

A computational framework expedites the identification and prediction of T cell antigen recognition.
Abstract
T cell receptor (TCR) antigen–specific recognition is essential for the adaptive immune system. However, building a TCR-antigen interaction map has been challenging due to the staggering diversity of TCRs and antigens. Accordingly, highly multiplexed dextramer-TCR binding assays have been recently developed, but the utility of the ensuing large datasets is limited by the lack of robust computational methods for normalization and interpretation. Here, we present a computational framework comprising a novel method, ICON (Integrative COntext-specific Normalization), for identifying reliable TCR-pMHC (peptide–major histocompatibility complex) interactions and a neural network–based classifier TCRAI that outperforms other state-of-the-art methods for TCR-antigen specificity prediction. We further demonstrated that by combining ICON and TCRAI, we are able to discover novel subgroups of TCRs that bind to a given pMHC via different mechanisms. Our framework facilitates the identification and understanding of TCR-antigen–specific interactions for basic immunological research and clinical immune monitoring.
INTRODUCTION
T cell antigen specificity, mediated via T cell receptors (TCRs), is a hallmark of cellular immunity. TCRs are heterodimeric proteins found on the T cell surface, commonly composed of an α and β chain. The TCR α- and β-chain genes are composed of discrete V, D (β chain only), and J segments that are joined by somatic recombination during T cell development (1–5). This genetic rearrangement generates a highly diverse TCR repertoire (estimated to range from 1015 to 1061 possible receptors in humans) (6–8) to ensure efficient control of viral infections and other pathogen-induced diseases. TCR diversity is primarily exhibited in complementarity determining region (CDR) loops (CDR1, CDR2, and CDR3), which engage peptides that are presented by major histocompatibility complex (MHC) proteins, and therefore directly determine the specificity of T cell peptide-MHC (pMHC) binding (9–12).
Although we do not fully understand the factors underlying TCR-pMHC recognition, recent studies have shown that T cells binding to a particular pMHC share common TCR sequence features and, in select cases, it is possible to predict the binding probability of an unseen TCR sequence based on learned TCR sequence features (13–21). However, these studies either were limited by the quantity and diversity of training data generated by traditional single multimer sorting or antigen reexposure assays or suffered from the challenging normalization issues associated with multi-omic characterization of T cells (22).
10x Genomics recently developed a highly multiplexed dextramer binding immune profiling platform that couples feature-barcoded dextramers and single-cell TCR sequencing (22). This approach makes it feasible to generate high-dimensional pMHC-specific binding data at the single-cell level with paired T cell αβ-chain sequences, whereas other large-scale pooled multimer approaches only estimate the composition of pMHC-specific binding cells (23, 24). As with other high-throughput technology, the binding data are associated with low signal-to-noise ratio. This makes it bioinformatically challenging to reliably identify TCR-pMHC binding events using these datasets and necessitates the development of new computational methods to discriminate true TCR-pMHC binding signal from nonspecific background noise.
As next-generation screening technologies have increased the volume of available TCR-pMHC binding data, robust classifiers to computationally validate and subsequently predict TCR-pMHC–specific recognition are desired. While the results from initial TCR-pMHC binding classifiers are encouraging, most of them were only trained using CDR loop sequences or β-chain sequences alone and thus unable to learn the overall complex sequence patterns from full-length TCR sequences, resulting in suboptimal prediction accuracy for highly diverse pMHC binding TCRs (13, 14, 19, 20). Leveraging the ability of deep learning algorithms to learn complex patterns, a couple of neural network–based classifiers were recently proposed (17, 18) to uncover binding patterns in large, highly complex pMHC binding TCR sequences. However, the ability to characterize and understand TCR-antigen interaction in sequence space is still in its infancy, and the predictive performance and flexibility of these models can be refined.
In this study, we report a computational framework for mapping TCR-antigen specificity. Our framework includes a novel method for identifying reliable TCR-pMHC interactions from high-throughput pMHC binding data and a neural network–based classifier for subsequently validating, characterizing, and predicting TCR-pMHC–specific recognition.
RESULTS
Identification of pMHC binding TCRs from high-throughput dextramer binding data
10x Genomics recently generated an expansive, publicly available TCR-pMHC binding dataset. In their initial report, the binding profile of CD8+ T cells from four human leukocyte antigen (HLA) haplotyped healthy donors (table S1, donors 1 to 4) was assessed across 44 dextramers using a single cell–based immune profiling platform, Immune Map, to directly detect antigen binding to T cells while simultaneously sequencing T cell αβ-chain pairs and transcriptomes (Fig. 1A). The dextramer pool consists of epitopes with known common viral and cancer reactivities across eight HLA alleles (table S2).
Fig. 1. Identification of pMHC binding T cells from the high-throughput dextramer binding data.
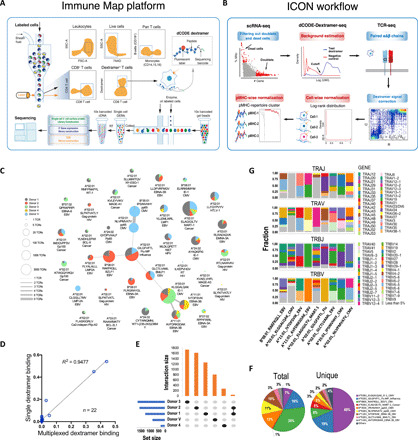
(A) Schema of the Immune Map platform. PBMC CD8+ T cells were enriched and stained with a pool of 50 dCODE dextramer antibodies. Dextramer-positive CD8+ T cells were sorted and then captured individually as inputs for 10x Genomics single-cell sequencing. (B) ICON workflow. Please see Materials and Methods for details. (C) Network of ICON identified pMHC binding unique TCRs. Each node represents a pMHC repertoire and is displayed as a pie chart of pMHC binding TCRs for each donor. The node size denotes the total number of unique TCRs. The thickness of an edge represents the number of shared unique TCR(s). (D) Correlation of the fraction of T cells binding to a given dextramer between the result from flow sorting on single dextramer binding and the relative abundance of pMHC binding T cells identified by ICON from the multiplexed dextramer binding data. (E) Uniqueness and overlap of pMHC binding unique TCRs among the five donors. (F) Pie charts of ICON identified pMHC binding TCRs. (G) V and J gene usage of the nine most abundant pMHC repertoires. The gene usage with less than 5% was combined and indicated in gray.
To our knowledge, this is the first reported highly multiplexed dextramer binding dataset generated at the single-cell level with paired αβ-chain sequences. 10x Genomics applied global cutoffs for nonspecific dextramer bindings to all donors, cells, and dextramers to identify pMHC binding TCRs (22). We found an unexpectedly high number of cross-reactive TCRs in the binders that 10x Genomics identified (fig. S1). To robustly identify reliable binding events from these high-throughput TCR-pMHC binding data, we developed ICON, an Integrative COntext-specific Normalization method. The ICON data process is performed in a donor-, cell-, and dextramer-specific manner. In brief, we used single-cell transcriptome data to select good quality cells (live and singleton). Then, negative control dextramers (n = 6) were used to empirically estimate the background binding noise for each donor. Raw dextramer binding signals were subsequently corrected by subtracting the estimated background noise for each donor separately. T cells with paired αβ chains were selected as the candidates of pMHC binding T cells, as previous studies have demonstrated that αβ pairing synergistically drive TCR-pMHC recognition (14, 16, 17, 25–27). We further corrected T cell dextramer binding signals by penalizing dextramers simultaneously binding to the same T cell/clone. Last, dextramer binding signals were normalized across pMHCs and cells to make them directly comparable (Fig. 1B, fig. S2, and Materials and Methods). To evaluate ICON performance, we assessed the pMHC binding specificities of CD8+ T cells from another healthy donor (donor V) using the same dextramer panel, as dextramer signal distributions of negative control and test dextramers suggest that 10x Genomics used a loose fluorescence-activated cell sorting (FACS) gating strategy for enriching dextramer-positive T cells from peripheral blood mononuclear cells (PBMCs) of donors 1 to 4 (figs. S2 and S3 and Materials and Methods). ICON was able to link 89% of sequenced 15,821 T cells with paired αβ chains to their antigen targets. To further validate ICON, we also conducted 22 individual dextramer binding assays using the T cells from the same donor, donor V (fig. S4 and Materials and Methods). The flow cytometry results from the single dextramer binding assays show agreement with the relative abundance of ICON-identified binders to these 22 dextramers (Fig. 1D).
Applying ICON, we identified a total of 53,062 CD8+ T cells belonging to 5722 unique T cell clones that bind to 37 pMHCs from five donors (Fig. 1C and table S3). The identified pMHC binding TCRs show high antigen specificity. Unique TCRs (99.6%) bind to one specific pMHC, and the remaining TCRs interact with two pMHCs. In addition, these TCR-pMHC interactions generally follow an HLA type–specific pattern. Ninety-four percent of binding events are HLA type matched, of which 6% involve cross-recognition between HLA A*03-supertype family members HLA A*03: 01 and A*11:01 that share similar main anchor positions of the presented peptide (28). Donors 1 and 2, who have the most common HLA haplotype (A*02:01) in the dextramer pool (tables S1 and S2), share a substantial fraction (n = 44) of unique TCR-pMHC interactions (Fig. 1E), supporting the dogma that TCR-pMHC binding patterns are most likely to be HLA type–restricted (29, 30). However, we also observed that 6% of binding events are cross-HLA type interactions.
Among all pMHC binding TCRs, 99% of total TCRs or 96% of unique TCRs bind to nine pMHCs: B*08:01_RAKFKQLL_BZLF1_EBV, A*02:01_GILGFVFTL_Flu-MP_Influenza, A*11:01_IVTDFSVIK_EBNA-3B_EBV, A*03:01_KLGGALQAK_IE-1_CMV, A*11:01_AVFDRKSDAK_EBNA-3B_EBV, A*02:01_GLCTLVAML_BMLF1_EBV, A*02:01_ELAGIGILTV_MART-1_Cancer, B*35:01_IPSINVHHY_pp65_CMV, and A*02:01_NLVPMVATV_pp65_CMV (Fig. 1F). To further understand the conserved TCR sequence features underlying the specific binding, we examined TCR VJ gene usages for these nine pMHC repertoires. In addition to the enrichment that previous studies reported, such as TRBV19 and TRAV27 in the Influenza repertoire, TRAV5 and TRBV20-1 in the BMLF1_EBV repertoire, and TRBV6-5 in the NLVPMVATV_pp65_CMV repertoire (14, 31, 32), we also found abundant usage of TRAV12-2 in the MART-1_Cancer repertoire, TRAV21, TRAV35, TRBV11-2, and TRBV6-6 in the IVTDFSVIK_EBNA-3B_EBV repertoire, TRAV8-3, TRAV13-1, and TRBV28 in the AVFDRKSDAK_EBNA-3B_EBV repertoire, TRAV13-1, TRAV13-2, and TRBV12-3 in the BZLF1_EBV repertoire, TRAV12-1, TRAV41, TRBV2, and TRBV20-1 in the IPSINVHHY_pp65_CMV repertoire, and TRAV23/D6 and TRBV12-4 in the NLVPMVATV_pp65_CMV repertoire (Fig. 1G). Consistent with the observed gene usage, Shannon diversity indexes and TCR clone size distributions also suggested that each pMHC binding T cell repertoire experienced different degrees of expansion in responding to their target peptides (fig. S5).
TCRAI: A neural network–based classifier of T cell antigen specificity
With many TCR-pMHC binding events identified, robust classifiers for rapidly validating these binders are needed. Recent work demonstrated that neural networks can learn high-dimensional information from TCR sequences and thus may robustly predict TCR-pMHC interaction (17, 18, 33). Here, we present the Python package TCRAI, using Tensorflow 2 (34), that provides a flexible framework for the study of TCR-pMHC specificity (Fig. 2A). The highly modularized TCRAI package allows one to easily adjust the architecture of the model. In brief, the TCRAI framework works as follows. One can define any number of the V(D)J genes, and CDR regions of the TCR as inputs to the model in their textual form. One next selects “processor” objects that convert text to numerical representations in a non-learnable way (not optimized during training). These numerical inputs are then passed to “extractor” objects, which are neural network blocks that give their output as lower-dimensional vector representations of the inputs, which we call fingerprints. These fingerprints are concatenated into a single TCRAI fingerprint describing the input TCR as a single numerical vector. This TCRAI fingerprint is then passed through a “closer” object, which forms the final block of the neural network architecture, producing a prediction on the input TCR. The TCRAI package provides a range of predesigned processors, extractors, and closers and is easily extensible to new variants. It also allows one to perform binomial, multinomial, regression, or other tasks by simply choosing to construct a different closer object (Materials and Methods).
Fig. 2. The framework and performance of TCRAI.
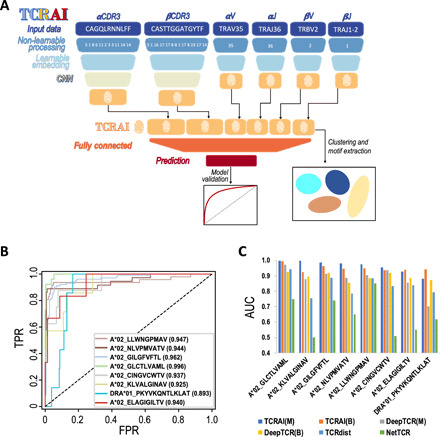
(A) Schematic of the TCRAI framework for a model receiving input of CDR3 and VJ genes of both the α and β chains. A trained TCRAI model creates a numerical fingerprint and prediction for a given TCR. CNN, convolutional neural network. Please see Materials and Methods for more details. (B) ROC curves for TCRAI (in binomial mode) classification performance using the eight curated public TCR-pMHC binding repertoires. Binders are unique TCRs that bind to a particular pMHC, and nonbinders are unique TCRs that bind to other pMHCs. Paired αβ TCR sequences were used as input data. FPR, false positive rate; TPR, true positive rate. (C) Comparison of classification performance. TCRAI was compared with predictive classifiers NetTCR, TCRdist, and DeepTCR. The AUC for NetTCR and TCRdist was generated using the original classifiers with default parameters. To compare with these two binomial classifiers (NetTCR and TCRdist), the AUC for DeepTCR (originally designed as a multinomial classifier) was derived from a slightly modified and hyperparameter optimized version of DeepTCR (Materials and Methods). TCRAI(M), TCRAI in multinomial mode; TCRAI(B), TCRAI in binary mode; DeepTCR(M), DeepTCR in multinomial mode; DeepTCR(B), DeepTCR in binary mode.
To evaluate the performance of TCRAI, we searched the literature for currently available methods (table S4) and compared our classifier to four major methods in this field: GLIPH2, DeepTCR, NetTCR, and TCRdist (14, 15, 17, 18). For the comparison, we collated eight pMHC-specific binding T cell repertoires with at least 50 unique paired αβ-chain TCRs generated by traditional single multimer binding or antigen reexposure assays as a gold-standard dataset (table S5 and Materials and Methods). Three of the methods—DeepTCR, NetTCR, and TCRdist—are, like TCRAI, predictive models. To compare TCRAI prediction performance with these methods, we used scripts and default parameters that the previous studies provided (detailed in Materials and Methods). The area under the ROC (receiver operator characteristic) curve (AUROC/AUC), a standard measure of classification success, of these prediction models indicates that TCRAI and DeepTCR, with similar neural network frameworks, perform better than TCRdist and NetTCR. Overall, TCRAI has more consistent and better performance than DeepTCR (Fig. 2, B and C, fig. S6, and Materials and Methods). Because GLIPH2 was designed for clustering TCR sequences into distinct groups of shared sequence features, we also measured sensitivity and specificity of these four prediction models to compare with GLIPH2 (detailed in Materials and Methods). The comparison result demonstrated that TCRAI has the best-balanced sensitivity and specificity (Table 1). A couple of methods listed in table S4 were not included in the comparison, as they have different functions/purposes from that of TCRAI. For example, ALICE is for detecting groups of homologous/expanded TCRs (13). TcellMatch uses cell-specific covariates (e.g., gene expression) but not TCR sequence alone as input, and its performance was tested on the noisy 10x Genomics Immune Map data without further cleanup (20).
Table 1. Comparison of TCR-antigen specificity classifiers.
The binary mode was used for both TCRAI and DeepTCR. Please see Materials and Methods for sensitivity and specificity calculation for each classifier.
| pMHC | GLIPH2 | NetTCR | TCRdist | DeepTCR | TCRAI | |||||
| Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity | Sensitivity | Specificity | |
| A*02_NLVPMVATV | 0.246 | 0.997 | 0.500 | 0.767 | 0.937 | 0.606 | 0.885 | 0.697 | 0.889 | 0.988 |
| A*02_ELAGIGILTV | 0.000 | 1.000 | 0.263 | 0.888 | 0.878 | 0.721 | 0.870 | 0.795 | 0.833 | 0.912 |
| A*02_GILGFVFTL | 0.867 | 0.988 | 0.562 | 0.845 | 0.955 | 0.751 | 0.928 | 0.763 | 0.902 | 0.947 |
| A*02_LLWNGPMAV | 0.119 | 1.000 | 0.738 | 0.843 | 0.920 | 0.770 | 0.874 | 0.757 | 0.936 | 0.951 |
| A*02_KLVALGINAV | 0.000 | 1.000 | 0.784 | 0.316 | 0.935 | 0.556 | 0.920 | 0.720 | 0.857 | 0.873 |
| A*02_GLCTLVAML | 0.646 | 0.999 | 0.559 | 0.885 | 0.957 | 0.830 | 0.928 | 0.806 | 1.000 | 0.947 |
| A*02_CINGVCWTV | 0.191 | 1.000 | 0.257 | 0.86 | 0.895 | 0.701 | 0.890 | 0.825 | 0.875 | 0.922 |
| DRA*01_ PKYVKQNTLKLAT |
0.000 | 1.000 | 1.000 | 0.006 | 0.048 | 1.000 | 0.630 | 0.660 | 1.000 | 0.831 |
| Average of HLA-A*02 binders |
0.296 | 0.998 | 0.523 | 0.772 | 0.925 | 0.705 | 0.899 | 0.766 | 0.899 | 0.934 |
| Average of all binders |
0.259 | 0.998 | 0.583 | 0.676 | 0.816 | 0.742 | 0.866 | 0.753 | 0.912 | 0.922 |
Classification of pMHC binding TCRs identified from the high-throughput data
We next applied TCRAI to the nine most abundant pMHC binding repertoires that ICON identified from the high-throughput data (Fig. 1F). TCRAI (in binomial mode) was able to classify the TCRs of these nine pMHC repertoires with an average AUC of 0.88 (Fig. 3A). We also saw similar prediction performance using TCRAI multinomial mode (fig. S7). For ease of interpretation, binomial classifiers for each pMHC are used hereinafter. Historically, TCR β-chain sequences were often used to infer T cell antigen binding specificity because of its higher combinatorial potential compared to α chains (35). To quantitatively evaluate the contribution of TCR α and β chains in predicting TCR-pMHC interaction, we used either the α chain or β chain in lieu of paired αβ chains as input to TCRAI. The performance with paired αβ chains is better than either chain alone, with an average increase of about 0.2 in the AUC (Fig. 3B). Consistent with previous studies (16, 25–27), our results demonstrate the importance of αβ pairing for accurate inference of TCR-pMHC interactions. We also observed that the predictive performance for β chains is not always better than α chains, suggesting the importance of α chains in TCR-pMHC–specific recognition, which has previously been overlooked.
Fig. 3. TCRAI prediction on the high-throughput dataset.
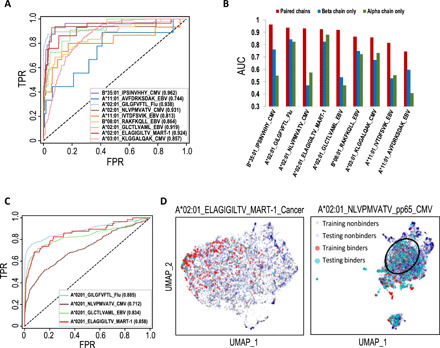
(A) ROC curves for TCRAI prediction on the nine most abundant pMHC binding repertoires. Binders are unique TCRs that bind to a particular pMHC, and nonbinders are unique TCRs that bind to other pMHCs. Paired αβ TCR sequences were used as input data. (B) Comparisons of TCRAI prediction on TCRα only, TCRβ only, and paired αβ chains as input data. (C) ROC curves for the independent tests of four overlapping pMHC repertoires between the curated public dataset and the high-throughput dataset. TCRAI was trained using pMHC repertoires identified from the high-throughput dataset and was tested on the curated public dataset. (D) UMAPs of both the training (high-throughput data) and testing (the gold-standard data) TCRAI fingerprints extracted from the models trained by the high-throughput data. The left panel shows the strong overlap between MART-1_cancer training and testing sets, while the poor overlap of NLVPMVATV_pp65_CMV training and testing datasets is shown in the right panel. The black circle highlights the region with almost no overlapping fingerprints of training and testing binders. UMAP, Uniform Manifold Approximation and Projection.
To further evaluate the performance of TCRAI, we used four pMHC repertoires (A*02:01_ELAGIGILTV_MART-1, A*02:01_GILGFVFTL_Flu-MP, A*02:01_GLCTLVAML_BMLF1_EBV, and A*02:01_NLVPMVATV_pp65_CMV) that also have binding TCRs available in the curated public dataset. We trained TCRAI using the four repertoires identified from the high-throughput dataset to predict the corresponding four curated repertoires. Figure 3C shows that the prediction results are generally comparable to the performance on the training set. However, we found that the performance of TCRAI when inferring on A*02:01_NLVPMVATV_pp65_CMV was significantly worse than the other three pMHCs. To understand the performance difference, we investigated the TCRAI fingerprint space of the model (Materials and Methods). In the case of A*02:01_ELAGIGILTV_MART-1_Cancer (Fig. 3D) and the other two pMHCs (fig. S8), binding TCRs from the high-throughput dataset and the curated dataset overlap spatially in fingerprint space, whereas the overlap is significantly worse for the case of pp65_CMV. We attribute this poor overlap to 98.2% of pp65_CMV binding TCRs in the high-throughput dataset coming from a single donor (table S3), thereby representing a small subspace of possible binding TCRs, whereas the public data contain TCRs from a range of donors representing a larger area of the TCR space. This result also highlights the importance of large diverse datasets for training a robust TCR-antigen prediction model.
Characterization of pMHC binding TCRs
To investigate the properties of TCRs that bind a given pMHC, we analyzed how TCRAI classifier models arrange TCRs within their fingerprint space (Materials and Methods). We show that TCR fingerprints from a classifier model allow the discovery of specific groups of TCRs with conserved gene usage and CDR3 motifs. These groups often exhibit different binding abilities and divergent structural binding modalities.
Clustering TCRs that bind to A*02:01_GILGFVFTL_Flu-MP_Influenza leads to two well-separated clusters, clusters 0 and 1, in the TCRAI fingerprint space (Fig. 4A). In cluster 0, we identified strongly conserved motifs xRSx (β chain) and xSxGx (α chain) that Dash et al. (14), Ishizuka et al. (36), and Song et al. (37) have also reported. In the smaller group cluster 1, we found the strong enrichment of the α-chain motif AGGTSYGKLT, which is consistent with the findings of Song et al. (37) (Fig. 4C). Related to the motif enrichment, we observed that the gene usage of TRB19 and TRAJ42 is highly enriched in cluster 0, and cluster 1 has very highly conserved usage of TRBV19/TRBJ1-2/TRAV38-1/TRAJ52 (Fig. 4C). This result is consistent with the well-known strong conservation of TCRBV19 gene usage in A*02:01_GILGFVFTL_Flu responsive T cells thought to be connected to its “featureless” pMHC complex (14, 36, 37). The dextramer signal in unique molecular identifier (UMI) distributions suggested that TCRs in cluster 0 have stronger binding to the flu dextramer than those in cluster 1 (Fig. 4B). Comparing to the classes of A*02:01_GILGFVFTL_Flu binding TCRs that Song et al. (37) recently identified, we were able to link our clusters 0 and 1 to their groups I (canonical) and II (novel), respectively. In line with our observation, they also found that their group I TCRs have stronger binding than those in group II (37). The three-dimensional (3D) structures of the TCR-pMHC binding complexes for cluster 0 {TCR [Protein Data Bank (PDB) 2VLJ]} (36) and cluster 1 [TCR (PDB 5JHD)] (37) suggest that because of different highly conserved motifs/residues, these two groups of TCRs have distinct binding modalities, which cause difference in rotation of the structurally key Phe-5 ring of the flu peptide in these two complexes (Fig. 4D) (37).
Fig. 4. Characterization of pMHC binding TCRs.
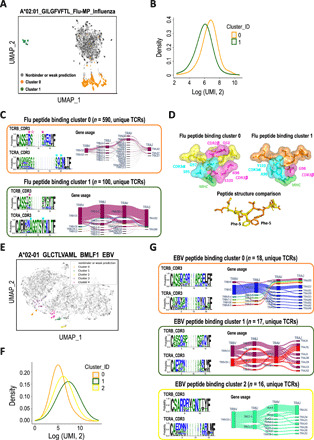
(A) Clustering TCRAI fingerprints of high-confidence TCRs from a model trained by A*02:01_GILGFVFTL_Flu binders identified from the high-throughput dataset. (B) Dextramer signal (in UMI) distributions of the flu peptide binding clusters 0 and 1. (C) Conserved CDR3 motifs and gene usage in flu peptide binding TCR clusters. Structurally important residues are highlighted by filled stars and also shown in (D). The residue with an unfilled star missed the cutoff for inclusion in (D) but is nevertheless in close proximity (4.18 Å to the Phe-5 ring) and strongly conserved. Only the 30 most common unique quadruplets of gene usage are shown for cluster 0 to highlight the key variabilities. For motif construction, please see Materials and Methods for details. (D) 3D structures of flu peptide binding TCR-pMHC complexes for cluster 0 TCR (PDB 2VLJ) and cluster 1 TCR (PDB 5JHD). In the top panels, only nonpeptide residues within 4 Å of the Phe-5 ring are shown. (E) Clustering TCRAI fingerprints of high-confidence TCRs from a model trained by A*02-01_GLCTLVAML_BMLF1_EBV binders identified from the high-throughput dataset. (F) Dextramer signal distributions of the EBV peptide binding clusters. (G) Conserved CDR3 motifs and gene usage in the three EBV peptide binding clusters.
We also characterized the TCRs binding to A*02:01_GLCTLVAML_BMLF1_EBV. In previous studies, a dominant public TCR constructed from TRBV20-1/TRBJ1-2/TRAV5/TRAJ31 has been observed for this pMHC repertoire (38). However, previous analyses of the TCRs binding to the BMLF1 peptide have focused on TRV5 TCRs (14, 38–40), toward which the population is heavily shewed. We unbiasedly identified five clusters of TCRs in the TCRAI fingerprint space (Fig. 4E). Clusters 1 and 2 represent the classic BMLF1 public TCRs, albeit split into two clusters based on their β-chain gene usage (Fig. 4G and fig. S9). The conserved motifs SARDRxGNTTY (β chain) and AEDNN (α chain) in cluster 2 are very similar to the canonical motifs that Dash et al. (14) and Kamga et al. (39) identified from the TCRs sharing the gene usage of TRAV5. The β-chain motif of cluster 1 (Fig. 4G) shows similarities to another motif reported by Kamga et al. (39). Cluster 0 contains TCRs following a gene usage (TRBV2/TRBJ2-2) and a highly conserved β-chain CDR3 motif, GxRxxAPGEL, that have not been previously presented. TCRs belonging to this novel group have fewer dextramer UMI counts than the canonical TCR clusters (Fig. 4F), which suggests a lower affinity and could explain why this group of TCRs has not yet been noted.
Immune phenotypes of pMHC binding CD8+ T cells
The combined information of antigen specificity and T cell phenotype has been reported to be important for the clinical success of immunotherapies, such as vaccination (32, 33, 35). The multi-omics data generated by the Immune Map platform enable the association of T cell antigen specificity with T cell phenotypes. Using gene [single-cell RNA sequencing (RNA-seq)] and surface protein [cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq)] expression from this multi-omics dataset, we grouped pMHC binding CD8+ T cells into subpopulations (Fig. 5A and Materials and Methods). The identified subpopulations were then annotated according to CD8+ T cell subtype marker genes described previously (41): naïve cells (CD45RA+CD62LhiCD127hi), central memory cells (Tcm, CD45RA−CD62L+CD127+EOMEShiTBETlo), T effector memory cells (Tem, CD45RA−CD62LloCD127+GZMB+), peripheral memory cells (Tpm, CD62L+CD127hiGZMB+), terminally differentiated effector cells (Temra, CD45RA+CD127loGZMBhi), and other memory cells (CD43loCD127loKLRG1hi) (Fig. 5, A and B).
Fig. 5. Immune phenotypes of pMHC binding CD8+ T cells.
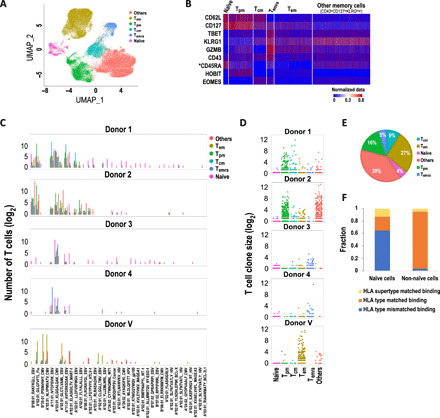
(A) Classification of pMHC binding cells. (B) Heatmap of the expression of CD8+ T cell subtype marker genes and proteins. *: protein expression measured by CITE-seq. (C) pMHC binding landscape by CD8+ T cell immune subtypes. Bars indicate the number of pMHC binding T cells in log2 scale. (D) Expanded clonotypes are enriched in the non-naïve compartment. Each dot represents a unique TCR clone. (E) Pie chart of subpopulations of pMHC binding CD8+ T cells. (F) Fraction of HLA type matched and mismatched binding naïve and non-naïve T cells. Tpm, peripheral memory cells; Tcm, central memory cells; Tem, effector memory cells; Temra, terminally differentiated effector memory cells; Others, other memory cells with the marker gene expression pattern CD43loCD127loKLRG1high.
We found that 96% of pMHC binding T cells were memory cells and were enriched in expanded T cell clones (Fig. 5, D and E, and Supplementary Materials), suggesting that these T cells were selected by specific immune responses and thus are likely to be responsive and reliable binders. Most of these memory T cells bound to common viral epitopes [e.g., influenza, Epstein-Barr virus (EBV), and cytomegalovirus (CMV)], and pMHC binding T cells from each donor demonstrated different distributions of memory cell subsets. For example, we observed that donors 1 and 2 had primarily Tpm, whereas donor V had Tem, and donors 3 and 4 had mostly Temra cells (Fig. 5, C and D).
Although most of the pMHC binding T cells expressed a memory phenotype, 4% of them were naïve cells. These naïve cells had more diverse pMHC interactions than non-naïve cells and were often bound to tumor-associated antigens (e.g., MART-1), endogenous antigens, or antigens derived from viruses for which the donor was purportedly seronegative [e.g., human papillomavirus (HPV)] (Fig. 5C). The fraction of naïve T cells with cross-HLA type binding was significantly higher than that of non-naïve cells (Fig. 5F). These results suggest that healthy donor T cell repertoires—particularly naïve cells—have the potential to respond to not-yet encountered or rare antigens and to retain cross-reactivity. Additional assays are required to assess whether these cells could mount a functional T cell response.
DISCUSSION
In this study, we present a computational framework to characterize and predict TCR-antigen associations. It includes the novel method ICON for reliably identifying TCR-antigen interactions from high-throughput pMHC binding data and TCRAI, a novel neural network architecture for accurate TCR-antigen classification, which outperforms other state-of-the-art methods and groups pMHC binding TCRs to reveal their conserved motifs and binding mechanisms.
High-throughput TCR-pMHC binding data present an attractive pathway for furthering our understanding of TCR antigen recognition. However, this type of data is often associated with low signal-to-noise ratio. The lack of a robust data normalization method for effectively increasing the signal-to-noise ratio has limited the broad application of highly multiplexed multimer binding assays, such as Immune Map. ICON was developed to meet this need. It uses multi-omics data generated by Immune Map for sequencing quality control and empirically identifying background dextramer signals, which was subsequently used to correct raw dextramer signals to increase the signal-to-noise ratio in the high-throughput data. We experimentally demonstrated the high specificity and sensitivity of ICON in identifying TCR-pMHC interactions, although we cannot completely rule out the possibility that a small number of weak or promiscuous binders could be underestimated. ICON computes the noise-corrected dextramer signal in a parameter-free manner, making it easily generalizable to pMHC-TCR binding data from a broader range of pMHC dextramer pools and potentially extensible to the normalization of protein binding signals in single-cell space, such as CITE-seq.
In this study, we also developed the Python package TCRAI, with which we demonstrated the robustness of deep-learning classifiers in predicting TCR-pMHC–specific binding. We showed that TCRAI can not only perform state-of-the-art classification of TCR-pMHC–specific binding but also identify groups of TCRs with differing binding profiles. Partnering the dextramer UMI counts with TCR sequence information allowed us to investigate differing binding abilities between these groups. Our findings suggest that as the volume of high-throughput TCR pMHC binding data grows, so will the ability to discover new TCR motifs and pair these with not only dextramer binding signal (in UMI) but also wider multi-omics data. The ability to investigate, for example, different transcriptional regulation of TCR signaling between groups of TCRs with different binding mechanisms (42) would be very exciting not only for broad scientific questions but also for the development of T cell therapeutics.
TCRAI can also potentially be applied to study T cell antigen–specific recognition in silico. Immune monitoring of T cell antigen–specific interaction has been used to determine the immune responses against specific antigens [e.g., severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), tumor-specific antigens, and peptide vaccines] and their possible correlation with disease severity and clinical outcome in patients receiving immunotherapies. However, experimentally mapping TCR sequences to antigen specificity is costly and labor intensive. With adequate training data for a particular pMHC, TCRAI can assign probabilities of pMHC binding to each TCR sequence of interest without conducting binding assays as shown in fig. S10.
We found that TCRAI requires a certain number of unique TCRs binding to a given pMHC to truly learn which sequence features of the CDR3 regions are essential for TCR-antigen–specific recognition. Because of the importance of the CDR3 regions in determining the specificity of TCRs to a given antigen, it is tempting to build a predictive model harnessing only this information, as has been done previously (18). However, because of highly conserved gene usage for many pMHCs, we find that the VJ gene usage is an important predictive element of TCRAI, particularly in the case of a particular subset of pMHC binding TCRs in the dataset. We observed that the predictive performance of models that receive CDR3 information outperforms gene-level only models when there are more than at least on the order of 100 pMHC binding unique TCRs (fig. S11), suggesting that this volume of data is necessary for this model to be able to extract useful sequence motifs from CDR3 regions.
MATERIALS AND METHODS
The 10x Genomics single-cell immune profiling datasets
10x Genomics data used for this study were downloaded from: https://support.10xgenomics.com/single-cell-vdj/datasets
Identification of pMHC binding T cell phenotypes
Seurat V3 single-cell sequencing analysis R package (43, 44) was used for the classification analysis based on single-cell RNA-seq data. Because we observed significant enrichment of TCR VJ gene usages in pMHC binding T cells, we took out the TCR genes from the classification, so cell clusters will not be dominated by their shared VJ gene usage. Gene expression was normalized and scaled using Seurat V3 with default parameters. Principal Component Analysis (PCA) was run on normalized and transformed UMI counts of variably expressed genes. The top 10 principal components (PCs) were used for the cell classification. Uniform Manifold Approximation and Projection (UMAP) (45) was used for visualization.
Curation of publicly available pMHC-specific binding TCRs
We downloaded raw files from VDJdb (46) (https://vdjdb.cdr3.net/) and the pathology-associated TCR database (47) (http://friedmanlab.weizmann.ac.il/McPAS-TCR/). We processed the data to get pMHC binding TCRs following the criteria: for VDJdb, we required paired αβ-chain CDR3 amino acid sequences for each “complex.id”; we removed TCRs annotated with “source” from 10x Genomics; we filtered for “Species” = “Human.” For McPAS-TCR, we required known “Epitope.ID” in the full data and having “CDR3.alpha.aa” and “CDR3.beta.aa.” Similarly, for VDJdb, we selected human TCRs.
Normalization of high-throughput TCR-pMHC binding data
We developed ICON to reliably identify TCR-pMHC interactions. It takes multi-omics single-cell sequencing data generated from a multiplexed multimer binding platform (e.g., 10x Genomics Immune Map) as inputs, including single-cell RNA-seq, paired αβ-chain single-cell TCR-seq, dCODE-Dextramer-seq, and cell surface protein expression sequencing—also named CITE-seq (40). ICON includes the following major steps (Fig. 1B and fig. S2).
Step 1: Single-cell RNA-seq–based filtering of low-quality cells. ICON filters out low-quality cells such as doublets and dead cells. The T cells with an unexpectedly high number of genes (e.g., >2500 genes per cell) were categorized as doublets, and cells with a high fraction of mitochondrial gene expression (e.g., ratio of mitochondrial gene expression to the total gene expression >0.2) or too few genes detected (<200 genes per cell) were classified as dead cells (fig. S2A).
Step 2: Single-cell dCODE-Dextramer-seq–based background noise estimation. Six negative control dextramers were designed for estimating the background noise from the multiplexed dextramer binding assay. To inspect signal and noise distributions, the maximum dextramer signals in UMI of negative control dextramers and test dextramers for each cell were used to represent the worst noise and best dextramer binding of each T cell. The density distributions of these two types of dextramer signals are shown in fig. S2B. The background cutoffs (the gray dashed lines in fig. S2B) were empirically chosen for each donor.
Step 3: Selecting T cells with paired αβ chains based on single-cell TCR-seq data. We removed T cells that have only a single chain. For T cells with multiple α or β chains detected, the ones with highest UMI counts were assigned to each T cell.
Step 4: Dextramer signal correction. Each dextramer has its own optimal binding condition; however, it is impossible to arrange the experimental conditions such that a multiplexed dextramer binding assay is optimal for every dextramer. This results in multiple dextramers possibly binding to the same T cell/clone, as we observed in this high-throughput dataset (fig. S2C). To correct for this effect, dextramer signals were penalized if multiple dextramers simultaneously bind to the same T cell/clone, using the following technique.
Defining the background noise subtracted dextramer signal for the ith T cell binding the jth dextramer as Eij, we further denote the fraction of dextramer signal because of binding of the jth dextramer for the ith T cell as Cij
| (1) |
Denoting the TCR clonotype of the ith T cell as ki and the number of T cells belonging to clonotype ki that bind dextramer j as Tkij, we denote the fraction of T cells that belong to clonotype ki that bind the jth dextramer as Rkij
| (2) |
Using these quantities, we calculate the corrected dextramer signal for the ith T cell binding the jth dextramer as Sij
| (3) |
Step 5: Cell- and pMHC-wise dextramer signal normalization and identification of dextramer-specific binding TCRs. To make all the dextramer binding signals comparable, the corrected dextramer binding signals were log ratio–normalized across 44 testing dextramers within a cell. pMHC-wise normalization was subsequently conducted on the basis of log-rank distribution. Normalized dextramer signal >0 was empirically chosen as the cutoff to define dextramer-specific binders.
Regeneron oligo-tagged dextramer staining and sorting
CD8+ T cells were enriched from a healthy donor’s PBMCs using Miltenyi CD8+ T cell negative enrichment (Miltenyi). The cells were then incubated for 45 min with benzonase (Millipore) and dasatinib (Axon) before being stained with oligo-tagged dextramer pools (Immudex, table S2) for 30 min at room temperature. Cells were then stained with fluorescently labeled CD3 (BD Biosciences, catalog no. 612750), CD4 (BD Biosciences, catalog no. 563919), CD8 (BD Biosciences, catalog no. 612889), CCR7 (BioLegend, catalog no. 353218), and CD45RA (BioLegend, catalog no. 304238) and CITE-seq antibodies for an additional 30 min on ice. Using an Astrios cell sorter (Beckman Coulter), FACS gating on forward scatter plot, side scatter plot, and fluorescent channels was set to select live cells while excluding debris and doublets. We used a 100-μm nozzle to sort single CD3+CD8+dextramer+ cells for further processing.
Building a neural network–based classifier TCRAI
We designed a flexible framework for building TCR-antigen specificity classifiers, TCRAI. We used TCRAI to build a specific and consistent architecture throughout this study. In addition to its flexible architecture, some key differences from the DeepTCR (17) architecture are the use of 1D convolutions and batch normalization for the CDR3 sequences and lower dimensional representations for the genes. These changes improved model regularization and forced the model to learn stronger gene associations.
To process the input information of the TCR into numerical format, we apply the following method. For each CDR3 sequence, we first convert amino acids to integers and subsequently encode these integer vectors into a one-hot representation. For the V and J genes, we separately build a dictionary of gene type to integer for each V and J gene and use these to convert each gene to an integer.
The neural network architecture applied to the processed input information includes embedding layers and convolutional networks. Specifically, processed CDR3 residues were embedded into a 16D space via a learned embedding, and the resulting numeric CDR3s are fed through three 1D convolutional layers, with filters of dimensions [64,128,256], kernel widths [4, 5], and strides [1, 3]. Each convolution is activated by an exponential linear unit activation and is followed by dropout (48) and batch normalization (49). Following these three convolutional blocks, global max pooling is applied to the final features; this process encodes each CDR3 by a vector of length 256, a “CDR3 fingerprint.” The processed gene input for each gene is one-hot encoded and embedded into a reduced dimensional space (16 for V genes and 8 for J genes) via a learned embedding, giving a “fingerprint” of each gene as a vector. The fingerprints of all selected CDR3s and genes are concatenated together into a single vector, the “TCRAI fingerprint.” The TCRAI fingerprint is passed through one final full-connected layer to give binomial predictions (single output value, sigmoid activation), regression predictions (single output, no activation), or multinomial predictions (multiple output values, softmax activation). We focus on binomial and multinomial predictions in this work.
TCR sequencing files were collected as a raw csv formatted file from 10x Genomics. Sequencing files were parsed to take the amino acid sequence of the CDR3 after removing unproductive sequences. Clones with different nucleotide sequences but the same matched amino acid sequence from CDR3s and the V, D, J genes were aggregated together under one TCR. Thus, each TCR record we used here includes single-paired α and β TCR chains, with CDR3 amino acid sequence and V, J genes for each chain.
The data are split into training (76.5%), validation (13.5%), and left-out test set (10%) for each model, and subsequently, a fivefold Monte-Carlo cross-validation is performed on the training set. The model is trained by minimizing the cross-entropy loss via the Adam optimizer, and the cross-entropy loss is weighted by weights 1/(number of classes * fraction of samples in that class) for each class. Early stopping is engaged, via a left-out validation dataset, to prevent overfitting, in which the model ceases training if the validation loss increases for more than five epochs and the weights of the model with minimal validation loss are restored. Because of the large number of models being trained here, only the learning rate and batch size are tuned during cross-validation. After cross-validation, the optimally performing hyperparameters are chosen and the model is retrained on the full training set, using the validation set to control early stopping. The retrained model is then evaluated on the left-out test set.
TCRAI fingerprint analysis
TCRAI models produce both a prediction for a TCR to bind a specific pMHC (or one of many pMHCs in the multinomial case) and a numerical vector fingerprint that describes that TCR within the context of the question of whether it can bind that pMHC. To gain an understanding of how the model works, and to identify groups of TCRs with different binding modalities, we analyze the distribution of these fingerprints. We use UMAP (45) to reduce the fingerprints to a 2D space. When using a model trained on one dataset and inferring fingerprints on another unseen dataset, we fit the UMAP projector with TCRs from the training dataset and transform the TCRs from the unseen set using that projector.
When clustering TCR fingerprints, we project the fingerprints of all TCRs of the dataset into 2D space as described above and then select those TCRs that are strong true positives (STPs; binomial prediction >0.95). We then cluster these STPs using a k-means classifier in the 2D space. TCRs from within in each cluster are then collected and used to construct CDR3 motif logos (using WebLogo) (50), gene usage, and UMI distributions by pairing the unique TCR clonotypes within the cluster with all repeated clonotypes in the high-throughput data.
Motif construction
To construct a motif from a set of CDR3s of different lengths, it is necessary to apply a gapped alignment to map them all to be sequences of the same length. For a set of CDR3s, we define the CDR3 motif length L as the longest CDR3 in the set. Then, we align each CDR3 to the L-length motif via the introduction of gaps in the middle of the sequence. This allows one to see common structure in a set of CDR3s with similar gene usage and conserved motifs but with slight variation in length due to the addition of one or more residues in the junction, without having to restrict oneself to a subset of CDR3s with the same length. WebLogo was used to construct motif logos (50). The width of the residue stack at a position indicates how often a gap appears at that position: A narrower stack means gaps appear at that location more frequently. The height of each residue in a stack at a position indicates how often that residue appears at that position.
Comparison of classifiers for TCR-antigen specificity
TCRdist
Dash et al. (14) recently reported a weighted hamming distance–based method, TCRdist, to predict TCR-pMHC binding specificity based on the sequence space of TCR CDR regions guided by structural information on pMHC binding. Nearest-neighbor (NN) distance (the average TCRdist between a receptor and its NN receptors within the repertoire) was calculated to measure receptor density within repertoires (14). We applied their method in this study. For each pMHC repertoire, binders were defined to be TCRs that bind to the given pMHC. NN distances were calculated between each binding TCR and each set of pMHC binders with the given TCR removed. The NN distances were separated on the basis of the known specificity of each TCR. ROC curves and AUC were calculated for the binary classifier of each pMHC using the plotROC R package (51). In brief, ROC curves were generated by calculating sensitivity and specificity at several NN distance thresholds for each classifier—classifying TCRs as binding to a given pMHC if their NN distance falls below the given threshold. The final prediction sensitivity and specificity were calculated at the model threshold that maximized the geometric mean of the two.
GLIPH2
The TCR-pMHC binding dataset was used as the input data to GLIPH2. The analysis was performed using GLIPH2 online portal (50.255.35.37:8080) with configurations: reference = “CD8” and default parameters. GLIPH2 classified TCRs into different groups based on their shared TCR sequence features. After the classification, the eight filtering criteria as stated in GLIPH2 paper (15) were applied to each TCR group to reduce the noise. After filtering, the most enriched binding pMHC for a given TCR group was assigned to all TCRs that belong to this particular group as their antigen specificity. Comparing the true pMHC label and the assigned pMHC binding specificity, TCRs were classified as true positive (TP), false positive (FP), true negative (TN), and false negative (FN). Sensitivity and specificity for each pMHC repertoire that GLIPH2 identified using the eight different filtering criteria were calculated as follows: Sensitivity = TP/(TP + FN) and Specificity = TN/(TN + FP). This resulted in eight sensitivity/specificity values for each pMHC repertoire. We used the maximum sensitivity and specificity of these eight values to represent the best performance of GLIPH2 to a given pMHC repertoire.
NetTCR
We used the scripts with default parameters as described by Jurtz et al. (18) to train and test the data of eight pMHC repertoires from the gold-standard dataset through fivefold cross-validation. ROC curves and AUC were calculated for each pMHC repertoire using the plotROC R package (51). Sensitivity and specificity were calculated at the model threshold that maximized the geometric mean of the two.
DeepTCR
We adapted the DeepTCR method (17) to construct a binary classifier with the minor adjustments as described below. For each TCR record, we used the single paired α and β TCR chains, with CDR3 amino acid sequence and V, J genes for each chain only, in line with the inputs we provide to the TCRAI package. That is, we did not include clonality, MHC, and D gene usage to the DeepTCR model. The final output layer was adjusted to give a single binomial output, and hyperparameters of the model were optimized for the problem at hand in the context of the DeepTCR framework. Sensitivity and specificity were calculated at the model threshold that maximized the geometric mean of the two.
Acknowledgments
We thank M. Stubbington, W. Mcdonnell, V. Reid, and M. Schnall-Levin at 10x Genomics for method discussion. Funding: The authors acknowledge that they received no funding in support of this research. Author contributions: Conceptualization: G.S.A. and W.Z.; methodology: G.S.A., W.Z., P.G.H., J.H., N.T.G., R.D., and M.D.; software: W.Z. and P.G.H.; investigation: G.S.A., W.Z., P.G.H., J.H., and N.T.G.; formal analysis: W.Z., P.G.H., J.H., N.T.G., J.L., G.C., R.D., M.D., S.W.J., C.R.C., and A.D.; writing (review and editing): W.Z., P.G.H., J.H., N.T.G., R.D., G.T., and G.S.A.; writing (original draft): W.Z. and P.G.H.; supervision: G.S.A., W.Z., L.E.M., and G.T. Competing Interests: The authors have stock options in Regeneron Pharmaceuticals Inc. G.S.A., L.E.M., and G.T. are officers of Regeneron Pharmaceuticals Inc. W.Z., P.G.H., J.H., N.T.G., and G.S.A. are inventors on a patent application related to this work filed by Regeneron Pharmaceuticals Inc. (no. 63/013,480, filed on 21 April 2020, which is pending and unpublished). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The source code for ICON and TCRAI is available in public repositories under Regeneron’s License for Noncommercial Use:https://github.com/regeneron-mpds/TCRAI; https://github.com/regeneron-mpds/ICON. Patent application: W.Z., J.H., N.T.G., G.S.A., and P.G.H. are named inventors on U.S. Provisional Application nos. 63/013,480, 63/090,498, and 63/111,395. IRB and/or IACUC guidelines were followed with human or animal subjects.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf5835/DC1
REFERENCES AND NOTES
- 1.Krangel M. S., Mechanics of T cell receptor gene rearrangement. Curr. Opin. Immunol. 21, 133–139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahe E., Pugh T., Kamel-Reid S., T cell clonality assessment: Past, present and future. J. Clin. Pathol. 71, 195–200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gascoigne N. R. J., Rybakin V., Acuto O., Brzostek J., TCR signal strength and T cell development. Annu. Rev. Cell Dev. Biol. 32, 327–348 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Jung D., Alt F. W., Unraveling V(D)J recombination; insights into gene regulation. Cell 116, 299–311 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Jackson K. J. L., Kidd M. J., Wang Y., Collins A. M., The shape of the lymphocyte receptor repertoire: Lessons from the B cell receptor. Front. Immunol. 4, 263 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis M. M., Bjorkman P. J., T-cell antigen receptor genes and T-cell recognition. Nature 334, 395–402 (1988). [DOI] [PubMed] [Google Scholar]
- 7.Elhanati Y., Marcou Q., Mora T., Walczak A. M., repgenHMM: A dynamic programming tool to infer the rules of immune receptor generation from sequence data. Bioinformatics 32, 1943–1951 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zarnitsyna V. I., Evavold B. D., Schoettle L. N., Blattman J. N., Antia R., Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front. Immunol. 4, 485 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrack P., Rubtsova K., Scott-Browne J., Kappler J. W., T cell receptor specificity for major histocompatibility complex proteins. Curr. Opin. Immunol. 20, 203–207 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennecke J., Wiley D. C., T cell receptor-MHC interactions up close. Cell 104, 1–4 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Wieczorek M., Abualrous E. T., Sticht J., Álvaro-Benito M., Stolzenberg S., Noé F., Freund C., Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 8, 292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.La Gruta N. L., Gras S., Daley S. R., Thomas P. G., Rossjohn J., Understanding the drivers of MHC restriction of T cell receptors. Nat. Rev. Immunol. 18, 467–478 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Pogorelyy M. V., Minervina A. A., Shugay M., Chudakov D. M., Lebedev Y. B., Mora T., Walczak A. M., Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLOS Biol. 17, e3000314 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dash P., Fiore-Gartland A. J., Hertz T., Wang G. C., Sharma S., Souquette A., Crawford J. C., Clemens E. B., Nguyen T. H. O., Kedzierska K., la Gruta N. L., Bradley P., Thomas P. G., Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glanville J., Huang H., Nau A., Hatton O., Wagar L. E., Rubelt F., Ji X., Han A., Krams S. M., Pettus C., Haas N., Arlehamn C. S. L., Sette A., Boyd S. D., Scriba T. J., Martinez O. M., Davis M. M., Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanzarotti E., Marcatili P., Nielsen M., T-cell receptor cognate target prediction based on paired α and β chain sequence and structural CDR loop similarities. Front. Immunol. 10, 2080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.J.-W. Sidhom, H. B. Larman, P. Ross-MacDonald, M. Wind-Rotolo, D. M. Pardoll, A. S. Baras, DeepTCR: A deep learning framework for understanding T-cell receptor sequence signatures within complex T-cell repertoires. bioRxiv 464107 [Preprint]. 23 December 2019. 10.1101/464107. [DOI]
- 18.V. I. Jurtz, L. E. Jessen, A. K. Bentzen, M. C. Jespersen, S. Mahajan, R. Vita, K. K. Jensen, P. Marcatili, S. R. Hadrup, B. Peters, M. Nielsen, NetTCR: Sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks. bioRxiv 433706 [Preprint]. 3 October 2018. 10.1101/433706. [DOI]
- 19.S. Gielis, P. Moris, N. De Neuter, W. Bittremieux, B. Ogunjimi, K. Laukens, P. Meysman, TCRex: A webtool for the prediction of T-cell receptor sequence epitope specificity. bioRxiv 373472 [Preprint]. 22 July 2018. 10.1101/373472. [DOI]
- 20.Fischer D. S., Wu Y., Schubert B., Theis F. J., Predicting antigen specificity of single T cells based on TCR CDR3 regions. Mol. Syst. Biol. 16, e9416 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H., Liu L., Zhang J., Chen J., Ye J., Shukla S., Qiao J., Zhan X., Chen H., Wu C. J., Fu Y.-X., Li B., Investigation of antigen-specific T-cell receptor clusters in human cancers. Clin. Cancer Res. 26, 1359–1371 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Boutet S. C., Walter D., Stubbington M. J. T., Pfeiffer K. A., Lee J. Y., Taylor S. E. B., Montesclaros L., Lau J. K., Riordan D. P., Barrio A. M., Brix L., Jacobsen K., Yeung B., Zhao X., Mikkelsen T. S., Scalable and comprehensive characterization of antigen-specific CD8 T cells using multi-omics single cell analysis. J. Immunol. 202, 131.4 (2019).30518569 [Google Scholar]
- 23.Bentzen A. K., Marquard A. M., Lyngaa R., Saini S. K., Ramskov S., Donia M., Such L., Furness A. J. S., McGranahan N., Rosenthal R., Straten P. t., Szallasi Z., Svane I. M., Swanton C., Quezada S. A., Jakobsen S. N., Eklund A. C., Hadrup S. R., Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 34, 1037–1045 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Kula T., Dezfulian M. H., Wang C. I., Abdelfattah N. S., Hartman Z. C., Wucherpfennig K. W., Lyerly H. K., Elledge S. J., T-Scan: A genome-wide method for the systematic discovery of T cell epitopes. Cell 178, 1016–1028.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carter J. A., Preall J. B., Grigaityte K., Goldfless S. J., Jeffery E., Briggs A. W., Vigneault F., Atwal G. S., Single T cell sequencing demonstrates the functional role of αβ TCR pairing in cell lineage and antigen specificity. Front. Immunol. 10, 1516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas S., Stauss H. J., Morris E. C., Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology 129, 170–177 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stadinski B. D., Trenh P., Smith R. L., Bautista B., Huseby P. G., Li G., Stern L. J., Huseby E. S., A role for differential variable gene pairing in creating T cell receptors specific for unique major histocompatibility ligands. Immunity 35, 694–704 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sidney J., Peters B., Frahm N., Brander C., Sette A., HLA class I supertypes: A revised and updated classification. BMC Immunol. 9, 1 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schendel D. J., Frankenberger B., Limitations for TCR gene therapy by MHC-restricted fratricide and TCR-mediated hematopoietic stem cell toxicity. Oncoimmunology 2, e22410 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Veken L. T., Hoogeboom M., de Paus R. A., Willemze R., Falkenburg J. H. F., Heemskerk M. H. M., HLA class II restricted T cell receptor gene transfer generates CD4+ T cells with helper activity as well as cytotoxic capacity. Gene Ther. 12, 1686–1695 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Chen G., Yang X., Ko A., Sun X., Gao M., Zhang Y., Shi A., Mariuzza R. A., Weng N.-p., Sequence and structural analyses reveal distinct and highly diverse human CD8+ TCR repertoires to immunodominant viral antigens. Cell Rep. 19, 569–583 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sant S., Grzelak L., Wang Z., Pizzolla A., Koutsakos M., Crowe J., Loudovaris T., Mannering S. I., Westall G. P., Wakim L. M., Rossjohn J., Gras S., Richards M., Xu J., Thomas P. G., Loh L., Nguyen T. H. O., Kedzierska K., Single-cell approach to influenza-specific CD8+ T cell receptor repertoires across different age groups, tissues, and following influenza virus infection. Front. Immunol. 9, 1453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davidsen K., Olson B. J., DeWitt W. S. III, Feng J., Harkins E., Bradley P., Matsen F. A. IV, Deep generative models for T cell receptor protein sequences. eLife 8, e46935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean, M. Devin, S. Ghemawat, I. Goodfellow, A. Harp, G. Irving, M. Isard, Y. Jia, R. Jozefowicz, L. Kaiser, M. Kudlur, J. Levenberg, D. Mane, R. Monga, S. Moore, D. Murray, C. Olah, M. Schuster, J. Shlens, B. Steiner, I. Sutskever, K. Talwar, P. Tucker, V. Vanhoucke, V. Vasudevan, F. Viegas, O. Vinyals, P. Warden, M. Wattenberg, M. Wicke, Y. Yu, X. Zheng, TensorFlow: Large-scale machine learning on heterogeneous distributed systems. arXiv:1603.04467 [cs.DC] (14 March 2016).
- 35.Woodsworth D. J., Castellarin M., Holt R. A., Sequence analysis of T cell repertoires in health and disease. Genome Med. 5, 98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishizuka J., Stewart-Jones G. B. E., van der Merwe A., Bell J. I., McMichael A. J., Jones E. Y., The structural dynamics and energetics of an immunodominant T cell receptor are programmed by its Vβ domain. Immunity 28, 171–182 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Song I., Gil A., Mishra R., Ghersi D., Selin L. K., Stern L. J., Broad TCR repertoire and diverse structural solutions for recognition of an immunodominant CD8+ T cell epitope. Nat. Struct. Mol. Biol. 24, 395–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miles J. J., Bulek A. M., Cole D. K., Gostick E., Schauenburg A. J. A., Dolton G., Venturi V., Davenport M. P., Tan M. P., Burrows S. R., Wooldridge L., Price D. A., Rizkallah P. J., Sewell A. K., Genetic and structural basis for selection of a ubiquitous T cell receptor deployed in Epstein-Barr virus infection. PLOS Pathog. 6, e1001198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamga L., Gil A., Song I., Brody R., Ghersi D., Aslan N., Stern L. J., Selin L. K., Luzuriaga K., CDR3α drives selection of the immunodominant Epstein Barr virus (EBV) BRLF1-specific CD8 T cell receptor repertoire in primary infection. PLOS Pathog. 15, e1008122 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen T. H. O., Bird N. L., Grant E. J., Miles J. J., Thomas P. G., Kotsimbos T. C., Mifsud N. A., Kedzierska K., Maintenance of the EBV-specific CD8+ TCRαβ repertoire in immunosuppressed lung transplant recipients. Immunol. Cell Biol. 95, 77–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin M. D., Badovinac V. P., Defining memory CD8 T cell. Front. Immunol. 9, 2692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tu A. A., Gierahn T. M., Monian B., Morgan D. M., Mehta N. K., Ruiter B., Shreffler W. G., Shalek A. K., Love J. C., TCR sequencing paired with massively parallel 3′ RNA-seq reveals clonotypic T cell signatures. Nat. Immunol. 20, 1692–1699 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.L. McInnes, J. Healy, J. Melville, UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv:1802.03426 [stat.ML] (9 February 2018).
- 46.Bagaev D. V., Vroomans R. M. A., Samir J., Stervbo U., Rius C., Dolton G., Greenshields-Watson A., Attaf M., Egorov E. S., Zvyagin I. V., Babel N., Cole D. K., Godkin A. J., Sewell A. K., Kesmir C., Chudakov D. M., Luciani F., Shugay M., VDJdb in 2019: Database extension, new analysis infrastructure, and a T-cell receptor motif compendium. Nucleic Acids Res. 48, D1057–D1062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tickotsky N., Sagiv T., Prilusky J., Shifrut E., Friedman N., McPAS-TCR: A manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929 (2017). [DOI] [PubMed] [Google Scholar]
- 48.G. E. Hinton, N. Srivastava, A. Krizhevsky, I. Sutskever, R. R. Salakhutdinov, Improving neural networks by preventing co-adaptation of feature detectors. arXiv:1207.0580 [cs.NE] (3 July 2012).
- 49.S. Ioffe, C. Szegedy, Batch normalization: Accelerating deep network training by reducing internal covariate shift. arXiv:1502.03167 (11 February 2015).
- 50.Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E., WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sachs M. C., plotROC: A tool for plotting ROC curves. J. Stat. Softw. 79, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf5835/DC1