

Abstract
Objectives
While myeloid-derived suppressor cells (MDSCs) were previously shown to promote a proinflammatory T helper (Th) 17 response in autoimmune conditions, a potential impact of the MDSC-Th17 immune axis on abnormal bone destruction in RA remains largely unknown.
Methods
We investigated the correlation between the frequency of MDSCs or its subsets and joint destruction in RA patients. The reciprocal actions of patient-derived MDSCs and Th17 cells were studied using osteoclast (OC) differentiation and bone resorption assays in vitro, which were further validated using mouse models of RA. Contribution of MDSCs to osteoclastogenesis and bone erosion in vivo was determined by depletion or transfer of MDSCs.
Results
Human MDSCs, particularly monocytic MDSCs (M-MDSCs), exhibit inherent OC-differentiating capacity and positively correlate with clinical bone erosion in RA patients. Strikingly, patient-derived M-MDSCs can program Th17 cells towards a pro-osteoclastogenic phenotype, which in return potentiates OC differentiation via the receptor activator of nuclear factor κΒ ligand (RANK-L)-RANK signalling. This enhanced osteolysis driven by the reciprocal actions of M-MDSCs and Th17 cells is further confirmed using mouse models of RA. Selective depletion of M-MDSCs significantly ameliorates osteoclastogenesis and disease severity in arthritic mice, whereas transfer of M-MDSCs aggravates bone erosion associated with increased OCs in recipient mice.
Conclusion
Our findings highlight the functional plasticity of MDSCs and identify a novel pro-osteoclastogenic pathway governed by interplay between myeloid cells and T lymphocytes in autoimmune RA.
Keywords: rheumatoid arthritis, bone erosion, osteoclast, myeloid-derived suppressor cell, Th17
Graphical Abstract
Rheumatology key messages
Monocytic MDSCs from patients with rheumatoid arthritis can differentiate into osteoclasts with bone resorbing activity.
MDSC-Th17 interaction upregulates the pro-osteoclastogenic signal RANK-L on Th17 cells.
MDSC-‘conditioned’ Th17 cells reprogram MDSCs into functional osteoclasts.
Introduction
RA is a chronic autoimmune disorder that is characterized by exacerbated synovial inflammation, progressive joint destruction and physical disability [1]. It has been well documented that uncontrolled differentiation and activation of osteoclasts (OCs), the primary somatic cell type capable of resorbing bone matrices, contribute to bone erosion in inflammatory arthritis [2, 3]. In vitro and in vivo studies have shown that the receptor activator of NF-κB ligand (RANK-L) is indispensable for the differentiation and activation of OCs [4–6]. While an important role of the RANK-L-induced OC formation and bone destruction in RA has been recently established, a better understanding of the underlying cellular and molecular mechanisms will provide new therapeutic targets for effective disease intervention.
Myeloid-derived suppressor cells (MDSCs) are a population of immune cells that have emerged as a critical regulator of many pathologic conditions, including autoimmune disorders [7, 8]. MDSCs in human are typically defined as CD11b+CD33+HLA-DRlow/- cells and divided into CD14+ monocytic and CD15+ granulocytic populations. MDSCs in mice, characterized by the expression of myeloid cells markers CD11b and Gr-1 (Ly6G/Ly6C), consist of two major subsets: CD11b+Ly6G-Ly6Chigh monocytic MDSCs (M-MDSCs) and CD11b+Ly6G+Ly6Clow granulocytic MDSCs (G-MDSCs) [9]. Although the immunosuppressive function of MDSCs are well established in cancer, the emerging evidence reveals previously under-appreciated features of these myeloid cells in aggravation of inflammation in cancer [10, 11] and autoimmune disorders [12–15]. Recently, preclinical and clinical studies from our and other groups showed that MDSCs, substantially expanded under autoimmune pathological conditions (e.g. RA), can promote T helper (Th) 17 polarization and IL-17 production, thereby driving excessive inflammation, tissue injury, and disease pathogenesis [12–15]. However, the potential involvement of this MDSC-Th17 immune axis in RA-associated osteoclastogenesis and bone metabolism remains to be elucidated.
In this study, we investigate the functional interactions of MDSCs and its subsets with Th17 cells in the context of osteoclastogenesis using specimen from patients with RA and two mouse models of autoimmune arthritis. For the first time, we report that human MDSCs, particularly M-MDSCs, exhibit inherent capability to differentiate into OCs inducing bone resorption, further underscoring the functional plasticity and diversity of these myeloid cells in RA. More importantly, our findings uncover a novel immuno-osteoclastogenic pathway involving MDSC-Th17 interplay and the pro-osteoclastogenic factor RANK-L.
Methods
Patient samples
Whole blood samples were collected from 69 patients fulfilling the 1987 ACR revised criteria for the classification of RA or 2010 ACR/EULAR classification criteria for RA from Nanfang Hospital, Southern Medical University. Clinical and laboratory data were collected for evaluation of disease activity score in 28 joints (DAS28). RA disease activity were defined based on DAS28-ESR: ≤3.2, remission or low disease activity; 3.2 ∼ 5.1, moderate disease activity; >5.1, high disease activity [16].
Bone erosion scores were quantified based on finger joints (total 20 joints, including proximal interphalangeal joints and metacarpophalangeal joints) ultrasonography [17, 18]. Each joint was scored according to a semiquantitative scoring system (grades 0–3) [18]: 0, regular bone surface; 1, irregularity of the bone surface without formation of a defect; 2, formation of a defect in the surface of the bone; 3, bone defect creating extensive bone destruction. Bone erosion score was the sum of the grades for each joint. All participants were given written informed consent, and all procedures in this study were approved by the ethics committee of Nanfang Hospital, Southern Medical University.
Mice
C57BL/6 mice were purchased from National Cancer Institute (Bethesda, MD, USA). SKG mice were kindly provided by Dr. Abul Abbas, University of California, San Francisco (UCSF), USA. All experiments and procedures involving mice have been reviewed and approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University, USA.
Induction and evaluation of experimental arthritis
Collagen-induced arthritis (CIA) was conducted as previously described [12]. Chicken type II collagen (Sigma-Aldrich, MO, USA) was emulsified with CFA containing 4 mg/ml heat-killed M. tuberculosis H37 (BD Biosciences, CA, USA) at 1:1 ratio (vol:vol). Male C57BL/6 mice at 8–10 weeks old were subjected to a single intradermal injection of the emulsion (100 µl) at the base of the tail. For in vivo depletion of MDSCs [19], mice were administrated intraperitoneally with 50 mg/kg 5-Fluorouracil (5-Fu, Sigma-Aldrich, MO, USA) or PBS every 3 days from day 30 after the first immunization (n = 8 per group). Signs of arthritis in mice were monitored according to the swelling and redness of each limbs, the severity of which was scored in a blinded manner [20]. For SKG mice, 8-week-old female mice were intraperitoneally (i.p.)injected 2 mg zymosan (Sigma-Aldrich, MO, USA) to facilitate the onset of disease.
Statistical analysis
The probability-probability plot (P-P plot) was used to examine data normality. For normally distributed data, differences between various groups were evaluated using unpaired two-tailed Student’s t test or one-way ANOVA with Fisher Least Significant Difference (LSD) test. Mann–Whitney U or Kruskal–Wallis followed by Dunn’s multiple comparisons test were used for non-normally distributed data. Spearman rank test was used for analysing correlation of non-normally distributed data. Generalized Estimating Equations was used for comparisons of repeated measurement data (i.e. arthritic scores). Fisher’s exact test was used for categorical variables. Statistical analyses were performed using SPSS 20.0 (SPSS Inc., IL, USA) or SigmaPlot 12.5 (Systat Software Inc., CA, USA). P < 0.05 were considered statistically significant.
Detailed experimental procedures are described in supplementary materials, available at Rheumatology online
Results
M-MDSCs correlate with bone destruction in RA and promote a pro-osteoclastogenic phenotype of Th17 cells
Our previous study reported that human MDSCs were significantly elevated in RA patients and correlated with the level of inflammatory cytokine IL-17A [12]. Given that IL-17-producing Th17 cells can promote RANK-L signalling dependent OC differentiation [21, 22], we analysed the frequency of MDSCs and joint destruction in RA patients to determine a potential association between these cells and bone erosion. Detailed clinical and laboratory characteristics and treatment information of included patients are presented in Supplementary Tables S2 and S3, available at Rheumatology online. We showed that the frequency of MDSCs or M-MDSCs, not G-MDSCs, positively correlated with bone erosion scores as assessed by joint ultrasound in RA patients (Supplementary Table S1, available at Rheumatology online). This correlation was also supported by significantly higher levels of TRAP5b and β-CTX, two bone destruction markers [23, 24], in RA patients with more circulating M-MDSCs (Supplementary Table S1, available at Rheumatology online). We further showed that CD11b+CD33+HLA-DR-/lowCD14+CD15- M-MDSCs, isolated from peripheral blood of RA patients, were able to differentiate into TRAP+ OCs that can induce bone resorption (Fig. 1A). Intriguingly, M-MDSCs from RA patients with high disease activity were more readily to differentiate into OCs as compared with those from patients with low disease activity (Fig. 1A), suggesting that the intrinsic OC-differentiating potential of M-MDSCs may be determined by the disease status.
Fig. 1.

Human M-MDSCs from RA patients promote osteolysis
(A) M-MDSCs from peripheral blood of RA patients with low disease activity (28-joint disease activity score, DAS28 ≤ 3.2) or high disease activity (DAS28 > 5.1) were cultured in the presence of M-CSF and RANK-L in vitro for 14 days. TRAP staining and pit assay were performed to assess differentiation and biological activity of OCs. Scale bars in the images of TRAP and pit assay represent 50 µm and 200 µm, respectively (TRAP, P =0.024; Pit, P =0.002, unpaired t test). (B) Naïve CD4+ T cells from RA patients were co-cultured with or without autologous M-MDSCs under Th17–polarizing conditions for 5 days. Membrane-bound RANK-L expression on Th17 cells was analysed by flow cytometry. Percentage of RANK-L positive Th17 cells and mean fluorescence intensity (MFI, difference between the fluoresce intensity and isotype control) of RNAK-L are shown (P =0.01, unpaired t test). (C) Relative mRNA levels of Tnfsf11 gene were analysed by qRT-PCR (P =0.032, unpaired t test). (D) Naïve CD4+ T cells from RA patients were co-cultured with autologous M-MDSCs under Th17–polarizing conditions in vitro to generate M-MDSC-conditioned Th17 cells. M-MDSCs were then cultured with or without these Th17 cells in the presence of M-CSF, IL-2 and RANK-L for OC differentiation (P=0.025, unpaired t test). Scale bar, 200 µm. Data are representative of three independent experiments.
During the study of human MDSC-Th17 interaction, we made a surprising observation that M-MDSCs from RA patients upregulated membrane-bound RANK-L on autologous Th17 cells during co-culture (Fig. 1B). Upregulation of RANK-L was also indicated by increased gene transcription of Tnfsf11 (i.e. rankl) in Th17 cells, assessed by quantitative real-time PCR (qRT-PCR) analysis (Fig. 1C). Our result suggests that, in addition to its intrinsic OC-differentiating capability, M-MDSCs in RA has acquired the capability to skew Th17 cells towards a pro-osteoclastogenic phenotype. Indeed, M-MDSC-conditioned human Th17 cells were highly efficient in supporting OC differentiation from M-MDSCs (Fig. 1D), suggesting that reciprocal actions of MDSCs and Th17 cells during their interaction may contribute to bone destruction in RA patients.
MDSCs induce the expression of RANK-L preferentially by Th17 cells in mouse arthritis model
We next sought to use mouse model of RA, i.e. collagen-induced arthritis (CIA) that resembles the pathogenesis of human RA, to critically understand this previously unrecognized immune-osteoclastogenic pathway. We showed that the presence of MDSCs, especially those from arthritic mice (MDSC-CIA), significantly enhanced the expression of RANK-L on Th17 cells (Fig. 2A and B). Upregulation of RANK-L was also indicated by increased transcription of Tnfsf11 gene in Th17 cells (Fig. 2C). Compared with G-MDSCs, M-MDSCs from arthritic mice are more efficient in stimulating RANK-L expression by Th17 cells (Fig. 2D and E), which is consistent with our previously reported superior Th17-promoting capacity of this subpopulation [12, 14]. Intriguingly, MDSCs failed to induce T-cell-associated RANK-L expression when co-cultured with Th1, Th2 or Treg cells (Fig. 2F and G), suggesting that arthritis-expanded MDSCs selectively stimulate the pro-osteoclastogenic activity in Th17 cells.
Fig. 2.

MDSCs from arthritic mice augment expression of pro-osteoclastogenic RANK-L on Th17 cells
(A) and (B) MDSCs derived from naïve or CIA mice were co-cultured with naïve CD4+CD62L+ T cells under Th17–polarizing conditions for 48 h. Membrane-bound RANK-L expression on Th17 cells was analysed by flow cytometry. Representative flow charts showing the fluorescence intensity of RANK-L staining are presented (A) (Naïve vs -MDSC, P =0.014; CIA vs Naïve, P=0.004. One-way ANOVA with Fisher LSD test). (C) Relative mRNA levels of Tnfsf11 gene were analysed by qRT-PCR (Naïve vs -MDSC, P =0.025; CIA vs Naïve, P =0.002. One-way ANOVA with Fisher LSD test). (D) and (E) M-MDSCs or G-MDSCs were cultured with naïve CD4+CD62L+ T cells under Th17–polarizing conditions (G-MDSC vs -MDSC, P =0.303; M-MDSC vs G-MDSC, P <0.001. One-way ANOVA with Fisher LSD test). (F) and (G) In vitro polarized Th17, Th1, Th2 or Treg cells were co-cultured with or without MDSCs in the presence of plate-bound anti-CD3 Ab for 24 h. RANK-L expression on T cells was analysed by flow cytometry (Th17, P=0.002; Th1, P=0.005; Th2, P=0.496; Treg, P=0.152. Unpaired t test). The results shown are representative of three independent experiments. N.S., not significant.
Arginase 1 is required for MDSC-enhanced RANK-L expression on Th17 cells
We examined the potential molecular factors that are responsible for MDSC-enhanced RANK-L expression. Considering that IL-1β is critical for Th17 polarization driven by MDSCs under autoimmune conditions [12, 14], we first assessed the role of IL-1β using either IL-1β mAbs or IL-1 receptor antagonist. However, the activity of MDSC-CIA in promoting RANK-L production from Th17 cells was not impaired by blockade of IL-1β signalling (Fig. 3A), suggesting that IL-1β is not required for MDSC-CIA enhanced RANK-L expression. In light of our recent report that the MDSC-associated effector molecule arginase 1 mediates the MDSC-Th17 interplay in SLE [13], we examined the expression of arginase 1 (Arg1) in CIA-associated MDSCs by qRT-PCR. Gene transcription of Arg1 was only detectable in MDSCs derived from arthritic mice, not those from naïve mice (Fig. 3B). The mRNA levels of Arg1 were higher in M-MDSCs than in G-MDSCs in arthritic mice (Fig. 3C). To test the contribution of arginase 1 to MDSC-enhanced RANK-L expression, we blocked arginase 1 activity in MDSCs using the arginase inhibitor N-omega-hydroxy-nor-L-arginine (nor-NOHA) [25] during cell co-culture. The presence of nor-NOHA, not L-NIL that inhibits inducible nitric oxide synthase [26], abolished MDSC-CIA facilitated RANK-L upregulation (Fig. 3D and E). Additionally, nor-NOHA reduced the capability of M-MDSCs from patients with RA to induce RANK-L production. However, it did not affect RANK-L expression on Th17 cells in the absence of MDSCs (Fig. 3F and G). Therefore, MDSCs enhance the pro-inflammatory and pro-osteoclastogenic phenotypes of Th17 cells in RA through independent pathways.
Fig. 3.

Arginase 1 is involved in MDSC-enhanced RANK-L expression on Th17 cells
(A) Naïve CD4+CD62L+ T cells were co-cultured with MDSCs from arthritic mice (i.e. CIA) under Th17 polarizing conditions in the presence of IL-1 receptor antagonist (IL-1ra) or IL-1β neutralizing antibodies (IL-1 mAb) for 48 h. RANK-L expression was examined by flow cytometry (Vehicle vs None, P<0.001; IL-1 ra vs Vehicle, P=0.247; IL-1 mAb vs Vehicle, P=0.123. One-way ANOVA with Fisher LSD test). (B) Relative mRNA levels of Arg1 in MDSCs purified from indicated mice were analysed by qRT-PCR. (N.D., not detected). (C) Transcription of Arg1 gene in M-MDSCs or G-MDSCs was determined by qRT-PCR. (D) and (E) RANK-L expression was examined after mouse Th17 polarization in the presence of L-NIL or nor-NOHA (Vehicle vs None, P<0.001; nor-NOHA vs Vehicle, P<0.001; L-NIL vs Vehicle, P=0.131. One-way ANOVA with Fisher LSD test). (F) and (G) Naïve CD4+ T cells from RA patients were co-cultured in the presence of M-MDSCs and/or nor-NOHA under Th17–polarizing conditions for 3 days. RANK-L expression on Th17 cells was examined (nor-NOHA vs Vehicle: No MDSC, P=0.986; +M-MDSC, P<0.001. One-way ANOVA with Fisher LSD test). Data are representative of three independent experiments. N.S., not significant.
Arthritis-associated M-MDSCs not G-MDSCs exhibit the OC-differentiating capacity
Given that human MDSCs from patients with RA can differentiate into fully functional OCs, we also compared the OC-differentiating capacity of MDSCs from naïve or CIA mice in the presence of M-CSF and RANK-L. CIA-associated MDSCs displayed an increase in its potential of differentiation into OCs, indicated by elevated numbers of OCs (Fig. 4A), heightened transcription of Acp5 (tartrate-resistant acid phosphatase), Ctsk (cathepsin k) or Mmp9 (matrix metallopeptidase 9) genes (Fig. 4B), as well as enhanced activity in bone degradation (Fig. 4C). SKG mice, a genetic model of autoimmune arthritis [27], were also used to examine MDSC-initiated osteoclastogenesis and bone absorption. Consistent with our study in CIA model [12], we showed a substantial expansion of MDSCs including CD11b+Ly6G-Ly6Chigh M-MDSCs and CD11b+Ly6G+Ly6Clow G-MDSCs in arthritic SKG mice (Supplementary Fig. S2A, available at Rheumatology online). MDSCs from arthritic SKG mice inhibited the proliferation of and IFN-γ production by T cells upon stimulation with anti-CD3/CD28 antibodies (Supplementary Fig. S2B and C, available at Rheumatology online), indicating these cells retain immunosuppressive activity. Similarly, MDSCs from arthritic SKG mice were also more readily to differentiate into TRAP-positive OCs and to induce bone resorption (Fig. 4D), which was associated with increased gene transcription of the OC signature genes Acp5, Ctsk or Mmp9 (Supplementary Fig. S3, available at Rheumatology online).
Fig. 4.

MDSCs from arthritic mice display enhanced capacity in differentiation into functional OCs
(A) MDSCs from naïve or CIA mice were cultured in the presence of M-CSF and RANK-L for OCs differentiation. Representative images of TRAP staining and OCs numbers are shown (P=0.005. Unpaired t test). (B) Relative mRNA levels of Acp5, Ctsk, and Mmp9 genes were assessed using qRT-PCR (Acp5, P=0.001; Ctsk, P=0.007; Mmp9, P=0.007. Unpaired t test). (C) MDSCs were seeded on bone slices in the presence of M-CSF and RANK-L. Bone resorption pit was examined using pit assays (P=0.029, Mann–Whitney U test). (D) MDSCs sorted from naïve or arthritic SKG mice were differentiated into OCs as described above (P=0.029, Mann–Whitney U test). (E) and (F) OC-differentiating potential of M-MDSCs or G-MDSCs and bone resorption activity were assessed. (G) Expression of CD115 on MDSCs subsets was assayed by flow cytometry. (H) Transcription of Tnfrsf11a (RANK) was determined by qRT-PCR. Scale bar, 200 µm. Results shown are representative of three independent experiments. N.D., not detected.
Considering that M-MDSCs and G-MDSCs not only are phenotypically and morphologically distinct, but also have different functional characteristics [28], we further examined the OC-differentiating capacity of these two MDSC subsets. TRAP staining and pit assays showed that M-MDSCs derived from CIA mice or arthritic SKG mice, not G-MDSCs, were able to differentiate into OCs (Fig. 4E and F). Consistent with their differential OC-differentiating capacities, G-MDSCs displayed significantly lower levels of the M-CSF receptor CD115 (Fig. 4G) and the RANK-L receptor RANK (Fig. 4H), as compared with M-MDSCs. These suggest that only arthritis-associated M-MDSCs exhibit a unique potential of differentiating into functionally active OCs.
MDSC-Th17 interaction potentiates MDSC differentiation into OCs
Because RANK-L expression on Th17 cells is upregulated by MDSCs with the intrinsic OC-differentiating compacity, we address the question of whether the MDSC-Th17 interplay further promotes the differentiation of MDSCs into OCs in RA. To test this, Th17 cells were polarized in the presence of MDSC-CIA to produce MDSC-conditioned Th17 cells, which were then co-cultured with MDSCs that have been primed with M-CSF for induction of RANK expression (Supplementary Fig. S4, available at Rheumatology online). Differentiation of MDSCs into TRAP+ OCs was barely detected when co-culturing with non-conditioned Th17 cells. In sharp contrast, Th17 cells that had been pre-conditioned with MDSC-CIA significantly enhanced MDSC-derived OC differentiation (Fig. 5A–C). This increased OC formation was independent of IL-17A, as addition of exogenous IL-17A failed to induce OC differentiation from MDSCs (Fig. 5A–C). However, use of osteoprotegerin (OPG), a soluble decoy RANK-L receptor that inhibits the RANK-L/RANK signalling, abrogated the pro-osteoclastogenic activity of MDSC-CIA-conditioned Th17 cells (Fig. 5D), supporting a requirement of the RANK-L signal that is derived from Th17 cells. Additionally, this pro-osteoclastogenic activity of MDSC-conditioned Th17 cells was further validated using bone marrow-derived macrophages as OC precursors (Fig. 5E–G). Therefore, interaction with MDSCs confers Th17 cells with a pro-osteoclastogenic phenotype that in return supports RANK-L-dependent differentiation of MDSCs or other OC precursors into OCs.
Fig. 5.

MDSC-Th17 interplay facilitates MDSC differentiation into OCs
(A)–(C) Th17 cells were polarized from naïve CD4+CD62L+ T cells in the presence or absence of MDSCs from CIA mice. Th17 cells were then co-cultured with MDSCs in the presence of M-CSF and IL-2 without addition of exogenous RANK-L for 5 days. IL-17A was added as a control. Representative images of TRAP staining (A) and statistical analysis of OCs number (B) are shown (Th17 vs Conditioned Th17, P =0.029, Mann–Whitney U test). (C) Relative mRNA level of Mmp9 gene was determined by qRT-PCR (Th17 vs Conditioned Th17, P=0.006, Unpaired t test). (D) MDSCs were co-cultured with MDSC-conditioned Th17 cells with or without osteoprotegerin (OPG) (P =0.029, Mann–Whitney U test). (E)–(G) Bone marrow derived macrophages (BMMφ) were co-cultured with indicated Th17 cells in the presence of M-CSF and IL-2 for 5 days. OCs number (F) and relative Mmp9 level (G) were determined [Th17 vs Conditioned Th17: P=0.004 (F); P=0.004 (G). Unpaired t test]. Scale bar, 200 µm. Data shown represent three independent experiments. N.D., not detected.
M-MDSCs are involved in osteoclastogenesis and osteolysis in arthritic mice
We next examined the pathogenic role of MDSCs in inflammatory arthritis using 5-Fluorouracil (5-Fu) that can selectively remove MDSCs without affecting other immune cell types such as T cells, natural killer cells, dendritic cells or B cells [19]. Arthritic mice were administrated with 5-Fu at a dose of 50 mg/kg every 3 days starting at the disease onset. We showed that 5-Fu treatment selectively depleted M-MDSCs, not G-MDSCs or other immune cells (Fig. 6A, Supplementary Fig. S5, available at Rheumatology online). Removal of M-MDSCs significantly reduced the severity of arthritis, indicated by decreased swelling of inflamed paws (Fig. 6B). Consistent with our previous report [12], absence of M-MDSCs resulted in decreased infiltration of Th17 cells (Supplementary Fig. S6, available at Rheumatology online). Ablation of M-MDSCs also caused a significant reduction of Tnfsf11 gene transcription in inflamed paws (Fig. 6C). Histology analysis of the paws in M-MDSC-depleted mice showed reduced infiltration of inflammatory cells, ameliorated cartilage damage, and decreased numbers of OCs (Fig. 6D–F). The reduced osteoclastogenesis in the absence of M-MDSCs was also supported by decreased transcription of Acp5, Ctsk or Mmp9 genes in the inflamed paws (Supplementary Fig. S7, available at Rheumatology online).
Fig. 6.

Arthritis-associated M-MDSCs differentiate into functional OCs in vivo
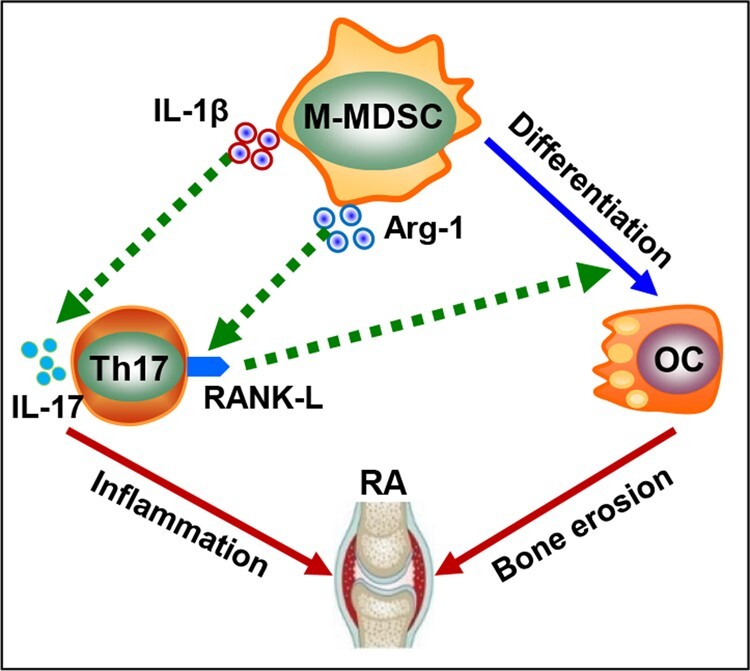
(A) CIA mice (n = 8) were administrated i.p. with 50 mg/kg 5-Fu or PBS every 3 days from day 30 after the collagen immunization. Frequency of MDSC subsets in peripheral blood was assessed on day 50. N.S., not significant (M-MDSC, P<0.001; G-MDSC, P=0.505. Mann–Whitney U test). (B) Clinical scores during arthritic development were recorded (Vehicle vs 5-Fu, P=0.003, Generalized Estimating Equations). (C) mRNA levels of Tnfsf11 gene in inflamed paws were analysed by qRT-PCR (Vehicle vs Naive, P<0.001; 5-Fu vs Vehicle, P<0.001. One-way ANOVA with Fisher LSD test). (D) Pathology of paws was assayed by H&E, toluidine blue or TRAP staining. Representative images of TRAP staining in proximal metatarsal epiphysis are shown. Arrows indicate infiltrated inflammatory cells (top panel, H&E), cartilage damage (middle panel, toluidine blue) and TRAP+ OCs (bottom panel, TRAP+), respectively. (E) Pathology scores of bone destruction were assessed. (F) Numbers of TRAP+ cells in the sections were quantified (vehicle vs 5-Fu, P=0.007. Mann–Whitney U test). (G) Arthritic mice depleted of MDSCs by 5-Fu were injected with PBS, M-MDSCs or G-MDSCs isolated from CIA mice into the tibia (n = 5). TRAP or toluidine blue staining were performed to evaluate OC differentiation and cartilage damage, respectively. Representative images of TRAP staining in proximal tibial epiphysis are shown. Arrow indicates cartilage damage as shown by toluidine blue staining. (H) Number of TRAP+ cells in G was quantified. Scale bar = 100 µm. (PBS vs M-MDSC, P=0.017; PBS vs G-MDSC, P=0.865. Kruskal-Wallis test followed by Dunn’s multiple comparisons). Data represent three independent experiments. (I) Proposed model for the role of MDSC-Th17 interplay in RA. M-MDSCs with inherent OC-differentiating capability display versatile functions during their interaction with Th17 cells. While promoting Th17 differentiation via pro-inflammatory IL-1β signalling, M-MDSCs can also upregulate RANK-L on Th17 cells in an arginase 1-dependent fashion. Th17-derived RANK-L, in return, further drives programing of M-MDSCs into bone-destructing OCs.
Lastly, we performed adoptive transfer of MDSCs to confirm MDSC-promoted osteoclastogenesis and bone resorption in vivo. Type II collagen/CFA immunized mice were first treated with 5-Fu to remove endogenous MDSCs, followed by intramedullary injection of CIA mice-derived M-MDSCs or G-MDSCs into the tibia of recipient mice. Elevation in the numbers of OCs were only seen in the tibia of mice that had been transferred with M-MDSCs, not G-MDSCs (Fig. 6G and H). This increased osteoclastogenesis in mice receiving M-MDSCs correlated with significantly aggravated cartilage damage (Fig. 6G).
Discussion
Despite their intrinsic immunosuppressive activity, our previous work established a highly pro-inflammatory feature of MDSCs in sustaining inflammation by amplifying a Th17 response in autoimmune disorders including RA [12–14]. Our research in the present study reveals a novel pro-osteoclastogenic activity of MDSCs in RA, highlighting their functional plasticity and versatility in disease pathogenesis. More importantly, we uncover a previously undefined signalling feedback loop engaged during the MDSC-Th17 interplay, which contributes to the dysregulation of osteoclastogenesis and bone destruction in RA.
To the best of our knowledge, this is the first report that MDSCs or M-MDSCs in patients with RA not only positively correlate with clinical bone erosion scores or markers, but also acquire the increased OC-differentiating capacity. This is further supported by our finding that M-MDSCs from patients with high disease activity were more readily to differentiate into bone resorption capable OCs than those from patients with low disease activity. It is likely that the inflammatory milieu governs the OC-differentiating potential of MDSCs. Our clinical data are consistent with the recent reports of MDSCs as OC progenitors in mouse models of cancer [29] and arthritis [30]. Furthermore, using two mouse arthritic models (i.e. CIA, SKG), we demonstrate that M-MDSCs display a superior OC-differentiating capacity as compared with G-MDSCs, which may be attributed to the high expression of CD115 and RANK required for OC programing. These results advance the understanding of the functional distinctions of these two MDSC subsets in RA-associated osteoclastogenesis.
In addition to the inherent OC-differentiating property of RA-associated human MDSCs, we have made an important finding in this work that MDSCs, particularly M-MDSCs, can program human Th17 cells towards a pro-osteoclastogenic phenotype during their interaction, indicated by upregulation of RANK-L in autologous Th17 cells. Moreover, this Th17 cell-derived RANK-L signal is functionally significant, because Th17 cells conditioned by RA-associated MDSCs greatly potentiate the MDSC differentiation into OCs. Our extensive studies in arthritic mice (i.e. CIA, SKG) also similarly elucidate MDSC-enhanced production of RANK-L by Th17 cells, which can further promote the MDSC-derived OC differentiation dependent on the RANK signalling. Intriguingly, we show that the elevated pro-osteoclastogenetic signal on Th17 cells appears to drive the differentiation of OCs from their other known precursors, such as macrophages [12]. Given that Th17 cells have been shown to promote arthritic development by exaggerating inflammation [31–33] and promoting bone erosion [21, 22], the results from this study may have broad implications in understanding of an intimate relationship between inflammation and osteoclastogenesis in RA or other autoimmune conditions.
While retaining immunosuppressive activity, mouse MDSCs display reduced suppressive activity on T cells during arthritic progression (i.e. CIA) [12]. These MDSCs associated with autoimmune arthritis [12] or other inflammatory disorder [13, 14] have acquired a pro-inflammatory phenotype indicated by their production of cytokines (e.g. IL-1β, TNF-α) as well as pro-osteoclastogenic activities as reported in our current study. This functional plasticity is likely to be dictated by the distinct inflammatory signals present in the inflamed sites. We postulate that these pro-inflammatory and pro-osteoclastogenic functions become dominant over their intrinsic T-cell suppressive activity, and collaboratively perpetuate multiple pro-arthritic processes.
The IL-1β signalling was previously shown by us and others to primarily mediate MDSC-enhanced Th17 response under several autoimmune conditions [12, 14, 15]. Intriguingly, the ability of MDSCs to enhance the pro-osteoclastogenic activity of Th17 cells involves arginase 1 not IL-1β signalling. Although arginase 1 has been reported to mediate the immune suppressive function of cancer-associated MDSCs, the immune suppressive activity of MDSCs-CIA does not appear to be arginase 1 dependent [12]. The functional significance of arginase 1 is also supported by the fact that arginase 1 is mostly expressed by arthritis-associated MDSCs, not by those from naïve mice, which corresponds to their differential RANK-L-promoting capabilities. In view of the involvement of arginase 1 in regulating pro-osteoclastogenic and/or pro-inflammatory effect of MDSCs, therapeutically targeting arginase 1 may help alleviate joint inflammation and bone erosion in patients with RA. Given the well-documented role of the IL-6 signalling in pathogenesis of human RA [34] and IL-6-induced up-regulation of arginase 1 in MDSCs from the SLE patients [13], it is possible that the therapeutic benefits of the IL-6 inhibitor in RA treatment may also be partially attributed to inhibition of arginase activity in MDSCs.
In the present study, we have also experimentally established a functional linkage of M-MDSCs with osteoclastogenesis and bone erosion in arthritic mice using both gain- and loss-of-function approaches. Depletion of M-MDSCs reduces numbers of OCs, cartilage damage and immune infiltration. However, transfer of M-MDSCs, not G-MDSCs, aggravates cartilage damage associated with elevation of OCs in inflamed paws. These striking results together with our recent reports [12–14] indicate that MDSCs, often expanded in autoimmune arthritis, not only promote polarization of inflammatory Th17 cells, but also define their pro-osteoclastogenic phenotype. However, Fujii et al. reported that adoptive transfer of MDSCs suppressed progression of collagen-induced arthritis in DBA/1 mice [35]. This discrepancy may be caused by the methodological differences (e.g. timing or number of cells for transfer). Indeed, the pathogenic role of MDSCs in promoting inflammation and arthritic progression has been confirmed by an independent study [15].
Our previous work established a highly pro-inflammatory role of MDSCs for exacerbating inflammation in autoimmune disorders [12–14]. Our current work identifies novel pro-osteoclastogenic function of MDSCs acquired in RA and elucidates an under-appreciated osteoimmune pathway involving complex interactions between M-MDSCs and Th17 cells (Fig. 6I). In addition to driving Th17 polarization and IL-17 production, as we previously documented [12], MDSCs or M-MDSCs skew Th17 cells towards a pro-osteoclastogenic phenotype. Production of RANK-L by Th17 cells can serve as a feedback loop to program M-MDSCs or other OC precursors into OCs. These reciprocal and distinctive actions of MDSCs and Th17 cells collaboratively perpetuate multiple pathogenic processes, including but not limited to aggravated inflammation and osteolysis. Targeting the pro-arthritic MDSC-Th17 axis based on a precise understanding of the nature of this cellular crosstalk may help alleviate disease symptoms by concurrently antagonizing abnormal inflammation as well as osteoclastogenesis in RA.
Supplementary Material
Acknowledgements
S.C., C.G., J.L. and X-Y.W. designed the experiments; X-Y.W. and J.L. supervised the study; S.C., C.G., R.W., Z.F., D.Z. and Z.L. performed experiments. R.W., L.W., S.Z., J.Z., D.Z., F.C. and J.X. collected patient samples and clinical data. S.C., C.G., R.W., Z.L., X.C., C.M.W., X-Y.W. and J.L. analysed data. S.C., C.G., X-Y.W. and J.L. wrote the manuscript.
Funding: This work was supported in part by funding from the National Natural Science Foundation of China (Nos. 81774091, 81973650, 81573730, 81703783 and 81803932), Virginia Commonwealth University Research Development Funds and US Department of Defense Award (W81XWH1910538). Histology service and products in support of the research project were generated by the Virginia Commonwealth University Cancer Mouse Models Core Laboratory, supported, in part, with funding to the Massey Cancer Center from NIH-NCI Cancer Center Support Grant P30 CA016059. Flow cytometry service from VCU Massey Cancer Center Flow Cytometry Resource Core was supported in part by NIH Grant P30CA16059.
Disclosure statement: The authors have declared no conflicts of interest.
Data availability statement
All data relevant to the present study are included in the article or uploaded as online supplementary information. All data are available upon request.
Supplementary data
Supplementary data are available at Rheumatology online.
References
- 1. McInnes IB, Schett G.. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017;389:2328–37. [DOI] [PubMed] [Google Scholar]
- 2. Udagawa N, Kotake S, Kamatani N, Takahashi N, Suda T.. The molecular mechanism of osteoclastogenesis in rheumatoid arthritis. Arthritis Res 2002;4:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanaka S. Emerging anti-osteoclast therapy for rheumatoid arthritis. J Orthop Sci 2018;23:717–21. [DOI] [PubMed] [Google Scholar]
- 4. Udagawa N, Takahashi N, Akatsu T. et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci USA 1990;87:7260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J, Sarosi I, Yan XQ. et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA 2000;97:1566–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanaka S, Nakamura K, Takahasi N, Suda T.. Role of RANKL in physiological and pathological bone resorption and therapeutics targeting the RANKL-RANK signaling system. Immunol Rev 2005;208:30–49. [DOI] [PubMed] [Google Scholar]
- 7. Manjili MH, Wang XY, Abrams S.. Evolution of our understanding of myeloid regulatory cells: from MDSCs to MREGS. Front Immunol 2014;5:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Veglia F, Perego M, Gabrilovich D.. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Motallebnezhad M, Jadidi-Niaragh F, Qamsari ES. et al. The immunobiology of myeloid-derived suppressor cells in cancer. Tumour Biol 2016;37:1387–406. [DOI] [PubMed] [Google Scholar]
- 10. Bruchard M, Mignot G, Derangere V. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 2013;19:57–64. [DOI] [PubMed] [Google Scholar]
- 11. Obermajer N, Wong JL, Edwards RP. et al. Induction and stability of human Th17 cells require endogenous NOS2 and cGMP-dependent NO signaling. J Exp Med 2013;210:1433–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guo C, Hu F, Yi H. et al. Myeloid-derived suppressor cells have a proinflammatory role in the pathogenesis of autoimmune arthritis. Ann Rheum Dis 2016;75:278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu H, Zhen Y, Ma Z. et al. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci Transl Med 2016;8:331ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yi H, Guo C, Yu X, Zuo D, Wang XY.. Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol 2012;189:4295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang H, Wang S, Huang Y. et al. Myeloid-derived suppressor cells are proinflammatory and regulate collagen-induced arthritis through manipulating Th17 cell differentiation. Clin Immunol 2015;157:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prevoo ML, van 't Hof MA, Kuper HH. et al. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 17. Szkudlarek M, Klarlund M, Narvestad E. et al. Ultrasonography of the metacarpophalangeal and proximal interphalangeal joints in rheumatoid arthritis: a comparison with magnetic resonance imaging, conventional radiography and clinical examination. Arthritis Res Ther 2006;8:R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Szkudlarek M, Court-Payen M, Jacobsen S. et al. Interobserver agreement in ultrasonography of the finger and toe joints in rheumatoid arthritis. Arthritis Rheum 2003;48:955–62. [DOI] [PubMed] [Google Scholar]
- 19. Vincent J, Mignot G, Chalmin F. et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 2010;70:3052–61. [DOI] [PubMed] [Google Scholar]
- 20. Inglis JJ, Simelyte E, McCann FE, Criado G, Williams RO.. Protocol for the induction of arthritis in C57BL/6 mice. Nat Protoc 2008;3:612–8. [DOI] [PubMed] [Google Scholar]
- 21. Kotake S, Udagawa N, Takahashi N. et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 1999;103:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato K, Suematsu A, Okamoto K. et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006;203:2673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Janckila AJ, Neustadt DH, Yam LT.. Significance of serum TRACP in rheumatoid arthritis. J Bone Miner Res 2008;23:1287–95. [DOI] [PubMed] [Google Scholar]
- 24. Nagase Y, Tanaka S.. [Biomarker of bone destruction]. Clin Cal 2012;22:199–204. [PubMed] [Google Scholar]
- 25. Tenu JP, Lepoivre M, Moali C. et al. Effects of the new arginase inhibitor N(omega)-hydroxy-nor-L-arginine on NO synthase activity in murine macrophages. Nitric Oxide 1999;3:427–38. [DOI] [PubMed] [Google Scholar]
- 26. Moore WM, Webber RK, Jerome GM. et al. L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J Med Chem 1994;37:3886–8. [DOI] [PubMed] [Google Scholar]
- 27. Sakaguchi N, Takahashi T, Hata H. et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 2003;426:454–60. [DOI] [PubMed] [Google Scholar]
- 28. Bronte V, Brandau S, Chen SH. et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sawant A, Deshane J, Jules J. et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 2013;73:672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang H, Huang Y, Wang S. et al. Myeloid-derived suppressor cells contribute to bone erosion in collagen-induced arthritis by differentiating to osteoclasts. J Autoimmun 2015;65:82–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakae S, Nambu A, Sudo K, Iwakura Y.. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 2003;171:6173–7. [DOI] [PubMed] [Google Scholar]
- 32. Tesmer LA, Lundy SK, Sarkar S, Fox DA.. Th17 cells in human disease. Immunol Rev 2008;223:87–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Korn T, Bettelli E, Oukka M, Kuchroo VK.. IL-17 and Th17 cells. Annu Rev Immunol 2009;27:485–517. [DOI] [PubMed] [Google Scholar]
- 34. Kang S, Tanaka T, Narazaki M, Kishimoto T.. Targeting interleukin-6 signaling in clinic. Immunity 2019;50:1007–23. [DOI] [PubMed] [Google Scholar]
- 35. Fujii W, Ashihara E, Hirai H. et al. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J Immunol 2013;191:1073–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the present study are included in the article or uploaded as online supplementary information. All data are available upon request.