

Abstract
The SAMPL challenges focus on testing and driving progress of computational methods to help guide pharmaceutical drug discovery. However, assessment of methods for predicting binding affinities is often hampered by computational challenges such as conformational sampling, protonation state uncertainties, variation in test sets selected, and even lack of high quality experimental data. SAMPL blind challenges have thus frequently included a component focusing on host-guest binding, which removes some of these challenges while still focusing on molecular recognition. Here, we report on the results of the SAMPL7 blind prediction challenge for host-guest affinity prediction. In this study, we focused on three different host-guest categories – a familiar deep cavity cavitand series which has been featured in several prior challenges (where we examine binding of a series of guests to two hosts), a new series of cyclodextrin derivatives which are monofunctionalized around the rim to add amino acid-like functionality (where we examine binding of two guests to a series of hosts), and binding of a series of guests to a new acyclic TrimerTrip host which is related to previous cucurbituril hosts. Many predictions used methods based on molecular simulations, and overall success was mixed, though several methods stood out. As in SAMPL6, we find that one strategy for achieving reasonable accuracy here was to make empirical corrections to binding predictions based on previous data for host categories which have been studied well before, though this can be of limited value when new systems are included. Additionally, we found that alchemical free energy methods using the AMOEBA polarizable force field had considerable success for the two host categories in which they participated. The new TrimerTrip system was also found to introduce some sampling problems, because multiple conformations may be relevant to binding and interconvert only slowly. Overall, results in this challenge tentatively suggest that further investigation of polarizable force fields for these challenges may be warranted.
Keywords: host-guest binding, free energy, binding affinity, SAMPL, blind challenge, OctaAcid, cyclodextrin, cucurbituril
1. Introduction
Docking and scoring methods have long been used to assist with hit identification and optimization in computer-aided drug design (CADD) [3]. More recently, efforts to improve the reliability of CADD methodologies have gone beyond qualitative docking and scoring towards quantitative modeling [4] via molecular simulations, which can be used to estimate a variety of physical properties of interest [3, 4]. In this area, predictions of protein-ligand binding free energies have gained much attention for a few decades for their potential to help accelerate small-molecule drug discovery [5], but have received increasing attention recently as this potential begins to be realized [6, 7]. The long-term goal is to use computational techniques to aid and direct small molecule design to more rapidly and efficiently produce new therapeutics [4]. Right now, much discovery works via a slow cycle of experimental trial and error, but accurate enough free energy methods could dramatically accelerate early stage discovery [3, 5].
The accuracy of free energy calculations is dependent on and limited by inaccuracy in the energy model used (i.e., force field used, finite-size effects, and water model) [8], sampling, and the protein-ligand system set up, which can include aspects such as protonation state, chosen tautomer state, and buffer, to name a few [3, 5]. Although sources of systematic error in free energy calculations are known, it is difficult to analyze errors when modeling protein-ligand systems due to their flexibility and complexity; such challenges mean that simulations of a few nanoseconds to microseconds may not always adequately sample the relevant conformations of the protein, ligand and environment [3, 5]. For this reason, host-guest systems are a great substitute for protein-ligand systems in evaluations of computational methods for predicting free energies of binding [3], as conformational sampling can be less of a challenge.
Host-guest systems are similar to protein-ligand systems in that they also involve binding of a small molecule to a pocket in a receptor, though they have certain differences. We can think of a host as resembling a very small protein molecule (of different chemistry) which has a binding cavity or pocket. A guest is a small molecule which can bind non-covalently to the host. Supramolecular host families such as the cucurbiturils, cavitands, and cyclodextrins have diverse binding affinities and the ability to bind small drug-like compounds with protein-ligand like affinities [4]. Unlike proteins, the hosts are smaller, simpler, and often more rigid [9], removing some of the challenges facing computational modeling of proteins. These characteristics make host-guest systems an ideal substitute to test current computational methodologies used to predict physical properties of interest and investigate issues including binding, receptor flexibility, solvation, hydrogen bonding, the hydrophobic effect, protonation, and tautomers [3]. That is, while prediction of protein-ligand binding is still of interest, host-guest systems can serve to help focus on the accuracy of computational methods themselves, without conflating as many other challenges.
In this work, we describe the recent SAMPL7 host-guest challenge, which allowed participants using diverse methods to predict host-guest binding free energies for a variety of guests to three different host families. Here, we give the challenge background, describe the hosts, survey participants’ results, and highlight key lessons learned.
2. SAMPL Challenge Background, History, and Expectations
2.1. SAMPL fields blind challenges to provide fair tests
The SAMPL (Statistical Assessment of the Modeling of Proteins and Ligands) challenges focus efforts on improving and advancing computational methods through crowdsourcing. Blind challenges, like SAMPL and companion challenges such the the Drug Design Data Resource (D3R) Grand Challenges, ensure participants do not know experimental values when running calculations [10], ensuring that method comparisons are fair and performance is hopefully indicative of what could be expected in real-world applications to related problems. Host-guest systems form the basis of one category of the SAMPL challenges (with others focusing on predicting physical properties, and on protein-ligand binding) and typical challenge performance indicates such host-guest systems still pose challenges to contemporary methods [10]. Occasionally, host(s) or guest(s) are revisited, so related experimental results are available, but we avoid cases where the experimental value being predicted is already available in the literature. A wealth of experimental data is already available, so SAMPL focuses on predictive tests rather than retrospective analysis. The SAMPL challenges are organized in this manner to ensure no participant, even accidentally, adjusts their method to agree with “correct” values thereby introducing bias. For example, when experimental values are known, a naive participant could stop calculations when they agree with the experimental value because they have “converged”. Or more subtly, a participant could run calculations with several different sets of settings in the simulation package used and conclude that the settings which gave the best results were optimal, whereas in fact they might be just observing random fluctuations. Blind challenges avoid such opportunities for bias.
In general, SAMPL blind challenges typically involve a host-guest component that provides the community an opportunity to test and compare performance of a variety of computational methods on the same diverse data. The subsequent release of experimental data allows accuracy to be compared relative to experimental results which were not known when predictions were made, and the subsequent statistical assessment compares methods on equal footing. Upon evaluation, participants and organizers can assess the lessons learned and the potential value of different methods. Subsequently, computational methods and their algorithms can be calibrated and optimized for application in future blind challenges and in the real world [11].
2.2. Host-guest systems
2.2.1. What are host guest systems?
As described briefly in Section 1, host-guest systems are similar to protein-ligand systems in that they both involve the binding of a small molecule to a pocket in a receptor. Hosts often contain less than 100 non-hydrogen atoms, but are slightly larger than small molecules [9], so the broader field of such chemistry is often called supramolecular chemistry. Usually, hosts don’t have large number of possible folds or conformational structures like a protein [9]. Eventually some host-guest systems are well characterized and become part of the driving force behind methodology improvement, with the ultimate goal of transferability to protein-ligand systems [11].
2.2.2. Why use host guest systems?
Despite their apparent relative simplicity, host-guest binding has proved a difficult challenge for computation. Large-scale protein-ligand binding free energy studies often report RMS errors in the 1–2 kcal/mol range [6, 7, 12–14], which is considerably better than typical performance in SAMPL host-guest challenges [3, 4, 15–17]. It may be that host-guest systems are “simple” enough that there is essentially nowhere for problems to hide, or confounding factors like polarizability and force field limitations may be more profound in these simple mini-receptors. Alternatively, performance of protein-ligand binding free energy calculations has often been worse in blind challenges like the SAMPL [18] and D3R [19–22] blind challenges than in the large-scale tests cited above, so it may be that typical retrospective tests simply benefit from participants utilizing additional knowledge which is not available prospectively or in blind challenges. This is supported to some extent by recent benchmarking work from Merck KGaA [13], and by an earlier industry perspective [23]. Moreover, binding affinities for protein-ligand systems are usually predicted via relative binding free energy calculations for similar ligands. On the other hand, host-guest systems are typically absolute binding free energy calculations and perhaps a reason for the increased difficulty.
3. Some aspects can pose particular challenges for free energy calculations
Several different issues arise in the context of binding free energy calculations that can cause particular difficulties or challenges. Here, we survey several major categories of issues which affect some methods participating in SAMPL7.
3.1. Guests bearing a formal charge can pose methodological challenges
Molecules with formal charges can pose challenges for molecular simulations, and especially for binding free energy calculations. These challenges, and differences in how they are handled, can be particularly important when studying binding in systems like those considered here.
In general, conducting efficient molecular simulations requires making approximations and simplifications of electrostatic interactions. For example, typically we are interested in bulk or bulk-like behavior, but simulating macroscale systems is cost prohibitive, so we may instead choose to simulate a microscopic box under periodic boundary conditions (PBCs) to minimize edge effects. Alternatively, a modeler might choose to apply effective electrostatic interaction functions.
To effectively treat electrostatic interactions, functions involving cutoff truncation schemes combined with reaction-field (RF) contribution or lattice-summation (LS) methods may be employed [8, 24, 25]. These methods cause the charging component of the calculated free energies to be sensitive to important system parameters like the cutoff radius or the box size [25]. In addition, the raw single-ion solvation free energies from explicit-solvent simulations are extremely sensitive to the boundary conditions and electrostatic interaction treatment [24].
The approximations described above may also introduce bias or offset in the electrostatic potential during the simulation. System-dependent artifacts can also arise from system parameters (such as cutoff radius, box shape and/or size). The artifacts are due to finite-size effects which impact computed charging free energies/binding free energies. While such errors do not have a major effect on computed free energies as long as systems remain net neutral or have a consistent formal charge, they become particularly pronounced when the formal charge of a system changes, such as during an alchemical binding free energy calculation [8] as employed by many SAMPL participants. For this reason methods may need to account and correct for artifacts that may not cancel when a formal charge is alchemically inserted in the system. The sign and magnitude of artifacts depend on the methods used to calculate electrostatic interactions.
The exact sources of such finite-size errors have been described previously. Briefly, the finite-size error in ligand/guest charging (and by extension, binding) free energies originates from at least four different physical effects in periodic systems: (a) Periodicity-induced net-charge interactions; (b) Periodicity-induced net charge undersolvation; (c) Discrete solvent effects; and (d) Residual integrated potential effects [8].
There have been some attempts to address these issues; particularly, both instantaneous and post-simulation correction strategies have been proposed [8, 24, 26]. One approach is to apply various after-the-fact corrections to computed free energies [8, 24, 26]. Alternatively, others have proposed applying a correction strategy during simulations, which has been called a co-alchemical ion approach, wherein an alchemical perturbation of a charged moiety is simultaneously performed with a counter-alchemical charge perturbation of a remote molecule (i.e. a counter-ion) [8, 25]. In other words, in this approach, the system is maintained net neutral by offsetting a charge change in one portion of a system with a compensating change in another portion of the system. The goal in this approach is to ensure that errors from finite-size effects are negligible. Post simulation strategies include charge-correction terms which have been shown to work for LS and RF, and can be evaluated via numerical and analytical methods [8, 24, 27, 28].
3.2. Polarization can potentially pose particular challenges
Charged molecules — like those frequent in SAMPL7 – can also pose particular challenges because of strong electrostatic interactions with their immediate surroundings. This poses two challenges which are particularly relevant here – first, any polarization of the surroundings may be particularly important. Second, other electrostatic interactions are quite strong, including interactions with surrounding ions. These can include screening effects, but also relatively more specific interactions.
Polarization is a phenomenon where atoms and molecules induce changes in the electron distributions of other atoms and molecules they interact with [29]. This effect grows stronger the stronger the electrostatic interactions and/or the more polarizable the atoms involved. Because of their strong electrostatics, then, the electrostatic interactions of charged groups can be particularly affected by polarization. Additionally, anions such as iodide and bromide are highly polarizable, including anions with phosphate or sulfate moieties which are present in a wide range of biomolecules [30, 31]. Phosphates and sulfates play important roles in biological functions, interactions, and are present in drug-like molecules [31]. On the other hand, small cations have low polarizability but can still strongly polarize their environment when it is polarizable.
Much molecular modeling uses classical fixed-charge force fields without an explicit accounting for polarization [31]. Such two-body additive force fields are implicitly polarized to hopefully match a level of polarization appropriate on average for condensed-phase simulations [32–34]. This is true for common force fields in the AMBER, CHARMM, GROMOS and OPLS-AA families (e.g. GAFF [35, 36], OpenFF [37], CGenFF [38–40], and OPLS-AA [41, 42]); these neglect polarization for computational efficiency. It’s possible that the approximations made by these fixed-charge force fields may result in particularly large errors in systems like those examined here [43].
Polarizability may also be particularly important for these systems due to the water model. Particularly, with fixed-charge force fields, the water model is also non-polarizable, which may be an especially bad approximation for systems like these where water interactions with a buried hydrophobic cavity are at play [43]. The expectation is that binding in host-guest systems like those examined here are heavily influenced by the hydrophobic effect, and the hydrophobic effect will certainly be strongly influenced by properties like polarizability.
Fixed point charge water models are limited in some ways by their use of the same partial charges to empirically fit the potential energy landscape and dipole moment, two distinct water properties [44, 45]. Inevitably, the choice in water model (many listed in [46, 47]) may also dictate the accuracy in (a) solvation, (b) dielectric constant, and (c) dipole moment [44], and affect ionic behavior along with many other properties. Previous work in the Gilson lab indicated that even fixed-charge water models can vary dramatically in water placement and orientation around hosts as well as in thermodynamic properties like the enthalpy of binding [48, 49], and it seems likely that polarizable models may exhibit even larger differences.
Polarizable force fields potentially help address some of these concerns and challenges. The first general purpose polarizable model was introduced by Arieh Warshel for a water model suitable for biomolecular simulations [50], building on his work with early QM/MM based approaches. Peter Kollman [51] and Berne and Friesner [52] developed early polarizable variants of AMBER in the 1980s and 1990s, respectively. More recently, the AMOEBA force field has been in development since the early 2000s and was first published by Ren and Ponder around 2002 [53]. Polarizable force fields, and their importance for such systems, are explained in Section 2. In addition, popular general force fields such as AMBER, OPLS-AA, GROMOS, and CHARMM are continuously evolving and polarizable versions of some of these are available [46]. One example of the latter is a recent release of CHARMM’s balanced Drude polarizable force field [31]. However, polarizable force fields have been applied relatively seldom in SAMPL challenges; the AMOEBA force field was used in some prior host-guest challenges [11], but the Drude polarizable force field has yet to be used in a SAMPL challenge.
In other words, polarizable force fields add additional complexity to the physical model used in describing these systems, potentially providing additional accuracy but with additional computational cost. However, for some host-guest systems, this may be particularly important for several physical reasons. First, these systems often exhibit strong electrostatic interactions in a buried, relatively hydrophobic environment, meaning that the precise degree of polarization and environmental shielding may be a key determinant. Polarizability may affect the strength of charge-charge interactions, and may strongly modulate the shielding effect of the environment. Additionally, the hydrophobic effect can be a key determinant of binding, and this is also likely strongly modulated by polarization of the water and host.
Polarizable force fields have shown some promise in prior SAMPL challenges. In the SAMPL6 host-guest challenge, a method using the AMOEBA force field was employed on CB8 with 14 guests ranging from small organic molecules to larger drug-like compounds, including approved drugs. The initial results had an ME and RMSE of 2.63 and 3.62 kcal/mol respectively, and interestingly, this method was able to correctly identify questionable host-guest complex ratios of CB8 with guests 11 and 12 [11]. The correct respective ratios for these systems were 1:1 and 1:2, and these were a bonus challenge in SAMPL6. Binding free energies were predicted to be too favorable for guests 2 and 3 (Palonosetron and Quinine) which was presumed to be due to (a) AMOEBA parameters for the host resulting in single and/or double indentation of the macrocycle and (b) conformers of flexible guests locked during solvation in water vs binding in solvated complex [11]. In subsequent studies, revised AMOEBA results reported the improved ME and RMSE to 1.20 and 1.68 kcal/mol respectively, though this was after challenge results were released. In total 8 of the 15 predicted free energies were within 0.65 kcal/mol of experiment while the predictions for Palonosetron and Quinine guests were in better agreement with experiments. The improvements were attributed to two factors: (a) the value of key torsion parameters for C(N)-C-amide N-carbonyl carbons of CB8 and CB7 were adjusted to improve the flexibility description of the host ring system and (b) a double annihilation scheme of electrostatics and van der Waals with annihilation of key guest torsions yielded much better conformational sampling and hence predictive accuracy. However, through the SAMPL6 challenge we had not yet seen methods using the AMOEBA force field dramatically outperform other methods prospectively.
3.3. The type and concentration of salt could play an important role in some cases
Empirical force fields’ predictive power can be limited by the quality of their parameters. Parameters are not always available for all relevant chemistry, or may not be of equal quality for all chemistry of interest. For example, experiments for all components of the SAMPL7 host-guest challenge were done in sodium phosphate buffer (of varying concentration and pH). However, because of concerns about the quality of phosphate force field parameters, we conducted our reference calculations in sodium chloride (of the same ionic strength) instead. While this choice seems reasonable and is not uncommon in molecular modeling, it might affect computed free energies.
Particularly, the type of salt and its concentration can alter the solubility of a solute (e.g. in what is known as the Hofmeister effect) [54, 55]. Such salt dependence also interacts with the choice of water model. Particularly, one computational study reported surprising differences in the salt dependency of binding enthalpy (comparing TIP3P, SPC/E, TIP4P-Ew, and OPC water models) during MD simulations for cucurbit[7]uril host with a neutral guest [56]. Despite the system being non-ionized, the salt concentration (and the choice of sodium and chloride parameters) affected the behavior and thermodynamics of water, raising issues regarding selection and adjustment of water models for charged groups [56]. Incorrect ionic behavior (i.e. dielectric constant, dipole moment, solvation, and excessive ion-pairing and/or ion pairing strength) has been shown to be due to unbalanced force field parameters [31, 44].
In the present SAMPL challenge, some participants did not use any ions beyond counter ions to neutralize their systems. However, salt concentration is known to play a significant role in modulating host-guest binding affinities experimentally in some cases [9, 57]. Thus, if salt concentration proves important here, such differences in protocol could produce a systematic difference between methods.
3.4. Some methods require considerable expertise to use successfully
Some methods for binding prediction require extensive knowledge and expertise. For example, a person with little computational experience may not be able to conduct a successful free energy calculation given the historical difficulty of setting up such simulations. Few available software tools are user-friendly enough that one might be able to simply insert receptor and ligand files and obtain an accurate estimate of a property like a binding free energy. This likely affects accuracy; it’s conceivable that users providing the same input files to the same package could obtain dramatically different results because of different choices of protocol.
Some tools provide a relatively straightforward interface for free energy calculations, at least, like YANK, but even YANK still requires a command-line interface and a wide variety of settings can affect computed values. Other tools like those from Schrödinger and the Chemical Computing Group allow free energy calculations from a GUI (Graphical User Interface), and the Schrödinger tools remove many key choices from the hands of users. However, we are not yet aware of a successful application of these tools to host-guest binding.
3.5. We avoid multimeric systems which introduce additional complications
Binding which involves stoichiometries other than 1:1 can be considered multimeric association. Some proteins exhibit this behavior, where a single protein molecule co-assembles with other proteins to form a complex; in other cases, a protein might oligomerize only on binding of a ligand or ligands. The reverse can also happen, with multiple ligands binding to a single protein, etc. The same holds true for some host-guest systems, with these systems exhibiting binding that is not 1:1 [58, 59], complicating both experimental measurement of binding and computational prediction thereof. In SAMPL7, we worked with experimental collaborators to deliberately ensure the challenge focuses on systems exhibiting 1:1 binding. However, the formation of host-guest multimeric complexes can even depend on the guest identity [59].
With multimeric host-guest complexes, cooperativity may play a role. Cooperativity occurs when a binding event can either increase or decrease the strength of subsequent binding events [60]. In the presence of ions, electrostatic attractions can also lead to cooperativity [61]. Indeed, experiments must verify 1:1 binding (as was done here) otherwise computation would need to consider other possibilities.
4. Previous SAMPL host-guest challenges used similar hosts
Previously SAMPL challenges have included a variety of host-guest systems, but the majority of SAMPL hosts have been in the cucurbituril [62] and Gibb deep cavity cavitand (often called “OctaAcid”) families [63] thanks to the contributions of Lyle Isaacs and Bruce Gibb’s labs. There have been several analogs of these two families since host-guest systems first appeared in SAMPL3. SAMPL7 includes several analogs in the cyclodextrin [64] family thanks to Michael Gilson’s lab.
Study of these various systems, in SAMPL and elsewhere, can help provide insight into the particular challenges each system presents. However, conclusions are not always clear; sometimes, performance remains highly variable across several challenges.
Particularly, performance in prior SAMPL challenges was highly variable by method and target, and no clear method emerged as reliable across all systems or most systems. Both SAMPL3 and SAMPL4 included some guests in cucurbituril family [15, 65–68], with the best RMS errors typically being around 2.5 kcal/mol unless empirical corrections were included [65, 69], and no method stood out across both challenges [17]. SAMPL4 also included cavitands. In SAMPL5, the best RMS error was closer to 3 kcal/mol [65], but correlation with experiment for this approach was not good. Methods based on explicit solvent and electronic structure calculations were noted to appear relatively consistent and generally provide the greatest reliability across all SAMPL challenges [70], but also had considerable room for improvement. In general, predictions for cavitands seemed to be modestly more accurate whereas clip-based hosts have been more challenging in prior challenges (like CB-Clip in SAMPL5 [70]). Thus, in the present challenge, we hoped to learn whether we might see a method or methods with significantly improved accuracy relative to prior challenges, and whether one might emerge that performs reasonably well (e.g. RMS error under 3 kcal/mol) across multiple host classes, as this has not typically been the case in prior challenges.
5. SAMPL7 Host-Guest Systems and Challenge Organization
The SAMPL7 host-guest challenge involved three different systems or categories which we explain here – one focusing on cucurbituril-derivatives, one focusing on Gibb deep cavity cavitands (GDCCs), and one focusing on modified cyclodextrins.
5.1. Cucurbiturils and derivatives (CB[n], CB-Clip and TrimerTrip)
Cucubiturils are a common and relatively well-studied system for host-guest binding [9] which have been featured in some prior SAMPL challenges.
Many cucurbiturils (CB[n]s) have been synthesized by the Isaacs Lab, and several featured in previous SAMPL challenges. The potential applications of cucurbiturils include use as solublizing excipients for insoluble drugs, sequestrants for drugs of abuse and neuromuscular blockers, and pH triggered delivery agents [62]. This family of hosts typically have a molecular structure containing n glycoluril units connected via 2n methylene bridges, forming a barrel shaped macrocycle with a central hydrophobic cavity. In addition, cucurbiturils contain electrostatic carbonyls protruding out from the hydrophobic cavity.
In the SAMPL7 challenge, the host is not a classic cucurbituril, as instead of being a macrocycle, it is a clip-shaped molecule based on similar chemistry. Particularly, the host is an acyclic cucurbituril clip composed of a glycoluril trimer capped with aromatic triptycene sidewalls at both ends (here called TrimerTrip, as it is a trimer of glycoluril units with triptycenes), and four sulfonate solubilizing groups protruding out from the sidewalls (Figure 1) [62]. The sulfonate groups also enhance ion-ion interactions with cationic guests [71], which are typical cucurbituril binders. Acyclic CB[n]-type receptors often take on a C-shape due to their increase in flexibility [62, 71, 72]. Experimentalists synthesized acyclic cucurbiturils with the idea to help increase the binding strength and capacity for different guests, including macrocyclic guests.
Figure 1. Structures of the TrimerTrip host and guest molecules for the SAMPL7 Host-Guest Blind Challenge.
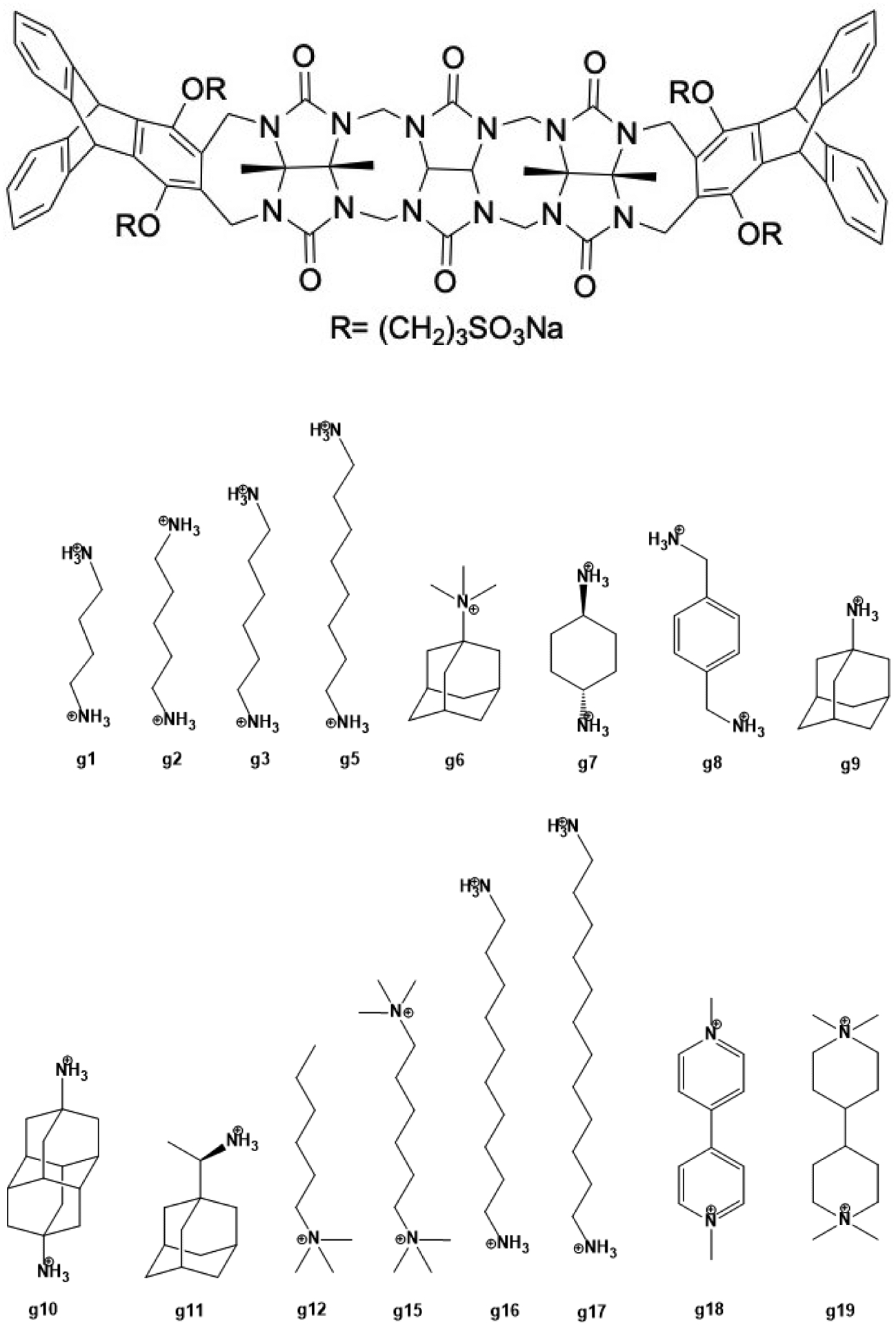
The acyclic CB[n]-type receptor, TrimerTrip, is shown on the top. It is composed of a glycoluril trimer with aromatic triptycene sidewalls at both ends, and four sulfonate groups to increase its solubility. The host can take on a C-shape (though other conformers can be possible) and binds guests inside the cavity. The guests for the SAMPL7 challenge have the characteristics of typical CB[n] binders. The guests are named g1 through g19 (g4, g13, g14 were not included in the challenge).
Typically, CB[n]-guest complexes have very high affinity, especially for charged hydrophobic ammonium guests similar to those of the SAMPL7 challenge (Figure 1). This high affinity is due to the presence of intracavity waters lacking a full complement of hydrogen bonds. The lack of hydrogen bonds is known to provide an enthalpic driving force for binding to macrocyclic CB[n] complexes [73]. In terms of CB[n]-guest complex interactions, the charged nitrogen group on guests interacts with oxygens from the carbonyl portal of the host. The latter contributes to limiting the number of poses that need to be considered [11], at least in cyclic hosts.
CB7 was used as a basis for host-guest benchmarking (including on binding of guests with adamantane and aromatic ring cores) since some of its properties and characteristics made it a convenient host both computationally and experimentally [9]. Four insights and challenges for CB7 are described [9] and some may be transferable to a clip type cucurbituril. (1) The tight exit portal of CB7 makes it difficult for guests with bulky hydrophobic cores such as adamantyl to fit through the portal and hence lead to convergence problems. (2) The timescales of wetting and dewetting events may be large compared to typical simulation timescales. In CB7, when gradually decoupling a guest there is a large fluctuation of waters in the host cavity. The latter occurs when the guest is partially decoupled and may also lead to convergence problems. (3) Experimental and computational binding thermodynamics are sensitive to the salt composition and concentration (for buffer conditions). (4) Guests with formal charges can pose challenges for binding free energy calculations (Section 3.1).
Previous studies of cucurbiturils, including CB7, have highlighted the importance of host and guest sampling, salt effects, and water model. Sampling of the CB7 host is thought to be straightforward because it is fairly rigid. However, guest binding modes might be challenging to adequately sample, especially for the more flexible guests. In the presence of buffer and/or salt, ions may compete with guests for the binding site. In addition, cationic guests could have interactions with counter-ions in solution, lowering affinity compared to zero-salt conditions [9]. One previous study showed a 6.4–6.8 kcal/mol dependence on salt concentration [9, 74]. The water structure around CB7 is sensitive to the choice of water model, and water is important in modulating binding in SAMPL7 systems. The choice of water model is also likely to have an impact on the number of sodium ions that must be displaced upon host-guest binding.
While these insights result from studies on CB7, some of them may carry over to the TrimerTrip host studied here. However, unlike its macrocyclic derivatives, TrimerTrip is acyclic and able to flex the methylene bridged glycoluril trimer backbone [72]. Hence, with more degrees of flexibility sampling of TrimerTrip may not be as straightforward. TrimerTrip, like the Calabadion “cousins” in the family of cucurbiturils, may allow guest cationic groups to interact with other regions of the host rather than the carbonyl portals as in CB[n] macrocycles [72], which may complicate guest sampling.
Previous acyclic CB[n]-type receptors contain a central glycoluril oligomer (monomer, dimer, and tetramer) with aromatic triptycene sidewalls, just like TrimerTrip. These clip-like receptors retain the essential molecular recognition properties of macrocyclic CB[n] [75]. The monomer [71], dimer [75], and tetramer [66, 75] clips are able to encapsulate typical hydrophobic cationic guests which also bind to macrocyclic CB[n]s. In addition, the dimer and tetramer display similar host-guest properties [75]. While TrimerTrip is a distinct host, it shares substantial similarity with these previous receptors and we expect it to exhibit relatively similar behaviors in binding to guests.
5.2. Gibb Deep Cavity Cavitands (GDCCs) – OctaAcid (OA) and exo-OctaAcid (exo-OA)
Of the several members in the GDCC host family [63], two have been used in several previous SAMPL challenges thanks to the Gibb group’s participation. Those featured in previous SAMPL challenges include OctaAcid (OA) and tetra-endomethyl OctaAcid (TEMOA). A newer exo-OctaAcid (exo-OA) along with OA are part of the SAMPL7 host-guest blind challenge (Figure 2). The guests for this system are diverse in their size and bulkiness, but typically have either a carboxylate or quaternary ammonium (Figure 2).
Figure 2. Structures of the GDCC host and guest molecules for the SAMPL7 Host-Guest Blind Challenge.
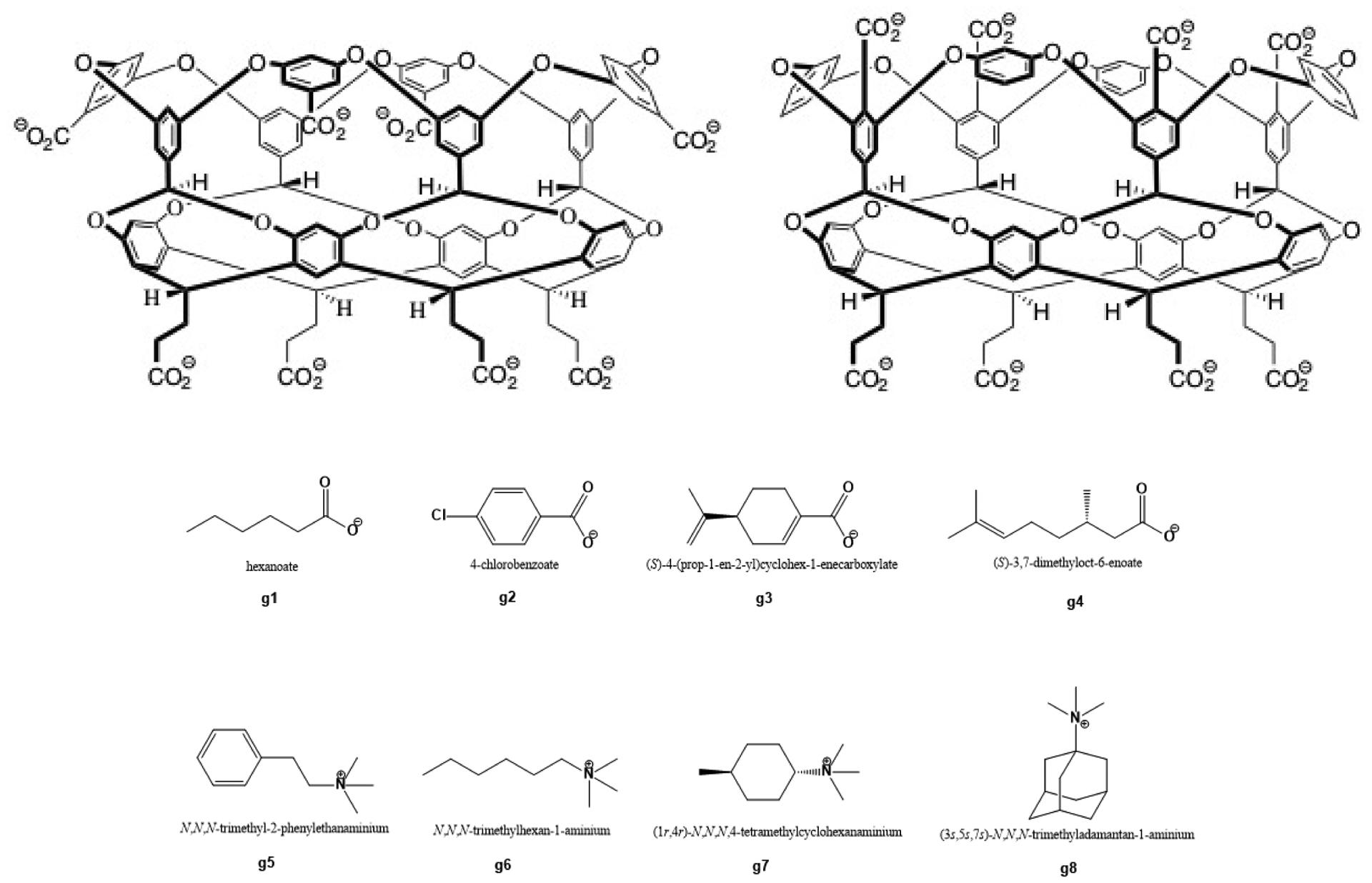
(top left) OctaAcid, (top right) exo-OctaAcid; (bottom) guests. The difference between the hosts is the placement of the carboxylate groups near the cavity opening. While the carboxylates protrude outward away from the cavity in OA, in exoOA they are at the rim of the cavity opening. The guests for SAMPL7 are named g1 - g8. Four guests have a carboxylate group, and four a quaternary ammonium group. For the OA host, guests g1 - g6 have binding free energies which were previously reported and thus calculation of values was made optional for participants.
OA and exo-OA have a deep and hydrophobic basket-shaped pocket, and are fairly rigid [9, 58]. In total there are eight carboxylate groups in both OA and exo-OA. The propionate groups at the exterior site of the cavity are the same in both hosts. The difference between the two hosts is the location of 4 carboxylates around the cavity opening. For OA the carboxylates are protruding out of the cavity while for exo-OA they are at the cavity entrance (Figure 2).
GDCCs have been used in SAMPL3–7 and there is much experimental data [9, 43, 63, 76] and insight available. This family of hosts bind guests with a hydrophobic moiety that fits the pocket and a hydrophilic group which points out towards the solvent [9].
The GDCCs have been shown to bind diverse guests varying in polarity, positively and negatively charged, as well as organic cations and anions [9, 77, 78]. The latter has been shown for OA, where binding thermodynamics is sensitive to the concentration and type of anions present. Shifts in binding enthalpies and free energies of approximately 10 kcal/mol and 2 kcal/mol respectively [54] have been observed and attributed to the competition between guests and anions leading to entropy-enthalpy trade-offs [9, 54]. In addition, experimental and computational simulation results show that de-wetting of GDCCs leads to increased guest affinity, because water cannot compete for the pocket [63, 76].
In the presence of elongated guests, such as a long aliphatic chain, two OA hosts can encapsulate a guest forming a ternary complex. This phenomena is more likely to occur as polarity decreases for the groups at both ends of the guest [77]. However, as described earlier in section 3.5, SAMPL7 was designed around systems which exhibit 1:1 binding. Isothermal titration calorimetry (ITC) experiments have shown that short-chain fatty acids, amphiphilic molecules, and large polarizable anions form 1:1 complexes [76], as do the guests reported here.
Previous work has proposed benchmarking free energy calculations on host-guest systems; for GDCCs, the proposed benchmark included OA binding to guests with adamantane, aromatic, and saturated cyclic carboxylates. These host-guest systems were chosen because of the broad range of binding free energy values produced, and because both host and guests are small and rigid enough to confidently converge binding free energy calculations [9]. Several key challenges were highlighted by prior work: (a) a tight entry/exit portal may create a barrier and prevent entry or exit of guests with bulky hydrophobic cores. Hence, this can hinder sampling of guests leading to convergence problems. (b) It is important to ensure adequate host conformational sampling (though the motions may be slow), particularly of the propionoic acid groups. Benzoic acid flips (at the rim of the cavity) have also been reported from several simulations [3, 9, 65], though these have not been verified experimentally and may be irrelevant to binding thermodynamics. However, it has been noted that the benzoic acid flips might be an important challenge in some force fields. (c) Waters move only slowly into and out of the cavity, with the number fluctuating over tens of nanoseconds [9, 79]. (d) Salt concentration and buffer conditions may modulate binding to GDCCs. Additionally, (e) charged guests may introduce finite-size artifacts. (f) Strong electrostatic interactions could result in modified protonation states of the host and/or guest. Acidic guests could be protonated, or two of the propionate groups could retain an acidic proton because they are in close proximity and can hydrogen bond. At the rim of the cavity a guest may also modulate protonation state of the neighboring carboxylates.
5.3. Cyclodextrins (CDs) and cyclodextrin derivatives
Cyclodextrin (CD) family hosts are composed of chiral glucose monomers linked to yield a cyclic polymer. The SAMPL7 challenge focused on modified CDs provided by the Gilson lab, which synthesized monofunctionalized derivatives differing by addition of a substituent projecting outward from a primary or secondary face hydroxyl of the cyclic oligosaccharide (Figure 3). The CD host derivatives and native (unmodified) CDs have a truncated cone shape (Figure 4) with a hydrophobic cavity and a hydrophilic surface, while the substituents are intended to alter the host’s chemical and physical properties. The new host substituents introduce new host-guest interactions, while retaining some of the same binding characteristics [80].
Figure 3. Structures of the cyclodextrin host derivatives and guests for the SAMPL7 Host-Guest Blind Challenge.
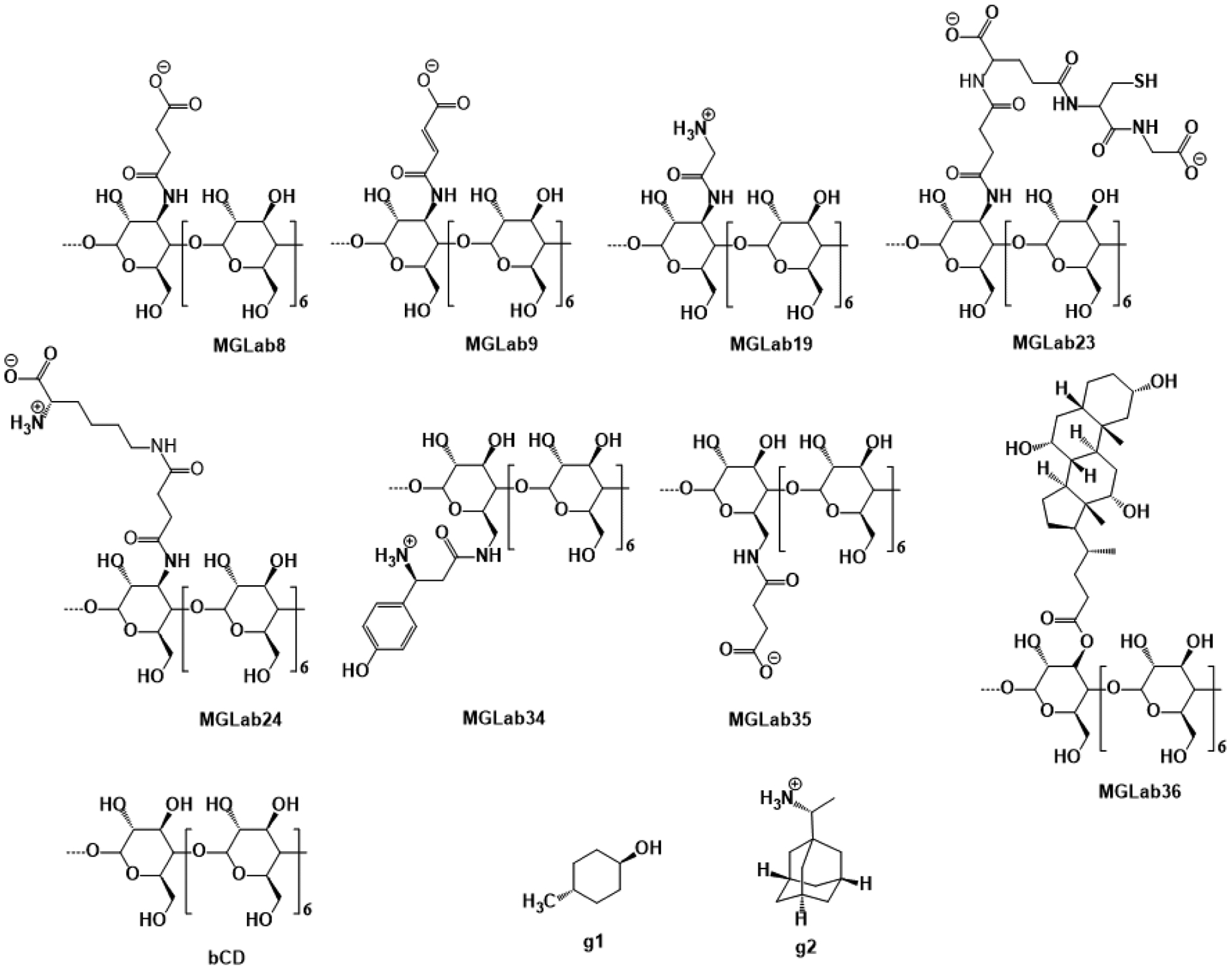
The cyclodextrin derivatives are a series of macrocycles composed of seven glucose subunits linked by 1,4 glycosidic bonds. The native β-cyclodextrin (bCD) contains the primary (2’OH) and secondary glucose subunit hydroxyls, while all of the cyclodextrin derivatives (MGLab#) differ by a substituent at either of these positions. MGLab8, MGLab9, MGLab19, MGLab23, MGLab24, and MGLab36 have substituents out from the top or primary face (wide opening), while MGLab34 and MGLab35 have the substituents out from the bottom or secondary face (narrow opening). The two guests are trans-4-methylcyclohexanol (g1) and cationic R-Rimantadine (g2).
Figure 4. βCD host structures.
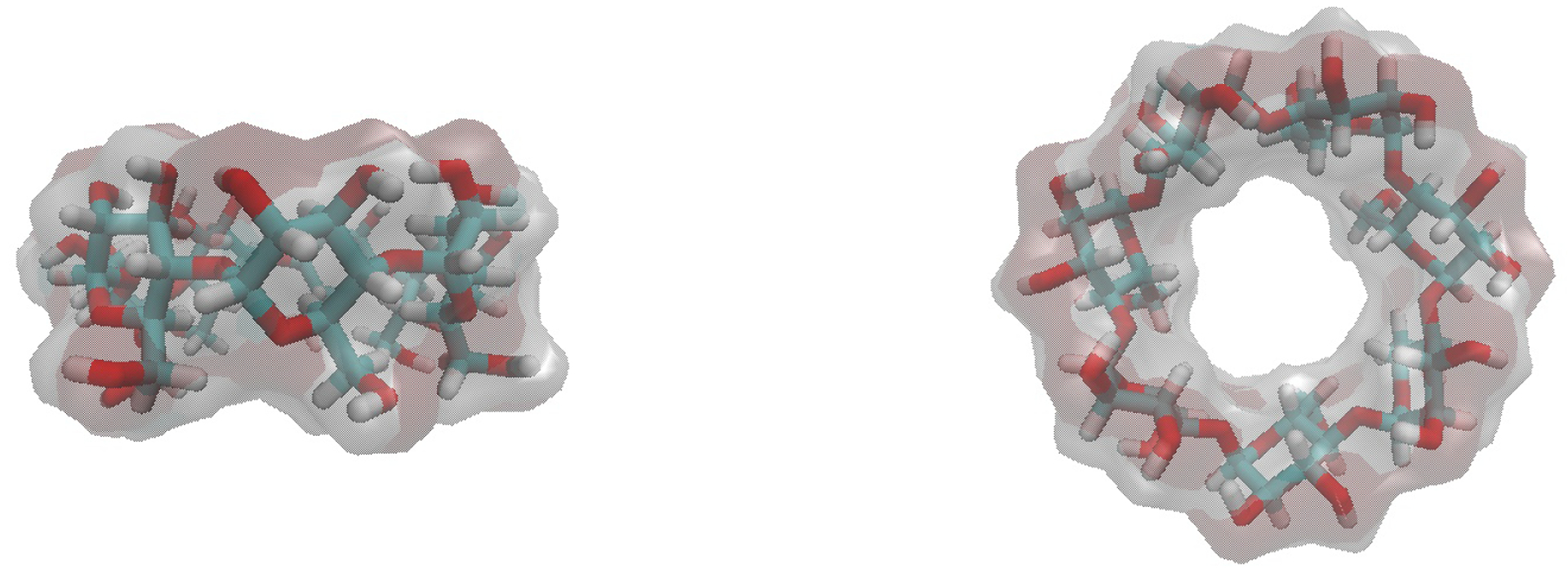
Shown are two views of βCD. It and its derivatives are known to bind guests in two orientations, primary and secondary. The primary binding orientation is when an asymmetric guest’s polar head group projects out towards the glucose primary alcohols or the smaller opening (down). The secondary binding orientation is when a guest’s polar head group projects towards the secondary alcohol or the larger opening (up).
While typical SAMPL host-guest challenges have focused on binding of a series of guests to one or two hosts, one unique aspect of this portion of the challenge is that it focuses on binding of just two guests to a series of related hosts.
Previous studies on CDs (α-CD, β-CD, and mono-3-carboxyproponamido-βCD) report two distinct bound states for each host-guest pair. The first bound state, called the “primary orientation”, has the guest polar group (i.e., alcohol, ammonium, carboxylate) towards the glucose subunits primary alcohols, while the “secondary orientation” has the guest polar group towards the secondary alcohol [80, 81] (Figure 4). Though a possible third “surface orientation”/binding mode has been speculated to exist, it may be this is a transition needed for the guest to flip from primary to secondary phase orientation or vice-versa [43]. The difference in binding free energy for the two main orientations has been reported as being about 2 kcal/mol and up to 5 kcal/mol using several different force fields [81], with this of course also depending on the guest. The same report suggested that using GAFF v2.1 better models the flexibility of β-CD compared to the SMIRNOOFF99Frosst and GAFF v1.7 force fields.
The guests proposed in SAMPL7 have been reported to bind native β-CD, mono-3-carboxyproponamido-β-CD, and β-CD substituted with an amine at the 3 position (secondary face). Rimantadine (Figure 3) binds beta-CD and mono-3-carboxyproponamido-βCD with its cationic ammonium group projecting out from the secondary face [80, 82]. On the other hand rimantadine prefers the primary orientation when binding β-CD with an amine at the 3 position. Both 4-methyl-cyclohexanol (g1) and rimantadine (g2) (Figure 3) may bind to the new β-CD derivative hosts (MGLab9 through MGLab36 Figure 3) in either of the three orientations. However, it was hypothesized that the rimantadine head group would be oriented towards a negatively charged substituent and away from a positively charged one [43].
Binding modes for the cyclodextrin dataset were determined using 2D NOESY NMR by the Gilson lab [64]. This experimental binding mode information can in turn be used to check if the selected binding mode(s) used in a particular method played a role in the accuracy (or lack thereof) of computed binding free energies. Table 1 summarizes the binding orientations for methylcyclohexanol and rimantadine with each host as determined by the Gilson lab (for specific details of the experimental methods see Ref [64]).
Table 1. Binding orientations of guests g1 (methylcyclohexanol) and g2 (rimantadine) with cyclodextrin hosts.
Binding orientations of guests complexed with hosts determined by NOESY NMR by the Gilson lab [64]. The orientations are summarized here to cross check with binding mode(s) used by SAMPL7 participants and ascertain the binding mode(s) which may contribute to accurate binding affinity predictions (or lack thereof). In some cases, experiments did not allow determination of a binding mode; such cases are labeled ND.
| Host | Location of Mono-substituent (Face) | Guest Binding Orientation |
|---|---|---|
| G1 - Methylcyclohexanol | ||
| β-CD | N/A | Primary and Secondary |
| MGLab8 | Secondary | Secondary |
| MGLab9 | Secondary | Primary and Secondary |
| MGLab19 | Secondary | Primary |
| MGLab23 | Secondary | Primary and Secondary |
| MGLab24 | Secondary | Primary |
| MGLab34 | Primary | Secondary |
| MGLab35 | Primary | Primary |
| MGLab36 | Secondary | Primary and Secondary |
| G2 - Rimantadine | ||
| β-CD | N/A | Secondary |
| MGLab8 | Secondary | Secondary |
| MGLab9 | Secondary | ND |
| MGLab19 | Secondary | Primary |
| MGLab23 | Secondary | Primary |
| MGLab24 | Secondary | Secondary |
| MGLab34 | Primary | Primary |
| MGLab35 | Primary | ND |
| MGLab36 | Secondary | Secondary |
5.4. Challenge Organization and Format
The SAMPL7 host-guest blind challenge was organized so participants may submit a ranked submission, a non-ranked submission, or both for any or all of the three host-guest systems. Participants were advised to submit their best method as their ranked submission since only one ranked submission is allowed, as detailed below.
Participants were provided with pre-prepared host and guest structures, with SMILES strings, mol2, PDB and sdf files provided for all compounds. We made an effort to provide reasonable protonation states, etc., but also provided disclaimers that participants should carefully consider the choice of protonation state, etc. All provided data/instructions are available in the SAMPL7 GitHub repository (https://github.com/samplchallenges/SAMPL7).
Participant submissions followed a prescribed template and included predicted values and uncertainties, as well as method and participant information and other details. All submission files are available in the GitHub repository. Predicted values were optionally allowed to include binding enthalpy.
Only ranked submissions were considered in challenge analysis. Groups were able to submit multiple submissions, but needed to designate additional submissions as non-ranked. Non-ranked submissions, or additional submissions, allow “benchmarking” of methods. For example, for a particular method a participant can change one parameter in their methodology (i.e. charging method, host conformer, guest pose, water model, etc.) to ascertain its impact on predictions. In previous challenges, participants were allowed multiple ranked submissions; the shift to a single ranked submission per participant is new to SAMPL7. This change was made to reduce the potential for multiple shots on goal to be more fair to groups which only submit one set of predictions.
In addition to the formal predictions, one member of our team (MA) conducted a set of blind reference calculations which were submitted informally in the non-ranked category. Data collection for TrimerTrip and its 16 guests (Figure 1) of this challenge was completed around August of 2019 and a challenge submission deadline of October 4, 2019 was set to avoid delaying the experimental publication. The GDCC dataset was finalized on May 25, 2019 and its submission deadline, along with that for the Cyclodextrin derivative challenge, was set to November 4, 2019. Submissions for OA with g1-g6 (Figure 2) guests were optional (and not part of rankings) since these have been reported in previous challenges and literature values are available. In addition, submitting binding enthalpies for GDCC predictions were optional. Similarly, for the Cyclodextrin derivatives dataset, predictions for g1 and g2 binding to β-cyclodextrin (Figure 3) were optional since literature values for these compounds are available.
As noted above, we provided input files in a variety of formats. Participants were advised that (a) further equilibration of the host with the guest might or might not be needed (for TrimerTrip, we pre-equilibrated the host structure as discussed in Methods) and (b) to exercise their best judgment on the state modeled (i.e protonation, conformer, binding mode, etc.). In essence, part of the host-guest challenge for some systems included binding mode prediction.
6. Methods
In this section we describe the details of our own reference calculations, give a general overview of methods used by participants’ submissions, summarize key experimental details and methodology (the experimental studies will be published elsewhere [63, 64, 72]), and describe our statistical analysis and evaluation approach.
6.1. Absolute Binding Free Energy Predictions
6.1.1. Reference Calculation Methodology
In this section we give details of our own reference calculations. These reference calculations were informally part of the challenge and used as additional methods for comparison. These calculations were also conducted blindly and were informally submitted as a “non-ranked” category, as they do not constitute a formal part of the challenge but are provided as a point of comparison.
Reference calculations were done using an alchemical free energy calculation toolkit known as YANK [10, 83]. YANK provides several schemes for sampling from multiple thermodynamic states. For reference calculations we applied the replica exchange sampler (also known as Hamiltonian Exchange) [83, 84], using the OpenMM simulation engine [85–89]. Free energies are estimated using the multistate Bennet acceptance-ratio (MBAR) [90]. (For details on the thermodynamic cycle used in YANK and the theory see http://getyank.org/latest/theory.html)
Initially, test simulations were done with the goal to determine if we could identify and apply a reasonable single protocol to run all host-guest systems. However, due to the guest formal charges and the diversity of the hosts and guests we guessed that successful protocols (especially lambda spacings) would be system dependent.
For the simulations, harmonic distance restraints (between the closest atom to the center of the host and the closest atom to the center of the guest from the initial geometries) were used to allow the guest to explore the cavity and different binding orientations since the binding mode of some guests were unknown. Restraints are needed to define the standard state and ensure the ligand remains near the host to avoid sampling problems. We chose harmonic center-of-mass restraints in particular to allow the ligand to sample alternate binding modes if needed. This may help reduce bias in free energy estimates if we start from an incorrect binding mode (especially in the cases where the binding mode is unknown).
We ended up choosing two protocols, varying in number of lambda windows (with all other simulation parameters kept consistent), with one being for systems with neutral guests and a second for guests with a formal charge. We expected that a second protocol for guests with a formal charge would be needed since electrostatic interactions would be much stronger with its environment and limit sampling. Indeed, after testing the “neutral” protocol on a charged guest we noticed insufficient replica mixing per an issued warning from a generated YANK simulation health report. The protocol for neutral guests had 31 lambda windows and was based on a previous protocol used on β-CD with cyclopentanol as the guest. This protocol was tested on β-CD with 4-methyl-cyclohexanol as the guest. For systems with a charged guest, we ran a test free energy calculation using YANK’s automatic pipeline to determine the best alchemical path (lambda windows and values) based on a β-CD and the positively charged rimantadine (g2) guest, resulting in 61 lambda windows. Both of the test calculations were within 4 kcal/mol of experimental values [80] upon completion, and simulation health reports showed reasonable exchange between replicas and exhibited apparently reasonable convergence. However, in the case of the charged guest, convergence was not as convincing at similar time scales. For example, the test calculation for the neutral guest showed reasonable convergence by 14 ns per window while in the case of a charged guest, simulations were run for 26 ns per window and convergence was still not as obvious.
The “neutral guest” protocol described above (31 lambda windows) was used to run all simulations in the cyclodextrin dataset with guest g1, for 16 ns per lambda window when free energy estimates appeared converged. On the other hand, the “charged guest” protocol (61 lambda windows) was used for the remaining host-guest systems across all datasets since all other guests bore a formal charge. In this case, simulations were run until free energy estimates apparently converged or up to 30 ns per lambda window, which ever came first. First, to determine feasible cross application of the “charged guest” protocol to different systems (GDCC and TrimerTrip datasets), the charged protocol was tested on OA-g2 and clip-g11. Experimental data for OA-g2 was available from a previous SAMPL challenge, so this was an ideal system to test the protocol. The OA-g2 test resulted in predicted free energy within 4 kcal/mol, after running the simulation to 26 ns per window. A health report for the OA-g2 simulation showed reasonable mixing between replicas, and there was apparent convergence. However sampling of replicas in individual states was not ideal. For the clip-g11 test simulation (for TrimerTrip dataset), the protocol was initially deemed reasonable based on YANK’s health report (with mixing_cutoff and mixing_warning_threshold options at default 0.05 and 0.9 settings, respectively) which can detect insufficient replica mixing or number of swaps between states and thus issue warnings. Warning messages were not issued in this test case. However, in this test case sampling of replicas in individual states was not ideal and the calculations apparently did not fully converge even after 30 ns per window. For this reason all simulations for TrimerTrip were run for 30 ns per window in an attempt to obtain reasonable convergence, though after the fact convergence was only apparent for clip-g1 of TrimerTrip dataset. In addition, an “open” host conformer was extracted from the clip-g11 test simulation trajectory, the guest was docked to the open host conformer, and simulation (found in Docking/GAFF/YANK_REF_2) was re-run in an attempt to allow the host to relax and adapt to the bulky guest. Still longer simulations, or protocol optimizations, might be needed for better converged results.
Reference calculations were conducted using GAFF parameters and AM1-BCC charges. GAFF parameters and guest AM1-BCC charges were assigned using Antechamber, and AM1-BCC charges for the host were assigned using the OpenEye toolkits because Antechamber could not charge the hosts. The starting poses were determined by docking via AutoDock Vina [91] and the top scoring pose was selected. If multiple orientations need to be considered, our Hamiltonian replica exchange based simulations, in theory, ought to sample them despite starting from a single orientation. A host-guest complex was manually created in tLeap and TIP3P was used to solvate the host-guest complex and the guest. In addition, sodium and chloride were manually added as counter ions, and additional ions were added to mimic experimental buffer conditions. Subsequently, AMBER restart, topology, and input coordinate files were generated with tLeap. The starting simulation files (AMBER restart/coordinate (rst7) and topology (prmtop)), workflow and methodology details, and yaml scripts (with protocol parameters) are available at SAMPL7 GitHub repository (see https://github.com/samplchallenges/SAMPL7/tree/master/host_guest).
6.1.2. Participant Calculation Methodologies
There were a total of 30 submissions (ranked and non-ranked) from 6 groups for the SAMPL7 host-guest challenge. A good number of methods used alchemical free energy calculations with classical fixed charge (GAFF [92], GAFF2 [93], CGenFF [92]) and polarizable force fields (AMOEBA) [94], different charging schemes (AM1BCC [92, 95], RESP [95]), several explicit water models (TIP3P [92], TIP4P-Ew [95], OPC [93]) and even implicit solvent [95]. Outside of simulation-based free energy methods, quantum mechanical (QM) and QM/MM (molecular mechanics) approaches were also used [95], and one group employed machine learning [96]. In addition, several groups submitted multiple predictions (particularly for the GDCCs) and the ensuing results are important to provide insight and give merit to the methods used here. Participants’ submissions with specific details on their methodologies are available in the relevant host-guest system directory in the SAMPL7 GitHub repo (https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Submissions) and methods are briefly summarized in Table 4.
Table 4. Summary of methods (ranked and non-ranked) used in the SAMPL7 host-guest blind challenge for binding free energy calculations.
Alchemical calculations are flagged by an (A), the use of explicit and/or implicit solvation is flagged by an (E) or (I) respectively, and a linear correction approach was taken on methods flagged with a (C). The Noneq/Alchemy/consensus method was an average of the energy models used in Noneq/Alchemy/CGENFF and Noneq/Alchemy/GAFF.
| ID | sid | Energy Model | Solvent Model | Sampling | Ranked | SAMPL7 Refs |
|---|---|---|---|---|---|---|
| TrimerTrip | ||||||
| AMOEBA/DDM/BAR/ALT-2 | 9 | AMOEBA | AMOEBA (E) | Replica Exchange | No | [94] |
| AMOEBA/DDM/BAR-ALT1 | 8 | AMOEBA | AMOEBA (E) | Replica Exchange | No | [94] |
| AMOEBA/DDM/BAR | 6 | AMOEBA | AMOEBA (E) | Replica Exchange | Yes | [94] |
| FSDAM/GAFF2/OPC3 | 4 | GAFF2/AM1-BCC | OPC3 (E) | RESP | Yes | [93] |
| MD/DOCKING/GAFF/xtb-GNF/ | 5 | GAFF/AM1-BCC | TIP3P (E) | MD/SA-MD | Yes | [96] |
| Docking/GAFF/YANK_REF | REF2 | GAFF/AM1-BCC | TIP3P (E) | Replica Exchange | No | |
| Docking/GAFF/YANK_REF_2 | REF3 | GAFF/AM1-BCC | TIP3P (E) | Replica Exchange | No | |
| GDCC-OA and exoOA | ||||||
| RESP/GAFF/MMPBSA-Cor (C) | 20 | GAFF/RESP | TIP4PEW/PBSA (I) | MD | Yes | [95] |
| AMOEBA/DDM/BAR | 29 | AMOEBA | AMOEBA (E) | Replica Exchange | Yes | [94] |
| AMOEBA/DDM/BAR_2 | 30 | AMOEBA | AMOEBA (E) | Replica Exchange | No | [94] |
| xtb-GNF/Machine Learning/CORINA MD | 28 | GAFF/AM1-BCC | TIP3P (E) | MD/SA-MD | Yes | [96] |
| AMOEBA/DDM/BAR_3 | 31 | AMOEBA | AMOEBA (E) | Replica Exchange | No | [94] |
| Docking/GAFF/YANK_REF | REF4 | GAFF/AM1-BCC | TIP3P (E) | Replica Exchange | No | |
| B2PLYPD3/SMD_QZ-R | 23 | DFT(B3PW91) | SMD (I) | MD | Yes | [95] |
| B2PLYPD3/SMD_QZ-NR | 24 | DFT(B3PW91) | SMD (I) | MD | No | [95] |
| FSDAM/GAFF2/OPC3 | 14 | GAFF2/AM1-BCC | OPC3 (E) | RESP | No | [93] |
| B2PLYPD3/SMD_TZ | 22 | DFT(B3PW91) | SMD (I) | MD | No | [95] |
| RESP/GAFF/MMPBSA/Nmode | 18 | GAFF/RESP | TIP4PEW/PBSA (I) | MD | No | [95] |
| RESP/GAFF/MMPBSA | 19 | GAFF/RESP | TIP4PEW/PBSA (I) | MD | No | [95] |
| B2PLYPD3/SMD_DZ | 21 | DFT(B3PW91) | SMD (I) | MD | No | [95] |
| AM1-BCC/GAFF/MMPBSA | 17 | GAFF/AM1-BCC | TIP4PEW/PBSA (I) | MD | No | [95] |
| RESP/GAFF/MMGBSA | 16 | GAFF/RESP | TIP4PEW/GBSA (I) | MD | No | [95] |
| Cyclodextrin derivatives | ||||||
| FSDAM/GAFF2/OPC3_ranked | 12 | GAFF2/AM1-BCC | OPC3 (E) | RESP | Yes | [93] |
| Noneq/Alchemy/CGENFF | 26 | CGENFF/AM1-BCC | TIP3P (E) | MD | No | [92] |
| Noneq/Alchemy/consensus | 27 | NA | NA | NA | NA | Yes [92] |
| FSDAM/GAFF2/OPC3_JB | 13 | GAFF2/AM1-BCC | OPC3 (E) | RESP | No | [93] |
| Noneq/Alchemy/GAFF | 25 | GAFF/AM1-BCC | TIP3P (E) | MD | No | [92] |
| Docking/GAFF/YANK_REF | REF1 | GAFF/AM1-BCC | TIP3P (E) | Replica Exchange | No | |
| AM1-BCC/MD/GAFF/TIP4PEW/QMMM | 15 | GAFF/AM1-BCC | TIP4PEW (E) | MD | Yes |
6.2. Experimental Measurements
The experimental binding data for all host-guest systems are listed in Table 2 and in the SAMPL7 GitHub repo (see https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/ExperimentalMeasurements); if there are any updates/changes, the GitHub version is the authoritative one. As mentioned in Section 3.5 a 1:1 binding stoichiometry was confirmed for all host-guest systems. The binding values were determined via ITC and/or NMR typically at 298K. Binding measurements for TrimerTrip were performed in 20 mM sodium phosphate at pH 7.4. Binding constants for GDCC systems were determined in 10 mM sodium phosphate buffer at pH 11.7. All binding for CD derivative systems were assayed in 25 mM pH 6.8 sodium phosphate buffer. Experimental results suggest all binding was inside the CD-derivative cavity so there is no surface binding. Specific experimental details can be found in the SAMPL7 github repository (see https://github.com/samplchallenges/SAMPL7/tree/master/host_guest) and in the relevant experimental papers [62–64], respectively. Binding of one guest (g1) to the GDCC exoOA was undetectable by ITC and NMR (Table 2).
Table 2.
Experimental binding details for all host-guest systems.
| ID | name | Ka (M−1) | ΔG (kcal/mol)(a) | ΔH (kcal/mol) | TΔS (kcal/mol)(b) | n |
|---|---|---|---|---|---|---|
| clip-g1 | 4-azaniumylbutylammonium | 31000.0 ± 9000.0 | −6.1 ± 0.2 | −6.1 ± 0.8 | 0.0 ± 0.8 | 0.86 |
| clip-g2 | 5-azaniumylpentylammonium | 1270000.0 ± 80000.0 | −8.32 ± 0.04 | −8.8 ± 0.3 | −0.4 ± 0.3 | 1.00 |
| clip-g3 | 6-azaniumylhexylammonium | 24000000.0 ± 3000000.0 | −10.05 ± 0.07 | −10.9 ± 0.3 | −0.8 ± 0.3 | 0.90 |
| clip-g15 | trimethyl-[6-(trimethylammonio)hexyl]ammonium | 52000000.0 ± 4000000.0 | −10.52 ± 0.05 | −12.8 ± 0.4 | −2.2 ± 0.4 | 0.97 |
| clip-g12 | hexyl(trimethyl)ammonium | 1210000.0 ± 70000.0 | −8.29 ± 0.03 | −8.4 ± 0.3 | −0.1 ± 0.3 | 0.94 |
| clip-g5 | 8-azaniumyloctylammonium | 150000000.0 ± 30000000.0 | −11.1 ± 0.1 | −11.4 ± 0.4 | −0.3 ± 0.4 | 0.89 |
| clip-g16 | 10-azaniumyldecylammonium | 300000000.0 ± 100000000.0 | −11.5 ± 0.2 | −11.2 ± 0.4 | 0.3 ± 0.4 | 0.89 |
| clip-g17 | 12-azaniumyldodecylammonium | 500000000.0 ± 300000000.0 | −11.8 ± 0.4 | −10.4 ± 0.3 | 1.4 ± 0.5 | 0.97 |
| clip-g9 | 1-adamantylammonium | 360000.0 ± 30000.0 | −7.57 ± 0.05 | −4.8 ± 0.2 | 2.8 ± 0.2 | 0.95 |
| clip-g6 | 1-adamantyl(trimethyl)ammonium | 11000000.0 ± 2000000.0 | −9.6 ± 0.1 | −10.2 ± 0.4 | −0.6 ± 0.4 | 0.83 |
| clip-g11 | 1-(1-adamantyl)ethanamine | 4100000.0 ± 600000.0 | −9.02 ± 0.08 | −7.4 ± 0.3 | 1.6 ± 0.3 | 0.85 |
| clip-g10 | decahydro-2, 8, 4, 6-(epibutane[1, 2, 3, 4]tetrayl)naphthalene-2, 6-diaminium | 1000000.0 ± 100000.0 | −8.17 ± 0.08 | −5.8 ± 0.2 | 2.3 ± 0.2 | 0.99 |
| clip-g8 | [4-(azaniumylmethyl)phenyl]methylammonium | 8500000.0 ± 700000.0 | −9.45 ± 0.05 | −10.6 ± 0.3 | −1.1 ± 0.3 | 0.90 |
| clip-g18 | 1-methyl-4-(1-methylpyridin-1-ium-4-yl)pyridin-1-ium | 54000000.0 ± 8000000.0 | −10.55 ± 0.09 | −12.4 ± 0.4 | −1.8 ± 0.4 | 0.95 |
| clip-g19 | 4-(1, 1-dimethylpiperidin-1-ium-4-yl)-1, 1-dimethyl-piperidin-1-ium | 360000000.0 ± 80000000.0 | −11.7 ± 0.1 | −13.6 ± 0.4 | −2.0 ± 0.5 | 0.79 |
| clip-g7 | (4-azaniumylcyclohexyl)ammonium | 59000.0 ± 5000.0 | −6.5 ± 0.05 | −6.7 ± 0.3 | −0.2 ± 0.3 | 0.83 |
| OA-g1 | hexanoate | 4400.0 ± 200.0 | −4.97 ± 0.02 | −5.54 ± 0.1 | −0.57 ± 0.07 | 1.00 |
| OA-g2 | 4-chlorobenzoate | 116000.0 ± 5000.0 | −6.91 ± 0.02 | −9.6 ± 0.3 | −2.6 ± 0.2 | 1.00 |
| OA-g3 | (4 S)-4-isopropenylcyclohexene-1-carboxylate | 870000.0 ± 40000.0 | −8.1 ± 0.02 | −12.0 ± 0.02 | −3.9 ± 0.02 | 1.00 |
| OA-g4 | (3 S)-3, 7-dimethyloct-6-enoate | 91000.0 ± 7000.0 | −6.76 ± 0.05 | −6.7 ± 0.2 | 0.1 ± 0.1 | 1.00 |
| OA-g5 | trimethyl-2-phenylethanaminium | 3000.0 ± 100.0 | −4.73 ± 0.02 | −7.48 ± 0.05 | −2.75 ± 0.05 | 1.00 |
| OA-g6 | hexyl(trimethyl)ammonium | 4400.0 ± 200.0 | −4.97 ± 0.02 | −7.3 ± 0.3 | −2.3 ± 0.3 | 1.00 |
| OA-g7 | trimethyl-(4-methylcyclohexyl)ammonium | 28000.0 ± 2000.0 | −6.07 ± 0.05 | −5.7 ± 0.2 | 0.3 ± 0.1 | 1.00 |
| OA-g8 | 1-adamantyl(trimethyl)ammonium | 1110000.0 ± 40000.0 | −8.25 ± 0.02 | −7.8 ± 0.2 | 0.4 ± 0.1 | 1.00 |
| exoOA-g1 | hexanoate | ND ± ND | ND ± ND | ND ± ND | ND ± ND | 1.00 |
| exoOA-g2 | 4-chlorobenzoate | 9.0 ± 4.0 | −1.3 ± 0.3 | ND ± ND | ND ± ND | 1.00 |
| exoOA-g3 | (4 S)-4-isopropenylcyclohexene-1-carboxylate | 300.0 ± 40.0 | −3.37 ± 0.07 | −6.0 ± 0.1 | −2.65 ± 0.07 | 1.00 |
| exoOA-g4 | (3 S)-3, 7-dimethyloct-6-enoate | 440.0 ± 20.0 | −3.61 ± 0.02 | −7.3 ± 0.7 | −3.7 ± 0.7 | 1.00 |
| exoOA-g5 | trimethyl-2-phenylethanaminium | 12100.0 ± 500.0 | −5.57 ± 0.02 | −6.17 ± 0.02 | −0.6 ± 0.02 | 1.00 |
| exoOA-g6 | hexyl(trimethyl)ammonium | 18900.0 ± 800.0 | −5.83 ± 0.02 | −3.25 ± 0.02 | 2.58 ± 0.02 | 1.00 |
| exoOA-g7 | trimethyl-(4-methylcyclohexyl)ammonium | 130000.0 ± 20000.0 | −6.98 ± 0.1 | −4.97 ± 0.07 | 2.01 ± 0.05 | 1.00 |
| exoOA-g8 | 1-adamantyl(trimethyl)ammonium | 420000.0 ± 20000.0 | −7.67 ± 0.02 | −5.04 ± 0.05 | 2.63 ± 0.02 | 1.00 |
| bCD-g1 | trans-4-methylcyclohexanol | 2100.0 ± 100.0 | −4.52 ± 0.03 | −2.6 ± 0.2 | 2.0 ± 0.2 | 0.88 |
| bCD-g2 | R-rimantadine | 35000.0 ± 3000.0 | −6.2 ± 0.04 | −10.4 ± 0.7 | −4.2 ± 0.7 | 1.00 |
| MGLab_8-g1 | trans-4-methylcyclohexanol | 260.0 ± 20.0 | −3.3 ± 0.05 | −1.8 ± 0.4 | 1.5 ± 0.4 | 0.89 |
| MGLab_8-g2 | R-rimantadine | 830.0 ± 50.0 | −3.98 ± 0.04 | −6.9 ± 0.5 | −2.9 ± 0.5 | 1.03 |
| MGLab_9-g1 | trans-4-methylcyclohexanol | 210.0 ± 20.0 | −3.17 ± 0.05 | −2.7 ± 0.8 | 0.4 ± 0.8 | 0.88 |
| MGLab_9-g2 | R-rimantadine | 700.0 ± 40.0 | −3.88 ± 0.03 | −9.0 ± 0.6 | −5.2 ± 0.6 | 1.00 |
| MGLab_19-g1 | trans-4-methylcyclohexanol | 210.0 ± 20.0 | −3.18 ± 0.04 | −2.1 ± 0.2 | 1.1 ± 0.2 | 0.83 |
| MGLab_19-g2 | R-rimantadine | 320.0 ± 20.0 | −3.41 ± 0.04 | −11.0 ± 1.0 | −8.0 ± 1.0 | 0.94 |
| MGLab_23-g1 | trans-4-methylcyclohexanol | 220.0 ± 20.0 | −3.2 ± 0.05 | −3.0 ± 1.0 | 0.0 ± 1.0 | 0.76 |
| MGLab_23-g2 | R-rimantadine | 1510.0 ± 90.0 | −4.33 ± 0.04 | −7.6 ± 0.5 | −3.3 ± 0.5 | 0.96 |
| MGLab_24-g1 | trans-4-methylcyclohexanol | 280.0 ± 20.0 | −3.34 ± 0.05 | −1.6 ± 0.2 | 1.7 ± 0.2 | 0.92 |
| MGLab_24-g2 | R-rimantadine | 1100.0 ± 70.0 | −4.15 ± 0.04 | −8.6 ± 0.6 | −4.5 ± 0.6 | 1.03 |
| MGLab_34-g1 | trans-4-methylcyclohexanol | 700.0 ± 100.0 | −3.85 ± 0.09 | −3.7 ± 0.3 | 0.1 ± 0.3 | 0.81 |
| MGLab_34-g2 | R-rimantadine | 11000.0 ± 7000.0 | −5.5 ± 0.4 | −9.0 ± 2.0 | −3.0 ± 2.0 | 0.99 |
| MGLab_35-g1 | trans-4-methylcyclohexanol | 2300.0 ± 200.0 | −4.58 ± 0.05 | −4.5 ± 0.3 | 0.1 ± 0.3 | 0.85 |
| MGLab_35-g2 | R-rimantadine | 27000.0 ± 2000.0 | −6.04 ± 0.04 | −7.3 ± 0.5 | −1.2 ± 0.5 | 0.78 |
| MGLab_36-g1 | trans-4-methylcyclohexanol | 200.0 ± 10.0 | −3.15 ± 0.04 | −3.0 ± 0.3 | 0.1 ± 0.3 | 0.87 |
| MGLab_36-g2 | R-rimantadine | 350.0 ± 20.0 | −3.48 ± 0.04 | −11.0 ± 1.0 | −7.0 ± 1.0 | 0.84 |
All quantities are reported as point estimate ± statistical error from the ITC data fitting procedure. The upper bound (1%) was used for errors reported to be < 1%. We also included a 3% relative uncertainty in the titrant concentration assuming the stoichiometry coefficient to be fitted to the ITC data [4] for the Isaacs (TrimerTrip) and Gilson (cyclodextrin derivatives) datasets, where concentration error had not been factored in to the original error estimates. For the OA/exo-OA sets, provided uncertainties already included concentration error. In some cases, exoOA-g1 binding constants were not detected (ND) by ITC or H NMR. Binding of guest g2 to exoOA was very weak so only H NMR spectroscopy could produce reliable free energy data. The stoichiometry for each host-guest system is defined in n
Statistical errors were propagated from the Ka measurements.
All experiments were performed at 298 K.
Units of M−2.
Units of M−3.
6.3. Statistical/Error Analysis of Challenge
In general, analysis was performed using Python scripts deposited in the SAMPL7 GitHub repository adapted from previous SAMPL challenges such as the SAMPL6 host-guest challenge, so analysis is extremely similar to what was performed there [97]. All binding free energy prediction sets were compared with experimental data via the following statistical measurements: RMSE (root mean-squared error), R2 (coefficient of determination), τ (Kendall Tau correlation coefficient), m (linear regression slope), ME (mean error), and MAE (mean absolute error). Any uncertainty in the error metrics was determined via bootstrapping with replacement, as described previously [3, 4]. Methods for each host-guest system dataset (TrimerTrip, GDCC, and CD derivatives) were only evaluated and compared within the same dataset. In addition, we computed RMSE and ME of methods to each individual host-guest system to ascertain problematic molecules.
The statistical evaluation was separated into two categories, ranked and non-ranked, as described in Section 5.4. All ranked submissions’ evaluation data, plots, and tables are available at the SAMPL7 GitHub repository (see https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Accuracy_ranked). Statistical analysis was carried out with and without optional guests. Optional guests were those for which experimental data was already available. In addition, one very poorly performing CD ranked method was not included in much of our analysis because its performance was so poor that it would have made most other methods appear virtually identical, but was included in the non-ranked analysis and in Table S1 and S2 (sid 15 or ID AM1-BCC/MD/GAFF/TIP4PEW/QMMM). All non-ranked evaluation data, plots, and tables are available in the SAMPL7 GitHub repository (see https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Reference/Accuracy), as is the raw data and the analysis tools.
7. Results and Discussion
We find that predictive accuracy of binding free energies for host-guests, in terms of RMSE, is comparable to previous SAMPL challenges. However, we do see moderate improvement in some cases. For instance, binding affinity predictions of hosts in the acyclic cucurbituril category such as H1 featured in SAMPL3 [15], CBClip from SAMPL5 [70], and TrimerTrip (SAMPL7) had a mean RMSE of 7.07, 5.87, and 4.15 kcal/mol, respectively. The best performing methods for acyclic cucurbiturils achieved RMSEs as low as 1.60, 3.40, and 1.58 kcal/mol. The accuracy of methods used for acyclic cucurbiturls similar to TrimerTrip show improvement across SAMPL challenges. On the other hand, methods used in predicting binding free energies for systems in the cavitand category OA/TEMOA (SAMPL5), OA/TEMOA (SAMPL6), and OA/exoOA (SAMPL7) show high variation from challenge to challenge. The RMSE across challenges shows similar or slightly poorer accuracy on average. However, the best performing methods in the cavitand category usually do better than methods in other categories, or at least as well, by RMSE, and achieve R2 values well above 0.7 kcal/mol. This is more apparent in SAMPL6 and SAMPL7, partly from methods using the extensive cavitand data available from previous challenges to apply corrections. Comparing accuracy of ranked and non-ranked methods, on average ranked methods performed better (Figure S4). In addition, we find participation in the SAMPL host-guest challenges to be fairly consistent over time with approximately 30 submissions (the exact submission amount shown in parenthesis next to the SAMPL challenge) each in SAMPL3 (29), SAMPL5 (31), and SAMPL7 (30), except the substantial increase to 80 submissions for SAMPL6.
Out of the 30 participant submissions in SAMPL7, 7 were for TrimerTrip, 16 for the GDCCs, and 9 for the CD derivatives. The TrimerTrip submissions included 3 ranked and 4 non-ranked, GDCCs included 4 ranked and 12 non-ranked, and CD-derivatives had 3 ranked and 6 non-ranked (Figure 5). For a large portion of methods submitted, docking was used to obtain starting structures, and one submission used self association molecular dynamics (SA-MD) [96]. General classical fixed charge force fields were commonly used, as has become common in SAMPL host-guest challenges (see Section 6.1.2 for methods submitted to SAMPL7 host-guest challenge). Alchemical free energy techniques were employed in many cases, with analysis done via thermodynamic integration (TI) and Bennett acceptance ratio (BAR) for equilibrium calculations. Nonequilibrium approaches were also employed, such as using the fast switching double annihilation method (FSDAM) approach [93, 98]. Here we introduce the results for all ranked methods separated by host-guest system dataset, give statistics for binding of individual host-guest systems averaged across methods, and lastly examine analysis of non-ranked methods including our own reference calculations.
Figure 5. SAMPL7 submission breakdown.
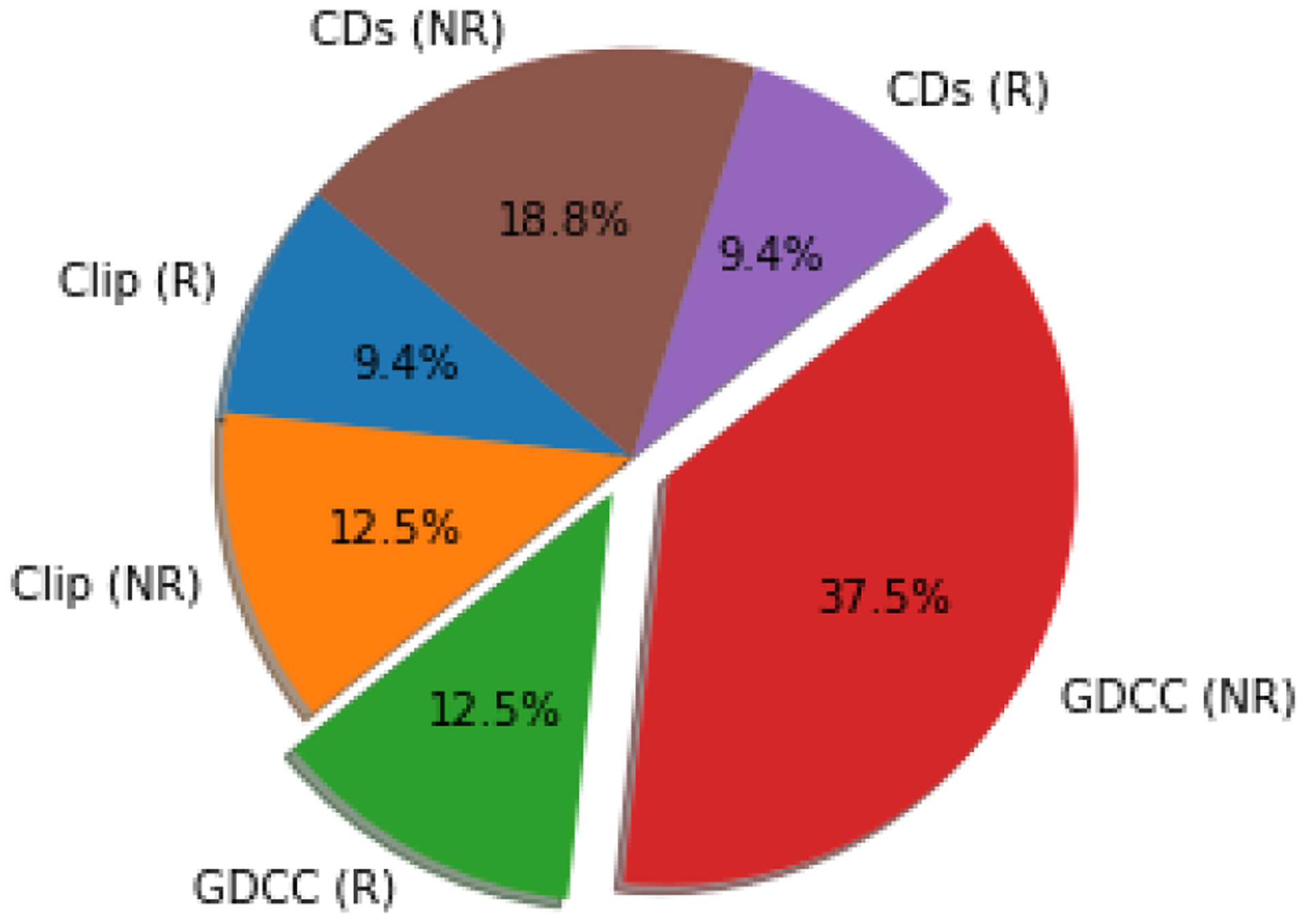
The SAMPL7 challenge saw 7 TrimerTrip submissions, of which 3 were ranked (blue) and 4 were non-ranked (orange). There were 16 GDCC submissions, with 4 ranked (green) and 12 nonranked (red), and 7 CD submissions, with 3 ranked (purple) and 4 nonranked (brown).
7.1. Ranked Submissions
7.1.1. TrimerTrip
Statistical analysis of the 3 sets of ranked absolute binding free energy predictions for the TrimerTrip dataset are summarized in Table S2 and Figure 6. All methods used explicit solvent. These submissions used nominally very similar free energy techniques (though with differences in protocol) but force fields were substantially different. Fixed-charge approaches used the GAFF and GAFF2 force fields, along with the TIP3P or OPC water models (the method called MD/DOCKING/GAFF/xtb-GNF/ used GAFF with TIP3P, while FSDAM/GAFF2/OPC3 used GAFF2 with OPC). The third submission in this category, AMOEBA/DDM/BAR, used the AMOEBA force field, which explicitly treats polarizability and includes multipoles; this AMOEBA-based approach was consistently the top performing method with values of 2.76 kcal/mol, 0.50, 1.25, and 0.47 in terms of RMSE, R2, slope (m), and τ respectively (Figure 6). The mean error (ME) for this AMOEBA submission was modestly larger in magnitude than one of the other ranked submissions, but in all other respects its performance was superior. Full statistics are in Table 3. AMOEBA-based approaches also perform well in the GDCC category, as we will see below.
Figure 6. TrimerTrip Error Metrics for Ranked Methods.
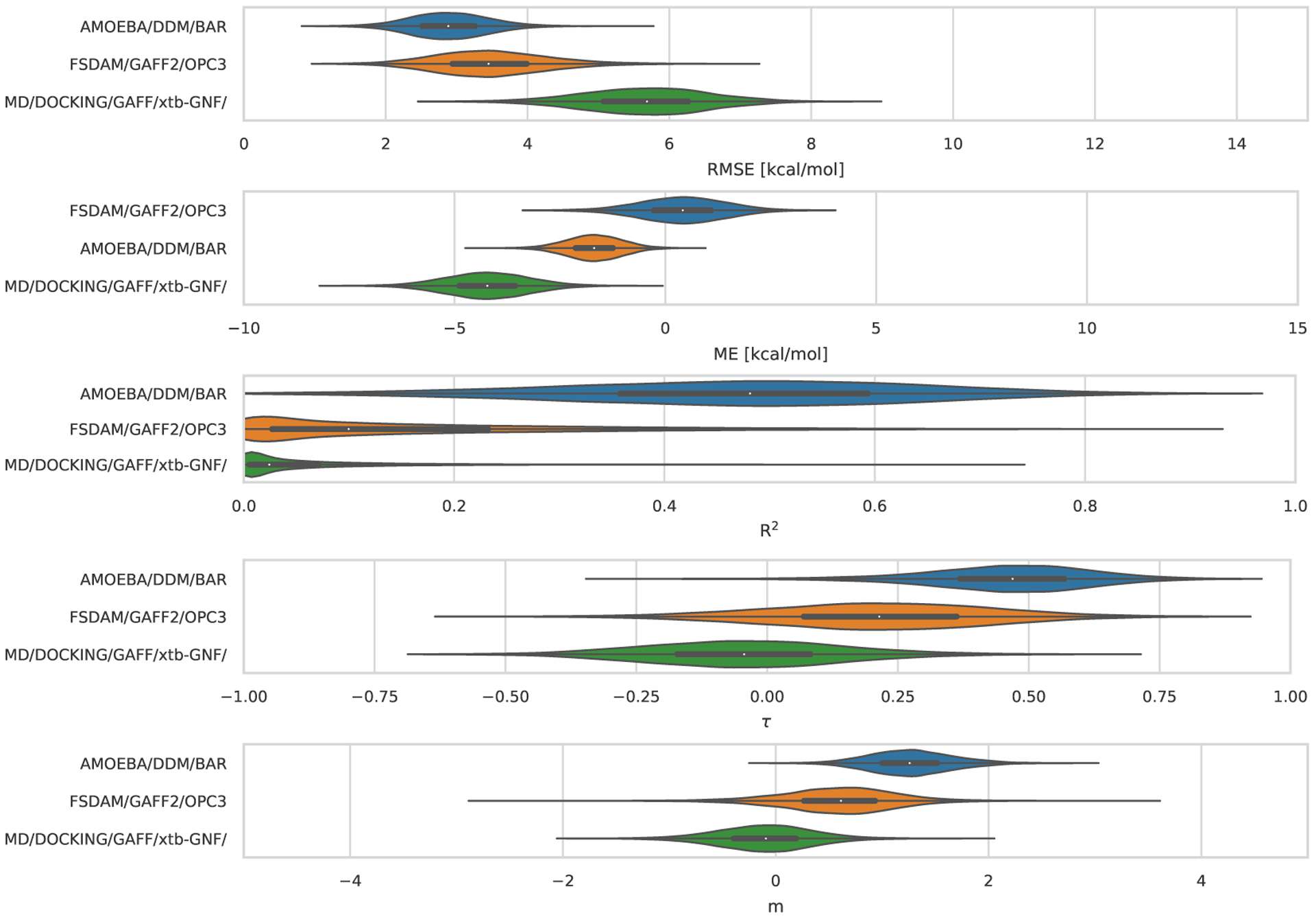
Shown is the distribution of performance for TrimerTrip submissions, ordered based on the median for each metric. The median is indicated by the white circle in the violin plots. The violin plots were generated by bootstrapping samples with replacement (including experimental uncertainties), and the plots describe the shape of the sampling distribution for each prediction. The black horizontal bar represents the first and third quartiles. From top to bottom the error metrics are RMSE, ME, R2, τ, and slope (m).
Table 3. Error metrics for all (ranked and non-ranked) SAMPL7 methods for all host-guest systems.
The root mean square error (RMSE), mean absolute error (MAE), signed mean error (ME), coefficient of correlation (R2), slope (m), and Kendall’s rank correlation coefficient (Tau) were computed via bootstrapping with replacement. Shown are results for individual host categories, as well as the artificially separated exoOA sub-dataset. Statistics do not include optional host-guest systems OA-g1, OA-g2, OA-g3 OA-g4, OA-g5, OA-g6, bCD-g1, and bCD-g2. Each method has an assigned unique submission ID (sid). Table S1 contains statistical data for submissions including optional system predictions.
| ID | sid | RMSE [kcal/mol] | MAE [kcal/mol] | ME [kcal/mol] | R2 | m | τ |
|---|---|---|---|---|---|---|---|
| TrimerTrip | |||||||
| AMOEBA/DDM/BAR/ALT-2 | 9 | 1.58 [1.19, 2.56] | 1.39 [0.95, 2.23] | −0.36 [−1.36, 0.68] | 0.63 [0.18, 0.83] | 1.14 [0.54, 1.76] | 0.60 [0.17, 0.80] |
| AMOEBA/DDM/BAR-ALT1 | 8 | 1.68 [1.28, 2.64] | 1.56 [1.03, 2.34] | −0.70 [−1.71, 0.32] | 0.70 [0.26, 0.88] | 1.28 [0.70, 1.88] | 0.67 [0.23, 0.85] |
| AMOEBA/DDM/BAR | 6 | 2.76 [1.83, 3.98] | 2.12 [1.35, 3.33] | −1.69 [−2.98, −0.44] | 0.50 [0.13, 0.77] | 1.25 [0.53, 2.06] | 0.47 [0.12, 0.74] |
| FSDAM/GAFF2/OPC3 | 4 | 2.97 [2.11, 5.13] | 2.24 [1.62, 4.22] | 0.43 [−1.59, 2.33] | 0.12 [0.00, 0.56] | 0.60 [−0.51, 1.60] | 0.24 [−0.23, 0.61] |
| MD/DOCKING/GAFF/xtb-GNF/ | 5 | 5.65 [3.87, 7.36] | 4.51 [3.01, 6.40] | −4.23 [−6.19, −2.23] | 0.00 [0.00, 0.26] | −0.10 [−1.02, 0.80] | −0.05 [−0.41, 0.35] |
| Docking/GAFF/YANK_REF | REF2 | 7.18 [5.63, 8.71] | 6.57 [5.16, 8.10] | −6.57 [−8.09, −5.16] | 0.11 [0.00, 0.59] | 0.57 [−0.56, 1.55] | 0.12 [−0.35, 0.56] |
| Docking/GAFF/YANK_REF_2 | REF3 | 7.21 [5.73, 8.75] | 6.63 [5.26, 8.13] | −6.63 [−8.12, −5.26] | 0.12 [0.00, 0.59] | 0.57 [−0.55, 1.54] | 0.12 [−0.34, 0.57] |
| GDCC-OA and exoOA | |||||||
| RESP/GAFF/MMPBSA-Cor | 20 | 1.24 [0.73, 2.45] | 0.95 [0.57, 2.13] | 0.94 [−0.12, 1.99] | 0.94 [0.10, 0.97] | 0.65 [0.18, 1.14] | 0.83 [0.03, 1.00] |
| AMOEBA/DDM/BAR | 29 | 1.25 [0.68, 2.55] | 0.92 [0.54, 2.13] | −0.36 [−1.59, 0.83] | 0.80 [0.36, 0.97] | 1.11 [0.58, 1.94] | 0.72 [0.17, 1.00] |
| AMOEBA/DDM/BAR_2 | 30 | 1.78 [0.86, 3.24] | 1.31 [0.67, 2.70] | −0.62 [−2.09, 0.77] | 0.55 [0.04, 0.96] | 0.87 [0.14, 1.92] | 0.50 [−0.09, 1.00] |
| xtb-GNF/Machine Learning/CORINA MD | 28 | 2.26 [1.38, 3.43] | 1.91 [1.09, 3.08] | 0.37 [−1.27, 2.13] | 0.01 [0.00, 0.78] | 0.04 [−0.58, 0.50] | 0.06 [−0.68, 0.78] |
| AMOEBA/DDM/BAR_3 | 31 | 2.32 [1.42, 3.58] | 2.05 [1.13, 3.22] | −0.29 [−1.95, 1.52] | 0.61 [0.21, 0.92] | 1.30 [0.54, 2.41] | 0.78 [0.24, 1.00] |
| Docking/GAFF/YANK_REF | REF4 | 4.05 [1.54, 5.88] | 2.90 [1.21, 4.93] | 2.40 [0.41, 4.67] | 0.12 [0.00, 0.65] | −0.30 [−1.06, 0.53] | −0.11 [−0.70, 0.60] |
| B2PLYPD3/SMD_QZ-R | 23 | 4.52 [2.55, 6.41] | 3.70 [1.95, 5.69] | 3.15 [0.85, 5.50] | 0.49 [0.02, 0.92] | 1.43 [−0.17, 2.92] | 0.37 [−0.33, 0.88] |
| B2PLYPD3/SMD_QZ-NR | 24 | 4.64 [2.77, 6.46] | 3.95 [2.23, 5.83] | 2.69 [0.06, 5.33] | 0.58 [0.03, 0.96] | 1.84 [−0.24, 3.28] | 0.39 [−0.31, 0.93] |
| FSDAM/GAFF2/OPC3 | 14 | 5.07 [3.12, 8.84] | 4.69 [2.53, 7.86] | −0.79 [−5.32, 3.40] | 0.77 [0.01, 0.94] | −1.26 [−2.65, 0.18] | −0.59 [−1.00, 0.24] |
| B2PLYPD3/SMD_TZ | 22 | 5.08 [3.04, 7.03] | 4.22 [2.39, 6.37] | 3.36 [0.67, 6.04] | 0.58 [0.02, 0.96] | 1.85 [−0.29, 3.31] | 0.39 [−0.33, 0.94] |
| RESP/GAFF/MMPBSA/Nmode | 18 | 5.84 [4.47, 7.31] | 5.60 [4.20, 7.03] | −5.60 [−7.03, −4.20] | 0.81 [0.44, 0.98] | 1.40 [0.79, 2.40] | 0.83 [0.31, 1.00] |
| RESP/GAFF/MMPBSA | 19 | 8.07 [6.96, 9.33] | 7.98 [6.81, 9.20] | 7.98 [6.81, 9.20] | 0.94 [0.54, 0.99] | 1.45 [0.96, 1.99] | 0.83 [0.39, 1.00] |
| B2PLYPD3/SMD_DZ | 21 | 8.13 [5.62, 10.34] | 7.17 [4.57, 9.75] | 7.17 [4.48, 9.75] | 0.55 [0.02, 0.96] | 1.80 [−0.36, 3.28] | 0.39 [−0.33, 0.94] |
| AM1-BCC/GAFF/MMPBSA | 17 | 10.96 [9.02, 12.80] | 10.61 [8.59, 12.59] | 10.61 [8.59, 12.59] | 0.91 [0.59, 0.99] | 2.12 [1.55, 2.83] | 0.89 [0.43, 1.00] |
| RESP/GAFF/MMGBSA | 16 | 11.85 [10.29, 13.47] | 11.68 [10.12, 13.29] | 11.68 [10.12, 13.29] | 0.88 [0.40, 0.99] | 1.69 [1.10, 2.36] | 0.78 [0.23, 1.00] |
| GDCC-exoOA | |||||||
| AMOEBA/DDM/BAR_2 | 30 | 1.23 [0.65, 2.53] | 1.02 [0.51, 2.25] | −0.13 [−1.47, 1.27] | 0.83 [0.39, 0.99] | 1.21 [0.56, 2.29] | 0.62 [0.16, 1.00] |
| AMOEBA/DDM/BAR | 29 | 1.27 [0.56, 2.72] | 0.91 [0.45, 2.31] | −0.66 [−1.98, 0.61] | 0.81 [0.30, 0.99] | 1.05 [0.45, 2.12] | 0.71 [0.05, 1.00] |
| RESP/GAFF/MMPBSA-Cor | 20 | 1.32 [0.68, 2.65] | 1.03 [0.54, 2.34] | 1.01 [−0.18, 2.20] | 0.95 [0.04, 0.99] | 0.61 [0.04, 1.20] | 0.81 [−0.14, 1.00] |
| AMOEBA/DDM/BAR_3 | 31 | 1.72 [0.93, 3.04] | 1.57 [0.75, 2.77] | −1.44 [−2.66, −0.19] | 0.79 [0.15, 0.99] | 0.80 [0.22, 1.72] | 0.81 [−0.05, 1.00] |
| xtb-GNF/Machine Learning/CORINA MD | 28 | 2.43 [1.40, 3.71] | 2.11 [1.10, 3.42] | 0.82 [−1.12, 2.77] | 0.00 [0.00, 0.91] | 0.01 [−0.81, 0.57] | 0.05 [−0.78, 1.00] |
| Docking/GAFF/YANK_REF | REF4 | 4.48 [1.56, 6.43] | 3.25 [1.10, 5.65] | 2.60 [0.06, 5.40] | 0.37 [0.03, 0.95] | −0.58 [−1.56, 0.08] | −0.43 [−1.00, 0.33] |
| B2PLYPD3/SMD_QZ-R | 23 | 4.76 [2.26, 6.93] | 3.90 [1.81, 6.26] | 3.50 [0.91, 6.12] | 0.72 [0.24, 0.99] | 1.97 [0.88, 3.77] | 0.59 [−0.06, 1.00] |
| FSDAM/GAFF2/OPC3 | 14 | 4.85 [2.61, 8.41] | 4.38 [2.13, 7.58] | 0.62 [−3.93, 5.08] | 0.82 [0.01, 0.99] | −1.24 [−2.89, 0.30] | −0.59 [−1.00, 0.33] |
| B2PLYPD3/SMD_QZ-NR | 24 | 4.90 [2.64, 6.93] | 4.23 [2.23, 6.33] | 2.91 [−0.26, 5.90] | 0.80 [0.26, 0.99] | 2.46 [0.99, 3.87] | 0.62 [0.00, 1.00] |
| B2PLYPD3/SMD_TZ | 22 | 5.36 [2.93, 7.56] | 4.57 [2.40, 6.98] | 3.60 [0.40, 6.66] | 0.81 [0.24, 0.99] | 2.48 [0.90, 3.84] | 0.62 [−0.05, 1.00] |
| RESP/GAFF/MMPBSA/Nmode | 18 | 6.28 [4.78, 7.92] | 6.09 [4.54, 7.71] | −6.09 [−7.71, −4.54] | 0.76 [0.26, 0.99] | 1.26 [0.47, 2.43] | 0.81 [0.11, 1.00] |
| RESP/GAFF/MMPBSA | 19 | 7.59 [6.37, 8.90] | 7.53 [6.25, 8.79] | 7.53 [6.25, 8.79] | 0.95 [0.48, 1.00] | 1.36 [0.74, 1.96] | 0.81 [0.29, 1.00] |
| B2PLYPD3/SMD_DZ | 21 | 8.41 [5.40, 10.95] | 7.42 [4.31, 10.43] | 7.42 [4.23, 10.42] | 0.79 [0.22, 0.99] | 2.44 [0.84, 3.81] | 0.62 [0.00, 1.00] |
| AM1-BCC/GAFF/MMPBSA | 17 | 10.05 [7.92, 12.08] | 9.73 [7.58, 11.84] | 9.73 [7.58, 11.84] | 0.93 [0.61, 1.00] | 2.06 [1.38, 2.94] | 0.90 [0.29, 1.00] |
| RESP/GAFF/MMGBSA | 16 | 11.11 [9.58, 12.68] | 11.00 [9.46, 12.56] | 11.00 [9.46, 12.56] | 0.96 [0.66, 1.00] | 1.67 [1.09, 2.38] | 0.90 [0.37, 1.00] |
| Cyclodextrin derivatives | |||||||
| FSDAM/GAFF2/OPC3_ranked | 12 | 1.28 [1.33, 3.48] | 1.04 [1.04, 2.96] | 0.63 [−0.83, 2.09] | 0.01 [0.00, 0.50] | 0.12 [−1.58, 2.32] | 0.21 [−0.44, 0.58] |
| Noneq/Alchemy/CGENFF | 26 | 1.62 [1.21, 2.39] | 1.44 [0.98, 2.13] | 1.12 [0.33, 1.88] | 0.05 [0.00, 0.41] | 0.26 [−0.67, 1.19] | 0.10 [−0.46, 0.51] |
| Noneq/Alchemy/consensus | 27 | 1.70 [1.28, 2.26] | 1.48 [1.03, 2.04] | 1.21 [0.52, 1.88] | 0.02 [0.00, 0.30] | 0.16 [−0.50, 0.96] | −0.02 [−0.43, 0.46] |
| FSDAM/GAFF2/OPC3_JB | 13 | 1.74 [1.50, 3.85] | 1.51 [1.18, 3.27] | 0.77 [−0.78, 2.34] | 0.00 [0.00, 0.48] | −0.08 [−1.92, 2.27] | 0.13 [−0.45, 0.56] |
| Noneq/Alchemy/GAFF | 25 | 1.94 [1.41, 2.69] | 1.66 [1.12, 2.38] | 1.30 [0.42, 2.15] | 0.00 [0.00, 0.29] | 0.06 [−0.73, 1.19] | 0.02 [−0.38, 0.44] |
| Docking/GAFF/YANK_REF | REF1 | 2.74 [1.88, 3.58] | 2.25 [1.49, 3.08] | 0.51 [−0.81, 1.88] | 0.17 [0.01, 0.52] | −1.11 [−2.14, −0.18] | −0.28 [−0.57, 0.05] |
| AM1-BCC/MD/GAFF/TIP4PEW/QMMM | 15 | 46.62 [22.85, 65.69] | 32.00 [17.92, 49.22] | 31.27 [16.89, 48.87] | 0.04 [0.00, 0.33] | 7.62 [−3.31, 30.72] | 0.24 [−0.13, 0.52] |
For this dataset, the AMOEBA/DDM/BAR method predicted 10/16 binding affinities within 2 kcal/mol, the majority of these being within 1 kcal/mol (as discussed in the SAMPL7 virtual workshop [43]; full data available in our GitHub repository). The outliers for this method were clip-g6, clip-g7, clip-g8, clip-g9, clip-g11, and clip-g17, of which binding affinities were predicted to be too unfavorable. The FSDAM/GAFF2/OPC3 method predicted 10/16 within 2 kcal/mol and host-guest system outliers were clip-g3, clip-g8, clip-g10, clip-g11, clip-g16, and clip-g18. The other ranked submission, MD/DOCKING/GAFF/xtb-GNF, predicted 5/16 binding affinities within 2 kcal/mol, with 4 of those 5 being within 1 kcal/mol. Interestingly, all of the predictions within 2 kcal/mol used starting poses generated not by docking, but by using SA-MD. The SA-MD approach makes the assumption that the host is not in its proper conformation and the host and guest are allowed to associate on their own [43, 96]. It would be interesting to see the predictive accuracy of this approach on the remaining TrimerTrip host-guest systems, and which systems if any prove to be troublesome.
Two of these methods, AMOEBA/DDM/BAR and MD/DOCKING/GAFF/xtb-GNF, tended to yield binding free energies which were too unfavorable while the FSDAM/GAFF2/OPC3 ranked method was too favorable (Figure 6). Thus, most predictions with errors larger in 2 kcal/mol in magnitude err in the direction of not predicting binding to be favorable enough, especially with AMOEBA/DDM/BAR and MD/DOCKING/GAFF/xtb-GNF are underpredicted (Figure 7). On the other hand, as shown by Figures 6 and 7, the FSDAM/GAFF2/OPC3 method errs in both directions more frequently.
Figure 7. Correlation plots for TrimerTrip ranked submissions.
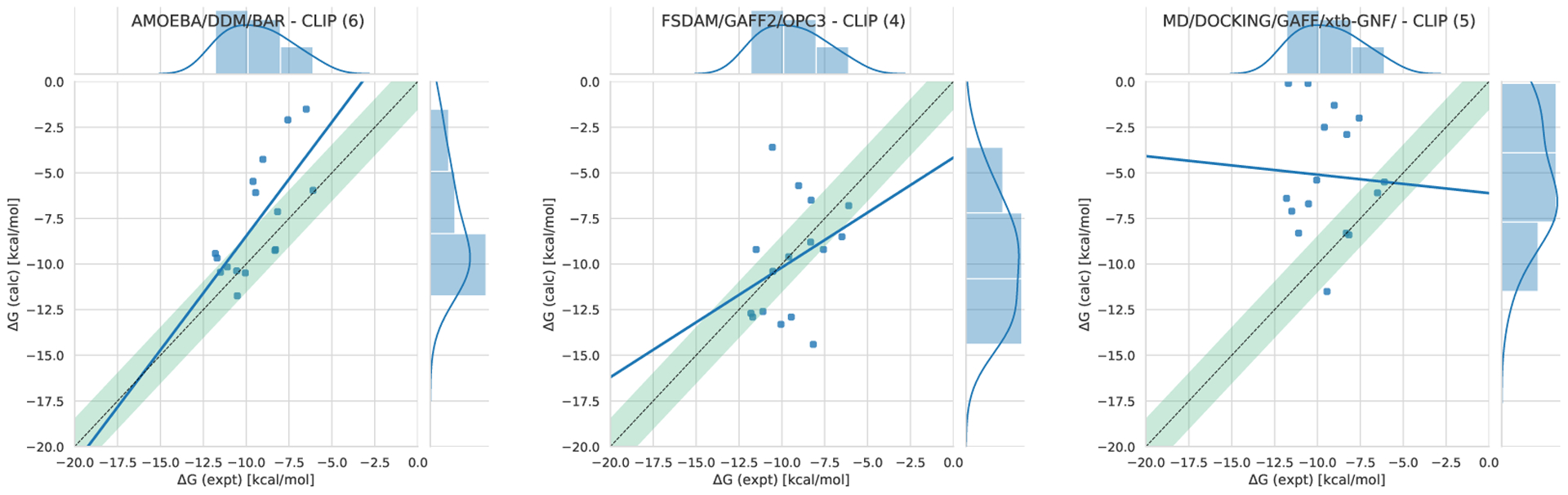
Shown are correlation plots comparing calculated versus experimental values for (Left to Right) AMOEBA/DDM/BAR, FSDAM/GAFF2/OPC3, and MD/DOCKING/GAFF/xtb-GNF ranked predictions for the TrimerTrip dataset. The R2 and slope for each ranked prediction were 0.50 and 1.25, 0.12 and 0.60, and 0.00 and −0.10 respectively.
We sought to determine whether some hosts/guests are particularly challenging to predict, across all ranked methods, so we examined the RMSE and ME by host and guest for ranked free energy predictions for all individual host-guest systems. This is shown in Figure 8. The ranked predictions of all methods for the TrimerTrip/”clip” host-guest systems (shown in blue in Figure 8) were in general the most problematic, especially clip-g6, clip-g9, clip-g10, clip-g11, clip-g18, and clip-g19 which had an RMSE of about 4 kcal/mol or greater. All of the guests with an adamantane moiety fall within this list of “problematic” molecules. The computed binding affinities for these host-guest systems are mostly too weak with ΔG ME of −2.5 kcal/mol or greater, the exception being clip-g10 which was predicted to be too favorable with a ΔG ME of 2 kcal/mol.
Figure 8. RMSE and ME statistics by host-guest system for ranked methods.
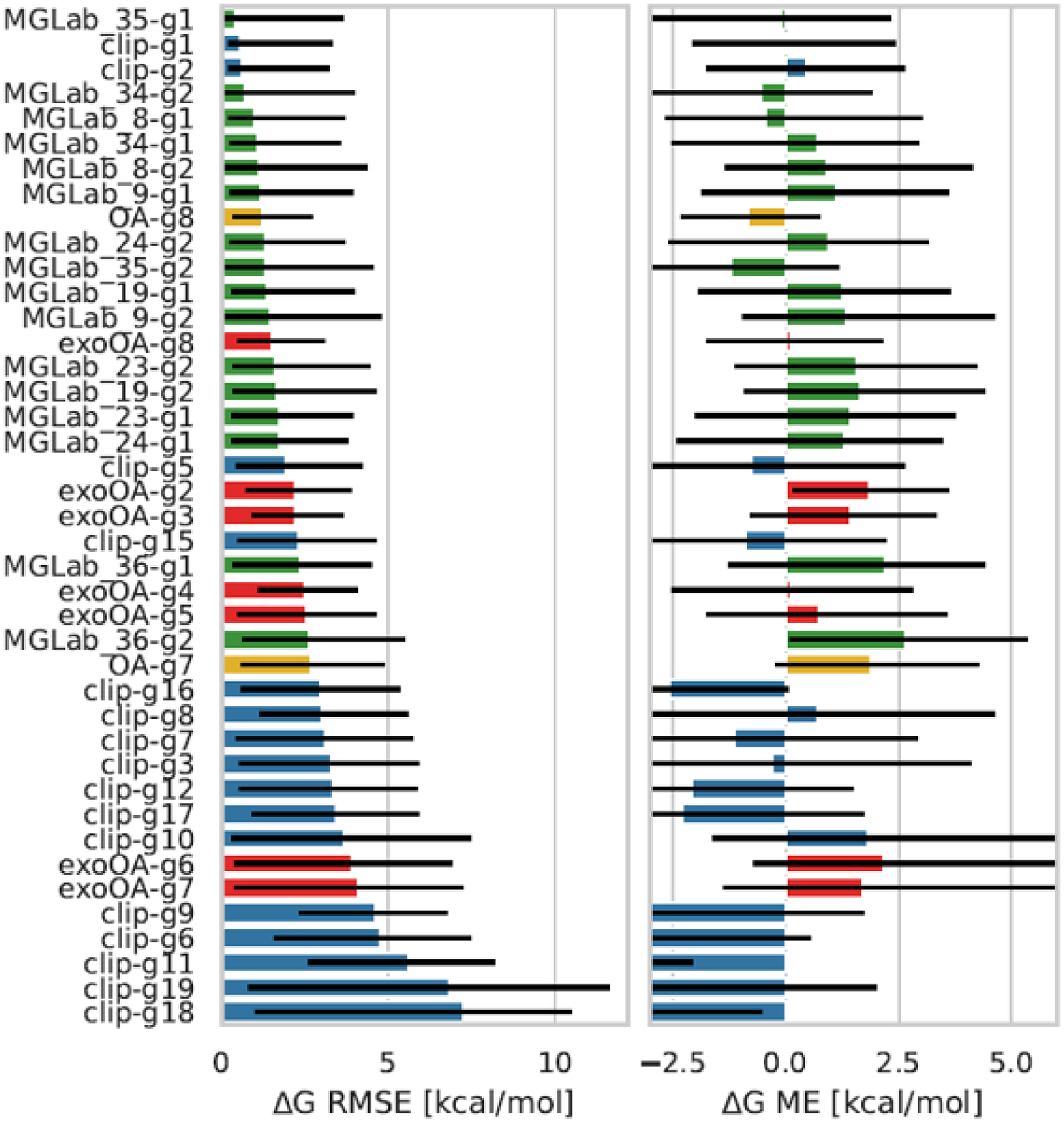
Shown are free energy error statistics by host-guest system, across methods/participants. The ΔG root mean square error (RMSE) and mean signed error (ME) were computed via bootstrapping with replacement (including experimental uncertainties) for all host-guest systems (except optional systems OA-g1, OA-g2, OA-g3, OA-g4, OA-g5, OA-g6, bCD-g1, and bCD-g2) and includes all ranked methods submitted (except the AM1-BCC/MD/GAFF/TIP4PEW/QMMM method for the cyclodextrin dataset which is omitted from this analysis because errors were so large for that method). The black error bars represent the 95-percentile bootstrap confidence intervals. The host-guest datasets for the SAMPL7 challenge were TrimerTrip (blue), GDCC (separated into OA (yellow) and exo-OA (red) sub-datasets to analyze each host-guest system), and cyclodextrin derivatives (green)
Overall for the TrimerTrip/clip-based systems, when we consider both ranked and non-ranked submissions, we believe the results suggest that any combination of the following may be limiting predictive accuracy: (a) chosen host conformer, (b) guest binding mode, (c) chosen energy model, and (d) water model. More specifically the general performance of the AMOEBA-based submissions pointed towards multipoles, polarization and/or shielding effects being important, especially as the guest becomes more hydrophobic, but the AMOEBA work (using multiple host conformers) [43, 94] also suggested host sampling could be an important issue since host conformers did not interconvert at nanosecond simulation timescales.
7.1.2. GDCC
The GDCC dataset, which includes OA and exo-OA host-guest systems, had the most submissions, probably because this host is familiar to many participants since it has formed part of a variety of previous SAMPL challenges. The statistical analysis of 4 sets of ranked methods are shown in Figure 9. For the entire GDCC dataset there was not a clear top performing method in terms of RMSE, R2, τ, and slope, but the RESP/GAFF/MMPBSA-Cor and AMOEBA/DDM/BAR methods were the two top performing methods. Again, the AMOEBA/DDM/BAR method emerged among the top performers, but unlike in the TrimerTrip challenge it is not the top method by all error metrics. The RESP/GAFF/MMPBSA-Cor method had the top ΔG RMSE, R2, and τ values of 1.24 kcal/mol, 0.94, and 0.83 respectively. Essentially, the latter approach seems to have done slightly better at ranking compounds for binding than the AMOEBA-based approach, but with a slope which is systematically incorrect. Full performance statistics are in Table S2.
Figure 9. GDCC Error Metrics for Ranked Methods.
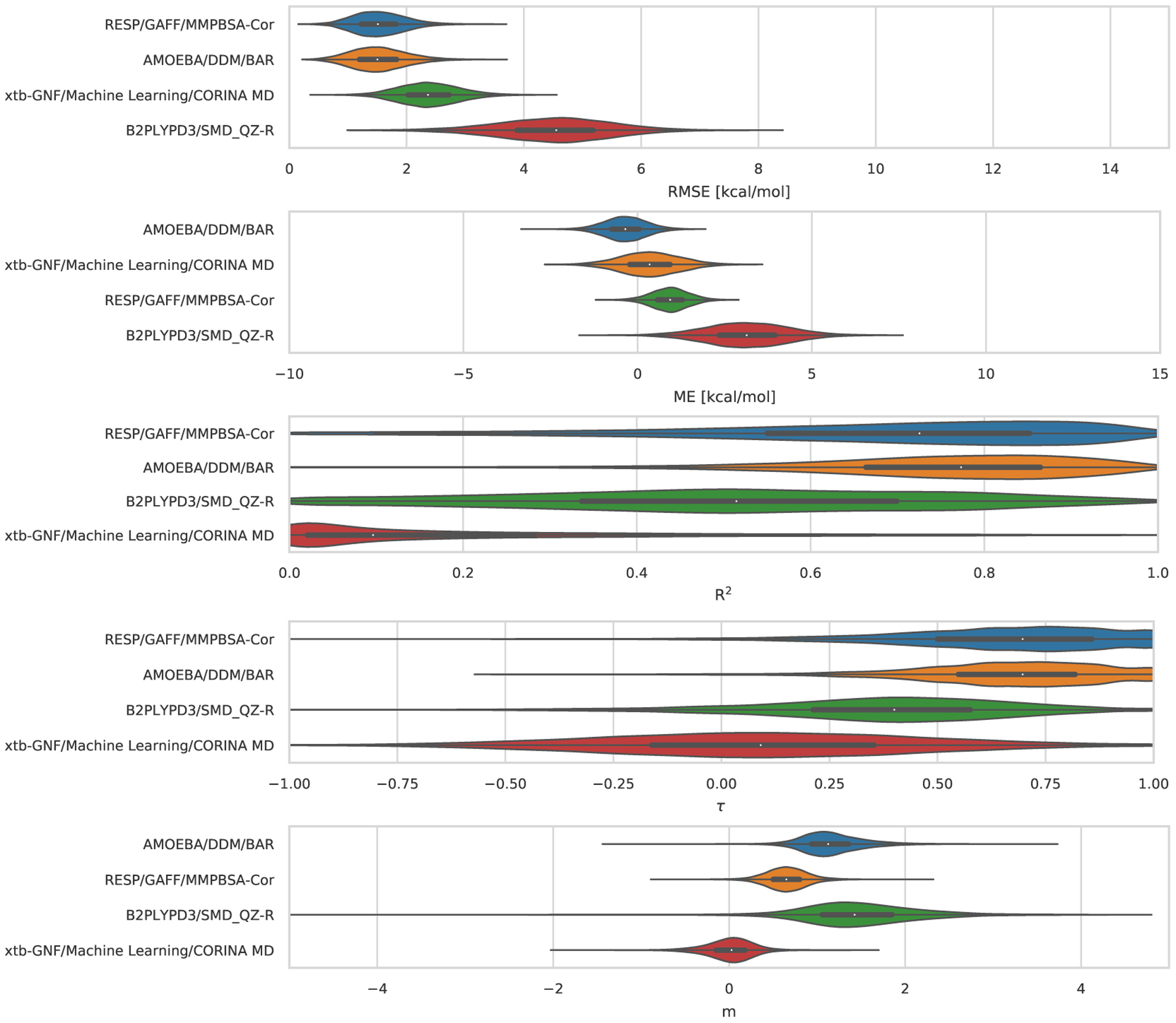
Shown is accuracy of GDCC submissions, with the median value for each metric indicated by the white circle in the violin plots. The violin plots were generated by bootstrapping samples with replacement, and the plots describe the shape of the sampling distribution for each prediction. The black horizontal bar represents the first and third quartiles. From top to bottom the error metrics are RMSE, ME, R2, τ, and slope (m).
Figure 10 shows performance of ranked methods relative to experiment. In general, the AMOEBA/DDM/BAR method tends to yield GDCC binding free energies which are too unfavorable, while all other ranked methods tend to predict binding free energies that are too favorable. The AMOEBA/DDM/BAR method gave calculated values that most directly correlated with experimental ones, as evidenced by a slope, m, of 1.11. With this approach, only exoOA-g4 had an error larger than 2 kcal/mol. The exoOA-g2 host-guest system was the only outlier for the RESP/GAFF/MMPBSA-Cor method, and the participants suggested this was likely due to guest g2 containing a chlorine atom. The QM-based method B2PLYPD3/SMD QZ-R had large prediction errors in more cases than any other method, in part because it overestimated the dynamic range of predictions and led to calculated binding free energies that were often far too negative. The xtb-GNF/MachineLearning/CORINA MD had smaller errors, but the correlation between calculated and experimental free energies was poor.
Figure 10. Correlation plots for GDCC (combined OA and exo-OA) and exo-OA ranked submissions.
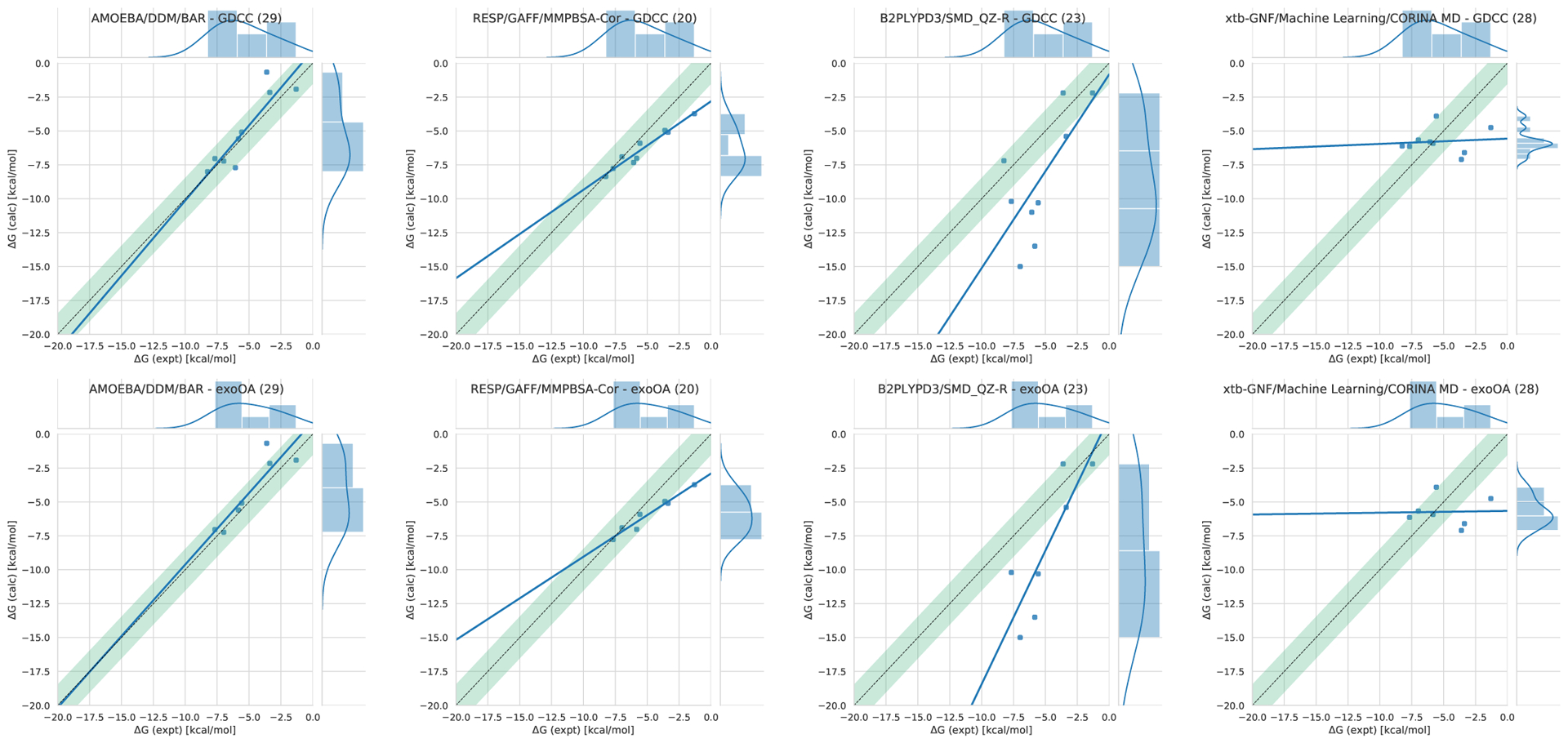
Shown are correlation plots comparing calculated and experimental values for (Left to Right) AMOEBA/DDM/BAR, RESP/GAFF/MMPBSA-Cor, B2PLYPD3/SMD_QZ-R, and xtb-GNF/Machine Learning/CORINA MD ranked predictions for GDCC (top row) and exo-OA (bottom row). The AMOEBA/DDM/BAR approach performed particularly well by a variety of metrics, as did RESP/GAFF/MMPBSA-Cor. The former had the slope closest to 1 and its RMS error was among the lowest, whereas the latter performed better on error and correlation metrics but had a slope which was systematically incorrect. (See Table 3)
The xtb-GNF/MachineLearning/CORINA MD, RESP/GAFF/MMPBSA-Cor, and AMOEBA/DDM/BAR methods have greater prediction errors for systems with negatively charged guests, which could potentially relate to the challenges alchemical methods have with charged guests (Section 3.1). Both xtb-GNF/MachineLearning/CORINA MD and RESP/GAFF/MMPBSA-Cor use the GAFF energy model, and its combination with explicit fixed charge water models typically results in predicted free energies that are too favorable (particularly, prior work has shown that GAFF with TIP3P leads to a consistent error in this direction for guests containing carboxylates and alcohols [49]). This is exactly the case here for systems with guests containing a carboxylate for the xtb-GNF/MachineLearning/CORINA MD method, where an AM1-BCC charging scheme, explicit TIP3P water, and GAFF energy model is used. The RESP/GAFF/MMPBSA-Cor method also used the GAFF energy model, but with implicit (PB/SA) water and a RESP charging scheme. During the SAMPL7 virtual workshop [43] the RESP/GAFF/MMPBSA-Cor participants noted that in their methodology comparison of RESP and AM1-BCC charging schemes, RESP resulted in better accuracy; it would be interesting to know if this holds true more generally. For the AMOEBA/DDM/BAR method, the single outlier was exoOA-g4, with a ΔG prediction error of 2.5 kcal/mol.
The Ponder group’s data suggests that the quality of torsional parameters for the upper rim’s diphenyl ether torsions can change predictions by 3 – 4 kcal/mol [43, 94]. In our reference calculations, we observe this guest folding in on itself and becoming effectively bulkier, which may mean host torsional parameters play a larger role for this particular guest.
On the other hand, the B2PLYPD3/SMD-QZ-R quantum method had larger prediction errors for guests with a positive charge. Particularly, the method’s ΔG prediction error for exoOA-g6 and exoOA-g7 was 10 kcal/mol, and 5 kcal/mol for exoOA-g5. Similarly, for the OA-g7 system which contains a positive guest, the method had a ΔG prediction error of 5 kcal/mol. These prediction errors substantially affected molecule statistics (Figure 8) for these systems.
7.1.3. Cyclodextrins
Method performance on the CD dataset is shown in Figure 12. Partly because of the narrow range of experimental binding free energies, we observe little difference in performance between the two better performing ranked methods. The third ranked submission AM1-BCC/MD/GAFF/TIP4PEW/QMMM method was not included in these plots because the range of binding free energies is so dramatically overestimated (Figure 13) that including it in the graph makes performance of the other two methods appear identical. In this analysis, optional systems bCD-g1 and bCD-g2 are not included, since these free energies have been previously reported. Of the two better performing techniques here (FSDAM/GAFF2/OPC3 and Noneq/Alchemy/consensus), performance was remarkably similar, as were the nonequilibrium free energy techniques employed. The third method – which typically predicted binding to be far too strong – was the AM1-BC/MD/GAFF/TIP4PEW/QMMM method, which had a ME and slope of 31.27 kcal/mol and 7.62 respectively. Since the GAFF force field is shared between this method and one of the more successful methods, it seems likely the larger error in this case is due to the QM/MM energy calculation approach.
Figure 12. Cyclodextrin derivatives error metrics for ranked methods.
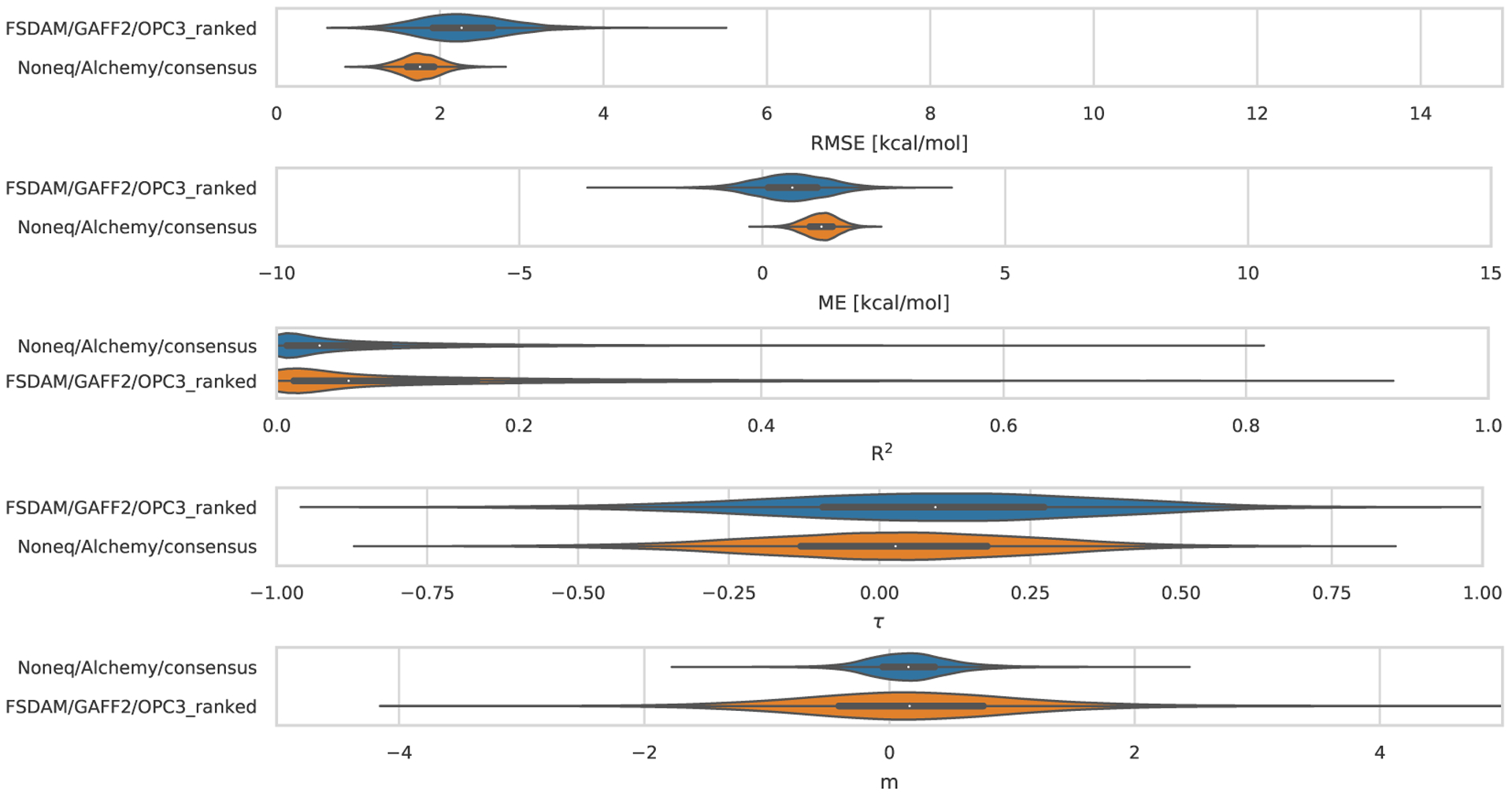
Shown are CD submissions ordered based on the median and is indicated by the white circle in the violin plots. The violin plots were generated by bootstrapping samples with replacement, and the plots describe the shape of the sampling distribution for each prediction. The black horizontal bar represents the first and third quartiles. From top to bottom the error metrics are RMSE, ME, R2, τ, and slope (m). AM1-BCC/GAFF/TIP4PEW/QMMM method was not included in these plots. In addition, the optional bCD-g1 and bCD-g2 host-guest systems are not included in this analysis.
Figure 13. Correlation plots for CD ranked submissions.
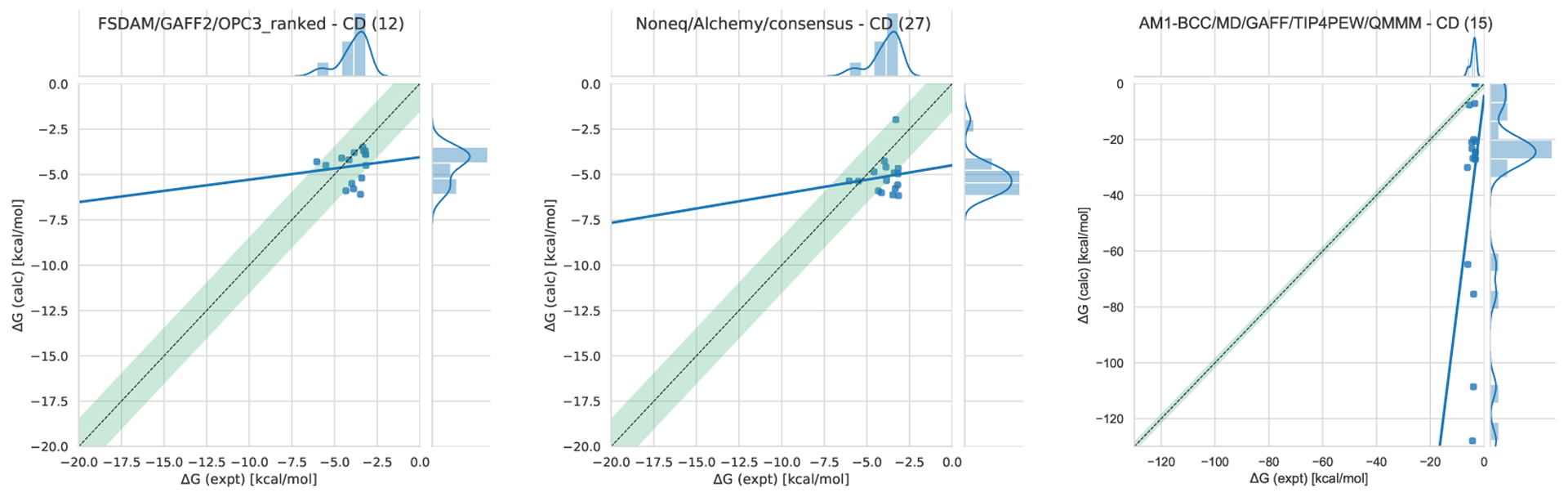
Shown are correlation plots comparing calculated versus experimental values for (Left to Right) FSDAM/GAFF2/OPC3, Noneq/Alchemy/consensus, and AM1-BCC/MD/GAFF/TIP4PEW ranked predictions for the CD dataset. The R2 and slope for each ranked predictions were 0.04 and 0.17, 0.03 and 0.18, and 0.04 and 7.62 respectively. Note: the optional bCD-g1 and bCD-g2 host-guest systems were not included in the analysis.
7.2. Non-Ranked Submissions
7.2.1. TrimerTrip
Several groups submitted multiple methods, often changing just one aspect of their approach. Such tests can help establish which aspects of an approach impact accuracy and how. Results for all submissions, ranked and non-ranked, are shown in Table 3. Results are listed in ascending order based on ΔG RMSE values. Here we discuss the analysis of these results and what we find that we can learn from them.
On TrimerTrip, two non-ranked submissions with the AMOEBA force field using the same approach, but alternate handling of host conformations (AMOEBA/DDM/BAR/ALT1 and AMOEBA/DDM/BAR/ALT2), were used to examine how the selected TrimerTrip conformer impacts calculated binding free energies. The submitters examined so-called “indented” and “overlapping” host conformers which they identified in exploratory simulations. They find that these do not interconvert on the timescale of typical free energy calculations. The indented conformer resembles the annealed structure we provided in the SAMPL7 GitHub repository, while the overlapping conformer is very similar to the previously published structure of the unligated clip analog with four glycoluril units [43, 94] and interconverts relatively rapidly with a so-called “spiral” conformer with staggered triptycene walls [43, 94].
Since some of these conformations interconvert slowly, this introduces a conformation-dependence in calculated binding free energies. Not only may guests bind differently to the different host conformations, but calculated binding free energies depend on the host conformation because different unligated host conformations have different free energies in solution (e.g. some will likely be more strained/less populated than others) and do not relax back on simulation timescales.
To address these issues, the Ponder group used a separate set of free energy calculations to compute the binding free energy to each host conformation (indented and overlapping). However, the resulting free energies are sensitive to the choice of host conformation, since it does not relax back on simulation timescales, so they needed to estimate the relative free energy of the two unligated host conformations. In their submissions, their ranked AMOEBA/DDM/BAR submission assumes the indented TrimerTrip conformer is 2.84 kcal/mol more stable than the overlapping conformer, while the AMOEBA/DDM/BAR/ALT1 method assumes the overlapping conformer is 2.41 kcal/mol more stable than the indented, and the AMOEBA/DDM/BAR/ALT2 assumes both conformers are equal in free energy. The non-ranked AMOEBA submissions performed better than their ranked counterpart by almost all of the error metrics (Table 3). Most of the improvement was attributed to better agreement for clip-g6, clip-g7, clip-g8, clip-g9, and clip-g11 when using an overlapping conformer. The Ponder group suggests that these results indicate that larger and bulkier guests prefer the overlap/spiral conformer(s), while the smaller guests prefer the indented conformer [43, 94]. TrimerTrip’s flexibility seems to allow it to alter its conformation when binding guests of various size – a feature we noticed in our reference calculations and one also reported by the Ponder group [43].
Overall, TrimerTrip predictions using the AMOEBA force field and alchemical absolute binding free energy calculations were consistently the best.
Our in-house reference calculations provided the only other non-ranked submissions for TrimerTrip. Our two sets of reference calculations (Docking/GAFF/YANK_REF and Docking/GAFF/YANK_REF_2) differed only in the choice of host conformer for clip-g11, where the latter submission used an alternate, relatively open host conformation to allow it to relax and adapt to the bulky cyclic guest in g11 (see Section 6.1.1), though this approach ended up not resulting in substantially different predicted binding free energies. Performance statistics for these reference calculations ended up being particularly poor in general (Tables 3 and S3). The reference method gives free energies for all TrimerTrip host-guest complexes which are too unfavorable, similar to ranked MD/DOCKING/GAFF/xtb-GNF predictions. Both submissions used docking (VINA) to obtain guest poses without any MD (except that the MD/DOCKING/GAFF/xtb-GNF technique used SA-MD to obtain poses for four guests), GAFF parameters, the TIP3P water model, and AM1-BCC charges, so it may not be surprising that performance was similar. However, the MD/DOCKING/GAFF/xtb-GNF approach performed better for the case of the four guests where starting poses were established by SA-MD, with errors under 1 kcal/mol in those cases.
We can perhaps learn a bit more from these non-ranked submissions by comparing to the ranked submission called FSDAM/GAFF2/OPC3, which uses the OPC3 classical 3-point rigid water model with the GAFF2 force field and performed better than methods using its TIP3P counter part with GAFF, though there were other methodological differences between these submissions. The prediction error values for this method were the closest to the top performing methods using free energy methods with the AMOEBA force field, however its correlation values were similar to the methods using GAFF/TIP3P. The OPC3 water model has been shown to be significantly more accurate for pure water properties compared to other popular 3-point water models (i.e TIP3P and SPCE) of the same class [99] which may be particularly important in this system. Given the results reported in ref [99] for OPC3 and ref [81] for GAFF2; it is tempting to attribute this method’s better performance to use of the OPC3 water model and GAFF2, though without comparison to other methods which differ by only small molecule force field or water model, it is difficult to know this for certain.
7.2.2. GDCC
There were 11 non-ranked submissions for the GDCC dataset in addition to the 4 ranked predictions (Table 3). Three of the four participants with ranked submissions included at least two non-ranked submissions which were different in only a single factor, allowing easy sensitivity analysis. For example, all three alchemical AMOEBA-based methods had RMSEs below 2 kcal/mol, including the ranked AMOEBA/DDM/BAR submission and the non-ranked AMOEBA/DDM/BAR_2 and AMOEBA/DDM/BAR_3 submissions. These methods differed by key AMOEBA torsional parameters describing the flexibility of the middle and upper rim of the cavity of OA and exo-OA. These differences appear to have substantially affected performance (Table 3). The non-ranked AMOEBA/DDM/BAR_2 RMSE was the best of all methods and all predicted binding free energies were within 2 kcal/mol of the experimental values, including those for exoOA-g4, which was poorly predicted by other AMOEBA methods. The host parameters used in AMOEBA/DDM/BAR_2 were similar to those used in previous SAMPL challenges, while the other predictions used modified parameters. Overall, these AMOEBA submissions suggest guest binding to GDCCs is particularly sensitive to the host’s diphenyl ether torsions, and especially so for guest g4 binding to exo-OA and guests g7 and g8 binding to OA.
Another ranked submission which performed well used MM/PBSA, and non-ranked variants of this explored variations based on both MM/PBSA and MM/GBSA. One variation assessed the charge model, and found that the RESP charge scheme led to improved performance compared to the AM1-BCC charge scheme (RESP/GAFF/MMPBSA vs AM1-BCC/GAFF/MMPBSA), as shown by RMS errors of 7.59 kcal/mol vs 10.96 kcal/mol. These methods predicted binding free energies to be too favorable, a common issue with such endpoint free energy methods, especially when entropy changes are neglected, as here. An additional variation assessed the difference between MM/PBSA and MM/GBSA by changing the solvent model; the use of PB solvation resulted in significantly lower RMS errors here (RESP/GAFF/MMPBSA vs RESP/GAFF/MMGBSA), though the correlation with the GB approach was modestly better. A further variation added an accounting for entropy via normal mode analysis (RESP/GAFF/MMPBSA/Nmode) while maintaining RESP charges and PB solvation. This improved typical errors, but hurt correlation and resulted in binding free energies often not being favorable enough. One other key difference between the ranked submission in this series, and many of the others was that it actually used an empirical correction to binding free energies. Particularly, RESP/GAFF/MMPBSA-Cor used a linear correction derived from an analysis of previous SAMPL challenges [43, 95]. Indeed, this correction led to much better agreement with experiments. With RMSE and ME values of 1.32 and 1.01 kcal/mol, the RESP/GAFF/MMPBSA-Cor performance was on par with alchemical AMOEBA results, and for some guests performed even slightly better. In terms of correlation, the ranked RESP/GAFF/MMPBSA-Cor was similar to that of RESP/GAFF/MMPBSA. However, such an approach could not be applied without prior binding studies for the specific system(s) of interest.
A series of density functional theory (DFT)-based methods, including ranked and non-ranked submissions, were also used here. In SAMPL6, a DFT-based approach yielded good quantitative results [4, 5, 43], though without geometry optimizations employed in the current challenge. Here, the QM DFT-based B2PLYPD3/SMD submissions use B3PW91 with GD3BJ [2, 95] to treat dispersion, B2PLYPD3 for single point energy calculations [1, 95], and the SMD implicit solvation model [95, 100]. Different submissions in this series differed in which basis set was chosen for geometry optimization [43, 95]. Overall, these methods were roughly in the middle of all submitted methods in terms of predictive accuracy. All of these QM methods yield binding free energies for most guests which are too negative, with ME values of 2.69 kcal/mol or greater, and this is especially true for cationic guests binding exo-OA. The participants also highlighted particular difficulties with chlorine-containing guest g2. In initial tests, the OA-g2 binding free energy was estimated to be close to −30 kcal/mol, while experimental value in literature was −6.91 kcal/mol [43]. In the combined GDCC dataset, the ranked B2PLYPD3/SMD_QZ-R method was within 2 kcal/mol of experiments in three of nine cases and correctly predicted exoOA-g1 to be a non-binder. Overall, it appears that QM methods are not yet competitive with the best other approaches for these systems, and potentially, that molecules containing halogens can be particularly problematic.
Two of the non-ranked methods do not allow for straightforward sensitivity analysis based on a single factor, because only a single version was submitted (FSDAM/GAFF2/OPC3 and reference calculation DOCKING/GAFF/YANK). Both of these methods were also used for TrimerTrip and the cyclodextrin challenge. The error metrics for both of the methods were relatively similar, although the DOCKING/GAFF/YANK method performed slightly better by a number of metrics. However, the ME for FSDAM/GAFF2/OPC3 is quite low – less than 1 kcal/mol – because the method tends to predict binding free energies for OctaAcids with guests bearing a carboxylate group which are too favorable, and too unfavorable for guests with cationic ammoniums. In comparison, DOCKING/GAFF/YANK errs for all guests with carboxylate group are too favorable. Still, enough things differ between these two submissions that it is difficult to attribute performance differences to any particular source. Such simple variations provide the greatest opportunity for the community to learn.
One exoOA guest posed a bit of a surprise, in that binding of g1 to exoOA was not detected experimentally (Section 6.2 and Figure 2). Since no clear evidence of binding was observed experimentally at the detection threshold via ITC or H-NMR, this indicates a binding constant (Ka) to be less than 5 M−1 or a ΔG more positive than −0.95. Of the 15 GDCC submissions, 7 predicted this correctly with computed free energies ranging between −0.98 and 6.40 kcal/mol. Of the methods which incorrectly predicted g1 to bind, computed binding free energies ranged from −2.45 to −11.54 kcal/mol. All of the QM based submissions (B2PLYPD3/SMD) predicted exoOA-g1 to be a nonbinder, with values between 2.70 and 6.40 kcal/mol, and the AMOEBA-based alchemical methods also correctly recognized this as a nonbinder. Most MM/PBSA and MM/GBSA submissions failed to recognize this as a nonbinder, except for the RESP/GAFF/MMPBSA/Nmode method utilizing empirical corrections. The other GAFF-based methods predicted exoOA-g1 to be a binder. Predicted binding free energies of xtb-GNF/MachineLearning/CORINA_MD, RESP/GAFF/MMPBSA-Cor, RESP/GAFF/MMGBSA, AM1-BCC/GAFF/MMPBSA, RESP/GAFF/MMPBSA, FSDAM/GAFF2/OPC3, and DOCKING/GAFF/YANK were all more favorable than −3.84 kcal/mol. Perhaps for this guest, the proximal carboxylates of the host and guest repel one another too strongly for binding. This guest has relatively less hydrophobic character than other guests, perhaps meaning that the hydrophobic effect is not enough to offset this potential electrostatic clash. Perhaps only the AMOEBA absolute binding free energy calculations and the QM based methods can capture the relevant polarization effects well enough to recognize this complex is unfavorable.
7.2.3. Cyclodextrins
The cyclodextrin challenge proved to be the least challenging of the SAMPL7 challenges as measured by RMS error, as all submissions except one had RMSE values less than 2.74 kcal/mol (the exception was the AM1-BCC/MD/GAFF/TIP4PEW/QMMM method, with RMSE and ME metrics over 30 kcal/mol). However, the dynamic range was particularly small for this challenge, with most host-guest complexes showing similar binding free energies. This meant that correlations between calculated and predicted values were typically quite poor (Table 3).
First we compare the ranked FSDAM/GAFF2/OPC3_ranked and non-ranked FSDAM/GAFF2/OPC3_JB methods, where the ranked method performed slightly better; these methods used the same simulation approach but differ in that the former used a Gaussian approximation for computing nonequilibrium free energies, whereas the latter used a “boosted Jarzynski” approach for analysis [93, 98]. Both analysis approaches ought to give equivalent binding free energies in certain limits, but their underlying assumptions and the amount of data available result in substantially different performance here. Here, despite its limitations in the SAMPL6 “SAMPLing” challenge [5], the Gaussian approximation was modestly superior, with 15 of 16 binding free energies predicted within 2 kcal/mol, versus 11 of 16 for FSDAM/GAFF2/OPC3_JB.
Three other nonequilibrium free energy methods participated for the cyclodextrin challenge – Noneq/Alchemy/CGENFF, Noneq/Alchemy/consensus, and Noneq/Alchemy/GAFF. All three methods used the TIP3P water model, included NaCl ions at 25mM, and considered multiple binding poses (primary and secondary orientation) and free energies reported as Boltzmann weighted averages across these poses. These methods differed only by force field – CGENFF (Noneq/Alchemy/CGENFF) or GAFF (Noneq/Alchemy/GAFF). The third submission, Noneq/Alchemy/consensus, gives “consensus” results obtained by averaging across both force fields. In this case the RMSE was under 2 kcal/mol for both methods, but CGENFF resulted in very slightly better performance by most metrics. Problematic systems for this method were MGLab23-g1, MGLab24-g1, MGLab24-g2, MGLab36-g1, and MGLab36-g2, and what they have in common is larger cyclodextrin side chains. Cyclodextrins with amino acid side chains tend to be the more accurately predicted systems for this method, suggesting the methods may be limited by forcefield parameters.
Our reference calculations (Docking/GAFF/YANK_REF performed reasonably well for this dataset (Table S3), and surprisingly had better correlation to experiments compared to other methods (Table 3). Predicted binding free energies were within 2 kcal/mol for 9 of 16 host-guest systems, and similar conditions and water model were used as for Noneq/Alchemy/GAFF, though different free energy estimation techniques were used. These submissions also differed in handling of binding modes; our reference calculations used only a single initial binding mode for each (determined by the top scoring pose from docking) whereas Noneq/Alchemy/consensus considered up to two poses whenever a second orientation was considered stable and was in better agreement with experiments. Thus, suggesting secondary guests orientations may need to be considered, and guest and/or host side chain sampling may be an issue.
When we compare the diverse methods submitted, some observations stand out. First, performance of Noneq/Alchemy/GAFF and Docking/GAFF/YANK_REF methods was quite similar with an RMS difference of 0.8 kcal/mol – likely due to use of the same force field (GAFF) and water model (TIP3P) despite the fact that the former used nonequilibrium free energy techniques and the latter used equilibrium, suggesting the force field played a larger role. Along the same lines, several nonequilibrium methods (FSDAM/GAFF2/OPC3, Noneq/Alchemy/CGENFF, and Noneq/Alchemy/GAFF) all used similar techniques but different force field/water model, and performance was thus reasonably similar with an RMS difference of at most 0.32 kcal/mol. In addition, binding free energy calculations have been shown to be more accurate using GAFF2 opposed to GAFF in previous computational studies [81]. Finally, the most challenging case seems to be binding of cyclodextrins with large side chains to rimantadine (g2), though the reason for this is not known.
7.2.4. Reference Calculations
In this section we survey additional retrospective tests with reference calculations and analyze the results. For most of the reference calculations, simulations which had the greatest error in binding affinity had poor sampling/mixing of the states within replicas. Moreover, many of the free energy estimates were not converged, or converged to a value which disagreed with experiment at timescales up to 30 ns per window. Convergence to a value which differs from experiment may indicate force field problems. These errors was particularly prevalent for TrimerTrip and Cyclodextrin derivatives while also in the presence of a guest with a formal charge (Figures S1 and S3). Interestingly, free energy estimates seemed to converge better for the GDCC dataset depending on whether the guest was positively or negatively charged (Figure S2). In addition, in simulations for exoOA with guests with a negatively charged carboxyl group had poor mixing of states within replicas, while with positively charged guests mixing of states was generally better. To check the contribution of the charged protocol in mixing of states between replicas and estimate error in reference calculations, additional calculations for exoOA-g1, exoOA-g3, and clip-g1 were done.
For the exoOAg3 and clip-g1 systems, the automatic pipeline in YANK (while additional simulation options remained the same) was used to determine individual and unique alchemical paths. Ideally, this should improve replica exchange overlap, thus improve sampling. Despite using unique alchemical protocols (with additional lambda windows) for these systems the sampling did not improve and the free energy was not convincingly converged or inaccurate even after simulations up to 30ns per iteration.
In addition, separate experiments were done with exoOA-g1 using the charged protocol, however this time changing simulation options. First, we added YANK’s ‘PME_treatment’ option meant to speed up and improve convergence of systems where a guest/ligand has a formal charge. Second, we tested a double annihilation scheme with soft core potentials rather than double decoupling. In both cases and with a combination of both we observed significant improvement in sampling of states between replicas for both the complex and solvent phase, and convergence of free energy estimates within a 10 ns timescale per iteration. However, agreement with experimental free energy for the exoOA-g1 test case did not improve. In retrospect, this is perhaps not surprising since only the AMOEBA and QM based methods predicted this with convincing accuracy. It would be interesting to see in future challenges how our methods simulation options affect the accuracy of the other systems of this challenge.
Our final test case involved changing the charge scheme option for the guest in exoOA-g1 from AM1-BCC to AM1-BCCELF10 with OpenEye Toolkits, otherwise retaining the same protocol. The change in charging scheme essentially made g1 slightly less polar, thus we thought this would result in less favorable binding to exoOA. However, that was not the case, the resulting binding free energy was slightly more negative at −8.813 ± 0.070 kcal/mol compared to our submitted prediction of −7.629 ± 0.090 kcal/mol.
In retrospect, perhaps the results of our follow up simulations should not be surprising since only the AMOEBA and QM based methods predicted binding of this guest accurately, perhaps indicating polarizability is particularly important in this case. Overall, these follow-up investigations did not find factors which dramatically affected the accuracy of the reference calculations on the exoOA-g1 system. It would be interesting to further assess this on the other systems considered.
8. Conclusions and Lessons Learned
The SAMPL7 host-guest blind challenge provided a platform to test the reliability of computational methods and tools to accurately predict binding free energies. Since hosts in the cucurbituril and cavitand families have been featured in previous SAMPL challenges (and likely in future challenges) these provide a mechanism to assess how the field progresses across a series of challenges. In addition, the amount of attention these have received helps us identify some potential lessons learned and give suggestions for improvement.
The TrimerTrip dataset of SAMPL7, like cucurbiturils from previous challenges, posed the largest challenge for participants, as judged by method performance. Specifically, most methods performed poorly at computing binding free energies for cationic guests with cyclic, aromatic, and adamantane based moieties. In addition, most methods were relatively inconsistent at predicting binding free energies of hydrocarbon chains of increasing length, but the AMOEBA alchemical binding free energy methods did very well predicting 7 of 8 within 2 kcal/mol. Both of the best performing methods here used alchemical free energy calculations. Predictions from the best fixed-charge force field submission, based on nonequilibrium free energy calculations (FSDAM/GAFF2/OPC3), had errors above 2 kcal/mol for 8/16 host-guest systems considered. In contrast, performance with the AMOEBA polarizable force field and alchemical methods was significantly better here, suggesting that one key source of error may be polarization effects and/or multipoles.
In the TrimerTrip case, participants also found evidence that binding free energies may be more accurate if different potential host conformations are considered, especially for bulkier guests such as those with adamantane moieties. This exploration of sensitivity to host conformation also provided insight into modeling the host’s flexibility; participants found binding free energies to be sensitive to the geometry of the triptycene rings [43, 94]. Our reference calculations showed poor sampling of interconversion between alchemical states in our simulations, despite use of Hamiltonian Replica Exchange.
Given these results, it appears that force field accuracy and choice of force field (e.g. GAFF, GAFF2, AMOEBA) may be a dominant factor limiting accurate binding affinity predictions.
On the Gibb deep cavity cavitands (GDCCs), OA and exoOA, as in previous SAMPL challenges, simulation based methods with empirical fixed charge energy models performed relatively well. Binding affinities for guests with adamantane, aromatic and saturated cyclic carboxlylates with OctaAcids were predicted with greater accuracy than TrimerTrip. Performance of methods within the GDCC dataset (OA and exoOA) demonstrates significant variation by guest, and especially when the formal charge of guest differs (negative vs positive).
In part because of the relatively extensive prior work on GDCCs, some submissions applied empirical corrections before making predictions, and/or utilized machine learning approaches. These tended to help performance, here, but rely on availability of training data on closely related systems – which is not always available for prospective applications.
On the GDCCs, as for TrimerTrip, submissions using the AMOEBA force field and absolute alchemical binding free energy techniques performed particularly well. Additionally, along with a QM based method, these AMOEBA-based approaches correctly predicted exoOA with g1 a non-binder. Perhaps only AMOEBA and QM methods capture relevant polarization effects well enough to accurately describe this particular complex well in general, though one MM/PBSA approach also recognized this as a nonbinder.
For the current challenge, the AMOEBA-based free energy calculations had the most consistent performance across the different host-guest complexes, and across datasets (TrimerTrip, OctaAcid, exoOA). Despite the lower variation for this method, guest g4 was particularly sensitive to diphenyl ether torsional parameters which worked very well in all other GDCC systems. The AMOEBA-based approach did rather well in SAMPL7, but improvements in the approach relative to prior SAMPL challenges were entirely in the sampling protocol and torsion values, indicating that these can provide gains in accuracy.
The cyclodextrin derivatives were new to SAMPL, and many methods achieved relatively low RMS errors – though this may partly be due to the low dynamic range of the set; a hypothetical method which predicted a constant binding free energy of −4 kcal/mol for all guests would achieve an RMS error of only 0.70 kcal/mol. This low dynamic range also meant that correlation metrics were typically poor. The force field used in this dataset played a role in computing accurate binding free energies, with GAFF2 seemingly giving more accurate results, followed by CGenFF and GAFF (a more detailed comparison of these force fields can be seen in Ref [92] and Ref [93]). In addition, nonequilibrium approaches appear to perform slightly better with cyclodextrin systems. The performance of methods for the cyclodextrin dataset varied across host-guest systems, but predicting reliable binding free energies for cyclodextrins with large side chains to rimantadine was frequently challenging. There were no AMOEBA submissions for this aspect of SAMPL7, but the use of a polarizable force field may help ameliorate agreement between computational methods with experiments and facilitate accurate modeling of cyclodextrin host-guest interactions.
Finally, note that two methods included predictions for all three datasets, DOCKING/GAFF/YANK and FSDAM/GAFF2/OPC, though not all of the submissions were ranked. The performance of these methods varied across different datasets and across different host-guest systems within the same dataset. For both methods, binding predictions for larger and more hydrophobic guests were apparently more difficult.
In terms of overall lessons learned in this challenge, we found that methods which only varied a single factor (such as force field or water model, with a fixed method) were particularly valuable in terms of providing insight into accuracy, thus we urge participants to continue with such explorations in the future. Another important area of work is to ensure that methods which ought to be equivalent do, in fact, give equivalent results across different simulation packages [5].
Overall, SAMPL7 showed marked progress in binding prediction relative to previous challenges, and in particular results with binding free energy calculations using the AMOEBA force field were particularly promising for two of the challenge components. For future challenges it will be interesting to continue investigations of host/guest sampling, polarization effects, and possibly salt behavior in similar systems. We look forward to continuing to work with the community to use the SAMPL challenge to drive accuracy improvements in binding predictions.
9. Code and Data Availability
All SAMPL7 host-guest challenge instructions, submissions, experimental data and analysis are available at https://github.com/samplchallenges/SAMPL7/tree/master/host_guest. An archive copy of SAMPL7 GitHub repository host-guest challenge directory is also available in the Supplementary Documents bundle (SAMPL7-supplementary-documents.tar.gz). Some useful files from this repository are highlighted below.
Table of participants submission filenames and their submission ID: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/SAMPL7-user-map-HG.csv
Submission files of prediction sets: https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Submissions
Python analysis scripts and outputs: https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Scripts
Table of performance statistics calculated for ranked methods for TrimerTrip dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Accuracy_ranked/TrimerTrip/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for TrimerTrip dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/TrimerTrip/StatisticsTables/statistics.csv
Table of performance statistics calculated for ranked methods for GDCC dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Accuracy_ranked/GDCC_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for GDCC (without optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/GDCC_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for GDCC (with optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/GDCC/StatisticsTables/statistics.csv
Table of performance statistics calculated for ranked methods for Cyclodextrin dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Accuracy_ranked/CD_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for Cyclodextrin (without optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/CD_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for Cyclodextrin (with optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/CD/StatisticsTables/statistics.csv
Supplementary Material
Figure 11. exo-OA Error Metrics for Ranked Methods.
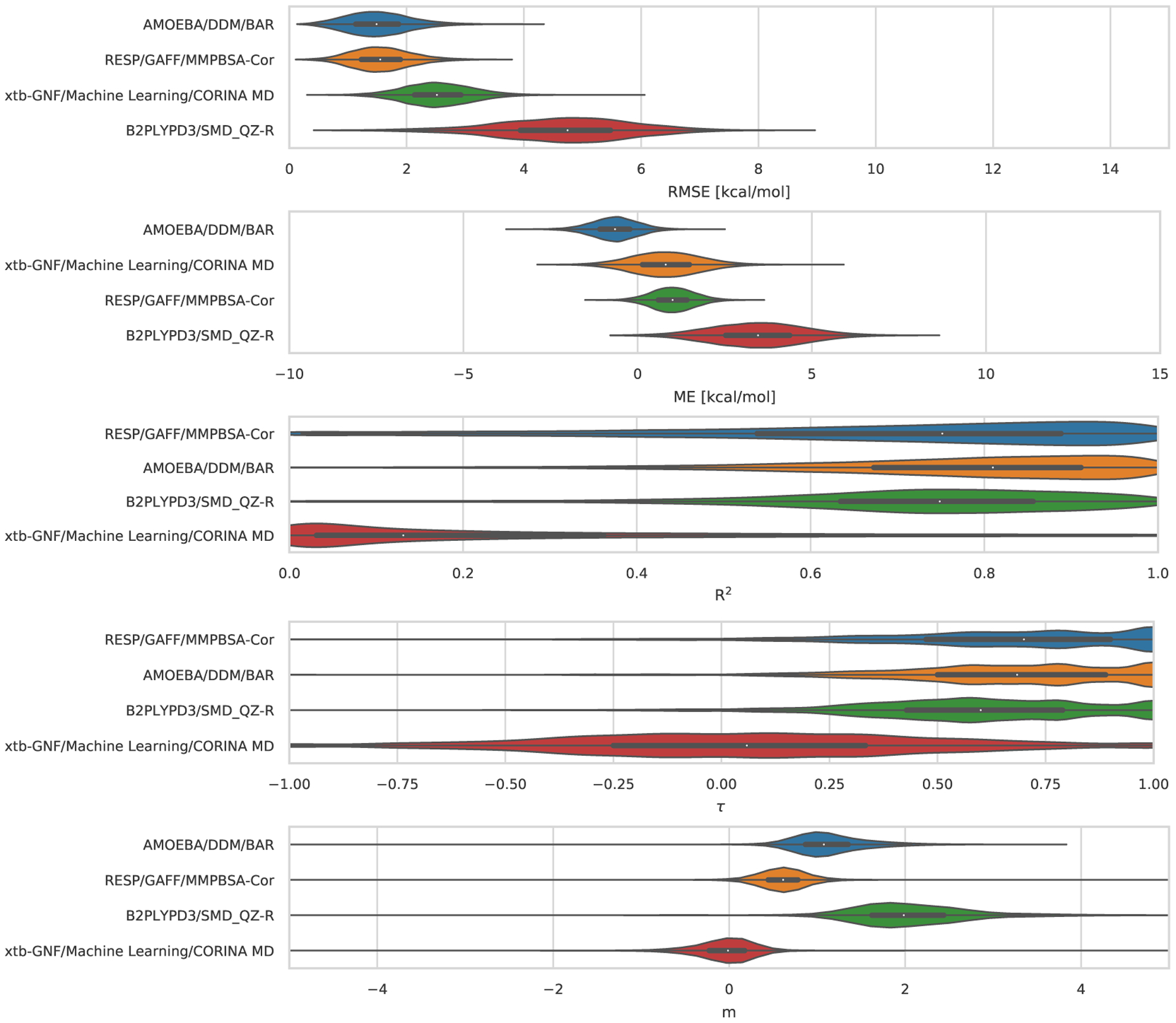
Shown are exo-OA methods, with the median indicated by the white circle in the violin plots. The violin plots for RMSE, ME, R2, τ, and slope describe the shape of the sampling distribution after bootstrapping for each method. The black horizontal bar represents the first and third quartiles. From top to bottom the error metrics are RMSE, ME, R2, τ, and slope (m).
10. Acknowledgments
MA and DLM gratefully acknowledge support from NIH grant R01GM124270 supporting the SAMPL Blind Challenges. We appreciate the laboratories of Michael K. Gilson (UCSD), Lyle Isaacs (Maryland) and Bruce Gibb (Tulane) for providing experimental data for the challenge. We are also grateful to OpenEye Scientific for providing a free academic software license for use in this work.
Abbreviations
- SAMPL
Statistical Assessment of the Modeling of Proteins and Ligands
- AM1-BCC
Austin model 1 bond charge correction
- RESP
Restrained electrostatic potential
- REST
Replica exchange with solute tempering
- FSDAM
Fast switching double annihilation method
- B2PLYPD3
Beck 2-parameter Lee-Yang-Parr D3 exchange-correlation functional [1]
- B3PW91
Becke 3-parameter Perdew-Wang 91 exchange-correlation functional [2]
- GAFF
Generalized AMBER force field
- CGenFF
CHARMM generalized force field
- AMOEBA
Atomic multipole optimized energetics for biomolecular simulations
- DDM
Double decoupling method
- DFT
Density functional theory
- QM/MM
Mixed quantum mechanics and molecular mechanics
- MMPBSA
Molecular mechanics Poisson Boltzmann/solvent accessible surface area
- MMGBSA
Molecular mechanics generalized born/solvent accessible surface area
- TIP3P
Transferable interaction potential three-point
- TIP4PEw
Traansferable interaction potential four-point Ewald
- OPC3
Optimal 3-point charge
- SEM
Standard error of the mean
- RMSE
Root mean squared error
- MAE
Mean absolute error
- ME
Mean signed error
- τ
Kendall’s rank correlation coefficient (Tau)
- R2
Coefficient of determination (R-Squared)
- QM
Quantum Mechanics
- MM
Molecular Mechanics
Footnotes
Publisher's Disclaimer: 11 Disclaimers
Publisher's Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
12 Disclosures
DLM is a member of the Scientific Advisory Board of OpenEye Scientific Software, and DLM is an Open Science Fellow with Silicon Therapeutics.
References
- [1].Goerigk L, Grimme S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals-Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. Journal of chemical theory and computation. 2011; doi: 10.1021/ct100466k. [DOI] [PubMed] [Google Scholar]
- [2].Grimme S, Ehrlich S, Goerigk L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J Comput Chem. 2011. May; 32(7):1456–1465. doi: 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- [3].Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK. Overview of the SAMPL5 Host–Guest Challenge: Are We Doing Better? J Comput Aided Mol Des. 2017. January; 31(1):1–19. doi: 10.1007/s10822-016-9974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL, Chodera JD. Overview of the SAMPL6 Host–Guest Binding Affinity Prediction Challenge. J Comput Aided Mol Des. 2018. October; 32(10):937–963. doi: 10.1007/s10822-018-0170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rizzi A, Jensen T, Slochower DR, Aldeghi M, Gapsys V, Ntekoumes D, Bosisio S, Papadourakis M, Henriksen NM, de Groot BL, Cournia Z, Dickson A, Michel J, Gilson MK, Shirts MR, Mobley DL, Chodera JD. The SAMPL6 SAMPLing Challenge: Assessing the Reliability and Efficiency of Binding Free Energy Calculations. J Comput Aided Mol Des. 2020. January; doi: 10.1007/s10822-020-00290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang L, Wu Y, Deng Y, Kim B, Pierce L, Krilov G, Lupyan D, Robinson S, Dahlgren MK, Greenwood J, Romero DL, Masse C, Knight JL, Steinbrecher T, Beuming T, Damm W, Harder E, Sherman W, Brewer M, Wester R, et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J Am Chem Soc. 2015. February; 137(7):2695–2703. doi: 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- [7].Cournia Z, Allen B, Sherman W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J Chem Inf Model. 2017. December; 57(12):2911–2937. doi: 10.1021/acs.jcim.7b00564. [DOI] [PubMed] [Google Scholar]
- [8].Rocklin GJ, Mobley DL, Dill KA, Hünenberger PH. Calculating the Binding Free Energies of Charged Species Based on Explicit-Solvent Simulations Employing Lattice-Sum Methods: An Accurate Correction Scheme for Electrostatic Finite-Size Effects. J Chem Phys. 2013. November; 139(18):184103. doi: 10.1063/1.4826261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mobley DL, Gilson MK. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu Rev Biophys. 2017. May; 46(1):531–558. doi: 10.1146/annurev-biophys-070816-033654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Işık M, Bergazin TD, Fox T, Rizzi A, Chodera JD, Mobley DL. Assessing the Accuracy of Octanol–Water Partition Coefficient Predictions in the SAMPL6 Part II Log P Challenge. J Comput Aided Mol Des. 2020. April; 34(4):335–370. doi: 10.1007/s10822-020-00295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Laury ML, Wang Z, Gordon AS, Ponder JW. Absolute Binding Free Energies for the SAMPL6 Cucurbit[8]Uril Host–Guest Challenge via the AMOEBA Polarizable Force Field. J Comput Aided Mol Des. 2018. October; 32(10):1087–1095. doi: 10.1007/s10822-018-0147-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gapsys V, de Groot BL. Pmx Webserver: A User Friendly Interface for Alchemistry. J Chem Inf Model. 2017. February; doi: 10.1021/acs.jcim.6b00498. [DOI] [PubMed] [Google Scholar]
- [13].Schindler C, Baumann H, Blum A, Böse D, Buchstaller HP, Burgdorf L, Cappel D, Chekler E, Czodrowski P, Dorsch D, Eguida M, Follows B, Fuchß T, Grädler U, Gunera J, Johnson T, Jorand Lebrun C, Karra S, Klein M, Kötzner L, et al. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. ChemRxiv. 2020. January; doi: 10.26434/chemrxiv.11364884.v1. [DOI] [PubMed] [Google Scholar]
- [14].Gapsys V, Pérez-Benito L, Aldeghi M, Seeliger D, van Vlijmen H, Tresadern G, de Groot BL. Large Scale Relative Protein Ligand Binding Affinities Using Non-Equilibrium Alchemy. Chemical Science. 2020; 11(4):1140–1152. doi: 10.1039/C9SC03754C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Muddana HS, Varnado CD, Bielawski CW, Urbach AR, Isaacs L, Geballe MT, Gilson MK. Blind Prediction of Host–Guest Binding Affinities: A New SAMPL3 Challenge. J Comput Aided Mol Des. 2012. February; 26(5):475–487. doi: 10.1007/s10822-012-9554-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Skillman AG. SAMPL3: Blinded Prediction of Host–Guest Binding Affinities, Hydration Free Energies, and Trypsin Inhibitors. J Comput Aided Mol Des. 2012. May; 26(5):473–474. doi: 10.1007/s10822-012-9580-z. [DOI] [PubMed] [Google Scholar]
- [17].Muddana HS, Fenley AT, Mobley DL, Gilson MK. The SAMPL4 Host–Guest Blind Prediction Challenge: An Overview. J Comput Aided Mol Des. 2014. March; 28(4):305–317. doi: 10.1007/s10822-014-9735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Peat TS, Dolezal O, Newman J, Mobley DL, Deadman JJ. Interrogating HIV Integrase for Compounds That Bind- a SAMPL Challenge. J Comput Aided Mol Des. 2014. February; 28(4):347–362. doi: 10.1007/s10822-014-9721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gathiaka S, Liu S, Chiu M, Yang H, Stuckey JA, Kang YN, Delproposto J, Kubish G, Dunbar JB, Carlson HA, Burley SK, Walters WP, Amaro RE, Feher VA, Gilson MK. D3R Grand Challenge 2015: Evaluation of Protein–Ligand Pose and Affinity Predictions. J Comput Aided Mol Des. 2016. September; 30(9):651–668. doi: 10.1007/s10822-016-9946-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gaieb Z, Liu S, Gathiaka S, Chiu M, Yang H, Shao C, Feher VA, Walters WP, Kuhn B, Rudolph MG, Burley SK, Gilson MK, Amaro RE. D3R Grand Challenge 2: Blind Prediction of Protein–Ligand Poses, Affinity Rankings, and Relative Binding Free Energies. J Comput Aided Mol Des. 2018. January; 32(1):1–20. doi: 10.1007/s10822-017-0088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gaieb Z, Parks CD, Chiu M, Yang H, Shao C, Walters WP, Lambert MH, Nevins N, Bembenek SD, Ameriks MK, Mirzadegan T, Burley SK, Amaro RE, Gilson MK. D3R Grand Challenge 3: Blind Prediction of Protein–Ligand Poses and Affinity Rankings. J Comput Aided Mol Des. 2019. January; 33(1):1–18. doi: 10.1007/s10822-018-0180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Parks CD, Gaieb Z, Chiu M, Yang H, Shao C, Walters WP, Jansen JM, McGaughey G, Lewis RA, Bembenek SD, Ameriks MK, Mirzadegan T, Burley SK, Amaro RE, Gilson MK. D3R Grand Challenge 4: Blind Prediction of Protein–Ligand Poses, Affinity Rankings, and Relative Binding Free Energies. J Comput Aided Mol Des. 2020. February; 34(2):99–119. doi: 10.1007/s10822-020-00289-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sherborne B, Shanmugasundaram V, Cheng AC, Christ CD, DesJarlais RL, Duca JS, Lewis RA, Loughney DA, Manas ES, McGaughey GB, Peishoff CE, van Vlijmen H. Collaborating to Improve the Use of Free-Energy and Other Quantitative Methods in Drug Discovery. J Comput Aided Mol Des. 2016. December; 30(12):1139–1141. doi: 10.1007/s10822-016-9996-y. [DOI] [PubMed] [Google Scholar]
- [24].Reif MM, Hünenberger PH. Computation of Methodology-Independent Single-Ion Solvation Properties from Molecular Simulations. III. Correction Terms for the Solvation Free Energies, Enthalpies, Entropies, Heat Capacities, Volumes, Compressibilities, and Expansivities of Solvated Ions. J Chem Phys. 2011. April; 134(14):144103. doi: 10.1063/1.3567020. [DOI] [PubMed] [Google Scholar]
- [25].Öhlknecht C, Lier B, Petrov D, Fuchs J, Oostenbrink C. Correcting Electrostatic Artifacts Due to Net-Charge Changes in the Calculation of Ligand Binding Free Energies. Journal of Computational Chemistry. 2020; 41(10):986–999. doi: 10.1002/jcc.26143. [DOI] [PubMed] [Google Scholar]
- [26].Hünenberger PH, McCammon JA. Ewald Artifacts in Computer Simulations of Ionic Solvation and Ion–Ion Interaction: A Continuum Electrostatics Study. J Chem Phys. 1999. January; 110(4):1856–1872. doi: 10.1063/1.477873. [DOI] [Google Scholar]
- [27].Lin YL, Aleksandrov A, Simonson T, Roux B. An Overview of Electrostatic Free Energy Computations for Solutions and Proteins. J Chem Theory Comput. 2014. July; 10(7):2690–2709. doi: 10.1021/ct500195p. [DOI] [PubMed] [Google Scholar]
- [28].Simonson T, Roux B. Concepts and Protocols for Electrostatic Free Energies. Molecular Simulation. 2016. September; 42(13):1090–1101. doi: 10.1080/08927022.2015.1121544. [DOI] [Google Scholar]
- [29].Ji C, Mei Y. Some Practical Approaches to Treating Electrostatic Polarization of Proteins. Acc Chem Res. 2014. September; 47(9):2795–2803. doi: 10.1021/ar500094n. [DOI] [PubMed] [Google Scholar]
- [30].Zhang C, Lu C, Wang Q, Ponder JW, Ren P. Polarizable Multipole-Based Force Field for Dimethyl and Trimethyl Phosphate. J Chem Theory Comput. 2015. November; 11(11):5326–5339. doi: 10.1021/acs.jctc.5b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kognole AA, Aytenfisu AH, MacKerell AD. Balanced Polarizable Drude Force Field Parameters for Molecular Anions: Phosphates, Sulfates, Sulfamates, and Oxides. J Mol Model. 2020. May; 26(6):152. doi: 10.1007/s00894-020-04399-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cerutti DS, Swope WC, Rice JE, Case DA. Ff14ipq: A Self-Consistent Force Field for Condensed-Phase Simulations of Proteins. J Chem Theory Comput. 2014. October; 10(10):4515–4534. doi: 10.1021/ct500643c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou A, Schauperl M, Nerenberg PS. Benchmarking Electronic Structure Methods for Accurate Fixed-Charge Electrostatic Models. J Chem Inf Model. 2020. January; 60(1):249–258. doi: 10.1021/acs.jcim.9b00962. [DOI] [PubMed] [Google Scholar]
- [34].Schauperl M, Nerenberg PS, Jang H, Wang LP, Bayly CI, Mobley DL, Gilson MK. Non-Bonded Force Field Model with Advanced Restrained Electrostatic Potential Charges (RESP2). Communications Chemistry. 2020. April; 3(1):1–11. doi: 10.1038/s42004-020-0291-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and Testing of a General Amber Force Field. J Comput Chem. 2004. July; 25(9):1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- [36].Wang J, Wang W, Kollman PA, Case DA. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J Mol Graph Model. 2006. October; 25(2):247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- [37].Mobley DL, Bannan CC, Rizzi A, Bayly CI, Chodera JD, Lim VT, Lim NM, Beauchamp KA, Slochower DR, Shirts MR, Gilson MK, Eastman PK. Escaping Atom Types in Force Fields Using Direct Chemical Perception. J Chem Theory Comput. 2018. October; doi: 10.1021/acs.jctc.8b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. Journal of Computational Chemistry. 2009; p. NA–NA. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J Chem Inf Model. 2012. December; 52(12):3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Vanommeslaeghe K, Raman EP, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J Chem Inf Model. 2012. December; 52(12):3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J Phys Chem B. 2001. July; 105(28):6474–6487. doi: 10.1021/jp003919d. [DOI] [Google Scholar]
- [42].Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput. 2016. January; 12(1):281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- [43].Mobley DL, Amezcua M, Ponder J, Khalak Y, Yigitkan Eken E, Almeida N, Isaacs L, Gibb B, Kellett K, Serrilon D, The SAMPL7 Host-Guest Challenge Virtual Workshop. Zenodo; 2020. doi: 10.5281/zenodo.3674155. [DOI] [Google Scholar]
- [44].Saric D, Kohns M, Vrabec J. Dielectric Constant and Density of Aqueous Alkali Halide Solutions by Molecular Dynamics: A Force Field Assessment. J Chem Phys. 2020. April; 152(16):164502. doi: 10.1063/1.5144991. [DOI] [PubMed] [Google Scholar]
- [45].Vega C Water: One Molecule, Two Surfaces, One Mistake. Molecular Physics. 2015. May; 113(9–10):1145–1163. doi: 10.1080/00268976.2015.1005191. [DOI] [Google Scholar]
- [46].González MA. Force Fields and Molecular Dynamics Simulations. JDN. 2011; 12:169–200. doi: 10.1051/sfn/201112009. [DOI] [Google Scholar]
- [47].Guillot B A Reappraisal of What We Have Learnt during Three Decades of Computer Simulations on Water. Journal of Molecular Liquids. 2002. November; 101(1):219–260. doi: 10.1016/S0167-7322(02)00094-6. [DOI] [Google Scholar]
- [48].Henriksen NM, Gilson MK. Evaluating Force Field Performance in Thermodynamic Calculations of Cyclodextrin Host–Guest Binding: Water Models, Partial Charges, and Host Force Field Parameters. J Chem Theory Comput. 2017. September; 13(9):4253–4269. doi: 10.1021/acs.jctc.7b00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yin J, Henriksen NM, Muddana HS, Gilson MK. Bind3P: Optimization of a Water Model Based on Host–Guest Binding Data. J Chem Theory Comput. 2018. July; 14(7):3621–3632. doi: 10.1021/acs.jctc.8b00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Warshel A Energetics of Enzyme Catalysis. PNAS. 1978. November; 75(11):5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Howard AE, Singh UC, Billeter M, Kollman PA. Many-Body Potential for Molecular Interactions. J Am Chem Soc. 1988. October; 110(21):6984–6991. doi: 10.1021/ja00229a009. [DOI] [Google Scholar]
- [52].Humphreys DD, Friesner RA, Berne BJ. Simulated Annealing of a Protein in a Continuum Solvent by Multiple-Time-Step Molecular Dynamics. J Phys Chem. 1995. June; 99(26):10674–10685. doi: 10.1021/j100026a035. [DOI] [Google Scholar]
- [53].Grossfield A, Ren P, Ponder JW. Ion Solvation Thermodynamics from Simulation with a Polarizable Force Field. J Am Chem Soc. 2003. December; 125(50):15671–15682. doi: 10.1021/ja037005r. [DOI] [PubMed] [Google Scholar]
- [54].Gibb CLD, Gibb BC. Anion Binding to Hydrophobic Concavity Is Central to the Salting-in Effects of Hofmeister Chaotropes. J Am Chem Soc. 2011. May; 133(19):7344–7347. doi: 10.1021/ja202308n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thormann E On Understanding of the Hofmeister Effect: How Addition of Salt Alters the Stability of Temperature Responsive Polymers in Aqueous Solutions | Request PDF. RSC Adv. 2012. July; p. 8297–8305. doi: DOI: 10.1039/c2ra20164j. [DOI] [Google Scholar]
- [56].Gao K, Yin J, Henriksen NM, Fenley AT, Gilson MK. Binding Enthalpy Calculations for a Neutral Host–Guest Pair Yield Widely Divergent Salt Effects across Water Models. J Chem Theory Comput. 2015. October; 11(10):4555–4564. doi: 10.1021/acs.jctc.5b00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Carnegie RS, Gibb CLD, Gibb BC. Anion Complexation and The Hofmeister Effect. Angew Chem. 2014. October; 126(43):11682–11684. doi: 10.1002/ange.201405796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gibb CLD, Gibb BC. Well-Defined, Organic Nanoenvironments in Water: The Hydrophobic Effect Drives a Capsular Assembly. J Am Chem Soc. 2004. September; 126(37):11408–11409. doi: 10.1021/ja0475611. [DOI] [PubMed] [Google Scholar]
- [59].Saltzman A, Tang D, Gibb BC, Ashbaugh HS. Emergence of Non-Monotonic Deep Cavity Cavitand Assembly with Increasing Portal Methylation. Mol Syst Des Eng. 2020. March; 5(3):656–665. doi: 10.1039/C9ME00076C. [DOI] [Google Scholar]
- [60].Brown A Analysis of Cooperativity by Isothermal Titration Calorimetry. Int J Mol Sci. 2009. August; 10(8):3457–3477. doi: 10.3390/ijms10083457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ma YL, Ke H, Valkonen A, Rissanen K, Jiang W. Achieving Strong Positive Cooperativity through Activating Weak Non-Covalent Interactions. Angewandte Chemie International Edition. 2018; 57(3):709–713. doi: 10.1002/anie.201711077. [DOI] [PubMed] [Google Scholar]
- [62].Ndendjio SZ, Liu W, Yvanez N, Meng Z, Zavalij PY, Isaacs L. Triptycene Walled Glycoluril Trimer: Synthesis and Recognition Properties. New J Chem. 2019. December; 44(2):338–345. doi: 10.1039/C9NJ05336K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Suating P, Nguyen TT, Ernst EN, Wang Y, Jordan HJ, Gibb DCL, Ashbaugh SH, Gibb CB. Proximal Charge Effects on Guest Binding to a Non-Polar Pocket. Chemical Science. 2020; 11(14):3656–3663. doi: 10.1039/C9SC06268H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kellett K, Slochower D, Schauperl M, Duggan BM, Gilson M. Experimental Characterization of the Association of Nine Novel Cyclodextrin Derivatives with Two Guest Compounds. chemRxiv. 2020. July; doi: 10.26434/chemrxiv.12663065.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lee J, Tofoleanu F, Pickard FC, König G, Huang J, Damjanovi¢ A, Baek M, Seok C, Brooks BR. Absolute Binding Free Energy Calculations of CBClip Host-Guest Systems in the SAMPL5 Blind Challenge. J Comput Aided Mol Des. 2017. January; 31(1):71–85. doi: 10.1007/s10822-016-9968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ma D, Zavalij PY, Isaacs L. Acyclic Cucurbit[n]Uril Congeners Are High Affinity Hosts. J Org Chem. 2010. July; 75(14):4786–4795. doi: 10.1021/jo100760g. [DOI] [PubMed] [Google Scholar]
- [67].Biedermann F, Rauwald U, Cziferszky M, Williams KA, Gann LD, Guo BY, Urbach AR, Bielawski CW, Scherman OA. Benzobis(Imidazolium)–Cucurbit[8]Uril Complexes for Binding and Sensing Aromatic Compounds in Aqueous Solution. Chemistry – A European Journal. 2010. December; 16(46):13716–13722. doi: 10.1002/chem.201002274. [DOI] [PubMed] [Google Scholar]
- [68].Gallicchio E, Levy RM. Prediction of SAMPL3 Host-Guest Affinities with the Binding Energy Distribution Analysis Method (BEDAM). J Comput Aided Mol Des. 2012. May; 26(5):505–516. doi: 10.1007/s10822-012-9552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Naïm M, Bhat S, Rankin KN, Dennis S, Chowdhury SF, Siddiqi I, Drabik P, Sulea T, Bayly CI, Jakalian A, Purisima EO. Solvated Interaction Energy (SIE) for Scoring Protein-Ligand Binding Affinities. 1. Exploring the Parameter Space. J Chem Inf Model. 2007. January; 47(1):122–133. doi: 10.1021/ci600406v. [DOI] [PubMed] [Google Scholar]
- [70].Yin J, Henriksen NM, Slochower DR, Shirts MR, Chiu MW, Mobley DL, Gilson MK. Overview of the SAMPL5 Host–Guest Challenge: Are We Doing Better? J Comput Aided Mol Des. 2017; 31(1):1–19. doi: 10.1007/s10822-016-9974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Liu W, Lu X, Xue W, Samanta SK, Zavalij PY, Meng Z, Isaacs L. Hybrid Molecular Container Based on Glycoluril and Triptycene: Synthesis, Binding Properties, and Triggered Release. Chem Eur J. 2018. September; 24(53):14101–14110. doi: 10.1002/chem.201802981. [DOI] [PubMed] [Google Scholar]
- [72].Ndendjio SAZ, Isaacs L. Molecular Recognition Properties of Acyclic Cucurbiturils toward Amino Acids, Peptides, and a Protein. Supramolecular Chemistry. 2019. July; 31(7):432–441. doi: 10.1080/10610278.2019.1619737. [DOI] [Google Scholar]
- [73].Biedermann F, Uzunova VD, Scherman OA, Nau WM, De Simone A. Release of High-Energy Water as an Essential Driving Force for the High-Affinity Binding of Cucurbit[n]Urils. J Am Chem Soc. 2012. September; 134(37):15318–15323. doi: 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]
- [74].Monroe JI, Shirts MR. Converging Free Energies of Binding in Cucurbit[7]Uril and Octa-Acid Host-Guest Systems from SAMPL4 Using Expanded Ensemble Simulations. J Comput Aided Mol Des. 2014. April; 28(4):401–415. doi: 10.1007/s10822-014-9716-4. [DOI] [PubMed] [Google Scholar]
- [75].Liu W, Lu X, Meng Z, Isaacs L. A Glycoluril Dimer–Triptycene Hybrid Receptor: Synthesis and Molecular Recognition Properties. Org Biomol Chem. 2018; 16(35):6499–6506. doi: 10.1039/C8OB01575A. [DOI] [PubMed] [Google Scholar]
- [76].Barnett JW, Sullivan MR, Long JA, Tang D, Nguyen T, Ben-Amotz D, Gibb BC, Ashbaugh HS. Spontaneous Drying of Non-Polar Deep-Cavity Cavitand Pockets in Aqueous Solution. Nature Chemistry. 2020. May; p. 1–6. doi: 10.1038/s41557-020-0458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gibb CLD, Gibb BC. Guests of Differing Polarities Provide Insight into Structural Requirements for Templates of Water-Soluble Nano-Capsules. Tetrahedron. 2009. August; 65(35):7240–7248. doi: 10.1016/j.tet.2009.01.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Gibb CLD, Gibb BC. Binding of Cyclic Carboxylates to Octa-Acid Deep-Cavity Cavitand. J Comput Aided Mol Des. 2014. April; 28(4):319–325. doi: 10.1007/s10822-013-9690-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ewell J, Gibb BC, Rick SW. Water Inside a Hydrophobic Cavitand Molecule. J Phys Chem B. 2008. August; 112(33):10272–10279. doi: 10.1021/jp804429n. [DOI] [PubMed] [Google Scholar]
- [80].Kellett K, Kantonen SA, Duggan BM, Gilson MK. Toward Expanded Diversity of Host–Guest Interactions via Synthesis and Characterization of Cyclodextrin Derivatives. J Solution Chem. 2018. November; 47(10):1597–1608. doi: 10.1007/s10953-018-0769-1. [DOI] [Google Scholar]
- [81].Slochower DR, Henriksen NM, Wang LP, Chodera JD, Mobley DL, Gilson MK. Binding Thermodynamics of Host–Guest Systems with SMIRNOFF99Frosst 1.0.5 from the Open Force Field Initiative. J Chem Theory Comput. 2019. November; 15(11):6225–6242. doi: 10.1021/acs.jctc.9b00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Carrazana J, Jover A, Meijide F, Soto VH, Vázquez Tato J. Complexation of Adamantyl Compounds by β-Cyclodextrin and Monoaminoderivatives. J Phys Chem B. 2005. May; 109(19):9719–9726. doi: 10.1021/jp0505781. [DOI] [PubMed] [Google Scholar]
- [83].Rizzi A, Grinaway P, Parton D, Shirts M, Wang K, Eastman P, Friedrichs M, Pande V, Branson K, Mobley D, Chodera J, YANK: A GPU-Accelerated Platform for Alchemical Free Energy Calculations.; 2020.
- [84].Wang K, Chodera JD, Yang Y, Shirts MR. Identifying Ligand Binding Sites and Poses Using GPU-Accelerated Hamiltonian Replica Exchange Molecular Dynamics. J Comput Aided Mol Des. 2013. December; 27(12):989–1007. doi: 10.1007/s10822-013-9689-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Friedrichs MS, Eastman P, Vaidyanathan V, Houston M, Legrand S, Beberg AL, Ensign DL, Bruns CM, Pande VS. Accelerating Molecular Dynamic Simulation on Graphics Processing Units. Journal of Computational Chemistry. 2009; 30(6):864–872. doi: 10.1002/jcc.21209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Eastman P, Pande V. OpenMM: A Hardware-Independent Framework for Molecular Simulations. Computing in Science Engineering. 2010. July; 12(4):34–39. doi: 10.1109/MCSE.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Eastman P, Pande VS. Constant Constraint Matrix Approximation: A Robust, Parallelizable Constraint Method for Molecular Simulations. J Chem Theory Comput. 2010. February; 6(2):434–437. doi: 10.1021/ct900463w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Eastman P, Pande VS. Efficient Nonbonded Interactions for Molecular Dynamics on a Graphics Processing Unit. Journal of Computational Chemistry. 2010; 31(6):1268–1272. doi: 10.1002/jcc.21413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Eastman P, Friedrichs MS, Chodera JD, Radmer RJ, Bruns CM, Ku JP, Beauchamp KA, Lane TJ, Wang LP, Shukla D, Tye T, Houston M, Stich T, Klein C, Shirts MR, Pande VS. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J Chem Theory Comput. 2013. Jan; 9(1):461–469. doi: 10.1021/ct300857j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shirts MR, Chodera JD. Statistically Optimal Analysis of Samples from Multiple Equilibrium States. J Chem Phys. 2008. September; 129(12):124105. doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Trott O, Olson AJ. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry. 2010; 31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Khalak Y, Tresadern G, de Groot BL, Gapsys V. Non-Equilibrium Approach for Binding Free Energies in Cyclodextrins in SAMPL7: Force Fields and Software. JComputAidedMolDes. 2020;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Procacci P, Guarnieri G. SAMPL7 Blind Predictions Using Nonequilibrium Alchemical Approaches. JComputAidedMolDes. 2020;. [DOI] [PubMed] [Google Scholar]
- [94].Shi Y, Laury ML, Wang Z, Ponder JW. AMOEBA Binding Free Energies for the SAMPL7 TrimerTrip Host-Guest Challenge. JComputAidedMolDes. 2020;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Eken Y, Almeida NMS, Wang C, Wilson AK. SAMPL7: Host-Guest Binding Prediction by Molecular Dynamics and Quantum Mechanics. JComp. 2020;. [DOI] [PubMed] [Google Scholar]
- [96].Serillon D, Barril X. Testing Automatic Methods to Predict Free Binding Energy of Host-Guest Complexes in SAMPL7 Challenge. JComputAidedMolDes. 2020;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rizzi A, Murkli S, McNeill JN, Yao W, Sullivan M, Gilson MK, Chiu MW, Isaacs L, Gibb BC, Mobley DL, Chodera JD. Overview of the SAMPL6 Host–Guest Binding Affinity Prediction Challenge. Journal of Computer-Aided Molecular Design. 2018. October; 32(10):937–963. doi: 10.1007/s10822-018-0170-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Procacci P. Precision and Computational Efficiency of Nonequilibrium Alchemical Methods for Computing Free Energies of Solvation. II. Unidirectional Estimates. J Chem Phys. 2019. October; 151(14):144115. doi: 10.1063/1.5120616. [DOI] [PubMed] [Google Scholar]
- [99].Izadi S, Onufriev AV. Accuracy Limit of Rigid 3-Point Water Models. J Chem Phys. 2016. August; 145(7). doi: 10.1063/1.4960175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Marenich AV, Cramer CJ, Truhlar DG. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J Phys Chem B. 2009. May; 113(18):6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All SAMPL7 host-guest challenge instructions, submissions, experimental data and analysis are available at https://github.com/samplchallenges/SAMPL7/tree/master/host_guest. An archive copy of SAMPL7 GitHub repository host-guest challenge directory is also available in the Supplementary Documents bundle (SAMPL7-supplementary-documents.tar.gz). Some useful files from this repository are highlighted below.
Table of participants submission filenames and their submission ID: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/SAMPL7-user-map-HG.csv
Submission files of prediction sets: https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Submissions
Python analysis scripts and outputs: https://github.com/samplchallenges/SAMPL7/tree/master/host_guest/Analysis/Scripts
Table of performance statistics calculated for ranked methods for TrimerTrip dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Accuracy_ranked/TrimerTrip/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for TrimerTrip dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/TrimerTrip/StatisticsTables/statistics.csv
Table of performance statistics calculated for ranked methods for GDCC dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Accuracy_ranked/GDCC_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for GDCC (without optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/GDCC_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for GDCC (with optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/GDCC/StatisticsTables/statistics.csv
Table of performance statistics calculated for ranked methods for Cyclodextrin dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Accuracy_ranked/CD_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for Cyclodextrin (without optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/CD_no_optional/StatisticsTables/statistics.csv
Table of performance statistics calculated for all methods for Cyclodextrin (with optionals) dataset: https://github.com/samplchallenges/SAMPL7/blob/master/host_guest/Analysis/Reference/Accuracy/CD/StatisticsTables/statistics.csv