction rates: a computational and theoretical study. Biophys J 96, 1333–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkow K, Odde DJ (2008). Model for protein concentration gradients in the cytoplasm. Cell Mol Bioeng 1, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (1985). A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc Natl Acad Sci USA 82, 3055–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TL, Upadhyayula S, Milkie DE, Singh V, Wang K, Swinburne IA, Mosaliganti KR, Collins ZM, Hiscock TW, Shea J, et al. (2018). Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 360, eaaq1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew LM, Schaff JC (2001). The Virtual Cell: a software environment for computational cell biology. Trends Biotechnol 19, 401–406. [DOI] [PubMed] [Google Scholar]
- Lopez CF, Muhlich JL, Bachman JA, Sorger PK (2013). Programming biological models in Python using PySB. Mol Syst Biol 9, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majarian TD, Murphy RF, Lakdawala SS (2019). Learning the sequence of influenza A genome assembly during viral replication using point process models and fluorescence in situ hybridization. PLoS Comput Biol 15, e1006199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masid M, Ataman M, Hatzimanikatis V (2020). Analysis of human metabolism by reducing the complexity of the genome-scale models using redHUMAN. Nat Commun 11, 2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matković K, Gračanin D, Hauser H (2018). Visual analytics for simulation ensembles. In 2018 Winter Simulation Conference (WSC), Gothenburg Sweden IEEE Press, 321–335, 10.1109/WSC.2018.8632312.
- Mattis DC, Glasser ML (1998). The uses of quantum field theory in diffusion-limited reactions. Rev Mod Phys 70, 979–1001. [Google Scholar]
- Maus C, Rybacki S, Uhrmacher AM (2011). Rule-based multi-level modeling of cell biological systems. BMC Syst Biol 5, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuarrie DA (1967). Stochastic Approach to Chemical Kinetics, London: Methuen. [Google Scholar]
- Meier-Schellersheim M, Varma R, Angermann BR (2019). Mechanistic models of cellular signaling, cytokine crosstalk, cell-cell communication in immunology. Front Immunol 10, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski PJ, Loew LM (2016). SpringSaLaD: A spatial, particle-based biochemical simulation platform with excluded volume. Biophys J 110, 523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R (2013). What is the total number of protein molecules per cell volume? A call to rethink some published values. BioEssays 35, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton AP (1995). Confinement as a determinant of macromolecular structure and reactivity. II. Effects of weakly attractive interactions between confined macrosolutes and confining structures. Biophys J 68, 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton AP (2006). How can biochemical reactions within cells differ from those in test tubes? J Cell Sci 119, 2863–2869. [DOI] [PubMed] [Google Scholar]
- Mitra ED, Dias R, Posner RG, Hlavacek WS (2018). Using both qualitative and quantitative data in parameter identification for systems biology models. Nat Commun 9, 3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra ED, Suderman R, Colvin J, Ionkov A, Hu A, Sauro HM, Posner RG, Hlavacek WS (2019). PyBioNetFit and the biological property specification language. iScience 19, 1012–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilner A, Rubinstein B (2005). The physics of filopodial protrusion. Biophys J 89, 782–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraru II, Schaff JC, Slepchenko BM, Blinov ML, Morgan F, Lakshminarayana A, Gao F, Li Y, Loew LM (2008). Virtual cell modelling and simulation software environment. Iet Systems Biology 2, 352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli MJ, Wolde PRT, Allen RJ (2009). DNA looping provides stability and robustness to the bacteriophage λ switch. Proc Natl Acad Sci 106, 8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Bieling P, Fletcher DA (2018). From solution to surface to filament: actin flux into branched networks. Biophys Rev 10, 1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsky B, Khammash M (2006). The finite state projection algorithm for the solution of the chemical master equation. J Chem Phys 124, 044104. [DOI] [PubMed] [Google Scholar]
- Nedelec F, Foethke D (2007). Collective Langevin dynamics of flexible cytoskeletal fibers. New J Physics 9, 427–427. [Google Scholar]
- Nickaeen M, Novak IL, Pulford S, Rumack A, Brandon J, Slepchenko BM, Mogilner A (2017). A free-boundary model of a motile cell explains turning behavior. PLoS Comput Biol 13, e1005862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrup SH, Allison SA, Mccammon JA (1984). Brownian dynamics simulation of diffusion-influenced bimolecular reactions. J Chem Phys 80, 1517–1526. [Google Scholar]
- Novak IL, Slepchenko BM (2014). A conservative algorithm for parabolic problems in domains with moving boundaries. J Comput Phys 270, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odell GM, Foe VE (2008). An agent-based model contrasts opposite effects of dynamic and stable microtubules on cleavage furrow positioning. J Cell Biol 183, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onsum M, Rao CV (2007). A mathematical model for neutrophil gradient sensing and polarization. PLoS Comput Biol 3, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz F, Schlange T, Asadullah K (2011). Believe it or not: how much can we rely on published data on potential drug targets? Nature Reviews Drug Discovery 10, 712–712. [DOI] [PubMed] [Google Scholar]
- Prüstel T, Meier-Schellersheim M (2012). Exact Green’s function of the reversible diffusion-influenced reaction for an isolated pair in two dimensions. J Chem Phys 137, 054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangamani P, Levy MG, Khan S, Oster G (2016). Paradoxical signaling regulates structural plasticity in dendritic spines. Proc Natl Acad Sci 113, E5298–E5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangamani P, Mandadap KK, Oster G (2014). Protein-induced membrane curvature alters local membrane tension. Biophys J 107, 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, van der Schaft A, van Eunen K, Bakker BM, Jayawardhana B (2014). A model reduction method for biochemical reaction networks. BMC Syst Biol 8, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin DM, de Boer PAJ (1999). Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc Natl Acad Sci USA 96, 4971–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raue A, Kreutz C, Maiwald T, Bachmann J, Schilling M, Klingmuller U, Timmer J (2009). Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood. Bioinformatics 25, 1923–1929. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A (2000). EMBOSS: The European molecular biology open software suite. Trends Genet 16, 276–277. [DOI] [PubMed] [Google Scholar]
- Richmond BG, Wright BW, Grosse L, Dechow PC, Ross CF, Spencer MA, Strait DS (2005). Finite element analysis in functional morphology. Anat Rec A Discov Mol Cell Evol Biol 283a, 259–274. [DOI] [PubMed] [Google Scholar]
- Roberts E, Stone JE, Luthey-Schulten Z (2013). Lattice microbes: high-performance stochastic simulation method for the reaction-diffusion master equation. J Comput Chem 34, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein B, Larripa K, Sommi P, Mogilner A (2009). The elasticity of motor-microtubule bundles and shape of the mitotic spindle. Phys Biol 6, 016005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha T, Rathmann I, Viplav A, Panzade S, Begemann I, Rasch C, Klingauf J, Matis M, Galic M (2016). Automated analysis of filopodial length and spatially resolved protein concentration via adaptive shape tracking. Mol Biol Cell 27, 3616–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samoilov MS, Arkin AP (2006). Deviant effects in molecular reaction pathways. Nat Biotechnol 24, 1235–1240. [DOI] [PubMed] [Google Scholar]
- Sanghvi JC, Regot S, Carrasco S, Karr JR, Gutschow MV, Bolival B Jr., Covert MW (2013). Accelerated discovery via a whole-cell model. Nat Methods 10, 1192–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaff JC, Slepchenko BM, Loew LM (2000). Physiological modeling with virtual cell framework. In: Numerical Computer Methods, Part C, Vol. 321, New York: Elsevier, 1–23. [DOI] [PubMed] [Google Scholar]
- Schaff JC, Vasilescu D, Moraru II, Loew LM, Blinov ML (2016). Rule-based modeling with Virtual Cell. Bioinformatics 32, 2880–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillings C, Sunnåker M, Stelling J, Schwab C (2015). Efficient Characterization of Parametric Uncertainty of Complex (Bio)chemical Networks. PLoS Comput Biol 11, e1004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoerr D, Sanguinetti G, Grima R (2017). Approximation and inference methods for stochastic biochemical kinetics-a tutorial review. J Phys A Math Theor 50, 1–60. [Google Scholar]
- Schoneberg J, Noe F (2013). ReaDDy - A software for particle-based reaction-diffusion dynamics in crowded cellular environments. Plos One 8, e74261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoneberg J, Ullrich A, Noe F (2014). Simulation tools for particle-based reaction-diffusion dynamics in continuous space. Bmc Biophysics 7, 10.1186/s13628-014-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G, Haran G, Zhou HX (2009). Fundamental aspects of protein-protein association kinetics. Chem Rev 109, 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar JAP, Tapia JJ, Faeder JR (2017). Automated visualization of rule-based models. PLoS Comput Biol 13, e1005857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, Bank JA, Jumper JM, Salmon JK, Shan Y, et al. (2010). Atomic-level characterization of the structural dynamics of proteins. Science 330, 341–346. [DOI] [PubMed] [Google Scholar]
- Shlemov A, Golyandina N, Holloway D, Spirov A (2015). Shaped singular spectrum analysis for quantifying gene expression, with application to the early drosophila embryo. BioMed Res Int 2015, Article ID 689745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockley EM, Vrugt JA, Lopez CF (2018). PyDREAM: high-dimensional parameter inference for biological models in python. Bioinformatics 34, 695–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoup D, Lipari G, Szabo A (1981). Diffusion-controlled bimolecular reaction-rates - the effect of rotational diffusion and orientation constraints. Biophys J 36, 697–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Grima R (2016). Breakdown of the reaction-diffusion master equation with nonelementary rates. Phys Rev E 93, 052135. [DOI] [PubMed] [Google Scholar]
- Smith S, Grima R (2019). Spatial Stochastic Intracellular Kinetics: A Review of Modelling Approaches. Bull Math Biol 81, 2960–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Xu W, Sun Y, Faeder JR, Marai GE (2012). RuleBender: integrated modeling, simulation and visualization for rule-based intracellular biochemistry. BMC Bioinformatics 13(Suppl 8), S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CD, Nguyen H, Pande VS, Gruebele M (2002). Absolute comparison of simulated and experimental protein-folding dynamics. Nature 420, 102–106. [DOI] [PubMed] [Google Scholar]
- Sokolowski TR, Paijmans J, Bossen L, Miedema T, Wehrens M, Becker NB, Kaizu K, Takahashi K, Dogterom M, Ten Wolde PR (2019). eGFRD in all dimensions. J Chem Phys 150, 054108. [DOI] [PubMed] [Google Scholar]
- Spagnolo JF, Rossignol E, Bullitt E, Kirkegaard K (2010). Enzymatic and nonenzymatic functions of viral RNA-dependent RNA polymerases within oligomeric arrays. RNA 16, 382–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan V, Kalappurakkal JM, Mehta SB, Nordenfelt P, Moore TI, Koga N, Baker DA, Oldenbourg R, Tani T, Mayor S, et al. (2017). Actin retrograde flow actively aligns and orients ligand-engaged integrins in focal adhesions. Proc Natl Acad Sci USA 114, 10648–10653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Schulten K, Schulten Z (1980). 1st passage time approach to diffusion controlled reactions. J Chem Phys 72, 4350–4357. [Google Scholar]
- Takahashi K, Arjunan SN, Tomita M (2005). Space in systems biology of signaling pathways—towards intracellular molecular crowding in silico. FEBS Lett 579, 1783–1788. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanase-Nicola S, ten Wolde PR (2010). Spatio-temporal correlations can drastically change the response of a MAPK pathway. Proc Natl Acad Sci USA 107, 2473–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Sichau D, Iber D (2015). LBIBCell: a cell-based simulation environment for morphogenetic problems. Bioinformatics 31, 2340–2347. [DOI] [PubMed] [Google Scholar]
- Tauber UC, Howard M, Vollmayr-Lee BP (2005). Applications of field-theoretic renormalization group methods to reaction-diffusion problems. J Phys A-Math Gen 38, R79–R131. [Google Scholar]
- Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW (2010). Single-cell NF-κB dynamics reveal digital activation and analogue information processing. Nature 466, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiger CF, Krause F, Cedersund G, Palmer R, Klipp E, Hohmann S, Kitano H, Krantz M (2012). A framework for mapping, visualisation and automatic model creation of signal-transduction networks. Mol Syst Biol 8, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita M, Hashimoto K, Takahashi K, Shimizu TS, Matsuzaki Y, Miyoshi F, Saito K, Tanida S, Yugi K, Venter JC, et al. (1999). E-CELL: software environment for whole-cell simulation. Bioinformatics 15, 72–84. [DOI] [PubMed] [Google Scholar]
- Torney DC, McConnell HM (1983). Diffusion-limited reaction-rate theory for two-dimensional systems. Proc. R. Soc. Lond. A 387, 147–170. [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turing AM (1952). The chemical basis of morphogenesis. Phil Trans R Soc Lond B 237, 37–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B (2003). Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol 15, 221–231. [DOI] [PubMed] [Google Scholar]
- Van Kampen NG (2007). Stochastic Processes in Physics and Chemistry, 3rd ed., Amsterdam: Elsevier. [Google Scholar]
- van Zon JS, ten Wolde PR (2005). Simulating biochemical networks at the particle level and in time and space: Green’s function reaction dynamics. Phys Rev Lett 94, 128103. [DOI] [PubMed] [Google Scholar]
- Varga M, Fu Y, Loggia S, Yogurtcu ON, Johnson ME (2020). NERDSS: a nonequilibrium simulator for multibody self-assembly at the cellular scale. Biophys J 118, P3026–P3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar JM, Kueh HY, Barkai N, Leibler S (2002). Mechanisms of noise-resistance in genetic oscillators. Proc Natl Acad Sci USA 99, 5988–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Dassow G, Meir E, Munro EM, Odell GM (2000). The segment polarity network is a robust developmental module. Nature 406, 188–192. [DOI] [PubMed] [Google Scholar]
- von Smoluchowski M (1917). Attempt to derive a mathematical theory of coagulation kinetics in colloidal solutions. Z Phys Chem 92, 129. [Google Scholar]
- Wedlich-Soldner R, Altschuler S, Wu L, Li R (2003). Spontaneous cell polarization through actomyosin-based delivery of the Cdc42 GTPase. Science 299, 1231–1235. [DOI] [PubMed] [Google Scholar]
- Wils S, De Schutter E (2009). STEPS: modeling and simulating complex reaction-diffusion systems with python. Front Neuroinform 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf V, Goel R, Mateescu M, Henzinger TA (2010). Solving the chemical master equation using sliding windows. BMC Syst Biol 4, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolgemuth CW, Zajac M (2010). The Moving Boundary Node Method: A level set-based, finite volume algorithm with applications to cell motility. J Comput Phys 229, 7287–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Su M, Tong C, Wu M, Liu J (2018). Membrane shape-mediated wave propagation of cortical protein dynamics. Nat Commun 9, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Vendome J, Shapiro L, Ben-Shaul A, Honig B (2011). Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature 475, 510–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga A, Murad Y, Li ITS (2019). Quantifying molecular tension—classifications, interpretations and limitations of force sensors. Phys Biol 17, 011001. [DOI] [PubMed] [Google Scholar]
- Yogurtcu ON, Johnson ME (2015). Theory of bi-molecular association dynamics in 2D for accurate model and experimental parameterization of binding rates. J Chem Phys 143, 084117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogurtcu ON, Johnson ME (2018). Cytosolic proteins can exploit membrane localization to trigger functional assembly. PLoS Comput Biol 14, e1006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Angermann BR, Meier-Schellersheim M (2013). The Simmune Modeler visual interface for creating signaling networks based on bi-molecular interactions. Bioinformatics 29, 1229–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX (1990). Kinetics of diffusion-influenced reactions studied by Brownian dynamics. J Phys Chem 94, 8794–8800. [Google Scholar]
- Zhou HX (1993). Brownian dynamics study of the influences of electrostatic interaction and diffusion on protein-protein association kinetics. Biophys J 64, 1711–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX, Rivas G, Minton AP (2008). Macromolecular crowding and confinement: biochemical, biophysical, potential physiological consequences. Annu Rev Biophys 37, 375–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HX, Szabo A (1996). Theory and simulation of the time-dependent rate coefficients of diffusion-influenced reactions. Biophys J 71, 2440–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman SB, Minton AP (1993). Macromolecular crowding: biochemical, biophysical, physiological consequences. Annu Rev Biophys Biomol Struct 22, 27–65. [DOI] [PubMed] [Google Scholar]
- Zuckerman DM (2011). Equilibrium sampling in biomolecular simulations. Annu Rev Biophys 40, 41–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.