

Abstract
Aims
Activity in the amygdala, a brain centre involved in the perception of and response to stressors, associates with: (i) heightened sympathetic nervous system and inflammatory output and (ii) risk of cardiovascular disease. We hypothesized that the amygdalar activity (AmygA) ratio is heightened among individuals who develop Takotsubo syndrome (TTS), a heart failure syndrome often triggered by acute stress. We tested the hypotheses that (i) heightened AmygA precedes development of TTS and (ii) those with the highest AmygA develop the syndrome earliest.
Methods and results
Individuals (N=104, median age 67.5 years, 72% female, 86% with malignancy) who underwent clinical 18 F-FDG-PET/CT imaging were retrospectively identified: 41 who subsequently developed TTS and 63 matched controls (median follow-up 2.5 years after imaging). AmygA was measured using validated methods. Individuals with (vs. without) subsequent TTS had higher baseline AmygA (P=0.038) after adjusting for TTS risk factors. Further, AmygA associated with the risk for subsequent TTS after adjustment for risk factors [standardized hazard ratio (95% confidence interval): 1.643 (1.189, 2.270), P=0.003]. Among the subset of individuals who developed TTS, those with the highest AmygA (>mean + 1 SD) developed TTS ∼2 years earlier after imaging vs. those with lower AmygA (P=0.028).
Conclusion
Higher AmygA associates with an increased risk for TTS among a retrospective population with a high rate of malignancy. This heightened neurobiological activity is present years before the onset of TTS and may impact the timing of the syndrome. Accordingly, heightened stress-associated neural activity may represent a therapeutic target to reduce stress-related diseases, including TTS.
Keywords: Takotsubo syndrome, Stress, 18F-FDG-PET/CT, Amygdalar activity
Graphical Abstract

Relationship between stress-associated neurobiological activity on 18F-FDG-PET/CT imaging and risk for subsequent Takotsubo syndrome. 18F-FDG-PET/CT, 18F-fluorodeoxyglucose positron emission tomography/computed tomography; TL, temporal lobe; TTS Takotsubo syndrome; vmPFC, ventromedial prefrontal cortex.
See page 1909 for the editorial comment on this article (doi: 10.1093/eurheartj/ehab026)
Introduction
Takotsubo syndrome (TTS) is an acute, usually reversible heart failure syndrome that is often triggered by acute emotional or physical stressors.1 Although an underlying link between the brain and heart has long been proposed as a critical factor in the development of TTS,2 , 3 the underlying mechanisms require further clarification.
The response to stressors, such as those that could trigger TTS, is governed by an ensemble of neural structures within the limbic system, which notably includes the amygdala.4 Heightened activity within such stress-associated neural tissues augments sympathetic nervous system (SNS) activity, increases leucopoietic activity, and has recently been shown to contribute to the pathogenesis of multiple chronic stress-related diseases, including atherosclerosis.4–8 Such alterations in stress-associated neural activity, along with the downstream physiologic consequences, have likewise been hypothesized to play a role in TTS pathogenesis.2 , 6 , 9 , 10 Within such a construct, chronically heightened stress-associated neural activity (including the amygdala as a key component) may hypothetically prime an individual to mount a more vigorous neurophysiologic response to subsequent stressors, thereby increasing TTS risk. In support of this hypothesis, functional magnetic resonance imaging (fMRI) studies have demonstrated altered neural connectivity in several stress-associated limbic brain regions (i.e. amygdala, anterior cingulate cortex, and hippocampus) among individuals with prior TTS.2 , 3 , 9 However, it remains unknown whether the observed alterations in neural activity predate TTS, or if they develop as a consequence of the syndrome. Accordingly, further study is needed to assess whether heightened stress-associated neural activity exists prior to the development of TTS and thus may represent a risk factor for the syndrome.
18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG-PET/CT) allows the assessment of stress-associated neurobiological activity, such as amygdalar activity (AmygA; a ratio of amygdalar metabolic activity divided by that of regulatory brain regions). AmygA is elevated in the setting of increased perceived stress and chronic stress conditions, is stable for up to 6 months, and associates with an increased risk of subsequent cardiovascular disease (CVD).4 , 5 , 11 Moreover, 18F-FDG-PET/CT imaging enables simultaneous assessment of extra-neural tissues, including bone marrow (BM) activity (an index of leucopoiesis), which partially mediates the relationship between increased AmygA and CVD.4 Thus, 18F-FDG-PET/CT provides a unique opportunity to evaluate the roles of neural and BM activities in linking stress to downstream TTS.
Accordingly, we leveraged 18F-FDG-PET/CT imaging in a retrospective cohort of individuals who underwent clinically indicated 18F-FDG-PET/CT imaging prior to TTS onset to test our hypotheses that (i) individuals who develop TTS have heightened AmygA prior to TTS onset, and (ii) among those with subsequent TTS, higher AmygA associates with a shorter duration between imaging and TTS.
Methods
Study design and population
The Partners HealthCare System Research Patient Data Registry (RPDR) was used to identify patients and controls for this retrospective case–control study. The RPDR is a centralized clinical data warehouse for >4.5 million patients from Massachusetts General Hospital (MGH) and Brigham and Women’s Hospital, as well as affiliated community and specialty hospitals in the Boston area (Figure 1). The data registry includes structured and unstructured electronic medical record information, including socio-demographic data, vital signs, laboratory and test results, problem list entries, prescribed medications, billing codes, and clinical notes for healthcare services provided within the system. Using the RPDR online query tool, we first identified individuals with a diagnosis of TTS who had undergone PET/CT imaging at MGH (Boston, MA, USA) between 2005 and 2019, by utilizing the diagnostic terms: ‘Takotsubo cardiomyopathy’, ‘Takotsubo syndrome’, ‘stress-induced cardiomyopathy’, ‘transient left ventricular apical ballooning syndrome’, and ‘reversible left ventricular dysfunction following sudden emotional stress’ among individuals who had undergone clinically indicated whole-body 18F-FDG-PET/CT imaging. The initial search identified 190 individuals. Next, individuals were manually excluded if (i) TTS occurred prior to imaging, (ii) they had any of the following clinical findings prior to imaging: acute atherosclerotic myocardial infarction, ischaemic cardiomyopathy, decompensated congestive heart failure, severe left-sided valvular heart disease, hypertrophic cardiomyopathy, intracranial tumours, posterior reversible encephalopathy syndrome, history of craniotomy, prior stroke or seizure disorder, or (iii) they had inadequate 18F-FDG-PET/CT imaging to measure AmygA. Further, two cardiologists blinded to imaging data (MTO and AT) adjudicated all potential cases of TTS using European Society of Cardiology–Heart Failure Association criteria using the available clinical data.12 If a diagnosis of TTS was not supported by those criteria after a detailed review, potential cases were excluded. The remaining cases were retained and were classified as probable, possible, or less likely TTS (Supplementary material online, Table S1A) to acknowledge uncertainty related to the retrospective nature of the evaluation and the fact that not all subjects had important clinical data (e.g. coronary angiography or follow-up functional assessment). Patients who met diagnostic criteria with a high clinical likelihood of TTS were classified as probable, those who met criteria for TTS with an alternative diagnosis that could not be fully excluded but could potentially explain the presentation were classified as possible, and patients who met criteria for TTS but carried another diagnosis that likely could have explained the presentation were classified as less likely. Of 190 TTS cases initially identified by the data search, ultimately 41 met inclusion criteria.
Figure 1.

Consort diagram for the selection of cases and controls. ESC-HFA, European Society of Cardiology-Heart Failure Association; FDG PET/CT, fluorodeoxyglucose positron emission tomography/computed tomography; HCM, hypertrophic cardiomyopathy; MGH, Massachusetts General Hospital; RPDR Research Patient Data Registry; TTS, Takotsubo syndrome.
Next, controls were matched to TTS cases in three steps. First, utilizing the RPDR online query tool, an initial group of 1368 individuals was identified via an automated matching algorithm. To do so, TTS patients were matched to cases using age, sex, race, and ‘comparative health’. The comparative health measure, which is provided by the RPDR tool, utilizes a weighted sum of all diagnoses, procedures, medications, and health history as a proxy for healthcare usage. Using the RPDR query, 1368 potential controls were identified. Then, from within this group, controls were manually matched to TTS subjects (up to 3:1) by matching oncologic history (presence or absence of malignancy, malignancy type, stage, and treatment). A cohort of 133 potential controls was identified. Of this group, 31 were excluded as they met the pre-defined exclusion criteria (as used for the TTS group) and 39 individuals lacked coverage of the amygdala, resulting in a final cohort of 63 controls. The study complies with the Declaration of Helsinki. The Partners Human Research Committee approved its protocol.
18F-FDG-PET/CT imaging protocol and measurement of amygdalar and bone marrow activity
18F-FDG-PET/CT imaging was performed using an integrated scanner (e.g. Biograph 64 Siemens Healthcare, Erlangen, Germany, or similar) in the resting state. Intravenous 18F-FDG (∼370 MBq) was given following an overnight fast. Three-dimensional PET imaging was performed after ∼1 h of quiet waiting. A non-gated, non-contrast CT was acquired for attenuation correction.
Analysis of stress-associated neural activity was performed in a retrospective analysis using validated research methods by investigators blinded to clinical data.4 , 5 Amygdalar 18F-FDG uptake (Figure 2A) was measured by placing circular regions of interest (ROIs) over the right and left amygdalae and measuring tracer accumulation as a standardized uptake value (SUV). The bilateral amygdalar mean SUVs were assessed both as an average and individually by dividing by regulatory regional neural uptake to generate a ratio of amygdalar to regulatory activity.13 The primary measure for AmygA was derived by averaging the SUVs of the right and left amygdalae, and dividing that value by the mean temporal lobe (TL) SUV.13 Additionally, because the ventromedial prefrontal cortex (vmPFC) serves as a regulatory centre under stressful conditions, AmygA was also adjusted for mean vmPFC SUV in a secondary analysis.14 The inter- and intra-reader variability of AmygA (as the ratio of amygdalar to TL activity) were assessed on a random sample of 30 18F-FDG-PET/CT images derived from the current cohort. The absolute mean differences (standard deviation [SD]) in AmygA were 0.07 (0.04) and 0.05 (0.07), for inter- and intra-reader assessments, respectively. The absolute per cent differences (SD) in AmygA were 6.88 (3.95) and 5.20 (6.03) for inter- and intra-reader assessments, respectively. All 18F-FDG measurements were made using the axial, sagittal, and coronal planes. Additional details are provided in Supplementary material online.
Figure 2.

Amygdalar activity among subjects with and without subsequent Takotsubo syndrome. (A) Axial 18F-FDG-PET/CT of the amygdala shows increased uptake in a patient with (right) vs. lower uptake in a patient without (left) subsequent TTS. Amygdalar activity is adjusted for regulatory TL or ventromedial prefrontal cortex (vmPFC) activity. (B) Between-group differences in amygdalar activity in models adjusted for TTS risk factors are shown. Individual values are shown. Mean and 95% confidence intervals (error bars) are depicted. SUV, standardized uptake value; TL, temporal lobe; TTS, Takotsubo syndrome; vmPFC, ventromedial prefrontal cortex.
BM 18F-FDG uptake, a measure of leucopoietic activity, was measured by placing ROIs over the axial sections of each vertebrae from T1 to L5. The mean of the maximum SUV from each vertebra was calculated.4
Statistical analyses
Statistical analyses were performed using SPSS version 26 (IBM Corporation, Armonk, NY, USA). Continuous variables are listed as mean and SD, or as median and interquartile range [interquartile range (IQR), 25th–75th percentile] when skewed. Independent sample t-tests were used to compare continuous variables between groups. Categorical variables were compared using Chi-square or Fisher’s exact tests as appropriate. Effect sizes were evaluated with Cohen’s d tests. Logistic regression was employed to evaluate odds ratios (ORs), as exponential β with 95% confidence intervals (CIs), between baseline variables and subsequent TTS. Linear regression was used to identify factors associated with AmygA and test the association between higher AmygA (defined as ≥mean AmygA + 1 SD) with timing of TTS (among those with subsequent TTS) and BM activity. Cox regression was used to calculate hazard ratios (HRs) for TTS events. Backward selection was used where appropriate. Additionally, post hoc stratified subset sensitivity analyses were performed. A two-sided P-value <0.05 was considered significant. Additional details about covariable selection for multivariable models are provided in Supplementary material online.
Results
Study population
Overall, 104 individuals were studied, including 41 cases and 63 controls (median age 67.5 years [IQR: 59.5–74.0], 72% female, 86% with malignancy). Individuals with TTS and controls had similar baseline characteristics, except autoimmune diseases were more common in individuals with TTS (Table 1). Among TTS patients and controls, the interval between baseline imaging and TTS onset, last follow-up, or death was 2.5 [0.5–5.3] years (median [IQR]). Among the subgroup of individuals with subsequent TTS, the interval between imaging and TTS was 0.9 [0.1–4.1] years. Among the subgroup of controls, the interval between imaging and last Follow-up, or death was 2.9 [1.0–6.0] years. Among all individuals, female sex, diabetes, and chronic kidney disease are associated with higher AmygA (Table 2). Among the 41 TTS cases, four patients had recurrent TTS. Additional clinical details, including an expanded description of the TTS cases, can be found in Supplementary material online, Tables S1A–C.
Table 1.
Baseline characteristics
| Variable | Overall (n = 104) | Cases (n = 41) | Controls (n = 63) | P-value |
|---|---|---|---|---|
| Demographics | ||||
| Median age, years (IQR) | 67.5 (59.5–74) | 66 (59–74.5) | 68 (61–74) | 0.983 |
| Female sex | 75 (72.1) | 29 (70.7) | 46 (73) | 0.800 |
| Race | 0.826 | |||
| Caucasian | 93 (89.4) | 37 (90.2) | 56 (88.9) | |
| African American | 5 (4.8) | 2 (4.9) | 3 (5.2) | |
| Asian | 6 (5.8) | 2 (4.9) | 4 (6.9) | |
| Vital status | ||||
| Death during follow-up | 48 (46.2) | 22 (53.7) | 26 (41.3) | 0.216 |
| Cardiovascular risk factors and medications | ||||
| Hypertension | 73 (70.2) | 29 (70.7) | 44 (69.8) | 0.923 |
| Hyperlipidaemia | 54 (51.9) | 22 (53.7) | 32 (50.8) | 0.775 |
| Current smoking | 13 (12.5) | 8 (19.5) | 5 (7.9) | 0.081 |
| Former smoker | 59 (56.7) | 27 (65.9) | 32 (50.8) | 0.130 |
| Diabetes | 20 (19.2) | 6 (14.6) | 14 (22.2) | 0.337 |
| Aspirin | 37 (35.6) | 14 (34.1) | 23 (36.5) | 0.806 |
| Statins | ||||
| Any | 41 (39.4) | 15 (36.6) | 26 (41.3) | 0.633 |
| High intensity | 9 (8.7) | 3 (7.3) | 6 (9.5) | 0.696 |
| Beta blockers | 29 (27.9) | 12 (29.3) | 17 (27) | 0.800 |
| ACE inhibitors/ARBs | 36 (34.6) | 11 (26.8) | 25 (39.7) | 0.178 |
| Diuretics | 19 (18.3) | 7 (17.1) | 12 (19) | 0.799 |
| Cardiovascular diseases | ||||
| Coronary artery disease | 10 (9.6) | 3 (7.3) | 7 (11.1) | 0.521 |
| Malignancy history | ||||
| Prior malignancy | 89 (85.6) | 35 (85.4) | 54 (85.7) | 0.961 |
| Prior chemotherapy/radiation | 48 (46.2) | 16 (39) | 32 (50.8) | 0.239 |
| Chemotherapy/radiation at the time of imaging | 11 (10.6) | 2 (4.9) | 9 (14.3) | 0.127 |
| Cardiotoxic drugs (anthracyclines or trastuzumab) | 17 (16.3) | 5 (12.2) | 12 (19) | 0.356 |
| Median time interval between cancer diagnosis and imaging date, years (IQR) | 1.8 (0.3, 5.4) | 3.2 (0.4, 5.7) | 1.4 (0.3, 5.1) | 0.366 |
| Psychiatric history | ||||
| History of depression/anxiety | 43 (41.3) | 18 (43.9) | 25 (39.7) | 0.669 |
| Antidepressant drugs | 36 (34.6) | 18 (43.9) | 18 (28.6) | 0.108 |
| Antipsychotic drugs | 6 (5.8) | 4 (9.8) | 2 (3.2) | 0.160 |
| Other diseases | ||||
| COPD/asthma | 31 (29.8) | 16 (39) | 15 (23.8) | 0.097 |
| Chronic kidney disease | 18 (17.3) | 8 (19.5) | 10 (15.9) | 0.632 |
| Autoimmune disease | 16 (15.4) | 12 (29.3) | 4 (6.3) | 0.002 |
| Chronic immunosuppressive drug therapy | 9 (8.7) | 4 (9.8) | 5 (8.1) | 0.515 |
Values are shown as n (%), unless noted.
ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; COPD, chronic obstructive pulmonary disease; IQR, interquartile range.
Table 2.
Univariable predictors of Takotsubo syndrome and amygdalar activity
| Predictors | Logistic Regression |
Linear Regression |
||||
|---|---|---|---|---|---|---|
| Takotsubo syndrome |
Amygdalar Activity |
|||||
| Amygdalar activity divided by temporal lobe activity |
Amygdalar activity divided by medial prefrontal cortex activity |
|||||
| OR (95%CI) | P-value | Standardized β (95% CI) | P-value | Standardized β (95% CI) | P-value | |
| Age (years) |
1.000 (0.969, 1.031) |
0.983 |
0.001 (−0.015, 0.017) |
0.903 |
−0.007 (−0.024, 0.01) |
0.403 |
| Female sex |
1.120 (0.468, 2.680) |
0.800 |
0.726 (0.309, 1.143) |
0.001 |
0.573 (0.128, 1.018) |
0.012 |
| Race |
1.156 (0.316, 4.229) |
0.826 |
−0.407 (−1.045, 0.230) |
0.208 |
−0.711 (−1.365, −0.058) |
0.033 |
| Cardiovascular risk factors | ||||||
| Hypertension |
1.044 (0.441, 2.470) |
0.923 |
0.161 (−0.270, 0.592) |
0.460 |
−0.091 (−0.553, 0.371) |
0.696 |
| Hyperlipidaemia |
1.122 (0.510, 2.466) |
0.775 |
0.230 (−0.163, 0.622) |
0.249 |
0.363 (−0.056, 0.782) |
0.089 |
| Current smoking |
2.812 (0.850, 9.302) |
0.090 |
0.168 (−0.429, 0.764) |
0.578 |
0.281 (−0.360, 0.923) |
0.386 |
| Former smoker |
1.868 (0.829, 4.212) |
0.132 |
−0.005 (−0.403, 0.394) |
0.982 |
0.280 (−0.143, 0.704) |
0.192 |
| Diabetes |
0.600 (0.210, 1.715) |
0.340 |
0.513 (0.022, 1.004) |
0.041 |
0.457 (−0.051, 0.966) |
0.077 |
| Aspirin |
0.902 (0.395, 2.057) |
0.806 |
0.060 (−0.353, 0.472) |
0.775 |
−0.005 (−0.455, 0.444) |
0.981 |
| Statin therapy |
0.821 (0.365, 1.845) |
0.633 |
0.132 (−0.271, 0.536) |
0.517 |
0.282 (−0.151, 0.715) |
0.199 |
| Beta blockers |
1.120 (0.468, 2.680) |
0.800 |
0.241 (−0.197, 0.679) |
0.278 |
0.304 (−0.170, 0.778) |
0.206 |
| ACE inhibitors/ARBs |
0.557 (0.237, 1.311) |
0.181 |
0.161 (−0.253, 0.575) |
0.443 |
−0.055 (−0.501, 0.391) |
0.807 |
| Diuretics |
0.875 (0.313, 2.447) |
0.799 |
0.065 (−0.447, 0.576) |
0.803 |
0.336 (−0.242, 0.914) |
0.251 |
| Psychiatric history | ||||||
| History of depression/anxiety |
1.190 (0.536, 2.639) |
0.669 |
0.009 (−0.392, 0.410) |
0.965 |
0.326 (−0.104, 0.756) |
0.135 |
| Antidepressant drug use |
1.957 (0.858, 4.461) |
0.110 |
0.105 (−0.310, 0.520) |
0.617 |
0.350 (−0.098, 0.797) |
0.124 |
| Antipsychotic drug use |
3.297 (0.575, 18.896) |
0.180 |
0.054 (−0.793, 0.902) |
0.899 |
0.268 (−0.575, 1.111) |
0.529 |
| Malignancy history | ||||||
| Prior malignancy |
0.972 (0.318, 2.971) |
0.961 |
−0.147 (−0.708, 0.415) |
0.606 |
0.015 (−0.606, 0.636) |
0.963 |
| Prior chemotherapy/radiation |
0.620 (0.279, 1.378) |
0.241 |
0.028 (−0.368, 0.424) |
0.888 |
−0.273 (−0.697, 0.151) |
0.204 |
| Current chemotherapy/radiation |
0.308 (0.063, 1.504) |
0.145 |
0.156 (−0.485, 0.798) |
0.630 |
−0.575 (−1.306, 0.156) |
0.121 |
| Anthracycline or trastuzumab exposure |
0.590 (0.191, 1.822) |
0.359 |
−0.031 (−0.565, 0.504) |
0.909 |
−0.280 (−0.921, 0.362) |
0.388 |
| Time interval between cancer diagnosis and imaging date (years) |
1.031 (0.964, 1.102) |
0.369 |
−0.019 (−0.053, 0.015) |
0.266 |
−0.025 (−0.062, 0.013) |
0.190 |
| Other diseases | ||||||
| COPD/asthma |
2.048 (0.872, 4.812) |
0.100 |
−0.097 (−0.529, 0.334) |
0.656 |
0.154 (−0.302, 0.610) |
0.505 |
| Chronic kidney disease |
1.285 (0.460, 3.586) |
0.632 |
0.925 (0.436, 1.415) |
<0.001 |
0.746 (0.230, 1.261) |
0.005 |
| Autoimmune disease |
6.103 (1.810, 20.586) |
0.004 |
−0.183 (−0.729, 0.364) |
0.509 |
−0.065 (−0.685, 0.556) |
0.836 |
| Chronic immunosuppressive drug therapy |
1.232 (0.311, 4.891) |
0.766 |
0.602 (−0.094, 1.298) |
0.089 |
0.415 (−0.324, 1.154) |
0.268 |
Bold values indicate P < 0.05. ACE, angiotensin-converting enzyme; ARB, angiotensin receptor blocker; CI, confidence interval; COPD, chronic obstructive pulmonary disease; OR, odds ratio.
Amygdalar activity in individuals with Takotsubo syndrome vs. controls
Among the clinical and demographic factors assessed, TTS is associated with a history of autoimmune disease (Table 2). In a model adjusted for TTS risk factors, AmygA adjusted for TL activity was higher among individuals who developed TTS vs. controls (meanSD: 0.98 0.10 vs. 0.95 0.07, P = 0.038, Figure 2, Table 3). In analyses wherein amygdalar SUV was adjusted for vmPFC activity, we also observed a significant between-group difference ([Amygdalar SUV÷vmPFC SUVSD: 0.97 0.10 vs. 0.91 0.10 for those with subsequent TTS vs. controls, P = 0.012, Figure 2, Table 3). The effect size of the ratio of amygdalar to regulatory activity between cases and controls was 0.37 and 0.53 when TL and vmPFC were used as adjustments, respectively.
Table 3.
Amygdalar activity and subsequent Takotsubo syndrome
| Covariables added to the model | Linear regression |
Cox regression |
||||||
|---|---|---|---|---|---|---|---|---|
| Amygdalar activity ÷ temporal lobe activity |
Amygdalar activity ÷ medial prefrontal cortex activity |
Amygdalar activity ÷ temporal lobe activity |
Amygdalar activity ÷ medial prefrontal cortex activity |
|||||
| Standardized ß (95% CI) | P-value | Standardized ß (95% CI) | P-value | Standardized HR (95% CI) | P-value | Standardized HR (95% CI) | P-value | |
| Univariable |
0.370 (−0.028, 0.768) |
0.068 |
0.535 (0.117, 0.953) |
0.013 |
1.493 (1.090, 2.045) |
0.013 |
1.580 (1.176, 2.125) |
0.002 |
| Takotsubo risk factorsa |
0.431 (0.024, 0.838) |
0.038 |
0.544 (0.121, 0.968) |
0.012 |
1.643 (1.189, 2.270) |
0.003 |
1.708 (1.240, 2.352) |
0.001 |
| Heart failure risk factorsb |
0.412 (0.021, 0.803) |
0.039 |
0.583 (0.158, 1.008) |
0.008 |
1.483 (1.072, 2.053) |
0.017 |
1.580 (1.176, 2.125) |
0.002 |
| CVD risk factorsc |
0.405 (0.015, 0.795) |
0.042 |
0.567 (0.151, 0.983) |
0.008 |
1.483 (1.072, 2.053) |
0.017 |
1.580 (1.176, 2.125) |
0.002 |
| Malignancy historyd |
0.495 (0.023, 0.967) |
0.040 |
0.640 (0.089, 1.190) |
0.024 |
1.823 (1.254, 2.652) |
0.002 |
1.711 (1.197, 2.445) |
0.003 |
| Characteristic that differed between cases and controls (i.e. autoimmune disease) |
0.429 (0.031, 0.828) |
0.035 |
0.574 (0.149, 0.998) |
0.009 |
1.643 (1.189, 2.270) |
0.003 |
1.708 (1.240, 2.352) |
0.001 |
| Characteristics that associated with amygdalar activity (see Table 2) |
0.362 (−0.009, 0.733) |
0.056 |
0.498 (0.104, 0.892) |
0.014 |
1.493 (1.090, 2.045) |
0.013 |
1.580 (1.176, 2.125) |
0.002 |
| Antidepressant drugs |
0.338 (−0.048, 0.724) |
0.085 |
0.475 (0.063, 0.887) |
0.024 |
1.493 (1.090, 2.045) |
0.013 |
1.580 (1.176, 2.125) |
0.002 |
All multivariable models included age and sex. Bold values indicate P < 0.05.
Takotsubo risk factors: hyperlipidaemia, autoimmune disorders, smoking, depression/anxiety, and malignancy.
Heart failure risk factors: hypertension, coronary artery disease, diabetes, and smoking.
CVD risk factors: hypertension, hyperlipidaemia, diabetes, and smoking.
Malignancy history: prior malignancy, prior chemotherapy/radiation, chemotherapy/radiation at the time of imaging, cardiotoxic drugs (anthracyclines or trastuzumab), and time interval between cancer diagnosis and imaging date.
CI, confidence interval; CVD, cardiovascular disease; HR, hazard ratio.
Amygdalar activity vs. Takotsubo risk
Two separate approaches were employed to evaluate associations between baseline AmygA and risk of TTS. First, in adjusted logistic regression analyses, higher AmygA associated with greater odds of developing TTS [standardized OR (95% CI) 1.64 (1.03, 2.61), P = 0.036] and remained significant in multivariable models, stratified Subgroup analyses, and when vmPFC was employed as the regulatory tissue correction of AmygA (Supplementary material online, Tables S2 and S3).
Secondly, time-adjusted Cox regression was performed. In that analysis, AmygA independently predicted subsequent TTS [standardized HR (95% CI) 1.643 (1.189, 2.270), P = 0.003] after adjustment for TTS risk factors. That is, each SD increase in AmygA is associated with a 64% increase in TTS risk. This association remained significant through other multivariable adjustments (Table 3), separate assessments of unilateral AmygA (Supplementary material online, Table S4), when vmPFC was used as the regulatory tissue correction of AmygA (Table 3), and in sensitivity analyses with follow-up limited to 5 years (Supplementary material online, Table S5).
Amygdalar activity vs. timing of Takotsubo
We additionally tested whether, among the subset of individuals with subsequent TTS, higher AmygA (i.e. ≥ mean + 1 SD) was associated with a shorter interval between imaging and TTS onset. Individuals with higher AmygA developed TTS ∼2 years before those with lower AmygA [β (95% CI) −2.72 (−5.12, −0.32), P = 0.028, Supplementary material online, Figure S1] after adjusting for TTS risk factors, remaining significant after alternatively adjusting for CVD (P = 0.029) and heart failure risk factors (P = 0.043).
Amygdalar activity vs. leucopoietic activity
AmygA (≥ vs. <mean + 1 SD) associated with BM (i.e. leucopoietic) activity [standardized β (95% CI) 0.65 (0.09, 1.21), P = 0.024, Supplementary material online, Figure S2] and remained significant after adjustment for age and sex (P = 0.038) and CVD risk factors (P = 0.027). Notably, BM activity did not associate with TTS (P = 0.640).
Discussion
This study is the first to employ 18F-FDG-PET/CT imaging to demonstrate that heightened stress-associated neurobiological activity precedes the development of TTS among a population with a high rate of malignancy. Furthermore, we observed that, among individuals who developed TTS, AmygA associated with the timing of subsequent TTS. We also identified a significant relationship between AmygA and leucopoietic activity. Together, the findings of the current study thus provide insights into one potential mechanism that may contribute to the ‘heart–brain connection’ in TTS by suggesting that chronically heightened stress-associated neurobiological activity (i.e. the ratio of amygdalar to regulatory activity) may potentially impact both the risk for and timing of subsequent TTS.
Amygdalar activity independently associates with subsequent Takotsubo syndrome
Patients with TTS commonly identify an acute stressor as the precipitating factor of the syndrome; thus, a mechanism linking the brain to the heart has long been suspected of playing a key role in its pathogenesis.2 , 3 A recent fMRI study showed that individuals with prior TTS manifest abnormal neural connectivity among regions associated with autonomic function and limbic system regulation (including the amygdala).15 However, those neural alterations were observed in individuals with prior TTS, raising the key question of whether such changes were the cause or result of TTS. This study findings provide important answers to that question by demonstrating that individuals who subsequently develop TTS manifest increased AmygA years before developing TTS. Such heightened AmygA may predispose individuals to TTS by potentiating the sympathetic, neurohormonal, and inflammatory consequences of future stressors.
Mechanistic insights
While a consensus on the pathophysiology of TTS is lacking, it is likely that stressors trigger a complex pathophysiologic cascade,16 including catecholamine toxicity, abnormal myocardial perfusion, myocardial stunning, and endothelial dysfunction.17 Several stress-associated neural centres in the limbic system (including the amygdala) play a critical role in responding to stressors and may initiate this cascade. Limbic system activation induces hormonal, autonomic, and immune changes associated with fear and stress.18 , 19 The amygdala, via its efferents, effects physiologic changes through the SNS.6 , 15 A perceived threat activates noradrenergic neurons in the brainstem and the sympatho-adrenomedullary axis, causing the release of catecholamines, likely contributing to TTS.6 These study findings suggest that increased AmygA predates the development of TTS and may effectively prime the limbic system to respond more vigorously to acute stressors, thus increasing TTS risk. The relationship between AmygA and timing of TTS (among TTS cases) provides further support for this priming hypothesis. That higher relative AmygA associates with earlier TTS development suggests that those with the highest AmygA are relatively more prone to develop TTS upon stress exposure and may develop the syndrome sooner.
Additionally, our results demonstrate an association between amygdalar and leucopoietic activities. This well-described association points to a possible neuro-immune mechanism.4 , 5 In mice, stress, through sympathetic signals, triggers leucopoietic activity that accelerates innate immune cell output and cytokine production.7 In humans, individuals with heightened AmygA have increased leucopoietic activity that also links to atherosclerosis and CVD events, thus delineating a neuro-immune link to CVD.4 , 5 In concert with this hypothesized neuro-immune mechanism, TTS involves myocardial macrophage infiltration, changes in the distribution of monocyte subsets, and increased systemic pro-inflammatory cytokines.20 Importantly, we did not observe a significant association between BM activity and TTS risk. Furthermore, consistent with prior studies,21–23 we observed a significant association between autoimmune disorders and TTS, suggesting a possible link between inflammation and TTS. Thus, additional work is needed to assess the potential relationship between heightened systemic inflammation (as well as leucopoietic activity) and TTS risk. Nevertheless, the relationship between AmygA and TTS remained robust to adjustment for autoimmune diseases and immunosuppressive medications in multivariable models. Taken together, these study results support a concept wherein individuals with chronically heightened stress-associated neural activity are predisposed to an exaggerated systemic response to stress that may prompt the development of TTS (Figure 3).
Figure 3.
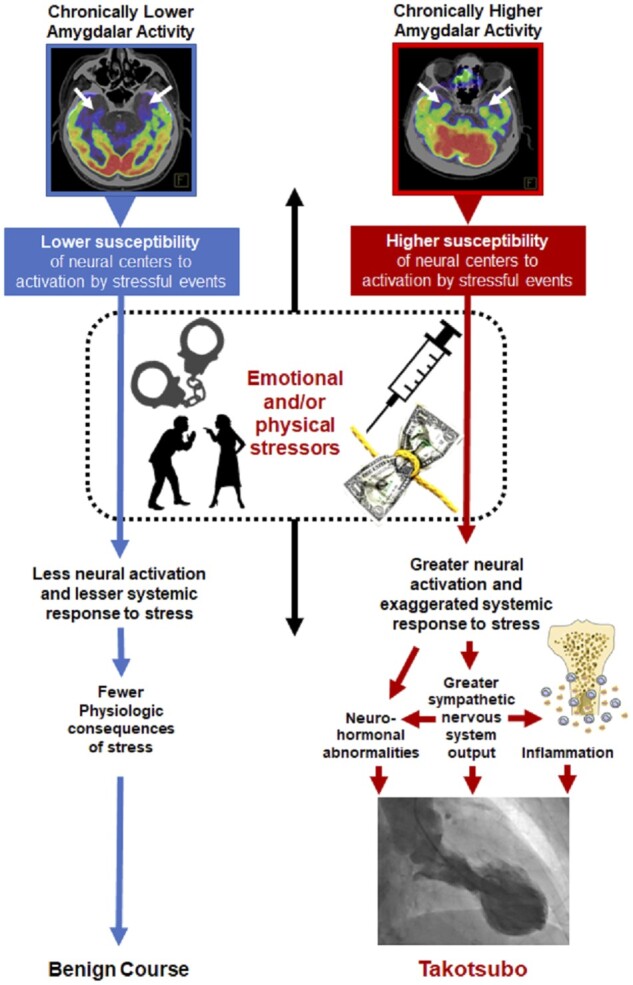
Hypothesized neurobiological pathway linking stress to Takotsubo syndrome. Individuals with a higher ratio of amygdalar to regulatory activity are primed to have a heightened response upon encountering subsequent acute stressors, resulting in greater activation of the sympathetic and immune system and increased Takotsubo risk.
Potential role of other brain regions
Assessment of AmygA involves dividing amygdalar metabolic activity by the activity of regulatory brain regions. This ratio (amygdalar to regulatory neural activity) is designed to reflect an interaction between functionally connected stress-associated neural regions that promote vs. regulate the stress response. The amygdala promotes a greater stress response, while regulatory regions (e.g. vmPFC) attenuate this response.24 In this study, we pre-specified the use of TL activity for background adjustment.4 However, we alternatively adjusted for vmPFC activity, given its important role in reducing the stress response14 and recent findings of altered prefrontal connectivity among individuals with TTS.4 Notably, using vmPFC activity to adjust for AmygA yielded even more robust relationships between stress-associated neural activity and TTS. These findings are consistent with the theory that the amygdala participates within a network of stress-responsive neural tissues, and that the balance of activity within that network (e.g. heightened AmygA and/or lowered regulatory activity) influences the risk for adverse physiological consequences that result from stress. This construct is also consistent with observations of hypo-connected sub-networks involving the amygdala3 and decreased vmPFC blood flow and connectivity among individuals with prior TTS.9 Additional brain regions (e.g. insular cortex, anterior cingulate cortex, and hippocampus) that were not evaluated in this study are involved in regulating the response to stress and have altered connectivity in individuals with prior TTS.2 , 3 Accordingly, while the ratio of amygdalar to regulatory activity appears to have a role in the risk for developing TTS, additional study is needed to clarify the roles of other important neural centres in TTS development.
Implications
Previous research has shown that increased AmygA may participate in the pathology of a number of stress-related cardiovascular and metabolic diseases.4 This study provides important insights into the link between AmygA and TTS that may improve the care of at-risk patients. Importantly, the ratio of amygdalar to regulatory activity may represent a useful target for both pharmacologic and non-pharmacologic behavioural interventions (e.g. stress reduction) in individuals at high risk for TTS and other stress-associated diseases. Because recurrent TTS is associated with considerable morbidity and mortality,1 heightened AmygA may be a particularly useful therapeutic target for secondary prevention. The downstream components of this pathway may also represent targets to attenuate TTS risk by using anti-inflammatory drugs to reduce heightened inflammation or using beta-blockers to reduce SNS discharge (though beta-blockers have not proven beneficial in TTS to date).25
Limitations
This study was a modestly sized single-centre retrospective case–control study and is subject to the inherent limitations. The population largely consisted of individuals with a diagnosis of malignancy (86%), a known TTS risk factor, which may limit the generalizability of our findings. Nevertheless, the controls in this study were carefully matched by type and stage of malignancy as well as malignancy treatment to those who developed TTS, which would serve to offset the frequency of malignancy among the TTS cases and address our inability to directly and uniformly measure cancer activity on 18F-FDG-PET/CT imaging. While imaging performed in the context of a possible or known diagnosis of malignancy is likely to induce stress, 18F-FDG-PET/CT imaging was performed under resting conditions in this study, and all individuals were subject to the same conditions. The results were robust to adjustment for clinically diagnosed depression and anxiety, but we were unable to assess perceived stress or evaluate for subclinical psychiatric symptoms in this retrospective study. As such, the relationship between subclinical stress conditions and AmygA may be incompletely evaluated. While our findings support the presence of chronically increased AmygA on resting imaging years before the development, we were unable to directly measure instantaneous changes in brain activity or metabolism in response to the acute stress during the TTS event and, therefore, could not directly assess a causal relationship. Accordingly, a subsequent study could also implement fMRI in conjunction with 18F-FDG-PET in a broader population to clarify the role of the stress response in triggering TTS. Due to limitations of clinical whole-body 18F-FDG-PET/CT imaging, we were unable to study all brain regions involved in the stress response, and there may have been a small degree of spillover in measurements from structures neighbouring the amygdala. These other important brain regions (e.g. insular cortex, anterior cingulate cortex, and hippocampus) should be evaluated in future PET/MRI studies. Finally, the study is potentially limited by ascertainment bias due to the use of diagnostic codes to identify cases of a rare disease in a retrospective cohort with strict inclusion criteria. To minimize this impact, each patient’s chart was carefully adjudicated by two blinded cardiologists; however, broader study in a general population is needed for confirmation. Nevertheless, these limitations are counterbalanced by unique and important insights provided by imaging performed before the development of TTS. Furthermore, given the rarity of TTS, this study would be extremely challenging to perform prospectively, and the fact that significant differences were still identified in a small study supports AmygA as a meaningful marker for TTS risk.
Conclusions
Among a population of individuals with a high frequency of malignancy, those who subsequently develop TTS have higher baseline ratio of amygdalar to regulatory activity compared with those who do not. This heightened stress-associated neurobiological activity is present years before disease onset and may represent a previously unrecognized TTS risk factor. Among individuals who later developed TTS, higher AmygA was associated with a shorter interval between imaging and development of TTS. Collectively, these findings shed light on a ‘heart–brain connection’ representing a neurobiological mechanism that contributes to TTS and suggests potential targets to reduce TTS risk.26
Supplementary material
Supplementary material is available at European Heart Journal online.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
Funding
This work was supported by National Institutes of Health (NIH) [T32HL076136, KL2TR002542, and K23HL151909] and American Heart Association [18CDA34110366] [M.T.O.], NIH P01HL131478, R33HL141047, R01HL149516, R01HL152957, R56AR077187, and R01HL137913 as well as support from the Harvard Medical School Osher Center [A.T.], and NIH [R01HL137562, R01HL130539, and K24HL113128] as well as support from A. Curtis Greer and Pamela Kohlberg [T.G.N.].
Conflict of interest: A.T. received institutional grants from Genentech for research outside the submitted work. T.G.N. reports acting as a consultant for Parexel, Bristol Myers-Squibb, Aprea Therapeutics, and Intrinsic Imaging unrelated to the current research. M.T.O. reports acting as a consultant for Intrinsic Imaging unrelated to the current research. The remaining authors have no disclosures.
Supplementary Material
Contributor Information
Azar Radfar, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
Shady Abohashem, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
Michael T Osborne, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
Ying Wang, Cardiovascular Imaging Research Center, Boston, MA, USA; Department of Nuclear Medicine, First Hospital of China Medical University, Shenyang, Liaoning Province, China.
Tawseef Dar, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
Malek Z O Hassan, Cardiovascular Imaging Research Center, Boston, MA, USA.
Ahmed Ghoneem, Cardiovascular Imaging Research Center, Boston, MA, USA.
Nicki Naddaf, Cardiovascular Imaging Research Center, Boston, MA, USA.
Tomas Patrich, Cardiovascular Imaging Research Center, Boston, MA, USA.
Taimur Abbasi, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
Hadil Zureigat, Cardiovascular Imaging Research Center, Boston, MA, USA.
James Jaffer, Cardiovascular Imaging Research Center, Boston, MA, USA.
Parastou Ghazi, Cardiovascular Imaging Research Center, Boston, MA, USA.
James A Scott, Department of Radiology, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Lisa M Shin, Department of Psychology, Tufts University, Medford, MA, USA; Department of Psychiatry, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Roger K Pitman, Department of Psychiatry, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Tomas G Neilan, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
Malissa J Wood, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA.
Ahmed Tawakol, Cardiology Division, Massachusetts General Hospital and Harvard Medical School, MA, USA; Cardiovascular Imaging Research Center, Boston, MA, USA.
References
- 1. Templin C, Ghadri JR, Diekmann J, Napp LC, Bataiosu DR, Jaguszewski M, Cammann VL, Sarcon A, Geyer V, Neumann CA, Seifert B, Hellermann J, Schwyzer M, Eisenhardt K, Jenewein J, Franke J, Katus HA, Burgdorf C, Schunkert H, Moeller C, Thiele H, Bauersachs J, Tschöpe C, Schultheiss H-P, Laney CA, Rajan L, Michels G, Pfister R, Ukena C, Böhm M, Erbel R, Cuneo A, Kuck K-H, Jacobshagen C, Hasenfuss G, Karakas M, Koenig W, Rottbauer W, Said SM, Braun-Dullaeus RC, Cuculi F, Banning A, Fischer TA, Vasankari T, Airaksinen KEJ, Fijalkowski M, Rynkiewicz A, Pawlak M, Opolski G, Dworakowski R, MacCarthy P, Kaiser C, Osswald S, Galiuto L, Crea F, Dichtl W, Franz WM, Empen K, Felix SB, Delmas C, Lairez O, Erne P, Bax JJ, Ford I, Ruschitzka F, Prasad A, Lüscher TF. Clinical features and outcomes of takotsubo (stress) cardiomyopathy. N Engl J Med 2015; 373: 929–938. [DOI] [PubMed] [Google Scholar]
- 2. Hiestand T, Hanggi J, Klein C, Topka MS, Jaguszewski M, Ghadri JR, Luscher TF, Jancke L, Templin C. Takotsubo syndrome associated with structural brain alterations of the limbic system. J Am Coll Cardiol 2018; 71: 809–811. [DOI] [PubMed] [Google Scholar]
- 3. Templin C, Hanggi J, Klein C, Topka MS, Hiestand T, Levinson RA, Jurisic S, Luscher TF, Ghadri JR, Jancke L. Altered limbic and autonomic processing supports brain-heart axis in Takotsubo syndrome. Eur Heart J 2019; 40: 1183–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tawakol A, Ishai A, Takx RA, Figueroa AL, Ali A, Kaiser Y, Truong QA, Solomon CJ, Calcagno C, Mani V, Tang CY, Mulder WJ, Murrough JW, Hoffmann U, Nahrendorf M, Shin LM, Fayad ZA, Pitman RK. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet 2017; 389: 834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goyal A, Dey AK, Chaturvedi A, Elnabawi YA, Aberra TM, Chung JH, Belur AD, Groenendyk JW, Lerman JB, Rivers JP, Rodante JA, Harrington CL, Varghese NJ, Sanda GE, Baumer Y, Sorokin AV, Teague HL, Genovese LD, Natarajan B, Joshi AA, Playford MP, Bluemke DA, Chen MY, Alavi A, Pitman RK, Powell-Wiley TM, Tawakol A, Gelfand JM, Mehta NN. Chronic stress-related neural activity associates with subclinical cardiovascular disease in psoriasis: a prospective cohort study. JACC Cardiovasc Imaging 2020; 13: 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pelliccia F, Kaski JC, Crea F, Camici PG. Pathophysiology of Takotsubo syndrome. Circulation 2017; 135: 2426–2441. [DOI] [PubMed] [Google Scholar]
- 7. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, von Zur Muhlen C, Bode C, Fricchione GL, Denninger J, Lin CP, Vinegoni C, Libby P, Swirski FK, Weissleder R, Nahrendorf M. Chronic variable stress activates hematopoietic stem cells. Nat Med 2014; 20: 754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dar T, Radfar A, Abohashem S, Pitman RK, Tawakol A, Osborne MT. Psychosocial stress and cardiovascular disease. Curr Treat Options Cardiovasc Med 2019; 21: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki H, Matsumoto Y, Kaneta T, Sugimura K, Takahashi J, Fukumoto Y, Takahashi S, Shimokawa H. Evidence for brain activation in patients with takotsubo cardiomyopathy. Circ J 2014; 78: 256–258. [DOI] [PubMed] [Google Scholar]
- 10. Scally C, Abbas H, Ahearn T, Srinivasan J, Mezincescu A, Rudd A, Spath N, Yucel-Finn A, Yuecel R, Oldroyd K, Dospinescu C, Horgan G, Broadhurst P, Henning A, Newby DE, Semple S, Wilson HM, Dawson DK. Myocardial and systemic inflammation in acute stress-induced (Takotsubo) cardiomyopathy. Circulation 2019; 139: 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schaefer SM, Abercrombie HC, Lindgren KA, Larson CL, Ward RT, Oakes TR, Holden JE, Perlman SB, Turski PA, Davidson RJ. Six-month test-retest reliability of MRI-defined PET measures of regional cerebral glucose metabolic rate in selected subcortical structures. Hum Brain Mapp 2000; 10: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lyon AR, Bossone E, Schneider B, Sechtem U, Citro R, Underwood SR, Sheppard MN, Figtree GA, Parodi G, Akashi YJ, Ruschitzka F, Filippatos G, Mebazaa A, Omerovic E. Current state of knowledge on Takotsubo syndrome: a Position Statement from the Taskforce on Takotsubo Syndrome of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2016; 18: 8–27. [DOI] [PubMed] [Google Scholar]
- 13. Britz-Cunningham SH, Millstine JW, Gerbaudo VH. Improved discrimination of benign and malignant lesions on FDG PET/CT, using comparative activity ratios to brain, basal ganglia, or cerebellum. Clin Nucl Med 2008; 33: 681–687. [DOI] [PubMed] [Google Scholar]
- 14. Felix-Ortiz AC, Burgos-Robles A, Bhagat ND, Leppla CA, Tye KM. Bidirectional modulation of anxiety-related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience 2016; 321: 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagai M, Dote K, Kato M, Sasaki S, Oda N, Kagawa E, Nakano Y, Yamane A, Higashihara T, Miyauchi S, Tsuchiya A, Harada W, Kario K. The insular cortex and takotsubo cardiomyopathy. Curr Pharm Des 2017; 23: 879–888. [DOI] [PubMed] [Google Scholar]
- 16. Ghadri J-R, Wittstein I S, Prasad A, Sharkey S, Dote K, Akashi Y J, Cammann V L, Crea F, Galiuto L, Desmet W, Yoshida T, Manfredini R, Eitel I, Kosuge M, Nef H M, Deshmukh A, Lerman A, Bossone E, Citro R, Ueyama T, Corrado D, Kurisu S, Ruschitzka F, Winchester D, Lyon A R, Omerovic E, Bax J J, Meimoun P, Tarantini G, Rihal C, Y.-Hassan S, Migliore F, Horowitz J D, Shimokawa H, Lüscher T F, Templin C. International Expert Consensus Document on Takotsubo Syndrome (Part I): Clinical Characteristics, Diagnostic Criteria, and Pathophysiology. Eur Heart J 2018;39:2032–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen W, Dilsizian V. Exploring the pathophysiology of takotsubo cardiomyopathy. Curr Cardiol Rep 2017; 19: 53. [DOI] [PubMed] [Google Scholar]
- 18. Wang SS, Yan XB, Hofman MA, Swaab DF, Zhou JN. Increased expression level of corticotropin-releasing hormone in the amygdala and in the hypothalamus in rats exposed to chronic unpredictable mild stress. Neurosci Bull 2010; 26: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lagraauw HM, Kuiper J, Bot I. Acute and chronic psychological stress as risk factors for cardiovascular disease: insights gained from epidemiological, clinical and experimental studies. Brain Behav Immun 2015; 50: 18–30. [DOI] [PubMed] [Google Scholar]
- 20. Scally C, Rudd A, Mezincescu A, Wilson H, Srivanasan J, Horgan G, Broadhurst P, Newby DE, Henning A, Dawson DK. Persistent long-term structural, functional, and metabolic changes after stress-induced (Takotsubo) cardiomyopathy. Circulation 2018; 137: 1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deshmukh A, Kumar G, Pant S, Rihal C, Murugiah K, Mehta JL. Prevalence of Takotsubo cardiomyopathy in the United States. Am Heart J 2012; 164: 66–71.e1. [DOI] [PubMed] [Google Scholar]
- 22. Ugurlucan M, Zorman Y, Ates G, Arslan AH, Yildiz Y, Karahan Zor A, Cicek S. Takotsubo cardiomyopathy in a patient with multiple autoimmune disorders and hyperthyroidism. Res Cardiovasc Med 2013; 2: 145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cakir M. Takotsubo cardiomyopathy in thyrotoxicosis. Int J Cardiol 2010; 145: 499–500. [DOI] [PubMed] [Google Scholar]
- 24. Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci 2009; 10: 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim H, Senecal C, Lewis B, Prasad A, Rajiv G, Lerman LO, Lerman A. Natural history and predictors of mortality of patients with Takotsubo syndrome. Int J Cardiol 2018; 267: 22–27. [DOI] [PubMed] [Google Scholar]
- 26. Ghadri J-R, Wittstein I S, Prasad A, Sharkey S, Dote K, Akashi Y J, Cammann V L, Crea F, Galiuto L, Desmet W, Yoshida T, Manfredini R, Eitel I, Kosuge M, Nef H M, Deshmukh A, Lerman A, Bossone E, Citro R, Ueyama T, Corrado D, Kurisu S, Ruschitzka F, Winchester D, Lyon A R, Omerovic E, Bax J J, Meimoun P, Tarantini G, Rihal C, Y-Hassan S, Migliore F, Horowitz J D, Shimokawa H, Lüscher T F, Templin C. International Expert Consensus Document on Takotsubo Syndrome (Part II): Diagnostic Workup, Outcome, and Management. Eur Heart J 2018;39:2047–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.