

Abstract
Sampling multiple binding modes of a ligand in a single molecular dynamics simulation is difficult. A given ligand may have many internal degrees of freedom, along with many different ways it might orient itself a binding site or across several binding sites, all of which might be separated by large energy barriers. We have developed a novel Monte Carlo move called Molecular Darting (MolDarting) to reversibly sample between predefined binding modes of a ligand. Here, we couple this with nonequilibrium candidate Monte Carlo (NCMC) to improve acceptance of moves. We apply this technique to a simple dipeptide system, a ligand binding to T4 Lysozyme L99A, and ligand binding to HIV integrase in order to test this new method. We observe significant increases in acceptance compared to uniformly sampling the internal, and rotational/translational degrees of freedom in these systems.
1. Introduction
Structure-based drug design allows for rational design of ligands, as computational methods can help predict desired qualities of a potential ligand prior to its synthesis.1–4 However, an understanding of ligand binding modes is often viewed as critical for structure-based design5–7 yet binding modes are not necessarily well known before compounds are made and tested.8–10
Thus, many computational methods seek to predict ligand binding modes. Several such methods for binding mode prediction are available, but overall computational prediction of binding modes is a difficult problem.8,11 One of the most commonly used methods for binding mode prediction is docking, which is able to sift through millions of compounds efficiently. Docking, however, does not tend to do well at predicting the true binding mode.9 On the other end of the spectrum of computational cost are free energy simulation-based methods, which are very promising for structure-based design and are attracting tremendous interest from industry.12–15
However, computational methods for studying binding have their limitations. Free energy methods for predicting binding affinity need to start close to, or sample the correct binding mode in order to offer accurate free energy predictions.13,16–18 This reliance on the starting position can cause issues; since the binding mode of a novel ligand has to be predicted and is typically slow to sample in a simulation,19 adequate sampling of the ligand’s motion in the binding site can be challenging. Even in the case of a congeneric series of molecules binding to the same target, the binding mode of the ligands can differ.8,20
In order to circumvent some of these short-comings of MD-based methods, we previously developed a mixed MD/nonequilibrium candidate Monte Carlo (NCMC) based method, and implemented it in a package called Binding modes of Ligands Using Enhanced Sampling (BLUES).21 Typically, Monte Carlo (MC) moves have difficulty achieving high acceptance rates in condensed-phase systems because of tight packing, allowing for only small perturbations to be performed on a system. NCMC provides a framework where a larger, instantaneous MC move can be broken up into a series of smaller perturbations. Between each perturbation the system is allowed to relax by applying dynamics. This process is repeated a number of times and the whole move is accepted or rejected based on the total work done during the perturbation steps. In BLUES we use NCMC moves to alchemically remove the interactions of a ligand and then reinstate them over the course of some number of steps (N). At the start of reinserting the ligand, a MC move can also be performed to further improve binding mode sampling. By slowly removing and regrowing the ligand, we can insert the ligand into a new binding mode and allow the rest of the system to slowly relax in response to the ligand’s motion, potentially leading to higher rates of acceptance compared to instantaneous MC moves.
As noted, an MC move can be performed at the midpoint of the NCMC protocol. In our original paper describing the BLUES method, the only such move offered was a center of mass rotation of the ligand. In subsequent work, the MC moves available were further expanded to include protein side-chain torsions22 as well as selected torsions of the ligand.22,23
These types of moves are helpful in generating small perturbations of the ligand’s binding mode, but ideally we would like to be able to generate binding mode predictions and sample between those directly. Generally, proposing reasonable candidate binding modes is a relatively easy task, since docking methods tend to do a good job at generating plausible binding modes, but are poor at ranking these binding modes.9,24,25 In many cases, such poses can be equilibrated via MD simulations to find a variety of different stable or metastable binding mode candidates.20,26–28
While some methods can improve sampling of a ligand’s internal degrees of freedom, we are not aware of any current MC method which can efficiently hop between potentially disparate predefined ligand binding modes in a way that preserves detailed balance.
Techniques such as Rosenbluth sampling,29 or configurational bias Monte Carlo30 are sampling methods originally applied to flexible molecules to grow and arrange polymers favorably, but these methods do not offer a way to directly sample between two specific conformations of a molecule.
Distance Geometry is another technique used to perform conformational analysis of ligands. methods.31 In this technique the atoms of a molecule are randomly placed and then minimized to generate a new structure. Like configurational bias MC, however, distance geometry methods do not satisfy detailed balance since they depend on a minimization step.
To more efficiently sample binding moves, we have developed a new Monte Carlo based method to directly sample transitions between candidate poses–which may even be in different binding sites. Furthermore, we have implemented this method in the BLUES package in connection with our previous BLUES NCMC-based method in an attempt to directly sample multiple binding modes in protein systems.
2. Theory and computational methods
Here, we first describe the background and motivation of the method we implement here, then move on to discuss technical details of its implementation and how it was tested.
2.1. Smart Darting allows for selective sampling between minima
Our novel Monte Carlo method is a logical descendant of another Monte Carlo sampling method called Smart Darting Monte Carlo.32 The general process of Smart Darting involves defining two key pieces of information. The first piece we need to specify is a set of “darts”, which represent different configurations of the system that are of interest. The second piece we need to specify is a set of parameters (and their ranges), which correspond to and define each of those darts, in order to specify the boundaries associated with each conformation.
To explain Smart Darting in more technical terms, a set of darts d0, d1…dj are first specified. Each of those darts corresponds to a particular set of microstates (i.e. a metastable binding mode which was given as input) each of which is defined by a set of parameters k0, k1,…kn, with each parameter ki having an associated range . Each parameter refers to a quantity that defines that microstate–such as a torsion angle, or some distance measurement, such as the distance between two atoms. The range should be the same for each parameter ki, (which is necessary to preserve detailed balance, or the acceptance criterion needs to be altered). When a given parameter is within its associated range, we refer to it as being within that parameter region. These parameters (and the size of the parameter range) are user-defined input and should be designed to cover the typical value ranges of those parameters, which can be determined for example by running short exploratory/equilibration simulations. When attempting to make a Smart Darting Monte Carlo move, the parameters are evaluated (the current value of that parameter is checked) for each dart. When the parameter is evaluated, if the current configuration is within the parameter regions for all rk of a given dart—which we refer to as being within the dart—then the system can jump to another set of parameters with equal probability. In the process of jumping to the new configuration, a new k0, k1,…kn are each generated–either uniformly between the ranges for a given or deterministically through some one-to-one mapping from the old k0 to the new k0. Additionally, to maintain detailed balance, no Smart Darting move can be performed on a system if the system is within the range of multiple darts.
2.2. Molecular darting moves use internal coordinates as part of move proposals
In our novel Smart Darting-inspired methodology, called Molecular Darting (MolDarting), the parameters that define a dart are defined by the internal torsions of the molecule, as well as a translational and rotational distance to a given configuration. The internal coordinates are described by a Z-matrix, which describes the molecule’s configuration in terms of internal bond distances, angles, and dihedrals. For this case of MolDarting, we assume the bond and angle internal coordinates are invariant between ligand conformations, and that the dihedral internal coordinates are independent of one another. The translational distance is defined by the Euclidean distance between the first atom of the Z-matrix of the current configuration and the corresponding atomic positions of the given dart. We used Chemcoords33 to generate the internal coordinates for our molecules of interest. The rotation matrix of the first three Z-matrix atoms of the ligand is calculated to each of the first three Z-matrix atoms of the references. The rotational distance is calculated by Eq 1, where R is the rotation matrix.
| (1) |
When using MolDarting on a protein-ligand system, it’s necessary to first account for the overall rotation and translational changes for the protein-ligand complex in regards to the reference darts. To account for those rotational and translational changes, heavy atoms of the residues around the binding site are chosen. When checking if the current configuration is within the rotational and translational regions, the chosen binding site residues of the selected dart are superposed to the same binding site residues of the current pose, then the rotational and translational distances are calculated.
When MolDarting between binding modes, the proposed internal coordinates from MolDarting are uniformly chosen anywhere inside the newly selected internal coordinate region (Figure 1). The rotational and translational motions are deterministically updated by assessing the displacement from the starting pose to the center of each of their respective regions and then applying those same displacements again after it is MolDarted (Figure 2, Figure 3).
Figure 1: Dihedrals are uniformly sampled during MolDarting.
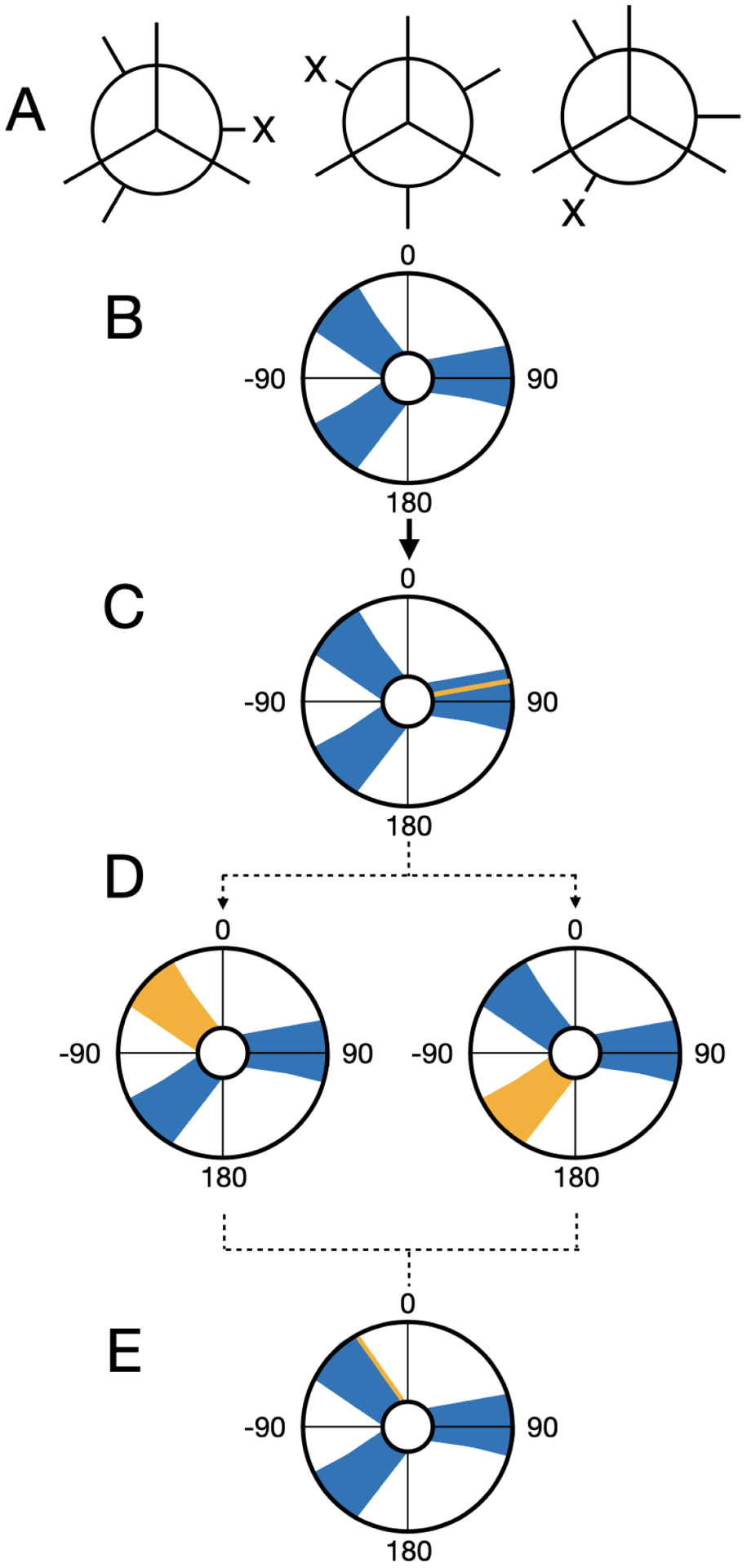
We illustrate how we perform our dihedral darting moves using a rose plot representation of a dihedral angle (in degrees) as an example. The dihedral regions are represented by the blue areas, and the current dihedral angle is represented by the yellow line/areas. In this example, there are three total darts, each with an associated region. (A) The Newman projection of a hypothetical ligand illustrating three different stable conformations. (B) A representation of the three dihedral regions for the three conformations. (C) When a particle is within a dihedral region then a darting move can be performed. (D) When MolDarting the dihedrals, the new dihedral is selected uniformly from a region the dihedral is not currently in (shown in yellow). The arrows refer to the two potential outcomes of the MolDarting move in which the ligand is darted to a new configuration. (E) One of the other dihedral regions are chosen randomly (with equal probability) to be MolDarted, and then a new dihedral is chosen randomly from the chosen region, resulting in a new configuration.
Figure 2: Translations are handled deterministically during MolDarting.
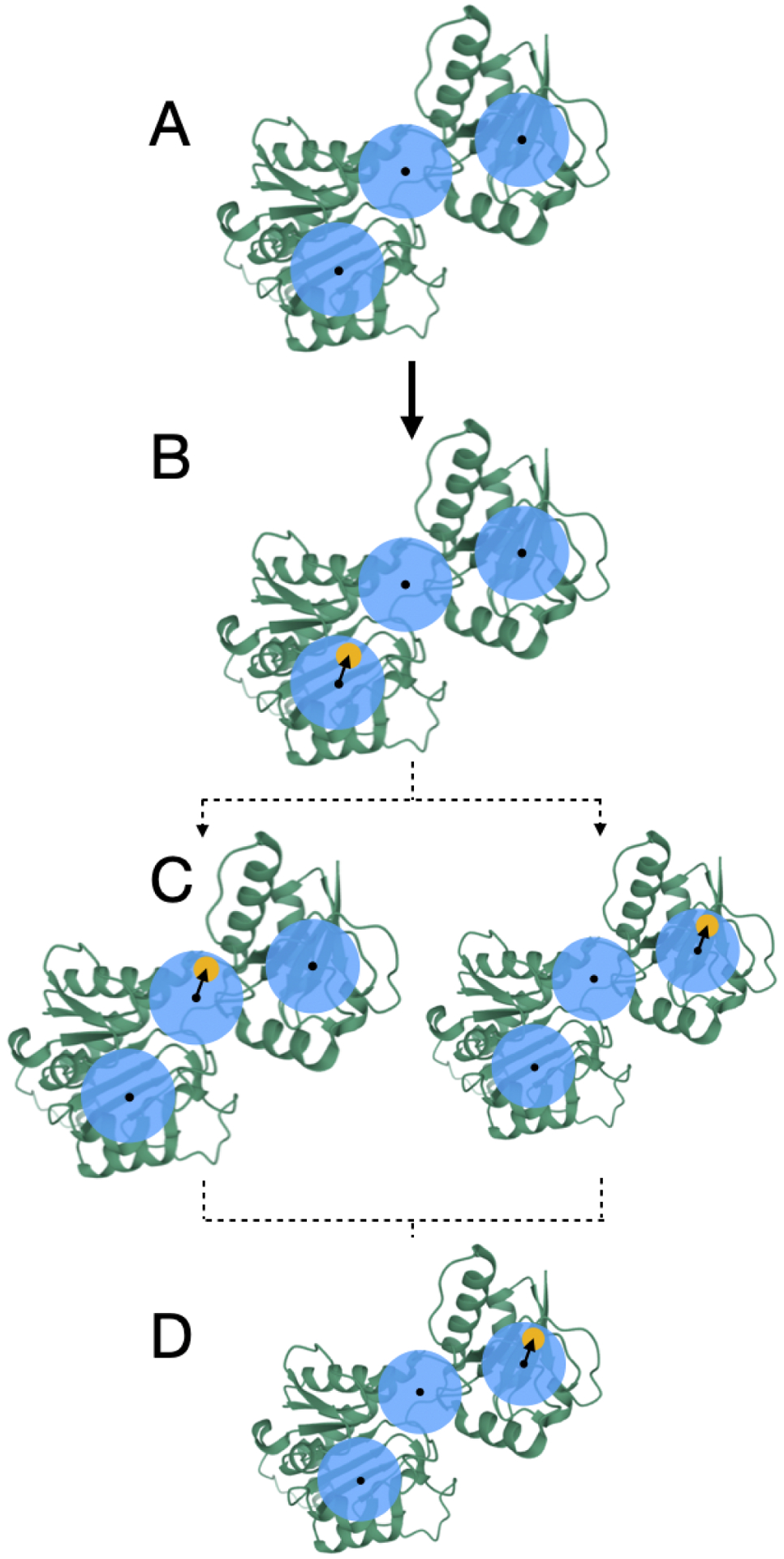
We illustrate how we perform our translational darting moves using a 2-dimensional translational region as an example, with a single particle, (that can represent an atom of a ligand, for example) that will be Moldarted. The translational regions are represented by the blue circle, with the center of each translational region represented by a black dot, and simplified molecule represented by yellow circles. In this example, there are three total darts. (A) A representation of the three rotational regions used. (B) When a particle is within a translational region, the vector from the particle’s center, to the translational region’s center is calculated (represented by the arrow). (C) When MolDarting the vector calculated in (B) is applied to the center of each other translational region to determine the particle’s new position. The dotted arrows refer to the two potential outcomes of the MolDarting move in which the ligand is darted to a new configuration. (D) One of the new reference regions are chosen randomly (with equal probability) to be MolDarted, resulting in a new configuration.
Figure 3: Rotations are handled deterministically during MolDarting.
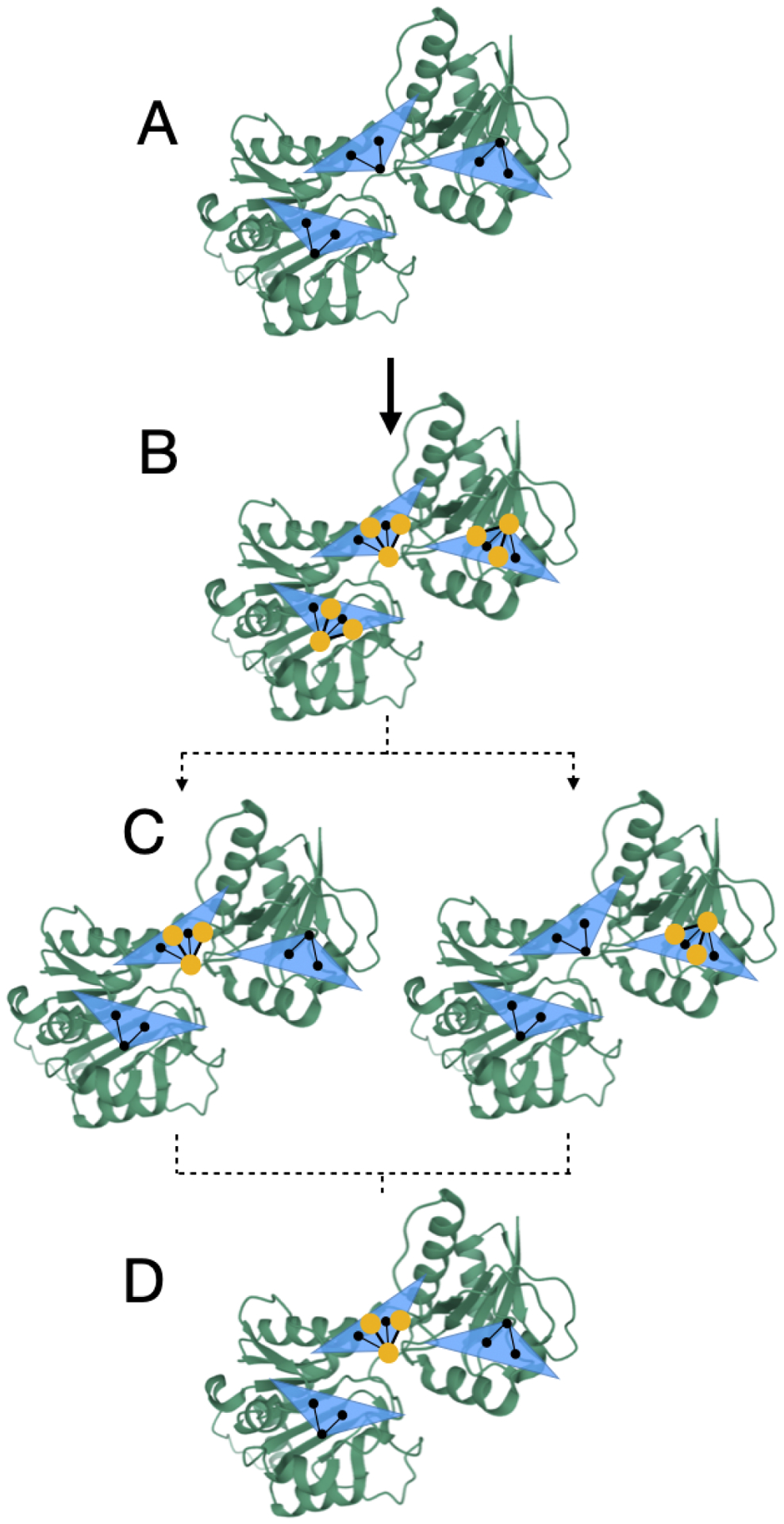
We illustrate how we perform our rotational darting moves using a 2-dimensional rotational region as an example, with a single molecule that will be moved via MolDarting. The rotational regions are represented by the blue triangle, with the center of each rotational region (which was defined by some reference pose) represented by the three black circles connected by black lines, and the ligand in our simulations represented by the yellow circles connected by yellow lines. In this example, there are three total darts, each with an associated rotational region. (A) A representation of the three rotational regions used. (B) When a particle is within a rotational region the rotation matrix is calculated from the current positions to the reference positions. (C) When MolDarting, the rotation matrix calculated in (B) is applied to the reference positions of each other rotational region to determine the molecule’s new position. The dotted arrows refer to the two potential outcomes of the MolDarting move in which the ligand is darted to a new configuration. (D) One of the new reference regions are chosen randomly (with equal probability) to be MolDarted, resulting in a new configuration.
When combining MolDarting with BLUES, an additional step is added to the MolDarting procedure. We found that these restraints were needed because when the ligand steric interactions are diminished, it is more labile inside the binding pocket and can frequently end up outside the darts. To reduce the lability of the ligand, an orientational restraint, also known as a Boresch-style restraint34 is applied to the first three ligand Z-matrix atoms, relative to three reference atoms in the protein. This restraint restricts the orientation relative to the binding site via restricting one distance, two angles and three torsions, and involves three reference atoms in the ligand and three in the receptor. Here, we scale this restraint with the lambda parameter that controls the electrostatics and sterics; when the ligand is fully non-interacting, the restraints are in full effect (Figure 4). To maintain detailed balance when applying restraints, before the NCMC move occurs the we check if the ligand is currently within a dart; if it is then the orientational restraints associated with that pose will be turned on over the first half of the NCMC move. If the ligand is not within the same dart as at the start of the move, then the move is rejected.
Figure 4:
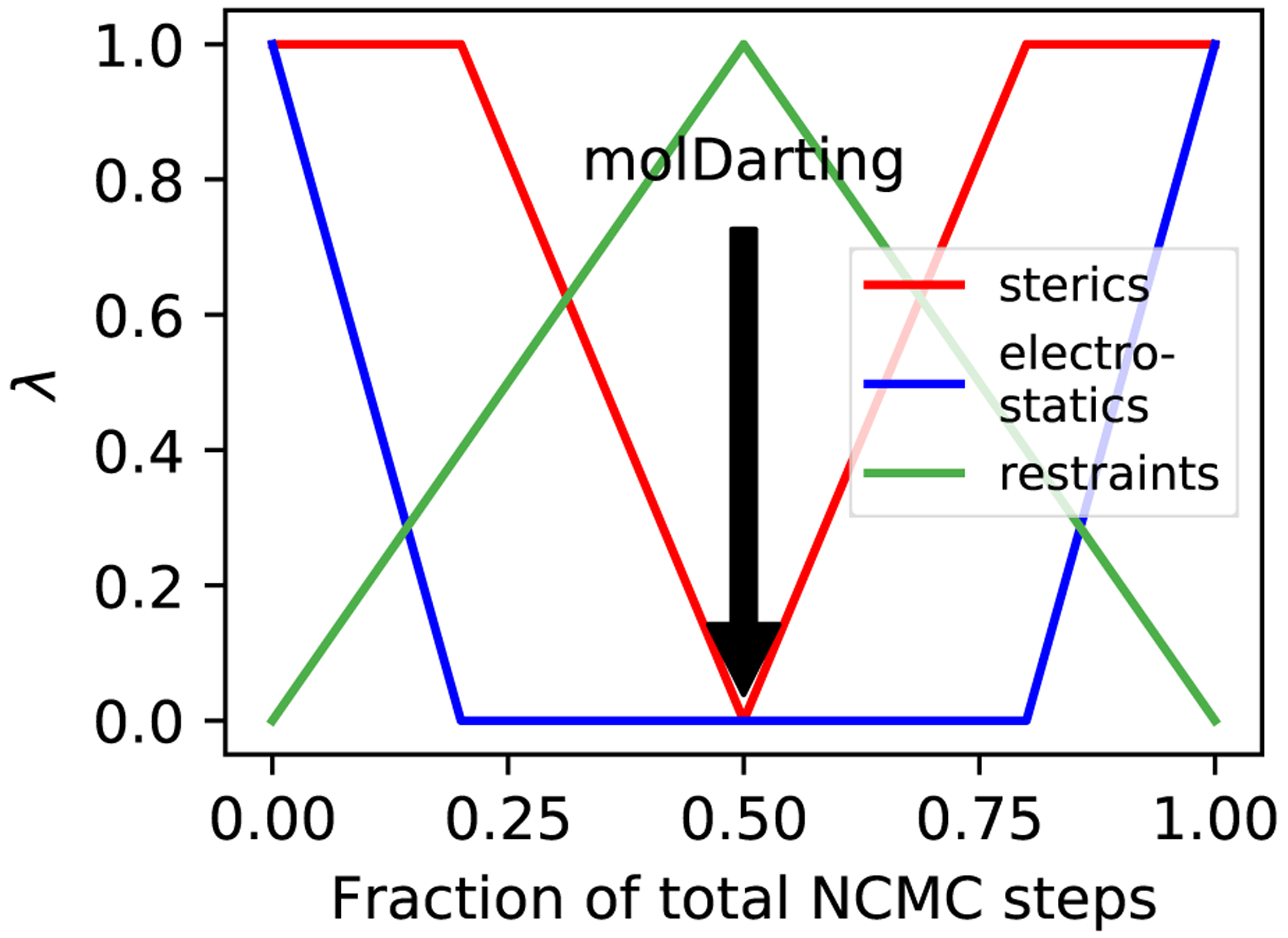
Restraints are included in the NCMC switching protocol. In order to keep the ligand in the binding site while the ligand’s interactions are off, an orientational restraint is used which corresponds to the dart that the ligand is in at the beginning of an NCMC move proposal. At the middle of the NCMC protocol, a MolDarting move is performed, and the restraint switches to a new orientational restraint corresponding to the new dart, which is subsequently turned off throughout the rest of the protocol.
Subsequently, after the MolDarting move is performed, the restraints corresponding to the new pose are turned on, and the previous pose’s restraints are turned off. Finally, after the NCMC move occurs, the parameters are evaluated again to see if they are within any dart. The modulation of steric, electrostatic and restraint interactions over the course of the NCMC move are illustrated in Figure 4, and the overall procedure is illustrated in Figure 5.
Figure 5:
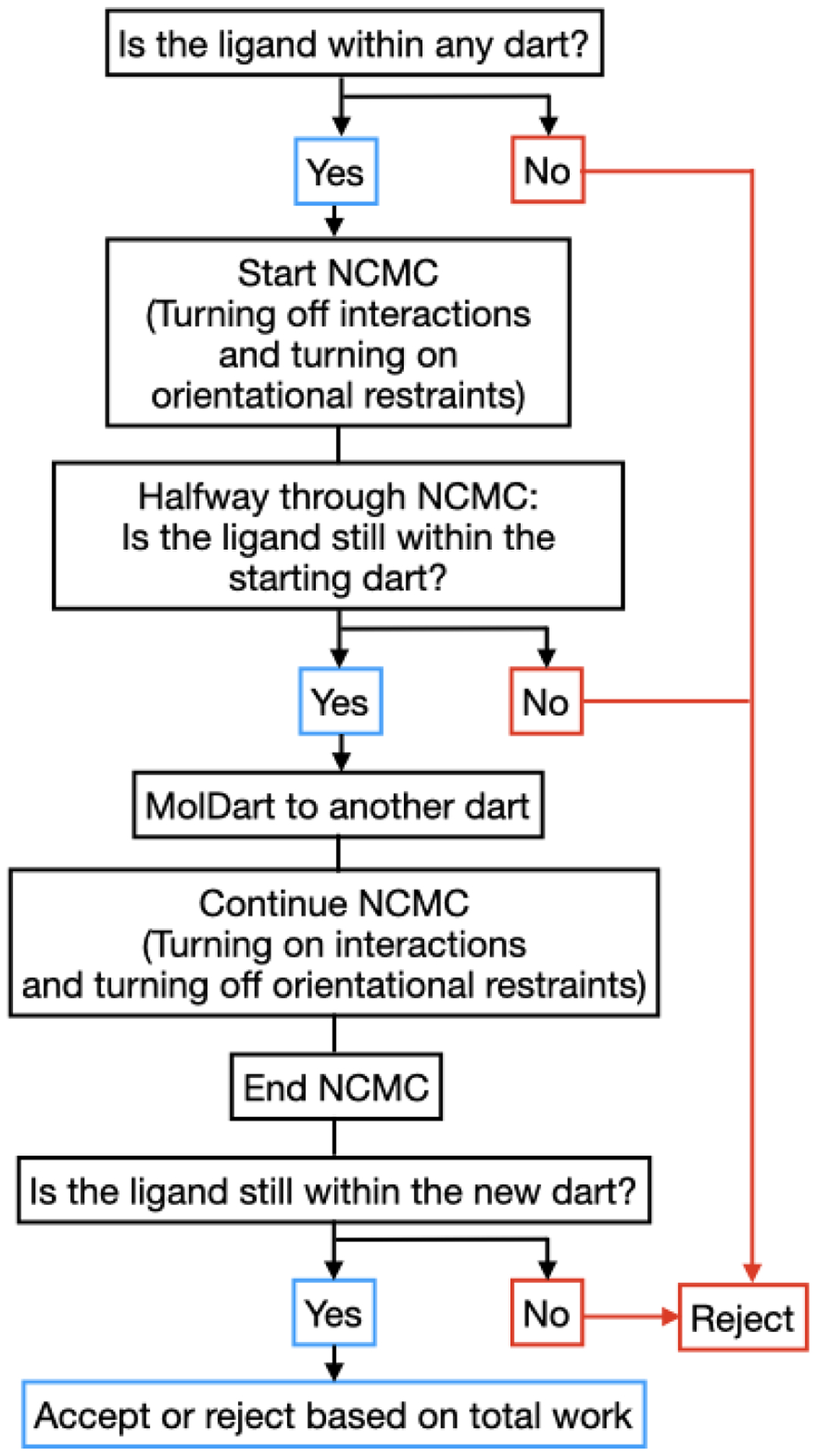
Adding restraints with NCMC and MolDarting requires additional consideration. When restraints are used alongside NCMC and MolDarting, it’s necessary to take into account several additional factors, which are illustrated by this flowchart and elaborated further in Section 2.2.
If the ligand is in a different pose than the pose the ending restraints were associated with, then the move is automatically rejected, since such a move would not be reversible. Otherwise the protocol work (the work that is done over the course of the NCMC move) determines whether the NCMC move is accepted or rejected. The application of the restraint is taken into account in the work done during the course of the NCMC move.
Taking into account the major degrees of freedom of the molecule allows reversible MolDarting moves between different potential ligand binding modes, not only with different ligand conformations, but potentially even in separate binding pockets.
2.3. We tested Molecular Darting on three different systems
To validate and explore the potential of MolDarting, we look at three different system with different requirements needed to sample binding modes. The first system explored is an alanine-valine dipeptide. While not typically considered a ligand, this peptide is a simple model system which exhibits three different stable conformations that vary by an internal torsion and can be slow to sample through plain MD.22 It also is a good test system for the darting approach we develop here, as MolDarting can be applied to any selected object in our system, not just a ligand. Here, since sidechains play an important role in ligand binding it is also important to be able to sample the rotamers in a binding site. The second system we look at with MolDarting is T4 lysozyme L99A with toluene bound, where the binding modes varies by rotation and translation. The final system we look at is HIV integrase with a variety of ligands bound. HIV integrase is an interesting test system because it has multiple binding sites where ligands can bind, and has proven difficult for binding mode predictions in a previous blind challenge,8 and we would like to test whether MolDarting can directly sample the binding modes in each binding site.
3. Methodology
3.1. System preparation
3.1.1. Alanine-valine dipeptide system setup
An alanine-valine dipeptide system was created using tleap from AmberTools 16.35 The amber99SBILDN forcefield was used for the protein parameters. Simulations were carried out at 300K with a Langevin integrator using a 0.002ps step size in implicit OBC2 solvent36 using OpenMM version 7.3.37 Nonperiodic cutoffs were used, with the hydrogen bonds constrained and a 1/ps friction applied. The peptide’s CA, N, and O backbone atoms were restrained using a restraint of 25 kcal/(mol·angstrom2) based on their starting conformation.
To prepare for MolDarting between the different stable rotameric states for this dipeptide, we initially ran a 100 ns simulation to identify the dihedral minima of the system. From this simulation, we found three stable valine rotamers, with dihedral maxima at approximately −170, −65, and 53 degrees. These dihedrals were calculated by measuring the dihedral angle between the CA, CB and CG1 atoms of valine on the alanine-valine dipeptide atom using MDTraj 1.9.3.38
From the three maxima, regions were chosen so that the region size encompassed 95% of the probability density associated with that dihedral maximum, estimated from a kernel density approximation with a 0.2 bandwidth and a Gaussian kernel.
Simulations of the alanine-valine system were performed using BLUES for 150000 iterations, with each iteration consisting of 1000 steps of MD and an instantaneous MC move consisting of either a sidechain rotation using the SideChainMove class or a MolDarting move using the MolDartMove class. The code used to run these simulations can be found in the SI.
Populations of the three dihedral maxima were separated based on the following bin definitions: from (−120, −40] defined one bin (with a maximum at 68 degrees), from [20,100] defined another bin (with a maximum at 68 degrees) and a third bin is discontinuous and is defined between [−180, −120] and [115,180] (with a maximum at 180 degrees).
3.1.2. T4 lysozyme/toluene system and simulation setup
Here, we used the same T4 lysozyme and toluene system and parameters for NCMC from our previous work.21 The only difference in our simulation protocol was that now a MolDarting move was performed instead a random center of mass rotation. For the MolDarting move, a rotational dart of 40 degrees was defined, using two poses of the non-symmetrically equivalent binding poses as a reference. A Boresch restraint with a force constant of 3kcal/(mol*angstrom2) for the radial component and 3kcal/(mol*rad2) for the angular and dihedral components was used with the first three internal coordinate atoms of toluene as chosen by ChemCoords (being the C6, C4, and C5 atoms respectively of the toluene molecule) and the CA atoms of PRO85, ALA98, and LEU117 using the Yank’s BoreschRestraint class to implement the restraints with the provided atoms from the receptor as the restrained_receptor_atoms and the ligand atoms as the restrained_ligand_atoms arguments for the class.39
3.1.3. HIV integrase system setup
We used the 4CHY pdb file as the basis structure for our study to serve as a uniform starting point for docking and equilibration. Omega from Openeye40 was used to generate the conformers for the 4 ligands from the PDB files of 4CHY, 4CGD, 4CHZ, and 4CJV,41 which are shown in Figure S1 in the SI, and Fred was used to dock the compounds in the three different binding sites.42
We then looked at the protocol work distributions that are accumulated throughout the NCMC MolDarting move attempts (Figure 11).
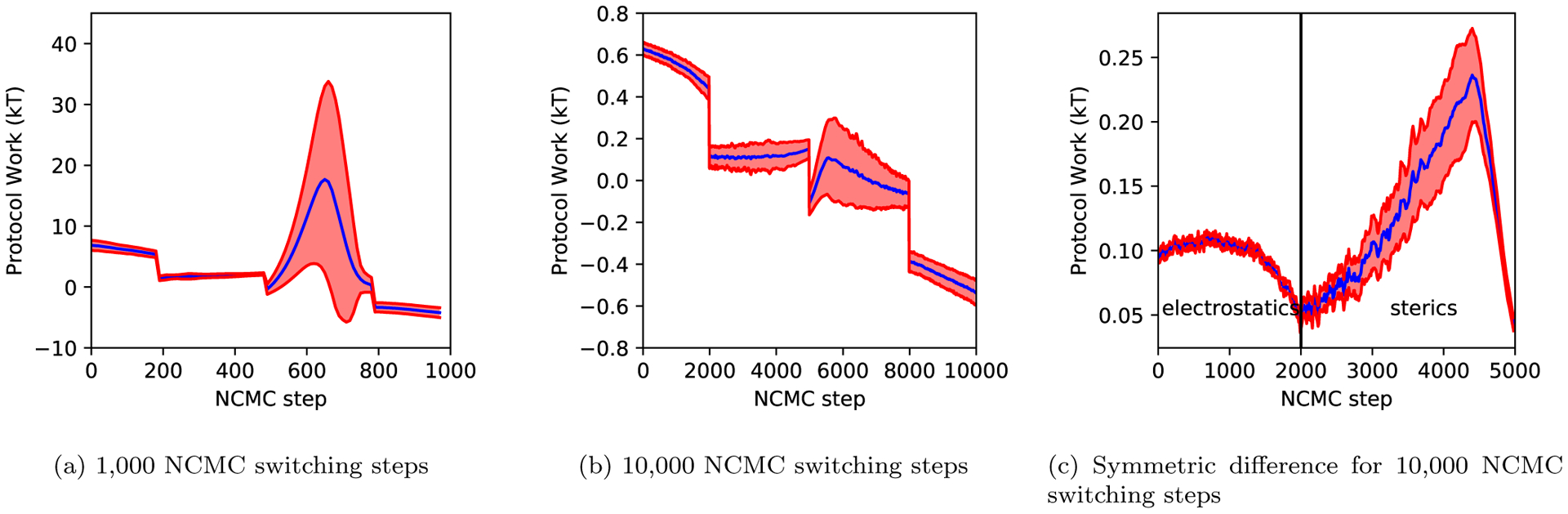
Figure 11: High protocol work leads to rejection for MolDarting moves.
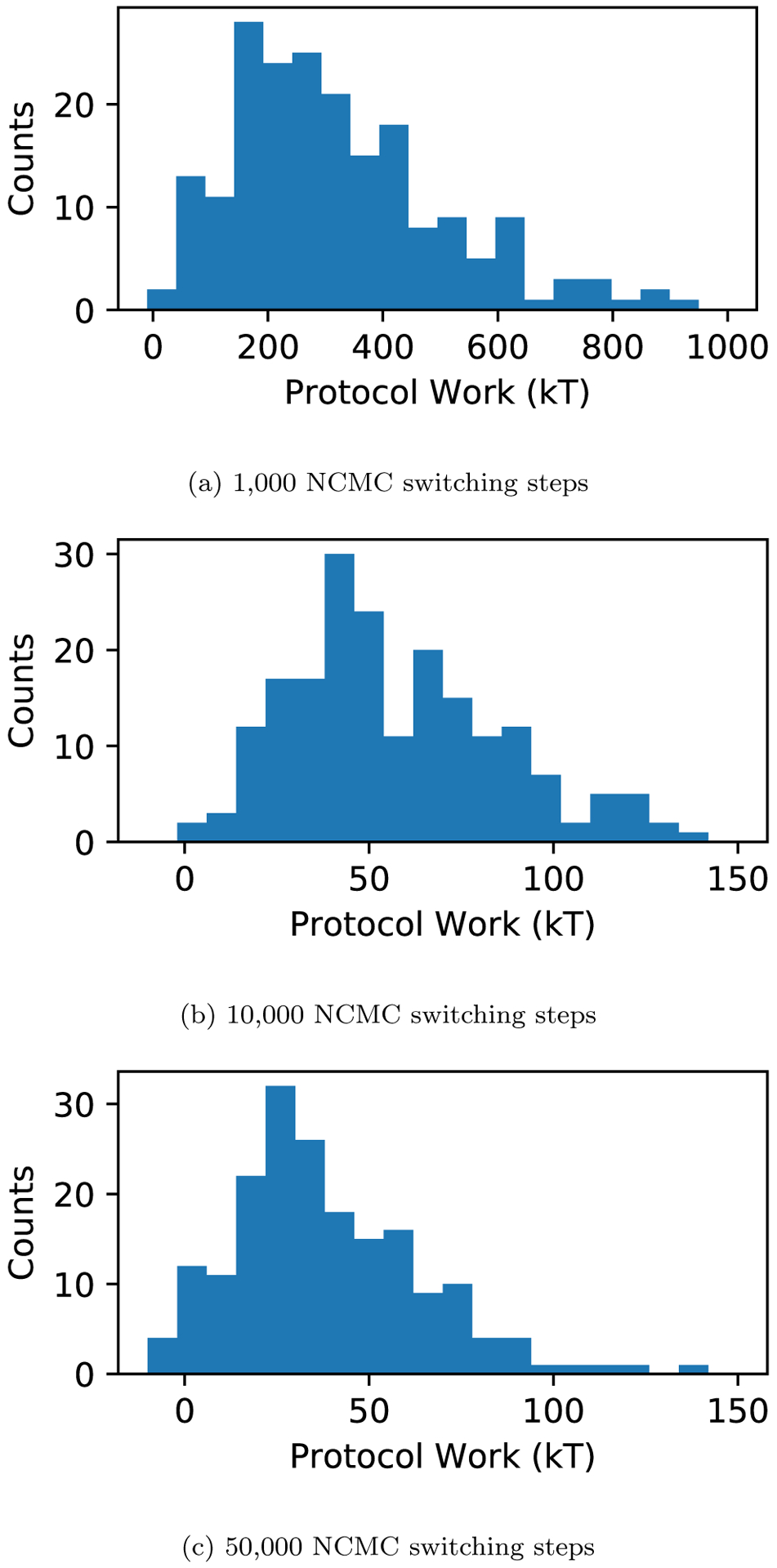
(a) The protocol work distribution of NCMC with MolDarting move attempts with 1,000 (a), 10,000 ((b), and 50,000 ((c) NCMC switching steps with the HIV integrase and the ligand found in 4CGD. The protocol work done over the course of the NCMC moves generally is highly positive (unfavorable), leading those moves to be rejected by the acceptance criteria. There are a small number of cases when the work values approach zero or are negative, but these were still rejected. In these cases, rejection was due to the ligand ending up outside the defined regions at one of the checks during the course of the move.
The highest scoring poses from docking were used, and to generate a diverse set of structures, root-mean-square deviation (RMSD) centroid clustering was performed on the poses, and the most diverse poses retained, to promote pose diversity. To further elaborate on the clustering procedure, the first centroid was defined using the top-scoring docking pose, and the subsequent centroids were chosen which were the greatest RMSD distance away from the other existing centroids for that binding site. Clustering of poses were done separately for each binding site, and the two poses with the centroids furthest from the top scoring pose were used as reference poses for use with MolDarting, for a total of three poses per binding site. Antechamber was then used with the AM1-BCC method35,43 to assign partial charges to the molecules.
Finally, Amber was used to add missing sidechains, heavy atoms, and hydrogens to the protein, with the parameter set from ff14SB used for the protein.35 Because the binding sites of HIV integrase are solvent exposed, we chose to use OBC2 implicit solvent model36 to bypass solvating and desolvating the binding sites in response to the ligand being MolDarted.
Equilibration MD simulations were performed at 300K for 1 ns for each binding pose. The positions of this equilibration trajectory were saved every 10,000 steps.
Unless otherwise noted, the simulation settings were the same as alanine-valine dipeptide system. The equilibration simulation trajectories were also used to define the dihedral regions. Kernel density estimation (KDE) was performed on the dihedral internal coordinates from the trajectory with a bandwidth of 0.5. From this, the maxima in the dihedral KDEs were identified. The maxima that the dihedral was closest to at the end of equilibration was used to determine the start of the region for that dihedral. The width of the dihedral regions was set so that these regions account for 95% of the probability density estimated by KDE. The width was calculated by first finding the total probability density contained within a maximum, and then expanding the width of the region starting at the maximum until 95% of that maximum’s probability density was covered by the region. During MolDarting simulations, restraint atoms were automatically chosen from the heavy atoms within 10 angstroms of the ligand using Yank.39 The production MolDarting simulations for each ligand were all started in the LEDGF binding site. For each ligand, a total of 4 production NCMC + MD simulations were run using BLUES. These simulation was run for 200 iterations and either 1,000, 10,000, or 50,000 NCMC steps for each iteration, with 10,000 MD steps after each iteration. We used MDTraj38 to analyze the proximity of the ligand to the protein before each MolDarting attempt where the ligand was within a dart, and after the MolDarting move.
4. Results
4.1. We validated the internal coordinate sampling of our method against uniform dihedral sampling of the valine-alanine dipeptide.
We assessed the ability of MolDarting to sample the sidechain torsion of the valine-alanine dipeptide in implicit solvent. We also the compare the sampling efficiency of Moldarting to that of uniform sampling of the torsion. We applied the MolDarting procedure described in Section 3.1.1 to validate this MC move correctly samples the correct population distributions, and to compare the sampling efficiency of MolDarting to a traditional MC method. Both methods converged to the same values for the three dihedral populations (Figure 7). Across seven simulation replicates using MolDarting, the acceptance rate of MolDarting moves was only 2.23%±0.6%, compared to the acceptance rate of uniform sampling at 8.04% ± 0.5%. Although the acceptance rate for molecular darting was lower, the number of transitions generated between dihedral populations was nearly doubled compared to uniform dihedral sampling, with an average of approximately 3400 transitions generated with MolDarting compared to approximately 1400 transitions on average with uniform dihedral sampling. Thus, because of the targeted nature of MolDarting, the number of transitions between conformations is higher than the uniform sampling case, despite the lower number of accepted moves.
Figure 7: MolDarting efficiently samples the conformations of valine-alanine.
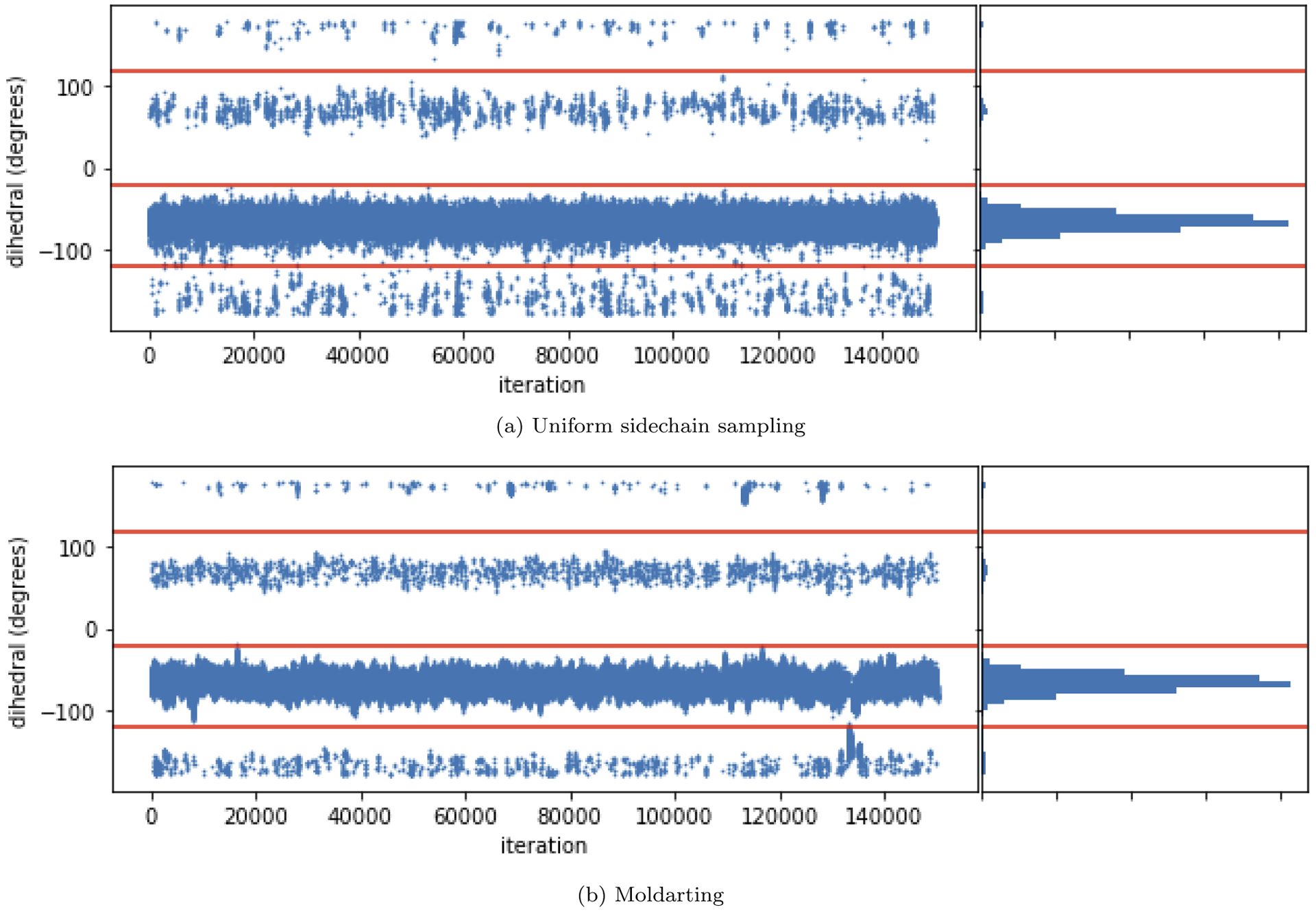
(a) (top) A trajectory consisting of MD+MC uniform rotations of the valine sidechain, with the histogram of the data (right). (b) (bottom) A trajectory consisting of MD+MC MolDarting moves of the valine sidechain. Molecular darting converges to the same distribution as uniform torsion rotations. However, MolDarting ends up being about twice as efficient at generating torsion transitions in this system. The red horizontal lines are included to help visually separate the three binding modes.
4.2. We applied Molecular Darting to sample the binding modes of toluene in T4 lyosozyme L99A
We further evaluated our method to sample rotational and translational degrees of freedom by applying MolDarting to sample the binding modes of toluene bound to T4 lysozme L99A. Toluene exhibits four binding modes when bound to T4 Lysozyme L99A. These binding modes vary by rotational and translational degrees of freedom; two are distinct and vary by a rotation, and the other two binding modes are symmetry-equivalent to the first pair.21 We applied MolDarting sampling with BLUES to the non-symmetric binding modes of toluene. The populations of the two binding modes were selectively sampled using MolDarting, without sampling the non-symmetric binding modes (Figure 8). MolDarting also was able to recover the correct populations of the binding modes, with the correct population split being 60:40, and our triplicate runs giving 58% ± 3% for the dominant binding mode and 42% ± 3% for the less populated binding mode. The acceptance rate for these moves over these trials was approximately 22%, which is roughly two times the acceptance rate for random center of mass moves we explored in the original BLUES paper,21 which further shows the benefit of targeted moves.
Figure 8: MolDarting generates selective transitions between binding modes.
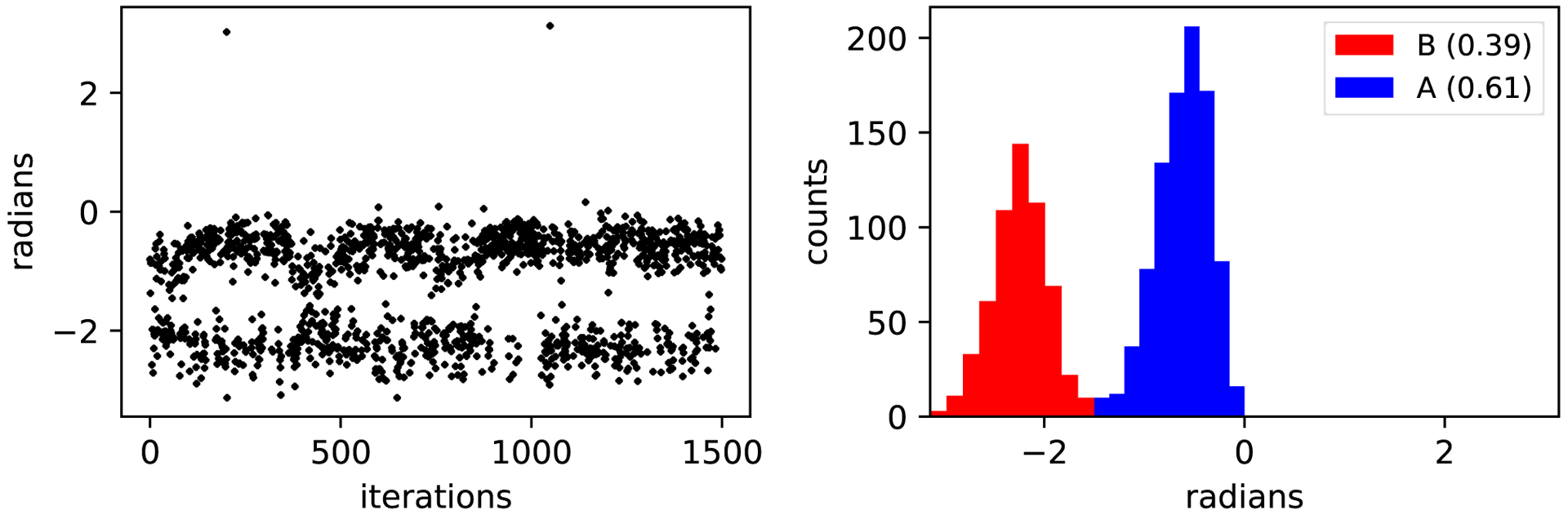
Toluene has four binding modes in the binding site, but only two of the binding modes are sampled here, due to the targeted nature of MolDarting. MolDarting is able to reproduce the correct relative probabilities of both binding modes, which are approximately 60% for binding mode A (the crystallographic binding mode), and 40% for the noncrystallographic pose.
4.3. Molecular Darting does not accelerate sampling when outside the dart
Sometimes, running longer simulations on the T4 lysozyme/toluene system resulted in toluene switching to the symmetry-equivalent binding mode (Figure 9). When this occurs, the ligand ends up being outside the pre-specified darts we defined in this test, and thus MolDarting moves cannot be attempted. We could have instead included all four ligand binding modes (two symmetry-equivalent pairs) as darts, but we elected not to here as we wanted to focus on non-redundant sampling. This issue highlights a key point: while MolDarting can be used to accelerate sampling, it is only effective when the system is within the selected darts; when outside the darts, we are effectively running plain MD. Thus, to maximize the applicability of MolDarting moves, care should be taken when defining the regions used for MolDarting.
Figure 9: MolDarting does not improve sampling when the simulation moves outside the darts.
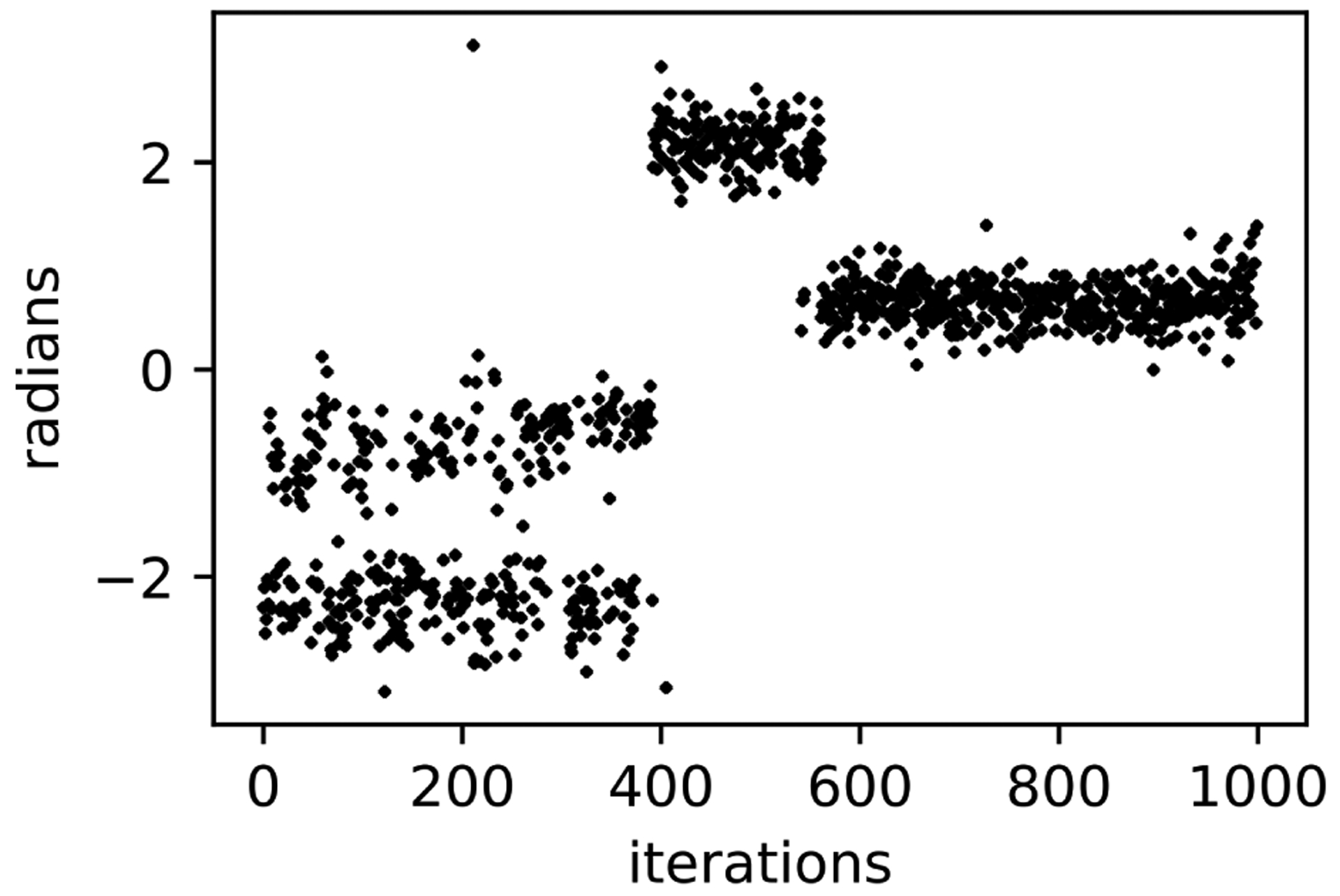
Here, the initial binding modes of toluene between 0 and π radians are well sampled (in the first 400 iterations), since these are covered by the rotational regions from MolDarting. However if the simulation leaves that region, then a MolDarting move cannot take place, and thus the simulation becomes just a normal MD simulation. In this particular simulation, around the 400th iteration toluene flips to the symmetric equivalent binding mode, which is not covered by the rotational regions, greatly reducing sampling.
Essentially, MolDarting attempts to trade bias for efficiency. More random procedures, like our initial translational moves in BLUES, allow enhanced exploration of binding mode transitions regardless of what pose the ligand is in, but do so rather inefficiently since so many proposed moves are to unfavorable binding modes. MolDarting requires more advance input or bias – selection of a set of potential binding modes to focus sampling on – and thus is able to ensure that proposed moves focus near those binding modes, potentially enhancing efficiency, but when the simulation strays from pre-defined binding modes, no enhanced sampling is possible.
4.4. We attempt to use Molecular Darting to explore multiple binding modes of HIV integrase Ligands
We applied Molecular Darting to an HIV integrase system with a set of diverse ligands. We chose HIV integrase in this study since this protein has three distinct binding sites ligands potentially bind to, leading to a plethora of potential binding modes that were hard for methods to discriminate between in a previous blind challenge.8 By using MolDarting we aimed to sample the various binding modes in the three binding sites in a single simulation.
The ligands we tested were chosen from the SAMPL4 dataset to include a diverse set of ligands as well as a diverse set of three poses in each binding site, for a total of 9 different binding modes (Section 3.1.3).
We attempted to use MolDarting to sample between binding sites. However, in all the cases with the ligands we studied, the acceptance rate for the moves was 0, thus no moves were accepted.
We looked at two possible sources that could lead to these MolDarting moves being rejected. One possible source of rejection is that the ligand falls outside the regions when MolDarting is being attempted, leading to these moves being rejected.
Another possible source of rejection is the protocol work produced during the move is high, so these moves are rejected by the acceptance criteria.
We first looked at the distribution of attempted MolDarting moves for the ligands (Figure 10). We found that although some moves did end up outside the defined regions (indicated by the ligand staying in the initial binding mode, shown in red), the majority of times, the ligand is being proposed to a new binding mode. While our handling of the regions could be improved, it does not appear to be the major cause of MolDarting moves being rejected.
Figure 10: MolDarting attempts sample all the defined binding modes.
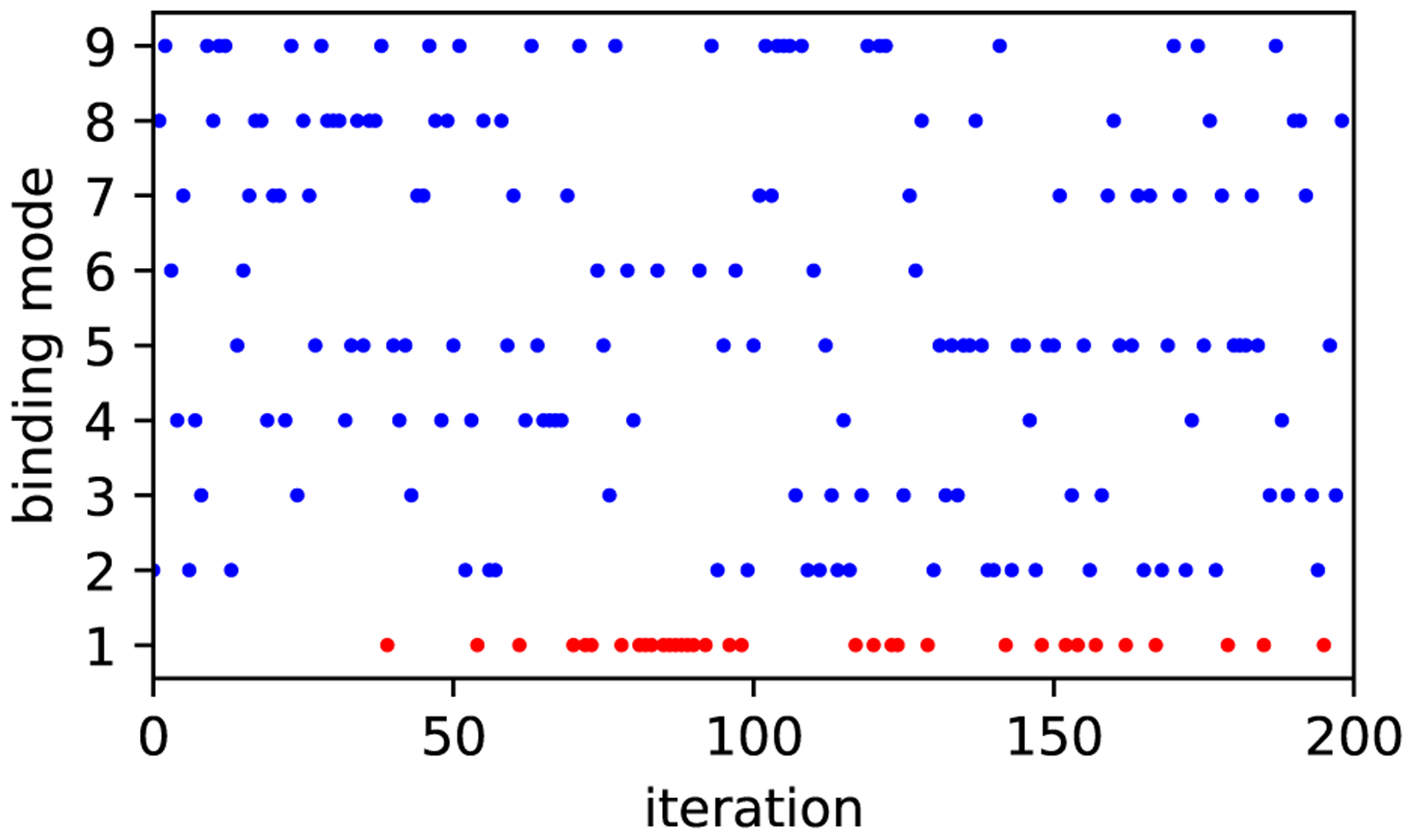
We looked at the binding modes sampled by MolDarting moves attempts. All 9 binding modes that were used for MolDarting with this ligand (4CGD) were sampled over the 200 iterations performed. The ligand started in binding mode 1. The points in blue indicate MolDarting move attempts which were successful at sampling new binding modes, while the red indicates that the ligand was outside the defined regions, so no darting move was attempted.
We then looked at the protocol work distributions that are accumulated throughout the NCMC MolDarting move attempts (Figure 11).
From the work distributions, we can see that there is that the protocol work accumulation is very large. Even for 50,000 NCMC switching steps, most of the moves attempted aren’t close to being favorable (near 0). To investigate further into these high protocol work values, we looked at the instantaneous derivative throughout the NCMC switching protocol (Figure 12). If there were infinite switching steps, then we would expect to see the instantaneous derivative being roughly inversely symmetric around the middle of the protocol. Instead, what we see is that when the ligand’s steric interactions are being turned back on, there is a huge spike of protocol work being accumulated. On the other hand, the electrostatics for the system are well-behaved when both turning off and turning on those interactions. This is illustrated by 12 (c), which looks at the directly at the differences between the protocol work accumulation of the forward and reverse directions of the NCMC move. These pieces of data suggest that the moves we propose introduce the steric interactions too quickly or in a way which causes clashes that are too severe. We therefore could potentially improve MolDarting move acceptance rates by altering our NCMC switching protocol. Specifically, one route we can take to improve the switching protocol is to increase the proportion of steric NCMC switching steps to the electrostatic NCMC switching steps. Another potential way to increase the acceptance rates is to minimize the variance of the protocol work.44 As seen in Figure 12, the protocol work variance is not constant and changes over the course of the switching steps, so modification of how we change the sterics and, to a lesser extent, the electrostatics (Figure 4) during our NCMC protocol could improve our acceptance rates of these MolDarting moves–and NCMC moves in general.
Figure 12: Turning on the steric interactions leads to unfavorable accumulation of protocol work.
(a) The instantaneous difference of protocol work accumulation over 1000 switching steps. (b) The instantaneous difference of protocol work accumulation over 10,000 switching steps. (c) The amount of asymmetry for the protocol work accumulation in ((b)), showing the difference between the process of turning the ligand off and that for turning it back on. From 200 iterations of NCMC and MolDarting simulation, we took the average values of the protocol work at each step for 1000 and 10,000 switching steps. From these average values, we calculated the instantaneous difference between the work values, shown by the blue line. The standard deviation of these differences are shown in red. We can see that there is a large accumulation of protocol work when the ligand’s interactions are being turned back on (after the halfway point of the NCMC steps). This difference is further illustrated in (c), which looks at the absolute difference between forward and reverse work values for equivalent points in the protocol shown in (b). Essentially, this shows how asymmetric the protocol is, and helps us identify that the largest asymmetry and greatest problems originate from the steric component of the protocol.
Another contribution to the high protocol work experienced during these simulations appears to come from proposing moves between binding pockets. We compared MolDarting moves proposing inter-pocket moves to intra-pocket moves (Table 2) and found that the inter-pocket move proposals resulted in more highly positive work (thus, were less likely to be accepted). This suggests improving the acceptance of these inter-pocket moves would also require additional consideration of the protein side-chains around the pocket the ligand is being moved to.
Table 2: Inter-pocket moves result in higher protocol work.
We tracked the protocol work across move attempts for MolDarting the various HIV integrase ligands and compared the work values between intra-pocket and inter-pocket moves. The first column specifies the PDB code corresponding to the ligand under study. The second column tracks the average protocol work (in units of kBT) and the standard deviation for 1,000 NCMC steps across all ligands studied. The third and fourth columns follow the same format as the second but give the average protocol work for 10,000 and 50,000 NCMC steps respectively. Here we see that the inter-pocket moves consistently result in higher protocol work values than their intra-pocket equivalents.
| 1,000 | 10,000 | 50,000 | |
| intra-pocket | 234 ± 167 | 43 ± 27 | 28 ± 26 |
| inter-pocket | 489 ± 509 | 64 ± 53 | 39 ± 28 |
To look at this issue further we looked at the inter-atomic distance between the ligand atoms and the protein atoms across all the MolDarting simulations for HIV integrase. We saw from the average distance between the ligand and protein before MolDarting (0.74 angstroms) was further than after MolDarting (0.57 angstroms). This suggests that another area of improvement might involve the translational and/or rotational darting regions; possibly the deterministic way these degrees of freedom are being handled could be resulting in them being inserted too close to the protein. We can see a situation where this occurs in Figure 13 (f), in which after MolDarting the ligand ends up being close to some of the sidechain residues.
Figure 13: MolDarting depends on the defined regions to jump to a new binding mode.
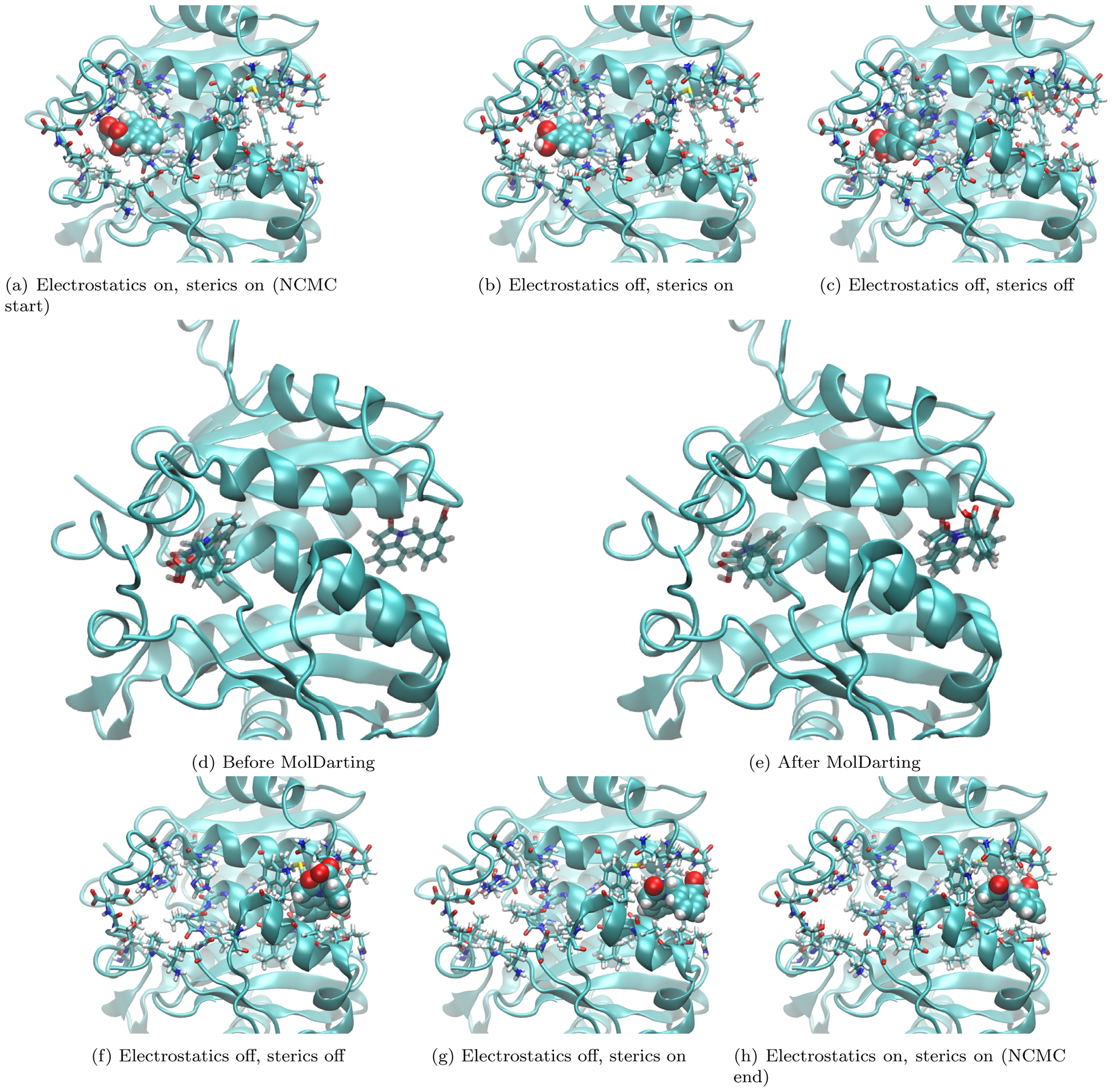
Here we show an example of a MolDarting Move attempt with NCMC. The ligand here is represented in VDW representation in (a-c,f-h), with the binding site sidechains shown in licorice representation, and the rest of the protein shown in cartoon representation. Panels (a-c) and (f-h) focus on turning the ligand off and back on, while panels (d) and (e) focus on a jump to a new binding mode. (a) shows the system at the start of the NCMC move, with the ligand fully interacting. (b) shows the system when the electrostatic interactions of the ligand are turned off, but the sterics are still fully interacting, while (c) shows the system after all the ligand steric and electrostatic interactions are turned off. (d) shows the same system in (c), but with only the ligand represented in licorice to illustrate the ligand positions before darting. Also the reference poses used to define the darting regions are shown in transparent licorice representation. We can see in this example that the ligand position is quite near the reference pose. (e) shows the same system in (d) but after the MolDarting move takes place. (f) shows the same system in (e) but using the previous representation found in (a-c). The VDW representation of the ligand in (f) helps visualize some new clashes present after MolDarting that will need to be resolved before the ligand is fully interacting. g shows the system after the ligand has full steric interactions. Here we can see there is a sizable reorientation of the ligand in the binding site. In (h) the ligand’s electrostatic and steric interactions are restored, and the protocol work accumulated is then used to accept or reject these new positions as shown here.
5. Conclusion/Discussion
5.1. MolDarting allows sampling of specific binding modes
We have shown that our newly developed Monte Carlo method — Molecular Darting — allows reversible sampling of specific binding modes/conformations by constructing darting moves based on the internal and external degrees of freedom of a ligand. This allows reversible hops between pre-defined metastable binding modes or conformations, opening up exciting new possibilities. Molecular Darting worked well in improving sampling of the different binding modes/conformations in the simpler model systems we considered, and notably showed marked improvements in sampling compared to uniform Monte Carlo sampling methods and plain Molecular Dynamics.
We did experience challenges, however, in getting acceptance of MolDarting moves in combination with NCMC in the HIV integrase system. Even though the NCMC/MolDarting moves were not accepted, we did find that the attempted MolDarting move proposals were into the intended binding sites/binding modes.
More work can be done in regards to improving move acceptance with NCMC. Potential areas to be explored could be to look into more efficient paths of turning off and on the electrostatics and sterics of the system. Different soft-core potentials could potentially be used as well, to further decrease the accumulated protocol work while turning on the ligand’s interactions by minimize the variance of this process.44,45
Molecular Darting also has potential applications in combination with other methods, which can be further explored. For instance, MolDarting could find use in equilibrium or expanded ensemble simulations to improve sampling. In the non-interacting states, MolDarting moves should have significant acceptance rates; since there are no clashes with the surrounding atoms of the ligand acceptance will just depend on the ligand’s internal degrees of freedom.
Further work can be also be done on generalizing Molecular Darting, as well as improving the move proposals to target favorable orientations of the ligand. One aspect of MolDarting to improve would be allowing regions of arbitrary sizes. While our original implementation of MolDarting only handles regions of the same size, different sized regions can be used instead if they are factored into the acceptance criterion.46 Similarly, instead of uniform sampling the dihedral regions, we could sample using a Gaussian distribution centered at the maximum of the dihedral, which would favor lower energy conformations of the ligand and thus potentially yield higher acceptance.
Another way in which we could improve MolDarting is better handling of the translational regions. Currently the deterministic way we handle the translations between darts can lead to unwanted clashes with the binding pocket since the available translational space within one darting region does not necessarily match the available space in another darting region. For example, if a ligand translates upwards within one binding pocket and darting region, proposed moves to another binding pocket will also involve an upwards translation – which may indeed result in steric clashes if the second site is sterically congested in that direction. A potential way to address this shortcoming would be to use the short trajectories we already run for defining the dihedral regions to define the translational regions. By doing this, we can better ensure that the ligand is translated to regions that are favorable by uniformly hopping to those translational regions.
An additional option to better target MolDarting to favorable areas of the binding site is to take into account the ligand’s center of mass for translational hopping. Currently in our approach, we use one atom from the ligand when defining the translational region, which can lead to large amounts of translational fluctuation. Using the center of mass, the translational center should vary less across the simulation and thus would better stay within favorable areas of the translational regions.
Another approach to making the protocol work more favorable could be to enhance sampling of the protein during the NCMC portion of the simulation. One way to accomplish this could be to incorporate a REST-like scheme47 in which the protein sidechain intramolecular potential energies are scaled down and then back up over the course of the NCMC protocol, which could allow the protein to respond more quickly to the insertion of the ligand.
Another aspect of MolDarting that we will need to address in future work is the use of implicit solvent. Here, we used implicit solovent because it offers a way to represent the effects of solvent without actually having solvent particles present. Thus, when using MolDarting with implicit solvent we avoided having to displace waters in the binding site when restoring the ligand’s interactions during NCMC, alleviating potential sampling problems. Implicit solvent simulations, however, are not without their limitations. Since implicit solvent aims to represent the bulk properties of water, interactions that depend on individual water molecules, like those often present in protein binding pockets, may not be well represented.48 However, there are a number of different circumstances that the implicit solvent representation could still yield reasonable results. One situation is when the ligand binding pocket is in an internal cavity that contains no water. While uncommon, these types of binding pockets do exist, such as in the model binding system T4 Lysozyme L99A and myoglobin.49–51 Another case would be when the binding site waters are bulk-like and are well described by an implicit solvent model. Still, for the best accuracy we expect to need to eventually move to explicit solvent and deal with any associated water sampling challenges.
Overall, we are excited of the potential applications of Molecular Darting, and its ability to sample phase space in combination with other sampling techniques.
Supplementary Material
Figure 6: The HSP90 ligands studied with MolDarting are chemically diverse.
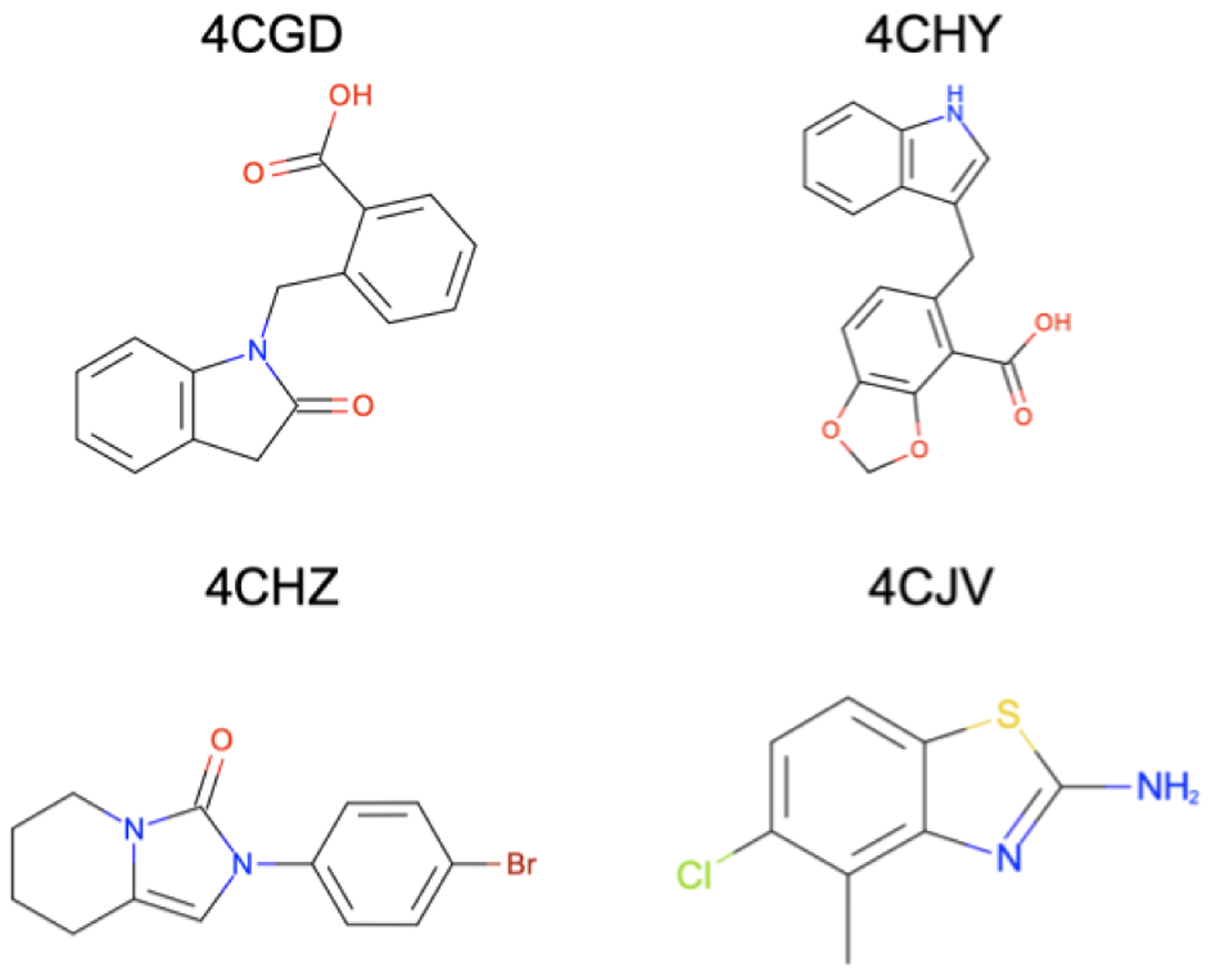
The ligands we used in this study were selected from the SAMPL4 challenge and were selected to cover a range of chemical diversity found in the challenge. These ligands cover a range of rotatable bonds and number of atoms. The ligands here are labeled by the PDB name that they were obtained from.
Table 1: Protocol work scales with ligand size.
We tracked the protocol work across move attempts for MolDarting the various HIV integrase ligands. The first column specifies the PDB code corresponding to the ligand under study. The second column tracks the average protocol work (in units of kBT) and the standard deviation for 1,000 NCMC steps. The third and fourth columns follow the same format as the second but give the average protocol work for 10,000 and 50,000 NCMC steps respectively. Here we see that the protocol work roughly scales with ligand size, with the smallest ligand, 4CJV, having the lowest protocol work values.
| PDB | 1,000 | 10,000 | 50,000 |
| 4CJV | 194 ± 244 | 40 ± 32 | 18 ± 8 |
| 4CHZ | 281 ± 255 | 68 ± 64 | 24 ± 19 |
| 4CHY | 701 ± 653 | 36 ± 19 | 50 ± 40 |
| 4CGD | 325 ± 194 | 58 ± 31 | 36 ± 28 |
6. Acknowledgments
D.L.M. and S.C.G. appreciate the financial support from the National Science Foundation (CHE 1352608) and the National Institutes of Health (1R01GM108889-01) and computing support from the UCI GreenPlanet cluster, supported in part by NSF Grant CHE-0840513. We would like to thank Nathan M. Lim for helping design and maintain the core BLUES code infrastructure and documentation. We also would like to acknowledge Christopher I. Bayly (OpenEye Scientific Software) and Ioan Andricioaei (UC Irvine) for their helpful scientific discussions and insights.
Footnotes
Disclosures
DLM is a member of the Scientific Advisory Board of OpenEye Scientific Software and an Open Science Fellow with Silicon Therapeutics.
Supporting information
- Set of scripts for running BLUES simulations with MolDarting,
- Parameter and coordinate files for the systems used
- Analysis scripts for interpreting the output
- A copy of the BLUES version used
- A README.md file detailing the layout of these files
BLUES is also available at https://github.com/mobleylab/BLUES.
References
- (1).Ferreira LG; Dos Santos RN; Oliva G; Andricopulo AD Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kalyaanamoorthy S; Chen Y-PP Modelling and Enhanced Molecular Dynamics to Steer Structure-Based Drug Discovery. Progress in Biophysics and Molecular Biology 2014, 114, 123–136. [DOI] [PubMed] [Google Scholar]
- (3).Sliwoski G; Kothiwale S; Meiler J; Lowe EW Computational Methods in Drug Discovery. Pharmacol Rev 2014, 66, 334–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lionta E; Spyrou G; Vassilatis K, D.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Current Topics in Medicinal Chemistry 2014, 14, 1923–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).‚led¹ P; Caflisch A Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Current Opinion in Structural Biology 2018, 48, 93–102. [DOI] [PubMed] [Google Scholar]
- (6).Michel J; Essex JW Prediction of Protein–Ligand Binding Affinity by Free Energy Simulations: Assumptions, Pitfalls and Expectations. J Comput Aided Mol Des 2010, 24, 639–658. [DOI] [PubMed] [Google Scholar]
- (7).Kalyaanamoorthy S; Chen Y-PP Structure-Based Drug Design to Augment Hit Discovery. Drug Discovery Today 2011, 16, 831–839. [DOI] [PubMed] [Google Scholar]
- (8).Mobley DL; Liu S; Lim NM; Wymer KL; Perryman AL; Forli S; Deng N; Su J; Branson K; Olson AJ Blind Prediction of HIV Integrase Binding from the SAMPL4 Challenge. J Comput Aided Mol Des 2014, 28, 327–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Warren GL; Andrews CW; Capelli A-M; Clarke B; LaLonde J; Lambert MH; Lindvall M; Nevins N; Semus SF; Senger S A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem 2006, 49, 5912.17004707 [Google Scholar]
- (10).Cross JB; Thompson DC; Rai BK; Baber JC; Fan KY; Hu Y; Humblet C Comparison of Several Molecular Docking Programs: Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model 2009, 49, 1455–1474. [DOI] [PubMed] [Google Scholar]
- (11).Koukos PI; Xue LC; Bonvin AMJJ Protein–Ligand Pose and Affinity Prediction: Lessons from D3R Grand Challenge 3. J Comput Aided Mol Des 2019, 33, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Michel J; Foloppe N; Essex JW Rigorous Free Energy Calculations in Structure-Based Drug Design. Molecular Informatics 2010, 29, 570–578. [DOI] [PubMed] [Google Scholar]
- (13).Cournia Z; Allen B; Sherman W Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model 2017, 57, 2911–2937. [DOI] [PubMed] [Google Scholar]
- (14).Schindler C et al. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. 2020, [DOI] [PubMed]
- (15).Wang E; Sun H; Wang J; Wang Z; Liu H; Zhang JZH; Hou T End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev 2019, 119, 9478–9508. [DOI] [PubMed] [Google Scholar]
- (16).Aldeghi M; Heifetz A; Bodkin MJ; Knapp S; Biggin PC Accurate Calculation of the Absolute Free Energy of Binding for Drug Molecules. Chem. Sci 2015, 7, 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Mobley DL; Chodera JD; Dill KA On the Use of Orientational Restraints and Symmetry Corrections in Alchemical Free Energy Calculations. The Journal of Chemical Physics 2006, 125, 084902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kellett K; Kantonen SA; Duggan BM; Gilson MK Toward Expanded Diversity of Host–Guest Interactions via Synthesis and Characterization of Cyclodextrin Derivatives. J Solution Chem 2018, 47, 1597–1608. [Google Scholar]
- (19).Shan Y; Kim ET; Eastwood MP; Dror RO; Seeliger MA; Shaw DE How Does a Drug Molecule Find Its Target Binding Site? Journal of the American Chemical Society 2011, 133, 9181–9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lim NM; Osato M; Warren GL; Mobley DL Fragment Pose Prediction Using Non-Equilibrium Candidate Monte Carlo and Molecular Dynamics Simulations. J. Chem. Theory Comput 2020, 16, 2778–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gill SC; Lim NM; Grinaway PB; Rustenburg AS; Fass J; Ross GA; Chodera JD; Mobley DL Binding Modes of Ligands Using Enhanced Sampling (BLUES): Rapid Decorrelation of Ligand Binding Modes via Nonequilibrium Candidate Monte Carlo. J. Phys. Chem. B 2018, 122, 5579–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Burley KH; Gill SC; Lim NM; Mobley DL Enhancing Side Chain Rotamer Sampling Using Nonequilibrium Candidate Monte Carlo. J. Chem. Theory Comput 2019, 5, 1848–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sasmal S; Gill SC; Lim NM; Mobley DL Sampling Conformational Changes of Bound Ligands Using Nonequilibrium Candidate Monte Carlo and Molecular Dynamics. J. Chem. Theory Comput 2020, 16, 1854–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen H; Lyne PD; Giordanetto F; Lovell T; Li J On Evaluating Molecular-Docking Methods for Pose Prediction and Enrichment Factors. J. Chem. Inf. Model 2006, 46, 401–415. [DOI] [PubMed] [Google Scholar]
- (25).Wang Z; Sun H; Yao X; Li D; Xu L; Li Y; Tian S; Hou T Comprehensive Evaluation of Ten Docking Programs on a Diverse Set of Protein–Ligand Complexes: The Prediction Accuracy of Sampling Power and Scoring Power. Phys. Chem. Chem. Phys 2016, 18, 12964–12975. [DOI] [PubMed] [Google Scholar]
- (26).Evoli S; Mobley DL; Guzzi R; Rizzuti B Multiple Binding Modes of Ibuprofen in Human Serum Albumin Identified by Absolute Binding Free Energy Calculations. Phys. Chem. Chem. Phys 2016, 18, 32358–32368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Sakano T; Mahamood MI; Yamashita T; Fujitani H Molecular Dynamics Analysis to Evaluate Docking Pose Prediction. BIOPHYSICS 2016, 13, 181–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liu K; Kokubo H Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-Docking Study. J. Chem. Inf. Model 2017, 57, 2514–2522. [DOI] [PubMed] [Google Scholar]
- (29).Rosenbluth MN; Rosenbluth AW Monte Carlo Calculation of the Average Extension of Molecular Chains. J. Chem. Phys 1955, 23, 356–359. [Google Scholar]
- (30).Escobedo FA; de Pablo JJ Extended Continuum Configurational Bias Monte Carlo Methods for Simulation of Flexible Molecules. J. Chem. Phys 1995, 102, 2636–2652. [Google Scholar]
- (31).Spellmeyer DC; Wong AK; Bower MJ; Blaney JM Conformational Analysis Using Distance Geometry Methods. Journal of Molecular Graphics and Modelling 1997, 15, 18–36. [DOI] [PubMed] [Google Scholar]
- (32).Andricioaei I; Straub JE; Voter AF Smart Darting Monte Carlo. J. Chem. Phys 2001, 114, 6994–7000. [Google Scholar]
- (33).Weser O An efficient and general library for the definition and use of internal coordinates in large molecular systems. M.Sc. thesis, Georg August Universität Göttingen, 2017. [Google Scholar]
- (34).Boresch S; Tettinger F; Leitgeb M; Karplus M Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B 2003, 107, 9535–9551. [Google Scholar]
- (35).Case DA; Cheatham TE; Darden T; Gohlke H; Luo R; Merz KM; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber Biomolecular Simulation Programs. J. Comp. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Onufriev A; Bashford D; Case DA Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins: Structure, Function, and Bioinformatics 2004, 55, 383–394. [DOI] [PubMed] [Google Scholar]
- (37).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang L-P; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLOS Computational Biology 2017, 13, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang L-P; Lane TJ; Pande VS MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).ajsilveira; Saladi Shyam; Boehm Kevin; Gmach Jürgen; Rodríguez-Guerra Jaime, A. R. J. C. L. N. K. B. S. A. P. G. D. P.-G. B. R. Yank https://github.com/choderalab/yank.
- (40).Hawkins PCD; Skillman AG; Warren GL; Ellingson BA; Stahl MT Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model 2010, 50, 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Peat TS; Dolezal O; Newman J; Mobley DL; Deadman JJ Interrogating HIV Integrase for Compounds That Bind- a SAMPL Challenge. J Comput Aided Mol Des 2014, 28, 347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).OEDOCKING. 2019; http://www.eyesopen.com.
- (43).Jakalian A; Jack DB; Bayly CI Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- (44).Pham TT; Shirts MR Optimal Pairwise and Non-Pairwise Alchemical Pathways for Free Energy Calculations of Molecular Transformation in Solution Phase. J. Chem. Phys 2012, 136, 124120. [DOI] [PubMed] [Google Scholar]
- (45).Pham TT; Shirts MR Identifying Low Variance Pathways for Free Energy Calculations of Molecular Transformations in Solution Phase. J. Chem. Phys 2011, 135, 034114. [DOI] [PubMed] [Google Scholar]
- (46).Sminchisescu C; Welling M Generalized Darting Monte Carlo. Pattern Recognition 2011, 44, 2738–2748. [Google Scholar]
- (47).Wang L; Friesner RA; Berne BJ Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Swanson JMJ; Mongan J; McCammon JA Limitations of Atom-Centered Dielectric Functions in Implicit Solvent Models. J. Phys. Chem. B 2005, 109, 14769–14772. [DOI] [PubMed] [Google Scholar]
- (49).Carugo O; Argos P Accessibility to Internal Cavities and Ligand Binding Sites Monitored by Protein Crystallographic Thermal Factors. Proteins: Structure, Function, and Bioinformatics 1998, 31, 201–213. [PubMed] [Google Scholar]
- (50).Key J; Scheuermann TH; Anderson PC; Daggett V; Gardner KH Principles of Ligand Binding within a Completely Buried Cavity in HIF2α PAS-B. J. Am. Chem. Soc 2009, 131, 17647–17654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tomita A; Sato T; Ichiyanagi K; Nozawa S; Ichikawa H; Chollet M; Kawai F; Park S-Y; Tsuduki T; Yamato T; Koshihara S.-y.; Adachi S.-i. Visualizing Breathing Motion of Internal Cavities in Concert with Ligand Migration in Myoglobin. PNAS 2009, 106, 2612–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.