

Graphical abstract
Keywords: Triple-negative breast cancer, CRISPR/Cas9, Tumorigenesis, Transcription factor, Anti-cancer drug resistance
Highlights
-
•
Triple-negative breast cancer (TNBC) is a breast cancer subtype characterized by the loss of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor--2.
-
•
Aberrant gene expression and protein degradation are considered to underlie the onset of tumorigenesis and metastasis. Early detection in asymptomatic patients is the key to improving survival outcomes.
-
•
Potential applications of CRISPR/Cas9 and its variants in deciphering or engineering intricate molecular and epigenetic mechanisms associated with TNBC.
-
•
CRISPR/Cas9 based editing is a potential application for diagnosis and Drug Resistance in of TNBC.
Abstract
Breast cancer (BC) is the most common type of cancer in women at the global level and the highest mortality rate has been observed with triple-negative breast cancer (TNBC). Accumulation of genetic lesions an aberrant gene expression and protein degradation are considered to underlie the onset of tumorigenesis and metastasis. Therefore, the challenge to identify the genes and molecules that could be potentially used as potent biomarkers for personalized medicine against TNBC with minimal or no associated side effects. Discovery of the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9) arrangement and an increasing repertoire of its new variants has provided a much-needed fillip towards editing TNBC genomes. In this review, we discuss the CRISPR/Cas9 genome editing, CRISPR Technology for diagnosis of (Triple-negative breast cancer) TNBC, Drug Resistance, and potential applications of CRISPR/Cas9 and its variants in deciphering or engineering intricate molecular and epigenetic mechanisms associated with TNBC. Furthermore, we have also explored the TNBC and CRISPR/Cas9 genome editing potential for repairing, genetic modifications in TNBC.
1. Introduction
Triple-negative breast cancer (TNBC) is an HER-2-negative (human epidermal growth factor receptor 2), PR (progesterone receptor)-negative, and estrogen receptor-negative (ER)-a negative molecular subtype of breast cancer (BC) as shown in Fig. 1 [1]. Approximately 15–20% of all types of breast cancer are TNBC, which is the most aggressive form of BC, growing and spreading faster than other types [2], and is more common in younger women less than 50 years [3]. Besides, TNBC has been reported to be more common in cases of inherited BRCA DNA repair-associated (BRCA) mutations. Common signs and symptoms of triple-negative breast cancer include the presence of a mass or lump of breast tissue, pain in the breast, skin in the nipple turning inwards, or discharge from the nipple. Sometimes, it has been shown to have spread to the lymph nodes near the collar bone, causing a swelling [4]. There are various methods available for the diagnosis of TNBC. For instance, 3D digital breast tomosynthesis is a technique used for 3D mammography with X -rays producing a 3D structure of the breast, which helps in clarifying any abnormalities detected in the breast mammogram [5]. Breast radio-imaging technique such as magnetic resonance imaging (MRI) is also helpful in the diagnosis of breast tumor, while the breast ultrasound uses sound waves to examine the breast structure in mammography [6]. A breast biopsy is performed to allow the histopathological analysis of the specimen under a microscope and is used to test for identification of the status of ER, PR, and HER-2 [7].
Fig. 1.

Classification and characteristic of Triple Negative breast cancer.
There are various stages of TNBC as shown in Fig. 1; more specifically, abnormal cells are being defined as stage zero, whereas stage one is identified as small specific localized cancer not spread to or near the lymph node [8], [9], [10] In stage two, cancer is in a moderate stage and not spread away from lymph nodes. The cancer spread from the breast but not metastasized to specific organs is considered stage four [11]. Accordingly, when metastasized malignancies are proliferating in isolated organs, and once the genetic and hormonal factors have been identified, then the clinician would recommend the most effective treatment [12]. This is significant because these hormone receptors, which are usually found inside and on the surface of healthy breast cells, are activated when exposed to the corresponding hormones and set off a cascade of reactions in the cells to help them grow and function properly [13]. In particular, ER, PR, and HER-2 hormones are known to be involved in accelerating tumorigenesis, and because TNBC lacks receptors for ER, PR, HER-2, most of the treatments are based on hormone therapy, chemotherapy, and radiation therapy [14]. For example, one such approach is the dimensional computerized radiation therapy (3DCRT), which is recommended for six week. Radiation therapy depends on the technical expert [15]. The treatment options for TNBC are very constrained due to hormone therapy and drug targets not being sensitive and specific [16]. However, some common chemotherapeutic agents, such as Cytoxan, taxol, fluorouracil, are typically used. Therefore, it is urgently required to develop therapeutic options for the prevention of TNBC [17]. A negative test report could be further evaluated for TNBC using markers, such as cytokeratin 14, cytokine 5/6, low expression of claudin‐low/mesenchymal‐like levels, high-level expression of epithelial to mesenchymal transition (EMT) cells[18]. Furthermore, in some cases, the expression of epidermal growth factor (EGFR) and transmembrane glycoprotein Nmb (GPNMB) has been shown in TNBC [19]. Hormone therapy for positive-tested tumors could be carried out using receptor-specific medicine for ERα+ and PR, whereas HRE-+ tested tumors would be treated with HER-2+ therapy [14]. Sometimes these treatments are ineffective in the case of TNBC [20]. As mentioned, TNBC is more aggressive than other types of BC tumors, such as other ER-negative BC, and has a very weak prognosis compared with other positive tested tumors. Patients with TNBC are usually treated with combination therapy or surgery [21]. Here, we discuss the CRISPR genome editing, CRISPR Technology for the diagnosis of TNBC. Drug Resistance in TNBC and CRISPR/Cas9 genome editing, and future prospective.
2. Heterogenicity of TNBC and genome-based targeted therapy
TNBC is possessed histopathological differences and showing heterogenicity at the transcriptomic level [1], [4], [13]. Early transcriptomic profiling of breast cancer is devoted to classifying TNBC into distinct clinical and molecular subtypes that could guide treatment decisions [13], [16]. Chemotherapy remains the only systemic therapeutic option for women with curable disease and was the only option for metastatic [4], [7]. Drug selection is dependent on factors such as tumor burden, drug treatment options available, and the presence of comorbidities [16], [18], [22]. Chemotherapy approvals for the treatment of metastatic TNBC were based on subgroup analysis of larger all-comer clinical trials (Table 2) [4], [19], [21], [23]. For metastatic breast cancer, taxanes (paclitaxel, docetaxel, and albumin-bound paclitaxel), anthracyclines (doxorubicin, pegylated liposomal doxorubicin, and epirubicin), anti-metabolites (capecitabine and gemcitabine), and nontaxane microtubule inhibitors (eribulin and vinorelbine) are preferred single-agent options based on the National Comprehensive Cancer Network guidelines. Other agents with activity are platinum salts (carboplatin and cisplatin) and other microtubule inhibitors, such as ixabepilone (Ixempra, Bristol-Myers Squibb) [23], [24], [25], [26], [27], [28], [29]. Some of these agents may appear to induce more favorable activity in TNBC populations [24], [25], [26], [27], [28], [29]. However, different types of therapies need to be developed through extensive clinical trials, which could work better than currently available treatment approaches [22]. Based on the gene expression studies of TNBC (Table 1) and the common characteristics of cancers, several potential targets have been identified in studies from in vitro, in vivo, and clinical trials are shown in Table 2, Table 3 [30], [31], [32], [33], [34], [35]. The efficacy of genomic-based targeted therapies in clinical trials in TNBC are shown in table 2 [16], [18], [22], [27], [30], [31], [32], [33], [34], [35]. In this context, we discuss here the gene regulation in TNBC and the therapeutic option of CRISPR/Cas9 mediated gene editing. Germline mutations in several genes involved in TNBC have been discussed, including those in BRCA1/2, BRIP1, PTEN, TP53, ATM, TERT, BARd1, and RAD51. BL-1 and BL-2 basal-like BC (BLBC) tumor sub-categories represent 80–90% of TNBC and are known to be involved in enhanced cell cycle progression and loss of checkpoint function due to increased proliferation. Further, the BL-2 subtype has been shown to have a role in the activation of EGFR and TP53 [36]. The subtype of mesenchyme stems-like is known to express the VEGFR2 (vascular endothelial growth factor receptor 2) and be responsive against inhibitors of mTOR and tyrosine kinase (TK) [37]. Patients with the luminal androgen receptor (LAR) subtype are known to be resistant to AR antagonists, such as bicalutamide [38]. In the case of basal-like breast cancer (BLBC), deregulation of BRCA1 along with overexpression of TP53 has also been reported [39]. Similarly, TP53 has been reported to be deleted or mutated in 60–80% of TNBC cases [40]. ER-negative TERT variants have also been detected in TNBC [30]. Besides, studies have shown that downregulation of the PTEN and TP53 genes are very common during the development of TNBC, with both genes being closely related due to the functional interaction of their proteins [31]. Loss of the expression of PTEN has been reported to promote tumorigenesis and inactivate the expression of TP53, leading to restricted mesenchymal features and poor clinical outcomes [41]. Experts from the field of breast cancer have made efforts to explore novel molecular therapeutic targets. However, TNBC remains very challenging, with the highest mortality rate among all types of breast cancer. Based on the protein expression of HER-2, BC tumors can be HER-2-positive or -negative [42].
Table 2.
Efficacy of genomic-based targeted therapies in clinical trials in TNBC.
| Drug | Targeted Mechanism | Targeted Patient | Status | Efficacy | Clinical trial ID |
|---|---|---|---|---|---|
| Buparlisib | at PI3K inhibitor | Patients with HER-2 Negative, Metastatic TNBC or Without PI3K Activation (BELLE-4) | Randomized phase III | PFS (full population): 8.0 vs. 9.2 (HR, 1.18. 95% CI, 0.82–1.68) |
NCT01572727 |
| Ipatasertib | AKT inhibitor | Metastatic TNBC | Randomized phase II | PFS (PIK3CA/AKT1/PTEN -altered): 9.0 vs. 4.9 (HR, 0.44; 95% CI, 0.20–0.99; P = 0.041) |
NCT02162719 |
| MK2206 | AKT inhibitor | Any tumor ER/PgR status, any HER-2/neu | Randomized phase II | pCR (all): 35.2 vs. 21.1 NCT01042379 pCR (TNBC): 40.2 vs. 22.4 |
NCT01042379 |
| Temsirolimus | mTORC1 inhibitor |
Metastatic metaplastic TNBC |
Phase I | ORR (PI3K-activated): 31 (95% CI, 16–50) |
NCT00761644 |
| Everolimus | mTORC1 inhibitor |
Patients With Stage II or Stage III TNBC | Phase II | pCR (all): 36 vs. 48 (P = 0.41) |
NCT00930930 |
| Panitumumab | K-ras and PI3K-activating mutations, EGFR, PTEN, and p53 |
Metastatic metaplastic TNBC |
Phase II | PFS (all): 4.4 (95% CI, 3.2–5.5) |
NCT00894504 |
| Cetuximab | EGFR/HER-2 inhibitor |
Locally advanced/ metastatic HER-2- negative |
Phase II | EFS (TNBC EGFR + ): 4.2 vs. 4.9 EFS (TNBC EGFR − ): 5.2 vs. 4.3 |
NCT00075270 |
| Cobimetinib | RAS/ MAPK; MEK1/2 inhibitor |
Metastatic TNBC | Phase II | PFS (intent-to-treat): 5.5 vs. 3.8 (HR, 0.73; 95% CI, 0.43–1.24; P = 0.25) |
NCT02322814 |
| Ruxolitinib | JAK1/2 inhibitor, JAK2 amplifi |
Metastatic TNBC or IBC of any subtype |
Phase II | PFS (all): 1.2 (95% CI, 0.97–1.84) |
NCT01562873 |
| PF-03084014 | NOTCH | Patients with Advanced Breast Cancer | Phase II | ORR: 16 (95% CI, 4.5–36.1) |
NCT01876251 |
Table 1.
Potentially therapeutic targetable genes in TNBC.
| Molecular subtype | Pathways | Potentially targetable genes | References |
|---|---|---|---|
| Basal-like | p53 pathway | TP53 mutant, a gain of MDM2 | [40], [46] |
| PI3K/PTEN pathway | PTEN mutant/loss, INPP4B loss, PIK3CA mutant | [28], [41], [46] | |
| RB1 pathway | RB1 mutant /loss, CCNE1 amplification (amp), high expression of CDKN2A, low RB1 expression | [46], [47], [48], [49] | |
| ER-negative | AKT signaling | PIK3CA, AKT1, PTEN, PIK3R1, FOXO3 | [47], [48], [49] |
| Cell-cycle regulation | RB1, CDKN2A | [46], [47] | |
| Chromatin function | KMT2C, ARID1A, NCOR1, PBRM1, KDM6A | ]50] | |
| DNA damage and apoptosis | TP53, BRCA1, BRCA2 | [50], [51] | |
| MAPK signaling | NF1, MAP3K1, MAP2K4, KRAS | [52], [53] | |
| Tissue organization | CDH1, MLLT4 | [2], [13], [18], [53] | |
| Transcription regulation | TBX3, RUNX1, GATA3, ZFP36L1, MEN1 | [33], [34], [35], [53] | |
| Ubiquitination | USP9X, BAP1 | [53] | |
| Other | ERBB2, SMAD4, AGTR2 | [53], [54] | |
| Neoadjuvant chemotherapy (triple-negative) Genomic alteration | Cell cycle | RB1 loss, CDKN2A loss, CDKN2B loss, CDK4 amp, CDK6 amp, CCND1 amp, CCND2 amp, CCN D3 amp, CCNE1 amp, AURKA amp | [30], [31], [32], [33], [34], [35], [37], [38], [39], [54] |
| PI3K/mTOR pathway | PTEN mut/loss, PIK3CA mut/amp, PIK3R1 mut/amp, AKT1 amp, AKT2 amp, AKT3 amp, RAPTOR amp, RICTOR amp, TSC1 truncations/mut | [28], [41], [47], [48] | |
| Growth factor receptor | IGF1R amp, EGFR amp, MET amp, KIT amp, FGFR1 amp, FGFR2 amp, FGFR4 amp | [26], [34], [37], [38], [49] | |
| RAS/MAPK pathway | KRAS amp/gain, BRAF amp/gain, RAF1 amp/gain, NF1 truncations | [47], [48], [49], [51] | |
| DNA repair | BRCA1 truncations/loss/mut , BRCA2 truncations/loss/mut, ATM mut | [25], [50] | |
| JAK2/STAT3 pathway | JAK2 amp | [50], [51], [52] |
Table 3.
Clinical trials that use CRISPR/Cas9 genome-editing technologies.
| Clinical status | Target | Interventions | Status | Clinical Trials Gov Identifier |
|---|---|---|---|---|
| Programmed Cell Death-1 Knockout Engineered T Cells in Patients With Previously Treated Advanced Esophageal Squamous Cell Carcinoma by CRISPR | To evaluate efficiency and safety of PD1 in regulating T cells | PD1-KO in regulating T cells immunity | Completed | NCT03081715 |
| Stem Cells in NF1 Patients With Tumors of the Central Nervous System | To screen and identify alleviating drugs of diseases | Collection of stem cells | Phase –I Ongoing | NCT03332030 |
| Mesothelin-positive solid tumors | To evaluate efficiency and safety of edited antimesothelin CAR-T cells | PD1-KO anti-mesothelin CAR-T cells | Completed | NCT03545815 |
| Muscle-invasive bladder | To evaluate efficiency of PD1-KO T cells | PD1-KO T cells | Phase -I Completed | NCT02863913 |
| Cell Therapy for High Risk T-Cell Malignancies Using CD7-specific CAR Expressed On Autologous T Cells (CRIMSON) | To evaluate efficiency and safety of CAR/28zeta CAR-T cells | CAR/28zeta CAR-T cells, Flu, CTX | Phase -I | NCT03690011 |
| CRISPR-Cas9 Gene-Editing CAR-T Cells | efficiency and safety of CD19 and CD20/CD22 CAR-T cells | CD19 and CD20 or CD22 CAR-T cells | Phase I/II | NCT03398967 |
| CRISPR-Cas9 Gene-Editing CAR-T Cells Targeting CD19(UCART019) in Patients With Relapsed or Refractory CD19 + Leukemia and Lymphoma | To monitor GVHD of allogeneic TCR- and B2M-disrupted CD19 CAR-T cells | TCR- and B2M-disrupted CD19 CAR-T cells | Phase I & II | NCT03166878 |
| CRISPR Gene Edited to Eliminate Endogenous TCR and PD-1 (NYCE T Cells) | To evaluate efficiency and safety of CAR-T cells | TCRendo and PD1, CAR-T cells. | Phase -I trial completed | NCT03399448 |
| CRISPR-Cas9 mediated PD-1 knockout-T cells from autologous origin | D-1 Knockout EBV-CTLs for Advanced Stage EBV Associated Malignancies | T cells | Phase -II and III trial completed | NCT03044743 |
| A Dose-escalation Phase I Trial of PD-1 Knockout Engineered T Cells for the Treatment of Metastatic Renal Cell Carcinoma | To evaluate efficiency and safety of PD1-KO T cells | IL-2, CTX, PD1-KO T cells | Phase -I COmpleted | NCT02867332 |
| CRISPR Cas9 in the laboratory (PD-1 Knockout T cells) | PD–1 | T cells | Phase-I Completed | NCT02867345 |
| CRISPR Cas9 in the laboratory (PD-1 Knockout T cells). | PD–1 | T cells | Phase-I Completed | NcT02793856 |
| CRISP-Cas9 system and EBV-CTL was generated in the laboratory (PD-1 Knockout EBV-CTL). | Stage IV gastric arcinoma; Stage IV nasopharyngeal carcinoma; Stage IV T cell lymphoma | Drug: fludarabine, cyclophosphamide, interleukin-2 | Phase-I Completed | NCT03044743 |
| Trial in Patients With Metastatic Gastrointestinal Epithelial Cancer Administering Tumor-Infiltrating Lymphocytes in Which the Gene Encoding CISH Was Inactivated Using the CRISPR/Cas9 System | CISH, inactivated TIL | Drug: cyclophosphamide, fludarabine, aldesleukin | Phase-I/II Completed | NCT04426669 |
| Genome-wide CRISPR Screen for Host Factors Associated With Norovirus Infections in Stem Cell-derived Human Intestinal Enteroid Model | Host factors of norovirus | Duodenal biopsy; saliva | Phase-I/II Completed | NCT03342547 |
| CRISPR/Cas9-HPV16 E6/E7T1 or CRISPR/Cas9-HPV18 E6/E7T2 | HPV-related cervical | TALEN, CRISPR/Cas9 | Phase-I/II Completed | NCT03057912 |
All types of BC are tested for estrogenic receptor α positive (ERα+), progesterone receptor (PR), and HER-2+ [43]. In case the pathological report comes out positive for all above types of receptor, this would suggest the involvement of many receptors in the development of the tumor, whereas, if it is negative, this would mean that only a few or none type of receptors had any involvement [44]. The impact of the combined inactivation of these tumor suppressors, which frequently occurs in BC, is made more severe in the case of TNBC as shown in Fig. 1. Furthermore, these BC tumors are resistant to targeted therapy due to the lack of expression of ER and HER-2.
The identification of the clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR associated protein 9 (Cas9) system and an increasing repertoire of its therapeutic application have provided a much-needed fillip towards editing of the genomes of BC tumors. CRISPR DNA sequences are known to be mainly found in bacteria. These sequences derived from bacteriophages have been reported to play a major role in destroying viruses. The Cas9 enzyme recognizes and cleaves specific target CRISPR sequences. This naturally occurring gene editing facility has been demonstrated to have a wide application in the treatment and early diagnosis of diseases [45].
3. CRISPR genome editing
The CRISPR/Cas9 system has three mechanisms; a single guide RNA (sgRNA), which is specific to a target sequence of DNA; Cas9 protein with DNA endonuclease activity; and a tracrRNA that interacts with Cas9 [55]. The Rec-I protein component is known to help in the binding of the gRNA [55], [56]. The PAM-Sequences (protospacer adjacent to motif) and arginine bridge helix play a significant role in the binding of the target DNA. The crRNA-tracrRNA (trans-acting CRISPR RNA) used for single guided RNA (sgRNA) genetic engineering [56]. The gRNA binds to the target site in the genome and directs the Cas9 protein. The Cas9 protein is an RNA-guided nuclease discovered in the CRISPR type II adaptive immunity system of Streptococcus pyogenes and it is responsible for cleaving double-strand DNA [57]. The efficiency of the CRISPR/Cas9 complex can be influenced by several factors, such as sequence, length, the genomic locus of the target, chromatin accessibility, nucleosomes, and other components around gRNA-binding sites and secondary structure, of gRNAs, can influence their efficiency and specificity [58]. The gRNA sequence has a crucial role in the efficiency, specificity, and accuracy of CRISPR/Cas9- mediated genome editing Truncated gRNAs with shorter complementary nucleotides (less than20) can reduce off-target effects by 5000-fold without sacrificing on-target efficiency. Moreover, extending the gRNA duplex by 5 base pairs can significantly improve the knockout efficiency [55], [56], [57], [58], [59]. Different versions of CRISPR-associated nucleases are currently under development, greatly expanding the CRISPR-based toolbox for genome editing [60] . Cpf1 is an RNA-guided endonuclease that belongs to the class 2 CRISPR-Cas’s system, the same as Cas9 The Cas 9 protein, which leads to recognition and cleavage of targeted DNA, contains six components: RuvC, HNH, PAM Interacting, Bridge, REC II, and REC I [55]. The RucC and HNH domains help in breaking the target sequences; the break-in target sequence mainly occurs via two mechanisms: the homologous direct repair mechanism (HDR) and nonhomologous end-joining (NHEJ) [58]. The TALEN (transcription activator-like effector nucleases) is a category of restriction enzymes used in genetic engineering to cleave target sequences; this is done through effector DNA-binding domains for specific HDR target sequences, to repair target DNA sequences for gene editing, whereas NHEJ is used for gene editing through insertions or deletions [59], [60], [61]. Usually, NHEJ can either repair the break sequences or induce a frameshift mutation by using sgRNA and Cas9; hence a highly sensitive and specific break of any target can be easily achieved, as shown in Fig. 2. The CRISPR/Cas9 system has several advantages over ZFN and TALENs in terms of its simplicity, flexibility, and affordability [64]. The most important difference is that the CRISPR system relies on RNA–DNA recognition, rather than on the protein–DNA-binding mechanism [64]. Therefore, it is more achievable and easier to construct a customized CRISPR/Cas9 complex by only changing the gRNA sequence instead of engineering a new protein. The target sequence needs to be immediately upstream of a PAM sequence (50-NGG-30) [56]. This short sequence occurs approximately once every eight base pairs in the human genome, which makes it possible to design several gRNAs for one specific target gene [61], [65]. Experts from the field have explored the possibilities of using CRISPR-Cas9 variants for diagnosis and treatment purposes [62]. The CRISPR-based system has been used to diagnose miRNA (microRNA), circulating tumor DNA, cell-free DNA (cfDNA), and tumor-specific biomarkers [65], [66]. Moreover, CRISPR could be altered for increased accuracy, sensitivity, and specificity to minimize off-target effects [63]. The CRISPR-Cas system is categorized into various types: type-I utilizes the Cas3 protein, type -II uses Cas-9, and type-III utilizes Cas-10 [64]. Further, the CRISPR system is also divided into 2 classes; class I contains the crRNA-effector complexes (multisubunit), whereas class II is the single-subunit protein system [65], [66]. Important TNBC genes have been knocked out in several studies to produce a TNBC model for the various subtypes of the disease [67].
Fig. 2.

Genome editing techniques CRISPR/Cas9.
Accordingly, TNBC treatment and resistance genes that might have been disrupted could be repaired by CRISPR [68]. These developments could assist with the existing challenging problems observed in TNBC, thereby furthering the current era of precision medicine. Experts from this field have generated various models for exploring the molecular basis of TNBC [69]. Furthermore, its clinical application has been challenged by the deficiency of sensitive and specific delivery approaches for in vivo therapeutic genome editing tools. As such, a CRISPR-based designed approach was shown to reduce the growth and development of TNBC, and reduce their growth rate in mice up to 77% without any toxic effect on healthy tissues [70]. The CRISPR barcoding technology has also been used to screen for tumor growth and development [71], while genome-wide CRISPR-Cas-9 analysis has been used to detect potential therapeutic targets in TNBC as shown in table 3. Therefore, we discussed new potential molecular targets for TNBC using CRISPR-Cas-9, which demands the need for investigations of other unknown targets of CRISPR/Cas9. The CRISPR/Cas9 system could be useful in editing hot mutated regions in genes, thus enhancing the efficacy of drugs for clinical applications against TNBC.
4. CRISPR technology for diagnosis of TNBC
The common methods used for the diagnosis of TNBC include mammography (gold standard), MRI, sonography, and ultrasound. These techniques are with their limitations, for example, mammograms are used for local diagnosis but are not useful for metastatic BC, while ultrasound has been reported to not be reliable in the identification of BC [72]. Similarly, MRI, which has a high sensitivity of over 94, has limited applicability. Therefore, more reliable methods are required to detect and diagnose breast cancer [73]. A new diagnostic tool for breast cancer could be developed by using the CRISPR/Cas system. The CRISPR/Cas-based approach could be applied for target ctDNA in the ultrasensitive detection of target DNA-PCR [52]. Gene-specific non-target DNA is removed using the Cas9 protein and cpf1, and then Cas9 and Cpf1 would recognize different PAM sequences [74]. Therefore, this PCR approach could detect different new oncogenic mutations. Experts from the field have developed a CRISPR-based PCR for the detection of oncogenic specific mutations. In this approach, CRISPR-based PCR oncogenic mutations are detectable at different PAM sites [56], [75]. Furthermore, this approach could reduce the requirement for invasive methods and help early diagnosis of genetic mutations [76]. TNBC tumors have been shown to change their genotypes in response to the regulation by hormone receptors [77]. These mutations could be monitored using a CRISPR/Cas-based diagnostic approach using a chip for point-of-care diagnosis [78]. This combination would be more sensitive and specific with their limitations [79]. For instance, extensive work is required before CRISPR/Cas-based diagnostics are applied in clinical settings [79]. These might also provide clinicians with a piece of more relevant information for a patient with TNBC, which could then help in providing more effective treatment. Therefore, CRISPR/Cas-based diagnostics tools could be useful for point-of-care diagnosis.
The Hart lab has developed an 18,000 CRISPR-based polled library for protein-coding genes by using cell lines, which has been demonstrated to be helpful in diagnosis and treatment [80]. The CERES (cut-elimination by resolution) method is used to analyze gene-specific levels from CRISPR-Cas9, for the copy number-specific effect, thus decreasing the false-positive results [81]. Experts from the field have also developed a computational approach to take part in CRISPR/Cas9 screens originating from various types of libraries. These libraries were used to analyze data from>80 types of CRISPR/Cas9 panels in human cancer cells [82]. The CRISPR system has allowed for analogous and combinatorial genome-wide screening in live animals, allowing for the identification of tumor-causing genes [83].
5. Triple-negative breast cancer and CRISPR/Cas9 genome editing
CRISPR/Cas9-based genetically engineered and cell-specific delivery systems have been employed to gain more advanced approaches for drug resistance mechanisms [84], [85]. In addition, several TNBC models have been used, either consisting of cell lines derived from TNBC xenografts or transgenic genetically modified mice [86], the clinical outcome and limitations of which have been extensively analyzed. Researchers have also developed the application of microfluidic devices for 3D models. So, currently, TNBC diagnosis and treatment models have been limited by the transformation of the TNBC genome that is comprised of mutations and genomic reorganizations that unavoidably result from the progress of TNBC through clonal selection [2]. Presently, considerable research is going on to identify various protein biomarkers for early diagnosis and treatment [87]. For instance, EGFR is known to play important role in angiogenesis, inhibition of apoptosis, metastatic spread, and its expression in TNBC has been shown to create challenges in diagnosis and treatment. The protein expression profile of TNBC (estrogen receptors, progesterone receptors, and excess HER-2 protein) and non-TNBC tumors have been compared by researchers, and several proteins were identified to be either upregulated or downregulated, with the upregulated proteins being more important for therapeutic interest [88]. Upregulated proteins in TNBC are classified into various groups, based on their activities, such as EGFR proteins (role in angiogenesis), metabolic proteins (glutathione S-transferase, pyruvate kinase, glucosidase, and fatty acid synthesis), transcription and translation controlling proteins, cell adhesive proteins (Catherine), heat shock proteins, and keratins [89]. These upregulated proteins were analyzed in TNBC tumors and shown to regulate the response to cellular stress [90]. VEGF (Vascular endothelial growth factor) is commonly found in six isoforms, with the VEGF165 isoform being more common, and showing affinity to VEGFR-1, VEGFR-2, VEGFR-3, VEGFR-1, resulting in endothelial cell survival, migration, and proliferation [91] In patients with TNBC, the level of VEGF, which is a target for bevacizumab, was found to be significantly higher. Many researchers have observed that the levels of cytokines are higher in TNBC than in other types of BC [92].
TP53 gene mutations are associated with low chemotherapy response, expressing TP63 and TP73 proteins in TNBC [93]. Overexpression of TOP2A in TNBC has been reported to cause reduced sensitivity to anthracyclines; this enzyme plays a major role in transcription by creating a DSB and rejoining them, altering the topology of DNA. As such, mutations in this gene lead to altered protein function [94]. Heat shock proteins (HSPd) are cellular chaperons that regulate posttranslational modifications and help maintain the levels of AKT, RAF-1, and cyclin-dependent kinase-4 (CDK-4). Once a mutation occurs in this important gene, the corresponding protein is disrupted by the proteasome. The PU-H71 anticancer drug is known to cause resistance in TNBC using a similar mechanism [95]. Proteasome deprivation of major cellular proteins could be a target for cancer therapy and drug resistance progression [96]. TNBC is an immensely combative form of BC with high levels of genetic heterogeneity affected by specific transcription and hormone signaling [97]. For example, ERα upon ligation with its estrogen ligand is activated and binds to the transcription regulatory element near to the promoters or enhancers of target genes [98]. Distant estrogen response elements (DEREs) regulate transcription of the target gene through chromatin interactions and act as a hot spot region for ER-α-positive luminal BC [99]. Therefore, ERα-DERE complex loci could be a potential target for transcriptional factor modulation by CRISPR/Cas9 [100]. The UBR5 E3-ubiquitin ligase is known to be a key regulator for the growth and metastasis of TNBC [101]. CRISPR/Cas9-based knockout of UBR5 was shown to diminish tumor progression and metastasis in a murine model of TNBC (Table 4) [102]. Very rare mutations have been identified in the poly (ADP-ribose) polymerase family member 3 (PARP3) gene in endocrine therapy [103]. The PARP3 protein is a major driver of BC, as activation of PARP3 has been linked with tumor progression [104]. Accordingly, TGFβ-induced epithelial-to-mesenchymal transition (EMT) could be used for CRISPR/Cas9-mediated editing in the absence of PARP3, to inhibit the tumor growth of TNBC cells as well EMT via the Rictor/mTORC2 signaling pathway, which might be a major drug target for BC [105]. The annexin protein was also observed to be highly expressed during the progression of TNBC tumors [17], [54], [77]. RICTOR plays a major regulator constituent of the mTORC2 complex, it can affect the cell proliferation and migration by over activation of AKT, which regulates the activity of Rac1 by triggering the Rac-GEF Tiam 1, Increase the expression level of RAC1 lead to metastatic and chemotaxis of the cell [28], [41]. An alternate signaling pathway led to the destruction of the Rac1 inhibitor RhoGDI2, after activation of AKT [47]. Researcher from the field investigated that mTORC2 acts as an important link between metabolization of glucose and EGFR-TKI resistance [52], [84]. Furthermore, the mTOR Downstream region of mNF-kB is linked with therapy resistance by blocking the apoptosis process [24], [28]. CRISPR/Cas 9 based reprogramming of mTORC2 could lead to reducing resistance therapy [106]. It is thought that this mechanism does not dissolve after inhibiting AKT phosphorylation, but this process can be altered by knocking down RICTOR. RICTOR-mediated Akt phosphorylation (s473) can maintain the survival of HER-2-amplified breast cancer cells [107], [108]. RICTOR ablation led to lapatinib-mediated apoptosis in HER-2-improved breast cancer cells, After the knockdown of RICTOR by using CRISPR/Cas9, the sensitivity of lapatinib-resistant cells to drugs can be improved in an extremely beautiful and delicate manner [108], [109]. This process will make it more sensitive to RICTOR/mTORC2 targeting and that the combined use of TKIs and dual mTOR the inhibitor is an effective therapeutic strategy [28], [107]. A researcher from the field has targeted both the complex mTORC1 and mTORC2 by including ATP competitive TKI, targeting PI3K [108]. Furthermore, most of these targeted drugs are under clinical trials, mTORC1 and mTORC2 combined regimen show more sensitive and specific effects. RICTOR act as an effector component of PI3K/AKT/mTOR, which is extremely confined for patient’s assistance from targeted drugs [41], [48], [107].
Table 4.
Possible target of CRISPR-Cas9 system against drug-resistant molecules in TNBC.
| Molecular Target | Mechanism | Regulation | Application | references |
|---|---|---|---|---|
| Cripto-1 | Receptor for the TGF signaling pathway | Mutated proteins are often found in TNBC | maturation of notch receptors | [110] |
| UBR5 | E3 ubiquitin-protein ligase is actively involved in BC | Regulation in proliferation | Common in TNBC and is associated with high risk | [111] |
| ETV6-NTRK3 combination gene | Transcriptional repressor | NTRK3 is a membrane anchored tyrosine kinase | Secretary breast carcinoma diagnostic biomarker | [112] |
| KDM(5A,5B,5C,6B) | KDM5A deletion inhibits cell growth in RB-negative human cancer cell lines | Association between expression of histone demethylases | Inhibits cell growth in RB-negative human cancer cell lines | [113] |
| BRD4 | Recognizes and binds acetylated histones to maintains epigenetic regulation | Sustains TNBC migration and invasion | Tumors of both TNBC subtypes | [114] |
| ATK1 | Substantially increased in tumor proliferation pathways | Play important role in metastases and observed among the differentially expressed genes, | AKT3 is a potential target for TNBC treatment as combination therapy | [115] |
| MAP3K1 | Phosphorylating kinase enzymes integrating cellular receptor responses | Regulates apoptosis | Allelic frequency Analysis for BC | [116] |
| SHCBP1 | Actively participate in cell signaling pathways and proliferation | malignant MCF-7 | Acts as a positive regulator of FGF signaling in neural progenitor cells. | [117] |
| BRM/BRG1 | BRG1 is the catalytic subunit that disrupts chromatin target promoters | Overexpressed in breast cancer | SWI/SNF complexes following the loss of a single subunit. | [118] |
| MDR1 | Encodes a drug efflux pump involved with drug resistance | Sensitize breast cancer cells to chemotherapy | MDR1 drug mutation | [119] |
| PIK3CA | Interaction with the AKT and mTOR pathways | regulates cell growth, survival | PI3K inhibition as part of a combination therapy | [120] |
| GATA3 | A pattern of GATA3 antibody reactivity in estrogen receptor | Regulates epithelial cell differentiation | Immunohistochemical evaluation of GATA3 expression | [121] |
| MAG13-AKT3 (fusion gene) | MAG13 regulates cell activate AKT activity. | AKT3 regulates proliferation, | Enriched in TNBC | [122] |
| PTEN | Phosphate and tensin homolog tumor suppressor gene | Cell Cycle Regulation | Mutational analysis | [123] |
| BRCA1/2 | Tumor suppressor genes and maintain genetics stability, actively involved in DNA damage response | Linked with oestrogen-receptor (ER) negative | Germline Mutational analysis | [124] |
| miR-21 | Interact with PTEN lead to drug resistance | Regulates cellular proliferation | Confers resistance to chemotherapy | [125] |
| LSD1 | Transcriptional corepressor through demethylation of histone 3 lysine 4 (H3K4) | Modulates the growth of breast cancer cells | targeting LSD1 is an emerging option for the treatment | [126] |
| KRAS | GTPase that signals messages between the extracellular space and the nucleus | control cell growth, cell maturation, and cell death | KRAS mutations found in breast cancer | [127] |
| Molecular targeted agents | ||||
| SAHA | p57 | Reverses drug resistance | Pancreatic ductal adenocarcinoma | [128] |
| Trastuzumab | HER-2 | Reverses drug resistance | Breast cancer Multiple myeloma | [129] |
| Bortezomib | Rpn13 | Inhibits proliferation | Breast cancer Multiple myeloma | [130] |
| Imatinib | ASXL1 | Enhances in differentiation ability | Chronic myelocytic leukemia | [131] |
| Ispinesib | Kinesin-5 A133P | Resistance mechanism | Cervical cancer | [132] |
| Cytotoxic agents | ||||
| Cisplatin | p53, CTR | Induces cell cycle arrest and inhibits cancer cells growth | Oesophageal adenocarcinoma | [133] |
| Epirubicin | MLL | Reverses drug resistance | Bladder cancer | [134] |
| Paclitaxel | Rsf-1 | Reverses drug resistance | Lung cancer | [135] |
| Doxorubicin | P-glycoprotein | Increases sensitivity to doxorubicin | Breast cancer | [136] |
| Immunotherapy | ||||
| T cell therapies | PBAF | Enhances sensitivity to immunotherapy | Melanoma | [137] |
The findings from these studies led to the foundation that highly proliferative and metastatic BC cells are likely using these genomic and transcriptional profiles; therefore, these programs acting as cancer drivers, which could be targeted by gene-editing technologies, constituting the future of personalized cancer medicine.
6. TNBC drug resistance and CRISPR/Cas9
The emergence of drug resistance leads to failure of drugs and poor prognosis, and thus needs development of new and effective modalities are urgently needed to overcome multidrug resistance of cancers and improve outcomes [138]. The treatment of breast cancer is also associated with drug resistance, which poses a major challenge to the control of diseases [138], [139]. Cell or tissue-specific drug delivery is very challenging. Multidrug resistance in BC is known to occur via multiple mechanisms, usually causing a genetic shift [139], [140], as shown in Fig. 2. Mutations in the wild-type alleles of HER-1 and HER-2 have been shown to lead to changes in signaling pathways. For instance, regulated ERα and ERγ have been reported to reduce the response against the therapy of TNBCs [106], while kinase domain mutations activate HER-2 and cause the progression of multiple solid cancers. Posttranslational modifications in EGFR and HER-2 activated by PI3K, AKT, mTOR, and MAPK cell signaling pathways have been shown to cause drug resistance as shown in Fig. 3 [141].
Fig. 3.

Chemotherapy Resistance in Triple-Negative Breast TNBC.
As such, the CRISPR/Cas9 mediated technology could be used to prevent the resistance factor, by targeting membrane transport proteins and enhancing DNA repair and the efflux mechanism [142]. Experts from the field have enhanced the efficacy of transporter resistance protein by using CRISPR/Cas9 in a mouse model [143]. Similarly, inactivation of PTEN was reported to increase the expression of ABCG2 [144]. Suppression of the function of PTEN which increased the chance of breast cancer sensitivity to mTOR inhibitors, as shown in Fig. 3, Fig. 4, could be a potential target for CRISPR/Cas9 targeted gene editing in breast cancer [145]. Persons with genetic mutations in the BRCA1 or BRCA2 genes have faulty DNA repair and an increased risk of developing BC and other cancers [146]. A researcher from the field observed that tamoxifen-resistant BC cells were resistant to DNA-damaging chemotherapy because of upregulated BRCA1 [147]. The expression of BRCA1 has also been associated with a worse prognosis of patients with early breast cancer as discussed earlier, indicating the potential use of CRISPR/Cas9 targeting PI3K to reverse chemoresistance [148] as shown in table 4.
Fig. 4.

Probable targets of CRISPR/Cas9 in Drug resistance mechanism in TNBC, the target is shown as.
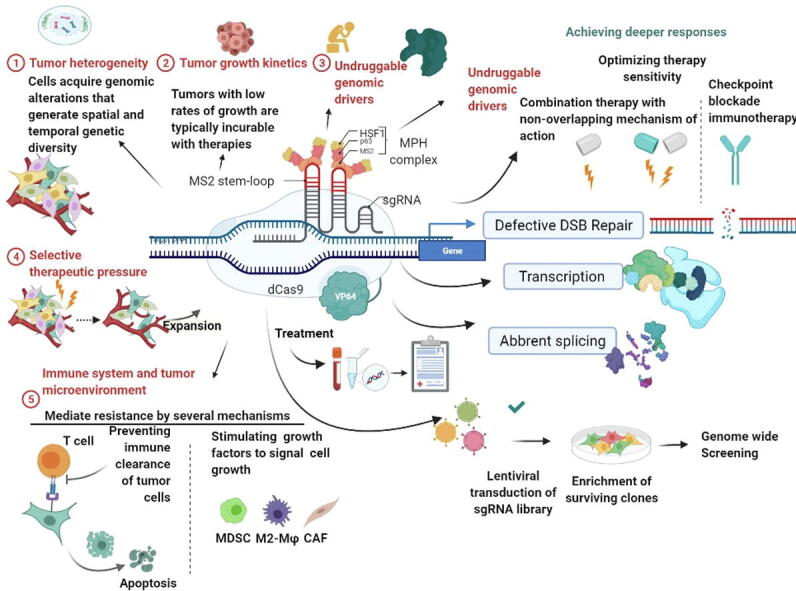
Genome-wide screening of CRISPR-Cas9 bases has provided information regarding mechanisms of drug resistance to protein kinase inhibitors in TNBC cell lines. Studies have identified the anaphase-promoting complex/cyclosome, which is closely linked with the TTK protein kinase and acts as an inhibitor in breast cancer cell lines [149]. Screening of cytotoxic T-cells using a CRISPR-Cas9 genome-wide analysis for resistance to tumor cells would also help to enhance the interpretation of metabolic activities based on tumor genetics networks [150]. High-throughput analysis of the CRISPR-Cas9-based library was used to analyze mutations for clinical drug resistance with high sensitivity and specificity [151]. Regarding cancer immunotherapy, a genome-scale CRISPR-Cas9 library comprised of about 123 000 sgRNAs was exploited to agitate genes in melanoma cells to mimic loss-of-function mutations implicated in the resistance to a T-cell-based therapeutic approach. The functional loss of the apelin receptor (APLNR) was shown to decrease the sensitivity and efficacy of the checkpoint blockade in animal models [152].
Furthermore, P-glycoproteins, also known as multidrug resistance protein 1 (MDR1), could be edited using CRISPR/Cas9 to recover drug sensitivity and specificity during the progression of BC [35]. Regulatory elements of AKT and ataxia-telangiectasia and Rad3-related (ATR), such as CDC25A have been reported to be significant components of drug resistance [29]. The VP64 transcriptional activator could be targeted using multiple sgRNA at the same time in the promoter region to identify the resistant gene [153], [154]. Various types of long noncoding RNAs (lncRNAs) have been demonstrated to be involved in cell signaling pathways and posttranscriptional regulation. For example, the HOX transcript antisense RNA (HOTAIR) and the increased expression of miR-200 were found to regulate metastasis in BC [155]. The loss of miR-200c, because of a p53 mutation, could upregulate the moesin oncogene and thus promote BC. Studies showed that moesin might play a role in metastasis and drug resistance of BC [156]. Bousquet et investigated pathological response after chemotherapy in TNBC observed that hypoxia increased drug resistance of autophagic TNBC stem-cells, and showed that molecular or chemical inhibition of the autophagic pathway was able to reverse chemoresistance [106].
CRISPR/Cas9-mediated MALAT1 promoter deletion in BT-549 TNBC model enhanced sensitivity to paclitaxel and doxorubicin, suggesting a role for MALAT1 in conferring resistance to the lncRNA transactional portrait and highlighted a complex regulatory network orchestrated by MALAT1 in the context of TNBC resistance to NAC therapy [157]. Screening of several TNBC cell lines showed altered Smad2 and Smad3 protein levels compared to normal mammary epithelial cells, suggesting the possibility that it could play an important role in the escape of cancer cells from TGF-β mediated growth inhibition [54]. Selective targeting of TGF-β-Smad3-TMEPAI axis by CRISPR-Cas9 system may be beneficial in triple-negative breast cancer therapy and prevention [54]. A researcher from the field investigated that the TMEPAI participated in the regulation of mRNA expression levels in drug efflux transporters (P-gp, BCRP, and multidrug resistance-associated protein. MEPAI knockout (KO) cells were previously developed from a TNBC cell line, Hs578T (wild-type/WT), using a CRISPR-Cas9 system [106], [109]. mRNA expression of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) was significantly increased in Hs578T-KO compared with that in Hs578T-WT cells [158]. Lian et al., investigated A genome-wide CRISPR screening to identify candidates involved in paclitaxel-resistant TNBCs. In vitro and in vivo genetic and cellular analyses explained the essential role of the MITR/MEF2A/IL11 axis in paclitaxel resistance and provided a novel therapeutic strategy for TNBC patients to overcome poor chemotherapy responses [159]. Lian et al. investigated A genome-wide CRISPR screening to identify candidates involved in paclitaxel-resistant TNBCs. in vitro and in vivo genetic and cellular analyses explained the essential role of the MITR/MEF2A/IL11 axis in paclitaxel resistance and provided a novel therapeutic strategy for TNBC patients to overcome poor chemotherapy responses [159]. Resistance to PARPi greatly hinders therapeutic effectiveness in TNBC, the mechanisms of PARPi resistance, including increased expression of MDR1, dissociation of PARP1 and PARG, HR restoration, and restoration of replication fork stalling, all reverse the DNA replication pressure and hinder the high sensitivity to PARPi treatment [160]. The CRISPR/Cas9 based approach could be useful in generating a knockout or knock-in TNBC genome and reverse the drug resistance as shown in Fig. 3, Fig. 4.
7. CRISPR and immunotherapy in TNBC
Immune destruction is one of the important mechanisms of the tumorigenesis process. Tumor cells can prevent immune effector cells via the secretion of extrinsic factors affecting the tumor microenvironment (TME) [137]. Tumour-specific CD8+ T cells are the main target, while immune checkpoint inhibitors have enhanced the discovery of novel targets [48], [49]. CRISPR/Cas-mediated genetic manipulation has strived to address some of the challenges regarding immune system misfunctioning from various prospects [68], [84], [161]. T cell-based immunotherapy is attributed to the implementation of ex vivo manipulated T lymphocytes aiming to eliminate tumors with TCR-engineered T lymphocytes and Chimeric Antigen Receptors T cells (CAR T Cells) as its main strategies [137]. In CAR T cell therapy, T cells can be derived from patients (autologous) or an allogeneic donor [152]. Using autologous T cells is a time-consuming process and largely depends on the quality and quantity of autologous T cells harvested from the patient [161], [162]. One of the substantial barriers in using allogeneic T cells in the presence of endogenous MHC class I and TCR on donor’s T lymphocytes, which cause alloreactivity and graft-versus-host disease (GVHD), respectively [137], [152], [162].
CRISPR/Cas9 is used to insert the CAR gene and remove the TCR gene concurrently by introducing the CAR gene into the TRAC locus [162]. They observed a regular CAR expression in T cells, increased potency of T cells, and decreased terminal differentiation and exhaustion in the mouse model of AML [161]. CRISPR/Cas9 technology was employed to knock out PDCD1, TRAC, and beta-2-microglobulin (β2M), which encodes the accessory chain of MHC class I in CD19 or PSCA CAR T cells [162]. In a similar study on EGFRvIII-targeted CAR T cells and their triple gene-edited CAR, T cells displayed an enhanced profile in preclinical glioblastoma models [48], [68]. Gene knock-out of PD-1 in Car T cells using CRISPR technology was also assessed against glioblastoma, hepatocellular, and K562 tumor cell lines and demonstrated enhanced anti-tumor activity, reduced exhaustion, and augmented killing power in Car T cells [163]. The role of programmed cell death 1 (PD-1) receptor-ligand (PD-L1 or PD-L2) interaction was highlighted as a major inhibitor pathway that may be hijacked by tumors to suppress immune control [164]. Atezolizumab is an engineered and humanized monoclonal antibody against PD-L1, which stimulates the T cell activity against cancer cells by inhibiting the binding to the PD-L1 receptors by activating an antitumor immune response without inducing antibody-dependent cellular cytotoxicity [163], [164].
8. CRISPR/Cas 9 delivery
The clinical outcome of CRISPR/Cas 9 delivery is mainly dependent on its effective potential to target more precise delivery methods [60], [143]. CRISPR/Cas9 system is the gRNA are delivered as DNA or RNA molecules associated with the other nucleases [143]. The Cas9 and sgRNA are delivered as ribonucleoprotein (RNP), a combination of Cas9 mRNA and sgRNA [60]. Plasmids are easily constructed, Delivery of the mRNA and sgRNA likewise evades the essential for nuclear localization and primes to transient Cas9 expression with condensed off-target effects [148], [156]. Viral and nonviral vector delivery systems are used. Electroporation, microinjection, hydrodynamic injection, and self-assembled nanoparticles (NPs), were used in ex vivo gene-editing as nonviral delivery techniques [148]. CRISPR/Cas9 systems have been used in investigations in vitro or in vivo by using combinational methods such as electroporation, injection, and nanoparticles [135]. Nonviral methods have unique advantages over viral vector delivery systems, due to their transient expression patterns [165], [166]. Viral vectors have the potential for the delivery of CRISPR/Cas9 in vivo in comparison to traditional non-integrated viral-based gene therapy and transgene expression is required for repeated dose, consistent genome editing is achieved by transient expression of CRISPR/Cas9 with a single administration [165]. The limited investigation has been carried out nonviral vectors in the clinical research stages [165]. Researchers from the field used lipids and gold nanoclusters as platforms for CRISPR/Cas9 system delivery to tumor cells [168]. Liposome-templated hydrogel nanoparticles are used by investigators to targeted delivery of CRISPR/ Cas9 in vivo for cancer, and which can penetrate the blood–brain barrier encouragingly [174]. Exosomes are used as a deliver CRISPR/Cas9 system in cancer cells effectively. Therefore, even a systemic delivery vector can efficiently deliver CRISPR/ Cas9 to cancer cells with minimum side effects. A considerate thoughtful of the cancer cell drug development and influences between diverse cellular trails in cancer cells is obligatory for emerging well-organized therapeutics, particularly because that each type of breast cancer has its cellular regulation and genetic influence [169]. CRISPR/Cas9 has shown high levels of efficiency, specificity, and stability, requirements of the viral vectors for delivery of CRISPR/Cas9 might not be as strict as previously required [165], [166]. The preferred viruses for the delivery of nucleases are AdVs (Adenovirus), integrase-defective lentiviruses (IDLV), and adeno-associated viruses (AAVs) [166]. More importantly, modified AdV vectors have shown more effective than IDLV for donor DNA delivery [165]. The disadvantage is the expression level of the coxsackie-adenovirus receptor (CAR) on the tumor cell surface, which controls the effectiveness of delivery using AdV vectors. The CAR expression level has a negative association with tumor cells [167]. IDLVs have been successfully used to deliver CRISPR/Cas9 and its donor template in vitro. Approximately 6–11% gene knock-in was achieved in hematopoietic stem and progenitor cells (HSPCs) [165]. Wrapping CRISPR/Cas9 into the suitable AAV (adeno-associated virus) serotype for targeted delivery is a proficient approach for cancer gene therapy. Various rAAV (Recombinant Adeno associated viruses) vectors have been industrialized for clinical trials for the treatment of an inherited retinal disease [168]. IDLVs have been used to co-deliver nucleases such as ZFNs, TALENs, and HDR donor templates into various types of cells with relatively high efficiency (>20%). Therefore, IDLV-mediated gene editing can cause off-target gene modifications [165], [167]. Reduce the expression of CAR on the tumor cell and nonappearance of precise receptors meaningfully confines the clinical application of AdV vectors in cancer cell therapy [167]. AdV vectors to tumor-specific cell surfaces such as EGFR and VEGFR can be selected for more specific targets [165], [166], [167], [168]. Nonviral vectors have been used for CRISPR/ Cas9 delivery in vitro and in vivo, cationic liposomes have been effectively used to deliver CRISPR/Cas9 to the inner ear in mouse models [165]. In another study, lipid-based vectors and AAVs accomplished > 6% gene repair in liver cells [168], [169]. Various studies have shown that CRISPR/Cas9 is proficient in editing genes with high precision and competence [171], [172], [173]. Precisely deliver CRISPR/Cas9 to TNBC is still the main impediment [171]. Delivery of CRISPR/Cas9 to tumor cells with such precision could accelerate the use of CRISPR-based cancer gene therapy from bench to bedside [168].
Nanoparticles are novel vehicles for CRISPR delivery compared to viral vectors and organic lipid and polymeric NPs [60], [174]. Nanoparticles are commonly used with other suitable cationic polyethyleneimines, which help in transfection efficiency [174]. Researchers are used polymeric nanoparticles in delivering siRNA to liver cancer cells [60]. Modification of cationic polylactides with PEG was done to prepare PEG to block cationic CPLA copolymer (PEG-b-CPLAs) for delivery of plasmid DNA into different cell lines [171], [174]. PEGylation of these nanocomplexes proficiently protected them from the extracellular environment and prohibited accumulation and consequent permission via RES [60], [168]. The investigator from the field explored that disulfide-modified hyaluronic acid (HA-SS-COOH) coated PEI/DNA complexes afforded a greater degree of shielding and higher transfection efficiency as compared to both DNA-PEI and DNA-PEI-HA, personalized nano-formulation has the potential to deliver the CRISPR/Cas9 cargo based on target [168], [174]. AuNPs (gold nanoparticles) can penetrate cells via a membrane synthesis procedure, avoiding potential lysosomal degradation [166], [170]. RNP binding has been achieved by binding a thiol-modified crRNA to the AuNP and reacting the resulting AuNP-crRNA with Cas9 to form the RNP [166,]. Transfer of the large CRISPR/Cas9 plasmid directing the gene has been designated using lipid-encapsulated TAT-coated AuNPs in vitro and in vivo melanoma tumors [166]. These Cas9-AuNCs possibly will simplify genome editing in systems where the Cas9, sgRNA, template for HDR is delivered separately. Nevertheless, the development of CRISPR delivery systems will benefit from the widespread clinical research that has already been conducted [166]. NPs have been established as delivery agents for gene therapy, and demonstrate excessively probable for the delivery of CRISPR/Cas9 systems [174]. Their dimensions for multi-functionalization permit for efficient delivery of all CRISPR/Cas9 systems. Before implementation of CRISPR/Cas9 systems delivery must be investigated in experimental animals based on delivery potential [169], [174].
9. Limitations of CRISPR/Cas9 system application
CRISPR/Cas9 systems have still some current limitations [156]. To enhance the sensitivity and specificity of a target sequence, thousands number of sgRNAs have been analyzed to create online tools to enable the identification of sensitive and specific guide RNAs for target sequences. The variants of Cas9 protein have been designed to increase the malleable and precision of genome editing [106]. Nevertheless, the molecular mechanisms underlying increase gRNA efficiency are not well known, there are some prediction values for guide activity in target genes [106], [156]. To overcome the limitations of CRISPR/Cas9 for some target loci, a researcher from the field has developed a method to improve the engineered clones, which can also be used for screening of target genes [175]. CRISPR/Cas9 system for clinical treatment is the existence of antigen-specific T-cells directed against Cas9 protein [156], [167]. To eliminate the effects of off-target, however, more scientific investigations are required to categorize and rule out the survival of SpCas9-specific T-cells through recombinant Cas9 [167]. There is a limitation in the CRISPR/Cas9 therapeutic approach due to target site recognition by PAM sequences. Researchers from the field had developed Cas 9 derivatives with modified Pam sequences by using advanced. SpCas9 PAM variants are more specific with fewer off-target effects [170]. The major challenge of CRISPR/Cas9 has to minimize off-target effects. Various approaches have been employed to test off-targets. Off-target effects should be identified with the help of advanced bioinformatics tools by searching the genome sequences contenting miss matches to the target, which are mainly followed by PAM sequences [172]. Furthermore, genome-wide analysis of off-target identification is provided vital insight into more specificity.
10. Future perspective
New targeted therapies are needed to improve the survival of TNBC patients. The molecular liabilities of subgroups of TNBC need to be identified to discover new therapeutic targets. The CRISPR/Cas9 technology has facilitated an attractive paradigm shift in gene editing for clinical and therapeutic intervention through the generation of various CRISPR/Cas9 based high-throughput screens. For instance, this technology was used for an efficient replacement of the hot spot region of the mutated sequence, which led to the suppression of tumor progression, reduction of drug resistance, and improvement of drug efficiency. CRISPR-based gene medicines, immunotherapy, or synthetic molecules have been used in emerging areas of cancer therapy; besides, recent advancements in the field have used CRISPR/Cas9 plasmids, sgRNAs, and mRNA for TNBC therapy. Although, CRISPR is highly specific, economically sustainable, and is a high throughput technique, on the other hand, its application involves measured risk of countering the toxic immune response of Cas protein, off-target effects, limitation of delivering the edited cells back into TNBC patients. Findlay et al., have investigated 4000 variants in a cancer-associated gene by using CRISPR/Cas9, which could help the identification of people at risk of BC [112]. Drost et al. have observed the mutation profiles of experimental models using CRISPR to modify human stem cells to have the genetic signature of cancer [171]. Moses et al. used a CRISPR/Cas9 based targeted activation of PTEN, which could substitute the current therapeutic approaches in BC [146]. Hu et al., utilized CRISPR/Cas9 based engineered the programmed cell death-1 (PD-1) gene locus in human primary T-cells, subsequent in a meaningfully abridged PD-1 inhabitant. The probable benefit of immune checkpoint mutation with CAR T-cells in monitoring solid tumors and delivering a substitute CAR T-cell policy for adoptive transfer therapy [173]. In conclusion, there is the potential application of the CRISPR/Cas9 system in the diagnosis and treatment of triple-negative breast cancer and its translation from the lab to the clinical setting.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
D. D. S. thankful to Amity University Rajasthan, Jaipur, India. Choi EH thankful for the Leading Foreign Research Institute Recruitment Program through the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (NRF-2016K1A4A3914113). D.K.Y. thankful to Gachon Institute of Pharmaceutical Science and the Department of Pharmacy, College of Pharmacy, Gachon University of Medicine and Science, Incheon, Korea.
Contributor Information
Eun-Ha Choi, Email: choipdp@gmail.com.
Dharmendra Kumar Yadav, Email: dharmendra30oct@gmail.com.
References
- 1.Anders, C. and L.A. Carey, Understanding and treating triple-negative breast cancer. Oncology (Williston Park), 2008. 22(11): p. 1233-9; discussion 1239-40, 1243. [PMC free article] [PubMed]
- 2.Fragomeni S.M., Sciallis A., Jeruss J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg Oncol Clin N Am. 2018;27(1):95–120. doi: 10.1016/j.soc.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assi H.A. Epidemiology and prognosis of breast cancer in young women. J Thorac Dis. 2013;5(Suppl 1):S2–S8. doi: 10.3978/j.issn.2072-1439.2013.05.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Mahmood S. Metastatic and triple-negative breast cancer: challenges and treatment options. Drug Deliv Transl Res. 2018;8(5):1483–1507. doi: 10.1007/s13346-018-0551-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo R. Ultrasound Imaging Technologies for Breast Cancer Detection and Management: A Review. Ultrasound Med Biol. 2018;44(1):37–70. doi: 10.1016/j.ultrasmedbio.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slanetz P.J., M., MPH, FACR, MRI of the breast and emerging technologies. UpToDate. 2020 [Google Scholar]
- 7.Joensuu K. ER, PR, HER-2, Ki-67 and CK5 in Early and Late Relapsing Breast Cancer-Reduced CK5 Expression in Metastases. Breast Cancer (Auckl) 2013;7:23–34. doi: 10.4137/BCBCR.S10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodriguez C. Concordance between HER-2 status determined by qPCR in Fine Needle Aspiration Cytology (FNAC) samples compared with IHC and FISH in Core Needle Biopsy (CNB) or surgical specimens in breast cancer patients. Mol Oncol. 2016;10(9):1430–1436. doi: 10.1016/j.molonc.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shyamala K., Girish H.C., Murgod S. Risk of tumor cell seeding through biopsy and aspiration cytology. J Int Soc Prev Community Dent. 2014;4(1):5–11. doi: 10.4103/2231-0762.129446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahba H.A., El-Hadaad H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12(2):106–116. doi: 10.7497/j.issn.2095-3941.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henry NL, S.P., Haider I, Freer PE, Jagsi R, Sabel MS, Cancer of the Breast, in Abeloff’s Clinical Oncology, A.J. Niederhuber JE, Doroshow JH, Kastan MB, Tepper JE, Editor. 2020, Elsevier: Philadelphia.
- 12.Zhang X.H. Metastasis dormancy in estrogen receptor-positive breast cancer. Clin Cancer Res. 2013;19(23):6389–6397. doi: 10.1158/1078-0432.CCR-13-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Y. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018;5(2):77–106. doi: 10.1016/j.gendis.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onitilo A.A. Breast cancer subtypes based on ER/PR and HER-2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7(1–2):4–13. doi: 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bakiu E. Comparison of 3D CRT and IMRT Tratment Plans. Acta Inform Med. 2013;21(3):211–212. doi: 10.5455/aim.2013.21.211-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isakoff S.J. Triple-negative breast cancer: role of specific chemotherapy agents. Cancer J. 2010;16(1):53–61. doi: 10.1097/PPO.0b013e3181d24ff7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberti M.P. Protein expression changes during human triple negative breast cancer cell line progression to lymph node metastasis in a xenografted model in nude mice. Cancer Biol Ther. 2012;13(11):1123–1140. doi: 10.4161/cbt.21187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Du F. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget. 2015;6(15):12890–12908. doi: 10.18632/oncotarget.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanematsu M. Clinical significance of glycoprotein nonmetastatic B and its association with HER-2 in breast cancer. Cancer Med. 2015;4(9):1344–1355. doi: 10.1002/cam4.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geyer F.C. The Spectrum of Triple-Negative Breast Disease: High- and Low-Grade Lesions. Am J Pathol. 2017;187(10):2139–2151. doi: 10.1016/j.ajpath.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao H. Triple-negative breast cancer: is there a treatment on the horizon? Oncotarget. 2017;8(1):1913–1924. doi: 10.18632/oncotarget.12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prat A. Predicting response and survival in chemotherapy-treated triple-negative breast cancer. Br J Cancer. 2014;8(111):1532–1541. doi: 10.1038/bjc.2014.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Molecular Cancer. 2019;18(1) doi: 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thu K.L. Disruption of the anaphase-promoting complex confers resistance to TTK inhibitors in triple-negative breast cancer. Proc Natl Acad Sci U S A. 2018;115(7):E1570–E1577. doi: 10.1073/pnas.1719577115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J.-M. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25(1):32–40. doi: 10.1093/annonc/mdt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thaler S. Proteasome inhibitors prevent bi-directional HER-2/estrogen-receptor cross-talk leading to cell death in endocrine and lapatinib-resistant HER-2+/ER+ breast cancer cells. Oncotarget. 2017;8(42):72281–72301. doi: 10.18632/oncotarget.20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myers S.M., Collins I. Recent findings and future directions for interpolar mitotic kinesin inhibitors in cancer therapy. Future Med Chem. 2016;8(4):463–489. doi: 10.4155/fmc.16.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steelman L.S. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008;27(29):4086–4095. doi: 10.1038/onc.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner J.M., Kaufmann S.H. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals. 2010;3(5):1311–1334. doi: 10.3390/ph3051311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens K.N., Vachon C.M., Couch F.J. Genetic susceptibility to triple-negative breast cancer. Cancer Res. 2013;73(7):2025–2030. doi: 10.1158/0008-5472.CAN-12-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu T. Cas9-based tools for targeted genome editing and transcriptional control. Appl Environ Microbiol. 2014;80(5):1544–1552. doi: 10.1128/AEM.03786-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bianchini, G., et al., Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nature Reviews Clinical Oncology, 2016. 13(11): p. 674-690. [79] [DOI] [PMC free article] [PubMed]
- 33.Stender J.D. The Estrogen-Regulated Transcription Factor PITX1 Coordinates Gene-Specific Regulation by Estrogen Receptor-Alpha in Breast Cancer Cells. Mol Endocrinol. 2011;25(10):1699–1709. doi: 10.1210/me.2011-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cicatiello L. Estrogen Receptor α Controls a Gene Network in Luminal-Like Breast Cancer Cells Comprising Multiple Transcription Factors and MicroRNAs. The American Journal of Pathology. 2010;176(5):2113–2130. doi: 10.2353/ajpath.2010.090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu P.Y. Spatiotemporal control of estrogen-responsive transcription in ERα-positive breast cancer cells. Oncogene. 2015;35(18):2379–2389. doi: 10.1038/onc.2015.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehmann B.D., Pietenpol J.A. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232(2):142–150. doi: 10.1002/path.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mina A., Yoder R., Sharma P. Targeting the androgen receptor in triple-negative breast cancer: current perspectives. Onco Targets Ther. 2017;10:4675–4685. doi: 10.2147/OTT.S126051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rampurwala M., Wisinski K.B., O'Regan R. Role of the androgen receptor in triple-negative breast cancer. Clin Adv Hematol Oncol. 2016;14(3):186–193. [PMC free article] [PubMed] [Google Scholar]
- 39.Abdeen S.K., Aqeilan R.I. Decoding the link between WWOX and p53 in aggressive breast cancer. Cell Cycle. 2019;18(11):1177–1186. doi: 10.1080/15384101.2019.1616998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varna M. TP53 status and response to treatment in breast cancers. J Biomed Biotechnol. 2011;2011 doi: 10.1155/2011/284584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haddadi N. PTEN/PTENP1: 'Regulating the regulator of RTK-dependent PI3K/Akt signalling', new targets for cancer therapy. Mol Cancer. 2018;17(1):37. doi: 10.1186/s12943-018-0803-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El Hachem G., Gombos A., Awada A. Recent advances in understanding breast cancer and emerging therapies with a focus on luminal and triple-negative breast cancer. F1000Res. 2019;8 doi: 10.12688/f1000research.17542.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang M.H. Estrogen receptor-positive breast cancer molecular signatures and therapeutic potentials (Review) Biomed Rep. 2014;2(1):41–52. doi: 10.3892/br.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siadati S. Correlation of ER, PR and HER-2/Neu with other Prognostic Factors in Infiltrating Ductal Carcinoma of Breast. Iran J Pathol. 2015;10(3):221–226. [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu Patrick D. Development and applications of CRISPR-Cas9 for genome engineering”. Cell. 2014;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alluri P., Newman L.A. Basal-like and triple-negative breast cancers: searching for positives among many negatives. Surg Oncol Clin N Am. 2014;23(3):567–577. doi: 10.1016/j.soc.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turner K.M. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc Natl Acad Sci. 2015;112(11):3421–3426. doi: 10.1073/pnas.1414573112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang F.F. Inactivation of PTEN increases ABCG2 expression and the side population through the PI3K/Akt pathway in adult acute leukemia. Cancer Lett. 2013;336(1):96–105. doi: 10.1016/j.canlet.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 49.Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers, 2017. 9(12). [DOI] [PMC free article] [PubMed]
- 50.Wu Q. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics. 2017;9(6):919–931. doi: 10.2217/epi-2017-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roy R., Chun J., Powell S.N. singn1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12(1):68–78. doi: 10.1038/nrc3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee S. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int J Mol Sci. 2020;21(3):1102. doi: 10.3390/ijms21031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rojas-Jiménez Ernesto. Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic. Genes. 2020;11(11):1367. doi: 10.3390/genes11111367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singha Prajjal K. Increased Smad3 and reduced Smad2 levels mediate the functional switch of TGF-β from growth suppressor to growth and metastasis promoter through TMEPAI/PMEPA1 in triple negative breast cancer. Genes & cancer. 2019;10(5–6):134–149. doi: 10.18632/genesandcancer.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Safari F. CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci. 2019;9:36. doi: 10.1186/s13578-019-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gleditzsch D. PAM identification by CRISPR-Cas effector complexes: diversified mechanisms and structures. RNA Biol. 2019;16(4):504–517. doi: 10.1080/15476286.2018.1504546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu X., Kriz A.J., Sharp P.A. Target specificity of the CRISPR-Cas9 system. Quant Biol. 2014;2(2):59–70. doi: 10.1007/s40484-014-0030-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang X.D. Methods for Enhancing Clustered Regularly Interspaced Short Palindromic Repeats/Cas9-Mediated Homology-Directed Repair Efficiency. Front Genet. 2019;10:551. doi: 10.3389/fgene.2019.00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh D.D. CRISPR/Cas9 guided genome and epigenome engineering and its therapeutic applications in immune mediated diseases. Semin Cell Dev Biol. 2019;96:32–43. doi: 10.1016/j.semcdb.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 60.Verma R. A CRISPR/Cas9 based polymeric nanoparticles to treat/inhibit microbial infections. Semin Cell Dev Biol. 2019;96:44–52. doi: 10.1016/j.semcdb.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 61.Singh D.D. Potential therapeutic relevance of CRISPR/Cas9 guided Epigenetic Regulations for Neuropsychiatric Disorders. Curr Top Med Chem. 2021;21 doi: 10.2174/1568026621666210317154502. [DOI] [PubMed] [Google Scholar]
- 62.Li H. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct Target Ther. Signal Transduct Target Ther. 2020;5(1):p. doi: 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tycko J., Myer V.E., Hsu P.D. Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity. Mol Cell. 2016;63(3):355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Norris A.D., Gracida X., Calarco J.A. CRISPR-mediated genetic interaction profiling identifies RNA binding proteins controlling metazoan fitness. Elife. 2017;6 doi: 10.7554/eLife.28129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chylinski K. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42(10):6091–6105. doi: 10.1093/nar/gku241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stewart C.M. The value of cell-free DNA for molecular pathology. J Pathol. 2018;244(5):616–627. doi: 10.1002/path.5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jitariu A. Triple negative breast cancer: the kiss of death. Oncotarget. 2017;8:46652–46662. doi: 10.18632/oncotarget.16938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mintz R.L. CRISPR/Cas9-mediated mutagenesis to validate the synergy between PARP1 inhibition and chemotherapy in BRCA1-mutated breast cancer cells. Bioeng Transl Med. 2020;5(1) doi: 10.1002/btm2.10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang X., Yang H., Zhang R. Challenges and future of precision medicine strategies for breast cancer based on a database on drug reactions. Biosci Rep. 2019;39(9) doi: 10.1042/BSR20190230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo P. Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel. Proc Natl Acad Sci U S A. 2019;116(37):18295–18303. doi: 10.1073/pnas.1904697116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guernet A. CRISPR-Barcoding for Intratumor Genetic Heterogeneity Modeling and Functional Analysis of Oncogenic Driver Mutations. Mol Cell. 2016;63(3):526–538. doi: 10.1016/j.molcel.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zubor P. Why the Gold Standard Approach by Mammography Demands Extension by Multiomics? Application of Liquid Biopsy miRNA Profiles to Breast Cancer Disease Management. Int J Mol Sci. 2019;20(12) doi: 10.3390/ijms20122878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vinnicombe S. How I report breast magnetic resonance imaging studies for breast cancer staging and screening. Cancer Imaging. 2016;16(1):17. doi: 10.1186/s40644-016-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee S.H. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene. 2017;36(49):6823–6829. doi: 10.1038/onc.2017.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wilkinson M. Structure of the DNA-Bound Spacer Capture Complex of a Type II CRISPR-Cas System. Mol Cell. 2019;75(1):90–101 e5. doi: 10.1016/j.molcel.2019.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCarthy M.W. Harnessing the potential of CRISPR-based platforms to advance the field of hospital medicine. Expert Rev Anti Infect Ther. 2020:1–7. doi: 10.1080/14787210.2020.1761333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen J.Q., Russo J. ERalpha-negative and triple negative breast cancer: molecular features and potential therapeutic approaches. Biochim Biophys Acta. 2009;1796(2):162–175. doi: 10.1016/j.bbcan.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hajian R. Detection of unamplified target genes via CRISPR-Cas9 immobilized on a graphene field-effect transistor. Nat Biomed Eng. 2019;3(6):427–437. doi: 10.1038/s41551-019-0371-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang M. Impact of CXCR4 and CXCR7 knockout by CRISPR/Cas9 on the function of triple-negative breast cancer cells. Onco Targets Ther. 2019;12:3849–3858. doi: 10.2147/OTT.S195661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.PICKLES. Pooled In-vitro CRISPR Knockout Library Essentiality Screens (PICKLES). 2017.
- 81.Meyers R.M. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rauscher B. Toward an integrated map of genetic interactions in cancer cells. Mol Syst Biol. 2018;14(2) doi: 10.15252/msb.20177656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wangensteen K.J. Combinatorial genetics in liver repopulation and carcinogenesis with a in vivo CRISPR activation platform. Hepatology. 2018;68(2):663–676. doi: 10.1002/hep.29626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nedeljkovic M., Damjanovic A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells. 2019;8(9) doi: 10.3390/cells8090957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park Choi, Nam, Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers. 2019;11(7) doi: 10.3390/cancers11070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Usary, J., et al., Overview of Genetically Engineered Mouse Models of Distinct Breast Cancer Subtypes. Curr Protoc Pharmacol, 2016. 72: p. 14 38 1-14 38 11. [DOI] [PMC free article] [PubMed]
- 87.Misek D.E., Kim E.H. Protein Biomarkers for the Early Detection of Breast Cancer. International Journal of Proteomics. 2011;2011:1–9. doi: 10.1155/2011/343582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jamdade V.S. Therapeutic targets of triple-negative breast cancer: a review. Br J Pharmacol. 2015;172(17):4228–4237. doi: 10.1111/bph.13211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shao F., Sun H., Deng C.-X. Potential therapeutic targets of triple-negative breast cancer based on its intrinsic subtype. Oncotarget. 2017;8(42):73329–73344. doi: 10.18632/oncotarget.20274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mittal L. Quantitative proteomic analysis of enhanced cellular effects of electrochemotherapy with Cisplatin in triple-negative breast cancer cells. Sci Rep. 2019;9(1) doi: 10.1038/s41598-019-50048-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes & Cancer. 2011;2(12):1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mancini P. Standard of Care and Promising New Agents for Triple Negative Metastatic Breast Cancer. Cancers. 2014;6(4):2187–2223. doi: 10.3390/cancers6042187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mukhopadhyay U.K. TP53 Status as a Determinant of Pro- vs Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. JNCI: Journal of the National Cancer Institute. 2019;111(11):1202–1215. doi: 10.1093/jnci/djz051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eltohamy M.I. Topoisomerase II α Gene alteration in Triple Negative Breast Cancer and Its Predictive Role for Anthracycline-Based Chemotherapy (Egyptian NCI Patients) Asian Pac J Cancer Prev. 2018;19(12):3581–3589. doi: 10.31557/APJCP.2018.19.12.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pernas S. CDK4/6 inhibition in breast cancer: current practice and future directions. Therapeutic Advances. Med Oncol. 2018;10 doi: 10.1177/1758835918786451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen D., Ping Dou Q. The Ubiquitin-Proteasome System as a Prospective Molecular Target for Cancer Treatment and Prevention. Curr Protein Pept Sci. 2010;11(6):459–470. doi: 10.2174/138920310791824057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim J. Genomic Characteristics of Triple-Negative Breast Cancer Nominate Molecular Subtypes That Predict Chemotherapy Response. Mol Cancer Res. 2020;18(2):253–263. doi: 10.1158/1541-7786.MCR-19-0453. [DOI] [PubMed] [Google Scholar]
- 98.Won Jeong K. Gene-specific patterns of coregulator requirements by estrogen receptor-α in breast cancer cells. Mol Endocrinol. 2012;26(6):955–966. doi: 10.1210/me.2012-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nassa G. The RNA-mediated estrogen receptor α interactome of hormone-dependent human breast cancer cell nuclei. Sci Data. 2019;6:173. doi: 10.1038/s41597-019-0179-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Treeck O. Estrogen Actions in Triple-Negative Breast Cancer. Cells. 2020;9(11):2358. doi: 10.3390/cells9112358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liao L. E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2017;77(8):2090–2101. doi: 10.1158/0008-5472.CAN-16-2409. [DOI] [PubMed] [Google Scholar]
- 102.Yang H. Break Breast Cancer Addiction by CRISPR/Cas9 Genome Editing. Journal of Cancer. 2018;9(2):219–231. doi: 10.7150/jca.22554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Faraoni I., Graziani G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers. 2018;10(12) doi: 10.3390/cancers10120487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karicheva O. PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget. 2016;7(39):64109–64123. doi: 10.18632/oncotarget.11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johansson J. TGF-Î21-Induced Epithelialâ€Mesenchymal Transition Promotes Monocyte/Macrophage Properties in Breast Cancer Cells. Frontiers. Oncology. 2015;5 doi: 10.3389/fonc.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen Y., Zhang Y. Application of the CRISPR/Cas9 System to Drug Resistance in Breast Cancer. Advanced. Science. 2018;5(6) doi: 10.1002/advs.201700964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Breuleux, M., et al., Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Molecular cancer therapeutics, Mol Oncol, 2019;13(10):p.2160-2177. [DOI] [PMC free article] [PubMed]
- 108.Ruicci, K. M., et al., Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition. Mol Oncol, 2019.13(10):p.2160-2177. [DOI] [PMC free article] [PubMed]
- 109.White M.K., Khalili K. CRISPR/Cas9 and cancer targets: future possibilities and present challenges. Oncotarget. 2016;7(11):12305–12317. doi: 10.18632/oncotarget.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Castro N.P. Cripto-1 as a novel therapeutic target for triple negative breast cancer. Oncotarget. 2014;6(14):11910–11929. doi: 10.18632/oncotarget.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xie Z. Significance of the E3 ubiquitin protein UBR5 as an oncogene and a prognostic biomarker in colorectal cancer. Oncotarget. 2017;8(64):108079–108092. doi: 10.18632/oncotarget.22531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Findlay G.M. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562(7726):217–222. doi: 10.1038/s41586-018-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Váraljai R. Increased mitochondrial function downstream from KDM5A histone demethylase rescues differentiation in pRB-deficient cells. Genes Dev. 2015;29(17):1817–1834. doi: 10.1101/gad.264036.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Z. Drug Discovery Targeting Bromodomain-Containing Protein 4. J Med Chem. 2017;60(11):4533–4558. doi: 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin F.-M. Differential gene expression and AKT targeting in triple negative breast cancer. Oncotarget. 2019;10(43):4356–4368. doi: 10.18632/oncotarget.27026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pham T.T., Angus S.P., Johnson G.L. MAP3K1: Genomic Alterations in Cancer and Function in Promoting Cell Survival or Apoptosis. Genes & Cancer. 2013;4(11–12):419–426. doi: 10.1177/1947601913513950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wei W. The Breast Cancer Susceptibility Gene Product Fibroblast Growth Factor Receptor 2 Serves as a Scaffold for Regulation of NF- B Signaling. Mol Cell Biol. 2012;32(22):4662–4673. doi: 10.1128/MCB.00935-12. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118.Qu Y. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics. 2017;9(6):919–931. doi: 10.2217/epi-2017-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bossennec M. MDR1 in immunity: friend or foe? OncoImmunology. 2018;7(12) doi: 10.1080/2162402X.2018.1499388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Costa Ricardo L B. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast Cancer Res Treat. 2018;169(3):397–406. doi: 10.1007/s10549-018-4697-y. [DOI] [PubMed] [Google Scholar]
- 121.Miettinen M. Gata3. Am J Surg Pathol. 2014;38(1):13–22. doi: 10.1097/PAS.0b013e3182a0218f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xia X. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol Cancer. 2019;18:131. doi: 10.1186/s12943-019-1056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abdulkareem I., Blair M. Phosphatase and tensin homologue deleted on chromosome 10. Nigerian Medical Journal. 2013;54(2) doi: 10.4103/0300-1652.110033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen H. et al., Association Between BRCA Status and Triple-Negative Breast Cancer: A Meta-Analysis. Front Pharmacol. 2018.9:p.909. [DOI] [PMC free article] [PubMed]
- 125.Si W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clinical. Epigenetics. 2019;11(1) doi: 10.1186/s13148-018-0587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Majello B. Expanding the Role of the Histone Lysine-Specific Demethylase LSD1 in Cancer. Cancers. 2019;11(3) doi: 10.3390/cancers11030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sexton R.E. Ras and exosome signaling. Semin Cancer Biol. 2019;54:131–137. doi: 10.1016/j.semcancer.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mazur P.K. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21(10):1163–1171. doi: 10.1038/nm.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nahta R. Targets and Therapy; Breast Cancer: 2012. New developments in the treatment of HER-2-positive breast cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stravodimou A. The Future of ER+/HER2- Metastatic Breast Cancer Therapy: Beyond PI3K Inhibitors. Anticancer Res. 2020;40(9):4829–4841. doi: 10.21873/anticanres.14486. [DOI] [PubMed] [Google Scholar]
- 131.Bavaro L. Mechanisms of Disease Progression and Resistance to Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukemia: An Update. Int J Mol Sci. 2019;20(24) doi: 10.3390/ijms20246141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang W. Discovery of a novel inhibitor of kinesin-like protein KIFC1. Biochem J. 2016;473(8):1027–1035. doi: 10.1042/BJ20150992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Qu K. Cisplatin induces cell cycle arrest and senescence via upregulating P53 and P21 expression in HepG2 cells. Nan Fang Yi Ke Da Xue Xue Bao. 2013;33(9):1253–1259. [PubMed] [Google Scholar]
- 134.Wu S. Novel variants in MLL confer to bladder cancer recurrence identified by whole-exome sequencing. Oncotarget. 2015;7(3):2629–2645. doi: 10.18632/oncotarget.6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang X. RSF-1 overexpression determines cancer progression and drug resistance in cervical cancer. BioMedicine. 2018;8(1) doi: 10.1051/bmdcn/2018080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bao L. Increased Expression of P-Glycoprotein and Doxorubicin Chemoresistance of Metastatic Breast Cancer Is Regulated by miR-298. The American Journal of Pathology. 2012;180(6):2490–2503. doi: 10.1016/j.ajpath.2012.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Grenier J.M., Yeung S.T., Khanna K.M. Combination Immunotherapy: Taking Cancer Vaccines to the Next Level. Front Immunol. 2018;9 doi: 10.3389/fimmu.2018.00610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Housman G. Drug resistance in cancer: an overview. Cancers. 2014;6(3):1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mansoori B. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 2017;7(3):339–348. doi: 10.15171/apb.2017.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Qu Y. Tumour microenvironment-driven non-cell-autonomous resistance to antineoplastic treatment. Mol Cancer. 2019;18(1):69. doi: 10.1186/s12943-019-0992-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gurdal H. Partial agonistic effect of cetuximab on epidermal growth factor receptor and Src kinase activation in triple–negative breast cancer cell lines. Int J Oncol. 2019;54(4):1345–1356. doi: 10.3892/ijo.2019.4697. [DOI] [PubMed] [Google Scholar]
- 142.Martinez-Lage M. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines. 2018;6(4):105. doi: 10.3390/biomedicines6040105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lino C.A. Delivering CRISPR: a review of the challenges and approaches. Drug Delivery. 2018;25(1):1234–1257. doi: 10.1080/10717544.2018.1474964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bleau A.M. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Phuah S.Y. Triple-negative breast cancer and PTEN (phosphatase and tensin homologue) loss are predictors of BRCA1 germline mutations in women with early-onset and familial breast cancer, but not in women with isolated late-onset breast cancer. Breast Cancer Res. 2012;14(6):R142. doi: 10.1186/bcr3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Moses C. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol Ther Nucleic Acids. 2019;14:287–300. doi: 10.1016/j.omtn.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhu Y. Tamoxifen-resistant breast cancer cells are resistant to DNA-damaging chemotherapy because of upregulated BARD1 and BRCA1. Nat Commun. 2018;9(1):1595. doi: 10.1038/s41467-018-03951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Biagioni A. Delivery systems of CRISPR/Cas9-based cancer gene therapy. J Biol Eng. 2018;12:33. doi: 10.1186/s13036-018-0127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.King J.L. TTK promotes mesenchymal signaling via multiple mechanisms in triple negative breast cancer. Oncogenesis. 2018;7(9):69. doi: 10.1038/s41389-018-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhao D. Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Mol Cell. 2018;69(4):699–708 e7. doi: 10.1016/j.molcel.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shalem O., Sanjana N.E., Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16(5):299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Patel S.J. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–542. doi: 10.1038/nature23477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ominguez A.A., Lim W.A., Qi L.S. Beyond editing: repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2015;17(1):5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lo A., Qi L. Genetic and epigenetic control of gene expression by CRISPR–Cas systems. F1000Research. 2017;6 doi: 10.12688/f1000research.11113.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huang Q.-Y. Long Non-Coding RNA: Dual Effects on Breast Cancer Metastasis and Clinical Applications. Cancers. 2019;11(11) doi: 10.3390/cancers11111802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liang X. Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol. 2017;241:136–146. doi: 10.1016/j.jbiotec.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 157.Shaath H. Single-cell long noncoding RNA (lncRNA) transcriptome implicates MALAT1 in triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy. Cell Death Discovery, Cell Death Discov. 2021;7(1):23. doi: 10.1038/s41420-020-00383-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wardhani B.W. TGF-β-Induced TMEPAI Attenuates the Response of Triple-Negative Breast Cancer Cells to Doxorubicin and Paclitaxel. J Exp Pharmacol. 2020;12:17–26. doi: 10.2147/JEP.S235233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lian B. et. al, Truncated HDAC9 identified by integrated genome-wide screen as the key modulator for paclitaxel resistance in triple-negative breast cancer. Theranostics, 2020.10(24):p.11092-11109. [DOI] [PMC free article] [PubMed]
- 160.Han Y. et al., New Perspectives for Resistance to PARP Inhibitors in Triple-Negative Breast Cancer, Front Oncol. 10, 578095. [DOI] [PMC free article] [PubMed]
- 161.Azangou-Khyavy M. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front Immunol. 2020;11:2062. doi: 10.3389/fimmu.2020.02062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Eyquem J. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rupp L.J. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737. doi: 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Han Y. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727–742. [PMC free article] [PubMed] [Google Scholar]
- 165.Li L., Hu S., Chen X. Non-viral delivery systems for CRISPR/Cas9-based genome editing: Challenges and opportunities. Biomaterials. 2018;171:207–218. doi: 10.1016/j.biomaterials.2018.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shim G. Therapeutic gene editing: delivery and regulatory perspectives. Acta Pharmacol Sin. 2017;38(6):738–753. doi: 10.1038/aps.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jung I.Y., Lee J. Unleashing the Therapeutic Potential of CAR-T Cell Therapy Using Gene-Editing Technologies. Mol Cells. 2018;41(8):717–723. doi: 10.14348/molcells.2018.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Liang C. Tumor cell-targeted delivery of CRISPR/Cas9 by aptamer-functionalized lipopolymer for therapeutic genome editing of VEGFA in osteosarcoma. Biomaterials. 2017;147:68–85. doi: 10.1016/j.biomaterials.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 169.Kim S.M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J Control Release. 2017;266:8–16. doi: 10.1016/j.jconrel.2017.09.013. [DOI] [PubMed] [Google Scholar]
- 170.Shin J., Oh J.-W. Development of CRISPR/Cas9 system for targeted DNA modifications and recent improvements in modification efficiency and specificity. BMB Reports. 2020;53(7):341–348. doi: 10.5483/BMBRep.2020.53.7.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Drost J. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science. 2017;358(6360):234–238. doi: 10.1126/science.aao3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Naeem M. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells. 2020;9(7) doi: 10.3390/cells9071608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hu W. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2018;68(3):365–377. doi: 10.1007/s00262-018-2281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Givens B.E. Nanoparticle-Based Delivery of CRISPR/Cas9 Genome-Editing Therapeutics. AAPS J. 2018;20(6):108. doi: 10.1208/s12248-018-0267-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Moon S.B. Recent advances in the CRISPR genome editing tool set. Exp Mol Med. 2019;51(11):1–11. doi: 10.1038/s12276-019-0339-7. [DOI] [PMC free article] [PubMed] [Google Scholar]