

Abstract
Multisystem inflammatory syndrome in children (MIS-C), a hyperinflammatory syndrome associated with SARS-CoV-2 infection, shares clinical features with toxic shock syndrome, which is triggered by bacterial superantigens. Superantigen specificity for different Vβ chains results in Vβ skewing, whereby T cells with specific Vβ chains and diverse antigen specificity are overrepresented in the T cell receptor (TCR) repertoire. Here, we characterized the TCR repertoire of MIS-C patients and found a profound expansion of TCRβ variable gene 11-2 (TRBV11-2), with up to 24% of clonal T cell space occupied by TRBV11-2 T cells, which correlated with MIS-C severity and serum cytokine levels. Analysis of TRBJ gene usage and complementarity-determining region 3 (CDR3) length distribution of MIS-C expanded TRBV11-2 clones revealed extensive junctional diversity. Patients with TRBV11-2 expansion shared HLA class I alleles A02, B35, and C04, indicating what we believe is a novel mechanism for CDR3-independent T cell expansion. In silico modeling indicated that polyacidic residues in the Vβ chain encoded by TRBV11-2 (Vβ21.3) strongly interact with the superantigen-like motif of SARS-CoV-2 spike glycoprotein, suggesting that unprocessed SARS-CoV-2 spike may directly mediate TRBV11-2 expansion. Overall, our data indicate that a CDR3-independent interaction between SARS-CoV-2 spike and TCR leads to T cell expansion and possibly activation, which may account for the clinical presentation of MIS-C.
Keywords: COVID-19, Immunology
Keywords: T cell receptor

Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 2019 (COVID-19), which started as an epidemic in China and culminated in a global pandemic. Typical COVID-19–related symptoms include fever, dry cough, breathing difficulties, and gastrointestinal (GI) symptoms (1). Although most infected adults develop a mild course of the disease, COVID-19 manifests as a severe interstitial pneumonia with hyperinflammation and many extrapulmonary complications in 20% of patients (2–4). Severe COVID-19 in adult patients is associated with overactivation of the immune system and increased release of proinflammatory cytokines, a process called “cytokine storm” (4). The percentage of pediatric cases diagnosed with SARS-CoV-2 infection has risen steadily since mid-April, 2020, from just 2% to 11% of all US COVID-19 cases (5). Children are less likely to suffer from life-threatening COVID-19 disease, and account for just 3.5% of current COVID-19 hospitalization in the United States (5). Although rare, children can develop severe disease caused by SARS-CoV-2 infection, presenting with high fever and acute respiratory disease. In addition, beginning in March and April 2020, there have also been reports of children admitted to hospital with a multisystem hyperinflammatory syndrome, presenting with fever, severe abdominal pain, diarrhea, myocardial dysfunction, and cardiogenic shock and rash, reminiscent of Kawasaki disease (KD) (6–11). The first reports of this new syndrome, now called multisystem inflammatory syndrome in children (MIS-C), were from the United Kingdom, and they later emerged from across Europe and the eastern part of North America (6–11). Recently, a similar disease in adults (MIS-A) has also been identified (12). Both MIS-C and MIS-A usually require intensive care; however, these two entities are not associated with severe respiratory symptoms (6–12). It is interesting to note that approximately one-third or fewer of MIS-C patients tested positive for SARS-CoV-2, but the majority had serologic evidence of infection or a history of exposure to COVID-19 (6–11). MIS-C may therefore be triggered by an extrapulmonary SARS-CoV-2 infection or be caused by a delayed, postinfectious inflammatory response.
Clinical and laboratory features of MIS-C are remarkably similar to those of toxic shock syndrome (TSS), including severe GI symptoms, neurological symptoms, myocardial involvement, lymphopenia, elevated levels of C-reactive protein, ferritin, and D-dimers, and increased expression of proinflammatory cytokines (6–8, 13–15). TSS is driven by bacterial superantigens (SAgs) such as staphylococcal enterotoxin B (SEB). SAgs are highly potent T cell activators able to engage both T cell receptors (TCRs) and MHC class II molecules (16). Typically, bacterial SAgs activate T cells by binding to specific β chains of TCRs at their variable domain in a complementarity-determining region 3–independent (CDR3-independent) manner. As such, SAgs can bypass the TCR antigen specificity and induce T cell activation and proliferation along with a cytokine storm that results in toxic shock (16, 17). SAg binding specificity for different TCR Vβ chains results in Vβ skewing, whereby T cells with specific Vβ chains and diverse antigen specificity are overrepresented in the TCR repertoire of patients upon exposure to SAgs.
Using structure-based computational models, we recently demonstrated that the SARS-CoV-2 spike glycoprotein has a unique insert that displays a SAg-like sequence motif that exhibits a high affinity for binding TCRs (18). Furthermore, we found that this SAg-like motif has high sequence and structural similarity to a motif in SEB (18). We further reported TCR Vβ skewing in adult COVID-19 patients with severe hyperinflammation, consistent with a SAg immune response (18).
Since MIS-C clinical features are quite similar to those of TSS, here we analyzed the TCR repertoire of MIS-C patients to determine if MIS-C is associated with Vβ skewing and junctional diversity as well as signs of SAg activation. Immunosequencing of the peripheral blood samples of MIS-C patients revealed a profound expansion of TCRβ variable gene 11-2 (TRBV11-2), which correlated with MIS-C severity and serum cytokine levels, consistent with SAg-triggered immune responses. Furthermore, HLA genotyping of MIS-C and control patients demonstrated that patients with TRBV11-2 expansion shared 3 HLA class I (HLA-I) alleles, suggesting what we believe is a novel T cell activation mechanism in MIS-C.
Results
The TCR repertoires of mild MIS-C patients are richer and more diverse than those of severe MIS-C patients
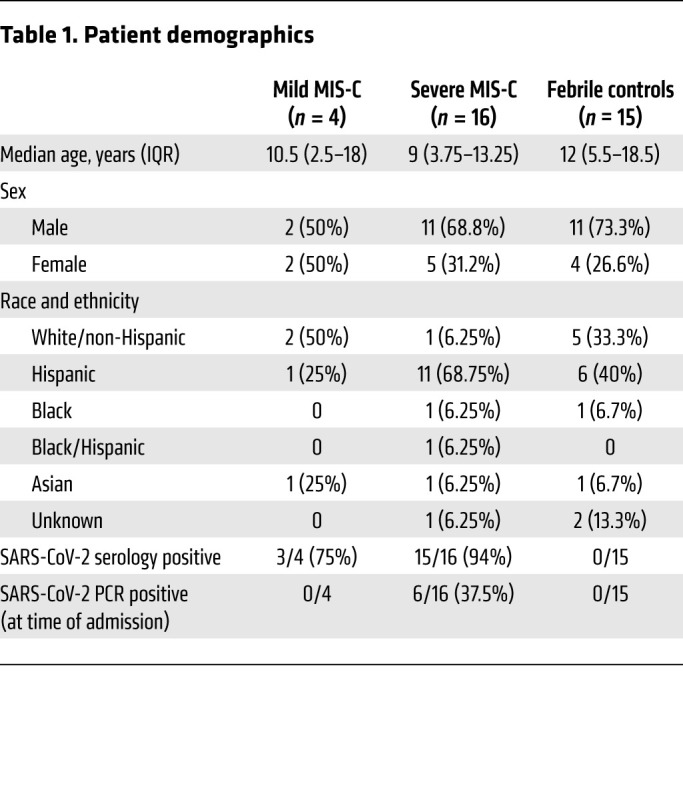
To determine if MIS-C is associated with TCR repertoire skewing, we collected blood samples from mild MIS-C (n = 4), severe MIS-C (n = 16), and age-matched febrile control patients (n = 15). Patient demographics are presented in Table 1. MIS-C diagnosis was performed according to the US Centers for Disease Control guidelines. TCR-sequencing analysis was performed on extracted RNA. Global T cell metrics showed no differences in basic repertoire metrics when comparing MIS-C patients (mild or severe) with febrile controls. However, we observed that 2 mild MIS-C patients were characterized by a generally richer TCR repertoire than severe MIS-C patients or febrile controls (Supplemental Figure 1, A–D; supplemental material available online with this article; https://doi.org/10.1172/JCI146614DS1). These findings were consistent with adult COVID-19, in which mild cases present with higher TCR richness than age-matched controls, indicating diversification of the repertoire in an antigen-dependent manner (19).
Table 1. Patient demographics.
Skewing of TRBV genes in MIS-C.
We next performed principal component analysis (PCA) of TCR V and J gene usage to determine their global distributions between cohorts. PCA of the TRBV gene repertoires revealed that MIS-C patients clustered apart from febrile controls, whereas the usages of TRAV, TRGV, and TRDV genes did not show any skewing (Figure 1A). PCA of TRBJ gene usage revealed no differences between febrile controls and MIS-C patients or among the severity groups (Supplemental Figure 2), indicating that a selective pressure was exclusively exerted on the V gene distribution.
Figure 1. TRBV-driven repertoire skewing in MIS-C patients.

(A) Principal component analysis (PCA) of differential TRBV, TRAV, TRGV, and TRDV gene usage in MIS-C patients (n = 20) versus febrile controls (n = 15), and between mild MIS-C (n = 4) and severe MIS-C (n = 16) patients. Statistical analysis: Pillai-Bartlett test in MANOVA of all principal components. (B) Frequencies of differentially used TRBV genes in MIS-C and febrile control patients. The boxes extend from the 25th to 75th percentiles, whiskers from min to max, and the line within the box indicates the median. Lines in dot plots indicate mean ± SEM. Individuals considered TRBV11-2 expanded are marked with a red box.
Severe MIS-C TCR repertories are characterized by a profound expansion of TRBV11-2.
Since MIS-C patients exhibit strong TRBV skewing, we next performed differential gene usage analysis between mild MIS-C, severe MIS-C, and febrile controls. Several TRBV genes were overrepresented in the overall MIS-C patient group. Specifically, TRBV11-2, TRBV24-1, and TRBV11-3 were expanded in MIS-C patients relative to febrile control patients, such that up to 24% of the clonal T cell space was taken up by clones using TRBV11-2 (Figure 1B). Further comparisons between subgroups showed that this enrichment was restricted to individuals with severe disease. In contrast, TRBV-28 was exclusively enriched in mild MIS-C patients. We previously found that TRBV24-1 was overrepresented in a cohort of adult COVID-19 patients with severe, hyperinflammatory courses compared with patients with mild COVID-19 (18). Figure 2A shows the distribution of TRBV11-2, TRBV24-1, and TRBV11-3 genes on an individual repertoire level. This analysis shows that expansion of the TRBV11-2 compartment predominantly occurs in individuals with severe disease. Furthermore, analysis of serum cytokines from matching samples (18 of 20 MIS-C patients) demonstrated significant correlation between TRBV11-2 usage and TNF-α, IFN-γ, IL-6, and IL-10 levels (Figure 2B), indicating that TRBV11-2 expansion is associated with the cytokine storm.
Figure 2. Clonal distribution in individual repertoires of MIS-C patients.

(A) Visualization of clonal richness and distribution in individual productive repertoires from mild (n = 4) and severe (n = 16) MIS-C patients and febrile controls (n = 15). One bubble represents one amino acid clonotype, and the area size of the bubble indicates its fraction within the repertoire. Clonotypes using TRBV11-2, TRBV24-1, or TRBV11-3 are highlighted. Individuals considered TRBV11-2 expanded are marked with a red box. (B) Correlation of serum cytokine levels with TRBV11-2 usage. Statistical analysis: Spearman’s r correlation test. (C) Distribution of nasopharyngeal SARS-CoV-2 PCR results among patients with or without TRBV11-2 expansion.
TRBV24-1 and TRBV11-3 expansions were less pronounced and restricted to fewer individuals. One patient (MIS-C_severe_14) showed massive expansion of TRBV24-1 (Figure 2A). This patient was 15 years old, which might explain why their TCR repertoire was closer to that previously observed in adult patients. Overall, children between 7 and 16 years of age showed strong TRBV11-2 expansions associated with the severe course of the disease (Supplemental Figure 3A). However, no age drifts in TRBV11-2, TRBV24-1, or TRBV11-3 distribution were found in the TCR repertoires of 254 individuals, indicating that age does not underlie TRBV11-2 skewing in severe MIS-C patients (Supplemental Figure 3B).
Of the MIS-C patients in this study, 95% were serology positive for SARS-CoV-2, while only 30% were PCR positive by nasopharyngeal swab. Interestingly, however, PCR positivity seemed to correlate with TRBV11-2 expansion. We found that 55% of patients with TRBV11-2 expansion were PCR positive for SARS-CoV-2, whereas none of the patients without expansion were PCR positive for SARS-CoV-2 (Figure 2C). These data suggest an association of TRBV11-2 expansion with the presence of SARS-CoV-2 RNA.
Junctional diversity in MIS-C patients with expanded TRBV11-2.
SAg interactions involve the V gene, but spare the CDR3 of the TCR (17, 20). If the TRBV11-2 expansions observed in patients with severe MIS-C were due to SARS-CoV-2 acting as a SAg, the diversity of the V(D)J junction in TCR with TRBV11-2 usage should be high. To investigate junctional diversity, we first searched for CDR3 overlaps between different MIS-C patients. This analysis showed no overlaps between patients with TRBV11-2 expansions, suggesting high CDR3 diversity in this cohort (Figure 3A). Repetitive blood samples taken from the same healthy donor were used as control, showing overlapping repertoires at the different sampling time points. In our global analysis, TRBV J gene usage was not biased in MIS-C patients (Supplemental Figure 2).
Figure 3. Junctional diversity in MIS-C patients with TRBV11-2 expansions.

(A) Overlap of CDR3 amino acid clonotypes per 200 sequences in repetitive samples of a healthy individual and between repertoires of MIS-C patients with TRBV11-2 expansion. (B) PCA and differential usage of J genes rearranged with TRBV11-2 in MIS-C patients and febrile controls. Bars indicate mean ± SD. Statistical analysis: Pillai-Bartlett test in MANOVA of all principal components. (C) Heatmap of productive CDR3 length distribution in the repertoires of MIS-C patients (n = 20) and febrile controls (n = 15). (D) CDR3 diversity in MIS-C patients with or without expanded TRBV11-2 and febrile controls displayed as positional weight matrix generated using GLAM2. (E) Unsupervised phylogenetic analysis of the amino acid sequences spanning FR2 to CDR3 versus CDR3 alone in the top 100 clones of MIS-C repertoires (n = 20), either comprising the complete TRBV sequence pool or TRBV11-2 sequences only.
To further investigate junctional diversity specifically for the V genes expanded in MIS-C cases, we extracted all the J genes rearranged with TRBV11-2 from the repertoires of severe and mild MIS-C patients, and compared these to J genes extracted from age-matched febrile controls. TRBJ2-1, 2-2, 2-3, and 2-7 were the most frequent combinations for TRBV11-2; however, we did not see a difference in TRBJ usage between the different subcohorts, suggesting a full diversity of J genes rearranged with TRBV11-2 (Figure 3B). Moreover, we found an even distribution of CDR3 lengths within the fraction of TCRs using TRBV11-2, suggesting no CDR3-driven clonal selection (Figure 3C). CDR3 diversity in TCRs using TRBV11-2 was also confirmed by gapped local alignment of motifs (GLAM) analysis (Figure 3D) as well as by unsupervised analysis of phylogenetic trees constructed from the top 100 clones of each repertoire (Figure 3E). The latter analysis showed clustering of expanded TRBV11-2 sequences only if the full sequence from framework 2 (FR2) to CDR3 was used for construction, but not if the clustering analysis was limited to the CDR3 sequence (Figure 3E). Together, our results suggest that patients with MIS-C show expansion of TCRs using distinct V genes, along with J gene and CDR3 diversity in these rearrangements, compatible with a SAg selection process.
Although the CDR3 of TRBV11-2 is heterogeneous, and its expansion is only observed in severe MIS-C and not in mild MIS-C, we asked whether TRBV11-2 could define a public clonotype that recognizes SARS-CoV-2 epitopes specifically in children. We analyzed publicly available databases including 8 TCR repertoires of patients aged 0–21 years with acute COVID-19 (21, 22). Compared with adult COVID-19 patients from the same data repository, there was no difference in mean TRBV11-2 usage (Supplemental Figure 4), further supporting our observations that TRBV11-2 expansion is exclusive to severe MIS-C and very unlikely the consequence of a shared public immune response to SARS-CoV-2.
TRAV8-4 is overrepresented in patients with expanded TRBV11-2
Although no global skewing was observed for TRA, TRG, or TRD V or J gene distributions, we reasoned that minor skewing, especially of the TRAV repertoire that is associated with TRBV, may have been missed due to the global PCA approach. Since some SAgs can interact with both the TCR Vβ and Vα chains (23), we searched for skewing in TRAV, TRGV, and TRDV genes associated with TRBV11-2 by investigating their distributions in MIS-C patients with and without TRBV11-2 expansions. While TRGV and TRDV genes did not show any skewed distribution, there was a significant TRAV skewing in MIS-C patients with and without TRBV11-2 expansions (Figure 4A). Further analyses suggested that no single TRAV gene was associated with expanded TRBV11-2, potentially reflecting the lack of skewing in mild versus severe MIS-C cases, but a number of different TRAVs were overrepresented in MIS-C patients with TRBV11-2 expansions and a severe clinical course (Figure 4B). TRAV8-4 was the most expanded gene, while expansions of TRAV17 and TRAV22 were less pronounced. These data suggest that in MIS-C patients, the interaction may also involve the TRAV genes.
Figure 4. TRAV repertoire skewing and differential gene usage in MIS-C patients.

(A) PCA of differential productive TRAV gene usage in MIS-C patients (n = 20) versus febrile controls (n = 15). MIS-C subgroups: mild MIS-C (n = 4), severe MIS-C (n = 16), MIS-C without TRBV11-2 shift (n = 9), and MIS-C with TRBV11-2 shift (n = 11). Statistical analysis: Pillai-Bartlett test in MANOVA of all principal components. (B) Frequencies of differentially used TRAV genes in MIS-C with or without TRBV11-2 shift as compared to febrile controls. The boxes extend from the 25th to 75th percentiles, whiskers from min to max, and the line within the box indicates the median. Lines in dot plots indicate mean ± SEM.
HLA-I allele–associated expansion of TRBV11-2 in severe MIS-C patients.
Specific HLA class II (HLA-II) haplotypes can enhance SAg-induced T cell proliferation and cytokine responses (24). To understand if certain HLA alleles are associated with TRBV11-2 expansion, we performed HLA genotyping on DNA samples from 10 febrile control patients, 3 severe MIS-C patients without TRBV11-2 expansion, and 4 severe MIS-C patients with TRBV11-2 expansion (Figure 5). While we did not find any association of MIS-C or TRBV11-2 expansion with HLA-II alleles (Figure 5A), all 4 severe MIS-C patients with TRBV11-2 expansion used HLA-I A02, B35, and C04 alleles (Figure 5, A–C). In contrast, the percentage of patients with all 3 of these HLA-I alleles (A02, B35, and C04) was 20% for febrile controls and 0% for MIS-C patients lacking TRBV11-2 expansion (Figure 5C). Although the numbers of patients analyzed was small, these data suggest that MHC class I may mediate TRBV11-2 expansion requiring a specific HLA-I allele combination. Furthermore, these analyses strongly indicate that a genetic component may contribute to MIS-C severity.
Figure 5. HLA class I alleles are associated with TRBV11-2 expansion in severe MIS-C patients.

(A) HLA genotypes of febrile controls (n = 10), severe MIS-C patients without TRBV11-2 expansion (n = 3), and severe MIS-C patients with TRBV11-2 expansion (n = 4). (B) Percentages of patient groups with HLA class I alleles. (C) Percentages of patient groups that share HLA-I A02, B35, and C04.
To determine if the HLA-I restriction with TRBV11-2 expansion was specific to MIS-C, we next analyzed a publicly available data set of 63 adult COVID-19 patients with matching TRB repertoires and HLA data from Adaptive Biotechnologies (22). Out of these 63 cases, we identified 6 patients with expanded TRBV11-2, which we defined as higher than the mean plus 1 SD of the entire cohort (Supplemental Figure 5). Within these 6 patients we did not observe HLA biases in HLA-I or -II (Supplemental Figure 5), indicating our findings are unique to MIS-C.
In silico modeling indicates the TCR Vβ chain encoded by TRBV11-2 engages in a CDR3-independent interaction with the SAg-like motif of SARS-CoV-2 spike.
We previously demonstrated through in silico modeling the presence of a SAg-like motif on the SARS-CoV-2 spike protein, distinguished by its high sequence and structural similarity to a segment of SEB (18). Furthermore, the Vβ chain encoded by TRBV24-1 was shown to have a high affinity for binding to the SAg-like motif. Given the extensive expansion of TRBV11-2 in MIS-C patients, we examined in silico whether the Vβ chain encoded by TRBV11-2 (also known as Vβ21.3) could engage in a strong interaction with the SAg-like motif on spike. To this end, we used a TCR structure resolved in a crystal structure of the monoclonal T cell line DD31 TCR complexed with HLA B*0801 (25), the β chain variable domain of which is sequentially identical to the Vβ chain encoded by TRBV11-2. Following our previous approach (18), we generated a series of structural models for the SARS-CoV-2 spike–TCR complex using the docking software ClusPro (26), and analyzed the models grouped in structurally similar clusters to detect recurrent binding patterns (Figure 6). The analysis led to 2 hot spots that bound the Vβ chain encoded by TRBV11-2 on the spike protein: (a) within the SAg-like motif (residues Q677 to R685) and (b) near the neurotoxin-like motif (residues E340 to R357) identified previously (18). Interestingly, both hot spots contained polybasic residues at their binding epitopes, P681RRAR685 and N355RKRI359. Notably, the amino acids D67 and D68 near the CDR2 of the Vβ chain encoded by TRBV11-2 played a significant role in binding to these 2 motifs on the spike, thus serving as a paratope in more than 90% of the generated models for the TCR-spike complex. While we do not exclude the potential role of other toxin-like motifs in stimulating hyperinflammatory responses, we focused on the SAg-like motif Q677–R685 reported in our previous study, whose deletion has been shown to attenuate the severity of the disease (27).
Figure 6. Complex formation between SARS-CoV-2 spike and TCR containing the Vβ chain encoded by TRBV11-2, and comparative analysis of TRBV sequences homologous to TRBV11-2.

(A) Binding of TCR (with Vβ chain sequentially identical to that of TRBV11-2 gene product) to the SAg-like region of SARS-CoV-2 spike (E661 to R685; colored yellow). The TCR α and β chains are shown in magenta and cyan, respectively. The spike subunits are colored dark red, beige, and gray, and the neurotoxin motif (299–356), green. (B and C) Overall and close-up view of the interactions between the SAg-like region and the TCR. Two basic residues, R682 and R683, on the SAg-like region of spike interact with 2 acidic residues, D67 and D68 in the Vβ domain, and D2 in the Vα domain. (D) Multiple sequence alignment (MSA) of the TCR Vβ chains distinguished by Vβ skewing in MIS-C (protein products of TRBV11-2, TRBV11-3, and TRBV24-1 with their corresponding UniProt IDs) and severe adult COVID-19 patients (protein products of TRBV24-1, TRBV14, and TRBV5-6). The MSA was generated by Clustal Omega (76). CDR1, CDR2, and CDR3 are indicated by orange shades. The regions enclosed in blue boxes indicate the highly conserved paratopes in Vβ chains corresponding to TRBV11-2 and TRBV14. See more extended MSA in Supplemental Figure 6.
Figure 6A illustrates the binding of the TCR containing the Vβ chain encoded by TRBV11-2 to the SARS-CoV-2 spike at the SAg-like region containing the polybasic insertion, P681RRAR685, unique to SARS-CoV-2 among all SARS family coronaviruses. Notably, the complex is stabilized by CDR3-independent interactions. Instead, D67-D68 in the Vβ chain encoded by TRBV11-2 (Figure 6, B and C) and Vα chain D2 tightly bind the polybasic insert of the spike via salt bridges between these oppositely charged pairs of residues. Modeling and simulations for the Vβ chain encoded by TRBV14 (a skewed TRBV gene, identified in adults of severe COVID-19 patients; ref. 18 and data not shown) also revealed the involvement of the corresponding acidic residues D67 and E68 in binding the same epitope. We also observed that D97-D98 in both the Vβ chains encoded by TRBV11-2 and TRBV14 were also able to bind to the P681RRAR685 region, albeit with lower probability than D67-D68.
To further assess the specificity of the polyacidic residues near CDR2, including D67 and D68 in the Vβ chain encoded by TRBV11-2, or D67 and E68 in the Vβ chain encoded by TRBV14, we searched the database of TRBV genes compiled in the IMGT Repertoire (28). We identified a total of 10 human TRBV genes encoding polyacidic residues near CDR2: TRBV7-3, TRBV11-1, TRBV11-2, TRBV11-3, TRBV12-3, TRBV12-4, TRBV12-5, TRBV14, TRBV16, and TRBV18. The corresponding multiple sequence alignment is presented in Supplemental Figure 5. With the exception of TRBV11-1, 11-2, and 11-3, all others have been identified as Vβ chains specific to SAgs secreted by Staphylococcus aureus and other select Staphylococcus species that lead to TRBV skewing in TSS patients (29). Notably, TRBV11-2 and TRBV14 share 3 homologous binding paratopes (blue boxes in Figure 6D and Supplemental Figure 6) and may share similar mechanisms of action.
Finally, although the amino acid sequences corresponding to TRBV11-1, -2, and -3 are highly similar (all being TRBV11 subtypes), their most important differences reside in their CDR2 sequence. Notably, TRBV11-2 CDR2 encodes a series of hydrophobic and polar residues that would favor interfacial contacts with the spike, which would bury the hydrophobic residues, whereas the CDR2s of TRBV11-1 and 11-3 each contain 2 or 3 acidic residues in addition to D67-D68. Such a cluster of 4 or 5 negatively charged residues that are closely neighboring in sequence (and space) may give rise to destabilizing effects at possible protein-protein interfaces, and would favor instead an exposure to the solution. Thus, the sequence difference between these TRBV11 subtypes seems to explain the significantly higher binding propensity of TRBV11-2, compared with TRBV11-1 and -3.
Discussion
Here, we report the identification of a TCR signature in two-thirds of patients with severe MIS-C that consists of (a) a massive CDR3-independent TCR Vβ skewing toward TRBV11-2 and (b) an association with HLA-I alleles A02, B35, and C04. In these children, up to 24% of the clonal T cell space is occupied by T cell clones with identical TRBV genes (but completely heterogeneous CDR3), suggesting that this massive expansion of a whole class of T cells may be an important pathophysiological mechanism underlying severe MIS-C. The elevated cytokine levels associated with this signature may indicate that this class of T cells is not only expanded, but also activated, thereby driving the cytokine storm in MIS-C. Moreover, the HLA association of these cases could explain why only a small proportion of children develop MIS-C after COVID-19 infection. Our structural modeling experiments suggest that unprocessed SARS-CoV-2 interacts with specific residues of the Vβ encoded by TRBV11-2 via its P681RRAR685 motif at the C-terminus of the S1 trimer, which has been previously reported as a superantigenic motif in adults with severe hyperinflammatory COVID-19 (18). Together, our data indicate that a CDR3-independent, HLA-I–associated, and hyperinflammatory T cell expansion is one of the essential features in the pathobiology of severe MIS-C, with an expansion pattern that is reminiscent of SAg-like stimulation.
While these data are intriguing, many questions remain unanswered. First, further examination is required to determine whether the CDR3-independent, HLA-I–associated T cell expansion occurs through a true superantigenic activity of the SARS-CoV-2 spike or through conventional presentation of processed SARS-CoV-2 antigenic peptides by the HLA-I binding groove to CD8+ T cells. Our structural modeling argues strongly in favor of a superantigenic interaction that relies on HLA-I restriction. This is at first glance counterintuitive, given that classical bacterial SAgs act via MHC class II interactions (16). However, it raises the possibility that MHC class I may act as a ligand to present antigen to TRBV11-2 in a mechanism independent of CDR3 recognition. This may occur in a similar way to that of MHC class II presentation of SAgs to TCR. Several studies have shown that MHC class II is not essential for SAg activation of T cells. Indeed, the MHC class II interaction of SAgs can be functionally replaced with an alternative receptor, as demonstrated with recombinant bacterial SAgs engineered as cancer immunotherapeutics to bind tumor-associated antigens (30, 31). In addition, the concept that MHC class I can act as a ligand for bacterial SAgs like SEB has been previously reported (32). It is noteworthy that while there is evidence that HLA-II allelic variation determines the severity of streptococcal SAg-mediated diseases (24), the HLA-I association observed in our cohort appears much stronger, potentially explaining the rarity of MIS-C. Moreover, the considerable CDR3 heterogeneity in the TRBV11-2 TCR repertoire of our MIS-C cohort also argues against conventional antigen presentation and TCR interaction. The V gene forms an essential part of CDR3, and in some instances public clonotypes recognizing viral epitopes share a specific TRBV gene. Yet, such public responses require some additional paratope similarity visible through CDR3 length restriction (33–36), TRBJ gene bias (33, 36–38), shared sequence motives (33, 36–39), or even identical CDR3 amino acid sequences (36, 37). In our analysis of MIS-C repertoires (Figure 3) we did not find any of these features. Furthermore, classical SAgs can induce both CD4+ and CD8+ T cell expansion. A recent preprint from an independent group also identified a TRBV11-2 expansion in MIS-C, and found this expansion in both CD4+ and CD8+ T cells (40). This would not be expected for HLA-I–associated conventional antigen activation, and may be further support of a SAg-like response.
Moreover, additional investigation is needed to further define the mechanism by which 3 specific MHC class I molecules are involved in the CDR3-independent TRBV11-2 expansion. While computational studies suggest the spike protein may possess SAg-like activity, in vitro functional studies are needed to support these findings. However, this awaits more advanced molecular tools, since expression of a stable recombinant spike protein currently still requires mutation of the R682RAR685 motif (41), which removes the putative SAg-like structure. Furthermore, exposure of the SARS-CoV-2 SAg motif depends on certain conformations of the spike protein, which are difficult to replicate in an in vitro system.
Studies examining the T cell compartment in MIS-C show an overall depletion of CD4+ and CD8+ T cells yet enhanced activation, particularly in a subset of vascular patrolling T cells (CX3CR1+ T cells; refs. 42–45). In addition, immune profiling of MIS-C patients has identified the presence of IgG and IgA autoantibodies that recognize endothelial cells, indicating a breakdown of B cell tolerance relevant for the endothelial compartment (42, 43). SAg-activated cytotoxic T cells have been shown to mediate killing of inflammatory endothelial cells (46). The HLA-I bias identified in our severe MIS-C patients raises the possibility that MHC class I expression on endothelial cells could mediate the SAg-like activation of T cells, thereby indirectly promoting endothelial cell damage and contributing to the endothelial dysfunction and multisystem inflammation typically observed in MIS-C patients. However, the manner in which these processes may be linked remains to be determined in future studies.
Approximately one-third or fewer of MIS-C patients were reported to test positive for SARS-CoV-2 by nasopharyngeal PCR (6–11). This suggests that SARS-CoV-2 causes a postinfectious, delayed hyperinflammatory immune response in certain children. Such a delayed response could be consistent with the effect of a viral SAg. Indeed, the ability of SAgs to induce a delayed immune response is supported by a number of studies (47–49), and delayed-onset TSS is frequently observed for nonmenstrual TSS (50, 51). SAgs have also been implicated in various autoimmune diseases (52). For example, the SAg-producing bacteria Streptococcus pyogenes can induce postinfectious acute rheumatic fever 2 to 4 weeks after pharyngeal infection (52). Interestingly, rheumatic fever primarily affects children ages 5 to 15 and is rare in adults, which is a similar demographic as MIS-C (53–55). One hypothesis is that SAgs induce autoimmunity by triggering self-reactive T cells (17, 52). As discussed above, SAg activation of autoantibodies in MIS-C may also contribute to a postinfectious autoimmune response. Another potential explanation could be that the delayed response results from insufficient control of the primary infection, for example by lack of neutralizing antibodies (56, 57), and lingering of virus in the GI tract (58).
Our structure-based in silico modeling indicated that the Vβ encoded by TRBV11-2 can engage in a CDR3-independent interaction with the SAg-like motif of SARS-CoV-2 spike protein. Interestingly, a number of very recent studies implicate the P681RRAR685 SAg-like motif we identified (18) in viral pathogenesis (27, 59–61). SARS-CoV-2 S1/S2 cleavage by proteases (including furin and TMPRSS2) is an important mechanism of cell infection and exposes the P681RRAR685 motif at the C-terminus of the S1 trimer. The same polybasic site has been recently shown to bind to the cell surface receptor neuropilin-1 (NRP1), which is important for SARS-CoV-2 cellular infectivity (60). Further, 2 independent studies show that deletion of the P681RRAR685 cleavage site (by natural deletion or mutagenesis) attenuates viral pathogenesis in hamster models (27, 61). Therefore, targeting this SAg-like motif might provide a new therapeutic avenue to treat and prevent severe COVID-19, MIS-C, and MIS-A.
Overall, our data show a unique TCR repertoire in MIS-C patients, characterized by a profound expansion of TRBV11-2, which is consistent with a SAg-like T cell skewing. Future investigations into the mechanism of how HLA-I may participate in TRBV11-2 expansion in a CDR3-independent manner are needed to unravel this mechanism and the potential that genetic predisposition plays a role in MIS-C. Furthermore, characterization of the phenotype and functional properties of MIS-C patient T cells utilizing TRBV11-2, and how this skewing relates to other pathogenic mechanisms in MIS-C, are needed to provide a complete understanding of the disease pathogenesis.
Methods
Patient samples.
Samples consisted of mild MIS-C (n = 4), severe MIS-C (n = 16), and age-matched febrile control patients (n = 15). MIS-C severity classification was based on admission to pediatric ICU requiring vasopressor use (severe) or no ICU admission (mild). Median ages of febrile controls, mild, and severe MIS-C were 12, 9, and 10.5, respectively. Ninety-five percent of patients were serology positive for SARS-CoV-2 and 30% of patients were PCR positive by nasopharyngeal swab for SARS-CoV-2. Eighty-five percent of samples were collected prior to IVIG administration.
TCR immunosequencing.
RNA was isolated form peripheral blood of febrile controls and MIS-C patients and assessed for quality with a bioanalyzer (Agilent). Sequencing of the TCR genes was performed using the QIAseq Immune Repertoire RNA Library Kit (QIAGEN) and the NovaSeq 6000 system (2 × 250 bp, 11 million average reads per sample).
In an additional cohort of 254 healthy individuals of all age groups, the TRB genetic locus was amplified using 500 ng DNA from peripheral blood mononuclear cells (PBMCs) in 2 consecutive PCR reactions using BIOMED2 TRB A/B primer pools (62). During the first PCR, fragments were tagged with Illumina-compatible adapters for hybridization to the flow cell. After bead purification (AMPure XP, Beckmann Coulter), each sample was tagged with a unique 7-nucleotide index during the second PCR. All PCRs were performed using Phusion HS II (Thermo Fisher Scientific). After the second PCR, amplicons were resolved by gel electrophoresis and purified using NucleoSpin Gel and PCR Clean-up (Macherey-Nagel). Library quantification and quality control were performed using Qubit (QIAGEN) and an Agilent 2100 Bioanalyzer, respectively.
TCR-immunosequencing analysis.
The MiXCR framework (3.0.8; ref. 63) was used for annotation of TCR rearrangements and clone construction, whereby the default MiXCR library served as reference for sequence alignment and each unique CDR3 nucleotide sequence was defined as 1 clone. Only productive sequences with a read count of 2 or greater were considered for further analysis. To account for differences in sequencing depth, TRB and TRA repertoires were normalized to 1.5 million reads; TRG and TRD were normalized to 50,000 reads per repertoire. All analyses and data plotting were performed using R version 3.5.1. Broad repertoire metrics (clonality, diversity, richness), gene usage, and repertoire overlap were analyzed using packages tcR (64) and ade4 (65) as previously described (66, 67). CDR3 length distributions were visualized with package pheatmap (68) and represent the clonal abundance within each repertoire.
Bubble diagrams, which depict the clonal distribution of repertoires, were generated using packages packcircles (69) and ggplot2 (70). For plotting purposes, repertoires were sampled down to 20,000 reads. The area size of the bubbles reflects the clonal fraction.
CDR3 amino acid sequence similarity was assessed using GLAM2 (71) and FastTreeMP (72). For GLAM2, the 50 most abundant CDR3 amino acid sequences of TRBV11-2 clones were used as input to generate the alignment LOGOs. Another alignment approach was to focus on the most frequent 100 clones of each repertoire, irrespective of V gene. The amino acid sequence covering FR2 to CDR3 or CDR3 only of the rearranged TRB locus was used to infer sequence similarities with FastTreeMP (72) and the data were visualized and analyzed using the Archaeopteryx viewer (0.9928 beta) (73). Fastq raw data are deposited in the European Nucleotide Archive (ENA) under accession number PRJEB43418.
HLA genotyping and analysis.
Patients were genotyped using a sequence-specific primer PCR–based low-resolution technique (Innotrain and Protrans.). HLA-I loci A, B, and C (Cw) and HLA-II loci DRB1 (DR), DRB3 (DR52), DRB4 (DR53), DRB5 (DR51), and DQB1 (DQ) were analyzed.
Serum cytokine analysis.
Quantification of cytokines in serum was performed with a highly sensitive multiplex enzyme-linked immunosorbent assay (ELISA) using electrochemiluminescence detection technology (Meso Scale Discovery). Cytokine correlation with TRBV11-2 usage was assessed by Spearman’s r correlation test.
In silico structural modeling.
The structural model for SARS-CoV-2 spike was generated using SwissModel (74) based on the resolved cryo-EM structures (PDB: 6VSB [ref. 41] and 6VXX [ref. 75]). The structure of the TCR with 100% Vβ domain sequence identity to TRBV11-2 was taken from the crystal structure of HLA B*0801 in complex with a peptide (HSKKKCDEL) and DD31 TCR (PDB: 4QRP) (25). Both α and β chains of the TCR (25) (respective chains D and E in PDB 4QRP) were adopted for docking.
Generation of the TCR-spike binary complexes: Using protein-protein docking software ClusPro (26), we constructed in silico a series of binary complexes for SARS-CoV-2 spike and TCR-TRBV11-2, following the protocol we used in our previous study (18). Given that the constant domain is proximal to the cell membrane and TCR employs the variable domain for binding SAgs and/or antigen-MHC complexes (23), we added restraints to our docking simulations to prevent the binding of the TCR constant domain. An ensemble of 460 models was generated for the complex, which were grouped in 30 clusters based on their structural similarities. Among them, 3 displayed binding between the spike and TRBV11-2 at the SAg-like region containing the polybasic insert PRRA.
Statistics.
Analysis of principal components was performed with the Pillai-Bartlett test in multivariate ANOVA (MANOVA). Cytokine correlation with TRBV11-2 usage was assessed by Spearman’s r correlation test. A P value of 0.05 or less was considered statistically significant.
Study approval.
Biobanked, deidentified, patient blood remnant samples as well as control samples were obtained from Massachusetts General Hospital, Cedars-Sinai Medical Center, Loma Linda University Hospital, and Martin-Luther-University Halle-Wittenberg under ethics committee approval and informed consent.
Author contributions
RAP, LP, MNR, IB, MB, and MA conceptualized the study. RAP, LP, DS, C Schultheiß, MHC, and C Santiskulvong conducted the investigation. LMY, HC, ML, JVE, and AF provided resources. RAP, LP, MNR, MHC, DS, C Schultheiß, IB, MB, JKM, and MA analyzed data. RAP, LP, MNR, MHC, IB, MB, JKM, and MA wrote the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from NIH awards P41 GM103712 (to IB) and R01 AI072726 and 3RO1AI072726-10S1 (to MA).
Version 1. 03/11/2021
In-Press Preview
Version 2. 05/17/2021
Electronic publication
Funding Statement
to Ivet Bahar
to Moshe Arditi
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(10):e146614.https://doi.org/10.1172/JCI146614.
See the related Commentary at SARS-CoV-2 as a superantigen in multisystem inflammatory syndrome in children.
Contributor Information
Rebecca A. Porritt, Email: Rebecca.Porritt@csmc.edu.
Lisa Paschold, Email: lisa.paschold@uk-halle.de.
Magali Noval Rivas, Email: Magali.novalrivas@cshs.org.
Mary Hongying Cheng, Email: hoc2@pitt.edu.
Lael M. Yonker, Email: LYONKER@mgh.harvard.edu.
Harsha Chandnani, Email: HChandnani@llu.edu.
Merrick Lopez, Email: MLopez@llu.edu.
Donjete Simnica, Email: donjete.simnica@uk-halle.de.
Christoph Schultheiß, Email: christoph.schultheiss@uk-halle.de.
Chintda Santiskulvong, Email: Chintda.Santiskulvong@cshs.org.
Jennifer Van Eyk, Email: Jennifer.VanEyk@cshs.org.
John K. McCormick, Email: john.mccormick@uwo.ca.
Alessio Fasano, Email: afasano@mgh.harvard.edu.
Ivet Bahar, Email: bahar@pitt.edu.
Mascha Binder, Email: mascha.binder@uk-halle.de.
Moshe Arditi, Email: moshe.arditi@cshs.org.
References
- 1.Huang C, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cristiani L, et al. Will children reveal their secret? The coronavirus dilemma. Eur Respir J. doi: 10.1183/13993003.00749-2020. [published online April 2, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tay MZ, et al. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vabret N, et al. Immunology of COVID-19: current state of the science. Immunity. 2020;52(6):910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. American Academy of Pediatrics. Children and COVID-19: state-level data report. Updated March 8, 2021. Accessed March 9, 2021. https://services.aap.org/en/pages/2019-novel-coronavirus-covid-19-infections/children-and-covid-19-state-level-data-report/
- 6.Riphagen S, et al. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395(10237):1607–1608. doi: 10.1016/S0140-6736(20)31094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verdoni L, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet. 2020;395(10239):1771–1778. doi: 10.1016/S0140-6736(20)31103-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belhadjer Z, et al. Acute heart failure in multisystem inflammatory syndrome in children (MIS-C) in the context of global SARS-CoV-2 pandemic. Circulation. 2020;142(5):429–436. doi: 10.1161/CIRCULATIONAHA.120.048360. [DOI] [PubMed] [Google Scholar]
- 9.Whittaker E, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. 2020;324(3):259–269. doi: 10.1001/jama.2020.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toubiana J, et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: prospective observational study. BMJ. 2020;369:m2094. doi: 10.1136/bmj.m2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung EW, et al. Multisystem inflammatory syndrome related to COVID-19 in previously healthy children and adolescents in New York City. JAMA. 2020;324(3):294–296. doi: 10.1001/jama.2020.10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris SB, et al. Case series of multisystem inflammatory syndrome in adults associated with SARS-CoV-2 infection — United Kingdom and United States, March-August 2020. MMWR Morb Mortal Wkly Rep. 2020;69(40):1450–1456. doi: 10.15585/mmwr.mm6940e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Low DE. Toxic shock syndrome: major advances in pathogenesis, but not treatment. Crit Care Clin. 2013;29(3):651–675. doi: 10.1016/j.ccc.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Cook A, et al. Manifestations of toxic shock syndrome in children, Columbus, Ohio, USA, 2010–2017. Emerg Infect Dis. 2020;26(6):1077–1083. doi: 10.3201/eid2606.190783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pierce CA, et al. Immune responses to SARS-CoV-2 infection in hospitalized pediatric and adult patients. Sci Transl Med. 2020;12(564):eabd5487. doi: 10.1126/scitranslmed.abd5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krakauer T. Staphylococcal superantigens: pyrogenic toxins induce toxic shock. Toxins (Basel) 2019;11(3):178. doi: 10.3390/toxins11030178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, et al. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17(1):435–466. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- 18.Cheng MH, et al. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Proc Natl Acad Sci U S A. 2020;117(41):25254–25262. doi: 10.1073/pnas.2010722117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schultheiss C, et al. Next-generation sequencing of T and B cell receptor repertoires from COVID-19 patients showed signatures associated with severity of disease. Immunity. 2020;53(2):442–455. doi: 10.1016/j.immuni.2020.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nur-ur Rahman AK, et al. The T cell receptor beta-chain second complementarity determining region loop (CDR2beta governs T cell activation and Vbeta specificity by bacterial superantigens. J Biol Chem. 2011;286(6):4871–4881. doi: 10.1074/jbc.M110.189068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corrie BD, et al. iReceptor: a platform for querying and analyzing antibody/B-cell and T-cell receptor repertoire data across federated repositories. Immunol Rev. 2018;284(1):24–41. doi: 10.1111/imr.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. doi: 10.21203/rs.3.rs-51964/v1. Nolan S, et al. A large-scale database of T-cell receptor beta (TCRbeta) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2 [preprint]. Posted on Research Square August 4, 2020. [DOI]
- 23.Saline M, et al. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat Commun. 2010;1(1):119. doi: 10.1038/ncomms1117. [DOI] [PubMed] [Google Scholar]
- 24.Kotb M, et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med. 2002;8(12):1398–1404. doi: 10.1038/nm1202-800. [DOI] [PubMed] [Google Scholar]
- 25.Nivarthi UK, et al. An extensive antigenic footprint underpins immunodominant TCR adaptability against a hypervariable viral determinant. J Immunol. 2014;193(11):5402–5413. doi: 10.4049/jimmunol.1401357. [DOI] [PubMed] [Google Scholar]
- 26.Kozakov D, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. doi: 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lau SY, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg Microbes Infect. 2020;9(1):837–842. doi: 10.1080/22221751.2020.1756700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefranc MP, et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37(suppl_1):D1006-D12. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuffs SW, et al. Manipulation of innate and adaptive immunity by staphylococcal superantigens. Pathogens. 2018;7(2):53. doi: 10.3390/pathogens7020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forsberg G, et al. Therapy of human non-small-cell lung carcinoma using antibody targeting of a modified superantigen. Br J Cancer. 2001;85(1):129–136. doi: 10.1054/bjoc.2001.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedlund G, et al. The tumor targeted superantigen ABR-217620 selectively engages TRBV7-9 and exploits TCR-pMHC affinity mimicry in mediating T cell cytotoxicity. PLoS One. 2013;8(10):e79082. doi: 10.1371/journal.pone.0079082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haffner AC, et al. Major histocompatibility complex class I molecule serves as a ligand for presentation of the superantigen staphylococcal enterotoxin B to T cells. Proc Natl Acad Sci U S A. 1996;93(7):3037–3042. doi: 10.1073/pnas.93.7.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miles JJ, et al. Antigen-driven patterns of TCR bias are shared across diverse outcomes of human hepatitis C virus infection. J Immunol. 2011;186(2):901–912. doi: 10.4049/jimmunol.1003167. [DOI] [PubMed] [Google Scholar]
- 34.Day EB, et al. Structural basis for enabling T-cell receptor diversity within biased virus-specific CD8+ T-cell responses. Proc Natl Acad Sci U S A. 2011;108(23):9536–9541. doi: 10.1073/pnas.1106851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sant S, et al. Single-cell approach to influenza-specific CD8+ T cell receptor repertoires across different age groups, tissues, and following influenza virus infection. Front Immunol. 2018;9:1453. doi: 10.3389/fimmu.2018.01453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miles JJ, et al. CTL recognition of a bulged viral peptide involves biased TCR selection. J Immunol. 2005;175(6):3826–3834. doi: 10.4049/jimmunol.175.6.3826. [DOI] [PubMed] [Google Scholar]
- 37.Price DA, et al. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity. 2004;21(6):793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, et al. Profiling the repertoire of T-cell receptor beta-chain variable genes in peripheral blood lymphocytes from subjects who have recovered from acute hepatitis B virus infection. Cell Mol Immunol. 2014;11(4):332–342. doi: 10.1038/cmi.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehner PJ, et al. Human HLA-A0201-restricted cytotoxic T lymphocyte recognition of influenza A is dominated by T cells bearing the V beta 17 gene segment. J Exp Med. 1995;181(1):79–91. doi: 10.1084/jem.181.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. doi: 10.1101/2020.12.01.20241364. Ramaswamy A, et al. Post-infectious inflammatory disease in MIS-C features elevated cytotoxicity signatures and autoreactivity that correlates with severity [preprint]. Posted on medRxiv December 4, 2020. [DOI]
- 41.Wrapp D, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consiglio CR, et al. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell. 2020;183(4):968–981. doi: 10.1016/j.cell.2020.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gruber CN, et al. Mapping systemic inflammation and antibody responses in multisystem inflammatory syndrome in children (MIS-C) Cell. 2020;183(4):982–995. doi: 10.1016/j.cell.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carter MJ, et al. Peripheral immunophenotypes in children with multisystem inflammatory syndrome associated with SARS-CoV-2 infection. Nat Med. 2020;26(11):1701–1707. doi: 10.1038/s41591-020-1054-6. [DOI] [PubMed] [Google Scholar]
- 45. doi: 10.1101/2020.09.25.20201863. Vella L, et al. Deep immune profiling of MIS-C demonstrates marked but transient immune activation compared to adult and pediatric COVID-19 [preprint]. Posted on medRxiv September 27, 2020. [DOI] [PMC free article] [PubMed]
- 46.Riesbeck K, et al. Endothelial cells expressing an inflammatory phenotype are lysed by superantigen-targeted cytotoxic T cells. Clin Diagn Lab Immunol. 1998;5(5):675–682. doi: 10.1128/CDLI.5.5.675-682.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Acha-Orbea H. Bacterial and viral superantigens: roles in autoimmunity? Ann Rheum Dis. 1993;52 suppl 1:S6–16. doi: 10.1136/ard.52.suppl_1.s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Posnett DN. Do superantigens play a role in autoimmunity? Semin Immunol. 1993;5(1):65–72. doi: 10.1006/smim.1993.1009. [DOI] [PubMed] [Google Scholar]
- 49.Schiffenbauer J. Superantigens and their role in autoimmune disorders. Arch Immunol Ther Exp (Warsz) 1999;47(1):17–24. [PubMed] [Google Scholar]
- 50.Kain KC, et al. Clinical spectrum of nonmenstrual toxic shock syndrome (TSS): comparison with menstrual TSS by multivariate discriminant analyses. Clin Infect Dis. 1993;16(1):100–106. doi: 10.1093/clinids/16.1.100. [DOI] [PubMed] [Google Scholar]
- 51.Reingold AL, et al. Nonmenstrual toxic shock syndrome: a review of 130 cases. Ann Intern Med. 1982;96(6 pt 2):871–874. doi: 10.7326/0003-4819-96-6-871. [DOI] [PubMed] [Google Scholar]
- 52.Hurst JR, et al. Streptococcal pharyngitis and rheumatic heart disease: the superantigen hypothesis revisited. Infect Genet Evol. 2018;61:160–175. doi: 10.1016/j.meegid.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 53.Lawrence JG, et al. Acute rheumatic fever and rheumatic heart disease: incidence and progression in the Northern Territory of Australia, 1997 to 2010. Circulation. 2013;128(5):492–501. doi: 10.1161/CIRCULATIONAHA.113.001477. [DOI] [PubMed] [Google Scholar]
- 54.Parnaby MG, Carapetis JR. Rheumatic fever in indigenous Australian children. J Paediatr Child Health. 2010;46(9):527–533. doi: 10.1111/j.1440-1754.2010.01841.x. [DOI] [PubMed] [Google Scholar]
- 55.Seckeler MD, et al. The persistent challenge of rheumatic fever in the Northern Mariana Islands. Int J Infect Dis. 2010;14(3):e226–e229. doi: 10.1016/j.ijid.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 56.Andrews MM, et al. Recurrent nonmenstrual toxic shock syndrome: clinical manifestations, diagnosis, and treatment. Clin Infect Dis. 2001;32(10):1470–1479. doi: 10.1086/320170. [DOI] [PubMed] [Google Scholar]
- 57.Weisberg SP, et al. Distinct antibody responses to SARS-CoV-2 in children and adults across the COVID-19 clinical spectrum. Nat Immunol. 2021;22(1):25–31. doi: 10.1038/s41590-020-00826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y, et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat Med. 2020;26(4):502–505. doi: 10.1038/s41591-020-0817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cantuti-Castelvetri L, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Daly JL, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370(6518):861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson BA, et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. doi: 10.1038/s41586-021-03237-4. [published online January 25, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van Dongen JJM, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 concerted action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 63.Bolotin DA, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12(5):380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 64.Nazarov VI, et al. tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics. 2015;16:175. doi: 10.1186/s12859-015-0613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dray S, Dufour AB. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22(4):10.18637/jss.v022.i04 [Google Scholar]
- 66.Simnica D, et al. T cell receptor next-generation sequencing reveals cancer-associated repertoire metrics and reconstitution after chemotherapy in patients with hematological and solid tumors. OncoImmunology. 2019;8(11):e1644110. doi: 10.1080/2162402X.2019.1644110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simnica D, et al. High-throughput immunogenetics reveals a lack of physiological T cell clusters in patients with autoimmune cytopenias. Front Immunol. 2019;10:1897. doi: 10.3389/fimmu.2019.01897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. pheatmap: Pretty Heatmaps. Version 1.0.12. Kolde R; 2019. Accessed March 9, 2021. https://rdrr.io/cran/pheatmap/
- 69. packcircles: Circle Packing. Version 0.3.3. Bedward M, et al; 2018. Accessed March 9, 2021. https://cran.r-project.org/web/packages/packcircles/index.html.
- 70. Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer; 2016. [Google Scholar]
- 71.Frith MC, et al. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput Biol. 2008;4(4):e1000071. doi: 10.1371/journal.pcbi.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Price MN, et al. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han MV, Zmasek CM. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics. 2009;10:356. doi: 10.1186/1471-2105-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Waterhouse A, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walls AC, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;180:281–292. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.