

Abstract
A major goal of current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccine efforts is to elicit antibody responses that confer protection. Mapping the epitope targets of the SARS-CoV-2 antibody response is critical for vaccine design, diagnostics, and development of therapeutics. Here, we develop a pan-coronavirus phage display library to map antibody binding sites at high resolution within the complete viral proteomes of all known human-infecting coronaviruses in patients with mild or moderate/severe coronavirus disease 2019 (COVID-19). We find that the majority of immune responses to SARS-CoV-2 are targeted to the spike protein, nucleocapsid, and ORF1ab and include sites of mutation in current variants of concern. Some epitopes are identified in the majority of samples, while others are rare, and we find variation in the number of epitopes targeted between individuals. We find low levels of SARS-CoV-2 cross-reactivity in individuals with no exposure to the virus and significant cross-reactivity with endemic human coronaviruses (CoVs) in convalescent sera from patients with COVID-19.
Keywords: SARS-CoV-2, COVID-19, epitopes, phage-display, cross-reactivity, variants, serology
Graphical abstract
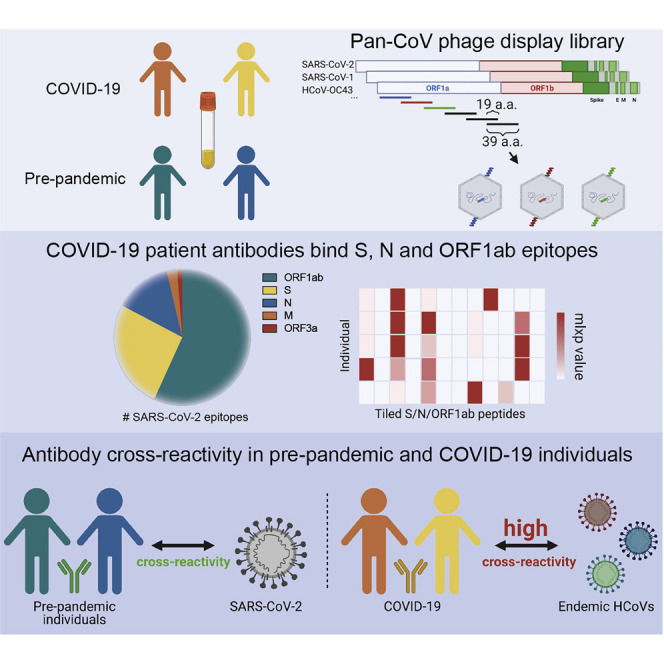
Stoddard et al. map common and rare coronavirus epitopes in individuals with mild or moderate/severe COVID-19 and healthy, pre-pandemic individuals. They find a subset of epitopes with residues that are mutated in SARS-CoV-2 variants of concern and show evidence for cross-reactivity between SARS-CoV-2 and commonly circulating human coronaviruses.
Introduction
A novel betacoronavirus, SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2), was transmitted into humans in late 2019 and has led to widespread infection, morbidity, and mortality across the globe (Wu et al., 2020). The disease caused by SARS-CoV-2 infection, coronavirus disease 2019 (COVID-19), is characterized by a striking diversity in clinical presentation, ranging from asymptomatic or mild disease to severe pneumonia and death. A number of studies have begun to address the role of the adaptive immune response in patients infected with SARS-CoV-2, but the repertoire of epitope targets linked to infection is only beginning to be comprehensively defined.
Coronaviruses (CoVs) have large, ∼30-kb non-segmented genomes consisting of virus-specific accessory proteins and several universal open reading frames (ORFs), including spike (S), membrane (M), nucleocapsid (N), envelope (E), and ORF1ab, which code for a multitude of nonstructural proteins (Chan et al., 2020; Cui et al., 2019). The S glycoprotein is highly immunogenic in SARS-CoV-2 infections, as well as for infections with the six other human CoVs (HCoVs) that are endemic and associated with the common cold (HCoV-OC43, HCoV-HKU1, HCoV-NL63, and HCoV-229E) or are the cause of more confined but highly pathogenic outbreaks in humans (SARS-CoV and MERS-CoV). The S protein decorates the surface of all CoVs and mediates viral entry (Shang et al., 2020). The SARS-CoV-2 S protein shares varying degrees of homology with other circulating CoVs, ranging from 28% amino acid identity with the endemic HCoV-OC43 to up to 76% identity with the highly virulent SARS-CoV (Walls et al., 2020; Wang et al., 2020a). Because of its surface exposure and role in infectivity, the S protein has been a major focus of vaccine development and recent efforts to isolate potent neutralizing antibodies targeting SARS-CoV-2 (Chi et al., 2020; Pinto et al., 2020).
While many vaccines are thought to protect by virus neutralization, antibodies that target viruses through mechanisms other than neutralization—often referred to as non-neutralizing antibodies—have been correlated with improved clinical outcomes for a variety of viruses, including HIV, influenza, and Ebola (Lee and Kent, 2018; Mayr et al., 2017; Padilla-Quirarte et al., 2019; Saphire et al., 2018). Antibody responses to non-S CoV proteins have been detected previously, including non-neutralizing responses to the N protein of SARS-CoV, which is involved in genome packaging and is found in the mature virion core (Dutta et al., 2020). Interestingly, immune responses to the N protein of SARS-CoV-2 have recently been linked to poor clinical outcomes (Atyeo et al., 2020). Despite mounting evidence that the SARS-CoV-2 N protein may be highly antigenic in the context of COVID-19, there has been limited effort to fully characterize antibody responses mediated by N or the other non-S ORFs that are expressed during SARS-CoV-2 infection.
Sequence homology between SARS-CoV-2 and other circulating HCoVs increases the likelihood for cross-reactive antibody responses resulting from prior infection or vaccination. The N protein and other nonstructural SARS-CoV-2 proteins are often more highly conserved than the S protein and thus may be targets for such cross-reactive non-neutralizing responses. Importantly, cross-reactive T cell responses stemming from exposure to low-pathogenic endemic HCoVs have been identified in SARS-CoV-2-unexposed individuals (Grifoni et al., 2020; Mateus et al., 2020). Additional studies aimed at the B cell immune response have identified cross-reactive antibody binding to the S proteins of SARS-CoV-2 and SARS-CoV, which share nearly 80% sequence identity genome-wide ((Lv et al., 2020); Shrock et al., 2020). Despite lower degrees of homology than to the highly pathogenic SARS-CoV, cross-reactivity against the S protein from the four commonly circulating HCoVs in COVID-19 patient sera has also been identified (Shrock et al., 2020; (Wölfel et al., 2020)). Importantly, cross-reactive viral immune responses can be either cross-protective, as in the case of influenza A and other viruses, or disease enhancing, as in the case of dengue virus and possibly SARS-CoV-2 (Arvin et al., 2020; Lee et al., 2020; Welsh et al., 2010). These divergent phenomena necessitate studies to evaluate SARS-CoV-2 antibody binding in unexposed individuals and measure sequence homology among prominent epitopes from the full genomes of all HCoVs.
In order to capture the complete repertoire of neutralizing and non-neutralizing linear epitopes targeted by antibodies generated in the presence and absence of SARS-CoV-2 infection, we used a phage display immunoprecipitation approach (Larman et al., 2011) to profile immune responses in a population of patients with COVID-19 and patients with no exposure to SARS-CoV-2. We developed a pan-CoV phage library encompassing the complete proteomes of all human-targeted CoVs and used it to immunoprecipitate antibodies from samples from patients with mild or moderate/severe COVID-19, as well as SARS-CoV-2-unexposed patients, all collected in Seattle, Washington. Notably, we detected a pool of significantly enriched peptides from the S, N, and ORF1ab polypeptides of SARS-CoV-2 in samples from patients with COVID-19, several of which contain amino acids that appear to be under selection. We also identified four cross-reactive SARS-CoV-2 peptides that were enriched in pre-pandemic, SARS-CoV-2-unexposed individuals. Finally, we found broad reactivity in patients with COVID-19 to other HCoVs and used local sequence alignment to identify three cross-reactive, minimal epitopes based on sequence homology.
Results
A pan-CoV bacteriophage library detects antibody responses across the SARS-CoV-2 proteome
We generated a phage display library composed of all seven HCoVs known to infect humans, a bat SARS-like CoV, and a set of control peptides derived from the HIV-1 envelope sequence (Figure 1 ). Oligonucleotide sequences were designed to cover complete CoV genomes in 39 amino acid tiles with 19 amino acid overlaps that were then cloned into T7 phage, amplified, and used in subsequent assays. This process was repeated to generate a replicate phage library from an independent oligonucleotide pool. We deeply sequenced both independent phage libraries and found similarly high coverage across the sequences included in the libraries, with >98% of expected sequences detected (Figure S1A).
Figure 1.

Summary of development of the pan-CoV T7 phage library and sample screening
Left panel: virus species and strains that comprise the pan-CoV phage library used in the study are listed. Right panel: summary of samples from COVID-19- or SARS-CoV-2-unexposed patients. The pan-CoV phage library and samples were combined in a plate-based immunoprecipitation assay, and phage DNA was isolated for downstream sequencing and analysis. Additional sample information can be found in Tables 1 and S1.
A total of 19 plasma or serum samples from patients with either mild or moderate/severe laboratory-confirmed SARS-CoV-2 infections (termed COVID-19 patients) were collected in Seattle, Washington as part of the Hospitalized and Ambulatory Adults with Respiratory Viral Infections (HAARVI) study or as residual clinical samples from hospital labs in Seattle. Samples from patients with mild COVID-19 were collected at ∼30 days post-symptom onset, and all moderate/severe samples were collected between 8 and 33 days post-symptom onset. Samples from patients with endemic (non-SARS-CoV-2) HCoV infections were collected as part of the HAARVI study or were residual clinical samples from Harborview Medical Center (Seattle, Washington, USA). Archived samples collected from Seattle individuals before the pandemic were used as unexposed SARS-CoV-2-negative samples. Additional sample demographic information is found in Tables 1 and S1.
Table 1.
Sample information and neutralization activity
| Sample ID | Patient status | Days PSO | Age (years) | Sex | NT50 |
|---|---|---|---|---|---|
| 32 | COVID-19 moderate/severe | 15 | 31 | F | 1,691 |
| 33 | COVID-19 moderate/severe | 15 | 56 | M | 2,536 |
| 34 | COVID-19 moderate/severe | 11 | 56 | M | 5,353 |
| 35 | COVID-19 moderate/severe | 8 | 76 | M | 1,701 |
| 53 | COVID-19 moderate/severe | 33 | 65 | F | 1,116 |
| 36 | COVID-19 mild | 27 | 47 | F | 237 |
| 37 | COVID-19 mild | 31 | 43 | F | 22 |
| 38 | COVID-19 mild | 29 | 65 | M | 248 |
| 39 | COVID-19 mild | 31 | 29 | M | 217 |
| 50 | COVID-19 mild | 31 | 48 | F | 251 |
| 56 | COVID-19 mild | 29 | 22 | M | 28 |
| 58 | COVID-19 mild | 31 | 31 | F | 104 |
| 64 | COVID-19 mild | 34 | 28 | F | 227 |
| 68 | COVID-19 mild | 34 | 30 | M | 205 |
| 70 | COVID-19 mild | 26 | 36 | F | 78 |
| 72 | COVID-19 mild | 28 | 65 | M | 400 |
| 74 | COVID-19 mild | 26 | 65 | F | 130 |
| 76 | COVID-19 mild | 48 | 52 | F | 208 |
| 82 | COVID-19 mild | 43 | 29 | M | 212 |
| 182 | endemic CoV+ | NA | 61 | M | <20 |
| 183 | endemic CoV+ | NA | NA | NA | <20 |
| 41 | healthy adult | NA | NA | NA | <20 |
| 42 | healthy adult | NA | NA | NA | <20 |
| 43 | healthy adult | NA | NA | NA | <20 |
| 44 | healthy adult | NA | NA | NA | 993 |
| 45 | healthy adult | NA | NA | NA | <20 |
Samples were deidentified. F, female; M, male; NA, not available; PSO, post-symptom onset; NT50, reciprocal dilution at which viral infection is inhibited by 50%.
To evaluate the neutralization capacity of the samples in our cohort, we measured neutralization titers for all samples using a pseudotyped lentiviral particle expressing SARS-CoV-2 S protein (Table 1, “NT50” column). Individuals with moderate/severe COVID-19 had the highest neutralizing antibody titers, which is consistent with other studies (Liu et al., 2020; Wang et al., 2020b). In individuals with mild COVID-19, there was a marked reduction in neutralization titer (NT50) compared to moderate/severe individuals, and neutralizing titers varied across an order of magnitude, suggesting that our cohort would represent a range of both neutralizing and non-neutralizing antibody responses. Surprisingly, one individual who was not diagnosed with COVID-19 did have neutralizing activity against SARS-CoV-2. Follow-up samples from this case were not available, and thus, we were unable to further assess their responses.
All samples were tested in a phage-based immunoprecipitation assay in which DNA from phage-antibody complexes was PCR amplified, and multiplexed samples were deep sequenced to determine enrichment of individual CoV peptides. We applied the following criteria to determine which samples to include in our downstream analyses: (1) a sample must have had a pairwise cross-correlation of at least 0.5 (Pearson’s R) between two technical (within-assay) replicates, and (2) a sample must have satisfied condition 1 in experiments conducted with both independent batches of the phage library (Figure S1A, libraries 1 and 2).
We performed a qualitative assessment of the SARS-CoV-2 epitope profile in COVID-19 patients by examining counts per million (CPM) from all SARS-CoV-2 ORFs. We detected signal for epitopes derived from all possible ORFs but found significantly higher magnitude in S, N, and ORF1ab (Figure 2 ). Signal was also detected for ORF3a and M, but at a much lower magnitude, with even lower signal for peptides enriched from the other proteins (note scale differences in Figure 2). In order to evaluate the significance of epitope enrichment quantitatively, we modeled enrichment for all peptides from all samples along with a pool of mock-immunoprecipitation samples to account for nonspecific peptide binding. We fit peptides to a gamma-Poisson model, in which each sample-peptide pairing was assigned a minus log10 p value (mlxp) (Figure 3 A). We exploited the HIV-1 envelope sequences in the pan-CoV library to estimate the false positive rate (FPR) of nonspecific binding peptides. Next, we determined the mlxp cutoff corresponding to a FPR of 0.05 and identified 2,689 and 4,604 sample-peptide pairs, or “hits,” from phage libraries 1 and 2, respectively (Figures 3A and S1B). Across the two replicate experiments, there were 933 intersecting hits, 456 of which were unique peptides, where non-unique peptides were a result of sequence overlap between CoVs.
Figure 2.

SARS-CoV-2 peptide enrichment based on raw counts per million (CPM)
Individual panels showing enrichment among all COVID-19 patient samples for peptides along the lengths of nine SARS-CoV-2 ORFs. Panel rows are in order of increasing maximum response from top to bottom. Note the scales also increase in each row, indicating higher enrichment of the identified peptides. Bars are segmented by color for each sample included in the analysis, as depicted in the legend.
Figure 3.

Results from global fit of all sample-peptide pairs with applied mlxp cutoff
(A) Data-processing scheme. Samples were tested with two separate phage libraries (library 1 and library 2; Figure S1). Peptide enrichment was scored using a gamma-Poisson model, and data were curated using a cutoff corresponding to FPR 0.05 (Figure S1).
(B) Proportion of SARS-CoV-2 epitopes derived from individual proteins in all patient samples tested. Numbers indicate the total enriched SARS-CoV-2 epitopes from each ORF.
(C) Proportions in (B) normalized with respect to polypeptide length.
(D) Epitope counts across COVID-19 patient samples for SARS-CoV-2 only. Bars are further sectioned by SARS-CoV-2 ORF, indicated to the right.
(E) Fraction of total epitopes arising from the S protein, calculated for moderate/severe and mild samples (number of S epitopes/number of total epitopes). p value was calculated using a two-tailed unpaired Welch’s t test (n = 5, moderate/severe COVID-19, n = 14 mild COVID-19; bars represent median and interquartile range).
Patterns of SARS-CoV-2 immune response at epitope resolution in patients with COVID-19 and unexposed individuals
After fitting enriched immunoprecipitated peptides, we detected significant responses in just five of the nine SARS-CoV-2 ORFs (Figure 3B). We identified the most reactivity in ORF1ab, but after normalizing the number of reactive sequences by the ORF length, we found that the S and N proteins had the highest density of epitopes (Figure 3C). Sparse responses to SARS-CoV-2 peptides were also detected within the M protein and ORF3a (Figures 3B and 3C). COVID-19 patient samples displayed variability in total abundance of reactive epitopes, ranging from 2 to 24 reactive peptides in a given sample (Figure 3D). The distribution of responses across the different ORFs also differed among individuals.
All COVID-19 patient samples were reactive to epitopes from the S protein, while two moderate/severe COVID-19 samples, both less than 12 days post-symptom onset, were reactive solely to the S protein and no other proteins (Figure 3D). The proportion of the total epitope response arising from the S protein was modestly higher in the five moderate/severe samples than in the mild samples (p = 0.07, Welch’s unpaired t test), suggesting that antibodies targeting the S protein are dominant in cases of moderate/severe COVID-19 in our cohort (Figure 3E).
To test whether the dominant S response was a function of sampling kinetics, we evaluated longitudinal samples from the individuals with moderate/severe COVID-19. In general, there was variation in the fraction of S epitopes targeted over time, with increases in some cases and decreases in others (Figure S2A). When we compared the number of positive epitopes from S versus N only, the number from S remained higher over time (Figure S2B). This result, which focused on the number of epitopes targeted within the N and S proteins, contrasts somewhat with studies showing total N protein antibody titers are higher than S protein antibody titers in individuals with severe COVID-19 (Atyeo et al., 2020; Röltgen et al., 2020). However, it is important to note that epitope number is a measure of the breadth of antibody responses to a given antigen and may not reflect the concentration of circulating antibody to that antigen.
We next examined the epitope profiles of all samples (both COVID-19 and SARS-CoV-2 unexposed) within the three dominant antigens, S, N, and ORF1ab. Within the S protein, we identified three key regions in which significant signal was detected, and these three regions were detected across the majority of individuals (Figure 4 A; Table 2 ). The most common epitope was S_1,121–1,179 (composed of two adjacent peptides in our library that overlap by 19 amino acids), which spans a portion of the second heptad repeat (HR2) in the S2 subunit. In total, 84% (16/19) of COVID-19 samples were reactive to this epitope. The second region, S_801–839, spans the fusion peptide (FP) and includes the S2′ cleavage site. This region was reactive in 78% (15/19) of the COVID-19 samples. The third region, S_541–579 is located at the C-terminal end of the S1 subunit, immediately after the receptor binding domain (RBD) and upstream of the S1/S2 cleavage site. This epitope was reactive in 68% (13/19) of SARS-CoV-2-positive samples. Interestingly, S_541–579 was reactive in 100% (5/5) of the moderate/severe COVID-19 samples tested but only 64% (9/14) of mild samples (Figure 4A). While not statistically significant within our current sample size (p = 0.68, Fisher’s exact test), this result may be suggestive of a correlation between COVID-19 severity and epitope patterning that should be explored in larger studies examining correlates of disease. Finally, we identified 13 additional dispersed S protein epitopes that were rarer among individuals, including a sequence spanning the S1/S2 cleavage site (S_661–699), suggesting antibody recognition of pre-processed S protein (Table 2). To visualize the three-dimensional positioning of significant S epitopes, we mapped all epitopes that were present in two or more individuals onto the cryoelectron microscopy structure of the pre-fusion S trimer in the closed conformation (Figure S3) (Walls et al., 2020). Significant epitopes we identified in this study are largely surface exposed on the pre-fusion S structure, suggesting they are readily accessible to antibodies before the virus interacts with host cells.
Figure 4.

SARS-CoV-2 epitope profiles for dominant antigens
(A–C) Location of significantly enriched epitopes across the S protein (A), N (B), and ORF1ab (C). Profiles for patients with COVID-19 are highlighted in gray (moderate/severe COVID-19) and purple (mild COVID-19). The remaining profiles are from SARS-CoV-2-unexposed individuals with confirmed endemic HCoV exposure (yellow) or healthy individuals (colorless). Log(mlxp) values are indicated by the red gradient, shown to the right of the maps. Protein domain architecture for each antigen is above the heatmap, with amino acid positions indicated.
Table 2.
Top SARS-CoV-2 epitopes present in two or more individuals
| Protein | Amino acids | Sequence | Number of COVID-19 samples/19 | Number of unexposed samples/7 | Mutation (variant) |
|---|---|---|---|---|---|
| Spike (S) | |||||
| S | 1,121–1,159a | FVSGNCDVVIGIVNNTVYDPLQPELDSFKEELDKYFKNH | 16 | 0 | |
| S | 1,141–1,179a | LQPELDSFKEELDKYFKNHTSPDVDLGDISGINASVVNI | 16 | 0 | |
| S | 801–839a | NFSQILPDPSKPSKRSFIEDLLFNKVTLADAGFIKQYGD | 15 | 0 | |
| S | 541–579 | FNFNGLTGTGVLTESNKKFLPFQQFGRDIADTTDAVRDP | 13 | 0 | A570D (B.1.1.7) |
| S | 621–659 | PVAIHADQLTPTWRVYSTGSNVFQTRAGCLIGAEHVNNS | 4 | 0 | H655Y (P.1) |
| S | 661–699 | ECDIPIGAGICASYQTQTNSPRRARSVASQSIIAYTMSL | 4 | 0 | P681H (B.1.1.7) |
| S | 761–799 | TQLNRALTGIAVEQDKNTQEVFAQVKQIYKTPPIKDFGG | 4 | 0 | T761I (B.1.1.7) |
| S | 281–319 | ENGTITDAVDCALDPLSETKCTLKSFTVEKGIYQTSNFR | 3 | 0 | |
| S | 521–559 | PATVCGPKKSTNLVKNKCVNFNFNGLTGTGVLTESNKKF | 3 | 0 | |
| S | 561–599 | PFQQFGRDIADTTDAVRDPQTLEILDITPCSFGGVSVIT | 3 | 0 | A570D (B.1.1.7) |
| S | 641–679 | NVFQTRAGCLIGAEHVNNSYECDIPIGAGICASYQTQTN | 3 | 0 | H655Y (P.1) |
| S | 781–819 | VFAQVKQIYKTPPIKDFGGFNFSQILPDPSKPSKRSFIE | 3 | 0 | |
| S | 1,161–1,199 | SPDVDLGDISGINASVVNIQKEIDRLNEVAKNLNESLID | 3 | 0 | |
| S | 1,235–1,273 | CCMTSCCSCLKGCCSCGSCCKFDEDDSEPVLKGVKLHYT | 3 | 0 | |
| S | 21–59b | RTQLPPAYTNSFTRGVYYPDKVFRSSVLHSTQDLFLPFF | 1 | 1 | P26S (P.1) |
| S | 261–299 | GAAAYYVGYLQPRTFLLKYNENGTITDAVDCALDPLSET | 2 | 0 | |
| S | 301–339 | CTLKSFTVEKGIYQTSNFRVQPTESIVRFPNITNLCPFG | 2 | 0 | |
| Nucleocapsid (N) | |||||
| N | 141–179 | TPKDHIGTRNPANNAAIVLQLPQGTTLPKGFYAEGSRGG | 8 | 0 | |
| N | 161–199 | LPQGTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTP | 8 | 0 | |
| N | 201–239 | SSRGTSPARMAGNGGDAALALLLLDRLNQLESKMSGKGQ | 7 | 0 | S235F (B.1.1.7) T205I (B.1.351) |
| N | 381–419 | ALPQRQKKQQTVTLLPAADLDDFSKQLQQSMSSADSTQA | 7 | 0 | |
| N | 1–39b | MSDNGPQNQRNAPRITFGGPSDSTGSNQNGERSGARSKQ | 4 | 1 | D3L (B.1.1.7) |
| N | 21–59 | SDSTGSNQNGERSGARSKQRRPQGLPNNTASWFTALTQH | 3 | 0 | |
| N | 221–259 | LLLLDRLNQLESKMSGKGQQQQGQTVTKKSAAEASKKPR | 3 | 0 | S235F (B.1.1.7) |
| N | 341–379 | DKDPNFKDQVILLNKHIDAYKTFPPTEPKKDKKKKADET | 3 | 0 | |
| N | 241–279a | QQGQTVTKKSAAEASKKPRQKRTATKAYNVTQAFGRRGP | 2 | 0 | |
| ORF1ab | |||||
| ORF1ab | 1,801–1,839b | ESPFVMMSAPPAQYELKHGTFTCASEYTGNYQCGHYKHI | 4 | 2 | |
| ORF1ab | 1,961–1,999 | PDLNGDVVAIDYKHYTPSFKKGAKLLHKPIVWHVNNATN | 4 | 0 | |
| ORF1ab | 741–779 | FLEGETLPTEVLTEEVVLKTGDLQPLEQPTSEAVEAPLV | 2 | 0 | |
| ORF1ab | 5,961–5,999 | EGLCVDIPGIPKDMTYRRLISMMGFKMNYQVNGYPNMFI | 2 | 0 | |
| ORF1ab | 4,481–4,519b | LLKDCPAVAKHDFFKFRIDGDMVPHISRQRLTKYTMADL | 1 | 1 | |
Predicted cross-reactive peptides with high homology to other HCoVs (Figure 6).
Cross-reactive peptides present in SARS-CoV-2-unexposed samples.
Peptides derived from the N protein also were bound by antibodies in many individuals. The most widely reactive region was N_141–199 (Figure 4B; Table 2). This region is composed of two overlapping peptides in the pan-CoV library, derived from the N protein RNA-binding domain. Both peptides were reactive in eight individuals (six of which were identical for both overlapping peptides). We identified two additional sequences that were enriched across seven samples each: N_201–239, found upstream of the dimerization domain, and N_381-419, which is located downstream of the dimerization domain at the C terminus of the N protein. Five more epitopes that were reactive in four or fewer individuals are listed in Table 2.
The SARS-CoV-2 replicase polyprotein 1ab (ORF1ab) is composed of two overlapping reading frames that code for 16 nonstructural proteins, including the viral-RNA-dependent RNA polymerase (RdRp), that are expected to be co- and post-translationally processed based on studies of SARS-CoV (Graham et al., 2008). We detected sequences with high mlxp values throughout ORF1ab, many of which were present in only a small fraction of individuals. The two most widely reactive regions, ORF1ab_1,801–1,839 and ORF1ab_1,961–1,999, were both located within the papain-like protease (PL-PRO) sequence. Both of these sequences were reactive in four individuals with COVID-19. The remaining peptides identified from ORF1ab that were reactive in two or more individuals, including sequences from nsp2, exonuclease (ExoN), and the RdRp, are listed in Table 2. Lastly, we identified two peptides from the M protein (M_161–199 and M_181–219, which overlap by 19 aa), each present in one individual, and one peptide from ORF3a (ORF3a_237–274), present in one individual.
At the time of writing, several concerning SARS-CoV-2 variants have emerged and threaten efforts to curb viral spread and escape from immunity. Mutations present in circulating variants could prevent antibodies produced by vaccination or natural infection from binding their cognate epitopes. To evaluate whether the epitopes identified in our phage display assay contained sites with high incidence of mutation, we harnessed existing mutation frequency data from the Nextstrain database to evaluate the median and maximum entropy across all amino acids in the epitopes listed in Table 2 (Hadfield et al., 2018). We identified one epitope from the S protein (S_661–699) and three from the N protein (N_161–199, N_201–239, and N_221–259) with an entropy > 0.2, which corresponds to the top 99th percentile (Figure S4). This suggests that viral evolution could impact antibody recognition of these epitopes. We also cross-referenced the top epitopes identified in our assay with known mutations in four SARS-CoV-2 variants that are currently of high concern: B.1.1.7, B.1.351, P.1, and CAL.20C (Table 2, “Variant mutation site” column) ((Faria et al., 2021); Galloway et al., 2021; Tegally et al., 2020; Zhang et al., 2021). Several sites of mutation from the S and N proteins found in B.1.1.7, B.1.351, and P.1 were present in common epitopes identified here, but no variant mutations from ORF1ab overlapped with key epitopes from our analysis. Additionally, no sites found in the CAL.20C variant were targeted by antibodies in our phage display assay.
SARS-CoV-2 cross-reactivity with other HCoVs in unexposed individuals and individuals with COVID-19
Cross-reactivity in the viral antibody response can drive host immunity, complicate diagnostics and surveillance, and potentiate negative outcomes such as antibody-dependent enhancement. We sought to identify cross-reactive sequences between SARS-CoV-2 and other HCoVs in two ways: (1) we examined whether SARS-CoV-2 sequences were enriched in pre-pandemic, SARS-CoV-2-unexposed individuals, and (2) we examined whether non-SARS-CoV-2 endemic HCoV sequences were enriched in individuals with COVID-19.
In pre-pandemic, unexposed individuals, we found four SARS-CoV-2 peptides with significant enrichment (Figure 5 A). One cross-reactive sequence, found in the N protein (N_1–39), was detected in a sample with RT-PCR-confirmed prior endemic CoV infection, although the precise CoV species at the time of sampling was unknown. We identified two cross-reactive peptides from ORF1ab (ORF1ab_1,801–1,839 from the PL-PRO protein and ORF1ab_6,481–6,520 from the RdRp protein), which shared ∼35%–47% and ∼46%–64% amino acid sequence identity with commonly circulating HCoVs, respectively (Figure 5A). Finally, we found a fourth cross-reactive peptide from the S1 subunit of the S protein, S_21–59, which shared only ∼17% identity with HCoV-NL63 but 35.7% identity with HCoV-OC43 (Figures 5A and S3). These peptides all exhibited significantly more homology with SARS-CoV and bat SARS-like coronavirus (bat-SL-CoV), but these viruses are unlikely to have been the source of the antibody response because of the demographics associated with these individuals. Importantly, our population of pre-pandemic individuals was small, and additional studies using larger cohorts will be critical in further defining and functionally characterizing cross-reactive responses with SARS-CoV-2 in unexposed individuals.
Figure 5.

SARS-CoV-2 cross-reactivity in two populations, SARS-CoV-2 unexposed and COVID-19
(A) Four SARS-CoV-2 epitopes that were reactive in unexposed/pre-pandemic individuals are shown on the vertical axis. The heatmap displays the percentage of amino acid conservation after local alignment with representative strains of the other circulating CoVs in the pan-CoV phage library.
(B) Stacked bar plots showing CPM (cpm) for peptides from each of the viral proteins on the y axis for each of the six non-SARS-CoV-2 HCoVs in the phage library. Colors represent individual samples, as indicated by the legend on the right. Representative endemic HCoV strains used in (A) and (B) are OC43_SC0776, HKU1_Caen1, 229E_SC0865, and NL63_ChinaGD01. Protein names on y axes are identical to GenBank entries for each viral protein (see STAR Methods).
Next, we considered non-SARS-CoV-2 sequences (those from endemic HCoVs) that were enriched in individuals with COVID-19. Interestingly, we found considerable reactivity among all of the HCoVs in COVID-19 samples, suggesting either cross-stimulation of memory B cells from a prior HCoV exposure or cross-reactivity to antibodies generated by SARS-CoV-2 infection (Figure 5B). Similar to SARS-CoV-2 epitopes, responses in other HCoVs in patients with COVID-19 were mainly found in S, N, and ORF1ab (Figure 5B). The strong responses to S, N, and ORF1ab in MERS-CoV and SARS-CoV suggest that a portion of the cross-reactive responses we observe are unlikely to result from restimulation of B cells given the low likelihood of prior infections with these HCoVs in our sample population.
Within the S protein of non-SARS-CoV-2 HCoVs, the majority of significant epitopes were localized to the S2 subunit, which is consistent with higher degrees of sequence homology in S2 versus S1 among HCoVs (Figure S5) (Jaimes et al., 2020). In individuals with COVID-19, responses near the FP and HR2 regions were common across HCoVs, similar to responses seen in SARS-CoV-2. The number of responses in these regions tended to reflect the phylogenetic distance between SARS-CoV-2 and the corresponding HCoV. For example, responses to both the FP and HR2 were common in SARS-CoV, MERS-CoV, HCoV-OC43, and HCoV-HKU1, which are all species from the Betacoronavirus genus (Figures S5A, S5B, S5E, and S5F). Conversely, S2 responses were mainly isolated to the FP region in alphacoronavirus species HCoV-229E and HCoV-NL63 among people with COVID-19 (Figures S4C and S4D).
To identify cross-reactive HCoV/SARS-CoV-2 sequence pairs with particularly high homology, we conducted local pairwise alignments using the top hits from all HCoVs (including SARS-CoV-2) in individuals with COVID-19 (Figures 6 A and S6). This approach served to (1) restrict assessment of sequence homology to only those sequences that were enriched in our cohort and (2) identify minimal epitopes among conserved sequences. Using an alignment score cutoff of 55, we identified multiple SARS-CoV-2 peptides with high sequence similarity to SARS-CoV, as expected, given the higher genome-wide sequence similarity between SARS-CoV and SARS-CoV-2 (Figure 6B). In the context of SARS-CoV-2, we found two HCoV/SARS-CoV-2 sequence pairs with high homology in the S protein. SARS-CoV-2 residues S_813–839 span the FP domain and shared 100% sequence identity across five amino acids found in the betacoronaviruses HCoV-HKU1 and HCoV-OC43. Residues S_1,143–1,158, just upstream of the SARS-CoV-2 HR2 region, shared 100% sequence identity across six amino acids found in HCoV-OC43 (Figures 6C and S3). Finally, we identified a pair of reactive sequences from the N protein (N_257–279 in SARS-CoV-2) with high homology to HCoV-OC43 (Figure 6C). Interestingly, none of the ORF1ab peptides that were significantly enriched among individuals with COVID-19 in our study were highly conserved between SARS-CoV-2 and the other commonly circulating CoVs, despite the higher degree of conservation between HCoV ORF1ab sequences (Figure 6B).
Figure 6.

Homology among significant HCoV/SARS-CoV-2 sequence pairs in individuals with COVID-19
(A) Unique peptide hits from all CoVs that were present in two or more COVID-19 patient samples were subjected to Smith-Waterman local alignment. Sequences that were 100% identical between SARS-CoV-2 and the other CoVs were not included in the analysis.
(B) Peptide pairs with alignment scores >55 (Figure S5) were plotted to show percent identity. Peptide start positions from SARS-CoV-2 are listed on the x axis, and peptide start positions from the other human-infecting CoVs are listed on the y axis. Green, blue, and purple outlines match with the corresponding peptides pairs shown in (C).
(C) Local sequence alignments for the high-scoring peptide pairs in (B).
Discussion
In this study, we profiled the humoral immune response to SARS-CoV-2 proteins in individuals with COVID-19 using phage display to capture linear immunogenic peptides spanning the entire viral proteome. By screening epitopes based on binding to SARS-CoV-2 protein sequences, we isolated epitopes with potential for neutralizing and non-neutralizing activity. We identified S, N, and ORF1ab from SARS-CoV-2 as highly immunogenic and isolated important regions at the epitope level.
SARS-CoV-2 epitopes stemming from the S protein were present in the highest density of patients with COVID-19. We identified 17 epitopes within the S protein that were present in two or more individuals, spanning both the S1 and S2 subunits, with some detected in >75% of individuals (S_1,121–1,179, S_801–839, and S_541–579). The breadth of antibody responses along the length of the S protein (and the other dominant ORFs) can be used to generate hypotheses about the SARS-CoV-2 immune response. For example, four individuals harbored antibodies targeting the S1/S2 cellular furin cleavage site, suggesting that this region of the S protein may be targeted when the SARS-CoV-2 virion is not yet mature (Hoffmann et al., 2020). Despite evidence for potently neutralizing antibodies targeting the S protein RBD, we did not identify epitopes in this region, possibly due to the tendency for RBD-directed antibodies to be conformational (Ju et al., 2020); such epitopes would only be detected with the phage display method if a large enough portion of the epitope was linear and not glycosylated. However, we identified strong antibody binding to a region flanking the RBD (S_541–579); antibody interactions at this region could influence RBD conformational changes during host cell binding. We found that epitopes from the S protein were dominant in moderate/severe COVID-19 samples versus mild COVID-19 samples, suggesting a correlation between COVID-19 severity and S protein epitope profile. This result coincides with recent evidence for stronger and more broad responses to both the S and N proteins in hospitalized COVID-19 patient samples (Shrock et al., 2020).
We identified nine epitopes within the N protein that were reactive in at least two individuals, four of which were present in at least 35% of patients. The two most reactive N protein epitopes were derived from the RNA-binding domain. Epitopes derived from the nonstructural N protein may be the results of exposure of the immune system to these antigens upon cell lysis, and if they have activity, they are more likely to be non-neutralizing, owing to their sequestration away from the viral surface. However, their reactivity in our assay implies that the epitopes are accessible to antibody binding and may be useful in informing the design of new diagnostics. Epitopes isolated from ORF1ab were the most variable across patients. Of the 46 unique ORF1ab epitopes we identified, only 5 were present in two or more individuals, suggesting that ORF1ab responses are highly individual specific. The two most reactive ORF1ab epitopes were positioned in the PL-PRO sub-protein, an enzyme implicated in attenuating the host innate immune response and viral replication (Shin et al., 2020). Finally, we identified epitopes from the S and N proteins that contain residues with exceptionally high mutational entropy and a number of significant epitopes containing sites of mutation in the currently concerning variants B.1.1.7, B.1.351, and P.1. These results suggest that there is evolving selection for mutations in the epitopes identified in this study. As additional variants of concern evolve, it will be critical to evaluate the functional consequences of mutations to residues in these regions.
Our pan-CoV phage library contained sequences from all seven human-infecting CoVs, allowing us to probe responses across CoV species in COVID-19- and SARS-CoV-2-unexposed individuals. Our small cohort of SARS-CoV-2-unexposed individuals showed cross-reactivity with four sequences from SARS-CoV-2. This relatively weak response is consistent with previous reports of minimal antibody responses that recognize SARS-CoV in individuals infected with HCoV-OC43 or HCoV-229E (Chan et al., 2005). Conversely, we found that patients with COVID-19 had several antibodies that were reactive with seasonal and highly pathogenic HCoVs and that these responses were focused to S, N, and ORF1ab sequences. Cross-reactive binding to the S protein was concentrated on the S2 subunit in individuals with COVID-19, and we identified two highly conserved minimal epitopes near the FP and HR2 regions of S2, both of which have been found to be neutralizing in other cohorts (Li et al., 2020; Poh et al., 2020). Whether cross-reactive antibodies affect immune responses during SARS-CoV-2 infection is a major outstanding question in the field that will require additional study. For example, the robust response to endemic HCoVs in patients with COVID-19 raises the question of whether the responses reflect stimulation of memory B cells from a prior HCoV infection or if these sequences are simply bound by newly generated SARS-CoV-2 antibodies. The potentially cross-reactive epitopes identified here will provide a focused group of epitopes for studies using larger, longitudinal cohorts of SARS-CoV-2-unexposed individuals or individuals with specific endemic CoV diagnoses to define the prevalence and function of these responses. These homologous sequences, along with the cross-reactive sequences from unexposed individuals, may be valuable in future diagnostic and surveillance efforts aimed at discrimination of HCoV serological profiles and may inform studies assessing the possibility of antibody-mediated cross-protection or disease enhancement.
Our study included a relatively small sample size, which limits our conclusions regarding the frequency of responses or whether there were differences in the response based on disease severity. Nevertheless, in light of the ongoing SARS-CoV-2 pandemic, our data reveal viral proteome-wide antibody binding signatures in patients with confirmed COVID-19. Given the urgency for targeted SARS-CoV-2 vaccine and therapeutic development and the importance of prospective surveillance to detect the emergence of potential antibody escape variants, it is absolutely essential that epitope-mapping studies be validated using multiple approaches. Indeed, our results nicely converge with a number of other studies aimed at mapping the epitope profiles of SARS-CoV-2 and together begin to provide a comprehensive picture of the responses to this pandemic virus (Amrun et al., 2020; Poh et al., 2020; Shrock et al., 2020). Our phage-based profiling method, coupled with robust and customized computational modeling of significantly enriched sequences, provides an important launch point for further characterization of neutralizing, non-neutralizing, and cross-reactive antibodies targeting SARS-CoV-2.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| T7 Select 10-3b bacteriophage | EMD Millipore Sigma | Cat. #70014-3 |
| Escherichia coli, strain BLT5403 | EMD Millipore Sigma | Cat. # 70548-3 |
| Biological samples | ||
| COVID-19 patient plasma and sera | This paper | See Tables 1 and S1 |
| Healthy human sera | This paper | See Table 1 |
| Endemic HCoV positive plasma and sera | This paper | See Table 1 |
| Chemicals, peptides, and recombinant proteins | ||
| FuGENE-6 | Promega | Cat. # E2692 |
| Protein A Dynabeads | Thermo Fisher | Cat. # 10002D |
| Protein G Dynabeads | Thermo Fisher | Cat. # 10004D |
| Q5 High Fidelity 2X Master Mix | NEB | Cat. # M0492S |
| Critical commercial assays | ||
| QIAquick Gel Extraction Kit | QIAGEN | Cat. # 28704 |
| AMPure XP | Beckman Coulter | Cat. # A63881 |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Fisher | Cat. # P7589 |
| Bright-Glo Luciferase Assay System | Promega | Cat. # E2620 |
| Deposited data | ||
| Demultiplexed Illumina sequencing reads | This paper | SRA: PRJNA72462 |
| Experimental models: Cell lines | ||
| Human Embryonic Kidney (HEK293T) | ATCC | Cat. # CRL-11268 |
| Human Embryonic Kidney cells expressing human ACE2 (HEK293T-hACE2) | BEI Resources | Cat. # NR-52511 |
| Oligonucleotides | ||
| Oligonucleotide library FWD primer: 5′ AATGATACGGCAGGAATTCTACGCTGAGT 3′ | Williams et al., 2019 | N/A |
| Oligonucleotide library REV primer: 5′ CGATCAGCAGAGGCAAGCTTGCTATCA 3′ | Williams et al., 2019 | N/A |
| Illumina library prep primer, Round 1 FWD: 5′ TCGTCGGCAGCGTCTCCAGTCAGGTG TGATGCTC 3′ |
Williams et al., 2019 | N/A |
| Illumina library prep primer, Round 2 REV: 5′ GTGGGCTCGGAGATGTGTATAAGAGAC AGCAAGACCCGTTTAGAGGCCC 3′ |
Williams et al., 2019 | N/A |
| Illumina library prep primer, Round 2 FWD: 5′ AATGATACGGCGACCACCGAGATCTAC ACNNNNNNNNTCGTCGGCAGCGTCTCCAGTC 3′ |
Williams et al., 2019 | N/A |
| Illumina library prep primer, Round 2 REV: 5′ CAAGCAGAAGACGGCATACGAGATNNN NNNNNGTCTCGTGGGCTCGGAGATGTGTA TAAGAGACAG 3′ |
Williams et al., 2019 | N/A |
| Custom Illumina sequencing primer: 5′ GCT CGGGGATCCGAATTCTACGCTGAGT 3′ |
Williams et al., 2019 | N/A |
| Recombinant DNA | ||
| HDM-SARS2-Spike-delta21 plasmid | Addgene | Cat. # 155130 |
| Luciferase-IRES-ZsGreen plasmid | BEI Resources | Cat. # NR-52516 |
| HDM-Hgpm2 gag/pol lentiviral helper plasmid | BEI Resources | Cat. # NR-52517 |
| pRC-CMV-Rev1b plasmid | BEI Resources | Cat. # NR-52519 |
| HDM-tat1b plasmid | BEI Resources | Cat. # NR-52518 |
| Software and algorithms | ||
| Geneious R11 | https://www.geneious.com | N/A |
| Prism 9 | https://www.graphpad.com | N/A |
| Nextflow | Di Tommaso et al., 2017 | https://github.com/matsengrp/pan-CoV-manuscript |
| Bowtie | Langmead et al., 2009 | N/A |
| samtools-idxstats | Li et al., 2009 | N/A |
| xarray | Hamman and Hoyer, 2017 | N/A |
| phip-stat | Laserson Lab | https://github.com/lasersonlab/phip-stat |
| scipy.optimize | Virtanen et al., 2020 | N/A |
| pairwise2 | Cock et al., 2009 | N/A |
| RStudio | https://www.rstudio.com | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Julie Overbaugh (joverbau@fredhutch.org).
Materials availability
The pan-CoV phage display library developed in this study is available on request. All other reagents have been previously deposited to Addgene or BEI (see key resources table).
Data and code availability
Original sequencing data have been deposited to SRA: PRJNA724692. The Nextflow pipeline used to align sequencing reads to the pan-CoV reference library and conduct downstream sample curation and analysis is available at https://github.com/matsengrp/pan-CoV-manuscript. The phip-stat Python package used to implement the Gamma-Poisson mixture model can be found at https://github.com/lasersonlab/phip-stat.
Experimental model and subject details
Human subjects
All patients with mild COVID-19 were outpatients, not requiring hospitalization. Patients with moderate/severe COVID-19 were hospitalized and a range of clinical outcomes were documented, ranging from supplemental oxygen, intubation and death. Additional sample demographic information is found in Tables 1 and S1. Prior to study initiation, the University of Washington IRB (Seattle, Washington) approved the protocol, and concurrent approvals were obtained from the Fred Hutchinson Cancer Research Center. All samples were heat-inactivated at 56°C for 60 minutes prior to short-term storage at 4°C or long-term storage at −80°C. Two additional endemic HCoV positive, and three mild COVID-19 samples were tested in the phage display assay but were not included in the global fit because of poor in-assay technical replicate correlation or poor correlation between experiments conducted using Library 1 and Library 2.
Cell lines
HEK293T (ATCC, Cat. # CRL-11268) and HEK293T-hACE2 (BEI, Cat. # NR-52511) cells were maintained in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and penicillin/streptomycin/fungizone. Seeding density and plating procedures for pseudovirus production and neutralization assays are described below.
Method details
Production of SARS-CoV-2 Spike protein pseudotyped lentivirus
Pseudotyped lentiviral particles expressing SARS-CoV-2 Spike protein were produced and titered as previously described (Crawford et al., 2020). HEK293T cells were seeded at a density of 5 × 105 cells per well in 6-well plates containing DMEM (supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and penicillin/streptomycin/fungizone). After incubating for 16-25 hours, cells were transfected using FuGENE-6 (Promega) with the Luciferase_IRES_ZsGreen backbone, the Gag/Pol-, Rev-, and Tat lentiviral helper plasmids, and a plasmid containing the codon-optimized Spike sequence from the Wuhan-Hu-1 strain. The Spike sequence contained a 21 amino acid deletion at the cytoplasmic tail (also known as HDM_Spikedelta21). After incubating for 24 hours, we replaced media with fresh supplemented DMEM. Between 50-60 hours post-transfection, viral supernatants were collected, filtered through a 0.22 m Steriflip filter and stored at −80°C. To titer pseudovirus, 1.25 × 104 HEK293T cells expressing ACE2 were plated in a volume of 50 uL in 96-well black-walled plates and incubated for 16-24 hours before addition of 100 uL viral supernatant to each well. Pseudovirus supernatants were diluted 1:10 in supplemented DMEM, followed by seven 2-fold serial dilutions. Each dilution was run in duplicate. Sixty hours after infection, 100 uL of media was removed from each well and 30 uL of Bright-Glo reagent (Promega) was added. Relative luciferase units (RLU) were then measured on a LUMIstar Omega plate reader.
Neutralization assays
Neutralization assays using pseudotyped lentivirus particles expressing wild-type SARS-CoV-2 Spike protein were carried out as previously described (Crawford et al., 2020). Briefly, 1.25e4 HEK293T cells expressing the ACE2 receptor were seeded into 96-well black walled plates. HEK293T cells with no ACE2 were also seeded into multiple wells as controls. After 12 hours, 3-fold plasma/serum dilutions were prepared in a separate plate with supplemented DMEM beginning at 1:20, and up to 1:14580 for a total of seven dilutions in a final volume of 60 uL each. Virus was diluted to a concentration corresponding to 2 × 105 RLU/well at a volume of 60 uL. 60 uL virus was added to plasma and preincubated for 1 hour at 37°C. 100 uL of virus/plasma mix was then transferred to the cell plate. 60 hours post-infection, 100 uL of media was removed from each well and 30 uL of Bright-Glo reagent was added. After a two minute incubation, RLU was measured on a LUMIstar Omega plate reader. Two plasma-free wells were included on each row of the assay plate and were used to calculate the fraction infectivity by dividing luciferase readings from wells with plasma by the average of the two plasma-free wells in the same row. Neutralization titers (NT50) were calculated with Prism (Graphpad) using an [Inhibitor] versus response curve with top and bottom parameters constrained to 1 and 0, respectively.
Pan-CoV phage display library construction and immunoprecipitation
Epitope mapping via phage display and immunoprecipitation was carried out essentially as previously described (Mohan et al., 2018; Williams et al., 2019). An oligonucleotide pool was generated from 17 CoV protein coding sequences retrieved from GenBank: OC43-SC0776 (MN310478), OC43-12689/2012 (KF923902), OC43-98204/1998 (KF530069), 229E-SC0865 (MN306046), 229E-0349 (JX503060), 229E-932-72/1993 (KF514432), NL63-ChinaGD01 (MK334046), NL63-Kilifi_HH-5709_19-May-2010 (MG428699), NL63-012-31/2001 (KF530105), NL63-911-56/1991 (KF530107), HKU1-SI17244 (MH90245), HKU1-N13 genotype A (DQ415909), HKU1-Caen1 (HM034837), MERS-KFMC-4 (KT121575), SARS-Urbani (AY278741), SARS-CoV-2-Wuhan-Hu-1 (MN908947), bat-SL-CoVZC45 (MG772933). Multiple strains of the endemic CoVs were selected to cover a wide range of circulation chronology using the Nextstrain database (Hadfield et al., 2018). A single HIV-1 envelope sequence was also included for controls (BG505.W6.C2, DQ208458).
Bacterial codon-optimized oligonucleotide libraries were designed using the Python script available at https://github.com/jbloomlab/phipseq_oligodesign. During the design process, viral protein coding sequences were reverse translated to DNA in 39 amino acid tiles with 19 amino acid overlaps. Adaptor sequences (5′: AGGAATTCTACGCTGAGT and 3′: TGATAGCAAGCTTGCC) were added. Two separate oligonucleotide pools with equivalent design were commercially synthesized (Twist Biosciences). The libraries were PCR amplified using in-house primers (FWD: AATGATACGGCAGGAATTCTACGCTGAGT and REV: CGATCAGCAGAGGCAAGCTTGCTATCA), digested, cloned into the T7Select 10-3b Vector, packaged in T7 phage and amplified according to manufacturer instructions (EMD Millipore).
For phage immunoprecipitation, 1.1 mL 96-deep-well plates (CoStar) were blocked with 3% BSA in TBST (Tris-buffered saline-Tween) by rocking overnight at 4°C. Amplified phage library was diluted in Phage Extraction Buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 6 mM MgSO4) to reach 2x105-fold phage representation (1.33x109 PFU/mL for a phage library containing 6,659 sequences) and added to each well at a volume of 1 mL. We estimated plasma and serum IgG concentrations to be 10 ug/uL (Mabuka et al., 2012) and added 10 ug of each sample to the diluted phage library in duplicate for a total of two technical (within-assay) replicates per experiment. Serum/plasma antibodies were allowed to bind to the phage library by rocking at 4°C for 20 hours. To account for non-specific interactions during the immunoprecipitation step, we prepared multiple wells with no serum and only phage library (“mock”-immunoprecipitations) and treated them to the same rocking procedure. To immunoprecipitate phage-antibody complexes, 40 uL of a 1:1 mixture of protein A and protein G magnetic Dynabeads (Invitrogen) was added to each well and rocked at 4°C for 4 hours. Dynabeads were then isolated using a magnetic plate, washed three times in 400 uL of wash buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl), 0.1% NP-40), and resuspended in 40 uL of water. Dynabead-bound phage were lysed by incubating samples at 95°C for 10 minutes and stored at −20°C prior to Illumina library preparation.
Illumina library preparation
Phage DNA was PCR-amplified with Q5 High-Fidelity DNA polymerase (New England Biolabs) in two rounds to produce Illumina libraries containing adaptor sequences and barcodes for multiplexing. Round 1 PCRs were performed using 10 uL of resuspended, lysed phage in a 25 uL reaction volume using primers R1_FWD (TCGTCGGCAGCGTCTCCAGTCAGGTGTGATGCTC) and R1_REV (GTGGGCTCGGAGATGTGTATAAGAGACAGCAAGACCCGTTTAGAGGCCC). Round 2 PCRs were performed using 2 uL of the Round 1 reaction in a 50 uL final volume with unique dual-indexed primers as previously described (Williams et al., 2019). Round 2 PCR products were quantified using Quant-iT PicoGreen according to manufacturer instructions (Thermo Fisher). Samples were pooled in equimolar quantities, gel purified, and submitted for sequencing on a MiSeq with 126 bp single-end reads.
Quantification and statistical analysis
Phage sequence alignment pipeline
An enrichment matrix was created by aligning all sequences to the pan-CoV reference library using a Nextflow data processing pipeline (https://github.com/matsengrp/pan-CoV-manuscript) (Di Tommaso et al., 2017). The pipeline was initiated with metadata for all samples (including a path to the fastq reads) as well as the metadata for all peptides in the library. The processing steps were as follows: (1) We built a Bowtie index from the peptide metadata by converting the metadata to fasta format and feeding it into the bowtie-build command (Langmead et al., 2009). (2) We aligned each of the samples to the library using end-to-end alignment allowing for up to two mismatches. Each read was 125 base pairs long, and the low-quality end of the read was trimmed to match the reference length, 117 base pairs, before alignment. (3) We extracted the peptide counts for each sample alignment using samtools-idxstats (Li et al., 2009). (4) All individual counts information for each sample were merged into an enrichment matrix. The resulting dataset containing the enrichment matrix, sample metadata, and peptide metadata was organized using the xarray package (Hamman and Hoyer, 2017).
Assessment of peptide significance using a Gamma-Poisson model
To determine significance of enriched peptides in the background of noise introduced by non-specific immunoprecipitation, curated sample sets from two separate phage libraries were fit to a Gamma-Poisson mixture model in the phip-stat Python package provided by the Laserson Lab (https://github.com/lasersonlab/phip-stat). For simplicity, we focused our downstream analyses on one strain from each of the CoV species included in the phage libraries. We required samples to have (1) high technical (in-assay) correlation and (2) high correlation in experiments conducted with phage libraries 1 and 2, in order to be included in the fit. For each model fit, data from mock-IP controls were included with the patient sample data to better account for the abundance and non-specific binding associated with each peptide in the phage library. To control for the variance in sequencing coverage between samples, we first normalized all samples using counts factor method (Anders and Huber, 2010). This resulted in a normalized raw counts matrix, M, with i peptides and j samples. The model assumes that each entry in the count matrix, for any given peptide i, is sampled from a Poisson distribution with rate, λi. Next, we assumed the prior distribution of any λi is a Gamma distribution defined by α and β parameters. We used the scipy.optimize package (Virtanen et al., 2020) to infer maximum likelihood estimates that would generate a set of mean normalized counts values across samples for each peptide, i. Given that the posterior of the rate is also Gamma distributed, the posterior hyperparameters for each peptide, i, are given by the formulas and , where n is the number of samples. Because the gamma distribution is a conjugate prior for the Poisson, we get for each peptide. Finally, -log10(pval) (mlxp) values are by computed using the value of the tail of the Poisson distribution for each normalized sample count at peptide, i.
False positive rate (FPR) estimation using HIV Env peptides
We observed quite extreme p values using the Gamma-Poisson approach (as well as using the Generalized Poisson approach in Larman et al.[ 2011]), indicating that these p values were not well calibrated. Thus, the selection of peptides with significant binding affinity was performed by applying a minimum threshold requirement on the mlxp, which was set based on using HIV peptides as a control. Specifically, peptides derived from the HIV-1 envelope were presumed not to truly bind with SARS-CoV-2 antibodies, so we used these HIV-1 peptides to estimate the FPR for a threshold under consideration: the number of HIV-1 sample-peptide pairs above this threshold divided by the total number of HIV-1 sample-peptide pairs in the library batch after the curation step. Hence, for each library batch, we set the threshold to be the value where 5% of the HIV-1 peptides have mlxp values above the threshold. In each library batch, there were a total 798 HIV-1 sample-peptide pairs. None of the HIV peptides have significant sequence homology with SARS-CoV-2 peptides.
Local sequence alignment
The similarity between SARS-CoV-2 and other CoV peptides was quantified by performing local alignment and then computing the identical fraction: the fraction of matching amino acids at each position of the aligned subsequence. The Smith-Waterman algorithm was applied with the BLOSUM62 cost matrix, a gap open penalty of 12, and a gap extension penalty of 3 (Smith and Waterman, 1981). We used the pairwise2 function of the Biopython software package to perform the alignment (Cock et al.).
Additional quantification and statistical analysis
Additional quantification and statistical analyses were performed in Prism (Graphpad). Plots were generated using R and Prism. Figure 1 was generated with BioRender. Additional sequence data analysis was conducted using Geneious R11.
Acknowledgments
We extend gratitude to all study participants who contributed samples. We thank members of the Overbaugh, Matsen, and Chu labs for thoughtful discussion and advice. We thank the Fred Hutchinson Cancer Research Center (FHCRC) Genomics Core, especially Cassie Sather, for assistance with Illumina sequencing. We thank Trevor Bedford (FHCRC) for advice on strains to include in the pan-CoV phage library, Theodore Gobillot (FHCRC) for advice on phage library construction and immunoprecipitation, and Haidyn Weight for assistance with neutralization assays. This work was funded by NIH grants AI138709 (principal investigator [PI] J.O.) and R01 AI146028 and U19 AI128914 (PI F.A.M.). J.O. received support as the Endowed Chair for Graduate Education (FHCRC). The research of F.A.M. was supported in part by a Faculty Scholar grant from the Howard Hughes Medical Institute and the Simons Foundation.
Author contributions
J.O., F.A.M., and C.I.S. conceived of the study and designed experiments. Samples were collected and curated by C.R.W., J.K.L., and A.M. under the supervision of H.Y.C. Experiments were conducted by C.I.S. with assistance from M.M.S., H.L.I., and M.E.G. Computational analyses were conducted by J.G. and K.S., with additional data analysis by C.I.S. Additional computational assistance was provided by K.H.D.C. and U.L. The manuscript was written by C.I.S. and J.O. with input from all authors.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Published: May 6, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109164.
Supplemental information
References
- Amrun S.N., Lee C.Y.-P., Lee B., Fong S.-W., Young B.E., Chee R.S.-L., Yeo N.K.-W., Torres-Ruesta A., Carissimo G., Poh C.M. Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS-CoV-2 exposure and disease severity. EBioMedicine. 2020;58:102911. doi: 10.1016/j.ebiom.2020.102911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106–R112. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvin A.M., Fink K., Schmid M.A., Cathcart A., Spreafico R., Havenar-Daughton C., Lanzavecchia A., Corti D., Virgin H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. 2020;584:353–363. doi: 10.1038/s41586-020-2538-8. [DOI] [PubMed] [Google Scholar]
- Atyeo C., Fischinger S., Zohar T., Slein M.D., Burke J., Loos C., McCulloch D.J., Newman K.L., Wolf C., Yu J. Distinct Early Serological Signatures Track with SARS-CoV-2 Survival. Immunity. 2020;53:524–532.e4. doi: 10.1016/j.immuni.2020.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.H., Cheng V.C.C., Woo P.C.Y., Lau S.K.P., Poon L.L.M., Guan Y., Seto W.H., Yuen K.Y., Peiris J.S.M. Serological responses in patients with severe acute respiratory syndrome coronavirus infection and cross-reactivity with human coronaviruses 229E, OC43, and NL63. Clin. Diagn. Lab. Immunol. 2005;12:1317–1321. doi: 10.1128/CDLI.12.11.1317-1321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.-W., Kok K.-H., Zhu Z., Chu H., To K.K.-W., Yuan S., Yuen K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020;9:221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369:650–655. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P., Antao T., Chang J.T., Chapman B.A. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K.H.D., Eguia R., Dingens A.S., Loes A.N., Malone K.D., Wolf C.R., Chu H.Y., Tortorici M.A., Veesler D., Murphy M. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses. 2020;12:513. doi: 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. doi: 10.1038/nbt.3820. [DOI] [PubMed] [Google Scholar]
- Dutta N.K., Mazumdar K., Gordy J.T. The Nucleocapsid Protein of SARS-CoV-2: a Target for Vaccine Development. J. Virol. 2020;94:270. doi: 10.1128/JVI.00647-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N.R., Mellan T.A., Whittaker C., Claro I.M., Candido D.D.S., Mishra S. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science. 2021 doi: 10.1126/science.abh2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway S.E., Paul P., MacCannell D.R., Johansson M.A., Brooks J.T., MacNeil A., Slayton R.B., Tong S., Silk B.J., Armstrong G.L. Emergence of SARS-CoV-2 B.1.1.7 Lineage - United States, December 29, 2020-January 12, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021;70:95–99. doi: 10.15585/mmwr.mm7003e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.L., Sparks J.S., Eckerle L.D., Sims A.C., Denison M.R. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008;133:88–100. doi: 10.1016/j.virusres.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181:1489–1501.e15. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamman J., Hoyer S. 2017. Xarray: N-D labeled arrays and datasets in Python.http://xarray.pydata.org/en/stable/ [Google Scholar]
- Hoffmann M., Kleine-Weber H., Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell. 2020;78:779–784.e5. doi: 10.1016/j.molcel.2020.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaimes J.A., André N.M., Chappie J.S., Millet J.K., Whittaker G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020;432:3309–3325. doi: 10.1016/j.jmb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B., Zhang Q., Ge J., Wang R., Sun J., Ge X., Yu J., Shan S., Zhou B., Song S. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584:115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larman H.B., Zhao Z., Laserson U., Li M.Z., Ciccia A., Gakidis M.A.M., Church G.M., Kesari S., Leproust E.M., Solimini N.L., Elledge S.J. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011;29:535–541. doi: 10.1038/nbt.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.S., Kent S.J. Anti-HIV-1 antibody-dependent cellular cytotoxicity: is there more to antibodies than neutralization? Curr. Opin. HIV AIDS. 2018;13:160–166. doi: 10.1097/COH.0000000000000439. [DOI] [PubMed] [Google Scholar]
- Lee W.S., Wheatley A.K., Kent S.J., DeKosky B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020;5:1185–1191. doi: 10.1038/s41564-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Lai D.-Y., Zhang H.-N., Jiang H.-W., Tian X., Ma M.-L., Qi H., Meng Q.-F., Guo S.-J., Wu Y. Linear epitopes of SARS-CoV-2 spike protein elicit neutralizing antibodies in COVID-19 patients. Cell. Mol. Immunol. 2020;17:1095–1097. doi: 10.1038/s41423-020-00523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., To K.K.-W., Chan K.-H., Wong Y.-C., Zhou R., Kwan K.-Y., Fong C.H.-Y., Chen L.-L., Choi C.Y.-K., Lu L. High neutralizing antibody titer in intensive care unit patients with COVID-19. Emerg. Microbes Infect. 2020;9:1664–1670. doi: 10.1080/22221751.2020.1791738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H., Wu N.C., Tsang O.T.-Y., Yuan M., Perera R.A.P.M., Leung W.S., So R.T.Y, Chan J.M.C., Yip G.K., Chik T.S.H. Cross-reactive Antibody Response between SARS-CoV-2 and SARS-CoV Infections. Cell Rep. 2020;31(107725) doi: 10.1016/j.celrep.2020.107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabuka J., Nduati R., Odem-Davis K., Peterson D., Overbaugh J. HIV-specific antibodies capable of ADCC are common in breastmilk and are associated with reduced risk of transmission in women with high viral loads. PLoS Pathog. 2012;8:e1002739. doi: 10.1371/journal.ppat.1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus J., Grifoni A., Tarke A., Sidney J., Ramirez S.I., Dan J.M., Burger Z.C., Rawlings S.A., Smith D.M., Phillips E. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science. 2020;370:89–94. doi: 10.1126/science.abd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr L.M., Su B., Moog C. Non-Neutralizing Antibodies Directed against HIV and Their Functions. Front. Immunol. 2017;8:1590. doi: 10.3389/fimmu.2017.01590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan D., Wansley D.L., Sie B.M., Noon M.S., Baer A.N., Laserson U., Larman H.B. PhIP-Seq characterization of serum antibodies using oligonucleotide-encoded peptidomes. Nat. Protoc. 2018;13:1958–1978. doi: 10.1038/s41596-018-0025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla-Quirarte H.O., Lopez-Guerrero D.V., Gutierrez-Xicotencatl L., Esquivel-Guadarrama F. Protective Antibodies Against Influenza Proteins. Front. Immunol. 2019;10:1677. doi: 10.3389/fimmu.2019.01677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.-J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Poh C.M., Carissimo G., Wang B., Amrun S.N., Lee C.Y.-P., Chee R.S.-L., Fong S.-W., Yeo N.K.-W., Lee W.-H., Torres-Ruesta A. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020;11:2806–2807. doi: 10.1038/s41467-020-16638-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röltgen K., Powell A.E., Wirz O.F., Stevens B.A., Hogan C.A., Najeeb J., Hunter M., Wang H., Sahoo M.K., Huang C. Defining the features and duration of antibody responses to SARS-CoV-2 infection associated with disease severity and outcome. Sci. Immunol. 2020;5:5. doi: 10.1126/sciimmunol.abe0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphire E.O., Schendel S.L., Gunn B.M., Milligan J.C., Alter G. Antibody-mediated protection against Ebola virus. Nat. Immunol. 2018;19:1169–1178. doi: 10.1038/s41590-018-0233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Wan Y., Luo C., Ye G., Geng Q., Auerbach A., Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA. 2020;117:11727–11734. doi: 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrock E., Fujimura E., Kula T., Timms R.T., Lee I.-H., Leng Y., Robinson M.L., Sie B.M., Li M.Z., Chen Y., MGH COVID-19 Collection & Processing Team Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science. 2020;370:eabd4250. doi: 10.1126/science.abd4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. 2020 2020.12.21.20248640. [Google Scholar]
- Virtanen P., Gommers R., Oliphant T.E., Haberland M., Reddy T., Cournapeau D., Burovski E., Peterson P., Weckesser W., Bright J., SciPy 1.0 Contributors SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17:261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Li X., Li T., Zhang S., Wang L., Wu X., Liu J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020;39:1629–1635. doi: 10.1007/s10096-020-03899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Zhang L., Sang L., Ye F., Ruan S., Zhong B., Song T., Alshukairi A.N., Chen R., Zhang Z. Kinetics of viral load and antibody response in relation to COVID-19 severity. J. Clin. Invest. 2020;130:5235–5244. doi: 10.1172/JCI138759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh R.M., Che J.W., Brehm M.A., Selin L.K. Heterologous immunity between viruses. Immunol. Rev. 2010;235:244–266. doi: 10.1111/j.0105-2896.2010.00897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K.L., Stumpf M., Naiman N.E., Ding S., Garrett M., Gobillot T., Vézina D., Dusenbury K., Ramadoss N.S., Basom R. Identification of HIV gp41-specific antibodies that mediate killing of infected cells. PLoS Pathog. 2019;15:e1007572. doi: 10.1371/journal.ppat.1007572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wölfel R., Corman V.M., Guggemos W., Seilmaier M., Zange S., Müller M.A., Niemeyer D., Jones T.C., Vollmar P., Rothe C. Virological assessment of hospitalized patients with COVID-2019. Nature. 2020;581:465–469. doi: 10.1038/s41586-020-2196-x. [DOI] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.-M., Wang W., Song Z.-G., Hu Y., Tao Z.-W., Tian J.-H., Pei Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Davis B.D., Chen S.S., Sincuir Martinez J.M., Plummer J.T., Vail E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA. 2021;325:1324–1326. doi: 10.1001/jama.2021.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original sequencing data have been deposited to SRA: PRJNA724692. The Nextflow pipeline used to align sequencing reads to the pan-CoV reference library and conduct downstream sample curation and analysis is available at https://github.com/matsengrp/pan-CoV-manuscript. The phip-stat Python package used to implement the Gamma-Poisson mixture model can be found at https://github.com/lasersonlab/phip-stat.