

A human heart cell type–specific cis-regulatory element atlas reveals gene regulatory mechanisms of cardiovascular diseases.
Abstract
Misregulated gene expression in human hearts can result in cardiovascular diseases that are leading causes of mortality worldwide. However, the limited information on the genomic location of candidate cis-regulatory elements (cCREs) such as enhancers and promoters in distinct cardiac cell types has restricted the understanding of these diseases. Here, we defined >287,000 cCREs in the four chambers of the human heart at single-cell resolution, which revealed cCREs and candidate transcription factors associated with cardiac cell types in a region-dependent manner and during heart failure. We further found cardiovascular disease–associated genetic variants enriched within these cCREs including 38 candidate causal atrial fibrillation variants localized to cardiomyocyte cCREs. Additional functional studies revealed that two of these variants affect a cCRE controlling KCNH2/HERG expression and action potential repolarization. Overall, this atlas of human cardiac cCREs provides the foundation for illuminating cell type–specific gene regulation in human hearts during health and disease.
INTRODUCTION
Disruption of gene regulation is an important contributor to cardiovascular disease, the leading cause of morbidity and mortality worldwide (1). Cis-regulatory elements such as enhancers and promoters are crucial for regulating gene expression (2–5). Mutations in transcription factors and chromatin regulators can result in heart disease (6, 7), and genetic variants associated with risk of cardiovascular disease are enriched within annotated candidate cis-regulatory elements (cCREs) in the human genome (8). However, a major barrier to understanding the genetic and molecular basis of cardiovascular diseases is the paucity of maps and tools to interrogate gene regulatory programs in the distinct cell types of the human heart. Recent single-cell/nucleus RNA sequencing (RNA-seq) (9–11) and spatial transcriptomic (12) studies have revealed gene expression patterns in distinct cardiac cell types across developmental and adulthood stages in the human heart, including some that display gene expression patterns that are cardiac chamber/region specific (10, 11). However, the transcriptional regulatory programs responsible for cell type–specific and chamber-specific gene expression, and their potential links to noncoding risk variants for cardiovascular diseases and traits, remain to be fully defined.
cCREs have been annotated in the human genome with the use of chromatin immunoprecipitation sequencing (ChIP-seq), deoxyribonuclease sequencing (DNase-seq), assay for transposase-accessible chromatin using sequencing (ATAC-seq), global run-on sequencing, etc. in a broad spectrum of human tissues including in bulk heart tissues and in purified cardiomyocytes (2–5, 13–18). These maps have provided important insights into dynamic gene regulation during heart failure (15, 16, 18) and begun to shed light on the function of noncoding cardiovascular disease variants (8, 13, 16, 18, 19). However, major limitations of these studies including their focus on particular chambers/regions of the heart and failure to interrogate cis-regulatory elements across all distinct cardiac cell types have restricted their utility in understanding how specific gene regulatory mechanisms may affect distinct cell types and regions of human hearts in health and disease. Although recent single-cell genomic tools provide the opportunity to interrogate cis-regulatory elements at single-cell resolution (20–23), their application to mammalian hearts has been limited to one single-cell ATAC-seq dataset from adult mouse heart (24), fewer than 200 total cells from mouse fetal hearts (25), and fetal human heart (26). Thus, to comprehensively investigate cis-regulatory elements in the specific cell types of the adult human heart, we profiled chromatin accessibility in ~80,000 heart cells using single-nucleus ATAC-seq (snATAC-seq) (21, 27) and created a comprehensive cardiac cell atlas of cCREs annotated by cell type and putative target genes. Integration of these data with single-nucleus RNA-seq (snRNA-seq) datasets from matched specimens revealed gene regulatory programs in nine major cardiac cell types. Using this human cardiac cCRE atlas, we further observed the remodeling of cell type–specific candidate enhancers during heart failure and the enrichment of cardiovascular disease–associated genetic variants in cCREs of specific cell types. Last, we showed that a cardiomyocyte-specific enhancer harboring risk variants for atrial fibrillation (AF) is necessary for cardiomyocyte KCNH2 expression and regulation of cardiac action potential repolarization.
RESULTS
Single nucleus analysis of chromatin accessibility and transcriptome in adult human hearts
To assess the accessible chromatin landscape of distinct cardiovascular cell types, we performed snATAC-seq (27), also known as sci-ATAC-seq (21), on all cardiac chambers from four adult human hearts without known cardiovascular disease (table S1). We obtained accessible chromatin profiles for 79,515 nuclei, with a median of 2682 fragments mapped per nucleus (Fig. 1, A and B; fig. S1; and table S2). We also performed snRNA-seq for a subset of the above heart samples to complement the accessible chromatin data and obtained 35,936 nuclear transcriptomes, with a median of 2184 unique molecular identifiers and 1286 genes detected per nucleus (Fig. 1, A and C; fig. S2, A to F; and table S3). Using SnapATAC (28) and Seurat (29), we identified nine clusters from snATAC-seq (Fig. 1B) and 12 major clusters from snRNA-seq (Fig. 1C and fig. S2, G and H), which were annotated on the basis of chromatin accessibility at promoter regions or expression of known lineage-specific marker genes, respectively (Fig. 1, D and E, and table S4) (10, 11). For example, chromatin accessibility and gene expression of atrial and ventricular cardiomyocyte markers such as NPPA and MYH7 (30) were used to classify these two cardiomyocyte subtypes (Fig. 1, D and E). Although gene expression patterns of lineage markers strongly correlated with accessibility at promoter regions across annotated cell types (Fig. 1F) and single-cell integration analysis (29) revealed 93% concordance in annotation between snATAC-seq and snRNA-seq datasets (fig. S3 and table S3), some cellular subtypes identified from snRNA-seq including endocardial cells and myofibroblasts were not detected by snATAC-seq (Fig. 1F). Notably, cluster correlation and integration analysis showed that these cell types are present within the snATAC-seq data as part of the endothelial and smooth muscle clusters, respectively (Fig. 1F, fig. S3, and table S3). The discrepancy in clustering may be attributable to the conservative snATAC-seq clustering parameters or the sparse nature of snATAC-seq data (31). In addition, atrial and ventricular cardiomyocyte nuclei from the left and right regions of the heart could be further clustered by transcriptome but not chromatin accessibility (fig. S2, I and J). We noted that cell type composition varied substantially between biospecimens and donors, highlighting the importance of single-cell approaches to limit biases due to cell proportion differences in bulk assays (fig. S4 and tables S2 and S3). In summary, we identified and annotated cardiac cell types using both chromatin accessibility and nuclear transcriptome profiles.
Fig. 1. Single-nucleus chromatin accessibility and transcriptome profiling of human hearts.
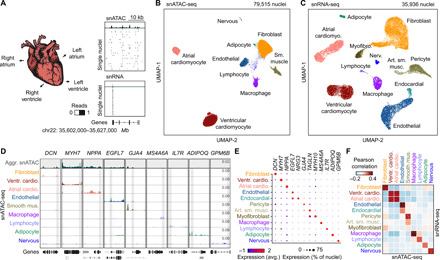
(A) snATAC-seq and snRNA-seq were performed on nuclei isolated from cardiac chambers from four human donors without cardiovascular pathology. snATAC-seq: n = 4 (left ventricle), n = 4 (right ventricle), n = 3 (left atrium), and n = 2 (right atrium); snRNA-seq: n = 2 (left ventricle), n = 2 (right ventricle), n = 2 (left atrium), and n = 1 (right atrium). (B) Uniform manifold approximation and projection (UMAP) (108) and clustering analysis of snATAC-seq data reveal nine clusters. Each dot represents a nucleus colored by cluster identity. (C) UMAP (108) and clustering analysis of snRNA-seq data reveal 12 major clusters. Each dot represents a nucleus colored by cluster identity. Nerv., nervous. Art. sm. musc., arterial smooth muscle. (D) Genome browser tracks (141) of aggregate chromatin accessibility profiles [scale: reads per million (RPM)] at selected representative marker gene examples for individual clusters and for all nuclei pooled together into an aggregated heart dataset (top track, gray). Black genes below tracks represent the indicated marker genes, nonmarker genes are grayed. (E) Dot plot illustrating expression of representative marker gene examples in individual snRNA-seq clusters. (F) Heatmap illustrating the correlation between clusters defined by chromatin accessibility and transcriptomes. Pearson correlation coefficients were calculated between chromatin accessibility at cCREs within 2 kbp of annotated promoter regions (76) and expression of the corresponding genes for each cluster.
Identification of cCREs in distinct cell types of the human heart
To discover the cCREs in each cell type of the human heart, we aggregated snATAC-seq data from nuclei comprising each cell cluster individually and determined accessible chromatin regions with MACS2 (32). We then merged the peaks from all nine cell clusters into a union of 287,415 cCREs, which covered 4.7% of the human genome (Fig. 2A and table S5). Sixty-seven percent of the cCREs identified in the current study overlapped previously annotated cCREs from a broad spectrum of human tissues and cell lines (fig. S5A) (5), and the union of heart cCREs captured 98.6 and 95.4% of candidate human heart enhancers reported in two previous bulk studies (fig. S5, B and C) (13, 15). Furthermore, 75% of cCREs in the union were at least 2 kilo–base pairs (kbp) away from annotated promoter regions, and 19,447 displayed high levels of cell type specificity [false discovery rate (FDR) < 0.01; Fig. 2B and table S6]. Gene ontology analysis (33) revealed that these cell type–specific cCREs were proximal to genes involved in relevant biological processes, including collagen fibril organization for cardiac fibroblast–specific cCREs (K1) and myofibril organization for ventricular cardiomyocyte–specific cCREs (K2; Fig. 2C and table S7). Using chromVAR (table S8) (34) and HOMER (table S9) (35), we detected cell type–dependent enrichment for 231 transcription factor–binding signatures, such as MEF2A/B, NKX2.5, and THR-β sequence motifs in cardiomyocyte-specific cCREs and TCF21 motifs in cardiac fibroblast–specific cCREs (Fig. 2D). To discover the transcription factors that may bind to these sites, we combined corresponding snRNA-seq data with sequence motif enrichments to correlate the expression of these transcription factors with motif enrichment patterns across cell types (Fig. 2E). As an example, we found strong enrichment of the binding motif for the macrophage transcription factor SPI1/PU.1 (36) in macrophage-specific cCREs, and SPI1 was exclusively expressed in macrophages (Fig. 2E and table S4). In addition, we observed that transcription factor family members were expressed in cell type–specific combinations. For instance, while GATA Family of transcription factors (GATA-binding factor) displayed similar motif enrichment patterns across sets of cell type–specific cCREs, we found that endothelial cells and cardiac fibroblasts expressed GATA2 and GATA6, respectively, whereas cardiomyocytes expressed both GATA4 and GATA6 and endocardial cells expressed GATA2, GATA4, and GATA6 (Fig. 2E and table S4). In summary, these results establish a resource of cCREs for interrogation of cardiac cell type–specific gene regulatory programs.
Fig. 2. Characterization of gene regulatory programs in cardiac cell types.
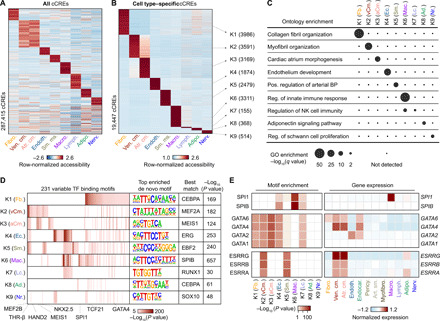
(A) Heatmap illustrating row-normalized chromatin accessibility values for the union of 287,415 cCREs. K-means clustering was performed to group cCREs on the basis of relative accessibility patterns. (B) Heatmap showing row-normalized chromatin accessibility of 19,447 cell type–specific cCREs (FDR < 0.01 after Benjamini-Hochberg correction; fold change > 1.2). K-means clustering was performed to group cCREs on the basis of relative accessibility patterns. Number of cCREs per K can be found in brackets. (C) Genomic Regions Enrichment of Annotations Tool (GREAT) ontology analysis (33) of cell type–specific cCREs. Q value for enrichment indicates Bonferroni-adjusted P value. NK, natural killer; GO, gene ontology; BP, blood pressure. (D) Transcription factor motif enrichment (35) for known and de novo motifs within cell type–specific cCREs. The heatmap in shows motifs with an enrichment P value of <10−5 in at least one cluster. For de novo transcription factor motifs, the best matches for the top motifs are displayed. Statistical test for motif enrichment: hypergeometric test. P values were not corrected for multiple testing. (E) Combination of transcription factor motif enrichment and gene expression shows cell type–specific roles for members of transcription factor families. Displayed are heatmaps for known motif enrichment in cell type–specific cCREs (left) and gene expression across clusters (right). (Fb., fibroblast; vCm., ventricular cardiomyocyte; aCm., atrial cardiomyocyte; Ec., endothelial; Sm., smooth muscle; Mac., macrophage; Lc., lymphocyte; Ad., adipocyte; Nr., nervous.)
Cardiac cell type–specific gene regulatory programs implicated in chamber-specific structure and function
Each cardiac chamber performs a unique role that is crucial to system-level heart function (37). To investigate the gene regulatory programs underlying chamber-specific gene expression and cellular functions in distinct cardiac cell types, we tested cCREs for differential accessibility across five of the most abundant cell types of the heart: cardiomyocytes, cardiac fibroblasts, endothelial cells, smooth muscle cells, and macrophages. We found 16,451 differentially accessible (DA) cCREs between pooled atria and ventricles, most of which were detected in cardiomyocytes (Fig. 3, A to C, and table S10). Specifically, 11,159 cCREs displayed differential accessibility between the right atrium and the right ventricle, and 12,962 cCREs exhibited differential accessibility between the left atrium and the left ventricle (fig. S6, A to C, and table S10). Comparing the left and right sides of the heart, we identified 101 DA cCREs between the right and left ventricles (fig. S6D) and 2687 DA cCREs between left and right atria, which, in contrast to comparisons between atria and ventricles, were found primarily in cardiac fibroblasts (fig. S6E and table S10).
Fig. 3. Cardiomyocyte cCREs display chamber-dependent differences in chromatin accessibility.
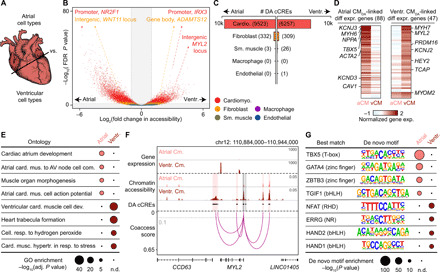
(A) Scheme for comparison of major cell types across heart chambers. (B) Volcano plot showing DA cCREs in each cell type between atria and ventricles. Each dot represents a cCRE, and the color indicates the cell type. cCREs with log2(fold change) > 1 and FDR < 0.05 after Benjamini-Hochberg correction (outside the shaded area) were considered as DA. (C) Number of DA cCREs between atria and ventricles by cell type. (D) Heatmaps showing normalized gene expression levels of differentially expressed genes between atrial (aCM) and ventricular cardiomyocytes (vCM) that were linked by coaccessibility to distal DA cCREs that were more accessible in atrial cardiomyocytes (Atrial CMDA) or ventricular cardiomyocytes (Ventr. CMDA), respectively. (E) GREAT ontology analysis (33) of DA cCREs between atrial and ventricular cardiomyocytes. AV, atrioventricular. P values shown are Bonferroni adjusted (n.d., not detected). (F) Genome browser tracks (141) showing chromatin accessibility (scale, RPM) and gene expression [scale, reads per kilobase per million mapped reads (RPKM)] in atrial and ventricular cardiomyocytes as well as DA cCREs that were coaccessible with the promoter of MYL2. Gray dotted line indicates coaccessibility threshold (>0.1). Red shaded areas, distal DA cCREs coaccessible with MYL2 promoter. Gray shaded areas, DA cCREs overlapping the promoter region of MYL2. (G) Transcription factor motif enrichment analysis (35) of DA cCREs between atrial and ventricular cardiomyocytes. The best matches for the top de novo motifs (score > 0.7) are shown. Statistical test for motif enrichment: hypergeometric test. P values were not corrected for multiple testing.
Using coaccessibility analysis (38) to link distal DA cCREs (~88% of all DA cCREs) to their putative target genes (table S11; median distance, 88.7 kbp), we observed that distal DA cCREs in cardiomyocytes between atria and ventricles were associated with chamber-specific gene expression of their putative target genes (Fig. 3D; fig. S6, B to E; and table S12), and genes near these DA cCREs were enriched for chamber-specific biological processes (Fig. 3E; fig. S6, B to E; and table S13). Specifically, distal DA cCREs with higher accessibility in atrial cardiomyocytes were associated with genes such as PITX2, a transcriptional regulator of cardiac atrial development (39), as well as the ion channel subunit SCN5A, which regulates cardiomyocyte action potential (Fig. 3E and table S13) (40). Furthermore, we found distal DA cCREs with higher accessibility in atrial cardiomyocytes at the HAMP gene locus, which encodes a key regulator of ion homeostasis and was recently described as a potential previously unknown cardiac gene in the right atrium by single-nucleus transcriptomic analysis (10, 11). Conversely, genes near distal DA cCREs with higher accessibility in ventricular cardiomyocytes were enriched for biological processes such as trabecula formation and ventricular cardiac muscle cell differentiation. For example, several distal DA cCREs with increased accessibility in ventricular cardiomyocytes compared to atrial cardiomyocytes were linked to the promoter region of MYL2, which encodes the ventricular isoform of myosin light chain 2 (Fig. 3F and table S4) (41), a regulator of ventricular cardiomyocyte sarcomere function.
In addition, analysis of distal DA cCREs in cardiac fibroblasts revealed that putative target genes were involved in distinct biological processes between right and left atria. In particular, we found that DA cCREs with higher accessibility in right atrial cardiac fibroblasts were proximal to genes involved in heart development, heart growth, and tube development, whereas DA cCREs with higher accessibility in left atrial cardiac fibroblasts were adjacent to genes involved in biological processes such as wound healing and vasculature development (fig. S6E and table S13). We further found a cardiac fibroblast–specific DA cCRE with higher accessibility in left atria at the fibrinogen FN1 gene locus, potentially indicating a more activated fibroblast state (10, 42). Supporting these findings, we identified several other DA cCREs with higher accessibility in left atrial cardiac fibroblasts adjacent to genes involved in generation of extracellular matrix (ECM) such as MMP2 and FBLN2 (table S13). These observations are consistent with previous findings that a higher fraction of ECM is produced in fibroblasts of the left atrium (10).
Using motif enrichment analysis, we inferred candidate transcriptional regulators involved in chamber-specific cellular specialization, including TBX5 (T-Box Transcription Factor 5), GATA4, and TGIF1 (TGFB Induced Factor Homeobox 1) for atrial cardiomyocytes and nuclear factor of activated T cells (NFAT), ERRG (Estrogen-Related Receptor Gamma), HAND1 (Heart- and Neural Crest Derivatives-Expressed Protein 1), and HAND2 for ventricular cardiomyocytes (Fig. 3G and table S14). While the TBX5 DNA binding motif was strongly enriched in both right and left atrial cardiomyocyte DA cCREs, the NFAT5 motif ranked highest in left ventricular cardiomyocyte DA cCREs and the TBX20 motif was strongly enriched in right ventricular cardiomyocyte DA cCREs (fig. S6, B and C, and table S14). Furthermore, cardiac fibroblast DA cCREs with higher accessibility in the right atrium were enriched for the binding motif of forkhead transcription factors (fig. S6E), whereas cardiac fibroblast DA cCREs with higher accessibility in the left atrium were enriched for the homeobox transcription factor CUX1 motif (fig. S6E and table S14). Together, we identified cCREs and candidate transcription factors associated with specific cardiac chambers, particularly within cardiomyocytes and cardiac fibroblasts.
Cell type specificity of candidate enhancers associated with heart failure
Recent large-scale studies profiling the H3K27ac histone modification in human hearts have uncovered candidate enhancers associated with heart failure (15, 18). However, because these studies either examined heterogeneous bulk heart tissue (15, 18) or focused solely on enriched cardiomyocytes (16), it remains unclear what role, if any, additional cardiac cell types and cCREs may contribute to heart failure pathogenesis. Using our cell atlas of cardiac cCREs, we revealed the cell type specificity of candidate enhancers showing differential H3K27ac signal strength between human hearts from healthy donors and donors with dilated cardiomyopathy (heart failure) (Fig. 4 and fig. S7, A to E) (15). We observed that a large fraction of candidate enhancers that displayed increased activity (45%) during heart failure were accessible primarily in cardiac fibroblasts (Fig. 4A, K2-4up, and table S15), whereas most of those exhibiting decreased activity (67%) were accessible primarily in cardiomyocytes (Fig. 4B, K1-3down, and table S15). Candidate enhancers with increased activity in cardiac fibroblasts were proximal to genes involved in ECM organization and connective tissue development (Fig. 4A, K2-4up, and table S16), whereas those exhibiting decreased activity in cardiomyocytes were proximal to genes involved in the regulation of heart contraction and cation transport (Fig. 4B, K1-3down, and table S16). For example, several of these cardiac fibroblast candidate enhancers were present at loci encoding the ECM proteins lumican (LUM) and decorin (DCN) and coaccessible with the promoters of these genes (Fig. 4C). Consistent with these findings, both genes were primarily expressed in cardiac fibroblasts (fig. S7F and table S4), and LUM has been reported to exhibit increased expression in failing hearts compared to control hearts (15). On the other hand, several cardiomyocyte candidate enhancers displaying decreased activity in heart failure were coaccessible with the promoter region of IRX4 (Fig. 4D), which encodes a ventricle-specific transcription factor (43) and is primarily expressed in cardiomyocytes of the left ventricle (fig. S7G and table S4).
Fig. 4. Cell type specificity of candidate enhancers associated with heart failure.
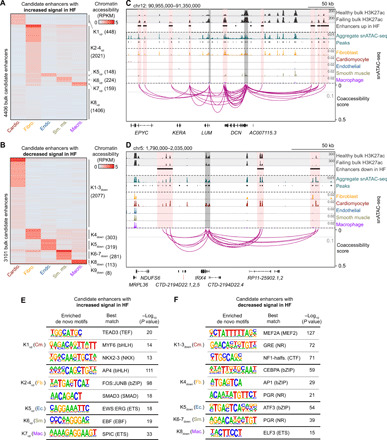
(A) Cell type specificity of 4406 candidate enhancers with increased H3K27ac signal in failing left ventricles (15). Heatmap displays cell type–resolved chromatin accessibility RPKM values for cell types from left ventricular snATAC-seq datasets. Candidate enhancers were grouped on the basis of chromatin accessibility patterns across cell clusters using K-means. HF, heart failure. (B) Cell type specificity of 3101 candidate enhancers with decreased H3K27ac signal in failing left ventricles (15). (C) Genome browser tracks (141) showing several candidate enhancers with increased activity during heart failure that were primarily accessible in fibroblasts and coaccessible with the promoters of LUM and/or DCN. For visualization, linkages between cCREs within candidate enhancers and all gene promoters are shown (coaccessibility > 0.1, gray dotted line). Candidate enhancers coaccessible with gene promoters are indicated by red shaded boxes and promoter regions are indicated by gray shaded boxes (scale, RPM). (D) Genome browser tracks (141) showing several bulk candidate enhancers with decreased activity in heart failure that were primarily accessible in cardiomyocytes and coaccessible with the promoter of IRX4 (scale, RPM). (E and F) Transcription factor motif enrichment (35) in the candidate enhancers with (E) increased and (F) decreased activity in failing left ventricles. Analysis was performed on the indicated K cluster(s) from (A) and (B), respectively. The best matches for selected de novo motifs (score > 0.7) are shown. Statistical test for motif enrichment: hypergeometric test. P values were not corrected for multiple testing.
To identify potential transcription factors regulating these pathologic responses during heart failure, we performed motif enrichment analysis in cell type–specific subsets of enhancers showing differential H3K27ac signal strength between healthy and failing hearts (table S17). For candidate enhancers exhibiting increased activity in heart failure, we identified enrichment of not only bHLH motifs such as AP4 in cardiac fibroblast candidate enhancers, which matched previous bulk analysis (Fig. 4E, K2-4up) (15), but also TEAD3 and MYF6 motifs in cardiomyocyte candidate enhancers (Fig. 4E, K1up). Conversely, for candidate enhancers displaying decreased activity in heart failure, we observed enrichment of nuclear receptor motifs such as glucocorticoid response element in cardiomyocyte candidate enhancers, which is consistent with previous findings (Fig. 4F, K1-3down) (15), as well as other motifs that were not detected in bulk analyses, such as the bZIP transcription factor CEBPA for cardiac fibroblast candidate enhancers (Fig. 4F, K4down). Thus, these results show that this cardiac cell atlas of cCREs may be used to assign disease-associated candidate enhancers from bulk assays to their affected cell types and infer transcriptional regulators involved in lineage-specific disease pathogenesis.
Interpreting noncoding risk variants of cardiac diseases and traits
Noncoding genetic variants contributing to risk of complex diseases are enriched within cCREs in a tissue and cell type–dependent manner (24, 44–47). To examine the enrichment of cardiovascular disease variants within cCREs active in cardiac cell types, we performed cell type–stratified LD (linkage disequilibrium) score regression analysis (48) using genome-wide association study (GWAS) summary statistics for cardiovascular diseases (Fig. 5A) (49–53) and control traits (fig. S8A and table S18) by measuring the enrichment of disease-associated variants within all cCREs identified for each cell type. This analysis revealed significant enrichment of AF-associated variants in both atrial (Z = 5.61, FDR = 1.9 × 10−6) and ventricular cardiomyocyte cCREs (Z = 6.80, FDR = 2.8 × 10−9), varicose vein-associated variants in endothelial cell cCREs (Z = 4.36, FDR = 3.9 × 10−4), and coronary artery disease–associated variants in cardiac fibroblast cCREs (Z = 3.29, FDR = 1.7 × 10−2; Fig. 5A). Notably, except for AF, these associations were not significant in a pseudo-bulk heart dataset created by combining chromatin accessibility profiles from all cardiac cell types (Fig. 5A). Furthermore, cardiovascular disease variants were not significantly enriched in accessible chromatin from noncardiac tissues (3, 5, 17) or human lung cell types (54), with the exception of a significant enrichment of varicose vein–associated variants in endothelial cells (fig. S8, B and C).
Fig. 5. Identification and characterization of AF-associated variants at the KCNH2 locus.
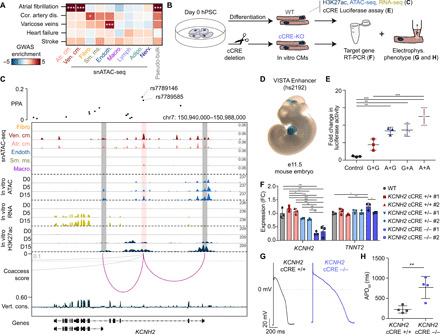
(A) Enrichment of cardiovascular disease variants within cardiac cell type cCREs. z scores are shown and were used to compute two-sided P values for enrichments. *FDR < 0.05 and ***FDR < 0.001. (B) Cardiomyocyte differentiation model schematic. (C) Fine mapping (55) and molecular characterization of two AF variants. Genome browser tracks (141) for snATAC-seq (top; scale, RPM) and indicated molecular features during hPSC-cardiomyocyte differentiation (scale, RPKM). Coaccessibility track shows linkages between the AF variant–containing cCRE and promoters (cutoff > 0.1). PPA, posterior probability of association (55). (D) Transgenic mouse embryo showing LacZ reporter expression under control of a genomic region [hs2192, VISTA database (58)] overlapping the variant-cCRE pair. (E) Luciferase reporter assay for the AF variant–harboring cCRE in D15 cardiomyocytes. Genotypes for rs7789146 and rs7789585 were either both G (risk), both A (nonrisk), or a combination. Each dot represents luciferase activity (average of two transfections) in independent replicates of D15 cardiomyocytes. Data are means ± SD. ***P < 0.001 and **P < 0.01 [one-way analysis of variance (ANOVA) and Tukey post hoc test]. Control: minimal promoter. (F) Expression of KCNH2 and TNNT2 in D25 cardiomyocytes after CRIPSR-Cas9–mediated cCRE deletion. Each dot represents an independent cardiomyocyte differentiation. Data are means ± SD. ***P < 0.001, **P < 0.01, and *P < 0.05 (one-way ANOVA and Tukey post hoc test); WT, unperturbed control. FC, fold change. (G) Action potential recordings in hPSC-derived cardiomyocytes with and without cCRE deletion at D25-35. (H) APD90 (action potential duration at 90% depolarization) at 1-Hz pacing for four independent hPSC-derived cardiomyocytes with and without cCRE deletion at D25-35. Data are means ± SD. **P < 0.01 (unpaired two-sided t test).
Next, to identify likely causal AF risk variants in cardiomyocyte cCREs, we first determined the probability that variants were causal for AF [posterior probability of association (PPA)] at 111 known loci using Bayesian fine mapping (55). We then intersected fine-mapped AF variants with cCREs detected in atrial and/or ventricular cardiomyocytes and identified 38 variants with PPA > 10% in cardiomyocyte cCREs, including previously reported variants at the HCN4 (13) and SCN10A/SCN5A (56) loci (table S19). We further prioritized AF variants for molecular characterization on the basis of their overlap with cCREs that were primarily accessible in cardiomyocytes, evolutionarily conserved, and coaccessible with promoters of genes expressed in cardiomyocytes. To experimentally validate the molecular functions of cCREs containing AF variants, we used a human pluripotent stem cell (hPSC)–derived cardiomyocyte differentiation model system (Fig. 5B) (57). From the variant prioritization analysis, we found a cCRE in the second intron of the potassium channel gene KCNH2 (HERG), which was coaccessible with the KCNH2 promoter (Fig. 5C) and harbored two variants, rs7789146 and rs7789585, with a combined PPA of 28% (Fig. 5C and fig. S9A). KCNH2 was primarily expressed in ventricular and atrial cardiomyocytes in the adult human heart (fig. S9B). The cCRE appeared to be activated during hPSC-cardiomyocyte differentiation, as evidenced by an increase in H3K27ac signal that correlated with KCNH2 expression (Fig. 5C). Supporting its in vivo role in regulating gene expression in mammalian hearts, a genomic region (hs2192) (58) containing this cCRE was previously shown to drive LacZ reporter expression in mouse embryonic hearts (Fig. 5D) (58).
A cardiomyocyte enhancer of KCNH2 is affected by noncoding risk variants associated with AF
To investigate whether these AF variants may affect enhancer activity and thereby regulate KCNH2 expression and cardiomyocyte electrophysiologic function, we initially carried out reporter assays using a hPSC cardiomyocyte model system. Results from these studies confirmed that in D15 hPSC cardiomyocytes, the KCNH2 enhancer carrying the rs7789146-G/rs7789585-G AF risk allele displayed significantly weaker enhancer activity than when containing the nonrisk variants (Fig. 5E and fig. S9C), thus supporting the functional significance of these AF variants. We next used CRISPR-Cas9 genome editing strategies to remove the enhancer and performed quantitative polymerase chain reaction (qPCR) and electrophysiologic assays to examine its role in KCNH2 expression and function. Supporting the aforementioned findings, CRISPR-Cas9 genome deletion of this cCRE in hPSC cardiomyocytes resulted in decreased KCNH2 expression in an enhancer dosage–dependent manner (Fig. 5F and fig. S9D). Similar to human cardiomyocytes with loss of KCNH2 function due to mutations in the KCNH2 coding sequence (59) or gene knockdown (60), cellular electrophysiologic studies demonstrated that these cCRE-deleted hPSC cardiomyocytes displayed a significantly prolonged action potential duration (Fig. 5, G and H), thus suggesting that cardiac repolarization abnormalities in atrial cardiomyocytes may lead to AF in an analogous manner to ventricular arrhythmias due to long QT syndrome (59). Together, these results highlight the utility of this single-cell atlas for assigning noncoding cardiovascular disease risk variants to distinct cell types and affected cCREs and functionally interrogating how these variants may contribute to cardiovascular disease risk.
DISCUSSION
The limited ability to interrogate cell type–specific gene regulation in the human heart has been a major barrier for understanding molecular mechanisms of cardiovascular traits and diseases. Here, we report a cell type–resolved atlas of cCREs in the human heart, which was ascertained by profiling accessible chromatin in individual nuclei from all four chambers of multiple human hearts and includes both cell type–specific and heart chamber–specific cCREs. In particular, we observed chamber-specific differences in chromatin accessibility between ventricles and atria as well as left and right atria but notably detected few differences between left and right ventricles. This finding is consistent with recent snRNA-seq analysis in human hearts, which found few differentially expressed genes between left and right ventricles (11). We note that the power to detect chamber-specific differences in chromatin accessibility depends on the number of samples assayed and the total nuclei used as input for differential analysis. Thus, future studies with larger cohorts will likely reveal additional chamber-specific differences between cardiac cell types.
We further highlight the utility of this atlas of heart cCREs to provide previously unidentified insight into aberrant gene regulation during cardiovascular pathology. To this end, we delineated the cell type specificity of enhancers that were differentially active between healthy and failing heart tissue (15) and identified additional transcription factors that may be involved in the pathogenesis of specific cell types during heart failure. Such cell type–specific analysis is particularly important in the context of heart failure because cellular composition can differ between diseased and control hearts (16, 61). This change in cellular composition may, in part, explain the cell type bias that we observed between candidate enhancers exhibiting increased and decreased activity during heart failure (i.e., cardiac fibroblasts and cardiomyocytes, respectively). However, because of the large differences in H3K27ac signal, we suspect that measured changes in candidate enhancer activity could be due to a combination of both enhancer remodeling and shift in cell type composition. Thus, future studies profiling snATAC-seq and H3K27ac in parallel from the same cardiac sample or novel approaches to profile histone modifications in single nuclei (62, 63) will provide greater insight into the extent of changes in chromatin accessibility and enhancer activity in individual cardiac cell types from diseased hearts.
Last, we show how this atlas can be used to not only assign noncoding genetic variants associated with cardiovascular disease risk to cCREs in specific cardiac cell types but also illuminate their cellular and molecular consequences. In particular, we found significant enrichment of AF-associated variants within cardiomyocyte cCREs and functionally interrogated one of these cCREs by demonstrating its role in regulating KCNH2 expression and cardiomyocyte repolarization. Similar to electrophysiologic phenotypes of human cardiomyocytes exhibiting KCNH2 loss of function (59, 60), hPSC cardiomyocytes harboring deletions of this cCRE displayed action potential prolongation, suggesting that cardiac repolarization abnormalities may contribute to AF, possibly through similar mechanisms as to how they may contribute ventricular arrhythmias (59). On the other hand, we found no enrichment of variants associated with heart failure in any cardiac cell type. This finding may reflect the heterogeneous etiologies of cardiovascular diseases and the limited number of currently known risk loci for heart failure (49). Future GWAS in large cohorts with detailed phenotyping, including biobanks such as the UK Biobank (64) and the BioBank Japan Project (65) and whole-genome sequencing efforts such as the National Heart, Lung, and Blood Institute Trans-Omics for Precision Medicine program (66), will help identify and refine disease association signals. Therefore, this atlas of cardiac cCREs will be a valuable resource for continued discovery of regulatory elements, target genes, and specific cell types that may be affected by noncoding cardiovascular genetic variants.
In summary, we created a human heart cell atlas of >287,000 cCREs, which may serve as a reference to further expand our knowledge of gene regulatory mechanisms underlying cardiovascular disease. To facilitate distribution of these data, we created a web portal at http://catlas.org/humanheart. Integrating this resource with genomic and epigenomic clinical cardiac datasets, we built a systematic framework to interrogate how cis-regulatory elements and genetic variants might contribute to cardiovascular diseases such as heart failure or AF. Overall, such information will have great potential to provide insight into the development of future cardiac therapies that are tailored to affected cell types and thus optimized for treating specific cardiovascular diseases.
MATERIALS AND METHODS
Experimental design
We performed snATAC-seq to define a comprehensive catalog of cCREs for the cell types in four regions of nonfailing human hearts and generated in parallel snRNA-seq datasets for a subset to delineate gene expression patterns. We used the cCRE catalog to computationally assign dynamic enhancers in failing hearts to cell types and to assign cardiovascular disease risk variants to cCREs in individual cardiac cell types. Last, we applied reporter assays, genome editing, and electrophysiological measurements in in vitro differentiated human cardiomyocytes to validate the molecular mechanisms of cardiovascular disease risk variants.
Human tissues
Adult human heart tissues were procured at the time of organ donation using an Institutional Review Board protocol (no. 101021) approved by the University of California, San Diego. Donated hearts were perfused with cold cardioplegia before cardiectomy and then explanted immediately into an ice-cold physiologic solution as we previously described (67). Full-thickness samples from each chamber were obtained, and epicardial fat was rapidly removed before immediately flash-freezing samples in liquid nitrogen. Samples were received from the United Network for Organ Sharing. Limited clinical data were obtained for each heart per approved Institutional Review Board protocol (table S1). All samples were stored at −80°C until processing.
Single-nucleus ATAC-seq
Combinatorial barcoding snATAC-seq was performed as described previously (21, 27, 28) with slight modifications. Nuclei were isolated in gentleMACS M tubes (Miltenyi) on a gentleMACS Octo dissociator (Miltenyi) using the “Protein_01_01” protocol in magnetic-activated cell sorting (MACS) buffer [5 mM CaCl2, 2 mM EDTA, 1× protease inhibitor (Roche, 05-892-970-001), 300 mM MgAc, 10 mM tris-HCl (pH 8), and 0.6 mM dithiothreitol (DTT)]. Nuclei were pelleted with a swinging-bucket centrifuge (500g, 5 min, 4°C; 5920R, Eppendorf) and resuspended in 1 ml of nuclear permeabilization buffer [1× phosphate-buffered saline (PBS), 5% bovine serum albumin (BSA), 0.2% IGEPAL CA-630 (Sigma-Aldrich), 1 mM DTT, and 1× protease inhibitor]. Nuclei were rotated at 4°C for 5 min before being pelleted again with a swinging-bucket centrifuge (500g, 5 min, 4°C; 5920R, Eppendorf). After centrifugation, permeabilized nuclei were resuspended in 500 μl of high-salt tagmentation buffer [36.3 mM tris acetate (pH 7.8), 72.6 mM potassium acetate, 11 mM Mg acetate, and 17.6% N,N′-dimethylformamide] and counted using a hemocytometer. Concentration was adjusted to 2000 nuclei/9 μl, and 2000 nuclei were dispensed into each well of a 96-well plate per sample (96 tagmentation wells per sample; samples were processed in batches of two to four samples). For tagmentation, 1 μl of barcoded Tn5 transposomes (table S20) was added using a BenchSmart 96 (Mettler Toledo), mixed five times, and incubated for 60 min at 37°C with shaking (500 rpm). To inhibit the Tn5 reaction, 10 μl of 40 mM EDTA (final 20 mM) was added to each well with a BenchSmart 96 (Mettler Toledo), and the plate was incubated at 37°C for 15 min with shaking (500 rpm). Next, 20 μl of 2× sort buffer (2% BSA and 2 mM EDTA in PBS) were added using a BenchSmart 96 (Mettler Toledo). All wells were combined into a separate fluorescence-activated cell sorter tube for each sample and stained with DRAQ7 at 1:150 dilution (Cell Signaling Technology). Using an SH800 (Sony), 20 nuclei per sample were sorted per well into eight 96-well plates (total of 768 wells) containing 10.5 μl of Elution Buffer (EB; Qiagen) [25 pmol of primer i7, 25 pmol of primer i5, and 200 ng of BSA (Sigma-Aldrich)]. During the sort, nuclei with two to eight copies of DNA (2-8n) were included because cardiomyocyte nuclei in human hearts are often polyploid (16). Preparation of sort plates and all downstream pipetting steps were performed on a Biomek i7 automated workstation (Beckman Coulter). After addition of 1 μl of 0.2% SDS, samples were incubated at 55°C for 7 min with shaking (500 rpm). One microliter of 12.5% Triton X was added to each well to quench the SDS. Next, 12.5 μl of NEBNext High-Fidelity 2× PCR Master Mix (New England Biolabs) were added, and samples were PCR-amplified [72°C 5 min, 98°C 30 s, (98°C 10 s, 63°C 30 s, 72°C 60 s) × 12 cycles, held at 12°C]. After PCR, all wells were combined. Libraries were purified according to the MinElute PCR Purification Kit manual (Qiagen) using a vacuum manifold (QIAvac 24 Plus, Qiagen), and size selection was performed with SPRISelect reagent (Beckman Coulter; 0.55× and 1.5×). Libraries were purified one more time with SPRISelect reagent (Beckman Coulter; 1.5×). Libraries were quantified using a Qubit fluorimeter (Life Technologies), and a nucleosomal pattern of fragment size distribution was verified using a TapeStation (High Sensitivity D1000, Agilent). Libraries were sequenced on a NextSeq 500 sequencer (Illumina) using custom sequencing primers with the following read lengths: 50 + 10 + 12 + 50 (read 1 + index 1 + index 2 + read 2). Primer and index sequences are listed in table S20.
Single-nucleus RNA-seq
Nuclei were isolated from heart tissue using a gentleMACS dissociator (Miltenyi). Frozen heart tissue (~40 mg) was suspended in 2 ml of MACS dissociation buffer [5 mM CaCl2 (G-Biosciences, R040), 2 mM EDTA (Invitrogen, 15575-038), 1× protease inhibitor (Roche, 05-892-970-001), 3 mM MgAc (Grow Cells, MRGF-B40), 10 mM tris-HCl (pH 8) (Invitrogen, 15568-075), 0.6 mM DTT (Sigma-Aldrich, D9779), and ribonuclease (RNase) inhibitor (0.2 U/μl; Promega, N251B) in water (Corning, 46-000-CV)] and placed on wet ice. Next, samples were homogenized using a gentleMACS dissociator (Miltenyi) with gentleMACS M tubes (Miltenyi, 130-096-335) and the Protein_01_01 protocol. Suspension was filtered through a 30 μM CellTrics filter (Sysmex, 04-0042-2316). M tube and filter were washed with 3 ml of MACS dissociation buffer and combined with the suspension. Suspension was centrifuged in a swinging-bucket centrifuge (Eppendorf, 5920R) at 500g for 5 min (4°C, ramp speed of 3/3). Supernatant was carefully removed and pellet was resuspended in 500 μl of nuclei permeabilization buffer[(0.1% Triton X-100 (Sigma-Aldrich, T8787), 1× protease inhibitor (Roche, 05-892-970-001), 1 mM DTT (Sigma-Aldrich, D9779), RNase inhibitor (0.2 U/μl; Promega, N251B), and 2% BSA (Sigma-Aldrich, SRE0036) in PBS]. Sample was incubated on a rotator for 5 min at 4°C and then centrifuged at 500g for 5 min (Eppendorf, 5920R; 4°C, ramp speed of 3/3). Supernatant was removed and pellet was resuspended in 600 to 1000 μl of sort buffer [1 mM EDTA and RNase inhibitor (0.2 U/μl) in 2% BSA (Sigma-Aldrich, SRE0036) in PBS] and stained with DRAQ7 (1:100; Cell Signaling Technology, 7406). Seventy-five thousand nuclei were sorted using an SH800 sorter (Sony) into 50 μl of collection buffer [RNase inhibitor (1 U/μl) and 5% BSA (Sigma-Aldrich, SRE0036) in PBS]. Sorted nuclei were then centrifuged at 1000g for 15 min (Eppendorf, 5920R; 4°C, ramp speed of 3/3), and supernatant was removed. Nuclei were resuspended in 18 to 25 μl of reaction buffer [RNase inhibitor (0.2 U/μl) and 1% BSA (Sigma-Aldrich, SRE0036) in PBS] and counted using a hemocytometer. Twelve thousand nuclei were loaded onto a Chromium controller (10x Genomics). Libraries were generated using the Chromium Single Cell 3′ Library Construction Kit v3 (10x Genomics, 1000078) according to the manufacturer specifications. Complementary DNA was amplified for 12 PCR cycles. SPRISelect reagent (Beckman Coulter) was used for size selection and cleanup steps. Final library concentration was assessed by the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific), and fragment size was checked using TapeStation High Sensitivity D1000 (Agilent) to ensure that fragment sizes were distributed normally around 500 bp. Libraries were sequenced using a NextSeq 500 or HiSeq 4000 (Illumina) using these read lengths: read 1, 28 cycles; read 2, 91 cycles; and index 1, 8 cycles.
hPSC culture
An engineered H9-hTnnTZ-pGZ-D2 hPSC transgenic reporter line was purchased from WiCell and maintained on Geltrex (Gibco) precoated tissue culture plates in E8 medium (68) containing Dulbecco’s modified Eagle’s medium/F12, l-ascorbic acid-2-phosphate magnesium (64 mg/liter), sodium selenium (14 μg/liter), fibroblast growth factor 2 (100 μg/liter), insulin (19.4 mg/liter), NaHCO3 (543 mg/liter), transferrin (10.7 mg/liter), and transforming growth factor–β1(2 μg/liter). Cells were passaged every 3 to 5 days upon reaching ~80% confluency. For single-cell passaging experiments, cells were incubated with prewarmed TrypLE Select Enzyme, no phenol red (1 ml per well of a six-well plate) for 2 to 3 min in a 37°C, 5% CO2 incubator. Following incubation, cells were triturated to create a single-cell suspension and cultured in E8 medium supplied with Rock inhibitor (69) for 18 to 24 hours after split, followed by daily feeding with E8 medium.
In vitro cardiomyocyte differentiation
The H9-hTnnTZ-pGZ-D2 cell line was differentiated into beating cardiomyocytes using a previously reported Wnt-based monolayer differentiation protocol (70). Briefly, the H9-hTnnTZ-pGZ-D2 cell line was cultured in E8 medium for 3 to 10 passages. Before differentiation, hPSCs were seeded at a density of 350,000 to 400,000 cells per well of a 12-well plate and cultured for 2 days. For direct differentiation, cells were treated with 10 μM CHIR99021 (Fisher Scientific, no. 442350) in RPMI/B-27 without insulin. Fresh RPMI/B-27 without insulin media was replaced at post 24 hours, and cells were then cultured for 2 days. At day 3, cells were treated with 5 μM IWP2 (TOCRIS, no. 353310) in conditional medium and RPMI/B-27 without insulin 1:1 mix medium for another 2 days. At day 5, cells were exposed to fresh RPMI/B-27 without insulin media again for 2 days. Then, fresh RPMI/B-27 with insulin media was used and replenished every 2 days. Contracting cardiomyocytes were usually observed at days 7 and 8. D25 in vitro cardiomyocytes were purified using a PSC-derived cardiomyocyte isolation kit, human (Miltenyi Biotec, 130-110-188) and used for real-time qPCR (RT-qPCR).
Luciferase reporter assay
A genomic region harboring the KCNH2 intronic enhancer (containing the risk allele rs7789146-G/rs7789585-G) was amplified by nested PCR (KCNH2-E-cF, CTGGCTGAAGACACCTTACTTT; KCNH2-E-cR, ACGGAGCAGTCAAGGAAAC and KCNH2-In-cF, CGGGGTACCCCTCCGTAAATGAGGTGCTATC; KCNH2-In-cR, CCCTCGAGACGGAGCAGTCAAGGAAAC) using the genomic DNA of H9-hTnnTZ-pGZ-D2 transgenic cells as a template and cloned into pGL4.23 [luc2/minP] (Promega, catalog no. E8411) luciferase reporter vector. Synthetic DNA containing the KCNH2 intronic 5′-half enhancer (rs7789045-rs7789690) with the nonrisk/nonrisk allele (rs7789146-A/rs7789585-A), nonrisk/risk allele (rs7789146-A/rs7789585-G), and risk/nonrisk allele (rs7789146-G/rs7789585-A) were purchased from Integrated DNA Technologies. KCNH2 intronic 3′-half enhancer (rs7789654-rs7790480) was amplified by PCR (KCNH2-R-In-cF, GCTGTGCAGTGTCAGGTTAT; KCNH2-In-cR, CCCTCGAGACGGAGCAGTCAAGGAAAC). Then, the whole KCNH2 intronic enhancer with the nonrisk/nonrisk allele (rs7789146-A/rs7789585-A), nonrisk/risk allele (rs7789146-A/rs7789585-G), and risk/nonrisk allele (rs7789146-G/rs7789585-A) were generated by second PCR (KCNH2-In-cF, CGGGGTACCCCTCCGTAAATGAGGTGCTATC; KCNH2-In-cR, CCCTCGAGACGGAGCAGTCAAGGAAAC). One day before transfection, 3 × 105 of D15 in vitro differentiated cardiomyocytes were plated in a Geltrex-coated 24-well plate. Cardiomyocytes were transfected with 500 ng of pGL4.23 plasmid (either empty, KCNH2 enhancer with G/G allele, A/A allele, or mix) and 10 ng of TK:Renilla-luc as an internal control using Lipofectamine Stem Transfection Reagent (Invitrogen, no. STEM00003). Media were replaced with fresh media at 24 hours after transfection. At 72 hours after transfection, media were removed and the cells were washed with PBS. Luminescence was measured using a Dual-Luciferase Reporter Assay System (Promega, no. E2920) according to the manufacturer’s protocol.
CRISPR-mediated genome editing experiments
To interrogate the functional significance of the AF-associated risk variant–containing cCRE at the KCNH2 locus, the cCRE sequence was genetically deleted in H9-hTnnTZ-pGZ-D2 transgenic hPSCs using an efficient CRISPR-Cas9–mediated knockout system (57, 71). Two adjacent guide RNAs (gRNAs) (KCNH2-enh gRNA-1, CTCATTTACGGAGGAGCGCA; KCNH2-enh gRNA-2, TACAGTGGCCTTCTAGACGA) targeting the cCRE were designed using a web-based software tool CRISPOR (72) based on targeting region of interest and minimizing potential off-target effects. The identified gRNAs were then synthesized in vitro using the GeneArt Precision gRNA Synthesis kit (Invitrogen) according to the manufacturer’s protocol. One day before transfection, 1.5 × 105 H9-hTnnTZ-pGZ-D2 hPSCs were seeded in 12-well plates. A pair of ribonucleoprotein complexes containing 1.2 μg of Cas9 protein (New England Biolabs) and 400 ng of in vitro transcribed gRNA was then transfected (73, 74) using Lipofectamine stem transfection reagent (Invitrogen). Seventy-two hours after the transfection, cells were diluted and clonally expanded another 7 days. Colonies were picked and lysates were prepared after the first passage for genotyping (75) (KCNH2-enh extended forward primer, ACACCTTACTTTGGGTGAGAAG; KCNH2-enh extended reverse primer, AGACAGAGCACAGACCTAGAA; KCNH2-enh internal forward primer, GCTGTGCAGTGTCAGGTTAT; KCNH2-enh internal reverse primer, TCTCCCTCCTTCTCTCTCATTC). After confirmation of genome-edited clones by Sanger sequencing, two transfected wild-type (WT) clones, two heterozygote clones, and two homozygote clones were selected for further functional analysis.
Real-time quantitative polymerase chain reaction
Total RNA was isolated from the cells using TRIzol reagent (Invitrogen). One microgram of total RNA was reverse-transcribed using the iScript Reverse Transcription Supermix kit (Bio-Rad) for RT-qPCR. RT-qPCR was performed using PowerUP SYBR Green Master Mix (Applied Biosystems) in the CFX Connect Real-Time System (Bio-Rad). The results were normalized to the TBP gene. The primers used for RT-qPCR are listed in table S21.
Electrophysiology of cardiomyocytes
Both WT and KCNH2 enhancer knockout D15 in vitro cardiomyocytes were purified using the PSC-derived cardiomyocyte isolation kit, human (Miltenyi Biotec, 130-110-188) and cultured for another 10 to 20 days in a low density before electrophysiological measurements. The single-pipette, whole-cell patch current-clamp technique was used for recordings. Action potentials were recorded with a patch-clamp amplifier (Axopatch 200B, Axon), and experiments were performed at a temperature of 35° ± 0.5°C. Current-clamp command pulses were generated by a digital-to-analog converter (DigiData 1440, Axon), which was controlled by the pCLAMP software (10.3; Axon). Pipettes (resistance, 3 to 5 megohm) were pulled using a micropipette puller (Model P-87, Sutter Instrument Co.). Several minutes after seal formation, the membrane was ruptured by gentle suction to establish the whole-cell configuration for voltage clamping. Subsequently, the amplifier was switched to the current-clamp mode. Cells were paced with 1 Hz and injected current stimuli from 3 to 15 nA for 5-ms duration. Cells were superfused with extracellular solution containing the following: 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.5 mM glucose, and 5.0 mM Hepes (pH 7.4 adjusted with NaOH). Pipette solution contained the following: 120 mM K-gluconate, 10 mM KCl, 5 mM NaCl, 10 mM Hepes, 5 mM phosphocreatine, 5 mM ATP-Mg2, and amphotericin (0.44 μM; pH 7.2 adjusted with KOH).
Data analysis
Demultiplexing of snATAC-seq reads
For each sequenced snATAC-Seq library, we obtained four FASTQ files, two for paired-end DNA reads as well as the combinatorial indexes for i5 (768 different PCR indices) and T7 (96 different tagmentation indices; table S20). We selected all reads with ≤2 mistakes per individual index (Hamming distance between each pair of indices is 4) and subsequently integrated the full barcode at the beginning of the read name in the demultiplexed FASTQ files (https://gitlab.com/Grouumf/ATACdemultiplex/).
Filtering of snATAC-seq profiles by transcriptional start site enrichment and unique fragments
TSS (transcriptional start site) positions were obtained from the GENCODE database v31 (76). Tn5-corrected insertions were aggregated ±2000 bp around each TSS genome wide. Then, this profile was normalized to the mean accessibility ± (1900 to 2000) bp from the TSS and smoothed every 11 bp. The maximum value of the smoothed profile was taken as the TSS enrichment. We selected all nuclei that had at least 1000 unique fragments and a TSS enrichment of at least 7 for all datasets.
Clustering strategy for snATAC-seq datasets
We used two rounds of clustering analysis to identify clusters. The first round of clustering analysis was performed on individual samples. We divided the genome into 5-kbp consecutive bins and then scored each nucleus for any insertions in these bins, generating a bin-by-cell binary matrix for each sample. We filtered out those bins that were generally accessible in all nuclei for each sample using a z score threshold of 1.65 (equivalent to a one-tailed P value < 0.05). On the basis of the filtered matrix, we then carried out dimensionality reduction followed by graph-based clustering to identify cell clusters. We called peaks using MACS2 (32) for each cluster using the aggregated profile of accessibility and then merged the peaks from all clusters to generate a union peak list. On the basis of the peak list, we generated a cell-by-peak count matrix and used Scrublet (77) to remove potential doublets with default parameters. Doublet scores returned by Scrublet (77) were then used to fit a two-component Gaussian mixture model using the BayesianGaussianMixture function from the python package scikit-learn (78). Nuclei in the component with the larger mean doublet score were removed from downstream analysis because they likely reflected doublets.
Next, to carry out the second round of clustering analysis, we merged peaks called from all samples to form a reference peak list. We generated a binary cell-by-peak matrix using nuclei from all samples and again performed dimensionality reduction followed by graph-based clustering to obtain the final cell clusters across the entire dataset.
Dimensionality reduction and batch correction of snATAC-seq data
For processing of snATAC-seq data, we adapted our previously published method, SnapATAC (28). To reduce the dimensionality of the peak by cell count matrix, SnapATAC uses spectral embedding for dimensionality reduction. To further increase the performance and scalability of spectral embedding, we applied the Nyström method (79) to enable handling of large datasets. Specifically, we first randomly sampled 35,000 nuclei as training data. We computed the matrix P = D−1S S, where D is the diagonal matrix such that . The eigendecomposition was performed on P, and the eigenvector with eigenvalue 1 was discarded. From the rest of the eigenvectors, we took k of them corresponding to the largest eigenvalues as the spectral embedding of the training data. We used the Nyström method (79) to extend the embedding to the data outside the training set. Given a set of unseen samples, we computed the similarity matrix S′ between the new samples and the training set. The embedding of the new samples is given by ′ = S′ UΛ−1, where U and Λ are the eigenvectors and eigenvalues of P obtained in the previous step. To correct for donor/batch-specific effects, after dimensionality reduction, we performed cell grouping on individual samples using k-mean clustering with k equal to 20. We then constructed k-NN graphs for each sample and used the MNN (Mutual Nearest Neighbor) correction method to identify mutual nearest neighbors (80). These mutual nearest neighbors were used as anchors to match the cells between different samples and correct for donor/batch effects as described previously (80).
Clustering of snATAC-seq data
We constructed the k-nearest neighbor graph (k-NNG) using low-dimensional embedding of the nuclei with k equal to 50. We then applied the Leiden algorithm (81) with constant Potts model to find communities in the k-NNG corresponding to the cell clusters. The Leiden algorithm can be configured to use different quality functions. The modularity model is a popular choice, but it is hampered by the resolution limit, particularly when the network is large (82). Therefore, we used the modularity model only in the first round of clustering analysis to identify initial clusters. In the final round of clustering, we chose the constant Potts model as the quality function because it is resolution limit free and is better suited for identifying rare populations in a large dataset (82). Nuclei from two small clusters (280 and 254 nuclei) with low reproducibility and stability were discarded from downstream analysis. Thirty-four nuclei that formed clusters of 1 and 2 nuclei were discarded as well.
Processing and clustering analysis of snRNA-seq datasets
Raw sequencing data were demultiplexed and preprocessed using the Cell Ranger software package v3.0.2 (10x Genomics). Raw sequencing files were first converted from Illumina BCL files to FASTQ files using cellranger mkfastq. Demultiplexed FASTQs were aligned to the GRCh38 reference genome (10x Genomics), and reads for exonic and intronic reads mapping to protein coding genes, long noncoding RNA, antisense RNA, and pseudo-genes were used to generate a counts matrix using cellranger count; expect-cells parameter was set to 5000. A separate counts matrix for each sample was also generated using only reads mapped to intronic regions.
Next, exon and intron count matrices for individual datasets were processed using the Seurat v3.1.4 R package (29) (https://satijalab.org/seurat/) to assess dataset quality. Features represented in at least three cells and barcodes with between 500 and 4000 genes were used for downstream processing. In addition, barcodes with mitochondrial read percentages greater than 5% were removed. Counts were log-normalized and scaled by a factor of 10,000 using NormalizeData. To identify variable genes, FindVariableFeatures was run with default parameters except for nfeatures = 3000 to return the top 3000 variable genes. All genes were then scaled using ScaleData, which transforms the expression values for downstream analysis. Next, principal components analysis was performed using RunPCA with default parameters and the top 3000 variable features as input. The first 20 principal components were used to run clustering using FindNeighbors and FindClusters (parameter res = 0.4). To generate uniform manifold approximation and projection (UMAP) coordinates, RunUMAP was run using the first 20 principal components and with parameters umap.method = “umap-learn” and metric = “correlation”. Doublet scores [pANN (proportion of Artificial Nearest Neighbors)] were generated for cell barcodes using DoubletFinder (https://github.com/chris-mcginnis-ucsf/DoubletFinder) (83) using the parameters pN = 0.15 and pK = 0.005; the anticipated collision rate was set by specifying 2% collisions per thousand nuclei for individual datasets.
Individual datasets were merged together using the merge function in Seurat to combine the count matrices and designate unique barcodes. Cell barcodes with pANN scores greater than 0 were removed from downstream analysis. Metadata were also encoded for each barcode, and the merged dataset was processed in a similar manner as described above; clusters were identified using FindNeighbors and FindClusters (res = 0.8). To generate the UMAP coordinates, the first 14 principal components were used in RunUMAP; the UMAP algorithm for Seurat v3.1.4 uses the uwot R package and that setting was used to generate the coordinates here. To regress out donor specific effects, the Harmony R package (https://github.com/immunogenomics/harmony) (84) was used, and the recomputed principal components were used to recluster the cells and rerun UMAP using the above parameters. For downstream analysis and comparison to snATAC-seq data, we manually combined ventricular cardiomyocyte clusters, atrial cardiomyocyte clusters, fibroblast clusters, and endothelial cell clusters on the basis of shared gene expression patterns (fig. S2, G and H). Cluster-specific genes in the all-transcripts dataset were identified in a global differential gene expression test using FindAllMarkers with parameters logFC = 0.25, min.pct = 0.25, and only.pos = FALSE.
Integration of snRNA-seq and snATAC-seq data
The snRNA-seq and snATAC-seq datasets were used to perform label transfer from the RNA cells onto the snATAC-seq dataset using the Seurat v3.1.4 R package (https://satijalab.org/seurat/) (29). Gene activity scores were calculated using chromatin accessibility in regions from the promoter up to 2 kb upstream for each ATAC nucleus. Activity scores were log-normalized and scaled using NormalizeData and ScaleData. To compare the snRNA and snATAC datasets and identify anchors, FindTransferAnchors was run considering the top 3000 variable features from the snRNA-seq dataset. Anchor pairs were used to assign RNA-seq labels to the snATAC-seq cells using TransferData, with the weight.reduction parameter set to the principal components used in snATAC-seq clustering. The efficacy of integration was assessed by examining the distribution of the maximum prediction scores output by TransferData and the distribution of annotated snATAC-seq identities to the corresponding predicted label.
Creation of a consensus list of heart cCREs
MACS2 (v2.1.2) (32) was used to identify accessible chromatin sites for each cluster with the following parameters: -q 0.01 --nomodel --shift -100 --extsize 200 -g 2789775646 --call-summits --keepdup-all. Estimated genome size was determined to be 2,789,775,646 bp and was indicated by the -g parameter. We next filtered out peaks overlapping with the ENCODE blacklist (hg38; https://github.com/Boyle-Lab/Blacklist/) (85). To generate the union of heart cCREs, we merged the blacklist-filtered peaks obtained for each cluster using the BEDtools merge command with default settings (v2.25.0) (86).
Computing relative accessibility scores for cCREs
To correct biases arising from differential read depth among cells and cell types, we derived a procedure that normalizes chromatin accessibility at cCREs identified by MACS2 peak calling (v2.1.2) (32). We define the set of accessible loci by L, and we define a peak p as a subset of related loci l from L. Let al be the accessibility of accessible locus l and P the set of nonoverlapping peaks used to define the loci. For a given cell type Si ∈ S, we computed the median medj number of reads sequenced per cell. For each feature pj ∈ P, we computed mij, the average number of reads sequenced from Si, and overlapping pj. We then defined the activity aij of loci pj in Si as . We then define the relative accessibility score (RAS) .
K-means clustering of cCREs
We clustered the union of 287,415 cCREs using a K-means clustering procedure. We first created a sparse cell × peak matrix that was transformed into a RAS-normalized cell type × peak matrix. We then performed K-means on the normalized matrix with K from 2 to 12 and computed the Davies-Bouldin (DB) index for each K (87). Let with sx the average distance of each cell of cluster x and dxy the distance between the centroids of clusters x and y. The DB index is defined as . We selected K = 9 because it resulted in the lowest DB index that indicates the best partition. We used the python library scikit-learn (78) to compute the K-means algorithm and the DB index (87).
Cell type annotation
We annotated snATAC-seq and snRNA-seq clusters on the basis of chromatin accessibility at promoter regions or expression of known lineage marker genes, respectively. We annotated atrial and ventricular cardiomyocytes on the basis of differential chromatin accessibility and gene expression at NPPA, MYH6, KCNJ3, MYL7, MYH7, HEY2, MYL2, and other reported markers of atrial and ventricular cardiomyocytes (30, 88, 89). We used, for example, the gene DCN to annotate cardiac fibroblasts (90), VWF and EGFL7 for endothelial cells (91, 92), GJA4 and TAGLN for smooth muscle cells (93, 94), CD163 and MS4A6A for macrophages (95, 96), IL7R and THEMIS for lymphocytes (97, 98), ADIPOQ and CIDEA for adipocytes (99, 100), and NRXN3 and GPM6B for a cluster of nervous cells with neuronal and Schwann-like gene expression and chromatin accessibility signatures (10, 11, 101). From snRNA-seq, we identified a population of endothelial-like cells with specific expression of endocardial cell markers NRG3 and NPR3 (102, 103). We also identified subtypes of mesenchymal cells that included myofibroblasts with characteristic expression of embryonic smooth muscle actin MYH10 (104, 105) as well as arterial smooth muscle cells with preferential expression of ACTA2 and TAGLN relative to a larger cluster of pericytes (table S4) (106). snRNA-seq annotations were consistent with recent single-cell transcriptomic analyses of adult human heart tissue (10, 11).
Identification of cell type–specific cCREs
We used edgeR (version 3.24) in R (107) to identify cell type–specific cCREs. For each cCRE, accessibility within a cell type was compared to average accessibility in all other clusters. For each cell type, we created a count table for each cCRE using the following strategy: Each sample was described with a donor and a chamber ID. For each sample ID, we reported read count within (i) the cell type and (ii) the rest of the cell types in aggregate. We used this count matrix as input for edgeR analysis (107) and performed a likelihood ratio test. We considered cCREs with fold change > 1.2 and FDR < 0.01 after Benjamini-Hochberg correction as cell type specific.
Coaccessibility analysis using Cicero
We used the R package Cicero (38) to infer coaccessible chromatin loci. For each chromosome, we used as input the corresponding peaks from our 287,415 cCRE union set and the coordinates of the snATAC-seq UMAP (108). We randomly subsampled 15,000 cells from our aggregate snATAC-seq dataset to construct input matrices for Cicero analysis. We used ±250 kbp as cutoff for coaccessibility interactions. All other settings were default.
Correlation of gene expression and promoter accessibility
We defined promoter regions as TSS ±2 kbp. TSS were extracted from annotation files from GENCODE release 33 (76). We identified promoter-overlapping peaks using BEDtools (86) and a custom script (see the “Code availability” section). For each overlapping pair (peak and promoter) identified, we kept only the open chromatin site closest to the TSS to obtain a 1:1 correspondence between genes and open chromatin peaks. We then used the RAS and the cluster-scaled gene expression (29) to create feature × cell type matrices for RNA-seq and ATAC-seq datasets. We then used these matrices to create heatmaps and to perform ATAC-seq/RNA-seq cluster correlation analysis using the Pearson similarity metric. For each cell type, we computed the Pearson correlation score between the RAS vector of the 7081 promoters and the scaled expression vector of the corresponding 7081 genes identified via the 1:1 correspondence method described above.
DA cCREs between heart chambers
Between-heart chamber differential accessibility analysis was performed for five cell types from our aggregated snATAC-seq dataset. We considered only cell types that had a representation of at least 50 nuclei per dataset and at least 300 nuclei across each tested condition. The cell types that met these inclusion criteria included cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, and macrophages. Within each cell type, a generalized linear model framework was used using the R package edgeR (107). All fragments for a given cell type were aggregated in the .bed format. MACS2 (32) was used to call peaks on the aggregate .bed file for each cell type with the parameters specified above. NarrowPeak output bed files were used for differential accessibility testing. The aggregate .bed file for each cell type was then partitioned on the basis of dataset of origin using nuclear barcodes. The “coverage” option of the BEDtools package (86) was applied with default settings to count the total number of chromatin fragments from each dataset overlapping narrowPeaks called on the aggregate .bed file for the corresponding cell type. This yielded a raw count matrix in the format of snATAC-seq datasets (columns) by narrowPeaks (rows) for each cell type. The raw count matrix was used as input for edgeR analysis. To filter low-coverage peaks from our analysis, we used the “filterByExpr” command within edgeR with default settings. We applied an average prior count of one during fitting of the generalized linear model to avoid inflated fold changes in instances for which peaks lacked coverage for one but not both tested conditions. We modeled chromatin accessibility at each peak as a function of heart chamber (group) with sex as a covariate. The generalized linear model was expressed as follows in edgeR notation:
Significance was tested using a likelihood ratio test. To account for testing multiple hypotheses, a Benjamini-Hochberg significance correction was applied for all cCREs tested within each considered cell type. Any cCRE with an absolute log2(fold change) > 1 and an FDR-corrected P value < 0.05 was considered significant.
Gene expression analysis of genes coaccessible with DA cCREs
To compare the expression of genes coaccessible with heart chamber–dependent distal DA cCREs (outside ±2 kb of TSS) in cardiomyocytes and fibroblasts, we performed differential expression testing for all genes between indicated heart chambers using Wilcoxon rank sum test in Seurat (29). Genes with an absolute fold change > 1.5 and an FDR-adjusted P value < 0.05 were considered differentially expressed. We then tested the resulting genes for coaccessibility (38) with distal DA cCREs at a coaccessibility score threshold of 0.1 and displayed scaled gene expression values from Seurat for the indicated differentially expressed genes linked to chamber-dependent distal DA cCREs.
Genomic Regions Enrichment of Annotations Tool ontology analysis
The Genomic Regions Enrichment of Annotations Tool (GREAT; http://great.stanford.edu/public/html/index.php) (33) was used with default settings for indicated cCREs or candidate enhancers in the .bed format. Biological process enrichments are reported. P values shown for enrichment are Bonferroni-corrected binomial P values.
Motif enrichment analysis
For de novo and known motif enrichment analysis of cluster-specific cCREs, the findMotifsGenome.pl utility of the HOMER package was used with default settings (35). For display of enrichment patterns for motifs from the JASPAR (109) database with evidence of enrichment in at least one set of cell type–specific cCREs, motifs with an enrichment P value < 10−5 in at least one set of cluster-specific cCREs were selected. For motif enrichment within DA cCREs, narrowPeak calls from MACS2 were used as input, with peaks called on the corresponding cell type (as described above) used as background. For enrichment of motifs within cell type–attributed bulk enhancers, snATAC-seq peaks from the union of snATAC-seq peaks were used. Summits were extracted from peaks that overlapped bulk enhancer annotations and extended by 250 bp on either side to obtain fixed-width peaks. We also computed motif enrichment scores at single-cell resolution using chromVAR (34).
For input to chromVAR, we used the summits of the 287,415 peaks in our consensus list extended by 250 bp in either direction and a set of 870 nonredundant motifs as input (https://github.com/GreenleafLab/chromVARmotifs). To identify differentially enriched motifs in each cell type, we used the following strategy: For each cell type and each motif, we computed a rank sum test between the chromVAR z score distributions from cells within the cell type and outside of the cell type. Tests were run using a random sampling of 40,000 cells. Then, for each cell type, we used 1 × 10−8 as P value cutoff. In addition, we applied a Bonferroni correction to account for multiple testing, which resulted in selection of significant motifs with P value < 1 × 10−11.
Measuring single-cell chromatin accessibility signal within bulk candidate heart enhancers
We obtained published candidate heart enhancers annotated by H3K27ac ChIP-seq from a recently reported bulk survey of healthy left ventricular tissue from 18 human donors (15). Candidate enhancers were defined per the study as H3K27ac ChIP-seq peaks that were at least 1 kb away from a TSS and present in two or more donors. Because these reference annotations were derived from bulk profiling of healthy left ventricles, we selected only left ventricular nuclei from our aggregate dataset for comparison. We limited our analysis to cell types that comprised at least 5% of nuclei by proportion in our aggregate dataset. These included cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, and macrophages. We first combined all fragments for each cell type from left ventricular datasets. The coverage option of BEDtools (86) was applied with default settings to count the total number of chromatin fragments from each ventricular cell type overlapping the candidate enhancer annotations. This yielded a raw count matrix in the format of snATAC-seq cell types (columns) by candidate enhancers (rows). The raw count matrix was normalized to RPKM (reads per kilobase per million mapped reads) for each candidate enhancer. We next used Cluster3.0 (110) to k-means cluster the 31,033 healthy heart candidate enhancers into K groups between 2 and 12 with the following settings (Method = k-Means, Similarity Metric = Euclidian distance, number of runs = 100). We calculated the DB index (87) as described above for each clustering using the index.DB function of the R package clusterSim (http://keii.ue.wroc.pl/clusterSim/). We selected a k-means of 8, which yielded the lowest DB index, indicating the best partitioning.
We repeated the above analysis for 4406 candidate enhancers reported to have increased bulk H3K27ac ChIP signal and 3101 candidate enhancers reported to have decreased signal in 18 late-stage idiopathic dilated cardiomyopathy (heart failure) left ventricles versus 18 healthy control left ventricles reported in the same study. We again clustered the candidate enhancers for both groups into k groups between 2 and 12 as above and selected the clustering that yielded the lowest DB index (87).
GWAS variant enrichment analysis
We used LD score regression (48, 111) to estimate genome-wide enrichment for GWAS traits using annotation sets from single-cell chromatin accessibility from the heart or lung (54) or bulk DNase hypersensitivity sites for tissues from ENCODE (3, 5, 17). For bulk DNase-seq datasets, peak annotations were merged across biological replicates from the same tissue type. We compiled published GWAS summary statistics for cardiovascular diseases (49–53), other diseases (112–122), and nondisease traits (123–132) using the European subset from transethnic studies where applicable. We created custom LD score files by using peaks from each cell type or tissue as a binary annotation. As background, we used baseline annotations included in the baseline-LD model v2.2. For each trait, we used LD score regression to estimate enrichment coefficient z scores for each annotation relative to the background. Using these z scores, we computed two-sided P values for enrichment and used the Benjamini-Hochberg procedure to correct for multiple tests within each set of annotations.
Fine mapping for AF
We obtained published AF GWAS summary statistics and index variants for 111 disease-associated loci (50). To construct credible sets of variants for each locus, we first extracted all variants in LD (r2 > 0.1 using the EUR (European) subset of 1000 Genomes phase 3) (133) in a large window (±2.5 Mb) around each index variant. We next calculated approximate Bayes factors (ABF) (55) for each variant using effect size and SE estimates. We then calculated PPAs for each variant by dividing its ABF by the sum of ABF for all variants within the locus. For each locus, we then defined 99% credible sets by sorting variants by descending PPA and retaining variants that added up to a cumulative PPA of >0.99. This resulted in an output of 6014 candidate causal variants.
Variant prioritization for functional validation
To prioritize variants for functional validation, we refined our list of candidate causal variants from fine mapping analysis to only those with a PPA > 0.1 (216 remaining of 6014). We used BEDtools (86) to intersect these variants with ATAC-seq peaks called on an aggregate .bed file for atrial and ventricular cardiomyocyte snATAC-seq clusters (cardiomyocyte cCREs). This resulted in 40 fine-mapped variants that resided within 38 candidate cardiomyocyte cCREs (38 cCRE-variant pairs).
We assessed each remaining cCRE-variant pair via the following criteria:
1) cCREs primarily accessible in cardiomyocytes
2) presence of a corresponding ATAC-seq peak at a testable time point in the in vitro hPSC-cardiomyocyte differentiation model system
3) sequence conservation in 100 vertebrates [genome browser track generated using phyloP of the PHAST5 package downloaded from UCSC genome browser (134), http://hgdownload.soe.ucsc.edu/goldenPath/hg38/phyloP100way/]
4) predicted coaccessibility of candidate enhancer with a gene promoter
5) expression of putative target gene associated with cCRE appearance (chromatin accessibility and H3K27ac) during hPSC-cardiomyocyte differentiation (57)
A candidate cCRE-variant pair at the KCNH2 locus was prioritized for functional experimentation.
ChIP-seq data processing
Reads were mapped to the human genome reference GRCh38 using Bowtie2 (version 2.2.6) (135) and reads with MAPQ (mapping quality) > 30 selected using SAMtools (version 1.3.1) (136). PCR duplicates were removed using the MarkDuplicates function of Picard tools (version 1.119) (137). RPKM-normalized signal tracks were generated using the BamCoverage function in deepTools (version 2.4.1) (138).
RNA-seq data processing
Reads were mapped to the human genome reference GRCh38 using STAR (version 020201) (139) and reads with MAPQ > 30 selected using SAMtools (version 1.3.1) (136). PCR duplicates were removed using the MarkDuplicates function of Picard tools (version 1.1.19) (137). RPKM-normalized signal tracks were generated using the BamCoverage function in deepTools (version 2.4.1) (138).
ATAC-seq data processing
Reads were mapped to the human genome reference GRCh38 using Bowtie2 (version 2.2.6) (135) and reads with MAPQ > 30 selected using SAMtools (version 1.3.1) (136). PCR duplicates were removed using SAMtools (version 1.3.1) (136). RPKM-normalized signal tracks were generated using the BamCoverage function in deepTools (version 2.4.1) (138).
Statistical analysis
No statistical methods were used to predetermine sample sizes. There was no randomization of the samples, and investigators were not blinded to the specimens being investigated. However, clustering of single nuclei based on chromatin accessibility was performed in an unbiased manner, and cell types were assigned after clustering. Low-quality nuclei and potential barcode collisions were excluded from downstream analysis as outlined above. Cluster specificity at each cCRE was tested using edgeR (107) as described above, with P values corrected via the Benjamini-Hochberg procedure. To identify DA sites between the heart chambers and for each cell type, a likelihood ratio test was used and the resulting P value was corrected using the Benjamini-Hochberg procedure. For significance of ontology enrichments using GREAT, Bonferroni-corrected binomial P values were used (33). For significance testing of enrichment of de novo and known motifs, a hypergeometric test was used without correction for multiple testing (35). For luciferase and qPCR data, we performed one-way ANOVA (analysis of variance) analysis with post hoc Tukey test using GraphPad Prism version 8.0.0 for Windows (GraphPad Software, San Diego, CA, USA; www.graphpad.com).
External datasets
Cardiomyocyte differentiation: RNA-seq, H3K27ac day 0 (hPSC); day 5 (cardiac mesoderm); and day 15 (primitive cardiomyocytes) were downloaded from GSE116862 (57). Signal tracks for heart H3K27ac ChIP-seq data were downloaded from https://portal.nersc.gov/dna/RD/heart/. List of candidate enhancers was downloaded from supplementary tables (15). H3K27ac ChIP-seq data for cardiomyocyte nuclei from nonfailing donors (NF1) were downloaded from National Center for Biotechnology Information Sequence Read Archive BioProject ID PRJNA353755 (140). We acquired snATAC-seq data for human lung from GSE161383 (54) and bulk DNase-seq datasets for human tissues from ENCODE (3, 5, 17) with the following identifiers: ENCSR053ZKP, ENCSR259GYP, ENCSR277KRY, ENCSR458AOS, ENCSR597NVK, ENCSR422IIZ, ENCSR968TPO, ENCSR788IZL, ENCSR859LTL, ENCSR060HPL, ENCSR783OCW, ENCSR171ADO, ENCSR171ETY, ENCSR520BAD, ENCSR686WJL, ENCSR791BHE, ENCSR856XLJ, ENCSR361DND, ENCSR579KDC, ENCSR693UHT, ENCSR365NDK, ENCSR712PYJ, ENCSR930PDT, ENCSR178JBL, ENCSR595HZQ, ENCSR261RWJ, ENCSR455GUW, ENCSR689DSM, ENCSR954AJK, ENCSR564FZH, ENCSR000ELO, ENCSR909HFI, ENCSR931UQB, ENCSR128GBN, ENCSR006IMH, ENCSR163PKT, ENCSR641ZPF, ENCSR782SSS, ENCSR101QXF, ENCSR709IYR, ENCSR866ODX, ENCSR195ONB, ENCSR450PWF, ENCSR549NRK, ENCSR749MUH, ENCSR080ISA, ENCSR090IDV, ENCSR102RSU, ENCSR401ESD, ENCSR484UAU, ENCSR508FVM, ENCSR340MRJ, ENCSR760QZM, ENCSR763AKE, ENCSR923JYH, ENCSR164WOF, ENCSR323UTX, ENCSR650FLQ, ENCSR702DPD, ENCSR129BZE, and ENCSR437AYW.
Code availability
The pipeline for processing snATAC-seq data is available as a part of the Taiji software: https://taiji-pipeline.github.io/. Custom code used for demultiplexing and downstream analysis for snATAC data is available here: https://gitlab.com/Grouumf/ATACdemultiplex/-/tree/master/ATACdemultiplex and https://gitlab.com/Grouumf/ATACdemultiplex/-/blob/master/scripts/.
Acknowledgments
We thank B. Li for bioinformatics support. We thank K. Jepsen and the UCSD IGM Genomics Center for sequencing the snRNA-seq libraries. We thank the QB3 Macrolab at UC Berkeley for purification of the Tn5 transposase. We thank A. R. Holman for support of validation experiments. We also thank the members of the Ren, Chi, Gaulton, and McCulloch laboratories and the Center for Epigenomics for feedback and discussions. Funding: This work was supported by the Ludwig Institute for Cancer Research (B.R.) and the National Institutes of Health (1UM1HL128773-01 to N.C.C. and B.R., and U01 HL126273 and R01 HL137100 to A.D.M.). J.D.H. was supported, in part, by Ruth L. Kirschstein Institutional National Research Service Award T32 GM008666 from the National Institute of General Medical Sciences. Work at the Center for Epigenomics was supported, in part, by the UC San Diego School of Medicine. Author contributions: J.D.H., S.P., N.C.C., and B.R. conceived the project. J.D.H. and J.B. carried out snATAC-seq and snRNA-seq library preparation with help from X.H. F.Z., Q.Z., and T.-M.W. performed validation experiments. J.D.H., O.B.P., J.B., K.Z., J.C., Y.E.L., and S.P. performed data analysis. O.B.P. created the web portal. X.H., E.N.F., Y.Z., A.W., A.D.M., K.J.G., and N.C.C. contributed to experimental design and computational analyses. J.D.H., S.P., N.C.C., and B.R. wrote the manuscript. All authors edited and approved the manuscript. Competing interests: B.R. is a shareholder and consultant of Arima Genomics Inc. and cofounder of Epigenome Technologies. K.J.G. is a consultant of Genentech and shareholder in Vertex Pharmaceuticals. A.D.M. is a cofounder and Scientific Advisor to Insilicomed Inc. and Vektor Medical Inc. These relationships have been disclosed to and approved by the UCSD Independent Review Committee. The authors declare that they have no other competing interests. Data and materials availability: Sequencing data are available from dbGaP (phs002204.v1.p1). Processed data are available from GEO (GSE165839) and can be explored using our publicly available web portal including a UCSC cell browser (https://github.com/maximilianh/cellBrowser) and genome browser track viewer (IGV.js: https://github.com/igvteam/igv.js#igvjs): http://catlas.org/humanheart. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf1444/DC1
REFERENCES AND NOTES
- 1.WHO, Cardiovascular Diseases (CVDs) (2017). The World Health Organization https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds).
- 2.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G. N., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thurman R. E., Rynes E., Humbert R., Vierstra J., Maurano M. T., Haugen E., Sheffield N. C., Stergachis A. B., Wang H., Vernot B., Garg K., John S., Sandstrom R., Bates D., Boatman L., Canfield T. K., Diegel M., Dunn D., Ebersol A. K., Frum T., Giste E., Johnson A. K., Johnson E. M., Kutyavin T., Lajoie B., Lee B.-K., Lee K., London D., Lotakis D., Neph S., Neri F., Nguyen E. D., Qu H., Reynolds A. P., Roach V., Safi A., Sanchez M. E., Sanyal A., Shafer A., Simon J. M., Song L., Vong S., Weaver M., Yan Y., Zhang Z., Zhang Z., Lenhard B., Tewari M., Dorschner M. O., Hansen R. S., Navas P. A., Stamatoyannopoulos G., Iyer V. R., Lieb J. D., Sunyaev S. R., Akey J. M., Sabo P. J., Kaul R., Furey T. S., Dekker J., Crawford G. E., Stamatoyannopoulos J. A., The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ENCODE Project Consortium, Moore J. E., Purcaro M. J., Pratt H. E., Epstein C. B., Shoresh N., Adrian J., Kawli T., Davis C. A., Dobin A., Kaul R., Halow J., Van Nostrand E. L., Freese P., Gorkin D. U., Shen Y., He Y., Mackiewicz M., Pauli-Behn F., Williams B. A., Mortazavi A., Keller C. A., Zhang X.-O., Elhajjajy S. I., Huey J., Dickel D. E., Snetkova V., Wei X., Wang X., Rivera-Mulia J. C., Rozowsky J., Zhang J., Chhetri S. B., Zhang J., Victorsen A., White K. P., Visel A., Yeo G. W., Burge C. B., Lécuyer E., Gilbert D. M., Dekker J., Rinn J., Mendenhall E. M., Ecker J. R., Kellis M., Klein R. J., Noble W. S., Kundaje A., Guigó R., Farnham P. J., Cherry J. M., Myers R. M., Ren B., Graveley B. R., Gerstein M. B., Pennacchio L. A., Snyder M. P., Bernstein B. E., Wold B., Hardison R. C., Gingeras T. R., Stamatoyannopoulos J. A., Weng Z., Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smemo S., Campos L. C., Moskowitz I. P., Krieger J. E., Pereira A. C., Nobrega M. A., Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Hum. Mol. Genet. 21, 3255–3263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaidi S., Brueckner M., Genetics and genomics of congenital heart disease. Circ. Res. 120, 923–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maurano M. T., Humbert R., Rynes E., Thurman R. E., Haugen E., Wang H., Reynolds A. P., Sandstrom R., Qu H., Brody J., Shafer A., Neri F., Lee K., Kutyavin T., Stehling-Sun S., Johnson A. K., Canfield T. K., Giste E., Diegel M., Bates D., Hansen R. S., Neph S., Sabo P. J., Heimfeld S., Raubitschek A., Ziegler S., Cotsapas C., Sotoodehnia N., Glass I., Sunyaev S. R., Kaul R., Stamatoyannopoulos J. A., Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui Y., Zheng Y., Liu X., Yan L., Fan X., Yong J., Hu Y., Dong J., Li Q., Wu X., Gao S., Li J., Wen L., Qiao J., Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 26, 1934–1950.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Litvinukova M., Talavera-López C., Maatz H., Reichart D., Worth C. L., Lindberg E. L., Kanda M., Polanski K., Heinig M., Lee M., Nadelmann E. R., Roberts K., Tuck L., Fasouli E. S., De Laughter D. M., Donough B. M., Wakimoto H., Gorham J. M., Samari S., Mahbubani K. T., Saeb-Parsy K., Patone G., Boyle J. J., Zhang H., Zhang H., Viveiros A., Oudit G. Y., Bayraktar O. A., Seidman J. G., Seidman C. E., Noseda M., Hubner N., Teichmann S. A., Cells of the adult human heart. Nature 588, 466–472 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker N. R., Chaffin M., Fleming S. J., Hall A. W., Parsons V. A., Bedi K. C. Jr., Akkad A. D., Herndon C. N., Arduini A., Papangeli I., Roselli C., Aguet F., Choi S. H., Ardlie K. G., Babadi M., Margulies K. B., Stegmann C. M., Ellinor P. T., Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asp M., Giacomello S., Larsson L., Wu C., Fürth D., Qian X., Wärdell E., Custodio J., Reimegård J., Salmén F., Österholm C., Ståhl P. L., Sundström E., Åkesson E., Bergmann O., Bienko M., Månsson-Broberg A., Nilsson M., Sylvén C., Lundeberg J., A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell 179, 1647–1660.e19 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Dickel D. E., Barozzi I., Zhu Y., Fukuda-Yuzawa Y., Osterwalder M., Mannion B. J., May D., Spurrell C. H., Plajzer-Frick I., Pickle C. S., Lee E., Garvin T. H., Kato M., Akiyama J. A., Afzal V., Lee A. Y., Gorkin D. U., Ren B., Rubin E. M., Visel A., Pennacchio L. A., Genome-wide compendium and functional assessment of in vivo heart enhancers. Nat. Commun. 7, 12923–12923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.May D., Blow M. J., Kaplan T., McCulley D., Jensen B. C., Akiyama J. A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Afzal V., Simpson P. C., Rubin E. M., Black B. L., Bristow J., Pennacchio L. A., Visel A., Large-scale discovery of enhancers from human heart tissue. Nat. Genet. 44, 89–93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spurrell C. H., Barozzi I., Mannion B. J., Blow M. J., Fukuda-Yuzawa Y., Afzal S. Y., Akiyama J. A., Afzal V., Tran S., Plajzer-Frick I., Novak C. S., Kato M., Lee E., Garvin T. H., Pham Q. T., Harrington A. N., Lisgo S., Bristow J., Cappola T. P., Morley M. P., Margulies K. B., Pennacchio L. A., Dickel D. E., Visel A., Genome-wide fetalization of enhancer architecture in heart disease. bioRxiv, 591362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilsbach R., Schwaderer M., Preissl S., Grüning B. A., Kranzhöfer D., Schneider P., Nührenberg T. G., Mulero-Navarro S., Weichenhan D., Braun C., Dreßen M., Jacobs A. R., Lahm H., Doenst T., Backofen R., Krane M., Gelb B. D., Hein L., Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 9, 391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vierstra J., Lazar J., Sandstrom R., Halow J., Lee K., Bates D., Diegel M., Dunn D., Neri F., Haugen E., Rynes E., Reynolds A., Nelson J., Johnson A., Frerker M., Buckley M., Kaul R., Meuleman W., Stamatoyannopoulos J. A., Global reference mapping of human transcription factor footprints. Nature 583, 729–736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan W. L. W., Anene-Nzelu C. G., Wong E., Lee C. J. M., Tan H. S., Tang S. J., Perrin A., Wu K. X., Zheng W., Ashburn R. J., Pan B., Lee M. Y., Autio M. I., Morley M. P., Tam W. L., Cheung C., Margulies K. B., Chen L., Cappola T. P., Loh M., Chambers J., Prabhakar S., Foo R. S.-Y., Assa S., Ellinor P., Felix J., van der Harst P., Meder B., Smith G. J., Tappu R., Epigenomes of human hearts reveal new genetic variants relevant for cardiac disease and phenotype. Circ. Res. 127, 761–777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anene-Nzelu C. G., Tan W. L. W., Lee C. J. M., Wenhao Z., Perrin A., Dashi A., Tiang Z., Autio M. I., Lim B., Wong E., Tan H. S., Pan B., Morley M. P., Margulies K. B., Cappola T. P., Foo R. S.-Y., Assigning distal genomic enhancers to cardiac disease-causing genes. Circulation 142, 910–912 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buenrostro J. D., Wu B., Litzenburger U. M., Ruff D., Gonzales M. L., Snyder M. P., Chang H. Y., Greenleaf W. J., Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cusanovich D. A., Daza R., Adey A., Pliner H. A., Christiansen L., Gunderson K. L., Steemers F. J., Trapnell C., Shendure J., Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satpathy A. T., Granja J. M., Yost K. E., Qi Y., Meschi F., McDermott G. P., Olsen B. N., Mumbach M. R., Pierce S. E., Corces M. R., Shah P., Bell J. C., Jhutty D., Nemec C. M., Wang J., Wang L., Yin Y., Giresi P. G., Chang A. L. S., Zheng G. X. Y., Greenleaf W. J., Chang H. Y., Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lareau C. A., Duarte F. M., Chew J. G., Kartha V. K., Burkett Z. D., Kohlway A. S., Pokholok D., Aryee M. J., Steemers F. J., Lebofsky R., Buenrostro J. D., Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 37, 916–924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cusanovich D. A., Hill A. J., Aghamirzaie D., Daza R. M., Pliner H. A., Berletch J. B., Filippova G. N., Huang X., Christiansen L., De Witt W. S., Lee C., Regalado S. G., Read D. F., Steemers F. J., Disteche C. M., Trapnell C., Shendure J., A single-cell atlas of in vivo mammalian chromatin accessibility. Cell 174, 1309–1324.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia G., Preussner J., Chen X., Guenther S., Yuan X., Yekelchyk M., Kuenne C., Looso M., Zhou Y., Teichmann S., Braun T., Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat. Commun. 9, 4877 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Domcke S., Hill A. J., Daza R. M., Cao J., O’Day D. R., Pliner H. A., Aldinger K. A., Pokholok D., Zhang F., Milbank J. H., Zager M. A., Glass I. A., Steemers F. J., Doherty D., Trapnell C., Cusanovich D. A., Shendure J., A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Preissl S., Fang R., Huang H., Zhao Y., Raviram R., Gorkin D. U., Zhang Y., Sos B. C., Afzal V., Dickel D. E., Kuan S., Visel A., Pennacchio L. A., Zhang K., Ren B., Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci. 21, 432–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang R., Preissl S., Li Y., Hou X., Lucero J., Wang X., Motamedi A., Shiau A. K., Zhou X., Xie F., Mukamel E. A., Zhang K., Zhang Y., Margarita Behrens M., Ecker J. R., Ren B., Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun. 21, 1337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W. M. III, Hao Y., Stoeckius M., Smibert P., Satija R., Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng S. Y., Wong C. K., Tsang S. Y., Differential gene expressions in atrial and ventricular myocytes: Insights into the road of applying embryonic stem cell-derived cardiomyocytes for future therapies. Am. J. Physiol. Cell Physiol. 299, C1234–C1249 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Chen H., Lareau C., Andreani T., Vinyard M. E., Garcia S. P., Clement K., Andrade-Navarro M. A., Buenrostro J. D., Pinello L., Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 20, 241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schep A. N., Wu B., Buenrostro J. D., Greenleaf W. J., chromVAR: Inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang D. E., Hetherington C. J., Chen H. M., Tenen D. G., The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol. Cell. Biol. 14, 373–381 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moorman A. F. M., Christoffels V. M., Cardiac chamber formation: Development, genes, and evolution. Physiol. Rev. 83, 1223–1267 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Pliner H. A., Packer J. S., McFaline-Figueroa J. L., Cusanovich D. A., Daza R. M., Aghamirzaie D., Srivatsan S., Qiu X., Jackson D., Minkina A., Adey A. C., Steemers F. J., Shendure J., Trapnell C., Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol. Cell 71, 858–871.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C., Liu W., Lu M. F., Brown N. A., Martin J. F., Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development 128, 2039–2048 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Rivaud M. R., Delmar M., Remme C. A., Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: Ionic and non-ionic mechanisms. Cardiovasc. Res. 116, 1557–1570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Veevers J., Farah E. N., Corselli M., Witty A. D., Palomares K., Vidal J. G., Emre N., Carson C. T., Ouyang K., Liu C., van Vliet P., Zhu M., Hegarty J. M., Deacon D. C., Grinstein J. D., Dirschinger R. J., Frazer K. A., Adler E. D., Knowlton K. U., Chi N. C., Martin J. C., Chen J., Evans S. M., Cell-surface marker signature for enrichment of ventricular cardiomyocytes derived from human embryonic stem cells. Stem Cell Rep. 11, 828–841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hortells L., Johansen A. K. Z., Yutzey K. E., Cardiac fibroblasts and the extracellular matrix in regenerative and nonregenerative hearts. J. Cardiovasc. Dev. Dis. 6, 29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruneau B. G., Bao Z.-Z., Tanaka M., Schott J. J., Izumo S., Cepko C. L., Seidman J. G., Seidman C. E., Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2-5 and dHand. Dev. Biol. 217, 266–277 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Corces M. R., Shcherbina A., Kundu S., Gloudemans M. J., Frésard L., Granja J. M., Louie B. H., Eulalio T., Shams S., Bagdatli S. T., Mumbach M. R., Liu B., Montine K. S., Greenleaf W. J., Kundaje A., Montgomery S. B., Chang H. Y., Montine T. J., Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hook P. W., McCallion A. S., Leveraging mouse chromatin data for heritability enrichment informs common disease architecture and reveals cortical layer contributions to schizophrenia. Genome Res. 30, 528–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chiou J., Zeng C., Cheng Z., Han J. Y., Schlichting M., Miller M., Mendez R., Huang S., Wang J., Sui Y., Deogaygay A., Okino M.-L., Qui Y., Sun Y., Kudtarkar P., Fang R., Preissl S., Sander M., Gorkin D. U., Gaulton K. J., Single-cell chromatin accessibility identifies pancreatic islet cell type– and state-specific regulatory programs of diabetes risk. Nat. Genet. 53, 455–466 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nott A., Holtman I. R., Coufal N. G., Schlachetzki J. C. M., Yu M., Hu R., Han C. Z., Pena M., Xiao J., Wu Y., Keulen Z., Pasillas M. P., O’Connor C., Nickl C. K., Schafer S. T., Shen Z., Rissman R. A., Brewer J. B., Gosselin D., Gonda D. D., Levy M. L., Rosenfeld M. G., McVicker G., Gage F. H., Ren B., Glass C. K., Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bulik-Sullivan B. K., Loh P.-R., Finucane H. K., Ripke S., Yang J.; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson N., Daly M. J., Price A. L., Neale B. M., LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arvanitis M., Tampakakis E., Zhang Y., Wang W., Auton A.; 23andMe Research Team, Dutta D., Glavaris S., Keramati A., Chatterjee N., Chi N. C., Ren B., Post W. S., Battle A., Genome-wide association and multi-omic analyses reveal ACTN2 as a gene linked to heart failure. Nat. Commun. 11, 1122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen J. B., Thorolfsdottir R. B., Fritsche L. G., Zhou W., Skov M. W., Graham S. E., Herron T. J., McCarthy S., Schmidt E. M., Sveinbjornsson G., Surakka I., Mathis M. R., Yamazaki M., Crawford R. D., Gabrielsen M. E., Skogholt A. H., Holmen O. L., Lin M., Wolford B. N., Dey R., Dalen H., Sulem P., Chung J. H., Backman J. D., Arnar D. O., Thorsteinsdottir U., Baras A., O’Dushlaine C., Holst A. G., Wen X., Hornsby W., Dewey F. E., Boehnke M., Kheterpal S., Mukherjee B., Lee S., Kang H. M., Holm H., Kitzman J., Shavit J. A., Jalife J., Brummett C. M., Teslovich T. M., Carey D. J., Gudbjartsson D. F., Stefansson K., Abecasis G. R., Hveem K., Willer C. J., Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 50, 1234–1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson C. P., Goel A., Butterworth A. S., Kanoni S., Webb T. R., Marouli E., Zeng L., Ntalla I., Lai F. Y., Hopewell J. C., Giannakopoulou O., Jiang T., Hamby S. E., Angelantonio E. D., Assimes T. L., Bottinger E. P., Chambers J. C., Clarke R., Palmer C. N. A., Cubbon R. M., Ellinor P., Ermel R., Evangelou E., Franks P. W., Grace C., Gu D., Hingorani A. D., Howson J. M. M., Ingelsson E., Kastrati A., Kessler T., Kyriakou T., Lehtimäki T., Lu X., Lu Y., März W., Pherson R. M., Metspalu A., Pujades-Rodriguez M., Ruusalepp A., Schadt E. E., Schmidt A. F., Sweeting M. J., Zalloua P. A., Ghalayini K. A., Keavney B. D., Kooner J. S., Loos R. J. F., Patel R. S., Rutter M. K., Tomaszewski M., Tzoulaki I., Zeggini E., Erdmann J., Dedoussis G., Björkegren J. L. M.; EPIC-CVD Consortium; CARDIoGRAMplusC4D; UK Biobank Cardio Metabolic Consortium CHD working group, Schunkert H., Farrall M., Danesh J., Samani N. J., Watkins H., Deloukas P., Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 49, 1385–1391 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Shadrina A. S., Sharapov S. Z., Shashkova T. I., Tsepilov Y. A., Varicose veins of lower extremities: Insights from the first large-scale genetic study. PLOS Genet. 15, e1008110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malik R., Chauhan G., Traylor M., Sargurupremraj M., Okada Y., Mishra A., Rutten-Jacobs L., Giese A.-K., van der Laan S. W., Gretarsdottir S., Anderson C. D., Chong M., Adams H. H. H., Ago T., Almgren P., Amouyel P., Ay H., Bartz T. M., Benavente O. R., Bevan S., Boncoraglio G. B., Brown R. D. Jr., Butterworth A. S., Carrera C., Carty C. L., Chasman D. I., Chen W.-M., Cole J. W., Correa A., Cotlarciuc I., Cruchaga C., Danesh J., de Bakker P. I. W., De Stefano A. L., den Hoed M., Duan Q., Engelter S. T., Falcone G. J., Gottesman R. F., Grewal R. P., Gudnason V., Gustafsson S., Haessler J., Harris T. B., Hassan A., Havulinna A. S., Heckbert S. R., Holliday E. G., Howard G., Hsu F.-C., Hyacinth H. I., Ikram M. A., Ingelsson E., Irvin M. R., Jian X., Jiménez-Conde J., Johnson J. A., Jukema J. W., Kanai M., Keene K. L., Kissela B. M., Kleindorfer D. O., Kooperberg C., Kubo M., Lange L. A., Langefeld C. D., Langenberg C., Launer L. J., Lee J.-M., Lemmens R., Leys D., Lewis C. M., Lin W.-Y., Lindgren A. G., Lorentzen E., Magnusson P. K., Maguire J., Manichaikul A., McArdle P. F., Meschia J. F., Mitchell B. D., Mosley T. H., Nalls M. A., Ninomiya T., O’Donnell M. J., Psaty B. M., Pulit S. L., Rannikmäe K., Reiner A. P., Rexrode K. M., Rice K., Rich S. S., Ridker P. M., Rost N. S., Rothwell P. M., Rotter J. I., Rundek T., Sacco R. L., Sakaue S., Sale M. M., Salomaa V., Sapkota B. R., Schmidt R., Schmidt C. O., Schminke U., Sharma P., Slowik A., Sudlow C. L. M., Tanislav C., Tatlisumak T., Taylor K. D., Thijs V. N. S., Thorleifsson G., Thorsteinsdottir U., Tiedt S., Trompet S., Tzourio C., van Duijn C. M., Walters M., Wareham N. J., Wassertheil-Smoller S., Wilson J. G., Wiggins K. L., Yang Q., Yusuf S.; AFGen Consortium; Cohorts for Heart; Aging Research in Genomic Epidemiology (CHARGE) Consortium; International Genomics of Blood Pressure (iGEN-BP) Consortium; INVENT Consortium; STARNET, Bis J. C., Pastinen T., Ruusalepp A., Schadt E. E., Koplev S., Björkegren J. L. M., Codoni V., Civelek M., Smith N. L., Trégouët D. A., Christophersen I. E., Roselli C., Lubitz S. A., Ellinor P. T., Tai E. S., Kooner J. S., Kato N., He J., van der Harst P., Elliott P., Chambers J. C., Takeuchi F., Johnson A. D.; BioBank Japan Cooperative Hospital Group; COMPASS Consortium; EPIC-CVD Consortium; EPIC-Inter ActConsortium; International Stroke Genetics Consortium (ISGC); METASTROKE Consortium; Neurology Working Group of the CHARGE Consortium; NINDS Stroke Genetics Network (SiGN); UK Young Lacunar DNA Study; MEGASTROKE Consortium, Sanghera D. K., Melander O., Jern C., Strbian D., Fernandez-Cadenas I., Longstreth W. T. Jr., Rolfs A., Hata J., Woo D., Rosand J., Pare G., Hopewell J. C., Saleheen D., Stefansson K., Worrall B. B., Kittner S. J., Seshadri S., Fornage M., Markus H. S., Howson J. M. M., Kamatani Y., Debette S., Dichgans M., Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat. Genet. 50, 524–537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang A., Chiou J., Poirion O. B., Buchanan J., Valdez M. J., Verheyden J. M., Hou X., Kudtarkar P., Narendra S., Newsome J. M., Guo M., Faddah D. A., Zhang K., Young R. E., Barr J., Sajti E., Misra R., Huyck H., Rogers L., Poole C., Whitsett J. A., Pryhuber G., Xu Y., Gaulton K. J., Preissl S., Sun X.; NHLBI LungMap Consortium , Single-cell multiomic profiling of human lungs reveals cell-type-specific and age-dynamic control of SARS-CoV2 host genes. eLife 9, e62522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wakefield J., Bayes factors for genome-wide association studies: Comparison with P-values. Genet. Epidemiol. 33, 79–86 (2009). [DOI] [PubMed] [Google Scholar]
- 56.van den Boogaard M., Smemo S., Burnicka-Turek O., Arnolds D. E., van de Werken H. J. G., Klous P., McKean D., Muehlschlegel J. D., Moosmann J., Toka O., Yang X. H., Koopmann T. T., Adriaens M. E., Bezzina C. R., de Laat W., Seidman C., Seidman J. G., Christoffels V. M., Nobrega M. A., Barnett P., Moskowitz I. P., A common genetic variant within SCN10A modulates cardiac SCN5A expression. J. Clin. Invest. 124, 1844–1852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y., Li T., Preissl S., Grinstein J., Farah E. N., Destici E., Lee A. Y., Chee S., Qiu Y., Ma K., Ye Z., Hu R., Zhu Q., Huang H., Fang R., Yu L., Belmonte J. C. I., Wu J., Evans S. M., Chi N. C., Ren B., 3D chromatin architecture remodeling during human cardiomyocyte differentiation reveals a role of HERV-H in demarcating chromatin domains. bioRxiv, 485961 (2019). [Google Scholar]
- 58.Visel A., Minovitsky S., Dubchak I., Pennacchio L. A., VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Curran M. E., Splawski I., Timothy K. W., Vincen G. M., Green E. D., Keating M. T., A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Jones D. K., Liu F., Vaidyanathan R., Eckhardt L. L., Trudeau M. C., Robertson G. A., hERG 1b is critical for human cardiac repolarization. Proc. Natl. Acad. Sci. U.S.A. 111, 18073–18077 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilsbach R., Preissl S., Grüning B. A., Schnick T., Burger L., Benes V., Würch A., Bönisch U., Günther S., Backofen R., Fleischmann B. K., Schübeler D., Hein L., Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 5, 5288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Q., Xiong H., Ai S., Yu X., Liu Y., Zhang J., He A., CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell 76, 206–216.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Kaya-Okur H. S., Wu S. J., Codomo C. A., Pledger E. S., Bryson T. D., Henikoff J. G., Ahmad K., Henikoff S., CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M., Liu B., Matthews P., Ong G., Pell J., Silman A., Young A., Sprosen T., Peakman T., Collins R., UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ishigaki K., Akiyama M., Kanai M., Takahashi A., Kawakami E., Sugishita H., Sakaue S., Matoba N., Low S. K., Okada Y., Terao C., Amariuta T., Gazal S., Kochi Y., Horikoshi M., Suzuki K., Ito K., Koyama S., Ozaki K., Niida S., Sakata Y., Sakata Y., Kohno T., Shiraishi K., Momozawa Y., Hirata M., Matsuda K., Ikeda M., Iwata N., Ikegawa S., Kou I., Tanaka T., Nakagawa H., Suzuki A., Hirota T., Tamari M., Chayama K., Miki D., Mori M., Nagayama S., Daigo Y., Miki Y., Katagiri T., Ogawa O., Obara W., Ito H., Yoshida T., Imoto I., Takahashi T., Tanikawa C., Suzuki T., Sinozaki N., Minami S., Yamaguchi H., Asai S., Takahashi Y., Yamaji K., Takahashi K., Fujioka T., Takata R., Yanai H., Masumoto A., Koretsune Y., Kutsumi H., Higashiyama M., Murayama S., Minegishi N., Suzuki K., Tanno K., Shimizu A., Yamaji T., Iwasaki M., Sawada N., Uemura H., Tanaka K., Naito M., Sasaki M., Wakai K., Tsugane S., Yamamoto M., Yamamoto K., Murakami Y., Nakamura Y., Raychaudhuri S., Inazawa J., Yamauchi T., Kadowaki T., Kubo M., Kamatani Y., Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 52, 669–679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.NHLBI, Trans-Omics for Precision Medicine (TOPMed) Program (2014).
- 67.Smyth J. W., Hong T.-T., Gao D., Vogan J. M., Jensen B. C., Fong T. S., Simpson P. C., Stainier D. Y.-R., Chi N. C., Shaw R. M., Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Invest. 120, 266–279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen G., Gulbranson D. R., Hou Z., Bolin J. M., Ruotti V., Probasco M. D., Smuga-Otto K., Howden S. E., Diol N. R., Propson N. E., Wagner R., Lee G. O., Antosiewicz-Bourget J., Teng J. M. C., Thomson J. A., Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 8, 424–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Watanabe K., Ueno M., Kamiya D., Nishiyama A., Matsumura M., Wataya T., Takahashi J. B., Nishikawa S., Nishikawa S. I., Muguruma K., Sasai Y., A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Lian X., Zhang J., Azarin S. M., Zhu K., Hazeltine L. B., Bao X., Hsiao C., Kamp T. J., Palecek S. P., Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Teumer A., Tin A., Sorice R., Gorski M., Yeo N. C., Chu A. Y., Li M., Li Y., Mijatovic V., Ko Y.-A., Taliun D., Luciani A., Chen M.-H., Yang Q., Foster M. C., Olden M., Hiraki L. T., Tayo B. O., Fuchsberger C., Dieffenbach A. K., Shuldiner A. R., Smith A. V., Zappa A. M., Lupo A., Kollerits B., Ponte B., Stengel B., Krämer B. K., Paulweber B., Mitchell B. D., Hayward C., Helmer C., Meisinger C., Gieger C., Shaffer C. M., Müller C., Langenberg C., Ackermann D., Siscovick D.; DCCT/EDIC, Boerwinkle E., Kronenberg F., Ehret G. B., Homuth G., Waeber G., Navis G., Gambaro G., Malerba G., Eiriksdottir G., Li G., Wichmann H. E., Grallert H., Wallaschofski H., Völzke H., Brenner H., Kramer H., Leach I. M., Rudan I., Hillege H. L., Beckmann J. S., Lambert J. C., Luan J., Zhao J. H., Chalmers J., Coresh J., Denny J. C., Butterbach K., Launer L. J., Ferrucci L., Kedenko L., Haun M., Metzger M., Woodward M., Hoffman M. J., Nauck M., Waldenberger M., Pruijm M., Bochud M., Rheinberger M., Verweij N., Wareham N. J., Endlich N., Soranzo N., Polasek O., van der Harst P., Pramstaller P. P., Vollenweider P., Wild P. S., Gansevoort R. T., Rettig R., Biffar R., Carroll R. J., Katz R., Loos R. J. F., Hwang S.-J., Coassin S., Bergmann S., Rosas S. E., Stracke S., Harris T. B., Corre T., Zeller T., Illig T., Aspelund T., Tanaka T., Lendeckel U., Völker U., Gudnason V., Chouraki V., Koenig W., Kutalik Z., O’Connell J. R., Parsa A., Heid I. M., Paterson A. D., de Boer I. H., Devuyst O., Lazar J., Endlich K., Susztak K., Tremblay J., Hamet P., Jacob H. J., Böger C. A., Fox C. S., Pattaro C., Köttgen A., Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes 65, 803–817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J. B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J. S., Concordet J. P., Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim S., Kim D., Cho S. W., Kim J., Kim J. S., Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zuris J. A., Thompson D. B., Shu Y., Guilinger J. P., Bessen J. L., Hu J. H., Maeder M. L., Joung J. K., Chen Z. Y., Liu D. R., Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Santos D. P., Kiskinis E., Eggan K., Merkle F. T., Comprehensive protocols for CRISPR/Cas9-based gene editing in human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 38, 5b.6.1–5b.6.60 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.-K., Chrast J., Lagarde J., Gilbert J. G. R., Storey R., Swarbreck D., Rossier C., Ucla C., Hubbard T., Antonarakis S. E., Guigo R., GENCODE: Producing a reference annotation for ENCODE. Genome Biol. 7 Suppl 1, S4–S54 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolock S. L., Lopez R., Klein A. M., Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fabian Pedregosa G. V., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Brucher M., Perrot M., Duchesnay É., Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12 (85), 2825–2830 (2011). [Google Scholar]
- 79.D. B. Bouneffouf, I, Theoretical analysis of the Minimum Sum of Squared Similarities sampling for Nyström-based spectral clustering, in 2016 International Joint Conference on Neural Networks (IJCNN) (2016), pp. 3856–3862. [Google Scholar]
- 80.Haghverdi L., Lun A. T. L., Morgan M. D., Marioni J. C., Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Traag V. A., Waltman L., van Eck N. J., From Louvain to Leiden: Guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Traag V. A., Van Dooren P., Nesterov Y., Narrow scope for resolution-limit-free community detection. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 84, 016114 (2011). [DOI] [PubMed] [Google Scholar]
- 83.McGinnis C. S., Murrow L. M., Gartner Z. J., DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P. R., Raychaudhuri S., Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amemiya H. M., Kundaje A., Boyle A. P., The ENCODE Blacklist: Identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davies D. L., Bouldin D. W., A cluster separation measure. IEEE Trans. Pattern Anal. Mach. Intell. PAMI-1, 224–227 (1979). [PubMed] [Google Scholar]
- 88.Sheikh F., Lyon R. C., Chen J., Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene 569, 14–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kubalak S. W., Miller-Hance W. C., O’Brien T. X., Dyson E., Chien K. R., Chamber specification of atrial myosin light chain-2 expression precedes septation during murine cardiogenesis. J. Biol. Chem. 269, 16961–16970 (1994). [PubMed] [Google Scholar]
- 90.Furtado M. B., Costa M. W., Pranoto E. A., Salimova E., Pinto A. R., Lam N. T., Park A., Snider P., Chandran A., Harvey R. P., Boyd R., Conway S. J., Pearson J., Kaye D. M., Rosenthal N. A., Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 114, 1422–1434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nichol D., Stuhlmann H., EGFL7: A unique angiogenic signaling factor in vascular development and disease. Blood 119, 1345–1352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalucka J., de Rooij L. P. M. H., Goveia J., Rohlenova K., Dumas S. J., Meta E., Conchinha N. V., Taverna F., Teuwen L.-A., Veys K., García-Caballero M., Khan S., Geldhof V., Sokol L., Chen R., Treps L., Borri M., Zeeuw P., Dubois C., Karakach T. K., Falkenberg K. D., Parys M., Yin X., Vinckier S., Du Y., Fenton R. A., Schoonjans L., Dewerchin M., Eelen G., Thienpont B., Lin L., Bolund L., Li X., Luo Y., Carmeliet P., Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 93.Shanahan C. M., Weissberg P. L., Metcalfe J. C., Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ. Res. 73, 193–204 (1993). [DOI] [PubMed] [Google Scholar]
- 94.The Tabula Muris Consortium; Overall coordination, Schaum N., Karkanias J., Neff N. F., May A. P., Quake S. R., Wyss-Coray T., Darmanis S., Batson J., Botvinnik O., Chen M. B., Chen S., Green F., Jones R. C., Maynard A., Penland L., Pisco A. O., Sit R. V., Stanley G. M., Webber J. T., Zanini F., Baghel A. S., Bakerman I., Bansal I., Berdnik D., Bilen B., Brownfield D., Cain C., Chen M. B., Chen S., Cho M., Cirolia G., Conley S. D., Darmanis S., Demers A., Demir K., de Morree A., Divita T., du Bois H., Dulgeroff L. B. T., Ebadi H., Espinoza F. H., Fish M., Gan Q., George B. M., Gillich A., Green F., Genetiano G., Gu X., Gulati G. S., Hang Y., Hosseinzadeh S., Huang A., Iram T., Isobe T., Ives F., Jones R. C., Kao K. S., Karnam G., Kershner A. M., Kiss B. M., Kong W., Kumar M. E., Lam J. Y., Lee D. P., Lee S. E., Li G., Li Q., Liu L., Lo A., Lu W.-J., Manjunath A., May A. P., May K. L., May O. L., Maynard A., McKay M., Metzger R. J., Mignardi M., Min D., Nabhan A. N., Neff N. F., Ng K. M., Noh J., Patkar R., Peng W. C., Penland L., Puccinelli R., Rulifson E. J., Schaum N., Sikandar S. S., Sinha R., Sit R. V., Szade K., Tan W., Tato C., Tellez K., Travaglini K. J., Tropini C., Waldburger L., van Weele L. J., Wosczyna M. N., Xiang J., Xue S., Youngyunpipatkul J., Zanini F., Zardeneta M. E., Zhang F., Zhou L., Bansal I., Chen S., Cho M., Cirolia G., Darmanis S., Demers A., Divita T., Ebadi H., Genetiano G., Green F., Hosseinzadeh S., Ives F., Lo A., May A. P., Maynard A., McKay M., Neff N. F., Penland L., Sit R. V., Tan W., Waldburger L., Youngyunpipatkul J., Batson J., Botvinnik O., Castro P., Croote D., Darmanis S., De Risi J. L., Karkanias J., Pisco A. O., Stanley G. M., Webber J. T., Zanini F., Baghel A. S., Bakerman I., Batson J., Bilen B., Botvinnik O., Brownfield D., Chen M. B., Darmanis S., Demir K., de Morree A., Ebadi H., Espinoza F. H., Fish M., Gan Q., George B. M., Gillich A., Gu X., Gulati G. S., Hang Y., Huang A., Iram T., Isobe T., Karnam G., Kershner A. M., Kiss B. M., Kong W., Kuo C. S., Lam J. Y., Lehallier B., Li G., Li Q., Liu L., Lu W.-J., Min D., Nabhan A. N., Ng K. M., Nguyen P. K., Patkar R., Peng W. C., Penland L., Rulifson E. J., Schaum N., Sikandar S. S., Sinha R., Szade K., Tan S. Y., Tellez K., Travaglini K. J., Tropini C., van Weele L. J., Wang B. M., Wosczyna M. N., Xiang J., Yousef H., Zhou L., Batson J., Botvinnik O., Chen S., Darmanis S., Green F., May A. P., Maynard A., Pisco A. O., Quake S. R., Schaum N., Stanley G. M., Webber J. T., Wyss-Coray T., Zanini F., Beachy P. A., Chan C. K. F., de Morree A., George B. M., Gulati G. S., Hang Y., Huang K. C., Iram T., Isobe T., Kershner A. M., Kiss B. M., Kong W., Li G., Li Q., Liu L., Lu W.-J., Nabhan A. N., Ng K. M., Nguyen P. K., Peng W. C., Rulifson E. J., Schaum N., Sikandar S. S., Sinha R., Szade K., Travaglini K. J., Tropini C., Wang B. M., Weinberg K., Wosczyna M. N., Wu S. M., Yousef H., Barres B. A., Beachy P. A., Chan C. K. F., Clarke M. F., Darmanis S., Huang K. C., Karkanias J., Kim S. K., Krasnow M. A., Kumar M. E., Kuo C. S., May A. P., Metzger R. J., Neff N. F., Nusse R., Nguyen P. K., Rando T. A., Sonnenburg J., Wang B. M., Weinberg K., Weissman I. L., Wu S. M., Quake S. R., Wyss-Coray T., Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fabriek B. O., Dijkstra C. D., van den Berg T. K., The macrophage scavenger receptor CD163. Immunobiology 210, 153–160 (2005). [DOI] [PubMed] [Google Scholar]
- 96.Martinez F. O., Gordon S., Locati M., Mantovani A., Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 (2006). [DOI] [PubMed] [Google Scholar]
- 97.Peschon J. J., Morrissey P. J., Grabstein K. H., Ramsdell F. J., Maraskovsky E., Gliniak B. C., Park L. S., Ziegler S. F., Williams D. E., Ware C. B., Meyer J. D., Davison B. L., Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 180, 1955–1960 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lesourne R., Uehara S., Lee J., Song K.-D., Li L.-Q., Pinkhasov J., Zhang Y., Weng N.-P., Wildt K. F., Wang L., Bosselut R., Love P. E., Themis, a T cell-specific protein important for late thymocyte development. Nat. Immunol. 10, 840–847 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Puri V., Ranjit S., Konda S., Nicoloro S. M. C., Straubhaar J., Chawla A., Chouinard M., Lin C., Burkart A., Corvera S., Perugini R. A., Czech M. P., Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc. Natl. Acad. Sci. 105, 7833–7838 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hu E., Liang P., Spiegelman B. M., AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 271, 10697–10703 (1996). [DOI] [PubMed] [Google Scholar]
- 101.Skelly D. A., Squiers G. T., McLellan M. A., Bolisetty M. T., Robson P., Rosenthal N. A., Pinto A. R., Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 22, 600–610 (2018). [DOI] [PubMed] [Google Scholar]
- 102.Zhao Y. Y., Sawyer D. R., Baliga R. R., Opel D. J., Han X., Marchionni M. A., Kelly R. A., Neuregulins promote survival and growth of cardiac myocytes: Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J. Biol. Chem. 273, 10261–10269 (1998). [DOI] [PubMed] [Google Scholar]
- 103.Tang J., Zhang H., He L., Huang X., Li Y., Pu W., Yu W., Zhang L., Cai D., Lui K. O., Zhou B., Genetic fate mapping defines the vascular potential of endocardial cells in the adult heart. Circ. Res. 122, 984–993 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Southern B. D., Grove L. M., Rahaman S. O., Abraham S., Scheraga R. G., Niese K. A., Sun H., Herzog E. L., Liu F., Tschumperlin D. J., Egelhoff T. T., Rosenfeld S. S., Olman M. A., Matrix-driven Myosin II mediates the pro-fibrotic fibroblast phenotype. J. Biol. Chem. 291, 6083–6095 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baum J., Duffy H. S., Fibroblasts and myofibroblasts: What are we talking about? J. Cardiovasc. Pharmacol. 57, 376–379 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vanlandewijck M., He L., Mäe M. A., Andrae J., Ando K., del Gaudio F., Nahar K., Lebouvier T., Laviña B., Gouveia L., Sun Y., Raschperger E., Räsänen M., Zarb Y., Mochizuki N., Keller A., Lendahl U., Betsholtz C., A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480 (2018). [DOI] [PubMed] [Google Scholar]
- 107.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McInnes L., Healy J., Saul N., Großberger L., UMAP: Uniform manifold approximation and projection. J. Open Source Softw. 3 (29), 861 (2018). [Google Scholar]
- 109.Mathelier A., Fornes O., Arenillas D. J., Chen C.-Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R., Zhang A. W., Parcy F., Lenhard B., Sandelin A., Wasserman W. W., JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44, D110–D115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Hoon M. J. L., Imoto S., Nolan J., Miyano S., Open source clustering software. Bioinformatics 20, 1453–1454 (2004). [DOI] [PubMed] [Google Scholar]
- 111.Finucane H. K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C., Farh K., Ripke S., Day F. R.; ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; RACI Consortium, Purcell S., Stahl E., Lindstrom S., Perry J. R. B., Okada Y., Raychaudhuri S., Daly M. J., Patterson N., Neale B. M., Price A. L., Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ripke S., Neale B. M., Corvin A., Walters J. T. R., Farh K.-H., Holmans P. A., Lee P., Bulik-Sullivan B., Collier D. A., Huang H., Pers T. H., Agartz I., Agerbo E., Albus M., Alexander M., Amin F., Bacanu S. A., Begemann M., Belliveau R. A. Jr., Bene J., Bergen S. E., Bevilacqua E., Bigdeli T. B., Black D. W., Bruggeman R., Buccola N. G., Buckner R. L., Byerley W., Cahn W., Cai G., Campion D., Cantor R. M., Carr V. J., Carrera N., Catts S. V., Chambert K. D., Chan R. C. K., Chen R. Y. L., Chen E. Y. H., Cheng W., Cheung E. F. C., Chong S. A., Cloninger C. R., Cohen D., Cohen N., Cormican P., Craddock N., Crowley J. J., Curtis D., Davidson M., Davis K. L., Degenhardt F., Favero J. D., Demontis D., Dikeos D., Dinan T., Djurovic S., Donohoe G., Drapeau E., Duan J., Dudbridge F., Durmishi N., Eichhammer P., Eriksson J., Escott-Price V., Essioux L., Fanous A. H., Farrell M. S., Frank J., Franke L., Freedman R., Freimer N. B., Friedl M., Friedman J. I., Fromer M., Genovese G., Georgieva L., Giegling I., Giusti-Rodríguez P., Godard S., Goldstein J. I., Golimbet V., Gopal S., Gratten J., de Haan L., Hammer C., Hamshere M. L., Hansen M., Hansen T., Haroutunian V., Hartmann A. M., Henskens F. A., Herms S., Hirschhorn J. N., Hoffmann P., Hofman A., Hollegaard M. V., Hougaard D. M., Ikeda M., Joa I., Julià A., Kahn R. S., Kalaydjieva L., Karachanak-Yankova S., Karjalainen J., Kavanagh D., Keller M. C., Kennedy J. L., Khrunin A., Kim Y., Klovins J., Knowles J. A., Konte B., Kucinskas V., Kucinskiene Z. A., Kuzelova-Ptackova H., Kähler A. K., Laurent C., Keong J. L. C., Lee S. H., Legge S. E., Lerer B., Li M., Li T., Liang K.-Y., Lieberman J., Limborska S., Loughland C. M., Lubinski J., Lönnqvist J., Macek M. Jr., Magnusson P. K. E., Maher B. S., Maier W., Mallet J., Marsal S., Mattheisen M., Mattingsdal M., McCarley R. W., Donald C. M., McIntosh A. M., Meier S., Meijer C. J., Melegh B., Melle I., Mesholam-Gately R. I., Metspalu A., Michie P. T., Milani L., Milanova V., Mokrab Y., Morris D. W., Mors O., Murphy K. C., Murray R. M., Myin-Germeys I., Müller-Myhsok B., Nelis M., Nenadic I., Nertney D. A., Nestadt G., Nicodemus K. K., Nikitina-Zake L., Nisenbaum L., Nordin A., O’Callaghan E., O’Dushlaine C., O’Neill F. A., Oh S.-Y., Olincy A., Olsen L., Van Os J.; Psychosis Endophenotypes International Consortium, Pantelis C., Papadimitriou G. N., Papiol S., Parkhomenko E., Pato M. T., Paunio T., Pejovic-Milovancevic M., Perkins D. O., Pietiläinen O., Pimm J., Pocklington A. J., Powell J., Price A., Pulver A. E., Purcell S. M., Quested D., Rasmussen H. B., Reichenberg A., Reimers M. A., Richards A. L., Roffman J. L., Roussos P., Ruderfer D. M., Salomaa V., Sanders A. R., Schall U., Schubert C. R., Schulze T. G., Schwab S. G., Scolnick E. M., Scott R. J., Seidman L. J., Shi J., Sigurdsson E., Silagadze T., Silverman J. M., Sim K., Slominsky P., Smoller J. W., So H.-C., Spencer C. C. A., Stahl E. A., Stefansson H., Steinberg S., Stogmann E., Straub R. E., Strengman E., Strohmaier J., Stroup T. S., Subramaniam M., Suvisaari J., Svrakic D. M., Szatkiewicz J. P., Söderman E., Thirumalai S., Toncheva D., Tosato S., Veijola J., Waddington J., Walsh D., Wang D., Wang Q., Webb B. T., Weiser M., Wildenauer D. B., Williams N. M., Williams S., Witt S. H., Wolen A. R., Wong E. H. M., Wormley B. K., Xi H. S., Zai C. C., Zheng X., Zimprich F., Wray N. R., Stefansson K., Visscher P. M.; Wellcome Trust Case-Control Consortium, Adolfsson R., Andreassen O. A., Blackwood D. H. R., Bramon E., Buxbaum J. D., Børglum A. D., Cichon S., Darvasi A., Domenici E., Ehrenreich H., Esko T., Gejman P. V., Gill M., Gurling H., Hultman C. M., Iwata N., Jablensky A. V., Jönsson E. G., Kendler K. S., Kirov G., Knight J., Lencz T., Levinson D. F., Li Q. S., Liu J., Malhotra A. K., McCarroll S. A., Quillin A. M., Moran J. L., Mortensen P. B., Mowry B. J., Nöthen M. M., Ophoff R. A., Owen M. J., Palotie A., Pato C. N., Petryshen T. L., Posthuma D., Rietschel M., Riley B. P., Rujescu D., Sham P. C., Sklar P., Clair D. S., Weinberger D. R., Wendland J. R., Werge T., Daly M. J., Sullivan P. F., O’Donovan M. C., Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wiberg A., Ng M., Schmid A. B., Smillie R. W., Baskozos G., Holmes M. V., Künnapuu K., Mägi R., Bennett D. L., Furniss D., A genome-wide association analysis identifies 16 novel susceptibility loci for carpal tunnel syndrome. Nat. Commun. 10, 1030 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grove J., Ripke S., Als T. D., Mattheisen M., Walters R. K., Won H., Pallesen J., Agerbo E., Andreassen O. A., Anney R., Awashti S., Belliveau R., Bettella F., Buxbaum J. D., Bybjerg-Grauholm J., Bækvad-Hansen M., Cerrato F., Chambert K., Christensen J. H., Churchhouse C., Dellenvall K., Demontis D., De Rubeis S., Devlin B., Djurovic S., Dumont A. L., Goldstein J. I., Hansen C. S., Hauberg M. E., Hollegaard M. V., Hope S., Howrigan D. P., Huang H., Hultman C. M., Klei L., Maller J., Martin J., Martin A. R., Moran J. L., Nyegaard M., Nærland T., Palmer D. S., Palotie A., Pedersen C. B., Pedersen M. G., dPoterba T., Poulsen J. B., Pourcain B. S., Qvist P., Rehnström K., Reichenberg A., Reichert J., Robinson E. B., Roeder K., Roussos P., Saemundsen E., Sandin S., Satterstrom F. K., Smith G. D., Stefansson H., Steinberg S., Stevens C. R., Sullivan P. F., Turley P., Walters G. B., Xu X.; Autism Spectrum Disorder Working Group of the Psychiatric Genomics Consortium; BUPGEN, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium; 23andMe Research Team, Stefansson K., Geschwind D. H., Nordentoft M., Hougaard D. M., Werge T., Mors O., Mortensen P. B., Neale B. M., Daly M. J., Børglum A. D., Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tachmazidou I., Hatzikotoulas K., Southam L., Esparza-Gordillo J., Haberland V., Zheng J., Johnson T., Koprulu M., Zengini E., Steinberg J., Wilkinson J. M., Bhatnagar S., Hoffman J. D., Buchan N., Süveges D.; arcOGEN Consortium, Yerges-Armstrong L., Smith G. D., Gaunt T. R., Scott R. A., McCarthy L. C., Zeggini E., Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 51, 230–236 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.de Lange K. M., Moutsianas L., Lee J. C., Lamb C. A., Luo Y., Kennedy N. A., Jostins L., Rice D. L., Gutierrez-Achury J., Ji S.-G., Heap G., Nimmo E. R., Edwards C., Henderson P., Mowat C., Sanderson J., Satsangi J., Simmons A., Wilson D. C., Tremelling M., Hart A., Mathew C. G., Newman W. G., Parkes M., Lees C. W., Uhlig H., Hawkey C., Prescott N. J., Ahmad T., Mansfield J. C., Anderson C. A., Barrett J. C., Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wray N. R., Ripke S., Mattheisen M., Trzaskowski M., Byrne E. M., Abdellaoui A., Adams M. J., Agerbo E., Air T. M., Andlauer T. M. F., Bacanu S.-A., Bækvad-Hansen M., Beekman A. F. T., Bigdeli T. B., Binder E. B., Blackwood D. R. H., Bryois J., Buttenschøn H. N., Bybjerg-Grauholm J., Cai N., Castelao E., Christensen J. H., Clarke T.-K., Coleman J. I. R., Colodro-Conde L., Couvy-Duchesne B., Craddock N., Crawford G. E., Crowley C. A., Dashti H. S., Davies G., Deary I. J., Degenhardt F., Derks E. M., Direk N., Dolan C. V., Dunn E. C., Eley T. C., Eriksson N., Escott-Price V., Kiadeh F. H. F., Finucane H. K., Forstner A. J., Frank J., Gaspar H. A., Gill M., Giusti-Rodríguez P., Goes F. S., Gordon S. D., Grove J., Hall L. S., Hannon E., Hansen C. S., Hansen T. F., Herms S., Hickie I. B., Hoffmann P., Homuth G., Horn C., Hottenga J.-J., Hougaard D. M., Hu M., Hyde C. L., Ising M., Jansen R., Jin F., Jorgenson E., Knowles J. A., Kohane I. S., Kraft J., Kretzschmar W. W., Krogh J., Kutalik Z., Lane J. M., Li Y., Li Y., Lind P. A., Liu X., Lu L., Mac Intyre D. J., Mac Kinnon D. F., Maier R. M., Maier W., Marchini J., Mbarek H., Grath P. M., Guffin P. M., Medland S. E., Mehta D., Middeldorp C. M., Mihailov E., Milaneschi Y., Milani L., Mill J., Mondimore F. M., Montgomery G. W., Mostafavi S., Mullins N., Nauck M., Ng B., Nivard M. G., Nyholt D. R., O’Reilly P. F., Oskarsson H., Owen M. J., Painter J. N., Pedersen C. B., Pedersen M. G., Peterson R. E., Pettersson E., Peyrot W. J., Pistis G., Posthuma D., Purcell S. M., Quiroz J. A., Qvist P., Rice J. P., Riley B. P., Rivera M., Mirza S. S., Saxena R., Schoevers R., Schulte E. C., Shen L., Shi J., Shyn S. I., Sigurdsson E., Sinnamon G. B. C., Smit J. H., Smith D. J., Stefansson H., Steinberg S., Stockmeier C. A., Streit F., Strohmaier J., Tansey K. E., Teismann H., Teumer A., Thompson W., Thomson P. A., Thorgeirsson T. E., Tian C., Traylor M., Treutlein J., Trubetskoy V., Uitterlinden A. G., Umbricht D., Van der Auwera S., van Hemert A. M., Viktorin A., Visscher P. M., Wang Y., Webb B. T., Weinsheimer S. M., Wellmann J., Willemsen G., Witt S. H., Wu Y., Xi H. S., Yang J., Zhang F.; eQTLGen; 23andMe, Arolt V., Baune B. T., Berger K., Boomsma D. I., Cichon S., Dannlowski U., de Geus E. C. J., De Paulo J. R., Domenici E., Domschke K., Esko T., Grabe H. J., Hamilton S. P., Hayward C., Heath A. C., Hinds D. A., Kendler K. S., Kloiber S., Lewis G., Li Q. S., Lucae S., Madden P. F. A., Magnusson P. K., Martin N. G., McIntosh A. M., Metspalu A., Mors O., Mortensen P. B., Müller-Myhsok B., Nordentoft M., Nöthen M. M., O’Donovan M. C., Paciga S. A., Pedersen N. L., Penninx B. W. J. H., Perlis R. H., Porteous D. J., Potash J. B., Preisig M., Rietschel M., Schaefer C., Schulze T. G., Smoller J. W., Stefansson K., Tiemeier H., Uher R., Völzke H., Weissman M. M., Werge T., Winslow A. R., Lewis C. M., Levinson D. F., Breen G., Børglum A. D., Sullivan P. F.; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium , Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mahajan A., Taliun D., Thurner M., Robertson N. R., Torres J. M., Rayner N. W., Payne A. J., Steinthorsdottir V., Scott R. A., Grarup N., Cook J. P., Schmidt E. M., Wuttke M., Sarnowski C., Mägi R., Nano J., Gieger C., Trompet S., Lecoeur C., Preuss M. H., Prins B. P., Guo X., Bielak L. F., Below J. E., Bowden D. W., Chambers J. C., Kim Y. J., Ng M. C. Y., Petty L. E., Sim X., Zhang W., Bennett A. J., Bork-Jensen J., Brummett C. M., Canouil M., Ec kardt K.-U., Fischer K., Kardia S. L. R., Kronenberg F., Läll K., Liu C.-T., Locke A. E., Luan J.’., Ntalla I., Nylander V., Schönherr S., Schurmann C., Yengo L., Bottinger E. P., Brandslund I., Christensen C., Dedoussis G., Florez J. C., Ford I., Franco O. H., Frayling T. M., Giedraitis V., Hackinger S., Hattersley A. T., Herder C., Ikram M. A., Ingelsson M., Jørgensen M. E., Jørgensen T., Kriebel J., Kuusisto J., Ligthart S., Lindgren C. M., Linneberg A., Lyssenko V., Mamakou V., Meitinger T., Mohlke K. L., Morris A. D., Nadkarni G., Pankow J. S., Peters A., Sattar N., Stančáková A., Strauch K., Taylor K. D., Thorand B., Thorleifsson G., Thorsteinsdottir U., Tuomilehto J., Witte D. R., Dupuis J., Peyser P. A., Zeggini E., Loos R. J. F., Froguel P., Ingelsson E., Lind L., Groop L., Laakso M., Collins F. S., Jukema J. W., Palmer C. N. A., Grallert H., Metspalu A., Dehghan A., Köttgen A., Abecasis G. R., Meigs J. B., Rotter J. I., Marchini J., Pedersen O., Hansen T., Langenberg C., Wareham N. J., Stefansson K., Gloyn A. L., Morris A. P., Boehnke M., McCarthy M. I., Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lambert J. C., Ibrahim-Verbaas C. A., Harold D., Naj A. C., Sims R., Bellenguez C., De Stafano A. L., Bis J. C., Beecham G. W., Grenier-Boley B., Russo G., Thorton-Wells T. A., Jones N., Smith A. V., Chouraki V., Thomas C., Ikram M. A., Zelenika D., Vardarajan B. N., Kamatani Y., Lin C. F., Gerrish A., Schmidt H., Kunkle B., Dunstan M. L., Ruiz A., Bihoreau M. T., Choi S. H., Reitz C., Pasquier F., Cruchaga C., Craig D., Amin N., Berr C., Lopez O. L., De Jager P. L., Deramecourt V., Johnston J. A., Evans D., Lovestone S., Letenneur L., Morón F. J., Rubinsztein D. C., Eiriksdottir G., Sleegers K., Goate A. M., Fiévet N., Huentelman M. W., Gill M., Brown K., Kamboh M. I., Keller L., Barberger-Gateau P., McGuiness B., Larson E. B., Green R., Myers A. J., Dufouil C., Todd S., Wallon D., Love S., Rogaeva E., Gallacher J., St George-Hyslop P., Clarimon J., Lleo A., Bayer A., Tsuang D. W., Yu L., Tsolaki M., Bossù P., Spalletta G., Proitsi P., Collinge J., Sorbi S., Sanchez-Garcia F., Fox N. C., Hardy J., Naranjo M. C. D., Bosco P., Clarke R., Brayne C., Galimberti D., Mancuso M., Matthews F.; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart, Aging Research in Genomic Epidemiology, Moebus S., Mecocci P., Del Zompo M., Maier W., Hampel H., Pilotto A., Bullido M., Panza F., Caffarra P., Nacmias B., Gilbert J. R., Mayhaus M., Lannefelt L., Hakonarson H., Pichler S., Carrasquillo M. M., Ingelsson M., Beekly D., Alvarez V., Zou F., Valladares O., Younkin S. G., Coto E., Hamilton-Nelson K. L., Gu W., Razquin C., Pastor P., Mateo I., Owen M. J., Faber K. M., Jonsson P. V., Combarros O., O’Donovan M. C., Cantwell L. B., Soininen H., Blacker D., Mead S., Mosley T. H. Jr., Bennett D. A., Harris T. B., Fratiglioni L., Holmes C., de Bruijn R. F., Passmore P., Montine T. J., Bettens K., Rotter J. I., Brice A., Morgan K., Foroud T. M., Kukull W. A., Hannequin D., Powell J. F., Nalls M. A., Ritchie K., Lunetta K. L., Kauwe J. S., Boerwinkle E., Riemenschneider M., Boada M., Hiltuenen M., Martin E. R., Schmidt R., Rujescu D., Wang L. S., Dartigues J. F., Mayeux R., Tzourio C., Hofman A., Nöthen M. M., Graff C., Psaty B. M., Jones L., Haines J. L., Holmans P. A., Lathrop M., Pericak-Vance M. A., Launer L. J., Farrer L. A., van Duijn C. M., Van Broeckhoven C., Moskvina V., Seshadri S., Williams J., Schellenberg G. D., Amouyel P., Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Okada Y., Wu D., Trynka G., Raj T., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Yoshida S., Graham R. R., Manoharan A., Ortmann W., Bhangale T., Denny J. C., Carroll R. J., Eyler A. E., Greenberg J. D., Kremer J. M., Pappas D. A., Jiang L., Yin J., Ye L., Su D.-F., Yang J., Xie G., Keystone E., Westra H.-J., Esko T., Metspalu A., Zhou X., Gupta N., Mirel D., Stahl E. A., Diogo D., Cui J., Liao K., Guo M. H., Myouzen K., Kawaguchi T., Coenen M. J. H., van Riel P. L. C. M., van de Laar M. A. F. J., Guchelaar H.-J., Huizinga T. W. J., Dieudé P., Mariette X., Louis Bridges S. Jr., Zhernakova A., Toes R. E. M., Tak P. P., Miceli-Richard C., Bang S.-Y., Lee H.-S., Martin J., Gonzalez-Gay M. A., Rodriguez-Rodriguez L., Rantapää-Dahlqvist S., Arlestig L., Choi H. K., Kamatani Y., Galan P., Lathrop M.; RACI consortium; GARNET consortium, Eyre S., Bowes J., Barton A., de Vries N., Moreland L. W., Criswell L. A., Karlson E. W., Taniguchi A., Yamada R., Kubo M., Liu J. S., Bae S.-C., Worthington J., Padyukov L., Klareskog L., Gregersen P. K., Raychaudhuri S., Stranger B. E., De Jager P. L., Franke L., Visscher P. M., Brown M. A., Yamanaka H., Mimori T., Takahashi A., Xu H., Behrens T. W., Siminovitch K. A., Momohara S., Matsuda F., Yamamoto K., Plenge R. M., Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bentham J., Morris D. L., Cunninghame Graham D. S., Pinder C. L., Tombleson P., Behrens T. W., Martín J., Fairfax B. P., Knight J. C., Chen L., Replogle J., Syvänen A. C., Rönnblom L., Graham R. R., Wither J. E., Rioux J. D., Alarcón-Riquelme M. E., Vyse T. J., Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aylward A., Chiou J., Okino M. L., Kadakia N., Gaulton K. J., Shared genetic risk contributes to type 1 and type 2 diabetes etiology. Hum. Mol. Genet., (2018). [DOI] [PubMed] [Google Scholar]
- 123.den Hoed M., Eijgelsheim M., Esko T., Brundel B. J. J. M., Peal D. S., Evans D. M., Nolte I. M., Segrè A. V., Holm H., Handsaker R. E., Westra H.-J., Johnson T., Isaacs A., Yang J., Lundby A., Zhao J. H., Kim Y. J., Go M. J., Almgren P., Bochud M., Boucher G., Cornelis M. C., Gudbjartsson D., Hadley D., van der Harst P., Hayward C., den Heijer M., Igl W., Jackson A. U., Kutalik Z., Luan J., Kemp J. P., Kristiansson K., Ladenvall C., Lorentzon M., Montasser M. E., Njajou O. T., O’Reilly P. F., Padmanabhan S., Pourcain B. S., Rankinen T., Salo P., Tanaka T., Timpson N. J., Vitart V., Waite L., Wheeler W., Zhang W., Draisma H. H. M., Feitosa M. F., Kerr K. F., Lind P. A., Mihailov E., Onland-Moret N. C., Song C., Weedon M. N., Xie W., Yengo L., Absher D., Albert C. M., Alonso A., Arking D. E., de Bakker P. I. W., Balkau B., Barlassina C., Benaglio P., Bis J. C., Bouatia-Naji N., Brage S., Chanock S. J., Chines P. S., Chung M., Darbar D., Dina C., Dörr M., Elliott P., Felix S. B., Fischer K., Fuchsberger C., de Geus E. J. C., Goyette P., Gudnason V., Harris T. B., Hartikainen A.-L., Havulinna A. S., Heckbert S. R., Hicks A. A., Hofman A., Holewijn S., Hoogstra-Berends F., Hottenga J.-J., Jensen M. K., Johansson A., Junttila J., Kääb S., Kanon B., Ketkar S., Khaw K.-T., Knowles J. W., Kooner A. S., Kors J. A., Kumari M., Milani L., Laiho P., Lakatta E. G., Langenberg C., Leusink M., Liu Y., Luben R. N., Lunetta K. L., Lynch S. N., Markus M. R. P., Marques-Vidal P., Leach I. M., McArdle W. L., McCarroll S. A., Medland S. E., Miller K. A., Montgomery G. W., Morrison A. C., Müller-Nurasyid M., Navarro P., Nelis M., O’Connell J. R., O’Donnell C. J., Ong K. K., Newman A. B., Peters A., Polasek O., Pouta A., Pramstaller P. P., Psaty B. M., Rao D. C., Ring S. M., Rossin E. J., Rudan D., Sanna S., Scott R. A., Sehmi J. S., Sharp S., Shin J. T., Singleton A. B., Smith A. V., Soranzo N., Spector T. D., Stewart C., Stringham H. M., Tarasov K. V., Uitterlinden A. G., Vandenput L., Hwang S.-J., Whitfield J. B., Wijmenga C., Wild S. H., Willemsen G., Wilson J. F., Witteman J. C. M., Wong A., Wong Q., Jamshidi Y., Zitting P., Boer J. M. A., Boomsma D. I., Borecki I. B., van Duijn C. M., Ekelund U., Forouhi N. G., Froguel P., Hingorani A., Ingelsson E., Kivimaki M., Kronmal R. A., Kuh D., Lind L., Martin N. G., Oostra B. A., Pedersen N. L., Quertermous T., Rotter J. I., van der Schouw Y. T., Verschuren W. M. M., Walker M., Albanes D., Arnar D. O., Assimes T. L., Bandinelli S., Boehnke M., de Boer R. A., Bouchard C., Caulfield W. L. M., Chambers J. C., Curhan G., Cusi D., Eriksson J., Ferrucci L., van Gilst W. H., Glorioso N., de Graaf J., Groop L., Gyllensten U., Hsueh W.-C., Hu F. B., Huikuri H. V., Hunter D. J., Iribarren C., Isomaa B., Jarvelin M.-R., Jula A., Kähönen M., Kiemeney L. A., van der Klauw M. M., Kooner J. S., Kraft P., Iacoviello L., Lehtimäki T., Lokki M.-L. L., Mitchell B. D., Navis G., Nieminen M. S., Ohlsson C., Poulter N. R., Qi L., Raitakari O. T., Rimm E. B., Rioux J. D., Rizzi F., Rudan I., Salomaa V., Sever P. S., Shields D. C., Shuldiner A. R., Sinisalo J., Stanton A. V., Stolk R. P., Strachan D. P., Tardif J.-C., Thorsteinsdottir U., Tuomilehto J., van Veldhuisen D. J., Virtamo J., Viikari J., Vollenweider P., Waeber G., Widen E., Cho Y. S., Olsen J. V., Visscher P. M., Willer C., Franke L.; Global BPgen Consortium; CARDIo GRAM Consortium, Erdmann J., Thompson J. R.; PR GWAS Consortium, Pfeufer A.; QRS GWAS Consortium, Sotoodehnia N.; QT-IGC Consortium, Newton-Cheh C.; CHARGE-AF Consortium, Ellinor P. T., Stricker B. H. C., Metspalu A., Perola M., Beckmann J. S., Smith G. D., Stefansson K., Wareham N. J., Munroe P. B., Sibon O. C. M., Milan D. J., Snieder H., Samani N. J., Loos R. J. F., Identification of heart rate–associated loci and their effects on cardiac conduction and rhythm disorders. Nat. Genet. 45, 621–631 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Okbay A., Beauchamp J. P., Fontana M. A., Lee J. J., Pers T. H., Rietveld C. A., Turley P., Chen G.-B., Emilsson V., Meddens S. F. W., Oskarsson S., Pickrell J. K., Thom K., Timshel P., de Vlaming R., Abdellaoui A., Ahluwalia T. S., Bacelis J., Baumbach C., Bjornsdottir G., Brandsma J. H., Pina Concas M., Derringer J., Furlotte N. A., Galesloot T. E., Girotto G., Gupta R., Hall L. M., Harris S. E., Hofer E., Horikoshi M., Huffman J. E., Kaasik K., Kalafati I. P., Karlsson R., Kong A., Lahti J., van der Lee S. J., deLeeuw C., Lind P. A., Lindgren K. O., Liu T., Mangino M., Marten J., Mihailov E., Miller M. B., van der Most P. J., Oldmeadow C., Payton A., Pervjakova N., Peyrot W. J., Qian Y., Raitakari O., Rueedi R., Salvi E., Schmidt B., Schraut K. E., Shi J., Smith A. V., Poot R. A., St Pourcain B., Teumer A., Thorleifsson G., Verweij N., Vuckovic D., Wellmann J., Westra H. J., Yang J., Zhao W., Zhu Z., Alizadeh B. Z., Amin N., Bakshi A., Baumeister S. E., Biino G., Bønnelykke K., Boyle P. A., Campbell H., Cappuccio F. P., Davies G., de Neve J. E., Deloukas P., Demuth I., Ding J., Eibich P., Eisele L., Eklund N., Evans D. M., Faul J. D., Feitosa M. F., Forstner A. J., Gandin I., Gunnarsson B., Halldórsson B. V., Harris T. B., Heath A. C., Hocking L. J., Holliday E. G., Homuth G., Horan M. A., Hottenga J. J., de Jager P. L., Joshi P. K., Jugessur A., Kaakinen M. A., Kähönen M., Kanoni S., Keltigangas-Järvinen L., Kiemeney L. A. L. M., Kolcic I., Koskinen S., Kraja A. T., Kroh M., Kutalik Z., Latvala A., Launer L. J., Lebreton M. P., Levinson D. F., Lichtenstein P., Lichtner P., Liewald D. C. M.; LifeLines Cohort Study, Loukola A., Madden P. A., Mägi R., Mäki-Opas T., Marioni R. E., Marques-Vidal P., Meddens G. A., McMahon G., Meisinger C., Meitinger T., Milaneschi Y., Milani L., Montgomery G. W., Myhre R., Nelson C. P., Nyholt D. R., Ollier W. E. R., Palotie A., Paternoster L., Pedersen N. L., Petrovic K. E., Porteous D. J., Räikkönen K., Ring S. M., Robino A., Rostapshova O., Rudan I., Rustichini A., Salomaa V., Sanders A. R., Sarin A. P., Schmidt H., Scott R. J., Smith B. H., Smith J. A., Staessen J. A., Steinhagen-Thiessen E., Strauch K., Terracciano A., Tobin M. D., Ulivi S., Vaccargiu S., Quaye L., van Rooij F. J. A., Venturini C., Vinkhuyzen A. A. E., Völker U., Völzke H., Vonk J. M., Vozzi D., Waage J., Ware E. B., Willemsen G., Attia J. R., Bennett D. A., Berger K., Bertram L., Bisgaard H., Boomsma D. I., Borecki I. B., Bültmann U., Chabris C. F., Cucca F., Cusi D., Deary I. J., Dedoussis G. V., van Duijn C. M., Eriksson J. G., Franke B., Franke L., Gasparini P., Gejman P. V., Gieger C., Grabe H. J., Gratten J., Groenen P. J. F., Gudnason V., van der Harst P., Hayward C., Hinds D. A., Hoffmann W., Hyppönen E., Iacono W. G., Jacobsson B., Järvelin M. R., Jöckel K. H., Kaprio J., Kardia S. L. R., Lehtimäki T., Lehrer S. F., Magnusson P. K. E., Martin N. G., McGue M., Metspalu A., Pendleton N., Penninx B. W. J. H., Perola M., Pirastu N., Pirastu M., Polasek O., Posthuma D., Power C., Province M. A., Samani N. J., Schlessinger D., Schmidt R., Sørensen T. I. A., Spector T. D., Stefansson K., Thorsteinsdottir U., Thurik A. R., Timpson N. J., Tiemeier H., Tung J. Y., Uitterlinden A. G., Vitart V., Vollenweider P., Weir D. R., Wilson J. F., Wright A. F., Conley D. C., Krueger R. F., Davey Smith G., Hofman A., Laibson D. I., Medland S. E., Meyer M. N., Yang J., Johannesson M., Visscher P. M., Esko T., Koellinger P. D., Cesarini D., Benjamin D. J., Genome-wide association study identifies 74 loci associated with educational attainment. Nature 533, 539–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Locke A. E., Kahali B., Berndt S. I., Justice A. E., Pers T. H., Day F. R., Powell C., Vedantam S., Buchkovich M. L., Yang J., Croteau-Chonka D. C., Esko T., Fall T., Ferreira T., Gustafsson S., Kutalik Z., Luan J., Mägi R., Randall J. C., Winkler T. W., Wood A. R., Workalemahu T., Faul J. D., Smith J. A., Zhao J. H., Zhao W., Chen J., Fehrmann R., Hedman Å. K., Karjalainen J., Schmidt E. M., Absher D., Amin N., Anderson D., Beekman M., Bolton J. L., Bragg-Gresham J. L., Buyske S., Demirkan A., Deng G., Ehret G. B., Feenstra B., Feitosa M. F., Fischer K., Goel A., Gong J., Jackson A. U., Kanoni S., Kleber M. E., Kristiansson K., Lim U., Lotay V., Mangino M., Leach I. M., Medina-Gomez C., Medland S. E., Nalls M. A., Palmer C. D., Pasko D., Pechlivanis S., Peters M. J., Prokopenko I., Shungin D., Stančáková A., Strawbridge R. J., Sung Y. J., Tanaka T., Teumer A., Trompet S., van der Laan S. W., van Setten J., Van Vliet-Ostaptchouk J. V., Wang Z., Yengo L., Zhang W., Isaacs A., Albrecht E., Ärnlöv J., Arscott G. M., Attwood A. P., Bandinelli S., Barrett A., Bas I. N., Bellis C., Bennett A. J., Berne C., Blagieva R., Blüher M., Böhringer S., Bonnycastle L. L., Böttcher Y., Boyd H. A., Bruinenberg M., Caspersen I. H., Chen Y.-D. I., Clarke R., Daw E. W., de Craen A. J. M., Delgado G., Dimitriou M., Doney A. S. F., Eklund N., Estrada K., Eury E., Folkersen L., Fraser R. M., Garcia M. E., Geller F., Giedraitis V., Gigante B., Go A. S., Golay A., Goodall A. H., Gordon S. D., Gorski M., Grabe H.-J., Grallert H., Grammer T. B., Gräßler J., Grönberg H., Groves C. J., Gusto G., Haessler J., Hall P., Haller T., Hallmans G., Hartman C. A., Hassinen M., Hayward C., Heard-Costa N. L., Helmer Q., Hengstenberg C., Holmen O., Hottenga J.-J., James A. L., Jeff J. M., Johansson Å., Jolley J., Juliusdottir T., Kinnunen L., Koenig W., Koskenvuo M., Kratzer W., Laitinen J., Lamina C., Leander K., Lee N. R., Lichtner P., Lind L., Lindström J., Lo K. S., Lobbens S., Lorbeer R., Lu Y., Mach F., Magnusson P. K. E., Mahajan A., McArdle W. L., Lachlan S. M., Menni C., Merger S., Mihailov E., Milani L., Moayyeri A., Monda K. L., Morken M. A., Mulas A., Müller G., Müller-Nurasyid M., Musk A. W., Nagaraja R., Nöthen M. M., Nolte I. M., Pilz S., Rayner N. W., Renstrom F., Rettig R., Ried J. S., Ripke S., Robertson N. R., Rose L. M., Sanna S., Scharnagl H., Scholtens S., Schumacher F. R., Scott W. R., Seufferlein T., Shi J., Smith A. V., Smolonska J., Stanton A. V., Steinthorsdottir V., Stirrups K., Stringham H. M., Sundström J., Swertz M. A., Swift A. J., Syvänen A.-C., Tan S.-T., Tayo B. O., Thorand B., Thorleifsson G., Tyrer J. P., Uh H.-W., Vandenput L., Verhulst F. C., Vermeulen S. H., Verweij N., Vonk J. M., Waite L. L., Warren H. R., Waterworth D., Weedon M. N., Wilkens L. R., Willenborg C., Wilsgaard T., Wojczynski M. K., Wong A., Wright A. F., Zhang Q.; Life Lines Cohort Study, Brennan E. P., Choi M., Dastani Z., Drong A. W., Eriksson P., Franco-Cereceda A., Gådin J. R., Gharavi A. G., Goddard M. E., Handsaker R. E., Huang J., Karpe F., Kathiresan S., Keildson S., Kiryluk K., Kubo M., Lee J.-Y., Liang L., Lifton R. P., Ma B., McCarroll S. A., McKnight A. J., Min J. L., Moffatt M. F., Montgomery G. W., Murabito J. M., Nicholson G., Nyholt D. R., Okada Y., Perry J. R. B., Dorajoo R., Reinmaa E., Salem R. M., Sandholm N., Scott R. A., Stolk L., Takahashi A., Tanaka T., van’t Hooft F. M., Vinkhuyzen A. A. E., Westra H.-J., Zheng W., Zondervan K. T.; ADIPOGen Consortium; AGEN-BMI Working Group; CARDIOGRAMplusCD Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium, Heath A. C., Arveiler D., Bakker S. J. L., Beilby J., Bergman R. N., Blangero J., Bovet P., Campbell H., Caulfield M. J., Cesana G., Chakravarti A., Chasman D. I., Chines P. S., Collins F. S., Crawford D. C., Cupples L. A., Cusi D., Danesh J., de Faire U., den Ruijter H. M., Dominiczak A. F., Erbel R., Erdmann J., Eriksson J. G., Farrall M., Felix S. B., Ferrannini E., Ferrières J., Ford I., Forouhi N. G., Forrester T., Franco O. H., Gansevoort R. T., Gejman P. V., Gieger C., Gottesman O., Gudnason V., Gyllensten U., Hall A. S., Harris T. B., Hattersley A. T., Hicks A. A., Hindorff L. A., Hingorani A. D., Hofman A., Homuth G., Hovingh G. K., Humphries S. E., Hunt S. C., Hyppönen E., Illig T., Jacobs K. B., Jarvelin M.-R., Jöckel K.-H., Johansen B., Jousilahti P., Jukema J. W., Jula A. M., Kaprio J., Kastelein J. J. P., Keinanen-Kiukaanniemi S. M., Kiemeney L. A., Knekt P., Kooner J. S., Kooperberg C., Kovacs P., Kraja A. T., Kumari M., Kuusisto J., Lakka T. A., Langenberg C., Marchand L. L., Lehtimäki T., Lyssenko V., Männistö S., Marette A., Matise T. C., McKenzie C. A., Knight B. M., Moll F. L., Morris A. D., Morris A. P., Murray J. C., Nelis M., Ohlsson C., Oldehinkel A. J., Ong K. K., Madden P. A. F., Pasterkamp G., Peden J. F., Peters A., Postma D. S., Pramstaller P. P., Price J. F., Qi L., Raitakari O. T., Rankinen T., Rao D. C., Rice T. K., Ridker P. M., Rioux J. D., Ritchie M. D., Rudan I., Salomaa V., Samani N. J., Saramies J., Sarzynski M. A., Schunkert H., Schwarz P. E. H., Sever P., Shuldiner A. R., Sinisalo J., Stolk R. P., Strauch K., Tönjes A., Trégouët D.-A., Tremblay A., Tremoli E., Virtamo J., Vohl M.-C., Völker U., Waeber G., Willemsen G., Witteman J. C., Zillikens M. C., Adair L. S., Amouyel P., Asselbergs F. W., Assimes T. L., Bochud M., Boehm B. O., Boerwinkle E., Bornstein S. R., Bottinger E. P., Bouchard C., Cauchi S., Chambers J. C., Chanock S. J., Cooper R. S., de Bakker P. I. W., Dedoussis G., Ferrucci L., Franks P. W., Froguel P., Groop L. C., Haiman C. A., Hamsten A., Hui J., Hunter D. J., Hveem K., Kaplan R. C., Kivimaki M., Kuh D., Laakso M., Liu Y., Martin N. G., März W., Melbye M., Metspalu A., Moebus S., Munroe P. B., Njølstad I., Oostra B. A., Palmer C. N. A., Pedersen N. L., Perola M., Pérusse L., Peters U., Power C., Quertermous T., Rauramaa R., Rivadeneira F., Saaristo T. E., Saleheen D., Sattar N., Schadt E. E., Schlessinger D., Slagboom P. E., Snieder H., Spector T. D., Thorsteinsdottir U., Stumvoll M., Tuomilehto J., Uitterlinden A. G., Uusitupa M., van der Harst P., Walker M., Wallaschofski H., Wareham N. J., Watkins H., Weir D. R., Wichmann H.-E., Wilson J. F., Zanen P., Borecki I. B., Deloukas P., Fox C. S., Heid I. M., O’Connell J. R., Strachan D. P., Stefansson K., van Duijn C. M., Abecasis G. R., Franke L., Frayling T. M., McCarthy M. I., Visscher P. M., Scherag A., Willer C. J., Boehnke M., Mohlke K. L., Lindgren C. M., Beckmann J. S., Barroso I., North K. E., Ingelsson E., Hirschhorn J. N., Loos R. J. F., Speliotes E. K., Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–I206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Day F. R., Thompson D. J., Helgason H., Chasman D. I., Finucane H., Sulem P., Ruth K. S., Whalen S., Sarkar A. K., Albrecht E., Altmaier E., Amini M., Barbieri C. M., Boutin T., Campbell A., Demerath E., Giri A., He C., Hottenga J. J., Karlsson R., Kolcic I., Loh P.-R., Lunetta K. L., Mangino M., Marco B., Mahon G. M., Medland S. E., Nolte I. M., Noordam R., Nutile T., Paternoster L., Perjakova N., Porcu E., Rose L. M., Schraut K. E., Segrè A. V., Smith A. V., Stolk L., Teumer A., Andrulis I. L., Bandinelli S., Beckmann M. W., Benitez J., Bergmann S., Bochud M., Boerwinkle E., Bojesen S. E., Bolla M. K., Brand J. S., Brauch H., Brenner H., Broer L., Brüning T., Buring J. E., Campbell H., Catamo E., Chanock S., Chenevix-Trench G., Corre T., Couch F. J., Cousminer D. L., Cox A., Crisponi L., Czene K., Smith G. D., de Geus E. J. C. N., de Mutsert R., De Vivo I., Dennis J., Devilee P., Dos-Santos-Silva I., Dunning A. M., Eriksson J. G., Fasching P. A., Fernández-Rhodes L., Ferrucci L., Flesch-Janys D., Franke L., Gabrielson M., Gandin I., Giles G. G., Grallert H., Gudbjartsson D. F., Guénel P., Hall P., Hallberg E., Hamann U., Harris T. B., Hartman C. A., Heiss G., Hooning M. J., Hopper J. L., Hu F., Hunter D. J., Ikram M. A., Im H. K., Järvelin M.-R., Joshi P. K., Karasik D., Kellis M., Kutalik Z., Chance G. L., Lambrechts D., Langenberg C., Launer L. J., Laven J. S. E., Lenarduzzi S., Li J., Lind P. A., Lindstrom S., Liu Y. M., Luan J., Mägi R., Mannermaa A., Mbarek H., McCarthy M. I., Meisinger C., Meitinger T., Menni C., Metspalu A., Michailidou K., Milani L., Milne R. L., Montgomery G. W., Mulligan A. M., Nalls M. A., Navarro P., Nevanlinna H., Nyholt D. R., Oldehinkel A. J., O’Mara T. A., Padmanabhan S., Palotie A., Pedersen N., Peters A., Peto J., Pharoah P. D. P., Pouta A., Radice P., Rahman I., Ring S. M., Robino A., Rosendaal F. R., Rudan I., Rueedi R., Ruggiero D., Sala C. F., Schmidt M. K., Scott R. A., Shah M., Sorice R., Southey M. C., Sovio U., Stampfer M., Steri M., Strauch K., Tanaka T., Tikkanen E., Timpson N. J., Traglia M., Truong T., Tyrer J. P., Uitterlinden A. G., Edwards D. R. V., Vitart V., Völker U., Vollenweider P., Wang Q., Widen E., van Dijk K. W., Willemsen G., Winqvist R., Wolffenbuttel B. H. R., Zhao J. H., Zoledziewska M., Zygmunt M., Alizadeh B. Z., Boomsma D. I., Ciullo M., Cucca F., Esko T., Franceschini N., Gieger C., Gudnason V., Hayward C., Kraft P., Lawlor D. A., Magnusson P. K. E., Martin N. G., Mook-Kanamori D. O., Nohr E. A., Polasek O., Porteous D., Price A. L., Ridker P. M., Snieder H., Spector T. D., Stöckl D., Toniolo D., Ulivi S., Visser J. A., Völzke H., Wareham N. J., Wilson J. F.; Life Lines Cohort Study; InterAct Consortium; kConFab/AOCS Investigators; Endometrial Cancer Association Consortium; Ovarian Cancer Association Consortium; PRACTICAL consortium, Spurdle A. B., Thorsteindottir U., Pollard K. S., Easton D. F., Tung J. Y., Chang-Claude J., Hinds D., Murray A., Murabito J. M., Stefansson K., Ong K. K., Perry J. R. B., Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat. Genet. 49, 834–841 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Day F. R., Ruth K. S., Thompson D. J., Lunetta K. L., Pervjakova N., Chasman D. I., Stolk L., Finucane H. K., Sulem P., Bulik-Sullivan B., Esko T., Johnson A. D., Elks C. E., Franceschini N., He C., Altmaier E., Brody J. A., Franke L. L., Huffman J. E., Keller M. F., McArdle P. F., Nutile T., Porcu E., Robino A., Rose L. M., Schick U. M., Smith J. A., Teumer A., Traglia M., Vuckovic D., Yao J., Zhao W., Albrecht E., Amin N., Corre T., Hottenga J.-J., Mangino M., Smith A. V., Tanaka T., Abecasis G., Andrulis I. L., Anton-Culver H., Antoniou A. C., Arndt V., Arnold A. M., Barbieri C., Beckmann M. W., Beeghly-Fadiel A., Benitez J., Bernstein L., Bielinski S. J., Blomqvist C., Boerwinkle E., Bogdanova N. V., Bojesen S. E., Bolla M. K., Borresen-Dale A.-L., Boutin T. S., Brauch H., Brenner H., Brüning T., Burwinkel B., Campbell A., Campbell H., Chanock S. J., Chapman J. R., Chen Y.-D. I., Chenevix-Trench G., Couch F. J., Coviello A. D., Cox A., Czene K., Darabi H., De Vivo I., Demerath E. W., Dennis J., Devilee P., Dörk T., Dos-Santos-Silva I., Dunning A. M., Eicher J. D., Fasching P. A., Faul J. D., Figueroa J., Flesch-Janys D., Gandin I., Garcia M. E., García-Closas M., Giles G. G., Girotto G. G., Goldberg M. S., González-Neira A., Goodarzi M. O., Grove M. L., Gudbjartsson D. F., Guénel P., Guo X., Haiman C. A., Hall P., Hamann U., Henderson B. E., Hocking L. J., Hofman A., Homuth G., Hooning M. J., Hopper J. L., Hu F. B., Huang J., Humphreys K., Hunter D. J., Jakubowska A., Jones S. E., Kabisch M., Karasik D., Knight J. A., Kolcic I., Kooperberg C., Kosma V.-M., Kriebel J., Kristensen V., Lambrechts D., Langenberg C., Li J., Li X., Lindström S., Liu Y., Luan J., Lubinski J., Mägi R., Mannermaa A., Manz J., Margolin S., Marten J., Martin N. G., Masciullo C., Meindl A., Michailidou K., Mihailov E., Milani L., Milne R. L., Müller-Nurasyid M., Nalls M., Neale B. M., Nevanlinna H., Neven P., Newman A. B., Nordestgaard B. G., Olson J. E., Padmanabhan S., Peterlongo P., Peters U., Petersmann A., Peto J., Pharoah P. D. P., Pirastu N. N., Pirie A., Pistis G., Polasek O., Porteous D., Psaty B. M., Pylkäs K., Radice P., Raffel L. J., Rivadeneira F., Rudan I., Rudolph A., Ruggiero D., Sala C. F., Sanna S., Sawyer E. J., Schlessinger D., Schmidt M. K., Schmidt F., Schmutzler R. K., Schoemaker M. J., Scott R. A., Seynaeve C. M., Simard J., Sorice R., Southey M. C., Stöckl D., Strauch K., Swerdlow A., Taylor K. D., Thorsteinsdottir U., Toland A. E., Tomlinson I., Truong T., Tryggvadottir L., Turner S. T., Vozzi D., Wang Q., Wellons M., Willemsen G., Wilson J. F., Winqvist R., Wolffenbuttel B. B. H. R., Wright A. F., Yannoukakos D., Zemunik T., Zheng W., Zygmunt M., Bergmann S., Boomsma D. I., Buring J. E., Ferrucci L., Montgomery G. W., Gudnason V., Spector T. D., van Duijn C. M., Alizadeh B. Z., Ciullo M., Crisponi L., Easton D. F., Gasparini P. P., Gieger C., Harris T. B., Hayward C., Kardia S. L. R., Kraft P., Knight B. M., Metspalu A., Morrison A. C., Reiner A. P., Ridker P. M., Rotter J. I., Toniolo D., Uitterlinden A. G., Ulivi S., Völzke H., Wareham N. J., Weir D. R., Yerges-Armstrong L. M.; PRACTICAL consortium; kConFab Investigators; AOCS Investigators; Generation Scotland; EPIC-InterAct Consortium; Life Lines Cohort Study, Price A. L., Stefansson K., Visser J. A., Ong K. K., Chang-Claude J., Murabito J. M., Perry J. R. B., Murray A., Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 47, 1294–1303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wood A. R., Esko T., Yang J., Vedantam S., Pers T. H., Gustafsson S., Chu A. Y., Estrada K., Luan J., Kutalik Z., Amin N., Buchkovich M. L., Croteau-Chonka D. C., Day F. R., Duan Y., Fall T., Fehrmann R., Ferreira T., Jackson A. U., Karjalainen J., Lo K. S., Locke A. E., Mägi R., Mihailov E., Porcu E., Randall J. C., Scherag A., Vinkhuyzen A. A. E., Westra H.-J., Winkler T. W., Workalemahu T., Zhao J. H., Absher D., Albrecht E., Anderson D., Baron J., Beekman M., Demirkan A., Ehret G. B., Feenstra B., Feitosa M. F., Fischer K., Fraser R. M., Goel A., Gong J., Justice A. E., Kanoni S., Kleber M. E., Kristiansson K., Lim U., Lotay V., Lui J. C., Mangino M., Leach I. M., Medina-Gomez C., Nalls M. A., Nyholt D. R., Palmer C. D., Pasko D., Pechlivanis S., Prokopenko I., Ried J. S., Ripke S., Shungin D., Stancáková A., Strawbridge R. J., Sung Y. J., Tanaka T., Teumer A., Trompet S., van der Laan S. W., van Setten J., Van Vliet-Ostaptchouk J. V., Wang Z., Yengo L., Zhang W., Afzal U., Arnlöv J., Arscott G. M., Bandinelli S., Barrett A., Bellis C., Bennett A. J., Berne C., Blüher M., Bolton J. L., Böttcher Y., Boyd H. A., Bruinenberg M., Buckley B. M., Buyske S., Caspersen I. H., Chines P. S., Clarke R., Claudi-Boehm S., Cooper M., Daw E. W., De Jong P. A., Deelen J., Delgado G., Denny J. C., Dhonukshe-Rutten R., Dimitriou M., Doney A. S. F., Dörr M., Eklund N., Eury E., Folkersen L., Garcia M. E., Geller F., Giedraitis V., Go A. S., Grallert H., Grammer T. B., Gräßler J., Grönberg H., de Groot L. C. P. G. M., Groves C. J., Haessler J., Hall P., Haller T., Hallmans G., Hannemann A., Hartman C. A., Hassinen M., Hayward C., Heard-Costa N. L., Helmer Q., Hemani G., Henders A. K., Hillege H. L., Hlatky M. A., Hoffmann W., Hoffmann P., Holmen O., Houwing-Duistermaat J. J., Illig T., Isaacs A., James A. L., Jeff J., Johansen B., Johansson Å., Jolley J., Juliusdottir T., Junttila J., Kho A. N., Kinnunen L., Klopp N., Kocher T., Kratzer W., Lichtner P., Lind L., Lindström J., Lobbens S., Lorentzon M., Lu Y., Lyssenko V., Magnusson P. K. E., Mahajan A., Maillard M., McArdle W. L., McKenzie C. A., Lachlan S. M., McLaren P. J., Menni C., Merger S., Milani L., Moayyeri A., Monda K. L., Morken M. A., Müller G., Müller-Nurasyid M., Musk A. W., Narisu N., Nauck M., Nolte I. M., Nöthen M. M., Oozageer L., Pilz S., Rayner N. W., Renstrom F., Robertson N. R., Rose L. M., Roussel R., Sanna S., Scharnagl H., Scholtens S., Schumacher F. R., Schunkert H., Scott R. A., Sehmi J., Seufferlein T., Shi J., Silventoinen K., Smit J. H., Smith A. V., Smolonska J., Stanton A. V., Stirrups K., Stott D. J., Stringham H. M., Sundström J., Swertz M. A., Syvänen A.-C., Tayo B. O., Thorleifsson G., Tyrer J. P., van Dijk S., van Schoor N. M., van der Velde N., van Heemst D., van Oort F. V. A., Vermeulen S. H., Verweij N., Vonk J. M., Waite L. L., Waldenberger M., Wennauer R., Wilkens L. R., Willenborg C., Wilsgaard T., Wojczynski M. K., Wong A., Wright A. F., Zhang Q., Arveiler D., Bakker S. J. L., Beilby J., Bergman R. N., Bergmann S., Biffar R., Blangero J., Boomsma D. I., Bornstein S. R., Bovet P., Brambilla P., Brown M. J., Campbell H., Caulfield M. J., Chakravarti A., Collins R., Collins F. S., Crawford D. C., Cupples L. A., Danesh J., de Faire U., den Ruijter H. M., Erbel R., Erdmann J., Eriksson J. G., Farrall M., Ferrannini E., Ferrières J., Ford I., Forouhi N. G., Forrester T., Gansevoort R. T., Gejman P. V., Gieger C., Golay A., Gottesman O., Gudnason V., Gyllensten U., Haas D. W., Hall A. S., Harris T. B., Hattersley A. T., Heath A. C., Hengstenberg C., Hicks A. A., Hindorff L. A., Hingorani A. D., Hofman A., Hovingh G. K., Humphries S. E., Hunt S. C., Hypponen E., Jacobs K. B., Jarvelin M.-R., Jousilahti P., Jula A. M., Kaprio J., Kastelein J. J. P., Kayser M., Kee F., Keinanen-Kiukaanniemi S. M., Kiemeney L. A., Kooner J. S., Kooperberg C., Koskinen S., Kovacs P., Kraja A. T., Kumari M., Kuusisto J., Lakka T. A., Langenberg C., Marchand L. L., Lehtimäki T., Lupoli S., Madden P. A. F., Männistö S., Manunta P., Marette A., Matise T. C., Knight B. M., Meitinger T., Moll F. L., Montgomery G. W., Morris A. D., Morris A. P., Murray J. C., Nelis M., Ohlsson C., Oldehinkel A. J., Ong K. K., Ouwehand W. H., Pasterkamp G., Peters A., Pramstaller P. P., Price J. F., Qi L., Raitakari O. T., Rankinen T., Rao D. C., Rice T. K., Ritchie M., Rudan I., Salomaa V., Samani N. J., Saramies J., Sarzynski M. A., Schwarz P. E. H., Sebert S., Sever P., Shuldiner A. R., Sinisalo J., Steinthorsdottir V., Stolk R. P., Tardif J.-C., Tönjes A., Tremblay A., Tremoli E., Virtamo J., Vohl M.-C.; Electronic Medical Records and Genomics (eMEMERGEGE) Consortium; MIGen Consortium; PAGEGE Consortium; Life Lines Cohort Study, Amouyel P., Asselbergs F. W., Assimes T. L., Bochud M., Boehm B. O., Boerwinkle E., Bottinger E. P., Bouchard C., Cauchi S., Chambers J. C., Chanock S. J., Cooper R. S., de Bakker P. I. W., Dedoussis G., Ferrucci L., Franks P. W., Froguel P., Groop L. C., Haiman C. A., Hamsten A., Hayes M. G., Hui J., Hunter D. J., Hveem K., Jukema J. W., Kaplan R. C., Kivimaki M., Kuh D., Laakso M., Liu Y., Martin N. G., März W., Melbye M., Moebus S., Munroe P. B., Njølstad I., Oostra B. A., Palmer C. N. A., Pedersen N. L., Perola M., Pérusse L., Peters U., Powell J. E., Power C., Quertermous T., Rauramaa R., Reinmaa E., Ridker P. M., Rivadeneira F., Rotter J. I., Saaristo T. E., Saleheen D., Schlessinger D., Slagboom P. E., Snieder H., Spector T. D., Strauch K., Stumvoll M., Tuomilehto J., Uusitupa M., van der Harst P., Völzke H., Walker M., Wareham N. J., Watkins H., Wichmann H.-E., Wilson J. F., Zanen P., Deloukas P., Heid I. M., Lindgren C. M., Mohlke K. L., Speliotes E. K., Thorsteinsdottir U., Barroso I., Fox C. S., North K. E., Strachan D. P., Beckmann J. S., Berndt S. I., Boehnke M., Borecki I. B., McCarthy M. I., Metspalu A., Stefansson K., Uitterlinden A. G., van Duijn C. M., Franke L., Willer C. J., Price A. L., Lettre G., Loos R. J. F., Weedon M. N., Ingelsson E., O’Connell J. R., Abecasis G. R., Chasman D. I., Goddard M. E., Visscher P. M., Hirschhorn J. N., Frayling T. M., Defining the role of common variation in the genomic and biological architecture of adult human height. Nat. Genet. 46, 1173–1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Horikoshi M., Beaumont R. N., Day F. R., Warrington N. M., Kooijman M. N., Fernandez-Tajes J., Feenstra B., van Zuydam N. R., Gaulton K. J., Grarup N., Bradfield J. P., Strachan D. P., Li-Gao R., Ahluwalia T. S., Kreiner E., Rueedi R., Lyytikäinen L.-P., Cousminer D. L., Wu Y., Thiering E., Wang C. A., Have C. T., Hottenga J.-J., Vilor-Tejedor N., Joshi P. K., Boh E. T. H., Ntalla I., Pitkänen N., Mahajan A., van Leeuwen E. M., Joro R., Lagou V., Nodzenski M., Diver L. A., Zondervan K. T., Bustamante M., Marques-Vidal P., Mercader J. M., Bennett A. J., Rahmioglu N., Nyholt D. R., Ma R. C. W., Tam C. H. T., Tam W. H.; CHARGE Consortium Hematology Working Group, Ganesh S. K., van Rooij F. J. A., Jones S. E., Loh P.-R., Ruth K. S., Tuke M. A., Tyrrell J., Wood A. R., Yaghootkar H., Scholtens D. M., Paternoster L., Prokopenko I., Kovacs P., Atalay M., Willems S. M., Panoutsopoulou K., Wang X., Carstensen L., Geller F., Schraut K. E., Murcia M., van Beijsterveldt C. E. M., Willemsen G., Appel E. V. R., Fonvig C. E., Trier C., Tiesler C. M. T., Standl M., Kutalik Z., Bonàs-Guarch S., Hougaard D. M., Sánchez F., Torrents D., Waage J., Hollegaard M. V., de Haan H. G., Rosendaal F. R., Medina-Gomez C., Ring S. M., Hemani G., Mahon G. M., Robertson N. R., Groves C. J., Langenberg C., Luan J., Scott R. A., Zhao J. H., Mentch F. D., MacKenzie S. M., Reynolds R. M.; Early Growth Genetics (EGG) Consortium, Lowe W. L., Tönjes A., Stumvoll M., Lindi V., Lakka T. A., van Duijn C. M., Kiess W., Körner A., Sørensen T. I. A., Niinikoski H., Pahkala K., Raitakari O. T., Zeggini E., Dedoussis G. V., Teo Y.-Y., Saw S.-M., Melbye M., Campbell H., Wilson J. F., Vrijheid M., de Geus E. J. C. N., Boomsma D. I., Kadarmideen H. N., Holm J.-C., Hansen T., Sebert S., Hattersley A. T., Beilin L. J., Newnham J. P., Pennell C. E., Heinrich J., Adair L. S., Borja J. B., Mohlke K. L., Eriksson J. G., Widén E., Kähönen M., Viikari J. S., Lehtimäki T., Vollenweider P., Bønnelykke K., Bisgaard H., Mook-Kanamori D. O., Hofman A., Rivadeneira F., Uitterlinden A. G., Pisinger C., Pedersen O., Power C., Hyppönen E., Wareham N. J., Hakonarson H., Davies E., Walker B. R., Jaddoe V. W. V., Järvelin M.-R., Grant S. F. A., Vaag A. A., Lawlor D. A., Frayling T. M., Smith G. D., Morris A. P., Ong K. K., Felix J. F., Timpson N. J., Perry J. R. B., Evans D. M., McCarthy M. I., Freathy R. M., Genome-wide associations for birth weight and correlations with adult disease. Nature 538, 248–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jiang X., O’Reilly P. F., Aschard H., Hsu Y. H., Richards J. B., Dupuis J., Ingelsson E., Karasik D., Pilz S., Berry D., Kestenbaum B., Zheng J., Luan J., Sofianopoulou E., Streeten E. A., Albanes D., Lutsey P. L., Yao L., Tang W., Econs M. J., Wallaschofski H., Völzke H., Zhou A., Power C., McCarthy M. I., Michos E. D., Boerwinkle E., Weinstein S. J., Freedman N. D., Huang W. Y., van Schoor N. M., van der Velde N., de Groot L. C. P. G. M., Enneman A., Cupples L. A., Booth S. L., Vasan R. S., Liu C. T., Zhou Y., Ripatti S., Ohlsson C., Vandenput L., Lorentzon M., Eriksson J. G., Shea M. K., Houston D. K., Kritchevsky S. B., Liu Y., Lohman K. K., Ferrucci L., Peacock M., Gieger C., Beekman M., Slagboom E., Deelen J., van Heemst D., Kleber M. E., März W., de Boer I. H., Wood A. C., Rotter J. I., Rich S. S., Robinson-Cohen C., den Heijer M., Jarvelin M. R., Cavadino A., Joshi P. K., Wilson J. F., Hayward C., Lind L., Michaëlsson K., Trompet S., Zillikens M. C., Uitterlinden A. G., Rivadeneira F., Broer L., Zgaga L., Campbell H., Theodoratou E., Farrington S. M., Timofeeva M., Dunlop M. G., Valdes A. M., Tikkanen E., Lehtimäki T., Lyytikäinen L. P., Kähönen M., Raitakari O. T., Mikkilä V., Ikram M. A., Sattar N., Jukema J. W., Wareham N. J., Langenberg C., Forouhi N. G., Gundersen T. E., Khaw K. T., Butterworth A. S., Danesh J., Spector T., Wang T. J., Hyppönen E., Kraft P., Kiel D. P., Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun. 9, 260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wittemans L. B. L., Lotta L. A., Oliver-Williams C., Stewart I. D., Surendran P., Karthikeyan S., Day F. R., Koulman A., Imamura F., Zeng L., Erdmann J., Schunkert H., Khaw K. T., Griffin J. L., Forouhi N. G., Scott R. A., Wood A. M., Burgess S., Howson J. M. M., Danesh J., Wareham N. J., Butterworth A. S., Langenberg C., Assessing the causal association of glycine with risk of cardio-metabolic diseases. Nat. Commun. 10, 1060 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jansen P. R., Watanabe K., Stringer S., Skene N., Bryois J., Hammerschlag A. R., de Leeuw C. A., Benjamins J. S., Muñoz-Manchado A. B., Nagel M., Savage J. E., Tiemeier H., White T.; 23andMe Research Team, Tung J. Y., Hinds D. A., Vacic V., Wang X., Sullivan P. F., van der Sluis S., Polderman T. J. C., Smit A. B., Hjerling-Leffler J., Van Someren E. J. W., Posthuma D., Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 51, 394–403 (2019). [DOI] [PubMed] [Google Scholar]
- 133.1000 Genomes Project Consortium, Auton A., Brooks L. D., Durbin R. M., Garrison E. P., Kang H. M., Korbel J. O., Marchini J. L., Carthy S. M., McVean G. A., Abecasis G. R., A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Siepel A., Bejerano G., Pedersen J. S., Hinrichs A. S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L. W., Richards S., Weinstock G. M., Wilson R. K., Gibbs R. A., Kent W. J., Miller W., Haussler D., Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.B. Institute, Picard Toolkit, Broad Institute, GitHub Repository (2019).
- 138.Ramirez F., Dundar F., Diehl S., Gruning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Deutsch M. A., Doppler S. A., Li X., Lahm H., Santamaria G., Cuda G., Eichhorn S., Ratschiller T., Dzilic E., Dreßen M., Eckart A., Stark K., Massberg S., Bartels A., Rischpler C., Gilsbach R., Hein L., Fleischmann B. K., Wu S. M., Lange R., Krane M., Reactivation of the Nkx2.5 cardiac enhancer after myocardial infarction does not presage myogenesis. Cardiovasc. Res. 114, 1098–1114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/20/eabf1444/DC1