

Abstract
Background:
DNA methylation may play a role in progression from normative to problematic drinking and underlie adverse health outcomes associated with alcohol misuse. In the current study, we examined the association between alcohol consumption and DNA methylation patterns using three approaches: a conventional epigenome-wide association study (EWAS); a co-twin comparison design, which controls for genetic and environmental influences that twins share; and a regression of age acceleration, defined as a discrepancy between chronological age and DNA methylation age, on alcohol consumption.
Methods:
Participants came from the Finnish Twin Cohorts (FinnTwin12/FinnTwin16; N = 1004; 55% female; average age = 23 years). Individuals reported the number of alcoholic beverages consumed in the past week, and epigenome-wide DNA methylation was assessed in whole-blood using the Infinium HumanMethylation450 BeadChip.
Results:
In the EWAS, alcohol consumption was significantly related to methylation at 24 CpG sites. When evaluating whether differences between twin siblings (185 monozygotic pairs) in alcohol consumption predicted differences in DNA methylation, co-twin comparisons replicated four CpG sites from the EWAS and identified 23 additional sites. However, when we examined qualitative differences in drinking patterns between twins (heavy drinker versus light drinker/abstainer or moderate drinker versus abstainer; 44 pairs), methylation patterns did not significantly differ within twin pairs. Finally, individuals who reported higher alcohol consumption also exhibited greater age acceleration, though results were no longer significant after controlling for genetic and environmental influences shared by co-twins.
Conclusions:
Our analyses offer insight into the associations between epigenetic variation and levels of alcohol consumption in young adulthood.
Keywords: age acceleration, alcohol, co-twin comparisons, epigenome-wide association study, FinnTwin12
Alcohol use and incidence of alcohol use disorder (AUD) increase across adolescence and peak during young adulthood (Lee and Sher, 2018). Research has extensively examined the interaction of genetic and environmental factors on alcohol consumption (Young-Wolff et al., 2011), as well as pathways from alcohol use to problems (Nestler, 2014). Yet, genetic variants identified through genome-wide association studies (GWAS) of alcohol use and dependence (Kranzler et al., 2019; Liu et al., 2019; Walters et al., 2018) explain only a modest proportion of the phenotypic variance (Zhang and Gelernter, 2017). Pre-existing epigenetic modifications, such as DNA methylation, may influence the initiation of alcohol use or liability to develop problems and dependence. Moreover, alcohol-induced DNA methylation changes may underlie functional consequences of alcohol misuse and play a significant role in progression from normative to problematic drinking (Nestler, 2014). In the current study, we investigated the association between alcohol consumption and epigenome-wide DNA methylation patterns using three different approaches: a conventional epigenome-wide association study; a co-twin comparison design, which controls for genetic and environmental influences that twins share; and a regression of age acceleration, defined as a discrepancy between chronological age and DNA methylation age, on alcohol consumption.
There has been significant interest in characterizing differential methylation patterns associated with alcohol use and problems. For instance, candidate gene studies have established AUD-associated DNA methylation changes in promoter regions of GDAP1 (Brückmann et al., 2016), MAOA (Philibert et al., 2008), and OPRM1 (Zhang et al., 2012). However, like candidate gene-environment research (Dick et al., 2015), epigenetic studies of candidate gene promoters are limited by small effect sizes of individual CpG sites and reliance on scientists’ a priori prediction of functionally relevant genes. Therefore, epigenome-wide association studies (EWAS) have emerged as a method to better understand molecular mechanisms and consequences of complex behavioral outcomes (Rakyan et al., 2011), such as alcohol dependence (Zhang et al., 2013) and alcohol consumption (Liu et al., 2018; Lohoff et al., 2017). A recent EWAS of alcohol consumption (measured in grams of ethanol per day) identified differential methylation at 361 CpG sites in whole blood among 6,926 adults of European ancestry. Significant CpG sites were mapped to 257 genes and enriched for 95 biological processes. Further, higher methylation levels were associated with lower mRNA expression at 78% of identified CpG-gene pairs, providing evidence for functional significance of alcohol-related methylation patterns (Liu et al., 2018). Similarly, Wilson et al. (2019) found that alcohol consumption was significantly related to methylation at 5,458 CpG sites; 677 of these sites were replicated in an independent set of blood DNA samples.
Yet, as with many correlational designs, EWAS are prone to confounding by genetic and environmental factors. Single nucleotide polymorphisms (SNPs) may be associated with the methylation status of CpG nucleotides, even across extended genomic regions (Zhang et al., 2014). A recent EWAS of alcohol consumption suggests methylation is influenced by cis-genetic variants at more than half of alcohol-related CpG sites, and SNPs explain 20-61% of interindividual differences in DNA methylation (Liu et al., 2018). Therefore, associations between alcohol consumption and CpG methylation may be confounded by differences in genetic variation between individuals with low versus high levels of alcohol use. Because DNA methylation changes in response to environmental stimuli, confounding environmental influences are also important to consider. For example, low socioeconomic status in childhood (Needham et al., 2015) and alcohol consumption are both associated with methylation of the FKBP5 promoter (Dogan et al., 2016). Similarly, smoking is significantly associated with both alcohol intake (Bobo and Husten, 2000) and genome-wide DNA methylation patterns (Kaur et al., 2019; Philibert et al., 2012).
Co-twin comparisons address potential confounding and enable stronger inferences by evaluating whether differences between co-twins in alcohol consumption predict within-pair differences in DNA methylation levels (D’Onofrio et al., 2013). The co-twin comparison design cannot fully resolve the direction of causality in cross sectional data: different patterns of DNA methylation between twins (presumably from prior exposure to different environments) may contribute to differences in their alcohol consumption, or different levels of alcohol consumption may produce within-pair epigenetic variation. However, the co-twin comparison design can account for heritable aspects of DNA methylation and for epigenetic variation induced by shared experiences between co-twins, facilitating stronger inferences when compared to an EWAS of unrelated individuals. For instance, Ruggeri et al. (2015) compared DNA methylation patterns among twin pairs discordant for AUD; 77 regions were differentially methylated within twin pairs, including PPM1G, SLC6A3, and OPRL1 (Ruggeri et al., 2015). These findings suggest that AUD-associated methylation changes in candidate gene studies are not merely attributable to factors that vary between families, underscoring the importance of using complementary methods, such as co-twin comparisons, to understand the robustness of correlational effects.
Finally, alcohol consumption may affect the biological aging of tissues. DNA methylation has been used as a robust estimate of biological age (Horvath, 2013; Horvath and Raj, 2018) and outperforms other such estimators, including telomere length and transcriptomic, proteomic, and metabolomics-based predictors (Jylhävä et al., 2017). Age acceleration, a discrepancy between DNA methylation-based biological age and chronological age (Rosen et al., 2018), is commonly observed in cancer and a number of other age-related conditions and diseases (Horvath and Raj, 2018). Age acceleration could therefore provide a mechanism through which alcohol misuse confers risk for chronic disease (Rehm et al., 2017). Very low and very high alcohol use are more closely associated with age acceleration than moderate use (Beach et al., 2015), and significant age acceleration has been observed among individuals with a current diagnosis of alcohol dependence. However, individuals with a lifetime diagnosis do not significantly differ from healthy controls in DNA methylation age, suggesting that age acceleration is more closely associated with recent alcohol consumption than prior misuse (Rosen et al., 2018).
Currently, we built upon recent EWAS of alcohol consumption (Liu et al., 2018; Wilson et al., 2019) by examining alcohol-related DNA methylation patterns among young adults, who are at heightened risk for alcohol use and problems (Lee and Sher, 2018). We also used a co-twin comparison design to investigate whether associations between alcohol consumption and CpG methylation remained significant after controlling for genetic and environmental influences shared by monozygotic (MZ) twins. As noted above, given the cross-sectional nature of the data, we cannot distinguish between DNA methylation as a cause or consequence of alcohol consumption. However, by effectively controlling for genetic and environmental influences that twins share, a co-twin comparison design can differentiate valuable biomarkers of alcohol use from markers of correlated genetic or environmental factors.
First, we evaluated whether differences between twins in continuous alcohol consumption predicted within-pair differences in DNA methylation. We then contrasted continuous differences between twins with more extreme qualitative differences in drinking patterns (i.e., heavy drinker versus light drinker/abstainer; moderate drinker versus abstainer), as associations between alcohol consumption and adverse health outcomes (e.g., risk of AUD, cardiovascular disease, and immune system deficiencies; Rehm et al. 2017) are heightened among heavy drinkers. Finally, we examined the association between alcohol consumption and age acceleration, an indicator of biological aging. By characterizing methylation patterns associated with alcohol use, our analyses inform efforts to develop a DNA methylation biomarker of alcohol consumption (Liu et al., 2018; Yousefi et al., 2019).
Materials and Methods
Study Population
The study sample is a subset of the FinnTwin12 and FinnTwin16 cohorts (Kaprio 2013). These population-based, longitudinal studies aim to examine genetic and environmental determinants of health-related behaviors and include extensive questionnaire and interview data on substance use. A total of 1004 individuals (227 monozygotic pairs, 263 dizygotic pairs, and 24 twin individuals without a co-twin) were included in the EWAS analysis and in the individual-level age acceleration analysis; 185 MZ pairs with within-pair differences in alcohol consumption in the within-pair analyses; and 44 discordant MZ pairs in the discordant analyses. The average age of the individuals was 23 years (SD = 1.8 years, range = 21-29 years), and 55% were females.
Measures
Alcohol consumption phenotype.
The alcohol consumption phenotype was derived from the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA; Bucholz et al. 1994) administered as in-person or telephone interviews (Kaprio, 2013). For each individual, we calculated the number of drinks (glass of beer, wine, grain alcohol, or other alcoholic beverage) consumed in the past week. Individuals who had not initiated alcohol use were coded as missing (n = 34). This measure was used for both the EWAS and within-pair models. For the discordant model, MZ individuals were assigned a drinking category according to the method described by Järvenpää et al. (2005) based on NIAAA guidelines, with abstainers consuming no alcohol, light drinkers consuming less than 3 standard drinks per week, moderate drinkers consuming between 3 and 7 drinks per week for women and between 3 and 14 drinks per week for men, and heavy drinkers including women who consumed more than 7 drinks per week and men who consumed more than 14 drinks per week. MZ pairs were classified as discordant if co-twins were separated by at least one drinking category, such as heavy versus light/abstainer (19 pairs) or moderate versus abstainer (25 pairs).
Methylation data.
Epigenome-wide DNA methylation was assessed with the Infinium HumanMethylation450 BeadChip in whole blood using standard protocols (Bibikova et al., 2011). Pre-processing of the resultant methylation data was based on the pipeline by van Dongen et al. (2016) using R packages “minfi” (Aryee et al., 2014), “MethylAid” (van Iterson et al., 2014), and “wateRmelon” (Pidsley et al., 2013). Quality control involved extensive sample and probe filtering. Initially, sample quality control was performed using MethylAid’s automatic assessment of quality based on five control probe metrics: bisulfite conversion, non-polymorphic sample-dependent control probes, median methylated versus unmethylated signal intensity, sample-independent hybridization control probes, and detection p-value of negative control probes (P > 0.05). The default thresholds were used for each metric. Samples were retained if they passed all five metrics. Following sample quality control, probe quality control was performed. We removed the following probes: ambiguously mapped and poor quality probes, as defined by Zhou et al. (2017); SNP probes located within 5 base pairs of a CpG site; and non-CpG targeting probes. Probes with an intensity value of exactly 0, a detection P > 0.01, or a bead count < 3 were set to ‘NA’. Only probes with a call rate of 95% or higher across all samples were retained. DNA methylation data were initially available for 1020 individuals. Following these sample and probe quality control steps, 432,081 probes and 1004 samples remained. Data were then normalized using functional normalization (Fortin et al., 2014) in “minfi” (Aryee et al., 2014), which accounts for systematic technical variation. Finally, probes within sex chromosomes were removed, leaving 421,811 autosomal probes for further analyses.
Covariates.
Participants were asked to report on their age, sex, and smoking status (current, former, or never smoker). Body mass index (BMI) was calculated from each participant’s measured height and weight. White blood cell composition was estimated using Houseman’s method (Houseman et al., 2015).
Statistical Analysis
EWAS.
To analyze the association between DNA methylation (beta values) at individual CpG sites and alcohol consumption (drinks per week), a linear mixed effects model was implemented through the “lmer” function in the “lme4” R package (Bates et al., 2015). DNA methylation was treated as the dependent variable and alcohol consumption as the independent variable. Age, sex, BMI, smoking status, and white blood cell composition were included as covariates (fixed effects), and zygosity and twin pair identifier were included as random effects to account for relatedness in the sample. Because the sample for this study came from Finland, where the genetic ancestry is relatively homogenous due to a history of genetic bottlenecks and geographic isolation (Kerminen et al., 2017), and a principal component analysis of the sample indicated a single dimension of ancestry (Meyers et al., 2012), ancestral principal components were not included as covariates.
Within-pair model.
A linear model was used to analyze the association between DNA methylation and alcohol consumption (drinks per week) within MZ twin pairs. Delta values for each pair were calculated for methylation at individual CpG sites (dependent variable), alcohol consumption (independent variable), and covariates (BMI, smoking status, and white blood cell composition). Because the within-pair model accounts for all characteristics shared by co-twins, age and sex were not included as covariates in these analyses. The linear model function “lm” from the base R package was employed to model within-pair differences.
Discordant model.
For MZ twin pairs discordant for alcohol consumption, a moderated paired t-test was computed using the “lmFit” function from the “limma” R package (Ritchie et al., 2015), allowing co-twin-pair effects to be included in a linear model. DNA methylation at individual CpG sites was treated as the dependent variable and alcohol status (lighter or heavier drinker) as the independent variable. The model included BMI, smoking status, and white blood cell composition as covariates.
In all the above models, p-values were adjusted for multiple testing correction using the FDR adjustment, with FDR-adjusted P < 0.05 considered statistically significant. QQ plots were generated with a custom R function, and lambda value estimates were calculated using the “estlambda” function from the “GenABEL” R package (Aulchenko et al., 2007).
Age acceleration analyses.
GrimAge (Lu et al., 2019) is the latest release of the epigenetic clocks and has been shown to outperform existing DNA methylation-based clocks. In addition, the GrimAge-derived age acceleration, AgeAccelGrim, shows robust and expected relationships with different lifestyle factors (Lu et al., 2019). GrimAge and AgeAccelGrim for our samples were obtained from the online calculator at https://dnamage.genetics.ucla.edu/home. AgeAccelGrim was then treated as the dependent variable in a linear mixed effects model using the “lmer” function in the “lme4” R package (Bates et al., 2015). Alcohol consumption (drinks per week) was treated as the independent variable, age and sex were included as fixed effects, and zygosity and twin pair identifier were included as random effects. In addition, within-pair analyses were performed for MZ twin pairs who differed in alcohol consumption. Delta values for each pair were calculated for AgeAccelGrim (dependent variable) and alcohol consumption (independent variable). BMI, smoking status, and white blood cell composition were included as covariates. The “lm” function from the base R package was used to fit the model. Discordant pair analyses were performed using the “limma” R package (Ritchie et al., 2015), with AgeAccelGrim as the dependent variable, alcohol status (lighter or heavier drinker) as the independent variable, and BMI, smoking status, and white blood cell composition as covariates.
Results
EWAS
Associations between alcohol consumption and epigenome-wide DNA methylation patterns were examined in a sample of 1004 young adults. A linear mixed effects model was implemented with DNA methylation (defined as the proportion of methylation in the sample for a given CpG site, or the beta value) as the dependent variable and alcohol consumption (drinks per week) as the independent variable. Age, sex, BMI, smoking status, and white blood cell composition were included as covariates (fixed effects), and zygosity and twin pair identifier were included as random effects. Descriptive statistics for alcohol consumption and covariates are shown separately by analysis type in Table 1. After multiple testing correction using the FDR adjustment, methylation levels at 24 CpG sites were significantly associated with alcohol consumption (Table 2). Most of these CpG sites were in open sea while only four were in CpG island core. Sixteen of the CpG sites mapped to 16 genes; methylation was negatively associated with alcohol consumption at nine of these sites and positively associated at the remaining seven CpG sites. Hypermethylation at all promoter CpG sites was associated with higher levels of alcohol consumption. The relationship between alcohol consumption and methylation at each CpG site is visualized in Figure 1. Figure S1a shows the QQ plot used to examine the observed versus expected –log10(p-values), with a Lambda estimate of 1.01.
Table 1.
Descriptive statistics for alcohol consumption and covariates.
| Analysis Type | Sample Size | Age M (SD) | Sex (Male/Female) | BMI M (SD) | Smoking Status N (%) | Drinks Per Week M (Range) | ||
|---|---|---|---|---|---|---|---|---|
| Current | Former | Never | ||||||
| EWAS/Individual-Level | 1004 | 23.28 (1.75) | 452 / 552 | 23.55 (4.09) | 391 (38.9%) | 154 (15.3%) | 459 (45.7%) | 7.41 (0-90) |
| Within-Pair | 370 (185 pairs) | 23.65 (1.96) | 168 / 202 | 23.26 (4.00) | 150 (40.5%) | 39 (10.5%) | 181 (48.9%) | 9.06 (0-90) |
| Discordant | 88 (44 pairs) | 23.43 (1.94) | 34 / 54 | 22.62 (3.17) | 28 (31.8%) | 8 (9.1%) | 52 (59.1%) | 9.95 (0-48) |
Abbreviations: EWAS, epigenome-wide association study; M, mean; SD, standard deviation.
Notes. Descriptive statistics are shown separately for the participants included within each model type: EWAS/individual-level analyses, within-pair models, and discordant pair models.
Table 2.
Significant CpGs associated with alcohol consumption in an EWAS of WBCs from the FinnTwin cohort.
| CpG | UCSC Gene | Chr | Position | P value | FDR adjusted P value | β | SE | Relation to UCSC CpG Island | Enhancer |
|---|---|---|---|---|---|---|---|---|---|
| cg11404486 | GOLGA8B | 15 | 34875930 | 0.00 | 0.00 | 0.00062 | 6.89E-05 | Island | |
| cg21851395 | 13 | 42108056 | 4.44E-16 | 9.37E-11 | −0.00093 | 1.15E-04 | OpenSea | TRUE | |
| cg08859215 | FAM20A | 17 | 66573573 | 1.32E-09 | 1.85E-04 | −0.00086 | 1.42E-04 | OpenSea | TRUE |
| cg17449759 | 1 | 168466986 | 4.42E-09 | 4.66E-04 | −0.00064 | 1.08E-04 | OpenSea | TRUE | |
| cg04393060 | KIAA0319L | 1 | 35919432 | 1.32E-08 | 1.11E-03 | −0.00074 | 1.30E-04 | OpenSea | TRUE |
| cg21757189 | 8 | 134462510 | 2.89E-08 | 2.03E-03 | −0.00064 | 1.15E-04 | OpenSea | TRUE | |
| cg26687120 | 2 | 9954121 | 4.54E-08 | 2.73E-03 | 0.00009 | 1.60E-05 | OpenSea | TRUE | |
| cg00288562 | TRA2A | 7 | 23571645 | 1.66E-07 | 8.74E-03 | 0.00044 | 8.48E-05 | Island | |
| cg23551720 | HOXB3 | 17 | 46633726 | 2.08E-07 | 9.76E-03 | −0.00047 | 9.14E-05 | S_Shore | TRUE |
| cg01940273 | 2 | 233284934 | 5.98E-07 | 2.52E-02 | −0.00078 | 1.56E-04 | Island | ||
| cg09109520 | GPR56 | 16 | 57673258 | 6.65E-07 | 2.55E-02 | 0.00055 | 1.10E-04 | OpenSea | |
| cg12103569 | SFRS8 | 12 | 132258560 | 7.38E-07 | 2.59E-02 | 0.00056 | 1.12E-04 | OpenSea | TRUE |
| cg13993469 | SLC24A4 | 14 | 92795592 | 9.58E-07 | 2.69E-02 | −0.00032 | 6.63E-05 | OpenSea | |
| cg16145216 | HIVEP3 | 1 | 42385662 | 8.85E-07 | 2.69E-02 | 0.00073 | 1.48E-04 | S_Shore | |
| cg24166814 | 2 | 56067277 | 9.15E-07 | 2.69E-02 | −0.00055 | 1.12E-04 | OpenSea | TRUE | |
| cg03028769 | CDH23 | 10 | 73421791 | 1.21E-06 | 3.19E-02 | −0.00035 | 7.30E-05 | OpenSea | |
| cg02208313 | CCDC57 | 17 | 80085332 | 1.40E-06 | 3.47E-02 | −0.00071 | 1.46E-04 | Island | |
| cg05575921 | AHRR | 5 | 373378 | 1.94E-06 | 3.89E-02 | −0.00125 | 2.63E-04 | N_Shore | TRUE |
| cg15451075 | 3 | 148966942 | 1.99E-06 | 3.89E-02 | −0.00041 | 8.56E-05 | OpenSea | TRUE | |
| cg19637821 | FBRSL1 | 12 | 133133572 | 2.00E-06 | 3.89E-02 | 0.00052 | 1.09E-04 | N_Shore | |
| cg21733098 | 12 | 127931219 | 1.91E-06 | 3.89E-02 | −0.00080 | 1.69E-04 | OpenSea | TRUE | |
| cg25246082 | TMEM156 | 4 | 39034284 | 2.03E-06 | 3.89E-02 | −0.00050 | 1.05E-04 | OpenSea | |
| cg04583842 | BANP | 16 | 88103117 | 2.79E-06 | 4.90E-02 | 0.00079 | 1.68E-04 | S_Shore | |
| cg08709672 | AVPR1B | 1 | 206224334 | 2.77E-06 | 4.90E-02 | −0.00046 | 9.74E-05 | S_Shore |
Abbreviations: EWAS, epigenome-wide association study; WBCs, whole blood cells; CpG, cytosine-phosphate-guanine dinucleotide; Chr, Chromosome; SE, standard error.
Notes. Epigenome-wide association of continuous alcohol consumption was performed using whole blood cell-derived DNA samples from the Finnish Twin Cohort. DNA methylation proportion was the outcome variable, alcoholic drinks per week was the predictor variable, and age, sex, BMI, smoking status, and white blood cell counts were included as covariates. A linear mixed-effects model was employed, with family ID and zygosity as the random effects. CpG loci were annotated to the human reference genome release GRCh37 with the “IlluminaHumanMethylation450kanno.ilmn12.hg19” R package.
Figure 1. Scatter plots of 24 significant CpGs associated with alcohol consumption in an EWAS of young adults.
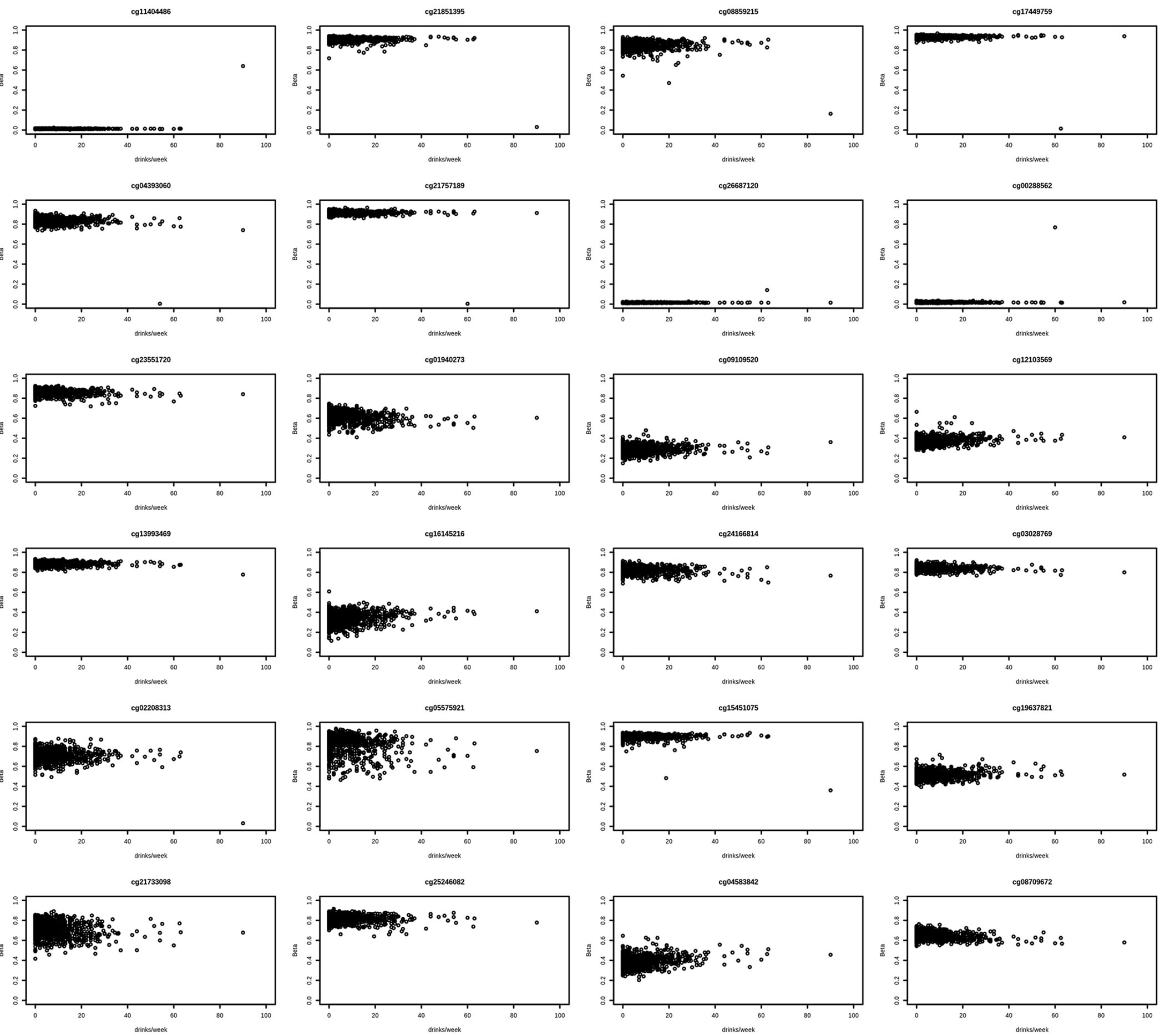
Figure 1 provides a visual representation of CpG sites significantly associated with alcohol consumption in the EWAS. Each plot represents a single CpG site with DNA methylation shown as beta (proportion of methylation at that site) on the y-axis and alcohol consumption shown as drinks per week on the x-axis.
Within-Pair Model
To further investigate the association between alcohol consumption and DNA methylation, we selected a subset of 185 MZ twin pairs who differed in their alcohol consumption. The design of the within-pair model evaluates whether differences between twin siblings in continuous alcohol consumption predict differences in DNA methylation, controlling for genetic and shared environmental factors. All significant CpGs identified in the EWAS showed the same direction of association in the within-pair model, and methylation at four of the CpG sites (cg02208313, cg11404486, cg21851395, and cg08859215) remained statistically significant. Three of these CpG sites are located in CCDC57, FAM20A, and GOLGA8B. Methylation levels at twenty-three additional CpG sites were significantly associated with alcohol consumption (Table 3), and the direction of effect for 16 of these sites was consistent across the EWAS and within-pair model. In total, of the 27 significant CpG sites, hypomethylation at 13 CpG sites and hypermethylation at 14 CpG sites were associated with increased alcohol consumption. The relationship between alcohol consumption and methylation at each CpG site is shown in Figure 2. Figure S1b demonstrates the QQ plot used to examine the observed versus expected –log10(p-values), with a Lambda estimate of 0.94.
Table 3.
Significant CpGs associated with alcohol consumption in a within-pair epigenome-wide analysis of WBCs from the FinnTwin cohort.
| CpG | UCSC Gene | Chr | Position | P value | FDR adjusted P value | β | SE | Relation to UCSC CpG Island | Enhancer |
|---|---|---|---|---|---|---|---|---|---|
| cg17625416 | ZMYM2 | 13 | 20533511 | 4.42E-14 | 1.86E-08 | −0.00142 | 1.89E-04 | Island | |
| cg02208313 | CCDC57 | 17 | 80085332 | 4.39E-12 | 9.26E-07 | −0.00252 | 3.64E-04 | Island | |
| cg13232357 | GABPB2 | 1 | 151042823 | 1.05E-11 | 1.47E-06 | −0.00236 | 3.48E-04 | N_Shore | |
| cg13319698 | 11 | 66176589 | 1.77E-11 | 1.70E-06 | 0.00065 | 9.72E-05 | Island | ||
| cg15083677 | 6 | 164758516 | 2.01E-11 | 1.70E-06 | 0.00265 | 3.95E-04 | S_Shore | ||
| cg11404486 | GOLGA8B | 15 | 34875930 | 2.87E-11 | 1.87E-06 | 0.00181 | 2.72E-04 | Island | |
| cg22396353 | GTSE1 | 22 | 46692687 | 3.10E-11 | 1.87E-06 | −0.00175 | 2.63E-04 | Island | |
| cg08758185 | SLC36A1 | 5 | 150849936 | 6.10E-11 | 3.22E-06 | 0.00257 | 3.92E-04 | OpenSea | TRUE |
| cg21851395 | 13 | 42108056 | 7.84E-11 | 3.67E-06 | −0.00256 | 3.94E-04 | OpenSea | TRUE | |
| cg20126775 | DEFB133 | 6 | 49917145 | 1.06E-10 | 4.46E-06 | −0.00237 | 3.67E-04 | OpenSea | |
| cg10177748 | HNRNPUL1 | 19 | 41770160 | 2.72E-10 | 1.04E-05 | −0.00065 | 1.02E-04 | Island | |
| cg17366225 | 4 | 11398983 | 7.68E-10 | 2.70E-05 | 0.00203 | 3.29E-04 | N_Shore | ||
| cg14863944 | 13 | 22740131 | 1.48E-09 | 4.47E-05 | 0.00244 | 4.03E-04 | OpenSea | ||
| cg24650913 | B3GNTL1 | 17 | 80970434 | 1.43E-09 | 4.47E-05 | 0.00244 | 4.03E-04 | S_Shore | |
| cg03180302 | TTR | 18 | 29171878 | 3.11E-09 | 8.74E-05 | 0.00226 | 3.81E-04 | OpenSea | |
| cg00699945 | NKX6–2 | 10 | 134600651 | 4.74E-09 | 1.25E-04 | −0.00020 | 3.44E-05 | Island | |
| cg13490575 | C9orf3 | 9 | 97735830 | 8.26E-09 | 2.05E-04 | 0.00227 | 3.94E-04 | OpenSea | TRUE |
| cg02583546 | C14orf4 | 14 | 77494451 | 9.84E-09 | 2.31E-04 | −0.00070 | 1.22E-04 | Island | |
| cg09168036 | MRPL13 | 8 | 121457072 | 2.45E-08 | 5.44E-04 | −0.00099 | 1.77E-04 | N_Shore | |
| cg18760251 | SEC62 | 3 | 169684683 | 5.93E-08 | 1.25E-03 | −0.00010 | 1.87E-05 | Island | |
| cg13496066 | TMEM231 | 16 | 75580831 | 8.13E-08 | 1.63E-03 | 0.00164 | 3.06E-04 | OpenSea | |
| cg16207974 | SLMO1 | 18 | 12431241 | 2.16E-07 | 4.13E-03 | 0.00037 | 7.05E-05 | Island | |
| cg08859215 | FAM20A | 17 | 66573573 | 3.37E-07 | 6.18E-03 | −0.00187 | 3.67E-04 | OpenSea | TRUE |
| cg07993036 | 22 | 46467577 | 6.75E-07 | 1.19E-02 | −0.00033 | 6.72E-05 | Island | ||
| cg18752774 | NLRP14 | 11 | 7060500 | 1.04E-06 | 1.75E-02 | 0.00249 | 5.11E-04 | OpenSea | |
| cg07886701 | FABP9 | 8 | 82375121 | 2.28E-06 | 3.57E-02 | 0.00212 | 4.48E-04 | OpenSea | |
| cg13620744 | 19 | 36478289 | 2.27E-06 | 3.57E-02 | 0.00015 | 3.10E-05 | Island |
Abbreviations: WBCs, whole blood cells; CpG, cytosine-phosphate-guanine dinucleotide; Chr, Chromosome; SE, standard error.
Notes. Within-pair epigenome-wide association of continuous alcohol consumption was performed using whole blood cell-derived DNA samples from the Finnish Twin Cohort. Within-pair differences were calculated and fit to a linear model, with DNA methylation proportion difference as the outcome variable and difference in alcoholic drinks consumed per week as the predictor variable. Within-pair differences in BMI, smoking status, and white blood cell counts were adjusted, and CpG loci were annotated to the human reference genome release GRCh37 with the “IlluminaHumanMethylation450kanno.ilmn12.hg19” R package.
Figure 2. Scatter plots of 27 significant CpGs associated with alcohol consumption in a within-pair model.

Figure 2 provides a visual representation of CpG sites significantly associated with alcohol consumption when examining differences within MZ twin pairs. Each plot represents a single CpG site with DNA methylation shown as beta (proportion of methylation at that site) on the y-axis and alcohol consumption shown as drinks per week on the x-axis.
Discordant Model
Following the within-pair analysis, a subset of 44 MZ twin pairs were classified as “discordant drinkers,” with co-twins separated by at least one drinking category (i.e., heavy versus light/abstainer or moderate versus abstainer). A moderated paired t-test was then computed, with co-twin-pair effects included in a linear model. After multiple testing correction using the FDR adjustment, no associations remained significant. Table S1 lists the top 50 CpG sites associated with alcohol consumption. Notably, none of the 24 significant CpG sites in the EWAS or the 27 significant CpG sites in the within-pair model were among the top 50 CpG sites associated with alcohol consumption in the discordant model. Figure S1c demonstrates the QQ plot used to examine the observed versus expected –log10(p-values), with a Lambda estimate of 0.86.
Age Acceleration Analyses
Using the latest release of the epigenetic clock (Lu et al., 2019), a measure of age acceleration was calculated, indicating biological age relative to chronological age. We then constructed a linear mixed effects model, with AgeAccelGrim as the dependent variable, alcohol consumption (drinks per week) as the independent variable, age and sex as fixed effects, and zygosity and twin pair identifier as random effects. Alcohol consumption was significantly associated with age acceleration (β = 0.053, SE = 0.009, P < 0.001), such that, if taken to be a causal effect, one additional drink consumed per week aged a person biologically by 0.053 years (equivalent to 19 days) more than their chronological age per year. However, in within-pair and discordant-pair analyses, within-pair differences in alcohol consumption did not predict differences in age acceleration (β = 0.020, SE = 0.017, P = 0.26 and B = −4.625, SE = 0.248, P = 0.50, respectively).
Discussion
In the current study, we investigated the association between a continuous measure of alcohol consumption and epigenome-wide DNA methylation patterns within a population-based sample of young adults. We then employed a co-twin comparison design to evaluate, first, the association between within-pair differences in alcohol consumption and within-pair differences in DNA methylation, and, second, differential DNA methylation patterns among twin pairs discordant for alcohol consumption. Finally, we assessed whether alcohol consumption was related to age acceleration, defined as a discrepancy between DNA methylation age and chronological age.
In a conventional EWAS, alcohol consumption (measured in drinks per week) was significantly associated with the proportion of methylation at 24 CpG sites (Table 2), of which 16 mapped to 16 different genes. Specifically, alcohol consumption was negatively associated with methylation in KIAA0319L, FAM20A, SLC24A4, HOXB3, CDH23, CCDC57, AHRR, TMEM156, and AVPR1B and positively associated with methylation in TRA2A, GPR56, GOLGA8B, SFRS8, HIVEP3, FBRSL1, and BANP. Prenatal alcohol exposure has been previously associated with decreased expression of Sfrs8 (Downing et al., 2012) and differential expression of Fam20a in the brain (Kleiber et al., 2013) among adult mice. In addition, we replicated one significant CpG site, cg01940273, from a recent EWAS of alcohol consumption among middle and older adults (Liu et al., 2018). Notably, candidate gene studies have identified AUD-associated DNA methylation changes in promoter regions of GDAP1 (Brückmann et al., 2016), MAOA (Philibert et al., 2008), and OPRM1 (Zhang et al., 2012). We did not observe methylation changes at these candidate gene promoters, which may reflect differential methylation patterns associated with alcohol use versus alcohol problems.
DNA methylation changes may offer insight into adverse health outcomes associated with alcohol misuse. For instance, GPR56 plays an important role in frontal cortex development, immune regulation, and tumor progression (Huang and Lin, 2018). Similarly, HIVEP3 binds enhancer regions with a κB motif, mediating changes in gene expression during an acute immune response or inflammation (Hicar et al., 2001). Therefore, alcohol-related DNA methylation changes observed in the EWAS could play a role in previously established associations between alcohol misuse, immune function, and cancer risk (Rehm et al., 2017).
Yet, epigenetic modifications can be environmentally-induced and are closely associated with cis-genetic variants (Liu et al., 2018). Therefore, associations between alcohol consumption and epigenome-wide DNA methylation may be driven by correlated genetic or environmental influences, such as socioeconomic status (McDade et al., 2019). Co-twin comparisons address potential confounding and complement conventional EWAS by evaluating whether differences in alcohol consumption between twins predict differences in the proportion of methylation at a given CpG site after controlling for genetic and environmental influences that twin siblings share. In the current study, we applied co-twin comparisons using two different methods. First, we evaluated whether within-pair differences in continuous alcohol consumption predicted within-pair differences in DNA methylation. The within-pair model replicated four CpG sites from the EWAS (cg02208313, cg11404486, cg21851395, and cg08859215) and identified differential methylation at 23 additional CpG sites (Table 3). Of the 27 significant CpG sites, four were mapped to enhancer regions where they may affect gene expression. Because we did not measure gene expression in the current study, future work should evaluate the functional impact of these alcohol-related methylation changes.
In light of evidence that alcohol consumption exhibits a curvilinear relationship with a variety of adverse health outcomes (e.g., AUD, cardiovascular disease, immune system deficiencies; Rehm et al. 2017), we also compared DNA methylation patterns among twins discordant for alcohol consumption. Twin pairs were defined as discordant if (1) one twin was categorized as a heavy drinker, and one twin was categorized as a light drinker/abstainer; or (2) one twin was categorized as a moderate drinker, and one twin was categorized as an abstainer. The discordant model did not identify any differentially methylated CpG sites associated with alcohol consumption (Table S1). Further, none of the 24 significant CpG sites in the EWAS or the 27 significant CpG sites in the within-pair model were among the top 50 CpG sites associated with alcohol consumption in the discordant model. This lack of replication within the discordant model should be considered in light of the limited number of discordant twin pairs (44 pairs), and replication is warranted.
To further investigate associations between alcohol intake, chronic disease, and mortality risk (Rehm et al., 2017), we evaluated the effects of alcohol consumption on age acceleration. We found that individuals who reported greater alcohol consumption also exhibited a greater discrepancy between their chronological age and DNA methylation age. These findings are consistent with prior research demonstrating that very high alcohol use (Beach et al., 2015) and a current diagnosis of alcohol dependence (Rosen et al., 2018) positively predict age acceleration. However, we did not identify alcohol-related differences in epigenetic age acceleration after controlling for genetic and environmental influences shared by MZ twins. Non-significant co-twin comparisons suggest that the association between alcohol consumption and age acceleration is attributable to family-level influences, though results should be considered in view of increased Type II error risk in within-pair and discordant analyses (Boardman and Fletcher, 2015).
Our results should be considered in light of several limitations. First, we evaluated cross-sectional DNA methylation data collected on a sample of young adults. Therefore, we could not characterize within-person changes in DNA methylation following alcohol use onset, nor could we evaluate whether alcohol-related DNA methylation patterns differ across developmental periods. Second, our measure of alcohol consumption referred to the number of drinks consumed in the past week, which may not reflect participants’ typical drinking patterns. However, it is encouraging that 89.2% of the study participants reported that their alcohol consumption within the past week matched their drinking habits over the last six months, and a narrow recall timeframe may provide a more accurate estimate of alcohol consumption in the week prior to DNA collection. Third, co-twin comparisons control only for genetic and environmental influences that twins share and do not account for unmeasured individual-level characteristics (e.g., trauma exposure), preventing a strong causal interpretation of results. Fourth, because the twin pair is the unit of analysis, co-twin comparisons involve an effective reduction in sample size when compared to conventional EWAS, increasing risk of Type II error (Boardman and Fletcher, 2015). The sample size in the present study was also relatively small (N = 1004), preventing us from evaluating whether differentially methylated CpG sites could reliably differentiate heavy drinkers from non-drinkers. Finally, effect sizes were quite small in both the EWAS and within-pair model and should be interpreted accordingly. Given the relatively small effect of CpG methylation on alcohol consumption observed in the present study, replication is warranted to distinguish between true-positive and false-positive CpG sites. Moreover, additional studies are needed to explore whether the nature and magnitude of associations differ across developmental periods and between population-based and clinically-ascertained samples, as well as to distinguish between DNA methylation as a cause or consequence of alcohol consumption.
The current study yields novel insights by using multiple approaches, including EWAS, co-twin comparisons, and age acceleration analyses, to evaluate DNA methylation patterns associated with alcohol consumption. Among 24 significant CpG sites in conventional EWAS, methylation levels at four CpG sites were also associated with alcohol consumption in co-twin comparisons, which control for genetic and environmental influences that twins share. Our analyses emphasize the utility of co-twin comparisons as a complementary approach to EWAS when developing a robust blood-based biomarker for alcohol consumption (Liu et al., 2018; Yousefi et al., 2019), as this type of design enables stronger inferences not possible in samples of unrelated individuals and differentiates valuable biomarkers from markers of correlated genetic or environmental liability. In addition, our results support a positive association between alcohol consumption and age acceleration in individual-level analyses, though alcohol consumption did not significantly predict age acceleration after controlling for genetic and environmental influences shared by co-twins. Although we did not assess the impact of DNA methylation changes on gene expression or protein structure and function, our analyses highlight targets for further research on the molecular mechanisms underlying the development of drinking patterns and pathways from alcohol use to dependence.
Supplementary Material
Acknowledgements
Preliminary data from this project were presented at the World Congress of Psychiatric Genetics and reported as a published abstract (doi:10.1016/j.euroneuro.2019.08.251).
This work was supported by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under award numbers R01AA012502, R01AA015416, K02AA018755, and K01AA024152; and the Academy of Finland (grants 100499, 205585, 118555, 141054, 265240, 263278, and 264146). JK and MO acknowledge support from the Academy of Finland (grants 263278, 308248, and 312073 to JK; 297908 to MO).
References
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA, 2014. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko YS, Ripke S, Isaacs A, van Duijn CM, 2007. GenABEL: An R library for genome-wide association analysis. Bioinformatics 23, 1294–1296. 10.1093/bioinformatics/btm108 [DOI] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, Walker S, 2015. Fitting linear mixed-effects models using lme4. Journal of Statistical Software 67, 1–48. 10.18637/jss.v067.i01 [DOI] [Google Scholar]
- Beach SRH, Dogan MV, Lei M-K, Cutrona CE, Gerrard M, Gibbons FX, Simons RL, Brody GH, Philibert RA, 2015. Methylomic aging as a window on lifestyle impact: Tobacco and alcohol use alter the rate of biological aging. J Am Geriatr Soc 63, 2519–2525. 10.1111/jgs.13830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, Fan J-B, Shen R, 2011. High density DNA methylation array with single CpG site resolution. Genomics, New Genomic Technologies and Applications 98, 288–295. 10.1016/j.ygeno.2011.07.007 [DOI] [PubMed] [Google Scholar]
- Boardman JD, Fletcher JM, 2015. To cause or not to cause? That is the question, but identical twins might not have all of the answers. Soc Sci Med 127, 198–200. 10.1016/j.socscimed.2014.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobo JK, Husten C, 2000. Sociocultural influences on smoking and drinking. Alcohol Res Health 24, 225–232. [PMC free article] [PubMed] [Google Scholar]
- Brückmann C, Di Santo A, Karle KN, Batra A, Nieratschker V, 2016. Validation of differential GDAP1 DNA methylation in alcohol dependence and its potential function as a biomarker for disease severity and therapy outcome. Epigenetics 11, 456–463. 10.1080/15592294.2016.1179411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI, Reich T, Schmidt I, Schuckit MA, 1994. A new, semi-structured psychiatric interview for use in genetic linkage studies: A report on the reliability of the SSAGA. J. Stud. Alcohol 55, 149–158. [DOI] [PubMed] [Google Scholar]
- Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S, Hewitt JK, Kendler KS, Sher KJ, 2015. Candidate gene-environment interaction research: Reflections and recommendations. Perspect Psychol Sci 10, 37–59. 10.1177/1745691614556682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan MV, Lei MK, Beach SRH, Brody G, Philibert RA, 2016. Alcohol and tobacco consumption alter hypothalamic pituitary adrenal axis DNA methylation. Psychoneuroendocrinology 66, 176–184. 10.1016/j.psyneuen.2016.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Onofrio BM, Lahey BB, Turkheimer E, Lichtenstein P, 2013. Critical need for family-based, quasi-experimental designs in integrating genetic and social science research. Am J Public Health 103, S46–S55. 10.2105/AJPH.2013.301252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing C, Flink S, Florez-McClure ML, Johnson TE, Tabakoff B, Kechris KJ, 2012. Gene expression changes in C57BL/6J and DBA/2J mice following prenatal alcohol exposure. Alcohol Clin Exp Res 36, 1519–1529. 10.1111/j.1530-0277.2012.01757.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin JP, Labbe A, Lemire M, Zanke BW, Hudson TJ, Fertig EJ, Greenwood CM, Hansen KD, 2014. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biology 15, 503. 10.1186/s13059-014-0503-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicar MD, Liu Y, Allen CE, Wu LC, 2001. Structure of the human zinc finger protein HIVEP3: Molecular cloning, expression, exon–intron structure, and comparison with paralogous genes HIVEP1 and HIVEP2. Genomics 71, 89–100. 10.1006/geno.2000.6425 [DOI] [PubMed] [Google Scholar]
- Horvath S, 2013. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, Raj K, 2018. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384. 10.1038/s41576-018-0004-3 [DOI] [PubMed] [Google Scholar]
- Houseman EA, Kelsey KT, Wiencke JK, Marsit CJ, 2015. Cell-composition effects in the analysis of DNA methylation array data: a mathematical perspective. BMC Bioinformatics 16. 10.1186/s12859-015-0527-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang KY, Lin H-H, 2018. The activation and signaling mechanisms of GPR56/ADGRG1 in melanoma cell. Front. Oncol. 8. 10.3389/fonc.2018.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvenpää T, Rinne J, Koskenvuo M, Räihä I, Kaprio J, 2005. Binge drinking in midlife and dementia risk. Epidemiology 16, 766–771. 10.1097/01.ede.0000181307.30826.6c [DOI] [PubMed] [Google Scholar]
- Jylhävä J, Pedersen NL, Hägg S, 2017. Biological age predictors. EBioMedicine 21, 29–36. 10.1016/j.ebiom.2017.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaprio J, 2013. The Finnish Twin Cohort Study: An update. Twin Res Hum Genet 16, 157–162. 10.1017/thg.2012.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Begum R, Thota S, Batra S, 2019. A systematic review of smoking-related epigenetic alterations. Arch. Toxicol. 93, 2715–2740. 10.1007/s00204-019-02562-y [DOI] [PubMed] [Google Scholar]
- Kerminen S, Havulinna AS, Hellenthal G, Martin AR, Sarin A-P, Perola M, Palotie A, Salomaa V, Daly MJ, Ripatti S, Pirinen M, 2017. Fine-scale genetic structure in Finland. G3 (Bethesda) 7(10), 3459–3468. 10.1534/g3.117.300217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiber ML, Mantha K, Stringer RL, Singh SM, 2013. Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. Journal of Neurodevelopmental Disorders 5, 6. 10.1186/1866-1955-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzler HR, Zhou H, Kember RL, Smith RV, Justice AC, Damrauer S, Tsao PS, Klarin D, Baras A, Reid J, Overton J, Rader DJ, Cheng Z, Tate JP, Becker WC, Concato J, Xu K, Polimanti R, Zhao H, Gelernter J, 2019. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nature Communications 10, 1499. 10.1038/s41467-019-09480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MR, Sher KJ, 2018. “Maturing out” of binge and problem drinking. Alcohol Res 39, 31–42. [PMC free article] [PubMed] [Google Scholar]
- Liu C, Marioni RE, Hedman ÅK, Pfeiffer L, Tsai P-C, Reynolds LM, Just AC, Duan Q, Boer CG, Tanaka T, Elks CE, Aslibekyan S, Brody JA, Kühnel B, Herder C, Almli LM, Zhi D, Wang Y, Huan T, Yao C, Mendelson MM, Joehanes R, Liang L, Love S-A, Guan W, Shah S, McRae AF, Kretschmer A, Prokisch H, Strauch K, Peters A, Visscher PM, Wray NR, Guo X, Wiggins KL, Smith AK, Binder EB, Ressler KJ, Irvin MR, Absher DM, Hernandez D, Ferrucci L, Bandinelli S, Lohman K, Ding J, Trevisi L, Gustafsson S, Sandling JH, Stolk L, Uitterlinden AG, Yet I, Castillo-Fernandez JE, Spector TD, Schwartz JD, Vokonas P, Lind L, Li Y, Fornage M, Arnett DK, Wareham NJ, Sotoodehnia N, Ong KK, Meurs JBJ, van, Conneely KN, Baccarelli AA, Deary IJ, Bell JT, North KE, Liu Y, Waldenberger M, London SJ, Ingelsson E, Levy D, 2018. A DNA methylation biomarker of alcohol consumption. Molecular Psychiatry 23, 422–433. 10.1038/mp.2016.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila-Velderrain J, McGuire D, Tian C, Zhan X, Choquet H, Docherty AR, Faul JD, Foerster JR, Fritsche LG, Gabrielsen ME, Gordon SD, Haessler J, Hottenga J-J, Huang H, Jang S-K, Jansen PR, Ling Y, Mägi R, Matoba N, McMahon G, Mulas A, Orrù V, Palviainen T, Pandit A, Reginsson GW, Skogholt AH, Smith JA, Taylor AE, Turman C, Willemsen G, Young H, Young KA, Zajac GJM, Zhao W, Zhou W, Bjornsdottir G, Boardman JD, Boehnke M, Boomsma DI, Chen C, Cucca F, Davies GE, Eaton CB, Ehringer MA, Esko T, Fiorillo E, Gillespie NA, Gudbjartsson DF, Haller T, Harris KM, Heath AC, Hewitt JK, Hickie IB, Hokanson JE, Hopfer CJ, Hunter DJ, Iacono WG, Johnson EO, Kamatani Y, Kardia SLR, Keller MC, Kellis M, Kooperberg C, Kraft P, Krauter KS, Laakso M, Lind PA, Loukola A, Lutz SM, Madden PAF, Martin NG, McGue M, McQueen MB, Medland SE, Metspalu A, Mohlke KL, Nielsen JB, Okada Y, Peters U, Polderman TJC, Posthuma D, Reiner AP, Rice JP, Rimm E, Rose RJ, Runarsdottir V, Stallings MC, Stančáková A, Stefansson H, Thai KK, Tindle HA, Tyrfingsson T, Wall TL, Weir DR, Weisner C, Whitfield JB, Winsvold BS, Yin J, Zuccolo L, Bierut LJ, Hveem K, Lee JJ, Munafò MR, Saccone NL, Willer CJ, Cornelis MC, David SP, Hinds DA, Jorgenson E, Kaprio J, Stitzel JA, Stefansson K, Thorgeirsson TE, Abecasis G, Liu DJ, Vrieze S, 2019. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nature Genetics 51, 237. 10.1038/s41588-018-0307-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohoff FW, Sorcher JL, Rosen AD, Mauro KL, Fanelli RR, Momenan R, Hodgkinson CA, Vendruscolo LF, Koob GF, Schwandt M, George DT, Jones IS, Holmes A, Zhou Z, Xu MJ, Gao B, Sun H, Phillips MJ, Muench C, Kaminsky ZA, 2017. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol. Psychiatry. 10.1038/mp.2017.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, Hou L, Baccarelli AA, Li Y, Stewart JD, Whitsel EA, Assimes TL, Ferrucci L, Horvath S, 2019. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 11, 303–327. 10.18632/aging.101684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDade TW, Ryan CP, Jones MJ, Hoke MK, Borja J, Miller GE, Kuzawa CW, Kobor MS, 2019. Genome-wide analysis of DNA methylation in relation to socioeconomic status during development and early adulthood. Am. J. Phys. Anthropol. 169, 3–11. 10.1002/ajpa.23800 [DOI] [PubMed] [Google Scholar]
- Meyers J, 2012. Elucidating genetic and environmental influences on alcohol-related phenotypes. ProQuest Dissertations & Theses. [Google Scholar]
- Needham BL, Smith JA, Zhao W, Wang X, Mukherjee B, Kardia SLR, Shively CA, Seeman TE, Liu Y, Diez Roux AV, 2015. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: The multi-ethnic study of atherosclerosis. Epigenetics 10, 958–969. 10.1080/15592294.2015.1085139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, 2014. Epigenetic mechanisms of drug addiction. Neuropharmacology 76, 259–268. 10.1016/j.neuropharm.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Gunter TD, Beach SR, Brody G, Madan A, 2008. MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet 147B, 565–570. 10.1002/ajmg.b.30778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert RA, Plume JM, Gibbons FX, Brody GH, Beach SRH, 2012. The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genet 3. 10.3389/fgene.2012.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC, 2013. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics 14, 293. 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Balding DJ, Beck S, 2011. Epigenome-wide association studies for common human diseases. Nat Rev Genet 12, 529–541. 10.1038/nrg3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm J, Gmel GE, Gmel G, Hasan OSM, Imtiaz S, Popova S, Probst C, Roerecke M, Room R, Samokhvalov AV, Shield KD, Shuper PA, 2017. The relationship between different dimensions of alcohol use and the burden of disease-An update. Addiction 112, 968–1001. 10.1111/add.13757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen AD, Robertson KD, Hlady RA, Muench C, Lee J, Philibert R, Horvath S, Kaminsky ZA, Lohoff FW, 2018. DNA methylation age is accelerated in alcohol dependence. Translational Psychiatry 8, 182. 10.1038/s41398-018-0233-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri B, Nymberg C, Vuoksimaa E, Lourdusamy A, Wong CP, Carvalho FM, Jia T, Cattrell A, Macare C, Banaschewski T, Barker GJ, Bokde ALW, Bromberg U, Büchel C, Conrod PJ, Fauth-Bühler M, Flor H, Frouin V, Gallinat J, Garavan H, Gowland P, Heinz A, Ittermann B, Martinot J-L, Nees F, Pausova Z, Paus T, Rietschel M, Robbins T, Smolka MN, Spanagel R, Bakalkin G, Mill J, Sommer WH, Rose RJ, Yan J, Aliev F, Dick D, Kaprio J, Desrivières S, Schumann G, 2015. Association of protein phosphatase PPM1G with alcohol use disorder and brain activity during behavioral control in a genome-wide methylation analysis. Am J Psychiatry 172, 543–552. 10.1176/appi.ajp.2014.14030382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen J, Nivard MG, Willemsen G, Hottenga J-J, Helmer Q, Dolan CV, Ehli EA, Davies GE, van Iterson M, Breeze CE, Beck S, BIOS Consortium, Suchiman HE, Jansen R, van Meurs JB, Heijmans BT, Slagboom PE, Boomsma DI, 2016. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat Commun 7, 11115. 10.1038/ncomms11115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Iterson M, Tobi EW, Slieker RC, den Hollander W, Luijk R, Slagboom PE, Heijmans BT, 2014. MethylAid: Visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 30, 3435–3437. 10.1093/bioinformatics/btu566 [DOI] [PubMed] [Google Scholar]
- Walters RK, Polimanti R, Johnson EC, McClintick JN, Adams MJ, Adkins AE, Aliev F, Bacanu S-A, Batzler A, Bertelsen S, Biernacka JM, Bigdeli TB, Chen L-S, Clarke T-K, Chou Y-L, Degenhardt F, Docherty AR, Edwards AC, Fontanillas P, Foo JC, Fox L, Frank J, Giegling I, Gordon S, Hack LM, Hartmann AM, Hartz SM, Heilmann-Heimbach S, Herms S, Hodgkinson C, Hoffmann P, Jan Hottenga J, Kennedy MA, Alanne-Kinnunen M, Konte B, Lahti J, Lahti-Pulkkinen M, Lai D, Ligthart L, Loukola A, Maher BS, Mbarek H, McIntosh AM, McQueen MB, Meyers JL, Milaneschi Y, Palviainen T, Pearson JF, Peterson RE, Ripatti S, Ryu E, Saccone NL, Salvatore JE, Sanchez-Roige S, Schwandt M, Sherva R, Streit F, Strohmaier J, Thomas N, Wang J-C, Webb BT, Wedow R, Wetherill L, Wills AG, 23andMe Research Team, Boardman JD, Chen D, Choi D-S, Copeland WE, Culverhouse RC, Dahmen N, Degenhardt L, Domingue BW, Elson SL, Frye MA, Gäbel W, Hayward C, Ising M, Keyes M, Kiefer F, Kramer J, Kuperman S, Lucae S, Lynskey MT, Maier W, Mann K, Männistö S, Müller-Myhsok B, Murray AD, Nurnberger JI, Palotie A, Preuss U, Räikkönen K, Reynolds MD, Ridinger M, Scherbaum N, Schuckit MA, Soyka M, Treutlein J, Witt S, Wodarz N, Zill P, Adkins DE, Boden JM, Boomsma DI, Bierut LJ, Brown SA, Bucholz KK, Cichon S, Costello EJ, de Wit H, Diazgranados N, Dick DM, Eriksson JG, Farrer LA, Foroud TM, Gillespie NA, Goate AM, Goldman D, Grucza RA, Hancock DB, Harris KM, Heath AC, Hesselbrock V, Hewitt JK, Hopfer CJ, Horwood J, Iacono W, Johnson EO, Kaprio JA, Karpyak VM, Kendler KS, Kranzler HR, Krauter K, Lichtenstein P, Lind PA, McGue M, MacKillop J, Madden PAF, Maes HH, Magnusson P, Martin NG, Medland SE, Montgomery GW, Nelson EC, Nöthen MM, Palmer AA, Pedersen NL, Penninx BWJH, Porjesz B, Rice JP, Rietschel M, Riley BP, Rose R, Rujescu D, Shen P-H, Silberg J, Stallings MC, Tarter RE, Vanyukov MM, Vrieze S, Wall TL, Whitfield JB, Zhao H, Neale BM, Gelernter J, Edenberg HJ, Agrawal A, 2018. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 21, 1656–1669. 10.1038/s41593-018-0275-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LE, Xu Z, Harlid S, White AJ, Troester MA, Sandler DP, Taylor JA, 2019. Alcohol and DNA methylation: An epigenome-wide association study in blood and normal breast tissue. Am J Epidemiol 188, 1055–1065. 10.1093/aje/kwz032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Wolff KC, Enoch MA, Prescott CA, 2011. The influence of gene-environment interactions on alcohol consumption and Alcohol Use Disorders: A comprehensive review. Clin Psychol Rev 31, 800–816. 10.1016/j.cpr.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi PD, Richmond R, Langdon R, Ness A, Liu C, Levy D, Relton C, Suderman M, Zuccolo L, 2019. Validation and characterisation of a DNA methylation alcohol biomarker across the life course. Clin Epigenetics 11, 163. 10.1186/s13148-019-0753-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gelernter J, 2017. Review: DNA methylation and Alcohol Use Disorders: Progress and challenges. Am J Addict 26, 502–515. 10.1111/ajad.12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Herman AI, Kranzler HR, Anton RF, Simen AA, Gelernter J, 2012. Hypermethylation of OPRM1 promoter region in European Americans with alcohol dependence. J Hum Genet 57, 670–675. 10.1038/jhg.2012.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wang F, Kranzler HR, Yang C, Xu H, Wang Z, Zhao H, Gelernter J, 2014. Identification of methylation quantitative trait loci (mQTLs) influencing promoter DNA methylation of alcohol dependence risk genes. Hum Genet 133, 1093–1104. 10.1007/s00439-014-1452-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Miao Q, Wang C, Zhao R, Li W, Haile CN, Hao W, Zhang XY, 2013. Genome-wide DNA methylation analysis in alcohol dependence. Addiction Biology 18, 392–403. 10.1111/adb.12037 [DOI] [PubMed] [Google Scholar]
- Zhou W, Laird PW, Shen H, 2017. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res 45, e22. 10.1093/nar/gkw967 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.