

Abstract
Objectives:
Ongoing adult sepsis clinical trials are assessing therapies that target three inflammation phenotypes including 1) immunoparalysis associated, 2) thrombotic microangiopathy driven thrombocytopenia associated, and 3) sequential liver failure associated multiple organ failure. These three phenotypes have not been assessed in the pediatric multicenter setting. We tested the hypothesis that these phenotypes are associated with increased macrophage activation syndrome and mortality in pediatric sepsis.
Design:
Prospective severe sepsis cohort study comparing children with multiple organ failure and any of these phenotypes to children with multiple organ failure without these phenotypes and children with single organ failure.
Setting:
Nine PICUs in the Eunice Kennedy Shriver National Institutes of Child Health and Human Development Collaborative Pediatric Critical Care Research Network.
Patients:
Children with severe sepsis and indwelling arterial or central venous catheters.
Interventions:
Clinical data collection and twice weekly blood sampling until PICU day 28 or discharge.
Measurements and Main Results:
Of 401 severe sepsis cases enrolled, 112 (28%) developed single organ failure (0% macrophage activation syndrome 0/112; < 1% mortality 1/112), whereas 289 (72%) developed multiple organ failure (9% macrophage activation syndrome 24/289; 15% mortality 43/289). Overall mortality was higher in children with multiple organ and the phenotypes (24/101 vs 20/300; relative risk, 3.56; 95% CI, 2.06–6.17). Compared to the 188 multiple organ failure patients without these inflammation phenotypes, the 101 multiple organ failure patients with these phenotypes had both increased macrophage activation syndrome (19% vs 3%; relative risk, 7.07; 95% CI, 2.72–18.38) and mortality (24% vs 10%; relative risk, 2.35; 95% CI, 1.35–4.08).
Conclusions:
These three inflammation phenotypes were associated with increased macrophage activation syndrome and mortality in pediatric sepsis-induced multiple organ failure. This study provides an impetus and essential baseline data for planning multicenter clinical trials targeting these inflammation phenotypes in children. (Pediatr Crit Care Med 2019; 20:1137–1146)
Keywords: immune paralysis, macrophage activation syndrome, sequential liver failure associated multiple organ failure, thrombocytopenia associated multiple organ failure
A contemporary global point prevalence study of sepsis in PICUs reported 25% mortality (1), with the highest rates found in children who developed new and progressive multiple organ failure (MOF) (2). In contrast to shock which causes death in less than 3 days (3), this condition leads to death after 3 days independent of timely administration of antibiotics and fluids in the emergency department (3–5). Present day treatment is directed to removing the infection source and supporting organ dysfunction without addressing inflammation pathobiology. It is unknown whether clinical trials that focus on inflammation pathobiology phenotypes could improve outcomes in these children who die late MOF deaths.
Three specific inflammation phenotypes related to abnormal immune and coagulation responses in sepsis include as follows: 1) immunoparalysis associated MOF (IPMOF) (6–10), characterized by prolonged depression of innate and adaptive immunity contributing to secondary infections; 2) thrombocytopenia associated MOF (TAMOF) (11–20), characterized by thrombotic microangiopathy with decreased a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity contributing to inability to resolve von Willebrand factor (vWF) multimer thrombi formation especially in the kidney, and 3) sequential liver failure associated MOF (SMOF) defined by high circulating soluble Factor-related Apoptosis Ligand (sFasL) contributing to subsequent liver injury (21–24). As a final common pathway of uncontrolled inflammation, macrophage activation syndrome (MAS) can occur in the setting of any of these sepsis phenotypes (25–28).
Adult clinical trials targeting these three inflammation phenotypes include studies of 1) septic immunoparalysis patients that use granulocyte-macrophage colony-stimulating factor (GM-CSF) and/or interferon-gamma to restore immune function and prevent secondary infection (NCT000252915, NCT02361528); 2) septic TAMOF patients with thrombotic microangiopathy that use plasma exchange (NCT02906345) or soluble thrombomodulin (NCT00487656) to restore coagulation hemostasis and resolve organ failure, as well as patients with bacterial toxin-mediated thrombotic microangiopathy that use C5A monoclonal antibody (NCT01194973, NCT01410916) to control overactivation of complement to liberate patients from renal replacement therapy dependence; and 3) septic post transplantation patients who develop Epstein-Barr virus-related SMOF that use CD20 monoclonal antibody to improve survival (NCT01058239). Differential modulation of uncontrolled inflammation manifest as MAS related to these phenotypes is being studied in septic adults in a trial which uses interferon γ for patients with immunoparalysis to restore immune function, but interleukin-1 receptor antagonist protein treatment for those with hyperferritinemia to reduce inflammation (NCT03332225).
The Eunice Kennedy Shriver National Institute of Child Health and Human Development Collaborative Pediatric Critical Care Research Network is interested in investigating U.S. Food and Drug Administration-approved treatments for new pediatric indications in these sepsis phenotypes (25, 26). In a single-center pediatric “proof of concept” study performed from 2009 to 2014, we found that these three inflammation phenotypes were present alone and in overlap in association with increased MAS and PICU mortality in sepsis-induced MOF (27). The purpose of our present study is to now assess these three phenotypes in the multicenter setting to inform plausibility for extending pertinent adult trials into our PICU network. We performed the present multicenter study from 2015 to 2017 testing the hypothesis that the three inflammation phenotypes are associated with increased MAS and mortality in pediatric sepsis-induced MOF by comparing children who developed MOF with these phenotypes to children who developed MOF without these phenotypes and children who never developed MOF.
MATERIALS AND METHODS
Based on analysis of the results of our previous single-center study, a sample size of 400 subjects was targeted (27). After obtaining Institutional Review Board approval in the nine participating PICUs at Children’s Hospital of Pittsburgh, Children’s Hospital of Philadelphia, Children’s National Medical Center, Children’s Hospital of Michigan, Nationwide Children’s Hospital, Children’s Hospital of Los Angeles, St. Louis Children’s Hospital, C. S. Mott Children’s Hospital, and Mattel Children’s Hospital at University of California Los Angeles, written parental permission was obtained and 401 children were enrolled between 2015 and 2017. Inclusion criteria were diagnosis of severe sepsis, an existing indwelling central venous or arterial catheter for blood drawing, an age greater than 44 weeks gestation and less than 18 years. The exclusion criterion was a lack of commitment to aggressive care. In addition, in order to reduce over-representation in the population sample from individual sites or seasonal epidemics, the total enrollments allowed per center were capped at 80 patients, and the total number of enrollments per quarter from all sites taken together were capped at 80 patients. In order to enrich for patients who survived their initial presentation but subsequently died after 3 days with MOF, we performed screening and enrollment for patients twice weekly every Monday/Tuesday and Thursday/Friday rather than daily. Subsequent biomarker and clinical data were assessed twice weekly (Monday/Thursday or Tuesday/Friday) up to 28 days in the PICU.
“Sepsis” was defined by the presence of two or more of the following four criteria: 1) tachycardia, 2) tachypnea, 3) abnormal temperature, and 4) abnormal WBC count, plus suspicion of infection (29). “Organ failure” was defined using modified organ failure index (OFI) criteria (Table 1) (22, 27, 30). “Severe sepsis” was defined by the presence of sepsis and one or more organ failures (27, 29, 30). “MOF” was defined by the development of two or more organ failures (27, 29, 30). The phenotypes IPMOF (6, 27), TAMOF (11, 27), and SMOF (22, 27); as well as the outcome MAS (27, 28, 31, 32), were defined according to clinical and confirmatory biomarker criteria used in our previously published single-center study (27) (Table 1). Justification for the confirmatory biomarker cutoff values for ex vivo tumor necrosis factor (TNF) response in IPMOF (6–10), ADAMTS13 in TAMOF (11, 15, 17–20), sFasL in SMOF (22–24), and ferritin in MAS (28, 31–35) was based on previous literature. Patients who met clinical criteria for TAMOF, SMOF, or MAS, but received plasma exchange before measurement of confirmatory biomarkers (SMOF n = 2; TAMOF n = 2), required only clinical criteria to be considered confirmed because plasma exchange restores ADAMTS13 activity and removes sFasL and ferritin. Mortality was defined a priori as PICU death rather than 28-day mortality because deaths related to MOF not uncommonly occur after 28 days in the PICU (27).
TABLE 1.
Clinical and Biomarker Criteria Used to Define Three Inflammation Phenotypes, Macrophage Activation Syndrome, and Mortality
| Group | Clinical Criteria | Confirmatory Biomarker |
|---|---|---|
| Thrombocytopenia associated MOF (thrombotic microangiopathy) | On at least one study day, the subject has ALL of the following: Platelet count of < 100,000/μL Renal failurea OFI of three or more |
A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 < 57% of normal at any time during the study |
| Sequential liver failure associated MOF (new hepatobiliary dysfunction [virus/lymphoproliferative disease]) | On at least one study day (consider day 3 or later only), the subject has both hepatic failurea and pulmonary failurea | On at least one study day on or after day 3, the subject has a soluble Fas ligand measurement > 200 pg/mL |
| Immunoparalysis associated MOF (immune depression) | On at least one study day, the subject has an OFI ≥ 2 | Ex vivo whole blood endotoxin-stimulated tumor necrosis factor-α response < 200 pg/mL at any time on day 3 or later |
| Macrophage activation syndrome (hyper inflammation common end pathway) | On at least one study day, the subject has both hepatic failurea and hematologic failurea | Ferritin > 500 ng/mL on any study day |
| Mortality | Death in PICU |
MOF = multiple organ failure, OFI = organ failure index.
OFI definitions: “Cardiovascular”: Inotrope OR vasopressor infusion requirement; “Pulmonary”: Pao2/Fio2 ratio of < 300 mm Hg AND mechanical ventilator requirement; “Hepatic”: alanine aminotransferase > 100 U/L AND either bilirubin > 1.0 mg/dL OR international normalized ratio (INR) > 1.5; “Renal”: Creatinine > 1 mg/dL with oliguria (urine output < 0.5 mL/kg/hr) or subject on renal replacement therapy; “Hematologic”: Platelet count < 100,000 cells/μL AND INR > 1.5; “CNS”: Glasgow Coma Score < 12 in absence of sedatives.
Two 4.7 mL blood samples were obtained twice weekly from an indwelling arterial or central venous catheter. The whole blood ex vivo lipopolysaccharide-stimulated TNF-α response was determined as previously described (6, 27). Stimulation kits were manufactured at Nationwide Children’s Hospital. The kits were quality controlled with a coefficient of variation less than 10%. TNF-α and sFasL assays were performed using enzyme-linked immunosorbent assay (R&D). ADAMTS13 activity was measured at the Institute for Transfusion Medicine Clinical Laboratory (Pittsburgh, PA), and C-reactive protein (CRP) and ferritin levels were measured at the University of Pittsburgh Medical Center Clinical Laboratory.
The children were classified as members of three investigational subgroups: those with MOF and one or more of the three phenotypes, those with MOF without any of the three phenotypes, and those without MOF (i.e., single organ failure). Cohort characteristics were summarized using counts and percentages for categorical variables. Continuous variables were summarized with the median and interquartile range (IQR). Mean ± sd, minimum, and maximum were also reported for maximum ferritin and CRP values in order to provide a more complete description of the distributions. Associations between cohort characteristics and the inflammation phenotypes were evaluated with Fisher exact test for categorical variables and the Wilcoxon rank-sum test for continuous variables.
Maximum CRP and ferritin values for subjects with an inflammation phenotype were compared with subjects with MOF that did not have any of the three inflammation phenotypes using the Wilcoxon rank-sum test. This comparison was made for each inflammation phenotype individually and as a group.
Poisson regression with robust error estimates was used throughout for all univariable and multivariable modeling. Relative risks (RRs) and the associated 95% CIs were calculated to investigate the risk of mortality associated with a condition of MOF and confirmed phenotype. Univariable models considering both mortality and the development of MAS as outcome are provided. Additionally, a multivariable Poisson regression model was constructed to further investigate risks associated with mortality considering children only with MOF (n = 289), using admission characteristics and development of inflammation phenotypes.
Analyses were performed using SAS 9.4 (SAS Institute, Cary, NC) and R Version 3.4.4 (The R Foundation for Statistical Computing, Vienna, Austria). p values are based on a two-sided alternative with p values of less than 0.05 considered significant.
RESULTS
Among the 401 enrolled PICU subjects (CONSORT; Supplemental Fig. 1, Supplemental Digital Content 1, http://linkslww.com/PCC/B75; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81), all had suspected infection with 225 having documented infection (141 bacterial infections, 112 viral infections, four fungal infections, and one protozoal infection) and 176 did not. The average age of the children was 6.8 years (sd ± 5.72 yr). There were 179 females (46%). Two-hundred twenty-three (56%) had a chronic illness. Forty-nine (12%) had a malignancy and 8% had a solid organ transplant (n = 8; 2%). The median Pediatric Risk of Mortality (PRISM)–III score was 8.0 with IQR (3.0–14.0). The median number of organ failures at enrollment was 2.0 IQR (1.0–2.0). The median study days were 11.0 with IQR (6.0–19.0). The median time from sepsis onset to first blood draw was 2 days IQR (1.0–2.0 d). The median blood draws were 2.0 with IQR (1.0–3.0).
Among 401 cases of severe sepsis, there were 112 with single organ failure, 188 with MOF without any of the phenotypes (OFI median [IQR] = 2.0 [2.0–3.0]) and 101 with MOF with one or more of the phenotypes (OFI median [IQR] = 3.0 [2.0–4.0]). Among the 101 patients with one or more phenotypes, 85 had IPMOF, 37 had TAMOF, and seven had SMOF. Figure 1 shows the degree of overlap between the three phenotypes. At admission patients with the IPMOF or TAMOF phenotypes were more likely to be older, have increased illness severity, and have malignancy (Table 2; p < 0.05).
Figure 1.
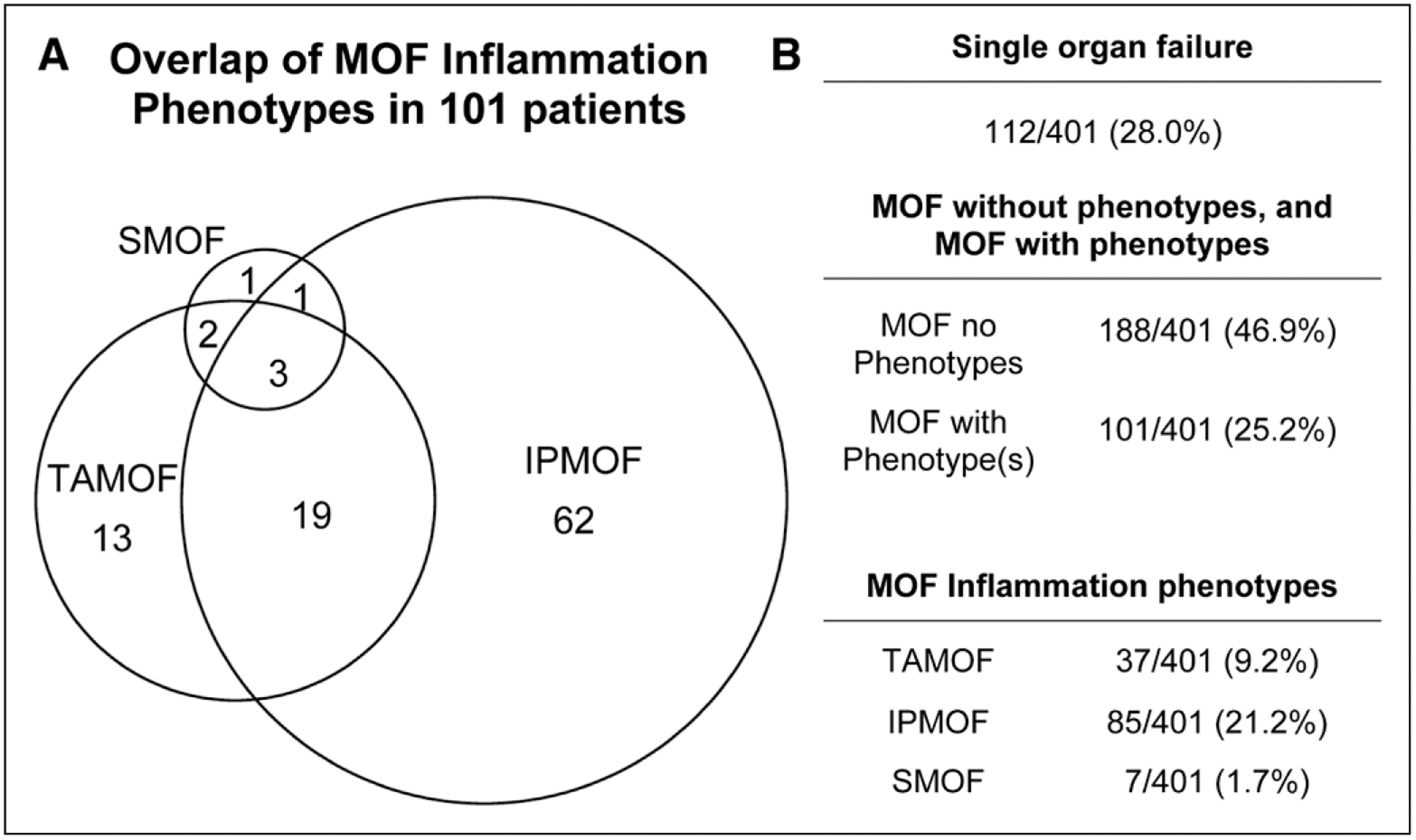
Distribution of organ failure groups in entire severe sepsis cohort. A, Distribution of immunoparalysis associated multiple organ failure (IPMOF), thrombocytopenia associated multiple organ failure (TAMOF), and sequential liver failure associated multiple organ failure (SMOF) among 101 patients with multiple organ failure (MOF) and one or more inflammation phenotypes. B, Numbers of patients with single organ failure, MOF without phenotypes, multiple organ with phenotypes, IPMOF, TAMOF, and SMOF.
TABLE 2.
Baseline Admission Characteristics of Each Inflammation Phenotype
| Cohort Characteristics | Overall (n = 401) | Confirmed Thrombocytopenia Associated MOFa | P | Confirmed Sequential Liver Failure Associated MOFa | P | Confirmed Immunoparalysis Associated MOFa | P | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Not Confirmed (n = 364) | Confirmed (n = 37) | Not Confirmed (n = 394) | Confirmed (n = 7) | Not Confirmed (n = 316) | Confirmed (n = 85) | |||||
| Age | 0.042b | 0.716b | 0.002b | |||||||
| Median | 5.6 | 5.5 | 7.9 | 5.6 | 5.5 | 5.0 | 7.7 | |||
| (Q1–Q3) | (1.4–12.3) | (1.3–11.9) | (2.7–15.0) | (1.4–12.3) | (2.3–15.6) | (1.0–11.7) | (3.2–14.0) | |||
| Race, n (%) | 0.051c | 1.000c | 0.233c | |||||||
| White | 270 (67) | 245 (67) | 25 (68) | 264 (67) | 6 (86) | 210 (66) | 60 (71) | |||
| Black or African American | 83 (21) | 77 (21) | 6 (16) | 82 (21) | 1 (14) | 72 (23) | 11 (13) | |||
| Multiracial | 3 (1) | 1 (0) | 2 (5) | 3 (1) | 0 (0) | 2 (1) | 1 (1) | |||
| Other | 19 (5) | 18 (5) | 1 (3) | 19 (5) | 0 (0) | 15 (5) | 4 (5) | |||
| Unknown or not reported | 26 (6) | 23 (6) | 3 (8) | 26 (7) | 0 (0) | 17 (5) | 9 (11) | |||
| Ethnicity, n (%) | 0.363c | 0.341c | 0.869c | |||||||
| Unknown or not reported | 14 (3) | 14 (4) | 0 (0) | 14 (4) | 0 (0) | 11 (3) | 3 (4) | |||
| Hispanic or Latino | 66 (16) | 62 (17) | 4 (11) | 64 (16) | 2 (29) | 53 (17) | 13 (15) | |||
| Not Hispanic or Latino | 321 (80) | 288 (79) | 33 (89) | 316 (80) | 5 (71) | 252 (80) | 69 (81) | |||
| Total Pediatric Risk of Mortality score | 0.005b | 0.937b | 0.004b | |||||||
| Median | 8.0 | 7.0 | 12.0 | 8.0 | 10.0 | 7.0 | 10.0 | |||
| (Q1–Q3) | (3.0–14.0) | (3.0–14.0) | (8.0–17.0) | (3.0–14.0) | (0.0–19.0) | (3.0–13.0) | (6.0–16.0) | |||
| Malignancy at ICU admission, n (%) | 49 (12) | 39 (11) | 10 (27) | 0.008c | 47 (12) | 2 (29) | 0.206c | 29 (9) | 20 (24) | 0.001c |
| Solid organ transplant, n (%) | 8 (2) | 6 (2) | 2 (5) | 0.163c | 8 (2) | 0 (0) | 1.000c | 4 (1) | 4 (5) | 0.066c |
| Infections at eligibilityd, n (%) | ||||||||||
| Bacterial | 141 (35) | 126 (35) | 15 (41) | 0.244c | 138 (35) | 3 (43) | 1.000c | 103 (33) | 38 (45) | 0.061c |
| Fungal | 4 (1) | 1 (0) | 3 (8) | 0.285c | 4 (1) | 0 (0) | 1.000c | 1 (0) | 3 (4) | 0.285c |
| Protozoal | 1 (0) | 1 (0) | 0 (0) | 1 (0) | 0 (0) | 1 (0) | 0 (0) | |||
| Viral | 112 (28) | 100 (27) | 12 (32) | 0.459c | 111 (28) | 1 (14) | 1.000c | 92 (29) | 20 (24) | 0.561c |
MOF = multiple organ failure.
All subjects had at least one study blood sample collected while in the ICU.
Wilcoxon rank-sum test.
Fisher exact test.
Documented infections at the time of study eligibility are reported.
In the overall population, MOF occurred on the first day of presentation with severe sepsis (median [IQR] days from sepsis onset to development of MOF = 0 [0.0]). In contrast, the onset of IPMOF (median [IQR] days from sepsis onset to diagnosis) was 5.0 d [4.0–6.0 d]; TAMOF was 4.0 d [4.0–8.0 d]; and SMOF was 6.0 d [6.0–12.0 d] [Supplemental Fig. 2, Supplemental Digital Content 2, http://links.lww.com/PCC/B76; legend, Supplemental Digital Content 7, http://links.lww/com/PCC/B81]). The PICU length of stay of patients with IPMOF (median days = 17.9 [9.2–29.6]), TAMOF (median days = 23.0 [15.9–42.4]), or SMOF (median days = 41.5 [14.4–42.4]) was longer than the PICU length of stay for patients with MOF without any of the inflammation phenotypes (median days [IQR] = 12.0 [7.1–19.8]) (Table 3).
TABLE 3.
Additional Clinical Details During PICU Stay for Single Organ Failure, Multiple Organ Failure With and Without Inflammation Phenotypes, and Each Multiple Organ Failure Inflammation Phenotype
| Clinical Details During PICU Stay | Single Organ Failure (n = 112) | MOF No Confirmed Phenotype (n = 188) | MOF With a Confirmed Phenotype (n = 101) | Confirmed Thrombocytopenia Associated MOF (n = 37) | Confirmed Sequential Liver Failure Associated MOF (n = 7) | Confirmed immunoparalysis Associated MOF (n = 85) |
|---|---|---|---|---|---|---|
| Extracorporeal membrane oxygenation on study | p = 0.318a,b | p = 0.041a,b | p = 0.025a,b | p = 0.210a,b | ||
| Yes, n (%) | 0 (0.0) | 17 (9.0) | 13 (12.9) | 8 (21.6) | 3 (42.9 | 12 (14.1) |
| Duration (calendar days) | p = 0.065a,c | p = 0.292a,c | p = 0.709a,c | p = 0.091a,c | ||
| Median | Not applicable | 7.0 | 12.0 | 10.0 | 11.0 | 11.5 |
| (Q1–Q3) | Not applicable | (5.0–11.0) | (8.0–15.0) | (7.0–14.5) | (5.0–12.0) | (8.0–17.0) |
| Mechanically ventilated on study | p = 0.133a,b | p = 0.190a,b | p = 1.000a,b | p = 0.113a,b | ||
| Yes, n (%) | 82 (73.2) | 185 (98.4) | 96 (95.0) | 35 (94.6) | 7 (100.0) | 80 (94.1) |
| Duration (calendar days) | p ≤ 0.001a,c | p ≤ 0.001a,c | p = 0.016a,c | p = 0.001a,c | ||
| Median | 6.0 | 10.0 | 13.0 | 17.0 | 27.0 | 12.5 |
| (Q1–Q3) | (4.0–11.0) | (6.0–16.0) | (8.0–26.5) | (10.0–29.0) | (10.0–29.0) | (8.0–26.5) |
| Continuous renal replacement therapy on study | p ≤ 0.001a,b | p ≤ 0.001a,b | p ≤ 0.001a,b | p ≤ 0.001a,b | ||
| Yes, n (%) | 2 (1.8) | 11 (5.9) | 38 (37.6) | 34 (91.9) | 5 (71.4) | 23 (27.1) |
| Duration (calendar days) | p = 0.140a,c | p = 0.074a,c | p = 0.040a,c | p = 0.284a,c | ||
| Median | 6.5 | 7.0 | 8.5 | 9.5 | 11.0 | 8.0 |
| (Q1–Q3) | (1.0–12.0) | (5.0–10.0) | (5.0–16.0) | (6.0–16.0) | (8.0–15.0) | (3.0–16.0) |
| ICU length of stay (calendar days) | p ≤ 0.001a,d | p ≤ 0.001a,d | p = 0.012a,d | p = 0.002a,d | ||
| Median | 7.7 | 12.0 | 18.5 | 23.0 | 41.5 | 17.9 |
| (Q1–Q3) | (3.8–15.6) | (7.1–19.8) | (10.0–33.7) | (15.9–42.4) | (14.4–42.4) | (9.2–29.6) |
MOF = multiple organ failure.
The statistical comparisons being made in this table are subjects with the specified confirmed inflammation phenotype status noted in the columns vs subjects with MOF who do not have any confirmed inflammation phenotype.
p values are calculated using the Fisher exact test comparing therapy initiation while on study.
p values are calculated using the Wilcoxon rank-sum test and compare calendar days on therapy for subjects who received each therapy at least once on study.
p values are calculated using the Wilcoxon rank-sum test. Length of stay comparisons consider all subjects, regardless of therapies used.
Children with MOF and any of the three inflammation phenotypes were as likely as children with MOF without the phenotypes to require mechanical ventilation (95.0% vs 98.40%) or extracorporeal membrane oxygenation (ECMO) (12.9% vs 9.0%); however, these children were more likely to receive continuous renal replacement therapy (CRRT) (37.6%) compared with children with MOF without any phenotype (5.9%). The highest utilization of CRRT was in children with TAMOF (91.9%), followed by children with SMOF (71.4%), and children with IPMOF (27.1%) (Table 3).
Children with MOF and any of the three inflammation phenotypes reached higher average maximum CRP levels than children with MOF without any phenotype (29.1 mg/dL [sd 40.76 mg/dL] vs 15.3 mg/dL [sd 12.61 mg/dL]; p < 0.001) and higher average maximum ferritin levels (15,751.9 ng/mL [sd 58,079.55 ng/mL] vs 950.3 ng/mL [sd 2,625.56 ng/mL]; p < 0.001) (Table 4). CRP and ferritin levels were higher in each of the three individual inflammation phenotypes compared with MOF patients without any of the phenotypes (p < 0.05) (Table 4). The median day (IQR) of maximum CRP was 3.0 days (1.0 to –5.0 d) and of maximum ferritin was 3.0 days [2.0–6.0 d).
TABLE 4.
Peak Ferritin and C-Reactive Protein Levels for Single Organ Failure, Multiple Organ Failure With and Without Inflammation Phenotypes, and Each Multiple Organ Failure Inflammation Phenotype
| Ferritin and CRP Descriptive Statistics | Single Organ Failure (n = 112) | MOF No Confirmed Phenotype (n = 188) | MOF With a Confirmed Phenotype (n = 101) | Confirmed Thrombocytopenia Associated MOF (n = 37) | Confirmed Sequential Liver Failure Associated MOF (n = 7) | Confirmed Immunoparalysis Associated MOF (n = 85) |
|---|---|---|---|---|---|---|
| Maximum ferritin | p ≤ 0.0011 | p ≤ 0.0011 | p = 0.002a | p ≤ 0.0011 | ||
| Mean (sd) | 2,374.4 (13085.06) | 950.3 (2,625.56) | 15,751.9 (58079.55) | 30,798.9 (86,659.62) | 11,043.3 (22,277.67) | 18,486.2 (62,989.84) |
| Minimum–maximum | 24.3–130,000.0 | 15.0–21,140.0 | 34.0–392,816.0 | 207.0–392,816.0 | 260.0–60,490.0 | 34.0–392,816.0 |
| Median | 184.0 | 215.9 | 694.0 | 1705.0 | 1097.1 | 663.6 |
| (Q1–Q3) | (80.1–581.0) | (113.7–509.0) | (272.5–3,106.0) | (694.0–13,000.0) | (634.0–13,000.0) | (260.7–4,049.0) |
| Maximum CRP | p ≤ 0.0011 | p ≤ 0.0011 | p = 0.006a | p ≤ 0.0011 | ||
| Mean (sd) | 14.3 (11.90) | 15.3 (12.61) | 29.1 (40.76) | 41.6 (57.85) | 35.7 (19.74) | 26.7 (28.54) |
| Minimum–maximum | 0.1–52.7 | 0.0–53.0 | 0.4–327.3 | 1.6–327.3 | 3.4–60.8 | 0.4–184.3 |
| Median | 11.8 | 12.6 | 21.6 | 28.8 | 40.7 | 21.9 |
| (Q1–Q3) | (4.8–21.6) | (4.8–23.5) | (10.6–32.9) | (17.0–40.7) | (19.9–51.3) | (10.6–33.4) |
CRP = C-reactive protein, MOF = multiple organ failure.
The statistical comparisons being made in this table are subjects with the specified confirmed inflammation phenotype status noted in the columns vs subjects with MOF who do not have any confirmed inflammation phenotype. p values are based on the Wilcoxon rank-sum test.
MAS occurred in 24 patients (6.0%) and was more commonly found in the MOF population with one or more of the phenotypes compared with the MOF population without any of the phenotypes (19/101 [18.8%] vs 5/188 [2.7%]; RR, 7.07; 95% CI, 2.72–18.38) (Table 5 and Supplemental Fig. 3 [Supplemental Digital Content 3, http://links.lww.com/PCC/B77; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81]). The time from onset of sepsis to development of MAS was 3.0 days (3.0–9.0 d) (median [IQR]) (Supplemental Fig. 2, Supplemental Digital Content 2, http://links.lww.com/PCC/B76; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81). MAS was most common in patients with SMOF (7/7; 100%), followed by patients with TAMOF (14/37; 37.8%) and patients with IPMOF (15/85; 17.6%) (p < 0.05 for each MOF pheno-type compared with MOF without any phenotype; Table 5 and Supplemental Fig. 4 [Supplemental Digital Content 4, http://links.lww.com/PCC/B78; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81]).
TABLE 5.
Relative Risks of Developing Macrophage Activation Syndrome by Inflammation Phenotype
| Groups | Macrophage Activation Syndrome, n = 24, n (%) | Relative Risk (95% CI) |
|---|---|---|
| Single organ failure | 0/112 (0) | NA |
| MOF | 24/289 (8.3) | NA |
| MOF no phenotype | 5/188 (2.7) | Reference |
| MOF with phenotype | 19/101 (18.8) | 7.07 (2.72–18.38) |
| Immunoparalysis associated MOF | 15/85 (17.6) | 6.74 (2.53–17.95) |
| Thrombocytopenia associated MOF | 14/37 (37.8) | 14.23 (5.46–37.10) |
| Sequential liver failure associated MOF | 7/7 (100) | 38.20 (16.08–90.72) |
MOF = multiple organ failure, NA = not applicable.
Results are based on individual univariable modified Poisson regression models.
There were 44 of 401 (10.9%) PICU deaths in the overall severe sepsis cohort; one of 112 (0.9%) in children with single organ failure and 43 of 289 (14.9%) in children with MOF (Table 6 and Supplemental Fig. 3 [Supplemental Digital Content 3, http://links.lww.com/PCC/B77; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81]) with 39% of these PICU deaths (17/44) occurring after 28 days. There were only two additional deaths in the hospital after discharge from the PICU. Mortality rates in the PICU were three-fold higher in children with MOF and one or more of these phenotypes compared with the remaining severe sepsis cohort (24/101 = 23.8% vs 20/300 = 6.7%; RR, 3.56; 95% CI, 2.06–6.17). Compared to the 188 MOF patients who did not develop any inflammation phenotypes, the 101 with the phenotypes had increased mortality (23.8% vs 10.1%; RR, 2.35; 95% CI, 1.36–4.08). Mortality was 17 of 85 (20.0%) in IPMOF, 16 of 37 (43.2%) in TAMOF, and three of seven (42.9%) in SMOF (p < 0.05 for each MOF phenotype; Table 6 and Supplemental Fig. 4 [Supplemental Digital Content 4, http://links.lww.com/PCC/B78; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81]). The median (IQR) time to PICU death in IPMOF was 16.0 days (8.0–35.0 d), in TAMOF was 22.0 days (13.5–44.0 d), and in SMOF was 34.0 days (14–42 d).
TABLE 6.
Relative Risks of PICU Mortality Among Groups of Interest
| Groups | Total Prevalence, n = 401, % | PICU Mortality, n = 44, n (%) | Relative Risk (95% CI) |
|---|---|---|---|
| Single organ failure | 27.9 | 1/112 (0.9) | 0.09 (0.01–0.65) |
| MOF | 72.1 | 43/289 (14.9) | Not applicable |
| MOF no phenotype | 46.9 | 19/188 (10.1) | Reference |
| MOF with phenotype | 25.2 | 24/101 (23.8) | 2.35 (1.36–4.08) |
| Immunoparalysis associated MOF | 21.2 | 17/85 (20.0) | 1.98 (1.08–3.61) |
| Thrombocytopenia associated MOF | 9.2 | 16/37 (43.2) | 4.28 (2.43–7.52) |
| Sequential liver failure associated MOF | 1.7 | 3/7 (42.9) | 4.24 (1.63–11.03) |
MOF = multiple organ failure.
Results are based on individual univariable modified Poisson regression models.
In multivariate analysis, the PICU mortality risk was over two-fold higher in children who developed MOF with any of the phenotypes compared with children who developed MOF without the phenotypes when controlling for admission characteristics including age, race, ethnicity, gender, illness severity, malignancy, organ transplantation, infection type, and previously healthy status (RR [95% CI] MOF with phenotype = 2.79 [1.62–4,83], p < 0.001; PRISM score = 1.04 [1.10–1.08], p = 0.004; and previously healthy status = 0.40 [0.20–0.81], p = 0.011). Organ failures and associated mortality in patients with MOF without any of the phenotypes are presented in Supplement Figure 5 (Supplemental Digital Content 5, http://links.lww.com/PCC/B79; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81).
Mortality risk for MOF patients with the phenotypes who did not develop MAS was increased compared with MOF without any phenotypes (16/82 [19.5%] vs 19/188 [10.1%]; RR, 1.93; 95% CI, 1.05–3.56); however, the development of MAS in patients with MOF and the phenotypes increased mortality risk even further (MOF with phenotypes plus MAS 8/19 [42.1%] vs MOF with phenotypes without MAS 16/82 [19.5%]; RR, 2.16; 95% CI, 1.09–4.29) (Supplemental Fig. 3, Supplemental Digital Content 3, http://links.lww.com/PCC/B77; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81). Development of MAS increased mortality risk in IPMOF patients (IPMOF with MAS 6/15 [40%] vs IPMOF without MAS 11/70 [15.7%]; RR, 2.55; 95% CI, 1.12–5.80); but not in TAMOF patients (TAMOF with MAS 7/14 [50%] vs TAMOF without MAS 9/23 [39.1%]; RR, 1.28; 95% CI, 0.61–2.65; p = 0.51) (Supplemental Fig. 4, Supplemental Digital Content 4, http://links.lww.com/PCC/B78; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81)
Specific inflammation phenotype biomarkers remained abnormal over time in the patients with MOF and the inflammation phenotypes (Supplemental Fig. 6, Supplemental Digital Content 6, http://links.lww.com/PCC/B80; legend, Supplemental Digital Content 7, http://links.lww.com/PCC/B81). Earlier recovery of the ex vivo TNF-α response to endotoxin greater than 200 pg/mL in IPMOF was observed in survivors. No significant differences were observed in ADAMTS13 threshold activity less than 57% over time for TAMOF survivors versus nonsurvivors, nor in ferritin threshold levels greater than 500 ng/mL in patients with MAS between survivors and nonsurvivors. There were too few SMOF patients to assess the relationship of sFasL thresholds and PICU survival over time.
DISCUSSION
Emergency department initiatives emphasizing early septic shock recognition and treatment can reduce pediatric mortality to less than 10% when administered in the first hour (5); however, most children with severe sepsis now die with new and progressive multiple organ dysfunction syndrome (MODS) and MOF after 3 days in the PICU, a condition seemingly unrelated to early resuscitation efforts (1–4). In lieu of failed “one silver bullet for all” sepsis trials, there is interest in designing “smarter” clinical trials that identify smaller groups of patients with recognizable phenotypes whom physicians think have biologic plausibility for benefitting from a therapy (25, 26); randomizing these patients to this phenotype targeting adjunctive therapy (25, 26); and then using post hoc whole genome/exome pathogenic variant (36), epigenetic modification (7), transcriptomic (37), and proteomic (38) subtype analyses to identify which individual patients within the phenotype responded best in order to inform potential precision medicine approaches in the PICU.
In our present multicenter study, we have sought and attained essential clinical data needed to justify and inform planning of “smarter” trials in children with sepsis-induced new and progressive MODS, by assessing the IPMOF, TAMOF, and SMOF phenotypes. We found that the pediatric burden of these three inflammation MOF phenotypes is substantial in our network severe sepsis cohort, developing in one of four children after a median of 3 days in the PICU, in association with greater use of extracorporeal support therapies, prolonged PICU length of stays, and increased risk of developing uncontrolled inflammation, MAS, and death. These data have enabled us to design trials targeting these phenotypes.
Because the immune paralysis phenotype was most prevalent, we are beginning our first multicenter trial targeting this pheno-type (6–10). We previously demonstrated that GM-CSF reverses immune paralysis, facilitates resolution of IPMOF, and prevents secondary infection (6). We have started the GM-CSF for Reversal of ImmunopAralysis in pediatriC sEpsis-induced MODS study (ClinicalTrials.gov Identifier: NCT03769844 principal investigator [PI] M.H.). Children with sepsis and MODS are enrolled at 48 hours, and if immune paralysis develops and their ferritin level is less than 500 ng/mL they are given GM-CSF. Post hoc assessment of pathogenic variant (36), epigenetic (7), and transcriptomic (37) analyses may inform future precision medicine approaches within this phenotype. Targeting the immune paralysis phenotype is important because it was the major contributor to mortality in children during the H1N1 influenza epidemic (8).
Dr. Hall chose not to give GM-CSF to patients with ferritin levels greater than 500 ng/mL because this MAS biomarker level has been associated with 60% mortality in pediatric sepsis (33) and there is concern that GM-CSF could worsen MAS in these children (10). We previously showed 100% survival in pediatric MAS with use of an anti-inflammatory regimen that includes the combination of methylprednisolone or IV immunoglobulin, plus plasma exchange (34). We also showed that the anti-inflammatory interleukin-1 receptor antagonist protein reduced mortality two-fold in adult patients with septic shock and MAS (39). Rajasekaran et al (35) showed that interleukin-1 receptor antagonist protein safely reverses MAS in critically ill children. In collaboration with our rheumatology colleagues (39), we are designing the Critical MAS in Sepsis (Critical MASS) trial (PI Rajasekaran et al [35]) to identify whether IV interleukin-1 receptor antagonist achieves reversal of MAS by 3 days in pediatric sepsis. Pathogenic variant (36), epigenetic (7), transcriptomic (37), and proteomic (38) analyses may inform future precision medicine approaches to uncontrolled inflammation and MAS. This is important because mortality among the three inflammation phenotypes was highest when hyperferritinemic MAS developed.
Because thrombotic microangiopathy driven TAMOF was the second most prevalent phenotype, we are also planning a clinical trial for these children (11–20). We previously demonstrated that plasma exchange removes thrombogenic ultra-large vWF multimers, restores ADAMTS13 activity that cleaves these multimers, and reverses renal failure and MOF in children with sepsis-induced TAMOF (11). Two pediatric TAMOF plasma exchange registries in Turkey (14) and in the United States (13) separately reported that plasma exchange therapy is associated with resolution of TAMOF and improved survival. Because we observed that more than nine of 10 children with TAMOF required CRRT in our study, we are in discussion with the pediatric CRRT research consortium to explore collaboration following Wong et al (12) recommendation to use the PERSEVERE II risk stratification panel and transcriptomics to identify which patients respond best to plasma exchange within this phenotype (37). It is important to target the TAMOF phenotype because it was associated with high use of ECMO and CRRT, longer PICU stays, and higher mortality.
The viral infection lymphoproliferative disease induced liver failure associated SMOF phenotype had the highest use of ECMO, the most prolonged length of stay, and the highest mortality, but the lowest prevalence among the three phenotypes (21–24). Because all seven patients with SMOF in our study developed MAS, they would be eligible for enrollment in the Critical MASS trial. We have decided to defer designing a specific trial for this phenotype until the above trial is completed.
There are limitations to consider. First, our study was designed to assess septic patients who die with late MOF after 3 days in the PICU seemingly independent of early resuscitative efforts, not children who die of refractory shock before 3 days in the PICU (1–5). Second, although the three empiric inflammation phenotypes were associated with increased need for extracorporeal therapies, prolonged ICU stay, and increased risk for developing uncontrolled inflammation, MAS, and death, there were other patients who died without these phenotypes. We are performing unbiased analyses of our clinical data using machine learning to explore any other phenotypes our empiric approach might have missed (40). Third, our article is limited to phenotype evaluation. Sepsis-induced MOF phenotypes may result from interactions between environmental triggers related to infection and microbiome (25, 26), and host genetic machinery (7, 36–38) related to immunity, inflammation, and coagulation (25, 26). Our laboratories are analyzing potential relationships between the three phenotypes and host pathogenic variants (36), epigenetic modifications (7), and proteomics (38). We did not collect RNA for transcriptomics but are doing so in future studies.
In conclusion, we have attained essential data needed to provide rationale for designing “smarter” clinical trials with the goal of reducing progression of these three inflammation phenotypes and ensuing uncontrolled inflammation and MAS in children with sepsis-induced MODS and MOF unresponsive to earlier resuscitation efforts. A synergistic role for the rapidly evolving field of gene expression, transcriptomics, and precision medicine within these phenotype directed studies is anticipated and carefully considered.
Supplementary Material
ACKNOWLEDGMENTS
Clinical Research Investigation and Systems Modeling of Acute illness center: Ali Smith, BS; Octavia Palmer, MD; Vanessa Jackson, AA; Renee Anderko, BS, MS. Children’s Hospital of Pittsburgh: Jennifer Jones, RN; Luther Springs. Children’s Hospital of Philadelphia: Carolanne Twelves, RN, BSN, CCRC; Mary Ann Diliberto, BS, RN, CCRC; Martha Sisko, BSN, RN, CCRC, MS; Pamela Diehl, BSN, RN; Janice Prodell, RN, BSN, CCRC; Jenny Bush, RNC, BSN; Kathryn Graham, BA; Kerry Costlow, BS; Sara Sanchez. Children’s National Medical Center: Elyse Tomanio, BSN, RN; Diane Hession, MSN, RN; Katherine Burke, BS. Children’s Hospital of Michigan: Ann Pawluszka, RN, BSN; Melanie Lulic, BS. Nationwide Children’s Hospital: Lisa Steele, RN, CCRC; Andrew R. Yates, MD; Josey Hensley, RN; Janet Cihla, RN; Jill Popelka, RN; Lisa Hanson-Huber, BS. Children’s Hospital of Los Angeles and Mattel Children’s Hospital: Jeni Kwok, JD; Amy Yamakawa, BS. Children’s Hospital of Washington University of Saint Louis: Michelle Eaton, RN. Mott Children’s Hospital: Frank Moler, MD; Chaandini Jayachandran, MS, CCRP. University of Utah Data Coordinating Center: Teresa Liu, MPH, CCRP; Jeri Burr, MS, RN-BC, CCRC, FACRP; Missy Ringwood, BS, CMC; Nael Abdelsamad, MD, CCRC; Whit Coleman, MSRA, BSN, RN, CCRC.
Supported, in part, by grant R01GM108618 (to Dr. Carcillo) from the National Institutes of General Medical Sciences, by 5U01HD049934-10S1 from the Eunice Kennedy Shriver National Institutes of Child Health and Human Development, National Institutes of Health, Department of Health and Human Services and the following cooperative agreements: U10HD049983, U10HD050096, U10HD049981, U10HD063108, U10HD63106, U10HD063114, U10HD050012, and U01HD049934.
Drs. Carcillo’s, Berg’s, Wessel’s, Pollack’s, Meert’s, Hall’s, Doctor’s, Cornell’s, Harrison’s, Zuppa’s, Reeder’s, Banks’s, and Holubkov’s institutions received funding from the National Institutes of Health (NIH). Drs. Carcillo’s, Newth’s, Shanley’s, and Dean’s institutions received funding from the National Institutes of Child Health and Human Development. Drs. Carcillo, Berg, Wessel, Polack, Meert, Hall, Newth, Doctor, Shanley, Cornell, Harrison, Zuppa, Reeder, Banks, Holubkov, Notterman, and Dean received support for article research from the NIH. Dr. Carcillo’s institution also received funding from the National Institutes of General Medical Sciences. Dr. Pollack disclosed that his research is supported by philanthropy from Mallinckrodt Pharmaceuticals. Dr. Hall received funding from Bristol Myers-Squibb (for service on an advisory board) and LaJolla Pharmaceuticals (service as a consultant), both unrelated to the current submission. Dr. Newth received funding from Philips Research North America. Dr. Doctor’s institution received funding from the Department of Defense and Kalocyte. Dr. Shanley received funding from Springer publishing, International Pediatric Research Foundation, and Pediatric Academic Societies. Dr. Cornell disclosed he is co-founder of Pre-Dixon Bio. Dr. Holubkov received funding from Pfizer (Data Safety Monitoring Board [DSMB] member), Medimmune (DSMB member), Physicians Committee for Responsible Medicine (biostatistical consulting), DURECT Corporation (biostatistical consulting), Armaron Bio (DSMB past member), and St Jude Medical (DSMB past member). The remaining authors have disclosed that they do not have any potential conflicts of interest.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (http://journals.lww.com/pccmjournal).
REFERENCES
- 1.Weiss SL, Fitzgerald JC, Pappachan J, et al. ; Sepsis Prevalence, Outcomes, and Therapies (SPROUT) Study Investigators and Pediatric Acute Lung Injury and Sepsis Investigators (PALISI) Network: Global epidemiology of pediatric severe sepsis: The sepsis prevalence, outcomes, and therapies study. Am J Respir Crit Care Med 2015; 191:1147–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin JC, Spinella PC, Fitzgerald JC, et al. ; Sepsis Prevalence, Outcomes, and Therapy Study Investigators: New or progressive multiple organ dysfunction syndrome in pediatric severe sepsis: A sepsis phenotype with higher morbidity and mortality. Pediatr Crit Care Med 2017; 18:8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss SL, Balamuth F, Hensley J, et al. : The epidemiology of hospital death following pediatric severe sepsis: When, why, and how children with sepsis die. Pediatr Crit Care Med 2017; 18:823–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Workman JK, Ames SG, Reeder RW, et al. : Treatment of pediatric septic shock with the surviving sepsis campaign guidelines and PICU patient outcomes. Pediatr Crit Care Med 2016; 17:e451–e458 [DOI] [PubMed] [Google Scholar]
- 5.Evans IVR, Phillips GS, Alpern ER, et al. : Association between the New York sepsis care mandate and in-hospital mortality for pediatric sepsis. JAMA 2018; 320:358–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hall MW, Knatz NL, Vetterly C, et al. : Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med 2011; 37:525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cornell TT, Sun L, Hall MW, et al. : Clinical implications and molecular mechanisms of immunoparalysis following cardiopulmonary bypass. J Thorac Cardiovasc Surg 2012; 143:1160–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall MW, Geyer SM, Guo CY, et al. ; Pediatric Acute Lung Injury and Sepsis Investigators (PALISI) Network PICFlu Study Investigators: Innate immune function and mortality in critically ill children with influenza: A multicenter study. Crit Care Med 2013; 41:224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall MW, Greathouse KC, Thakkar RK, et al. : Immunoparalysis in pediatric critical care. Pediatr Clin North Am 2017; 64:1089–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Höflich C, Volk HD: [Immunomodulation in sepsis]. Chirurg 2002; 73:1100–1104 [DOI] [PubMed] [Google Scholar]
- 11.Nguyen TC, Han YY, Kiss JE, et al. : Intensive plasma exchange increases a disintegrin and metalloprotease with thrombospondin motifs-13 activity and reverses organ dysfunction in children with thrombocytopenia-associated multiple organ failure. Crit Care Med 2008; 36:2878–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong HR, Cvijanovich NZ, Anas N, et al. : Pediatric sepsis biomarker risk model-II: Redefining the pediatric sepsis biomarker risk model with septic shock phenotype. Crit Care Med 2016; 44:2010–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fortenberry JD, Nguyen T, Grunwell JR, et al. ; Thrombocytopenia-Associated Multiple Organ Failure (TAMOF) Network Study Group: Therapeutic plasma exchange in children with thrombocytopenia-associated multiple organ failure: The thrombocytopenia-associated multiple organ failure network prospective experience. Crit Care Med 2019; 47:e173–e181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sevketoglu E, Yildizdas D, Horoz OO, et al. : Use of therapeutic plasma exchange in children with thrombocytopenia-associated multiple organ failure in the Turkish thrombocytopenia-associated multiple organ failure network. Pediatr Crit Care Med 2014; 15:e354–e359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen TC, Cruz MA, Carcillo JA: Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges BC, Hardison D, Pietsch J: A case series of the successful use of ECMO, continuous renal replacement therapy, and plasma exchange for thrombocytopenia-associated multiple organ failure. J Pediatr Surg 2013; 48:1114–1117 [DOI] [PubMed] [Google Scholar]
- 17.Nguyen TC, Liu A, Liu L, et al. : Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica 2007; 92:121–124 [DOI] [PubMed] [Google Scholar]
- 18.Bongers TN, Emonts M, de Maat MP, et al. : Reduced ADAMTS13 in children with severe meningococcal sepsis is associated with severity and outcome. Thromb Haemost 2010; 103:1181–1187 [DOI] [PubMed] [Google Scholar]
- 19.Tsai HM, Lian EC: Antibodies to von Willebrand factor-cleaving pro-tease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998; 339:1585–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy GG, Nichols WC, Lian EC, et al. : Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001; 413:488–494 [DOI] [PubMed] [Google Scholar]
- 21.Dharnidharka VR, Webster AC, Martinez OM, et al. : Post-transplant lymphoproliferative disorders. Nat Rev Dis Primers 2016; 2:15088. [DOI] [PubMed] [Google Scholar]
- 22.Doughty L, Clark RS, Kaplan SS, et al. : sFas and sFas ligand and pediatric sepsis-induced multiple organ failure syndrome. Pediatr Res 2002; 52:922–927 [DOI] [PubMed] [Google Scholar]
- 23.Nakae H, Narita K, Endo S: Soluble Fas and soluble Fas ligand levels in patients with acute hepatic failure. J Crit Care 2001; 16:59–63 [DOI] [PubMed] [Google Scholar]
- 24.Ryo K, Kamogawa Y, Ikeda I, et al. : Significance of Fas antigen-mediated apoptosis in human fulminant hepatic failure. Am J Gastroenterol 2000; 95:2047–2055 [DOI] [PubMed] [Google Scholar]
- 25.Podd BS, Simon DW, Lopez S, et al. : Rationale for adjunctive therapies for pediatric sepsis induced multiple organ failure. Pediatr Clin North Am 2017; 64:1071–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carcillo JA, Podd B, Aneja R, et al. : Pathophysiology of pediatric multiple organ dysfunction syndrome. Pediatr Crit Care Med 2017; 18:S32–S45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carcillo JA, Halstead ES, Hall MW, et al. ; Eunice Kennedy Shriver National Institute of Child Health and Human Development Collaborative Pediatric Critical Care Research Network Investigators: Three hypothetical inflammation pathobiology phenotypes and pediatric sepsis-induced multiple organ failure outcome. Pediatr Crit Care Med 2017; 18:513–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kyriazopoulou E, Leventogiannis K, Norrby-Teglund A, et al. ; Hellenic Sepsis Study Group: Macrophage activation-like syndrome: An immunological entity associated with rapid progression to death in sepsis. BMC Med 2017; 15:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldstein B, Giroir B, Randolph A; International Consensus Conference on Pediatric Sepsis: International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med 2005; 6:2–8 [DOI] [PubMed] [Google Scholar]
- 30.Villeneuve A, Joyal JS, Proulx F, et al. : Multiple organ dysfunction syndrome in critically ill children: Clinical value of two lists of diagnostic criteria. Ann Intensive Care 2016; 40:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cron RQ, Davi S, Minoia F, et al. : Clinical features and correct diagnosis of macrophage activation syndrome. Expert Rev Clin Immunol 2015; 11:1043–1053 [DOI] [PubMed] [Google Scholar]
- 32.Castillo L, Carcillo J: Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multi-organ dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med 2009; 10:387–392 [DOI] [PubMed] [Google Scholar]
- 33.Garcia PC, Longhi F, Branco RG, et al. : Ferritin levels in children with severe sepsis and septic shock. Acta Paediatr 2007; 96:1829–1831 [DOI] [PubMed] [Google Scholar]
- 34.Demirkol D, Yildizdas D, Bayrakci B, et al. ; Turkish Secondary HLH/MAS Critical Care Study Group: Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: What is the treatment? Crit Care 2012; 16:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajasekaran S, Kruse K, Kovey K, et al. : Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med 2014; 15:401–408 [DOI] [PubMed] [Google Scholar]
- 36.Kernan KF, Ghaloul-Gonzalez L, Shakoory B, et al. : Adults with septic shock and extreme hyperferritinemia exhibit pathogenic immune variation. Genes Immun 2019; 20:520–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sweeney TE, Azad TD, Donato M, et al. : Unsupervised analysis of transcriptomics in bacterial sepsis across multiple datasets reveals three robust clusters. Crit Care Med 2018; 46:915–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen P, Huang NT, Chung MT, et al. : Label-free cytokine micro- and nano-biosensing towards personalized medicine of systemic inflammatory disorders. Adv Drug Deliv Rev 2015; 95:90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shakoory B, Carcillo JA, Chatham WW, et al. : Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome: Reanalysis of a prior phase III trial. Crit Care Med 2016; 44:275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knox DB, Lanspa MJ, Kuttler KG, et al. : Phenotypic clusters within sepsis-associated multiple organ dysfunction syndrome. Intensive Care Med 2015; 41:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.