

Abstract
Mitochondrial dysfunction is an early feature of Alzheimer’s disease (AD), whereby accumulation of damaged mitochondria in conjunction with impaired mitophagy contributes to neurodegeneration. Various non-transcribed microRNAs (miRNAs) are involved in this process. In the present study, we aimed to decipher the participation of miR-204 in a murine AD model. Primary hippocampal neurons were isolated from mice and treated with β-amyloid 1-42 (Aβ1-42) to establish a cell model of AD. Dichloro-dihydro-fluorescein diacetate and dihydrorhodamine 123 staining assays were performed to measure total reactive oxygen species (ROS) and mitochondrial ROS production in neurons, and MitoSOX staining was done to analyze mitochondrial ROS production in hippocampus. Furthermore, mitochondrial autophagy was observed in hippocampus from amyloid precursor protein/pesenilin-1 AD modeled mice, and their cognitive function was assessed by Morris water maze. Mitochondrial damage, ROS production, and mitochondrial autophagy were observed in AD cell model induced by Aβ1-42. In AD, signal transducer and activator of transcription 3 (STAT3) and transient receptor potential mucolipin-1 (TRPML1) expression was downregulated, although miR-204 expression was upregulated. TRPML1 overexpression, downregulation of miR-204, or STAT3 pathway activation reduced the Aβ1-42-induced mitochondrial damage, along with ROS production and mitochondrial autophagy in vivo and in vitro. Silencing of miR-204 could upregulate TRPML1 expression, thus suppressing ROS production and mitochondrial autophagy in AD through STAT3 pathway.
Keywords: Alzheimer’s disease, signal transducer and activator of transcription 3, microRNA-204, transient receptor potential mucolipin-1, mitochondrial autophagy, reactive oxygen species, Aβ1-42, hippocampal neurons
Graphical Abstract
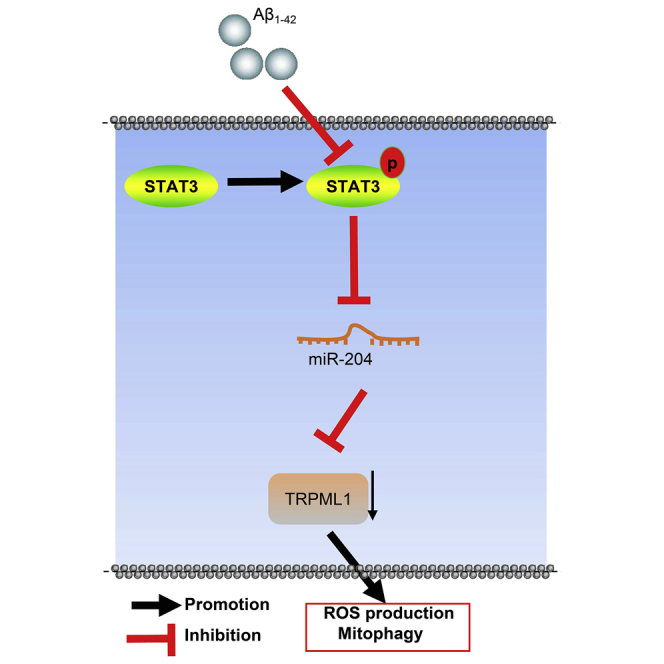
In our experiments, a cellular model of Alzheimer’s disease was induced by Aβ1-42, and APP/PS1 mice were used to explore the involvement of miR-204. It found that miR-204 silencing could upregulate TRPML1 expression, thus reducing mitochondrial damage, ROS production, and mitochondrial autophagy in Alzheimer’s disease through STAT3 pathway.
Introduction
As a progressive neurodegenerative disease commonly occurring in the elderly, Alzheimer’s disease (AD) is the main reason for dementia. AD is featured by impairment in memory and executive function, spatial awareness, situational memory, and other cognitive deficits; deterioration of language; social isolation and defective social cognition; as well as mental disorder, apathy, depression, and aggression. Although individual patients suffer from specific psychiatric symptoms, all will suffer from a decrease in daily activities and a gradual loss of self-sufficiency.1 Around the world, the number of people suffering from AD-related dementia is expected to reach 135.5 million by 2050, thus making AD (together with cancer) the major health problem for the 21st century, imposing an enormous social and economic burden.2 Studies have shown that many cellular changes are related to the development and progress of neurodegenerative diseases, including mitochondrial dysfunction, synaptic damage, hormonal imbalance, inflammatory responses, the formation and accumulation of hyperphosphorylated tau, and neuronal loss, among which mitochondrial dysfunction and synaptic damage are early changes in the pathogenesis of AD.3
Transient receptor potential mucolipin-1 (TRPML1), which is widely expressed in the lysosome or endosomal membrane of mammalian cells,4 is the main channel for release of Ca2+ from lysosomes and the main regulator of lysosomal storage and transport of metabolites. In addition, TRPML1 can regulate autophagy and accumulation of β-amyloid (Aβ) in neurons,5 both of which are pathological characteristics of AD. MicroRNAs (miRNAs) are a class of non-coding RNA molecules with 19–24 nt, which are involved in epigenetic regulation of gene expression at a post-transcriptional level.2 Current research has revealed that miRNAs in brain microvessels may promote a highly activated endothelial phenotype, thus promoting elimination of Aβ oligomers through perivascular drainage and interfering in the progression of AD.6 Moreover, transcription factor signal transducer and activator of transcription 3 (STAT3) is activated in reactive astrocytes in several murine and primate models of AD,7 although inhibition of the STAT3 pathway can prevent astrocyte reactivity and affect the progression of AD.
Of note, AD is characterized by mitochondrial dysfunction and synaptic damage, which have been recognized to be ameliorated by phosphatase and tensin homologue-induced putative kinase 1 (PINK1).8,9 Mitophagy, a selective autophagy to eliminate dysfunctional mitochondria, is now understood to be dependent on Parkin.10 In addition, Beclin1 is known as a molecular platform involved in the mediation of autophagosome formation in AD.11 Also, light-chain 3II (LC3II), an autophagosome membrane marker, has been observed to be upregulated in human neuroblastoma cells exposed to Aβ, whereas its downregulation was associated with reduced deposition of Aβ in a mouse model of AD.12,13 Given the aforementioned findings, we have investigated the miRNA miR-204 as a factor affecting mitochondrial autophagy in cell and murine AD models via upregulation of TRPML1 through activation of STAT3 pathway, manifesting in reduced levels of PINK1, Parkin, LC3II, and Beclin1 proteins.
Results
Mitochondrial damage, reactive oxygen species (ROS) production, and mitochondrial autophagy were observed in AD-model neurons induced by Aβ1-42 exposure
To verify whether mitochondrial autophagy is involved in AD, we first developed an AD cell model by treatment of cultured neurons with Aβ1-42. The expression of mitochondrial-autophagy-related proteins (PINK1, Parkin, LC3II, and Beclin1) was upregulated in the AD cells compared with that in the control cells (Figure 1A). Moreover, the binding of autophagosomes to lysosomes was detected by monomeric red fluorescent protein (mRFP)-green fluorescent protein (GFP)-LC3 staining, which suggested that autophagic lysosome formation was blocked in AD (Figure 1B).
Figure 1.

Aβ1-42 induces mitochondrial damage, ROS production, and mitochondrial autophagy in AD-modeled neurons
(A) Expression of mitochondrial-autophagy-related proteins normalized to β-actin in AD-modeled neurons detected by western blot analysis. (B) Binding of autophages to lysosomes in AD neurons by mRFP-GFP-LC3 staining is shown. (C) The mitochondrial membrane potential of AD-modeled neurons by JC-1 staining is shown. (D) Cell ATP production detected in AD neurons is shown. (E) Total ROS production in AD-modeled neurons detected by DCFH-DA staining is shown. DCFH-DA without intrinsic fluorescence can enter into cells through the membrane and then be hydrolyzed into DCFH by esterase. DCFH is incapable of crossing through the cell membrane, such that probes can be easily loaded into cells where active oxygen can oxidize non-fluorescent DCFH to generate DCF with green fluorescence, which indicates the ROS content. (F) ROS production in mitochondria in AD-modeled neurons detected by dihydrorhodamine (DHR) 123 staining is shown. DHR123 was oxidized into rhodamine123 by ROS in the mitochondria, and rhodamine123 was excited at 488 nm while green fluorescence was emitted at 515 nm. Measurement data were expressed in the form of mean ± standard deviation. The comparison between two groups was performed by independent sample t test. ∗p < 0.05. The experiment was repeated three times independently.
Compared with normal neurons, the mitochondrial membrane potential and mitochondrial adenosine triphosphate (ATP) production in AD-modeled hippocampal neurons were decreased, suggesting pervasive mitochondrial damage (Figures 1C and 1D). In addition, total ROS and mitochondrial ROS production in hippocampal neurons of AD were increased (Figures 1E and 1F). Therefore, Aβ1-42 treatment may induce mitochondrial damage, increase ROS production, and impair mitochondrial autophagy in cultured hippocampal neurons.
Overexpression of TRPML1 reduces mitochondrial damage, ROS production, and mitochondrial autophagy induced by Aβ1-42
Reverse transcription quantitative polymerase chain reaction (qRT-PCR) showed that TRPML1 expression was decreased in AD-modeled neurons (Figure 2A). Upon overexpressing TRPML1 in AD neurons (Figures 2A and 2B), western blot analysis revealed that this overexpression downregulated the expression of PINK1, Parkin, LC3II, and Beclin1 proteins (Figure 2B). Immunofluorescence showed that the number of co-localized mitochondria and lysosomes was increased after overexpressing TRPML1 in AD-modeled neurons (Figure S1A). Moreover, overexpression of TRPML1 increased the mitochondrial membrane potential and mitochondrial ATP production (Figures 2C and 2D), although reducing total ROS and mitochondrial ROS production in AD-modeled hippocampal neurons (Figures 2E and 2F). These results suggest that TRPML1 overexpression reduces mitochondrial damage, ROS production, and mitochondrial autophagy induced by Aβ1-42 toxicity.
Figure 2.

Restoring TRPML1 reduces Aβ1-42-induced mitochondrial damage, ROS production, and mitochondrial autophagy
(A) Expression of TRPML1 detected by qRT-PCR. (B) Expression of mitochondrial-autophagy-related proteins normalized to β-actin detected by western blot analysis is shown. (C) The mitochondrial membrane potential of AD-modeled neurons detected by JC-1 staining is shown, with green referring to JC-1 monomer and golden color referring to JC-1 polymer. (D) Cell ATP production is shown. (E) Total ROS production detected by DCFH-DA staining is shown. DCFH-DA without intrinsic fluorescence can enter into cells through the membrane and then be hydrolyzed into DCFH by esterase. DCFH is incapable of crossing through the membrane so that probes can be easily loaded into cells where active oxygen can oxidize non-fluorescent DCFH to generate DCF with green fluorescence, which indicates the ROS content. (F) ROS production in mitochondria detected by DHR123 staining is shown. DHR123 was oxidized into rhodamine123 by ROS in the mitochondria, and rhodamine123 was excited at 488 nm while green fluorescence was emitted at 515 nm. Measurement data were expressed in the form of mean ± standard deviation. The comparison between two groups was performed by independent sample t test. ∗p < 0.05. The experiment was repeated three times independently.
miR-204 targets TRPML1
To uncover the upstream regulation mechanism of TRPML1 in AD pathology, we searched in databases for miRNAs that may potentially target TRPML1. Our online analysis identified that miR-204 could bind to TRPML1, indicating that miR-204 may be an upstream miRNA of TRPML1 (Figure 3A). Compared with the normal neurons, miR-204 expression in AD-modeled neurons was increased (Figure 3B). Dual luciferase reporter gene assay confirmed that miR-204 mimic treatment could reduce the luciferase activity of TRPML1-wide type (WT) in a dose-dependent manner but had no significant effect on the luciferase activity of TRPML1 mutant (Mut) (Figure 3C). Western blot analysis showed that treatment with miR-204 inhibitor increased the expression of TRPML1 in AD-modeled neurons, and the promotion by miR-204 inhibitor had a dose effect (0.5 μM, 5 μM, 10 μM, and 15 μM; Figure 3D). These results suggest that TRPML1 expression can be specifically inhibited by miR-204 in AD-modeled neurons.
Figure 3.

TRPML1 is targeted by miR-204
(A) The binding sites of miR-204 on TRPML1-WT (left) and binding sites of miR-204 on TRPML1-Mut (right) predicted by online websites. (B) Expression of miR-204 in AD neurons detected by qRT-PCR is shown. (C) The binding relationship between miR-204 and TRPML1 verified by dual luciferase reporter gene assay is shown. (D) Expression of TRPML1 normalized to β-actin in AD neurons detected by western blot analysis is shown. Measurement data were expressed in the form of mean ± standard deviation. The comparison between two groups was performed by independent sample t test. ∗p < 0.05. The experiment was repeated three times independently.
Downregulating miR-204 reduces mitochondrial damage, ROS production, and mitochondrial autophagy by upregulating TRPML1 in AD-modeled neurons
To investigate the effects of miR-204 in AD neurons, we tested the consequences of treatment with miR-204 inhibitor in different doses (0.5 μM, 5 μM, 10 μM, and 15 μM) and short hairpin RNA (shRNA) against TRPML1 (sh-TRPML1) on mitochondrial damage, ROS production, and mitochondrial autophagy. According to the off-target efficiency results shown in Figure S2, sh-TRPML-3 was selected for subsequent experiments. miR-204 inhibitor could reduce PINK1, Parkin, LC3II, and Beclin1 expression with a dose-response relationship (Figure 4A). Also, miR-204 inhibitor was observed to enhance the degree of co-localization between mitochondria and lysosomes, but these effects could be reversed by co-transfection of miR-204 inhibitor + sh-TRPML1 (Figure S1B). Furthermore, miR-204 inhibitor increased mitochondrial membrane potential and mitochondrial ATP production and decreased total ROS and mitochondrial ROS production in AD-modeled neurons in a dose-effect manner, although co-transfection of sh-TRPML1 reversed the effects of miR-204 inhibitor (Figures 4B–4E). Thus, downregulation of miR-204 inhibits mitochondrial damage, ROS production, and mitochondrial autophagy induced by Aβ1-42 by increasing the expression of TRPML1.
Figure 4.

Downregulation of miR-204 inhibits mitochondrial damage, ROS production, and mitochondrial autophagy induced by Aβ1-42 through upregulating TRPML1
(A) Expression of mitochondrial-autophagy-related proteins normalized to β-actin in AD-modeled neurons detected by western blot analysis. (B) The mitochondrial membrane potential of neurons detected by JC-1 staining is shown, where green refers to JC-1 monomer and golden color refers to JC-1 polymer. (C) Cell ATP production is shown. (D) Total ROS production in AD neurons detected by DCFH-DA staining is shown. DCFH-DA without intrinsic fluorescence can enter into cells through the membrane and then be hydrolyzed into DCFH by esterase. DCFH is incapable of crossing through the membrane, so that probes can be easily loaded into cells where active oxygen can oxidize non-fluorescent DCFH to generate DCF with green fluorescence, which indicates ROS content. (E) ROS production in mitochondria in AD-modeled neurons detected by DHR123 staining is shown. DHR123 was oxidized into rhodamine123 by ROS in the mitochondria, and rhodamine123 was excited at 488 nm while green fluorescence was emitted at 515 nm. Measurement data were expressed in the form of mean ± standard deviation. The comparison between two groups was performed by independent sample t test. ∗p < 0.05. The experiment was repeated three times independently.
STAT3-mediated miR-204/TRPLM1 activity mediates mitochondrial damage, ROS production, and mitochondrial autophagy
We observed that, compared with the normal neurons, the AD-modeled neurons exhibited lower phosphorylation level of STAT3 (Figure 5A), suggesting the inactivation of STAT3 pathway. Next, AD neurons were treated with STAT3 pathway activator colivelin (CLN). qRT-PCR and western blot analysis showed that CLN treatment lowered the expression of miR-204 and increased the expression of TRPML1 in AD neurons (Figure 5B). Moreover, western blots showed that PINK1, Parkin, LC3II, and Beclin1 expression was downregulated after CLN treatment, which could be reversed by sh-TRPML1 (Figure 5C). Further, immunofluorescence showed an increased co-localization of mitochondria and lysosomes, along with higher mitochondrial membrane potential and ATP production, although total ROS and mitochondrial ROS production was decreased in AD neurons treated with CLN, all of which could be prevented by sh-TRPML1 (Figures 5D–5G and S1C). These results suggest that STAT3-mediated miR-204/TRPLM1 activity regulates mitochondrial damage, ROS production, and mitochondrial autophagy induced by Aβ1-42.
Figure 5.

STAT3 regulates miR-204/TRPLM1 activity to mediate mitochondrial damage, ROS production, and mitochondrial autophagy
(A) Expression of STAT3/p-STAT3 normalized to β-actin in AD-modeled neurons detected by western blot analysis. (B) Expression of miR-204 detected by qRT-PCR is shown. (C) The expression of PINK1, Parkin, LC3II, and Beclin1 normalized to β-actin in AD neurons detected by western blot analysis is shown. (D) The mitochondrial membrane potential of AD-modeled neurons detected by JC-1 staining is shown, with green referring to JC-1 monomer and golden color referring to JC-1 polymer. (E) Cell ATP production is shown. (F) Total ROS production in AD-modeled neurons detected by DCFH-DA staining is shown. DCFH-DA without inherent fluorescence can enter into cells through the membrane and then be hydrolyzed into DCFH by esterase. DCFH is incapable of crossing through the membrane, so that probes can be easily loaded into cells where active oxygen can oxidize non-fluorescent DCFH to generate DCF with green fluorescence, which indicates the ROS content. (G) ROS production in mitochondria in AD-modeled neurons detected by DHR123 staining is shown. DHR123 was oxidized into rhodamine123 by ROS in the mitochondria, and rhodamine123 was excited at 488 nm while green fluorescence was emitted at 515 nm. ∗p < 0.05. Measurement data were expressed as mean ± standard deviation. The comparison between two groups was performed by independent sample t test. The experiment was repeated three times independently.
STAT3/miR-204/TRPML1 axis mediates AD development in vivo through mitochondrial autophagy and ROS production
To verify whether the STAT3/miR-204/TRPML1 axis is involved in AD in vivo, amyloid precursor protein (APP)/presenilin-1 (PS1) transgenic AD mouse model was established. Compared with the normal mice, STAT3 pathway and TRPML1 expression in AD mice was downregulated, although miR-204 expression was upregulated (Figures 6A and 6B). Next, AD mode mice were injected with miR-204 angomir, miR-204 antagomir, or lentivirus overexpressing TRPML1. Western blot analysis showed that TRPML1 expression was reduced after miR-204 angomir treatment and elevated after miR-204 antagomir or TRPML1 overexpression treatment (Figure 6C). Further, western blot analysis showed that expression of PINK1, Parkin, LC3II, and Beclin1 in AD mice was higher than in the normal mice, whereas these increases could be further promoted by miR-204 angomir or blocked by miR-204 antagomir or overexpression of TRPML1 (Figure 6C). Moreover, the number of mitochondrial autophagy in hippocampus was reduced, the ROS level was enhanced, and hippocampal tissue damage was aggravated in AD mice, all of which could be further aggravated by miR-204 angomir or ameliorated by miR-204 antagomir or overexpression of TRPML1 (Figures 6D, 6E, and S3). These effects of miR-204 angomir showed a significant dose-response relationship.
Figure 6.

STAT3/miR-204/TRPML1 axis mediates the development of AD by mediating mitochondrial autophagy and ROS production in vivo
(A) Expression of TRPML1 and miR-204 in hippocampus of normal and APP/PS1 mice detected by qRT-PCR. (B) Expression of STAT3/p-STAT3 normalized to β-actin in hippocampus of mice in normal and APP/PS1 mice detected by western blot analysis is shown. (C) Expression of mitochondrial-autophagy-related proteins normalized to β-actin in hippocampus of normal and APP/PS1 mice detected by western blot analysis is shown. (D) Frequency of mitochondrial autophagy in hippocampus of mice in each group observed by electron microscopy is shown. (E) ROS production in hippocampus mitochondria of mice in each group detected by MitoSOX fluorescence is shown. MitoSOX was oxidized to generate red fluorescence, and the nucleus was stained in blue by DAPI. (F) Escape latency in the Morris water maze test is shown. (G) The time spent in the quadrant containing the hidden platform in the Morris water maze test is shown. (H) The times of crossing the maze platform detected by water maze test are shown. ∗p < 0.05. Measurement data were expressed in the form of mean ± standard deviation. The comparison between multiple groups was conducted by one-way ANOVA, followed by Tukey’s post-test, and the comparison between multiple groups at different time points was conducted by repeated-measurement ANOVA, with Bonferroni’s post-test. n = 5.
Cognitive function of mice was examined by Morris water maze experiments. We found that the escape latency of AD-modeled mice was higher than normal (Figure 6F), although time spent in the maze, quadrant time of containing the hidden platform, and the number of times crossing the platform were lower in AD mice (Figures 6G and 6H). Moreover, the escape latency was increased and time spent in the maze quadrant time of containing the hidden platform and the number times crossing the platform were decreased by miR-204 angomir in a dose-dependent manner, although reverse tendencies were observed by miR-204 antagomir or overexpression of TRPML1 (Figures 6G and 6H). These results suggested that the STAT3/miR-204/TRPML1 axis can mediate development of cellular and behavioral aspects of AD through mitochondrial autophagy and ROS level in vivo.
Discussion
Obtaining a deep understanding of the pathophysiology of AD is essential for development of new and sensitive diagnostic methods in the early and preclinical stages, when salvage might be possible.2 This study was designed to explore the effects of miR-204/TRPML1 on AD in cellular and mouse models. The collective results from this study demonstrated that suppressing miR-204 upregulated TRPML1 expression to reduce mitochondrial damage and ROS production and promote mitochondrial autophagy through STAT3 pathway in AD models.
TRPML1, which belongs to the transient receptor potential protein family, is an evolutionarily conserved cation channel located in the late endosome and lysosomal membrane. TRPML1 has a wide distribution in the brain and other tissues and plays an important role in signal transduction, maintenance of lysosomal ion homeostasis, membrane transport, and exocytosis.14 We found that TRPML1 was under-expressed in AD-modeled neurons, and the forced expression of TRPML1 could reduce mitochondrial damage and ROS production in the cell model. Previous studies have found that loss-of-function mutations of TRPML1 can induce type IV mucolipidosis, a devastating neurodegenerative disease that causes retinal degeneration and intellectual disability.4 A recent study showed that TRPML1 was downregulated in APP/PS1 transgenic AD-modeled mice and that overexpressing TRPML1 improved memory and spatial recognition deficits in the mice and reduced neuronal apoptosis in vivo, although decreasing neuronal viability and the altered lysosomal [Ca2+] ion concentration provoked by Aβ1-42 in vitro.15
miRNAs have been extensively studied and analyzed in blood, cerebrospinal fluid, and brain tissues as potential biomarkers of AD. However, due to high variability of reported data, lack of standard methods, and heterogeneity of AD, promising candidate mRNA biomarkers for AD have eluded discovery. A previous study revealed that miR-34a expression was high in affected brain regions of patients with AD and transgenic AD mice. Moreover, Aβ42 treatment increased the release of tumor necrosis factor-α, upregulated miR-34a expression, and thus induced mitochondrial dysfunction.16 Additionally, a specific set of miRNAs, including miR-9, miR-125b, and miR-128, were upregulated in AD brain.17 In our study, aiming to identify the upstream regulation mechanism of TRPML1 in AD pathology, we searched for miRNAs that may target TRPML1 through online analysis, which uncovered that miR-204 may be an upstream miRNA of TRPML1. We confirmed that TRPML1 was targeted by miR-204 in AD-modeled neurons and also found that downregulation of miR-204 inhibits mitochondrial damage, ROS production, and mitochondrial autophagy otherwise induced by Aβ1-42.
Previous studies have shown that phospho-STAT3 can bind directly to the STAT3-binding site lying near the promoter region of TRPM3 upstream of miR-204-5p.18 Thus, activation of STAT3 inhibited the expression of miR-204.19 A low expression of STAT3 can to some extent promote the occurrence of neurodegenerative diseases.20 Other studies have shown that Aβ42 overexpression causes memory impairment in mice by interfering with the JAK2/STAT3 axis in hippocampal neurons.21 We speculated that the STAT3 pathway may be involved in mitochondrial autophagy by inhibiting miR-204 expression and promoting TRPML1 expression. In our study, hippocampal neurons were treated with STAT3 activator (CLN) and analyzed by qRT-PCR, which showed that miR-204 expression was lowered and TRPML1 expression was enhanced, suggesting that STAT3 pathway indeed regulates the miR-204/TRPML1 axis to mediate mitochondrial damage, ROS production, and mitochondrial autophagy.
In conclusion, by conducting assays in vitro and in vivo, we may conclude that miR-204 targeted TRPML1, promoted ROS production, and inhibited mitochondrial autophagy, thereby promoting AD progression in cellular and murine models. Activation of STAT3 pathway inhibited miR-204 expression and enhanced TRPML1 expression, thus inhibiting ROS production and promoting mitochondrial autophagy and ultimately inhibiting AD progression (Figure 7). Of note, assessment on the markers of stemness can mimic certain cellular profiles, such as those undergoing autophagy, which is a concept to be explored in future investigations.
Figure 7.

STAT3/miR-204/TRPML1 mediates mitochondrial autophagy and ROS production in AD-modeled cells and mice
In AD models, high expression of miR-204 targets TRPML1, thus promoting ROS production and inhibiting mitochondrial autophagy, thereby promoting AD progression. Activation of STAT3 pathway can inhibit expression of miR-204 and advance expression of TRPML1, thus inhibiting ROS production and promoting mitochondrial autophagy and ultimately inhibiting the progression of AD.
Materials and methods
Culture of mice primary hippocampal neurons
Mixed cortical and hippocampal neurons were isolated from fetal rats of 14–16 days old. The isolated hippocampal neurons were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco, Grand Island, NY, USA) at 37°C with 5% CO2.
Cell treatment
A cell model of AD was established by treating hippocampal neurons with Aβ1-42 (5 μM) as described previously.15 Cells were divided into the following groups: control (normal hippocampal neurons); AD (AD neurons without treatment); control + GFP-RFP-LC3 (HanBio, Shanghai, P.R. China; normal hippocampal neurons infected with mRFP-GFP-LC3 autophagic double-labeled adenovirus); AD + GFP-RFP-LC3 (AD neurons infected with mRFP-GFP-LC3 autophagic double-labeled adenovirus); AD + oe-negative control (NC) (AD neurons infected with lentivirus harboring NC for overexpression plasmids); AD + oe-TRPML1 (AD neurons infected with lentivirus harboring overexpressed TRPML1); AD + inhibitor-NC (AD neurons treated with NC for miR-204 inhibitor); AD + miR-204 inhibitor (AD neurons treated with miR-204 inhibitor); AD + miR-204 inhibitor + sh-TRPML1 (AD neurons treated with miR-204 inhibitor + sh-TRPML1); AD + miR-204 inhibitor + sh-NC (AD neurons treated with miR-204 inhibitor + NC for sh-TRPML1); AD + veh (AD neurons treated with sterile deionized water containing 5% sefsol and 20% isopropanol); AD + CLN (AD neurons treated with CLN, a STAT3 activator); AD + CLN + sh-NC (AD neurons treated with CLN + NC for sh-TRPML1); and AD + CLN + sh-TRPML1 (AD neurons treated with CLN + sh-TRPML1). Lentiviruses were used to infect neurons, forcing them to overexpress or silence gene expression.
Co-localization of mitochondria and lysosomes
MitoTracker < Green FM (Thermo Fisher Scientific, Waltham, MA, USA) and Lyso Tracker Red DND-99 (Thermo Fisher Scientific) were used to label mitochondria and lysosomes, respectively. In short, the isolated hippocampal neurons were collected and stained for 30 min with 200 nM Mitotracker or 75 nM Lysotracker dissolved in anhydrous dimethyl sulfoxide. The neurons were then observed and photographed under a confocal laser microscope.
ROS generation
Hippocampal neurons were collected and inoculated in a 96-well plate at a density of 3 × 104 cells/well. Dichloro-dihydro-fluorescein diacetate (DCFH-DA) (STA-342, Cell Biolabs, San Diego, CA, USA) was used to measure the level of ROS according to the manufacturer’s instructions. Fluorescence microscopy (BX60, Olympus, Tokyo, Japan) was used for observation and photographing.
To measure the level of mitochondrial ROS in vivo, the hippocampus slices from the mice were incubated with MitoSOX (5 μm, Invitrogen, Carlsbad, CA, USA) for 30 min. Laser confocal microscopy (TCS-SP2) was used to detect the signal with the excitation and emission wavelengths at 498 and 522 nm, respectively. The ROS fluorescence value was analyzed by Leica SP2 software.
Detection of mitochondrial membrane potential
The mitochondrial membrane potential (ΔΨm) was detected by JC-1 staining (Beyotime Institute of Biotechnology, Jiangsu, P.R. China). In short, the cells were stained with 2.5 g/mL JC-1 for 30 min at 37°C. After washing with buffer solution, the cells were analyzed by fluorescence microscopy.
Detection of mitochondrial ATP production
As described previously,22 neuronal ATP levels were detected with a fluorescein ATP detection kit (Beyotime).
Dual luciferase reporter gene assay
TRPML1 3′ UTR fragment was introduced into the pGL3-reporter (Promega, Madison, WI, USA). The mutant form bearing mutation of the potential miR-204 binding sites was also constructed. The pGL3-TRPML1-WT and pGL3-TRPML1-Mut were co-transfected with miR-204 mimic or mimic-NC to HEK293 cells, respectively. Then, 48 h after transfection, the cells were collected and lysed. Luciferase activity was detected on Luminometer TD-20/20 (model: E5311, Promega) using Dual-Luciferase Reporter Assay System kit (Promega).
RNA isolation and quantitation
Total RNA was extracted by TRIzol reagents (Invitrogen). TaqMan MicroRNA Reverse Transcription Kit (4366596; Thermo Fisher Scientific) and High-Capacity cDNA Reverse Transcription Kit (4368813; Thermo Fisher Scientific) were used to reversely transcribe the extracted total RNA into cDNA. qRT-PCR was performed on ABI 7500 qPCR (Thermo Fisher Scientific) using SYBR Premix Ex TaqTM (Tli RNaseH Plus) kit (RR820A, TaKaRa, Dalian, Liaoning, P.R. China). With glyceraldehyde-3-phosphate dehydrogenase and U6 as internal references, the relative expression of target genes was analyzed by 2−ΔΔCt method. The primers (Table S1) were synthesized by TaKaRa.
Western blot analysis
Total protein was isolated using radio immunoprecipitation assay lysis buffer. The protein was dissolved in 2× sodium dodecyl sulfate sample buffer and boiled for 5 min. The protein samples were transferred to polyvinylidene fluoride membranes after separation by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The membrane was then blocked with 5% skimmed milk and incubated overnight with diluted primary antibodies of rabbit antibodies to PINK1 (ab23707; 1:2,000), Parkin (ab77924; 1:200), LC3 (ab51520; 1:3,000), Beclin1 (ab62557; 1:1,000), TRPML1 (ab28508; 1:2,000), and β-actin (ab8226; 1:1,000). The membrane was washed with Tris-buffered saline Tween and then incubated with horseradish-peroxidase-labeled secondary antibody (ab205718; 1:2,000; Abcam, Cambridge, USA) for 1 h. The membrane was developed with an enhanced chemiluminescence fluorescence test kit (BB-3501; Amersham, Little Chalfont, UK), imaged using Bio-Rad image analysis system (Bio-Rad, Hercules, CA, USA), and finally analyzed by Quantity One v4.6.2 software. The relative expression was expressed by the ratio of the gray value of corresponding protein bands to the gray value of β-actin protein bands.
Animal treatment
Twenty-five male APP/PS1 transgenic AD mice (aged 3 months; 28 ± 2 g) and 5 WT C57BL/6J mice of the same age (Animal Model Center of Nanjing University, Nanjing, Jiangsu, P.R. China) with an average body weight of 26 ± 4 g were used for the study. The mice were assigned randomly into the following groups:21,23 normal (C57BL/6J WT mice); APP/PS1 (APP/PS1 mice without treatment); APP/PS1 + CLN (APP/PS1 mice treated with intranasal injection of CLN); APP/PS1 + miR-204 antagomir (APP/PS1 mice treated with stereotactic injection of miR-204 antagomir in brain); APP/PS1/TRPML1+/+ (APP/PS1 mice treated with overexpressing TRPML1); and APP/PS1/TRPML1+/+ + miR-204 antagomir (APP/PS1 mice treated with overexpressing TRPML1 and stereotactic injection of miR-204 antagomir in brain). miR-204 antagomir was purchased from Shanghai GenePharma (Shanghai, P.R. China). At the end of the behavioral testing, the mice were euthanized by anesthesia overdose, blood samples were collected, and the brain tissues were isolated for histopathological analysis. The animal experiment was approved by the Animal Ethics Committee of the First Affiliated Hospital of Zhengzhou University. All experimental procedures were approved by the Committee on Laboratory Animals and implemented in accordance with the guidelines of the International Association for Pain Research.
The ultrastructure of mitochondria was visualized and imaged with transmission electron microscopy by a US-certified electron microscopist (J. Bernbaum). The images for neurons, astrocytes, and microglia were acquired independently. The percentages of damaged mitochondria as well as mitophagy-like events were calculated by cell type. All quantifications were performed in a double-bind fashion.
Morris water maze
Morris water maze was used to analyze the behavior of the mice in each group, following procedures described in detail previously.24
Statistical analysis
All data were processed by SPSS 21.0 statistical software (IBM, Armonk, NY, USA). Measurement data obeying normal distribution and homogeneity of variance were expressed in the form of mean ± standard deviation. The comparison between two groups was performed by independent sample t test. The comparison between multiple groups was conducted by one-way analysis of variance (ANOVA), followed by Tukey’s post hoc test, and the comparison between multiple groups at different time points was conducted by repeated-measures ANOVA, with Bonferroni’s post-test. p <0.05 showed significant difference.
Availability of Data and Material
The original contributions presented in the study are included in the article/supplemental materials.
Acknowledgments
We acknowledge and appreciate our colleagues for their valuable suggestions and technical assistance for this study. This work was supported by Natural Science Foundation of China (no. 81871033) and Key Scientific Research Projects of Colleges and Universities in Henan Province (no. 18B310038).
Author contributions
L.Z., Y.F., X.Z., Y.Z., and Y.M. contributed conception and design of the study, conducted the experiments, as well as drafted the manuscript. L.Z., S.L., Z.H., and L.L. contributed toward acquisition and analyses of data. Y.F. and X.Z. contributed toward interpretation of data and manuscript preparation. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2021.02.010.
Supplemental information
References
- 1.Millan M.J. Linking deregulation of non-coding RNA to the core pathophysiology of Alzheimer’s disease: an integrative review. Prog. Neurobiol. 2017;156:1–68. doi: 10.1016/j.pneurobio.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Nagaraj S., Zoltowska K.M., Laskowska-Kaszub K., Wojda U. microRNA diagnostic panel for Alzheimer’s disease and epigenetic trade-off between neurodegeneration and cancer. Ageing Res. Rev. 2019;49:125–143. doi: 10.1016/j.arr.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Reddy P.H., Oliver D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells. 2019;8:488. doi: 10.3390/cells8050488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng X., Shen D., Samie M., Xu H. Mucolipins: intracellular TRPML1-3 channels. FEBS Lett. 2010;584:2013–2021. doi: 10.1016/j.febslet.2009.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang L., Fang Y., Cheng X., Lian Y.-J., Xu H.-L., Zeng Z.-S., Zhu H.-C. Retraction: curcumin exerts effects on the pathophysiology of Alzheimer’s disease by regulating PI(3,5)P2 and transient receptor potential mucolipin-1 expression. Front. Neurol. 2019;10:255. doi: 10.3389/fneur.2017.00531. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Fu L., Jiang G., Weng H., Dick G.M., Chang Y., Kassab G.S. Cerebrovascular miRNAs correlate with the clearance of Aβ through perivascular route in younger 3xTg-AD mice. Brain Pathol. 2020;30:92–105. doi: 10.1111/bpa.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben Haim L., Ceyzériat K., Carrillo-de Sauvage M.A., Aubry F., Auregan G., Guillermier M., Ruiz M., Petit F., Houitte D., Faivre E. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015;35:2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Du F., Yu Q., Yan S., Hu G., Lue L.F., Walker D.G., Wu L., Yan S.F., Tieu K., Yan S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain. 2017;140:3233–3251. doi: 10.1093/brain/awx258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starling S. Alzheimer disease: PINK1 rescues pathology in Alzheimer disease. Nat. Rev. Neurol. 2018;14:4. doi: 10.1038/nrneurol.2017.160. [DOI] [PubMed] [Google Scholar]
- 10.Ye X., Sun X., Starovoytov V., Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015;24:2938–2951. doi: 10.1093/hmg/ddv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salminen A., Kaarniranta K., Kauppinen A., Ojala J., Haapasalo A., Soininen H., Hiltunen M. Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog. Neurobiol. 2013;106-107:33–54. doi: 10.1016/j.pneurobio.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Song G.L., Chen C., Wu Q.Y., Zhang Z.H., Zheng R., Chen Y., Jia S.Z., Ni J.Z. Selenium-enriched yeast inhibited β-amyloid production and modulated autophagy in a triple transgenic mouse model of Alzheimer’s disease. Metallomics. 2018;10:1107–1115. doi: 10.1039/c8mt00041g. [DOI] [PubMed] [Google Scholar]
- 13.Çelik H., Karahan H., Kelicen-Uğur P. Effect of atorvastatin on Aβ1-42 -induced alteration of SESN2, SIRT1, LC3II and TPP1 protein expressions in neuronal cell cultures. J. Pharm. Pharmacol. 2020;72:424–436. doi: 10.1111/jphp.13208. [DOI] [PubMed] [Google Scholar]
- 14.Di Paola S., Scotto-Rosato A., Medina D.L. TRPML1: the Ca(2+)retaker of the lysosome. Cell Calcium. 2018;69:112–121. doi: 10.1016/j.ceca.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L., Fang Y., Cheng X., Lian Y., Xu H., Zeng Z., Zhu H. TRPML1 participates in the progression of Alzheimer’s disease by regulating the PPARγ/AMPK/Mtor signalling pathway. Cell. Physiol. Biochem. 2017;43:2446–2456. doi: 10.1159/000484449. [DOI] [PubMed] [Google Scholar]
- 16.Russell A.E., Doll D.N., Sarkar S.N., Simpkins J.W. TNF-α and beyond: rapid mitochondrial dysfunction mediates TNF-α-induced neurotoxicity. J. Clin. Cell. Immunol. 2016;7:467. doi: 10.4172/2155-9899.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukiw W.J., Pogue A.I. Induction of specific micro RNA (miRNA) species by ROS-generating metal sulfates in primary human brain cells. J. Inorg. Biochem. 2007;101:1265–1269. doi: 10.1016/j.jinorgbio.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao W., Wang H.H., Tian F.J., He X.Y., Qiu M.T., Wang J.Y., Zhang H.J., Wang L.H., Wan X.P. A TrkB-STAT3-miR-204-5p regulatory circuitry controls proliferation and invasion of endometrial carcinoma cells. Mol. Cancer. 2013;12:155. doi: 10.1186/1476-4598-12-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma L., Deng X., Wu M., Zhang G., Huang J. Down-regulation of miRNA-204 by LMP-1 enhances CDC42 activity and facilitates invasion of EBV-associated nasopharyngeal carcinoma cells. FEBS Lett. 2014;588:1562–1570. doi: 10.1016/j.febslet.2014.02.039. [DOI] [PubMed] [Google Scholar]
- 20.Bai H., Zhang Q.F., Duan J.J., Yu D.J., Liu L.J. Downregulation of signal transduction and STAT3 expression exacerbates oxidative stress mediated by NLRP3 inflammasome. Neural Regen. Res. 2018;13:2147–2155. doi: 10.4103/1673-5374.241470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiba T., Yamada M., Sasabe J., Terashita K., Shimoda M., Matsuoka M., Aiso S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry. 2009;14:206–222. doi: 10.1038/mp.2008.105. [DOI] [PubMed] [Google Scholar]
- 22.Chen W., Zou P., Zhao Z., Chen X., Fan X., Vinothkumar R., Cui R., Wu F., Zhang Q., Liang G., Ji J. Synergistic antitumor activity of rapamycin and EF24 via increasing ROS for the treatment of gastric cancer. Redox Biol. 2016;10:78–89. doi: 10.1016/j.redox.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tóth M.E., Szegedi V., Varga E., Juhász G., Horváth J., Borbély E., Csibrány B., Alföldi R., Lénárt N., Penke B., Sántha M. Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones. 2013;18:759–771. doi: 10.1007/s12192-013-0428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leone S., Recinella L., Chiavaroli A., Ferrante C., Orlando G., Vacca M., Salvatori R., Brunetti L. Behavioural phenotyping, learning and memory in young and aged growth hormone-releasing hormone-knockout mice. Endocr. Connect. 2018;7:924–931. doi: 10.1530/EC-18-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/supplemental materials.