

Abstract
Objective:
Disease-modifying clinical trials to prevent disease onset are often limited in methods to assess the impact associated with experimental therapeutics. Although some measures demonstrate decline before onset, there are no outcomes accepted by FDA or EMA. This study suggests that sample enrichment can provide a robust method for prevention of disease onset in individuals with premanifest HD.
Methods:
Using HD onset prediction indexes, we calculated the receiver operating curve (ROC) analysis for HD diagnosis within a three-year time frame. We determined optimal cut-points for recruitment and conducted sample size and power calculations to detect varying effect sizes for treatment efficacy in reducing three-year rates of disease onset. Baseline and longitudinal MRI volumes were analyzed for concurrent and predictive biological validation of the sample enrichment algorithm.
Results:
Areas under the curve (AUC) for three HD onset prediction indexes all demonstrated excellent value with the best sensitivity and specificity shown for the multivariate risk score (MRS).
Conclusions:
The enriched target sample size required for a preventive trial in HD using the MRS was only 89 per arm with a study-wide rate of disease onset of over 37% in the untreated group. This approach makes possible a preventive clinical trial of an experimental therapeutic to delay onset of HD with feasible resource costs.
Trial Registration:
PREDICT-HD is registered with www.clinicaltrials.gov, number NCT00051324.
Keywords: Huntington disease, Clinical trials Methodology/study design, Genetics, Movement disorders, MRI
Introduction
Clinical trial successes in Huntington disease (HD) are symptomatic treatments1–6. Disease-modifying interventions face a number of challenges7,8. Two recent papers proposed endpoint measures to facilitate such trials for HD. One of the most frequently cited complexities in clinical trials involves the properties of inclusion criteria to capture heterogeneity1, 9–14 while maximizing efficiencies in statistical power, time and cost. The purpose of this investigation was to examine sample enrichment for preventive clinical trials with calculations of required sample sizes for the clinically-relevant outcome, diagnosis7, 8, 15.
Huntington disease (HD) is an autosomal dominant neurodegenerative disease caused by a triplet repeat expansion of cytosine-adenine-guanine (CAG) in the first exon of the huntingtin gene16. The CAG repeat expansion is associated with age of onset and leads to a progressive loss of function in motor, cognitive, and behavioral integrity resulting in death17. Given that the mutant gene (mHTT) and its resultant protein (huntingtin) are among the most proximal therapeutic targets, options at this level are intensifying18–22. However, clinical trial approaches to indicate when to treat or how to distinguish changes associated with interventions are lacking. Despite efforts to predict onset, estimated algorithms are imprecise and require large sample sizes to demonstrate delay of the conversion to disease diagnosis. Though many investigators have given up on the idea of preventing the onset of HD the purpose of this study is to examine possible clinical trial enrichment strategies to facilitate the design of feasible preventive clinical trials.
Methods
Study design and participants
The PREDICT-HD study43, 44 was a longitudinal natural history study of over 1100 premanifest HD participants conducted in 32 multinational sites from 2002 to 2014. All participants had prior and independent genetic testing and none had received a diagnosis of HD at study entry. Exclusion criteria included other CNS disease, injury, or developmental disorder, or evidence of an unstable medical or psychiatric illness. During the 12-year study, 225 subjects received a prospective diagnosis of HD according to traditional standards established using the Diagnostic Confidence Level (DCL) of the Unified HD Rating Scale45 (UHDRS).
Standard Protocol Approvals, Registrations, and Patient Consents
The research protocol was approved by each site’s respective institutional review board and ethics committee, and all participants gave written informed consent and were treated in accordance with ethical standards.
Procedures
Data collection and storage was described in the other reports of the PREDICT-HD study23, 43.
Primary measures used in this research include motor and cognitive sections of the UHDRS and brain imaging volumetrics.
Briefly, the Total Motor Scale (TMS) is a standardized 31-item assessment rated on a scale from 0 to 4 by an examiner certified by the European HD Network. The range for TMS is 0 – 124 with higher scores reflecting greater abnormality. Following completion of the TMS, the rater completes the UHDRS Diagnostic Confidence Level (DCL), a categorical scale ranging from 0 to 4, where 0 corresponds to non-impaired motor functioning, and 4 corresponds to HD diagnosis defined as unequivocal abnormality of extrapyramidal motor signs of HD with ≥ 99% confidence on the part of the motor examiner.
Cognitive assessment included the Stroop Color Word Test (SCWT)46 on which Individuals are invited to read words, name colors and then name the ink color of words as quickly as possible (each for 45 seconds). The test measures cognitive processing efficiency and inhibition of automatic cognitive processing. The Symbol Digit Modality Test (SDMT)47 is a 90-second task requiring individuals to fill in boxes with a matching number according to a set of symbol-number pairs presented on the same page. The total number of correct items measures speed of cognitive processing, psychomotor accuracy and working memory.
Onset prediction indexes: Despite widespread usage, age of onset is not completely explained by CAG repeat length25–31. Figure 132 confirms wide onset age variation for individuals with the same CAG repeat length. The PREDICT-HD investigators developed five statistical models to index the cumulative toxicity of mutant huntingtin at a given age. First, Langbehn35 used retrospective data from chart review to develop an estimate that was validated with 80 prospectively diagnosed patients with an estimated three-year diagnosis rate of 11.3 percent36. Many studies utilized this model24. Next, Zhang37 developed a revision to the model using 225 prospectively diagnosed HD patients and named it the CAG by Age Product, or CAP score. This formula had more widespread appeal since it didn’t involve the computation of an exponential and was easily calculated. Three-year diagnosis rates for this formula ranged by estimated proximity to diagnosis such that individuals with low CAP showed a conversion rate of 1.6% over three years; medium CAP=7.6%; and High CAP= 22.3%. Clinicians quickly observed that individuals’ CAP scores remained imprecise and did not always reflect the manifestation of clinical phenotype observed upon examination. The multivariate risk score (MRS) was developed by incorporating measures of subtle HD clinical phenotype (motor and cognitive impairments) into the CAP formula23, 38. Most recently, a large collaborative effort validated a normalized prognostic index (PIN) for HD using all available data from PREDICT23, TRACK39, REGISTRY40, and COHORT41, 42 . The higher these index values, the more progressive state a premanifest HD individual is considered.
Figure 1.
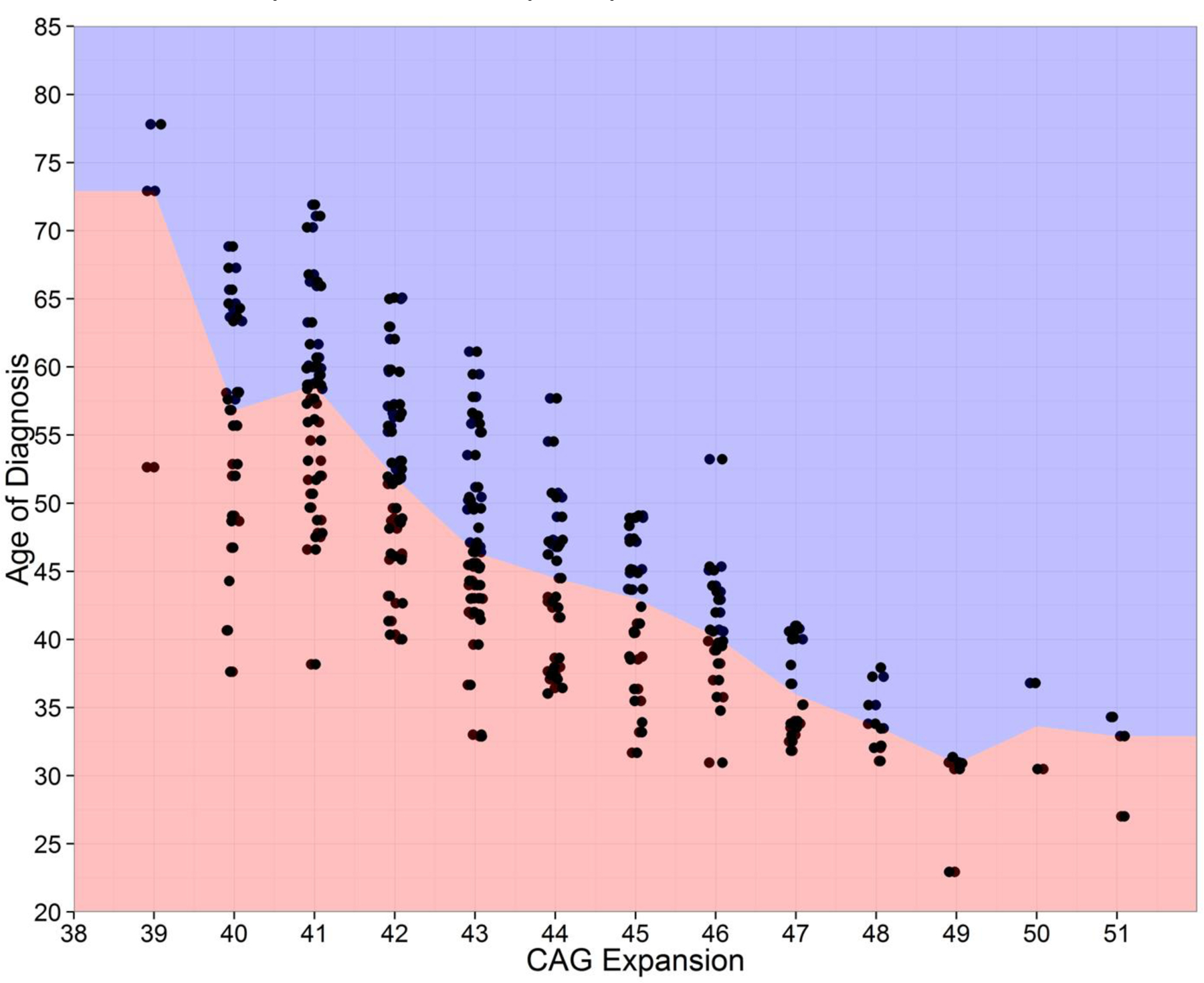
Age of HD onset by CAG repeat length for 225 prospectively diagnosed individuals in the 12-year natural history study entitled PREDICT-HD.
The three indexes (i.e., CAP, MRS and PIN) examined for a preventive trial enrichment algorithm are shown in Table 1. There was no pre-conceived notion about the strength of these formula for three-year prognosis so all three onset prediction indexes were utilized.
Table 1.
CAP, MRS, and PIN indexes and comparison among outcomes using the sample enrichment algorithm for RCT to delay age of onset in Huntington disease
| Prediction Onset Indexes | |||
|---|---|---|---|
|
Characteristics |
CAP37 AGE x (CAG – 33.66) |
MRS38 −0.282 x AGE + 0.140 x CAG + 0.565 x TMS −0.021 x SDMT + 0.347 x DCL1 + 0.542 x DCL2 + 1.086 x DCL3 – 0.004 x SC + 0.002 x SW – 0.023 x SI – 0.004 x TMS2 – 0.010 x TMS x CAG + 0.009 x AGE x CAG |
PIN41 (51 x TMS – 34 x SDMT + 7 x CAP - 883) / 1044 |
| AUC | 0.7897 | 0.8771 | 0.8559 |
| Optimal Cut-Point | >390.4 | >8.914 | >0.3757 |
| Sensitivity | 0.6778 | 0.8222 | 0.8000 |
| Specificity | 0.7734 | 0.8047 | 0.7578 |
| Three-Year Rate of HD diagnosis (%) | 29.5 | 37.2 | 31.7 |
| Required sample size per arm in RCT | 126 | 89 | 114 |
| Target Sample available in Predict | 262 | 248 | 286 |
CAP=CAG age product; MRS=Multivariate risk score; PIN=Prognostic index normalized; RCT=Randomized controlled trial; AUC=Area under the curve; CAG=cytosine, adenosine, guanine repeat length; TMS=Total motor score; SDMT=Symbol digit modalities test; DCL=Diagnostic confidence level; SC=Stroop color; SW=Stroop word; SI=Stroop interference
Brain imaging volumes obtained from Magnetic Resonance Imaging (MRI) are well known to show progressive worsening in concert with clinical and functional decline in premanifest HD48, 49. We chose to use MRI volumes of caudate, putamen, globus pallidus, total gray matter and total white matter for each of the four lobes of the cerebral cortex, total subcortical white matter as well as total cerebrospinal fluid (CSF) since these measurements have demonstrated good replicability across studies and reliability over time24, 48, 50.
Statistical analysis
Methods for the Sample Enrichment Algorithm consisted of three steps: 1. For any given onset prediction index for which a higher value indicates a more advanced premanifest HD state, a receiver operating curve (ROC) analysis51 for three-year HD diagnosis was conducted; 2. The optimal cut-point for each onset prediction index value (i.e., CAP, MRS, PIN) was determined placing equal importance on sensitivity and specificity; 3. The participants classified into the higher onset group for each of the onset prediction indexes suggested the enriched target sample for preventive clinical trials.
Sample sizes were calculated for conducting a randomized controlled trial (RCT) to test the treatment efficacy in reducing three-year rate of HD onset (i.e., conversion from gene-carrier to motor diagnosis) with a given effect size using a two-sided test for proportions at 0.05 significance level powered at 0.9.
Brain volume data were used as a biological disease parameter showing replication in premanifest HD across studies, sites and methods24, 52. First, a two-way analysis of variance (ANOVA) was conducted to determine whether differences were significant between the enriched and the traditional groups on mean baseline structural volume as a percentage of intra-cranial volume (ICV). This analysis was conducted to consider whether the enriched target sample for HD preventive clinical trials showed concurrent/content validity. Next, linear mixed models (LMM)53, 54 were fit to assess whether longitudinal progression rate differences (based on annual percentage of brain volume loss) were evident between the enriched groups adjusting for the effects of scanner strength. This analysis was conducted to consider whether the enriched sample for HD preventive clinical trials showed predictive validity with disease-related biological expectations using brain imaging.
Results
Findings of the sample enrichment algorithm to the three onset prediction indexes using PREDICT-HD data are summarized in Table 1. The AUC values for CAP, MRS, and PIN are 0.7897, 0.8771, and 0.8559 respectively, indicating excellent prognostic value for all prediction formulas. A representative ROC curve is shown in Figure 2. In this example, a RCT to delay HD onset using the MRS would require the targeted recruitment of premanifest gene-expansion carriers with a value above 8.914. Optimal cut-points are shown for each index with related sensitivity and specificity values.
Figure 2.
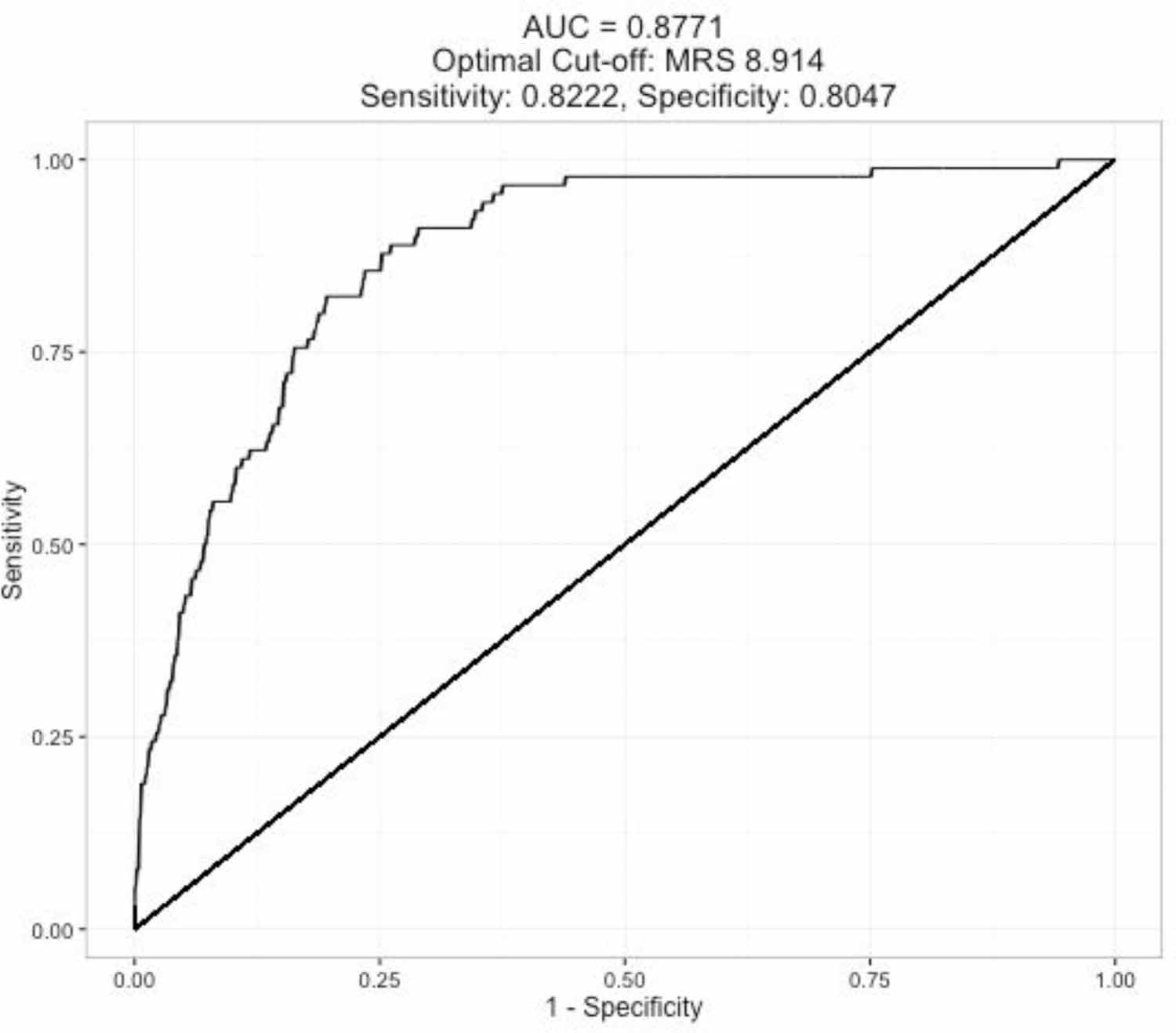
The ROC curve for the three-year HD diagnosis based on the prognostic marker MRS.
The three-year rate of HD diagnosis in the enriched Target samples (Table 1; 29.5, 37.2, 31.7) can be regarded as the baseline diagnosis rate for untreated participants. Figure 3 displays the sample sizes required for each arm of a RCT to determine treatment efficacy in reducing the three-year rate of HD diagnosis with various effect sizes (reduction rates range from 30% to 80%), based on a two-sided test for proportions at significance level 0.05 and powered at 0.90.
Figure 3.
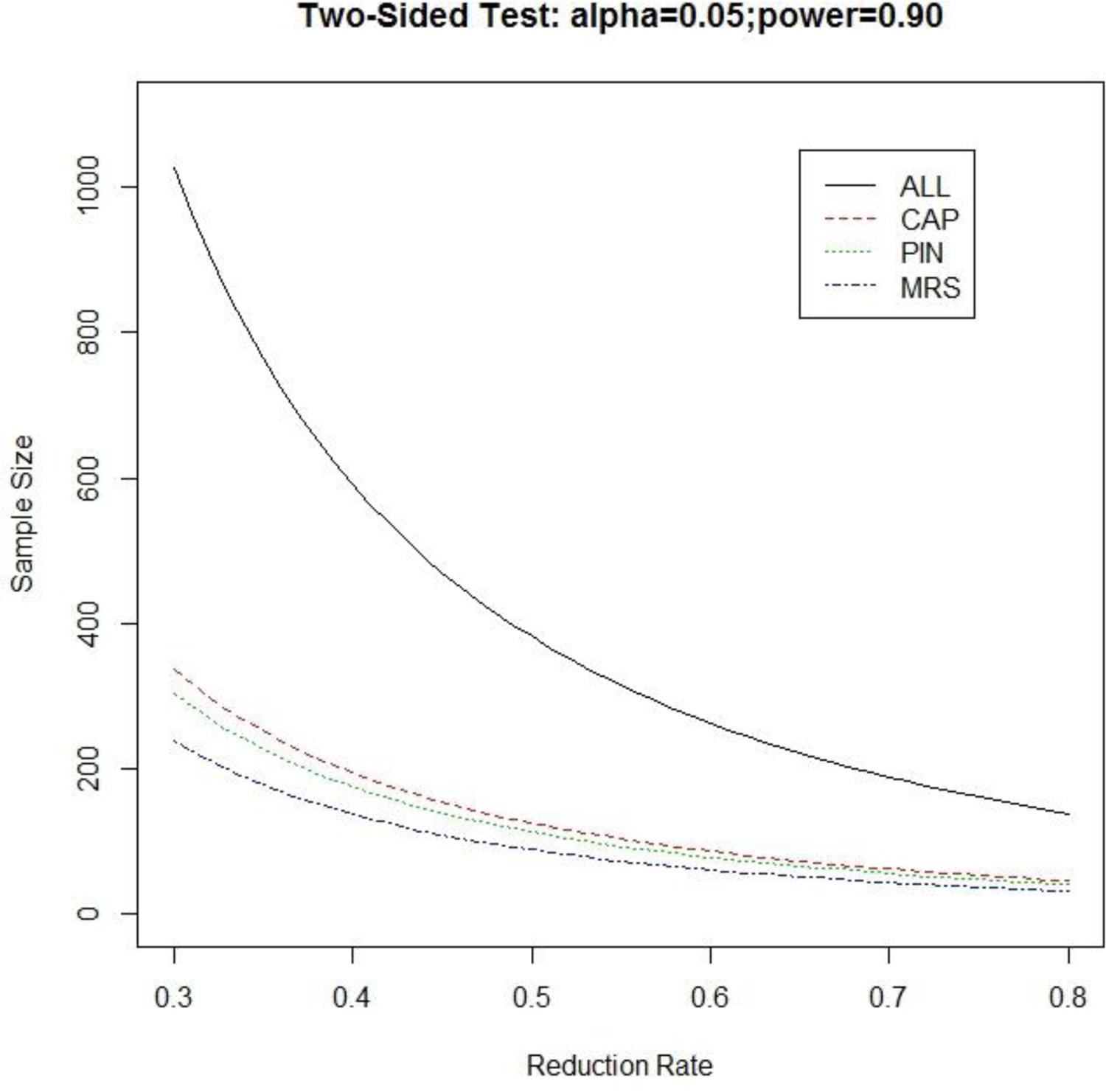
Sample size required in each arm for a randomized treatment-control clinical trial equipped with two-sided test for proportions at significance level 0.05 and powered at 0.9.
Using the most robust published index (i.e., MRS) comparisons of age, sex, CAG repeat length, CAP score, and the baseline clinical measures between an enriched target sample and the traditional premanifest sample recruited into clinical trials showed highly significant differences on all selected demographic, genetic, and clinical measures (ps<0.0001; see supplementary Table A). Participants in the enriched sample were older with greater CAG expansion, showed worsened motor and cognitive impairment, and were rated higher in diagnostic confidence level at study entry. Moreover, individuals in the high group of CAP were more likely to be in the enriched versus the traditional sample (83.2% vs 20.2%).
Baseline MRI ANOVA results with a total of 774 gene-expanded individuals whose data were available at study entry were compared to individuals in the traditional sample (See Table B in supplementary materials). Individuals in the enriched target sample had substantially smaller volumes for Putamen, Caudate and Globus (ps<0.0001) with relative differences of 30.95%, 31.03% and 27.06% respectively. The enriched sample also had 17.59% greater CSF volume on average, compared to the unselected traditional sample (p<0.0001). Although hypothesis testing demonstrated statistical differences between the two groups in many other brain regions, the relative difference between the groups was no more than three percent (See Table B).
The LMM results for MRI volume percentage annual change was conducted to examine brain atrophy over time between the enriched and the typical groups. Because 61 individuals only had baseline MRIs, 713 gene-expanded individuals were analyzed. MRI volumes decreased over time in both samples, though the annual decrease in the enriched sample was significantly greater than in the traditional sample. In particular, differences were highly significant (p<0.0001) in the Putamen, Thalamus and Globus, with relative differences being 27.87%, 40.79%, and 25.09%, respectively.
Discussion
Using data from the 12-year PREDICT-HD study we developed and validated a sample enrichment algorithm that can be used to study any experimental therapeutic to delay or prevent the onset of HD. To date we are unaware of any RCT design to feasibly test the efficacy of an intervention to delay or prevent the onset of HD. Indeed, some authors have dismissed the conversion rate comparison secondary to feasibility limitations in the number of research participants required55, 56. The proposed algorithm can be used with any index or variable the investigators choose to target the best premanifest/prodromal HD participants to recruit into a trial. The algorithm was applied to three disease prognostic markers: CAP, PIN, and MRS and demonstrated its proficiency in detecting the optimal cut point for RCT sample recruitment. Accurate and enriched selection of participants into studies for prevention efficacy is judicious to maximize statistical power, minimize required sample numbers and increase the probability of detecting therapeutic benefits within three years.
Previous clinical trials in premanifest HD were designed for safety and tolerability57 and currently active studies are enrolling premanifest participants whose three-year rate of onset is below 10%. Enriched target group selection based on MRS shows a conversion rate of over 37%. As illustrated in Figure 2, the sample size required for conducting a preventive trial for any given effect size in the two-sided 0.05 significant test of proportions powered at 0.9 is much reduced using the proposed algorithm. For example, if 50% reduction in three-year rate of HD onset is the expected therapeutic efficacy in a RCT, the required one-arm sample size using all HD gene-expansion carriers is about 382. Application of an enrichment algorithm to target recruitment, however, shows greatly reduced samples of 126 and 114 for algorithm-based enrichment using high CAP scores and PIN respectively, and shows the greatest benefit and lowest required sample size (n=89) when the algorithm is applied to MRS.
The use of the proposed enrichment algorithm to select study participants for a three-year preventive trial only requires acquisition of the prognostic variables. This study utilized the CAP, MRS, and PIN, which contain the participant’s current age, CAG repeat length and two to four scores from the UHDRS. The total time to obtain the cognitive and motor variables required for calculation of the MRS or PIN is less than 20 minutes. All measures can be obtained at home, clinic or bedside and no additional equipment is needed.
Since many investigators had dismissed the possibility of preventive trials using diagnosis as an endpoint, a great deal of attention has been directed to the development of other biological or clinical markers that can become surrogates58–60. In theory, for a surrogate endpoint to be an effective substitute for the clinical endpoint, effects of the intervention on the surrogate must reliably predict the overall effect on the clinical outcome. In practice, this requirement frequently fails. Among several explanations for this failure is the possibility that the disease process could affect the clinical outcome through pathways that are not mediated through the surrogate. Additionally, the intervention might affect the clinical outcome by unintended, unanticipated, and unrecognized mechanisms of action that operate independently of the disease process. Although the search for biomarkers, refined clinical markers and eventual surrogate markers for HD is beneficial (and necessary), there are no markers yet qualified for implementation in costly Phase III trials7–9, 61, 62. Excitement generated by novel treatments can lead to clinical trial designs making assumptions without adequate data-driven support. The FDA recently published guidelines for outcome qualification that are likely to increase the probability of the successful outcome of preventive trials for HD63.
Limitations of this research include the concern that onset/diagnosis of insidious neurodegenerative diseases can be arbitrarily dependent upon frequency of clinician visits, expertise of the diagnostician and reliability of the onset outcome. Comparisons of clinical, retrospective research with the prospectively-diagnosed studies using certified motor examiners (such as TRACK and PREDICT), however, demonstrate high consistency among research outcomes using this parameter. Some investigators might be concerned about the three-year limitation of the proposed enrichment sample, although the methods used here can easily be extended to five-year studies should the experimental therapeutics require longer observation. Benefits to longer studies is there will be higher rates of motor onset/diagnosis in five years than in three years, which will yield more power in terms of demonstrating the treatment efficacy in lowering the rate of conversion during the study period.
Conclusions
Recent natural history studies generated rich and essential information to evaluate the disease progression from premanifest healthy individuals through prodromal disease stages to a meaningful endpoint, motor diagnosis. This paper translates knowledge gained from observational natural history studies to critical decision making for preventive clinical trials.
Supplementary Material
Acknowledgements:
We thank the PREDICT-HD sites (investigators, coordinators, cognitive and motor raters), the study participants, the National Research Roster for Huntington Disease Patients and Families, the Huntington’s Disease Society of America, the Huntington Study Group and the European HD Network.
Funding Sources: National Center for Advancing Translational Sciences, National Institutes of Health (NIH) Grant NS040068 and CHDI Foundation, Inc.
Footnotes
Author Disclosures: No authors have any financial disclosures for research covered in this article.
Financial Disclosures of all authors (for the preceding 12 months):
Dr. Paulsen was a consultant for Roche, is a consultant for Wave Life Sciences and CHDI, Inc., is funded by NIH for research grants, was funded by CHDI for research grants, and has received honoraria from the Movement Disorder Society and the Huntington Disease Society of America for presentations and lay publications.
Dr. Lourens reports no disclosures.
Dr. Kieburtz consults for the United States National Institutes of Health, Acorda, Astellas Pharma, AstraZeneca, BioMarin Pharmaceutica, Biotie, Britannia, CHDI, Clearpoint Strategy Group, Clintrex, Corium International, Cynapsus, Forward Pharma, Genzyme, INC Research, Intec, Lundbeck, Medivation, Melior Discovery, Neuroderm.Neurmedix, Orion Pharma, Otsuka, Pfizer, Pharma2B, Prana Biotechnology, Prothena/Neotope/Elan Pharmaceutical, Raptor Pharmaceuticals, Remedy Pharmaceuticals Roche/Genetech, Sage Bionetworks, Sanofi, Serina, Sunovion, Synagile, Titan, UpsherSmith, USWorldMeds, Vaccinex, Vertex Pharmaceuticals, Weston Brain Institute Grants/Research Support, Michael J. Fox Foundation, Teva.
Dr. Zhang reports no disclosures.
Bibliography
- 1.Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, Mantua V, Mecocci P, Pani L, Winblad B, Kivipelto M. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med 2014;275(3):251–83. doi: 10.1111/joim.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner P, Piehl F. Fatigue and depression in multiple sclerosis: pharmacological and non-pharmacological interventions. Acta Neurol Scand 2016;134 Suppl 200:47–54. doi: 10.1111/ane.12648. [DOI] [PubMed] [Google Scholar]
- 3.Desamericq G, Youssov K, Charles P, Saleh N, Olivier A, Sherer-Gagou C, Verny C, multidisciplinary working g, Bachoud-Levi AC. Guidelines for clinical pharmacological practices in Huntington’s disease. Rev Neurol (Paris) 2016;172(8–9):423–32. doi: 10.1016/j.neurol.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Kaur N, Kumar P, Jamwal S, Deshmukh R, Gauttam V. Tetrabenazine: Spotlight on Drug Review. Annals of neurosciences 2016;23(3):176–85. doi: 10.1159/000449184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laurencin C, Danaila T, Broussolle E, Thobois S. Initial treatment of Parkinson’s disease in 2016: The 2000 consensus conference revisited. Rev Neurol (Paris) 2016;172(8–9):512–23. doi: 10.1016/j.neurol.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Orsini M, Oliveira AB, Nascimento OJ, Reis CH, Leite MA, de Souza JA, Pupe C, de Souza OG, Bastos VH, de Freitas MR, Teixeira S, Bruno C, Davidovich E, Smidt B. Amyotrophic Lateral Sclerosis: New Perpectives and Update. Neurol Int 2015;7(2):5885. doi: 10.4081/ni.2015.5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cedarbaum JM, Stephenson D, Rudick R, Carrillo MC, Stebbins G, Kerr D, Heemskerk J, Galpern WR, Kaufmann P, Cella D, Isaac M, Walton MK. Commonalities and challenges in the development of clinical trial measures in neurology. Neurotherapeutics 2015;12(1):151–69. doi: 10.1007/s13311-014-0310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szabo ST, Kinon BJ, Brannan SK, Krystal AK, van Gerven JM, Mahableshwarkar A, Sachs GS. Lessons Learned and Potentials for Improvement in CNS Drug Development: ISCTM Section on Designing the Right Series of Experiments. Innov Clin Neurosci 2015;12(3–4):11S–25S. [PMC free article] [PubMed] [Google Scholar]
- 9.Babbs CF. Choosing inclusion criteria that minimize the time and cost of clinical trials. World J Methodol 2014;4(2):109–22. doi: 10.5662/wjm.v4.i2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grill JD, Monsell SE. Choosing Alzheimer’s disease prevention clinical trial populations. Neurobiol Aging 2014;35(3):466–71. doi: 10.1016/j.neurobiolaging.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayward RA, Kent DM, Vijan S, Hofer TP. Multivariable risk prediction can greatly enhance the statistical power of clinical trial subgroup analysis. BMC Med Res Methodol 2006;6:18. doi: 10.1186/1471-2288-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent DM, Rothwell PM, Ioannidis JP, Altman DG, Hayward RA. Assessing and reporting heterogeneity in treatment effects in clinical trials: a proposal. Trials 2010;11:85. doi: 10.1186/1745-6215-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicholson KA, Cudkowicz ME, Berry JD. Clinical Trial Designs in Amyotrophic Lateral Sclerosis: Does One Design Fit All? Neurotherapeutics 2015;12(2):376–83. doi: 10.1007/s13311-015-0341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol 2015;14(2):208–23. doi: 10.1016/S1474-4422(14)70264-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai TL, Lavori PW. Innovative Clinical Trial Designs: Toward a 21st-Century Health Care System. Stat Biosci 2011;3(2):145–68. doi: 10.1007/s12561-011-9042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. . Cell 1993;72(6):971–83. [DOI] [PubMed] [Google Scholar]
- 17.Walker FO. Huntington’s Disease. Semin Neurol 2007;27(2):143–50. Epub 2007/03/29. doi: 10.1055/s-2007-971176. [DOI] [PubMed] [Google Scholar]
- 18.Aronin N, Kim M, Laforet G, DiFiglia M. Are there multiple pathways in the pathogenesis of Huntington’s disease? Philos Trans R Soc Lond B Biol Sci 1999;354(1386):995–1003. doi: 10.1098/rstb.1999.0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garriga-Canut M, Agustin-Pavon C, Herrmann F, Sanchez A, Dierssen M, Fillat C, Isalan M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci U S A 2012;109(45):E3136–45. doi: 10.1073/pnas.1206506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012;74(6):1031–44. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magen I, Hornstein E. Oligonucleotide-based therapy for neurodegenerative diseases. Brain Res 2014;1584:116–28. doi: 10.1016/j.brainres.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: What’s in the pipeline? Mov Disord 2014;29(11):1434–45. doi: 10.1002/mds.26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulsen JS, Long JD, Johnson HJ, Aylward EH, Ross CA, Williams JK, Nance MA, Erwin CJ, Westervelt HJ, Harrington DL, Bockholt HJ, Zhang Y, McCusker EA, Chiu EM, Panegyres PK, PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front Aging Neurosci 2014;6:78. doi: 10.3389/fnagi.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR, TRACK-HD Investigators. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol 2013;12(7):637–49. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- 25.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: mechanisms of dynamic mutations. Nature reviews Genetics 2005;6(10):729–42. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 26.Bates GP. History of genetic disease: the molecular genetics of Huntington disease - a history. Nature reviews Genetics 2005;6(10):766–73. Epub 2005/09/02. doi: nrg1686 [pii] 10.1038/nrg1686. [DOI] [PubMed] [Google Scholar]
- 27.Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. (vol 3, pg 291, 2004). Lancet Neurol 2004;3(7):443-. [DOI] [PubMed] [Google Scholar]
- 28.Squitieri F, Ciarmiello A, Di Donato S, Frati L. The search for cerebral biomarkers of Huntington’s disease: a review of genetic models of age at onset prediction. Eur J Neurol 2006;13(4):408–15. Epub 2006/04/29. doi: ENE1264 [pii] 10.1111/j.1468-1331.2006.01264.x. [DOI] [PubMed] [Google Scholar]
- 29.Antonini G, Giubilei F, Mammarella A, Amicucci P, Fiorelli M, Gragnani F, Morino S, Ceschin PV, Fragola PV, Gennarelli M. Natural history of cardiac involvement in myotonic dystrophy: correlation with CTG repeats. Neurology 2000;55(8):1207–9. [DOI] [PubMed] [Google Scholar]
- 30.Mateo I, Llorca J, Volpini V, Corral J, Berciano J, Combarros O. GAA expansion size and age at onset of Friedreich’s ataxia. Neurology 2003;61(2):274–5. [DOI] [PubMed] [Google Scholar]
- 31.Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet 1997;60(5):1202–10. [PMC free article] [PubMed] [Google Scholar]
- 32.Paulsen JS, Long JD, Ross CA, Harrington DL, Erwin CJ, Williams JK, Westervelt HJ, Johnson HJ, Aylward EH, Zhang Y, Bockholt HJ, Barker RA, PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Prediction of manifest Huntington’s disease with clinical and imaging measures: a prospective observational study. L ancet Neurol 2014;13(12):1193–201. Epub 3 November 2014. doi: 10.1016/S1474-4422(14)70238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li JL, Hayden MR, Warby SC, Durr A, Morrison PJ, Nance M, Ross CA, Margolis RL, Rosenblatt A, Squitieri F, Frati L, Gomez-Tortosa E, Garcia CA, Suchowersky O, Klimek ML, Trent RJ, McCusker E, Novelletto A, Frontali M, Paulsen JS, Jones R, Ashizawa T, Lazzarini A, Wheeler VC, Prakash R, Xu G, Djousse L, Mysore JS, Gillis T, Hakky M, Cupples LA, Saint-Hilaire MH, Cha JH, Hersch SM, Penney JB, Harrison MB, Perlman SL, Zanko A, Abramson RK, Lechich AJ, Duckett A, Marder K, Conneally PM, Gusella JF, MacDonald ME, Myers RH. Genome-wide significance for a modifier of age at neurological onset in Huntington’s disease at 6q23–24: the HD MAPS study. BMC Med Genet 2006;7:71. Epub 2006/08/18. doi: 1471–2350-7–71 [pii] 10.1186/1471-2350-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Genetic Modifiers of Huntington’s Disease C. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015;162(3):516–26. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR, International Huntington’s Disease Collaborative Group. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 2004;65(4):267–77. Epub 2004/03/18. doi: 10.1111/j.1399-0004.2004.00241.x CGE241 [pii]. [DOI] [PubMed] [Google Scholar]
- 36.Langbehn DR, Hayden MR, Paulsen JS, PREDICT-HD Investigators of the Huntington Study Group. CAG-repeat length and the age of onset in Huntington disease (HD): A review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet 2010;153B:397–408. Epub 2009/06/24. doi: 10.1002/ajmg.b.30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Long JD, Mills JA, Warner JH, Lu W, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet 2011;156B(7):751–63. doi: 10.1002/ajmg.b.31232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long JD, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Multivariate prediction of motor diagnosis in Huntington’s disease: 12 years of PREDICT-HD. Mov Disord 2015;30(12):1664–72. doi: 10.1002/mds.26364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC, TRACK-HD investigators. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: cross-sectional analysis of baseline data. Lancet Neurol 2009;8(9):791–801. doi: 10.1016/S1474-4422(09)70170-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orth M, Handley OJ, Schwenke C, Dunnett SB, Craufurd D, Ho AK, Wild E, Tabrizi SJ, Landwehrmeyer GB, Investigators of the European Huntington’s Disease N. Observing Huntington’s Disease: the European Huntington’s Disease Network’s REGISTRY. PLoS currents 2010;2:RRN1184. doi: 10.1371/currents.RRN1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Long JD, Langbehn DR, Tabrizi SJ, Landwehrmeyer BG, Paulsen JS, Warner J, Sampaio C. Validation of a prognostic index for Huntington’s disease. Mov Disord 2016. doi: 10.1002/mds.26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huntington Study Group CI, Dorsey E. Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study. PLoS One 2012;7(2):e29522. doi: 10.1371/journal.pone.0029522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, Guttman M, Nance M, Kieburtz K, Oakes D, Shoulson I, Kayson E, Johnson S, Penziner E, Predict-HD Investigators of the Huntington Study Group. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol 2006;63(6):883–90. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- 44.Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M, PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry 2008;79(8):874–80. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huntington Study Group. Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov Disord 1996;11(2):136–42. Epub 1996/03/01. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 46.Stroop JR. Studies of interference in serial verbal reactions. J Exp Psychol 1935;18:643–62. doi: 10.1037/0096-3445.121.1.15. [DOI] [Google Scholar]
- 47.Smith A. Symbol digit modalities test Los Angeles: Western Psychological Services; 1991. [Google Scholar]
- 48.Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, Brandt J, Gourley LM, Liang K, Zhou H, Margolis RL, Ross CA. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004;63(1):66–72. Epub 2004/07/14. doi: [DOI] [PubMed] [Google Scholar]
- 49.Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, Nopoulos PC. Brain structure in preclinical Huntington’s disease. Biol Psychiatry 2006;59(1):57–63. doi: 10.1016/j.biopsych.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Paulsen JS, Nopoulos PC, Aylward E, Ross CA, Johnson H, Magnotta VA, Juhl A, Pierson RK, Mills J, Langbehn D, Nance M. Striatal and white matter predictors of estimated diagnosis for Huntington disease. Brain Res Bull 2010;82(3–4):201–7. Epub 2010/04/14. doi: S0361–9230(10)00070–5 [pii] 10.1016/j.brainresbull.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hajian-Tilaki K. Receiver Operating Characteristic (ROC) Curve Analysis for Medical Diagnostic Test Evaluation. Caspian journal of internal medicine 2013;4(2):627–35. [PMC free article] [PubMed] [Google Scholar]
- 52.Aylward EH, Liu D, Nopoulos PC, Ross CA, Pierson RK, Mills JA, Long JD, Paulsen JS, PREDICT-HD Investigators Coordinators of the Huntington Study Group. Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol Psychiatry 2012;71(9):822–8. doi: 10.1016/j.biopsych.2011.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verbeke G, Molenberghs G. Linear mixed models for longitudinal data New York, NY: Springer-Verlag New York LLC; 2000. 568 p. [Google Scholar]
- 54.Cox DR. Regression Models and Life-Tables. Journal of the Royal Statistical Society, Series B 1972;34(2):187–220. [Google Scholar]
- 55.Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, Reilmann R, Unschuld PG, Wexler A, Margolis RL, Tabrizi SJ. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10(4):204–16. doi: 10.1038/nrneurol.2014.24. [DOI] [PubMed] [Google Scholar]
- 56.Huntington Study Group PI, Biglan KM, Shoulson I, Kieburtz K, Oakes D, Kayson E, Shinaman MA, Zhao H, Romer M, Young A, Hersch S, Penney J, Marder K, Paulsen J, Quaid K, Siemers E, Tanner C, Mallonee W, Suter G, Dubinsky R, Gray C, Nance M, Bundlie S, Radtke D, Kostyk S, Baic C, Caress J, Walker F, Hunt V, O’Neill C, Chouinard S, Factor S, Greenamyre T, Wood-Siverio C, Corey-Bloom J, Song D, Peavy G, Moskowitz C, Wesson M, Samii A, Bird T, Lipe H, Blindauer K, Marshall F, Zimmerman C, Goldstein J, Rosas D, Novak P, Caviness J, Adler C, Duffy A, Wheelock V, Tempkin T, Richman D, Seeberger L, Albin R, Chou KL, Racette B, Perlmutter JS, Perlman S, Bordelon Y, Martin W, Wieler M, Leavitt B, Raymond L, Decolongon J, Clarke L, Jankovic J, Hunter C, Hauser RA, Sanchez-Ramos J, Furtado S, Suchowersky O, Klimek ML, Guttman M, Sethna R, Feigin A, Cox M, Shannon B, Percy A, Dure L, Harrison M, Johnson W, Higgins D, Molho E, Nickerson C, Evans S, Hobson D, Singer C, Galvez-Jimenez N, Shannon K, Comella C, Ross C, Saint-Hilaire MH, Testa C, Rosenblatt A, Hogarth P, Weiner W, Como P, Kumar R, Cotto C, Stout J, Brocht A, Watts A, Eberly S, Weaver C, Foroud T, Gusella J, MacDonald M, Myers R, Fahn S, Shults C. Clinical-Genetic Associations in the Prospective Huntington at Risk Observational Study (PHAROS): Implications for Clinical Trials. JAMA neurology 2016;73(1):102–10. doi: 10.1001/jamaneurol.2015.2736. [DOI] [PubMed] [Google Scholar]
- 57.Rosas HD, Doros G, Gevorkian S, Malarick K, Reuter M, Coutu JP, Triggs TD, Wilkens PJ, Matson W, Salat DH, Hersch SM. PRECREST: a phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology 2014;82(10):850–7. doi: 10.1212/WNL.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carlozzi NE, Miciura A, Migliore N, Dayalu P. Understanding the Outcomes Measures used in Huntington Disease Pharmacological Trials: A Systematic Review. Journal of Huntington’s disease 2014;3(3):233–52. doi: 10.3233/JHD-140115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorsey ER, Venuto C, Venkataraman V, Harris DA, Kieburtz K. Novel Methods and Technologies for 21st-Century Clinical Trials: A Review. JAMA neurology 2015;72(5):582–8. doi: 10.1001/jamaneurol.2014.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sampaio C, Borowsky B, Reilmann R. Clinical trials in Huntington’s disease: Interventions in early clinical development and newer methodological approaches. Mov Disord 2014;29(11):1419–28. doi: 10.1002/mds.26021. [DOI] [PubMed] [Google Scholar]
- 61.Steeves JD, Zariffa J, Kramer JL. Are you “tilting at windmills” or undertaking a valid clinical trial? Yonsei Med J 2011;52(5):701–16. doi: 10.3349/ymj.2011.52.5.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tabrizi SJ, Reilmann R, Roos RA, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL, Kennard C, Landwehrmeyer B, Stout JC, Borowsky B, Scahill RI, Frost C, Langbehn DR, TRACK-HD investigators. Potential endpoints for clinical trials in premanifest and early Huntington’s disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol 2012;11(1):42–53. doi: 10.1016/S1474-4422(11)70263-0. [DOI] [PubMed] [Google Scholar]
- 63.Food and Drug Administration. Clinical Outcome Assessment Qualification Program 2016. [updated April 1, 2016; cited 2016 May 5]. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/ucm284077.htm. [Google Scholar]
- 64.Chen T, Wang Y, Ma Y, Marder K, Langbehn DR. Predicting Disease Onset from Mutation Status Using Proband and Relative Data with Applications to Huntington’s Disease. Journal of probability and statistics 2012;2012. doi: 10.1155/2012/375935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen T, Ma Y, Wang Y. Predicting cumulative risk of disease onset by redistributing weights. Stat Med 2015;34(16):2427–43. doi: 10.1002/sim.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Albin RL, Burke JF. Potential trade-offs in treatment of premanifest Huntington’s disease. Mov Disord 2015;30(10):1319–23. doi: 10.1002/mds.26318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.