

Abstract
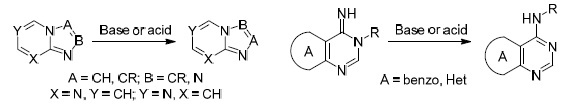
The review discusses the use of the Dimroth rearrangement in the synthesis of condensed pyrimidines which are key structural fragments of antiviral agents. The main attention is given to publications over the past 10 years. The bibliography includes 107 references.
Keywords: furo[2,3-d]pyrimidines; imidazo[1,2-a]pyrimidines; purines; pyrazolo[3,4-d]pyrimidines; pyrrolo[2,3-d]pyrimidines; quinazolin(on)es; thieno[2,3-d]pyrimidines; [1,2,4]triazolo[1,5-a]pyrimidines; [1,2,4]triazolo[1,5-c]pyrimidines; antiviral activity; Dimroth rearrangement
The Dimroth rearrangement represents the isomerization of heterocycles which involves relocation of two heteroatoms in heterocyclic systems or in their substituents via the processes of ring opening and ring closure. This rearrangement can be subdivided into two types: relocation of heteroatoms within the rings of condensed systems (Type I) and migration of exo- and endocyclic heteroatoms in heterocyclic systems (Type II) (Fig. 1). The second type of rearrangement, a particular case of which is the isomerization of 1-substituted 2-imino-1,2-dihydropyrimidines to 2-substituted aminopyrimidines by the action of bases (amidine rearrangement), is more common.
Figure 1.
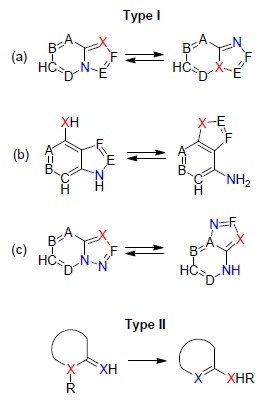
General schematic representation of the two types of the Dimroth rearrangement.
The rearrangement bearing the name of Dimroth was first observed by B. Rathke on a triazine derivative, but he did not provide any explanation for this phenomenon.1 In 1909, O. Dimroth proposed a mechanism for the rearrangement of triazole.2 The generality of this process for pyrimidines was recognized in the mid-1950s,3,4 and later it turned out that this is an even more general process characteristic of many nitrogen-containing heterocyclic systems.5 The term "Dimroth rearrangement" was introduced in 1963 by D. J. Brown and J. S. Harper.6
The Dimroth rearrangement is catalyzed by acids,7,8 bases (alkali),9,10 is accelerated by heat or light.11,12 Numerous factors affect the course of the Dimroth rearrangement in heterocyclic systems: 1) the degree of aza-substitution in rings (more nitrogen atoms in the ring facilitates a nucleophilic attack);13 2) pH of the reaction medium (affects the rate of the rearrangement);14 3) the presence of electronwithdrawing groups (facilitates the opening of the ring);13 4) the thermodynamic stability of the starting compound and the product.15 The nature of functional groups, electronic and steric effects also affect the possibility of the Dimroth rearrangement and its course.13,16–18
Despite the fact that the specific route by which the Dimroth rearrangement takes place depends on many factors, in general, three fundamentally different stages can be identified: 1) formation of an adduct by an attack of the heterocyclic ring by a nucleophile, 2) electrocyclic ring opening in the adduct followed by rotation around the single bond, and 3) closure of the ring with the participation of other structural units. In total, these stages are known as the ANRORC mechanism (addition of nucleophile, ring opening, and ring closure). If the rearrangement occurs as a result of heating or irradiation, the first step is electrocyclic opening of the ring followed by ring closure. The presented mechanism illustrates the rearrangement of 2-imino-1-methyl-1,2-dihydropyrimidine (1-methylpyrimidin-2(1H)-imine) into 2-(methylamino)-pyrimidine19 (Scheme 1).

Scheme 1
Primary information on the Dimroth rearrangement can be obtained from reference books on name reactions,20,21 whereas review articles devoted to its individual aspects provide more detailed information. There is, for example, a 1998 review article by Fujii and Itaya concerned with the rearrangement of adenine derivatives.22 Other reviews on this topic date from 1965–1998 and require substantial additions,23–27 as do sections in the review articles by L'аbbé28 and by Maiboroda and Babaev.29 More recent advances in the Dimroth rearrangement are reflected in relatively recent reviews.5,15,30 Their authors have clearly demonstrated that although the Dimroth rearrangement is old, it is not obsolete.
An analysis of the literature over the past 10 years has identified several studies that were not included in the 2017 review on the Dimroth rearrangement.30 In addition, in the last three years, a number of new publications have been published on the synthesis of a wide variety of heterocyclic systems, namely, 2-aminoimidazolotriazoles (2-substituted triazoles),31 [1,2,4]triazolo[1,5-a]pyridines,32 7,8,9,10-tetrahydro[1,2,4]triazolo[5,1-a][2,7]naphthyridines,33 [1,2,4]triazolo[1,5-d][1,2,4]triazines,34 4-diazo-1,4-dihydroisoquinolin-3(2H)-ones,35 2-sulfido-1,2,3,5-tetrahydro-4H[1,2]oxazolo-[4',5':5,6]pyrano[2,3-d][1,3,2]diazaphosphinines,36 and thieno-[2,3-d][1,3,2]diazaphosphorin-6-thione 2-sulfides37 relying the Dimroth rearrangement, indicating its enormous potential.
This review is devoted to methods for the synthesis of benzo- and hetero-annulated pyrimidine derivatives which are the structural basis of many biologically active compounds and drugs with antiviral activity based on the Dimroth rearrangement. The synthesis methods are grouped depending on the type of the starting heterocyclic systems undergoing rearrangement. Before proceeding to the methods of synthesis, let us briefly analyze condensed pyrimidine derivatives with antiviral activity.
Benzo-annulated pyrimidine derivatives shown in Figure 2, namely, 4-sulfanylquinazolines 1a,b exhibit an inhibitory effect against the tobacco mosaic virus (TMV),38 2,4-disubstituted quinazoline derivatives 2a,b containing amide fragments show high inhibitory activity against influenza A/WSN/33 virus (H1N1).39 Pyrimidine derivative 3 (BIX-01294), which is known as a methyltransferase inhibitor, entered the ranks of the most effective published Ebola virus inhibitors after a virtual screening.40
Figure 2.

Benzo-annulated pyrimidine derivatives (quinazolines) with antiviral activity.
Figure 3 shows the structures of a number of heteroannulated pyrimidine derivatives with antiviral activity: pyrrolo[2,3-d]pyrimidin(on)e(s), thieno[2,3-d]pyrimidines, purines, and pyrazolo[3,4-d]pyrimidines. Pyrrolo[2,3-d]-pyrimidines 4a–c show excellent activity as effective inhibitors of bovine viral diarrhea virus (BVDV),41 compounds 5–8 show high antiviral activity against rotavirus Wa strain and Coxsackievirus B4.42 Compound 7b is the most effective of all tested compounds against the Wa strain of rotavirus and Coxsackie B4 virus which makes it a lead compound in the search for antiviral drugs active against both viruses. Non-glutamate derivatives of 4-[2-(2-amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]pyrimidin-3-yl)ethyl]benzamide 9 (R = NH2) show antiviral activity 4–7 times higher than the structurally similar drug pemetrexed against Newcastle disease virus (NDV) which belongs to the paramyxoviruses family. Among the studied compounds 9, compounds 9a,b exhibit the highest antiviral activity.43 Thienopyrimidine derivatives 10a,b and 11a–c exhibit inhibitory activity against hepatitis C virus (HCV).44 Purine 12 exhibits an inhibitory effect (IC50 1.9 μM, selectivity index 58) against dengue virus (DENV),45 purine 13 possesses good activity (IC50 0.4–13 μM) against 80% of the 47 tested rhinovirus serotypes.46 Aristeromycin (14) and its 6'-fluorinated analogs 15a,b are active against RNA viruses such as Middle East respiratory syndrome coronavirus (MERS-CoV), severe acute respiratory syndrome coronavirus (SARS-CoV), Zika virus (ZIKV), and Chikungunya virus (CHIKV).47,48 6',6'-Difluoroaristeromycin (15c) exhibits strong antiviral effect against MERS-CoV.48 Phosphoramidate prodrug 16 demonstrates high broad-spectrum antiviral activity.48 6'-β-Fluorohomoaristeromycin (17) exhibits antiviral activity (IC50 0.12 μM) against Chikungunya virus.47 Nucleotide and nucleoside analog drugs famciclovir and vidarabine are used to treat herpes simplex viruses (HSV) and varizella zoster virus (VZV),49 while adefovir and tenofovir are medications against chronic viral hepatitis B.50 S-Acyclic nucleosides of pyrazolo[3,4-d]pyrimidine derivatives 18a,b are active against herpes simplex virus type 1 (HSV-1),51 pyrazolo[3,4-d]pyrimidines with the thiophene substituent 19a–d exhibit high inhibitory activity against Coxsackievirus B3 (IC50 0.063–0.089 μM) and moderate activity against enterovirus 71 (IC50 0.32–0.65 μM).
Figure 3.
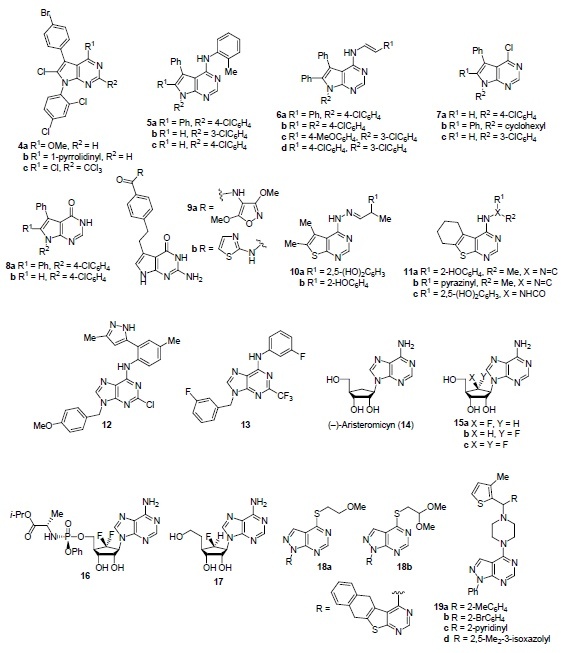
Hetero-annulated pyrimidine derivatives with antiviral activity.
Compounds with antiviral activity were also found among hetero-annulated pyrimidine derivatives containing a bridgehead nitrogen atom. For example, imidazo[1,2-a]-pyrimidine 20 exhibits specific activity against cytomegalovirus (CMV),54 [1,2,4]triazolo[1,5-a]pyrimidine 21 is active in inhibiting hepatitis B virus surface antigen HBsAg,50 preladenant (22) (Fig. 4), known as a selective inhibitor of 2α-adenosine receptors and used in the treatment of Parkinson's disease, exhibits high inhibitory activity against Zika virus.55
Figure 4.
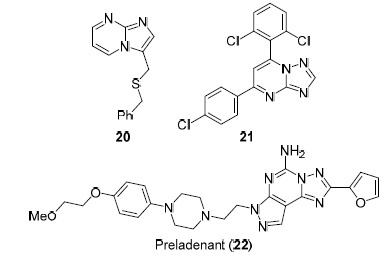
Hetero-annulated pyrimidine derivatives with a bridgehead nitrogen atom possessing antiviral activity.
Rearrangement of condensed heterocyclic systems containing five-membered rings with two nitrogen atoms
A special feature of aza-heterocycles such as imidazo-[1,2-a]pyrimidines is that they can undergo the Dimroth rearrangement under appropriate reaction conditions. This transformation is described as the migration of heteroatoms in heterocyclic system 23 with changes in the ring structure (compound 24) or without them (compound 23') (Fig. 5), and this is often an unwanted side reaction that usually occurs in basic media. Many factors influence the propensity of aza-heterocycles to undergo the Dimroth rearrangement. Typically, decreasing the π-electron density of the condensed 6-membered ring increases the rate of the rearrangement. Thus, aza-substitution in the imidazo[1,2-a]-pyrimidine system with the formation of the corresponding imidazo[1,2-a]pyrimidine 23 leads to an easier nucleophilic attack at position 5 (Fig. 5); the same is observed in the imidazo[1,2-a]pyridine system with electron-withdrawing substituents. As a result, 2-phenylimidazo[1,2-a]pyridine does not undergo rearrangement under alkaline conditions; however, the same ring system undergoes rearrangement in the presence of electron-withdrawing substituents such as the nitro group at the C-6 or C-8 position.56 The rearrangement rate depends on pH of the reaction medium, and the ratio of the products usually depends on the nature of the substituents.5,13 For the rearrangements described for the imidazo[1,2-a]pyrimidine system,13,57–60 the use of hydrolytic58–60 or haloform reaction conditions is typical.57,59 The Dimroth rearrangement can also occur under acidic conditions or upon photoactivation in other aza-heterocycles, especially in triazolo[4,3-a]pyrimidines and triazolo[4,3-c]pyrimidines, although such transformations were not observed in imidazo[1,2-a]pyrimidine system.
Figure 5.

The proposed mechanism of the Dimroth rearrangement in the imidazo[1,2-a]pyrimidine ring under basic conditions.
Mechanistic aspects of the rearrangement including some important kinetic parameters, electronic and steric factors have been described (Guerret et al.),13 identifying the minimum characteristics of aza-heterocycles to undergo the Dimroth rearrangement. In this study, the authors acknowledge the possibility of H2O recruitment by other mechanisms, such as 1,4-addition or tautomerism, but conclude that their data best support a mechanism involving a nucleophilic attack on the C-5 atom with the opening of the pyrimidine ring as shown in Figure 5.
Russell et al. has shown61 that the reactions of ethyl 6-arylimidazo[1,2-a]pyrimidine-2-carboxylates 25а–с obtained from 2-amino-5-iodopyrimidine (26a) by condensation with ethyl bromopyruvate to form 6-iodoimidazo[1,2-a]pyrimidine-2-carboxylate 27 at the first step and its subsequent Suzuki cross coupling with variously substituted arylboronic acid derivatives, depending on the amidation method, either lead to amides of imidazo[1,2-a]-pyrimidine-2-carboxylic acid 28a–c (Scheme 2, route a), or via intermediates 29a–c to isomeric imidazo[1,2-a]-pyrimidine-3-carboxylic acid amides 30a–e (route b). In this case, the direct amidation of the ethyl ester of 6-arylimidazo[1,2-a]pyrimidine-2-carboxylic acid 25a leads to the formation of the corresponding 2-carboxylic acid amides 28a–c (route a). However, when an alternative route was used for this purpose involving hydrolysis of esters 25a–c with subsequent amidation of the resulting carboxylic acids 29a–c, the formation of imidazo[1,2-a]-pyrimidine-3-carboxylic acid amides 30a–e (route b) as a result of the Dimroth rearrangement took place. Obviously, isomerization should occur either at the hydrolysis step or at the amide formation step, and the step of ester hydrolysis proceeding under aqueous basic conditions is more likely for this process.
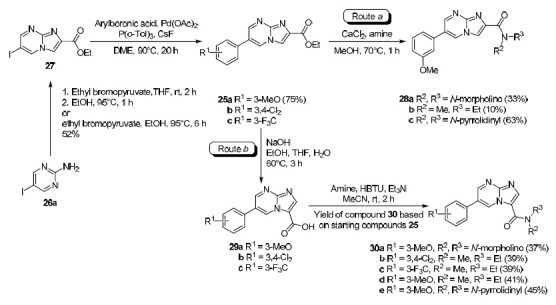
Scheme 2
The authors of a study61 performed a thorough analysis of structures 25a–c and 29a–с using a set of NMR methods, including 15N-labeled derivatives 34c,d, 35b, 36b and 14N-labeled derivatives 34а,b, 35a, 36a of imidazo[1,2-a]-pyrimidines specially synthesized from compounds 31–33 for this purpose (Schemes 3 and 4). As a result, it was shown that Dimroth rearrangement actually occurs at the hydrolysis step and not at the amidation step.
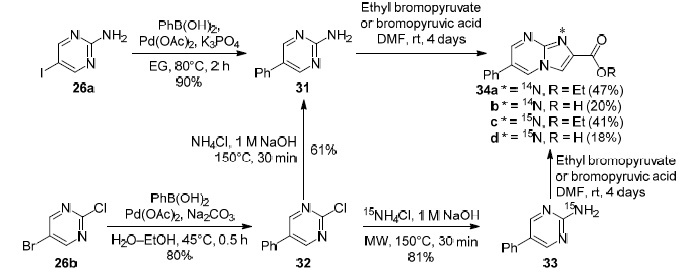
Scheme 3

Scheme 4
Rozentsveig et al.62 synthesized the products of nucleophilic addition to the azomethine group, N-[2,2-dichloro- 1-(hetarylamino)-2-phenylethyl]sulfonamides 39 and 40 in good yields by the reaction of 2-aminopyrimidines 37 with N-(2,2-dichloro-2-phenylethylidene)arenesulfonamides 38 (Scheme 5), as well as showed that the latter easily cyclize, including in situ, to form imidazo[1,2-a]pyrimidin-3-ylsulfonamides 41 and 42 in the presence of NaOH, while the expected isomeric imidazo[1,2-a]pyrimidin-2-ylsulfonamides 41' and 42' are not formed (Scheme 6). The formation of annulated heterocyclic imidazo[1,2-a]-pyrimidin-3-ylsulfonamide derivatives 41 and 42 is explained by the Dimroth rearrangement.
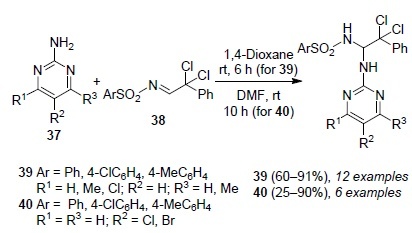
Scheme 5

Scheme 6
The formation of compounds 41 and 42 can proceed via the heterocyclization of adducts 39 and 40 to intermediate imidazopyrimidines 41' and 42' which undergo further isomerization to the final heterocycles 41 and 42 in accordance with the Dimroth rearrangement mechanism (Scheme 7). It should be noted that, based on the presented data, this type of isomerization proceeds in the opposite direction: in the presence of a base, 3-aminoimidazo-[1,2-a]pyrimidines undergo isomerization to 2-aminoimidazo[1,2-a]pyrimidines. This reaction is one of the methods for the preparation of 2-aminoimidazo[1,2-a]-pyrimidine derivatives.63

Scheme 7
Rearrangement of condensed heterocyclic systems containing five-membered rings with three nitrogen atoms
Wang's group showed64 that [1,2,4]triazolo[4,3-c]-pyrimidine derivatives 44 formed in situ as a result of oxidative cyclization of (6-chloropyrimidin-4-yl)hydrazones 43 by the action of iodobenzene diacetate in CH2Cl2 undergo Dimroth rearrangement with the formation of [1,2,4]triazolo[1,5-c]pyrimidines 45 in moderate (in the case of using propionaldehyde hydrazones) and high (in the case of using substituted benzaldehyde and furfural hydrazones) yields (Scheme 8). It was found that the rearrangement of triazole 44 into compound 45, although very slow, occurs spontaneously. The process can be catalyzed by HCl. In one case, pure [1,2,4]triazolo[4,3-c]-pyrimidine 44a (R1 = Me, R2 = Ph) was isolated in 90% yield, which was then dissolved in EtOH and stirred at room temperature. After a day, the formation of [1,2,4]-triazolo[1,5-c]pyrimidine 45a (R1 = Me, R2 = Ph) was observed, while isomerization was complete after 10 days, as evidenced by a shift in the signal of the methyl group in the 1H NMR spectrum from 2.39 ppm (compound 44a) to 3.04 ppm (product 45a). Since compounds 44 are usually unstable compared to their [1,5-c]-analogs 45, isolation of the starting [1,2,4]triazolo[4,3-c]pyrimidine intermediates had no preparative value.

Scheme 8
The accepted mechanism of the Dimroth rearrangement incorporates protonation of the nitrogen atom of pyrimidine derivative 44 with the formation of intermediate A (44·H+), ring opening (intermediate B), tautomerization with the H-shift (intermediate C) in the 1,2,4-triazole ring, ring closure (intermediate D), and deprotonation to isomeric [1,2,4]triazolo[1,5-c]pyrimidine 45 (Scheme 9).

Scheme 9
Thus, the authors of a study64 proposed a general and convenient method for the synthesis of new derivatives of [1,2,4]triazolo[1,5-c]pyrimidines. The process has several advantages, including good yields, ease of operation, environmental benignness, relatively short reaction times, and the possibility to use a wide range of substrates which makes it a useful and attractive process for the synthesis of structurally diverse triazolopyrimidines.
Chernyshev and Astakhov showed65 that 3-amino-2-benzyl[1,2,4]triazolo[4,3-a]pyrimidinium chlorides 46а–с obtained by brief heating of 1-benzyl[1,2,4]triazole-3,5-diamine (47) with 1,3-diketones 48а,b or 1,1,3,3-tetramethoxypropane (48с) undergo the Dimroth rearrangement with the formation of 2-amino[1,2,4]triazolo[1,5-a]-pyrimidines 49а–с (Scheme 10).
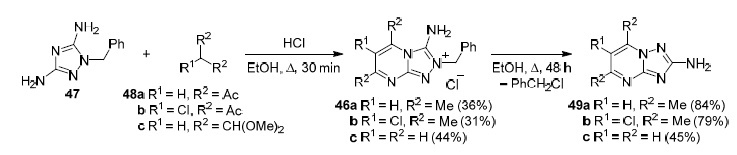
Scheme 10
Song and Son demonstrated66 that 3-arylthieno[3,2-e]-[1,2,4]triazolo[4,3-c]pyrimidines 52 obtained from compounds 50 and 51 by heating under reflux in EtOH in the presence of NaOAc isomerize into thermodynamically more stable compounds 53 via sequential ring opening and closure as a result of a Dimroth-type rearrangement (Scheme 11, method I). For example, the reaction of thieno-[3,2-e][1,2,4]triazolo[4,3-c]pyrimidine 52а (R = H, 1 equiv) with NaOAc (2 equiv) in EtOH under reflux for 5 h led to only a single product, compound 53а (R = H) in 76% yield. In particularly, each isomer 52 and 53 was distinguished by their 1H NMR spectra. For example, the most prominent peak in the spectrum of compound 52а was observed at 9.02 ppm as a singlet attributed to the pyrimidine proton, while a similar singlet in the spectrum of isomer 53а was observed downfield at 9.27 ppm. The relatively downfield region of the pyrimidine proton in the spectrum of thieno[3,2-e][1,2,4]triazolo[4,3-c]pyrimidine 52а can be explained by the proximity of the nitrogen atom rearranged in the triazole ring. In order to obtain convincing evidence of the exact structure of compounds 53, product 53а (R = H) was compared66 with an authentic sample obtained by an alternative method (Scheme 11, method II) developed for the synthesis of imidazole and pyrazole analogs of tricyclic compounds 5367 which confirmed the formation of products 53 from compounds 52 by the Dimroth rearrangement. The transformation of thieno[3,2-e][1,2,4]triazolo[4,3-c]pyrimidines 52 into isomers 53 is similar to the rearrangement of [1,2,4]triazolo[4,3-a]-pyrimidines in alkali into isomeric [1,2,4]triazolo[1,5-a]-pyrimidines.9,14,68 This rearrangement is also consistent with the rearrangement of thieno[3,2-е][1,2,4]triazolo[4,3-c]-pyrimidin-5(1H)-ones,69 pyrazolo[4,3-e][1,2,4]triazolo[4,3-c]-pyrimidines, and 1,4-disubstituted [1,2,4]triazolo[4,3-a]-quinazolin-5(4H)-ones.70,71 Thus, a convenient and reliable method for the synthesis of 2-arylthieno[3,2-е][1,2,4]triazolo[ 1,5-c]pyrimidines by the rearrangement of 3-arylthieno- [3,2-e][1,2,4]triazolo[4,3-c]pyrimidines was developed.

Scheme 11
Lauria's group showed72 that the reaction of 3-phenylbenzo[4,5]thieno[2,3-e][1,2,3]triazolo[1,5-a]pyrimidin-5(4H)-one (54a) with ethyl 4-bromobutyrate in DMF in the presence of K2CO3 proceeds with the formation of, in addition to the expected product 55 (in low yield), a linear isomer 56 as the main product73 (Scheme 12). This is not unexpected since the known rearrangement of (pyrrolo)-indolo[1,2,3]triazolo[1,5-a]pyrimidines 57 actually occurs. Pyrimidines 57 are converted under basic conditions into linear isomers 58 via the Dimroth rearrangement74,75 (Scheme 13 ).
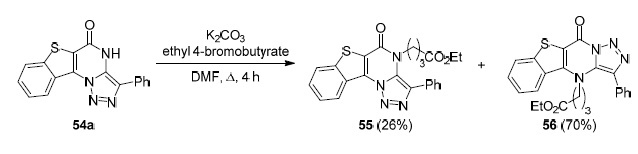
Scheme 12
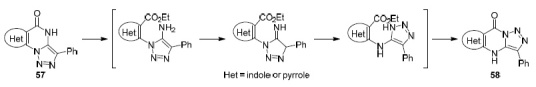
Scheme 13
The linear isomer 56 was hydrolyzed by the action of NaOH in EtOH–H2O to give the corresponding intermediate 59. Derivative 60 was obtained by the reaction of carboxylic acid 59 with histamine in the presence of 3-(3-dimethylaminopropyl)-1-ethylcarbodiimide and 4-dimethylaminopyridine73 (Scheme 14).

Scheme 14
In the reaction of the angular tetracyclic compound 54b with 1-bromo-3-chloropropane in DMF in the presence of K2CO3, along with the formation of the expected chloropropyl derivative 61, rearrangement was again observed with the competitive formation of the linear isomer 62 as the main product. As a result of heating compound 62 in 1-methylpiperazine under reflux, derivative 6373 was obtained (Scheme 15).

Scheme 15
In conclusion, the obtained experimental data contributed to the synthesis of new linear isomers of derivatives of benzo- and pyridine-annulated thieno[2,3-e]-[1,2,3]triazolo[1,5-a]pyrimidines 60 and 63 possessing antitumor activity, analogs of angular isomers 64 and 6573 (Fig. 6).
Figure 6.

Annulated thieno[2,3-e][1,2,3]triazolo[1,5-a]pyrimidines with good antitumor activity (for compound 64, the negative decimal logarithm of molar concentration which inhibits the growth of 50% of cells (pGI50) equals 4.73–6.74; for compound 65 pGI50 equals 5.03–6.80).
Rearrangement of condensed heterocyclic systems containing six-membered rings with nitrogen and oxygen atoms
Li et al.76 developed an efficient Lewis acid catalyzed condensation of aromatic o-aminonitriles 66 with aromatic aldehydes 67 in DMF under reflux as a convenient method for the synthesis of 1,2-dihydroquinazolin-4(3H)-ones 68.
In this case, derivatives of 1,2-dihydroquinazolin-4(3H)-one 68 were obtained in good yields. The position and nature of the substituent in the phenyl ring of arylaldehydes does not affect the yields of quinazolinones, despite the fact that benzaldehydes with nitro and methoxy substituents with radically different electronic effects on the aromatic system were used. Heteroaromatic aldehydes 69 also easily reacted with o-aminonitrile 66a (R1 = H, R2 = NO2) to form quinazolinones 70a,b in 72 and 77% yields, respectively (Scheme 16).

Scheme 16
A possible reaction mechanism involves the addition of the amino group of o-aminonitrile 66 to the carbonyl group of aldehyde 67 to form intermediate A. The hydroxy group in intermediate A intramolecularly attacks the nitrile group (the Pinner reaction)77 to form benzoxazine B which then undergoes the Dimroth rearrangement to form the final product 68 (Scheme 17).

Scheme 17
The use of iso- and terephthalaldehydes 71a,b in this reaction affords the corresponding bisquinozalinones 72a,b76 (Scheme 18).

Scheme 18
The group of Mansoor78 developed a novel simple, efficient and solvent-free method for the synthesis of derivatives of 5-aryl-7-methyl-2-(2-oxo-2H-chromen-3-yl)-4-oxo-4,5-dihydro-3H-chromeno[2,3-d]pyrimidine-6-carboxylic acid ethyl esters 73 which is based on the condensation of 2-amino-4-aryl-3-cyano-6-methyl-4H-pyran-5-carboxylic acid ethyl esters 74 with coumarin-3-carboxylic acid (75) in the presence of pentafluorophenylammonium triflate as an inexpensive organocatalyst (Scheme 19). This method is distinguished by high yields, environmental friendliness, simplicity of execution, short reaction time, and ease of product isolation.

Scheme 19
To explain the formation of compound 73a (R = H) by the condensation reaction, a mechanism was proposed that involves, first, the protonation of coumarin-3-carboxylic acid (75) by pentafluorophenylammonium triflate as a Brønsted acid to form cationic intermediate C. Next, as a result of amidation of intermediate C with 2-amino-4Hpyran derivative 74a (R = H), intermediate product D is formed. In the next step, the nitrile group of intermediate D is protonated, followed by a cycloaddition reaction with the formation of intermediate product E. Subsequently, the addition of pentafluorophenylamine with ring opening to intermediates F and G and the following ring closure of intermediate G leads to the formation of intermediate H. This intermediate is converted to product 73а (R = H) as a result of deprotonation (Scheme 20). Interestingly, the formation of compound 73а obtained as a result of condensation of coumarin-3-carboxylic acid (75) with 2-amino-4H-pyran derivative 74а confirms the reaction mechanism which is occasionally described in the literature as the Dimroth rearrangement.2,79

Scheme 20
Davoodnia's group revealed80 that the reaction of 2-amino-4-aryl-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4Hchromene-3-carbonitriles 76 with an excess of aliphatic carboxylic acids 77а,b in the presence of POCl3 leads to new 2-alkyl-5-aryl-8,8-dimethyl-8,9-dihydro-3Н-chromeno-[2,3-d]pyrimidine-4,6(5Н,7Н)-diones 78a–h in high yields (Scheme 21). The optimal conditions for the reaction are heating of 2-amino-4-aryl-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitriles 76 in an excess of AcOH (77а) under reflux in the presence of POCl3 as a chlorinating agent for 150 min. A decrease in the reaction temperature to 100°C led to a decrease in the product yield from 90 to 78%, all other parameters being equal. For comparison, the synthesis of compound 78а was also carried out using SOCl2. Under these conditions, product 78a was obtained in 82% yield. Therefore, all subsequent synthesis reactions of compounds 78b–h were carried out in the presence of POCl3 at reflux in AcOH (77а) or propanoic acid (77b).

Scheme 21
The proposed mechanism for the formation of compounds 78 includes the tandem intramolecular Pinner reaction and the Dimroth rearrangement. Chlorination of carboxylic acid 77 with POCl3 leads to the formation of acyl chloride I which reacts with starting compound 76 to form intermediate J. This compound undergoes an intramolecular Pinner reaction and subsequent Dimroth rearrangement, as a result of which the final product 7880 is formed via oxazine intermediate K (Scheme 22).

Scheme 22
Davoodnia's group81 also synthesized some 9-alkyl-12-aryl-10,12-dihydro-11H-benzo[f]chromeno[2,3-d]pyrimidin-11-ones 79 via the intramolecular Pinner reaction of 3-amino-1-aryl-1H-benzo[f]chromene-2-carbonitriles 80 with aliphatic carboxylic acids 77a,b in the presence of POCl3 followed by the Dimroth rearrangement (Scheme 23).

Scheme 23
In terms of the sequence of transformations, the mechanism of formation of tetracyclic compounds 79a–g is identical to the mechanism of formation of compounds 78a–h80 as shown in Scheme 22. The synthesized compounds 79a–g were tested for antibacterial activity against B. cereus, S. aureus, S. epidermidis, S. enterica subsp. enterica, and E. coli. All compounds inhibited the growth of the tested bacteria at a concentration of 5 mg/ml. Compound 79e with the lowest values of the minimum inhibitory concentration and the minimum bactericidal concentration against B. cereus was comparable to tetracycline and ampicillin, the current standards against this bacterium.81
Li et al.82 group developed a method for the synthesis of 2,3-dihydropyrimido[4,5-d]pyrimidine, catalyzed by N-heterocyclic carbene (NHC-PPIm – in this case, it was generated by concentrating an aqueous solution of 1,3-dipropylimidazolium hydroxide) method of synthesis of 2,3-dihydropyrimido[4,5-d]pyrimidin-4(1H)-ones 81 based on the three-component reaction of 2-(ethoxymethylene) malononitrile (82), guanidines 83 (or amidines 84), and ketones 85 (or aldehydes 67)83 (Scheme 24). This highly efficient method incorporates a cascade of transformations such as the Michael reaction, cyclization, isomerization, aromatization followed by nucleophilic attack and the Dimroth rearrangement. The method avoids the use of expensive reagents and multistep processes. A series of ketones 85 (or benzaldehydes 67) and guanidines 83 (or amidines 84) were investigated (Scheme 24). Theoretically, various carbonyl compounds could adversely affect this reaction due to steric hindrance and ring loading, but the reactions of all carbonyl compounds with guanidine 83a (R3 = NH2) led to products 81 in good and high yields (75–92%); reactions with N,N-dimethylguanidine 82b (R3 = NMe2) also led to the corresponding compounds 81 in good yields (79–86%). To broaden the scope of this onepot methodology, a specific series of guanidines 83 (compounds 83c (R3 = NHPh), 83d (R3 = NHMe), 83e (R3 = NHEt)) and amidines 84 (compounds 84a (R3 = Me), 84b (R3 = Ph)) was chosen, and the corresponding compounds 81 were obtained in good or high yields (75–92%). These results illustrate the versatility of the NHCPPIm catalyst and the advantages of this one-pot method.
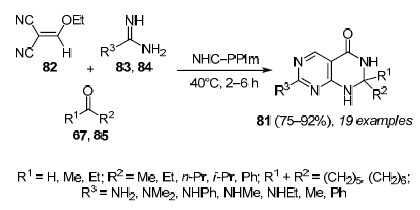
Scheme 24
As for the reaction mechanism, 2-(ethoxymethylene)-malononitrile (82) enters into the Michael addition reaction with guanidines 83 (or amidines 84) as the first step, followed by cyclization, isomerization, and aromatization to the intermediate compound 4-aminopyrimidine- 5-carbonitrile 86. Nucleophilic attack of the Breslow intermediate L at carbonitrile 86 leads to the formation of compound M. Then, intermediate M releases NHC-PPIm and forms 1,3-oxazine N which subsequently undergoes the Dimroth rearrangement with the formation of the final product 8183 (Scheme 25).

Scheme 25
Derivatives of the pyrimido[4',5':3,4]pyrazolo[1,2-b]-phthalazine-4,7,12-trionic cyclic system 88 were obtained by Davoodnia's group84 by means of the reaction of 3-amino-1-aryl-5,10-dioxo-5,10-dihydro-1H-pyrazolo[1,2-b]-phthalazine-2-carbonitriles 87 with aliphatic carboxylic acids 77a,b in the presence of POCl3, (Scheme 26).

Scheme 26
The reaction mechanism involves a cascade process similar to the synthesis of compounds 78a–h80 (Scheme 22) and 79a–g (Scheme 23)81 which is initiated by POCl3 and leads to the formation of acyl chloride I from the corresponding carboxylic acid. Further, the nucleophilic attack of the amino group in compounds 87 on the activated carbonyl group of acyl chloride I leads to the formation of intermediate O which then undergoes a Pinner-type intramolecular cyclization with the formation of the oxazine intermediate P. Intermediate P subsequently undergoes the Dimroth rearrangement via intermediate Q to form the final tetracyclic products 88 (Scheme 27). Under these conditions, attempts to isolate intermediate compounds were unsuccessful even after careful monitoring of the reactions.

Scheme 27
Rearrangement of condensed heterocyclic systems containing six-membered rings with two nitrogen atoms
Besson et al.79 developed microwave-initiated condensation of N-(4-aryl-2-cyano)-N,N-dimethylformamidines 89a,b with anilines 90 as a useful and fast tool for the synthesis of 4-anilinoquinazolines 91a–c and 92 (Scheme 28). In the case of the reaction of compound 89b with aromatic amine 90a (R1 = 4-NO2, R2 = H) with the strong electronwithdrawing nitro group, the process stops at the step of formation of amidine 93 (Scheme 28).
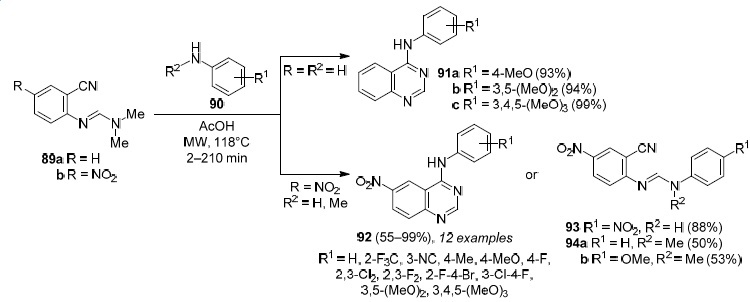
Scheme 28
The formation of compounds 94a,b upon condensation of N-methylanilines 90b (R1 = H, R2 = Me) and 90c (R1 = 4-MeO, R2 = Me) with imine 89b (Scheme 28) confirms the reaction mechanism that has so far rarely been described in the literature.85 The authors of the study79 suggest that aromatic amine 90 attacks the carbon atom of N,N-dimethylamidine 89 which leads to elimination of NHMe2. The intermediate aromatic amidine can then cyclize into a quinazoline structure in which the endocyclic and exocyclic nitrogen atoms are interchanged as a result of the Dimroth rearrangement leading to 4-anilinoquinazolines 91 and 92 (Scheme 29).

Scheme 29
Besson's group proposed a short and effective route to Azixa (EPi28495, MPC-6827), N-(4-methoxyphenylamino)-N,2-dimethylquinazoline (95) (Scheme 30), which is a low molecular weight microtubule formation inhibitor and has been identified as a potent inducer of apoptosis.86–88 Moreover, Azixa (95) is able to cross the blood-brain barrier and accumulate in the brain.88 This property makes Azixa (95) a good candidate for the treatment of primary and metastatic brain tumors the therapy of which is practically limited. The synthesis of 4-anilinoquinazoline 95 begins with the reaction of anthranilonitrile (66b) and N,N-dimethylacetamide dimethyl acetal (Scheme 30). Compared to the previously synthesized compounds 89a,b, the synthesis of amidine 96 requires more energy due to steric hindrance of the methyl substituents at the nitrogen atom. However, it was obtained in high yield (90%) after 2 min of microwave irradiation at 115°C. Condensation of 4-methoxyaniline (90d) with amidine 96 under the conditions described for the synthesis of products 91a–c and 92 required a longer reaction time (30 min) to obtain quinazoline 97 in 56% yield together with a significant amount of byproducts. However, heating amidine 96 by microwave irradiation in an MeCN–AcOH, 7:3 mixture leads to a high yield (88%) of N-(4-methoxyphenylamino)-2-methylquinazoline (97) which, after N-methylation, transforms into Azixa (95) in 55% yield based on the starting anthranilonitrile (66b) (Scheme 30).

Scheme 30
Utilizing the Dimroth rearrangement, Smith et al. proposed89 an alternative route involving sequential transformation of compounds 98–106 to obtain vandetanib (107) (Schemes 31–33). Vandetanib (107), discovered by AstraZeneca, is an orally available tyrosine kinase inhibitor with activity against VEGFR/EGER/RET receptors and is currently used for the treatment of medullary thyroid cancer.90 The 9-step method89 (Schemes 31–33) made it possible to synthesize vandetanib (107) in 7% yield compared to the previously described 12-14-step methods involving compounds 108–110 (Scheme 34), which afford vandetanib (107) in 4–20% yield.91–93 This method is easily carried out; chromatographic purification is required only at the fourth step for product 102 (Scheme 31).
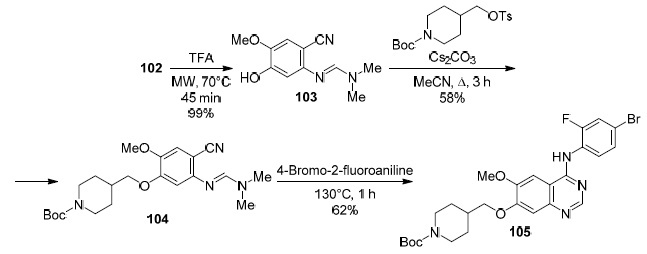
Scheme 32

Scheme 33

Scheme 31

Scheme 34
The proposed mechanism for the Dimroth rearrangement is shown in Scheme 35.89

Scheme 35
Proença's group has shown94 that the reactions of anthranilonitrile (66b) and triethyl orthoformate (TEOF), depending on the experimental conditions, lead to various quinazoline derivatives in high and low yields both as individual compounds and in the form of mixtures. For example, compound 111 was isolated after stirring for 5 days at room temperature of an anthranilonitrile (66b) – TEOF, 1:1 mixture in the presence of AcOH (Table 1, entry 1). The use of petroleum ether as a solvent led to the formation of compound 112а either by keeping at room temperature for 5 days (entry 2) or by heating under reflux for 30 min (entry 3). The formation of compound 112а occurs as a result of the Dimroth rearrangement of compound 111 (Scheme 36) which was confirmed by 1H NMR spectroscopy. The reaction of 2-amino-4-chlorobenzonitrile (66c) with TEOF in EtOH under reflux in the presence of AcOH as a catalyst proceeds within 3 days with the formation of product 112b in 4% yield (entry 6). In this case, 88% of the original compound remains unchanged. The reactions of anthranilonitrile (66b) and TEOF in the presence of AcOH in EtOH or in MeCN at 40°С for a long time (11 days) resulted in the formation of mixtures of compounds 111 and 112а in a 1:2.4 molar ratio with the total yield of 14% (entry 4) and in a 1:1 ratio with a total yield of 11% (entry 5), which in both cases were difficult to identify.
Table 1.
The reaction of 2-aminobenzonitriles 66b,c and TEOF under different conditions
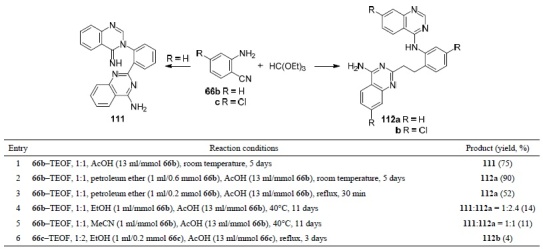

Scheme 36
The formation of compound 111 by the action of AcOH can be explained if one assumes that intramolecular cyclization of intermediate B (114·AcOH) obtained from the intermediate product A (113·H+) is hindered by the close ionic interaction between amidinium and acetate ions.94 Then, the cyano group becomes available for nucleophilic attack by another anthranilonitrile molecule (66b) leading to intermediate C. Intramolecular formation of a bond between the nitrogen atom of the imino group and the cyano group leads to intermediate D with a new more basic aminopyrimidine fragment. The acetate ion would now be preferentially stabilized by ionic interaction with the amidine moiety in intermediate D' which would lead to intramolecular cyclization with the formation of compound 111. The nonpolar solvent will promote the formation of a close ion pair ultimately responsible for the main route. This can explain the high yield of the Dimroth rearrangement product, compound 112а, obtained by heating under reflux or prolonged stirring at room temperature in petroleum ether of compound 111 in the presence of a catalytic amount of acid (Scheme 36).
Han et al.95 developed a new efficient and selective divergent synthesis of furo- and pyrrolo[2,3-d]pyrimidine- 4(3H)-imino derivatives 117a–e and furo- and thieno[2,3-d]-pyrimidin-4-amines 118a–n based on compounds 115a–d and 116a–f using microwave irradiation. The reaction used readily available amines 116a–f and substituted N,N-dimethylformamidines 115a–d. The optimal reaction conditions for the synthesis of furo- and pyrrolo[2,3-d]-pyrimidin-4-imino derivatives 117a–e were 110 or 140°C, 25–35 min, whereas for the preparation of structurally different furo- and thieno[2,3-d]pyrimidines 118a–n – 180°C, 35 min (Table 2).
Table 2.
The yields of furo- and pyrrolo[2,3-d]pyrimidine-4(3H)-imino derivatives 117a–e and furo- and thieno[2,3-d]pyrimidin-4-amines 118a–n obtained by the reaction of nitriles 115a–d and amines 116a–f

*Yields of isolated and characterized products.
**Reaction conditions for compounds 118a–n: 180°C, 35 min.
***Reaction conditions: 110°C, 25–35 min.
*4Reaction conditions: 140°C, 25–35 min.
The proposed reaction mechanism95 as shown in Scheme 37 involves the Dimroth rearrangement. First, the amino group of compound 116 attacks the carbon atom of formamidine 115 to produce intermediate E. Then, intramolecular ring closure takes place with the formation of intermediate F followed by removal of HNMe2 to give product 117 (imino product 117 is the kinetic product). After that, H2O attacks the pyrimidine ring as a nucleophile and opens it with the formation of compound G in which the amidine fragment is rotated by 180° in comparison with the tautomeric form G'. Subsequent electrocyclization and elimination of H2O from compound H leads to thermodynamically stable product 118 (preferred at high temperature). The same trend was observed even when N-(2,6-dichlorobenzyl)pyrrole derivative 115d was used as the starting reagent (Scheme 38).

Scheme 37

Scheme 38
Not only N,N-dimethylformamidines 115 but also their condensed analogs undergo a similar reaction. For example, Besson's group has shown96 that the reaction of N,N-dimethylformamidine 119, easily accessed from 3-aminofuro[3,2-b]pyridine-2-carbonitrile (120) by the action of N,N-dimethylformamide dimethyl acetal (DMF–DMA), with various aromatic amines and formamide (which plays the dual role of the solvent and reagent) under the conditions of microwave irradiation leads to pyrido-[2',3':4,5]furo[3,2-d]pyrimidines 121 and 122, respectively (Scheme 39).

Scheme 39
Besson's group was also the first to develop and optimize an efficient method for the synthesis of benzo[b]-thieno[3,2-d]pyrimidin-4-amines 125a–j, their pyrido analogs 126 and 127 a–j, and pyrazino analogs 128a–j based on compounds 123 and 124 a–d. N-Arylbenzo[b]-thieno[3,2-d]pyrimidin-4-amines 125–128 b–j were obtained by condensation of aromatic amines with N,N-dimethylformamidines 124a–d accelerated by microwave irradiation. The latter were synthesized by the reaction of thiophene derivatives 123 with DMF–DMA (Scheme 40). The inhibitory activity of the end products against five protein kinases (CDK5/p25, CK1δ/ε, GSK3α/β, DYRK1A, and CLK1) was evaluated. A series of pyrido- [3',2':4,5]thieno[3,2-d]pyrimidin-4-amine derivatives 126a–j proved to be especially promising for the development of new pharmacological inhibitors of kinases CK1 and CLK1.97

Scheme 40
Shikhaliev's group showed98 that the reaction of N'-(4-cyano-1-phenyl-1H-pyrazol-5-yl)-N,N-dimethylformamidine (129) with anilines 90 in AcOH under the conditions of microwave irradiation results in the formation of N-substituted 1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amines 130a–f (Scheme 41).

Scheme 41
Dias et al.99 developed novel and efficient methods for the synthesis of N(1)- and C(6)-substituted adenines from readily available 5-aminoimidazole-4-carboxamidines 131. Condensation of these compounds with TEOF in the presence of H2SO4 led to the selective synthesis of N(1)-substituted adenines 132. In this case, the reaction with neutral amidines 131 was preliminarily carried out at room temperature in the presence of 1–4 equiv of TEOF and a catalytic amount of H2SO4 but the reaction was very slow. When an excess of H2SO4 (4 equiv) was added, a fast reaction took place (reaction time 5–10 min) with the formation of white products which were easily isolated by filtration and were identified as salts 132a·H2SO4 (R1 = 4-FC6H4, R2 = 4-MeOC6H4, yields 64%) and 132b·H2SO4 (R1 = 4-MeOC6H4, R2 = 4-MeC6H4, yield 76%). The free bases, adenines 132, were obtained in situ by treatment with DBU of the corresponding salts 132·H2SO4 (Scheme 42).
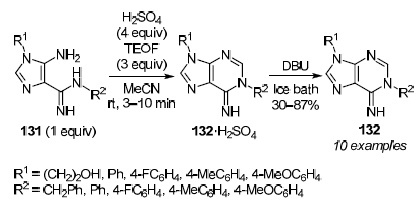
Scheme 42
The formation of adenines 132 is explained by the regioselective condensation of TEOF with the 5-amino group of imidazoles 131 as depicted in Scheme 43. The alternative condensation with the 4-carboxamidine group is unfavorable due to the formation of an amidinium salt in the presence of H2SO4.99

Scheme 43
Regioselective synthesis of С(6)-substituted adenines 133a–l occurs when the same precursors 131 are reacted with N,N-dimethylformamide diethyl acetal (DMF–DEA) in MeCN at 40°С. When the reaction was carried out for 1 day under these conditions, products 133a–i were isolated in good and high yields (61–93%). C(6)-Alkyladenines 133j–l were obtained using a one-pot two-step reaction of imidazoles 134 with benzylamine or 2-methoxyethylamine.
In the first step, imidazole 134 was reacted with an amine and 1 equiv TFA at room temperature. Then, DMF–DEA was added to the reaction mixture and the reaction was continued overnight which led to the formation of adenines 133j–l in 48–86% yields99 (Table 3).
Table 3.
The yields of adenines 133a–l, obtained from imidazoles 131 and 134
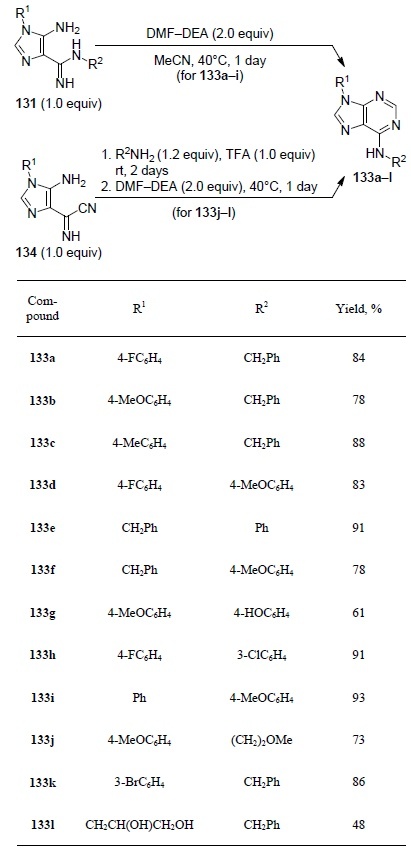
The formation of exclusively C(6)-isomer 133 by this route indicates regioselective condensation of DMF–DEA with the free amino group of the 4-carboxamidine substituent of imidazole 13199 as shown in Scheme 44.

Scheme 44
C(6)-Substituted adenines 133a–f can also be obtained from N(1)-substituted adenines by the Dimroth rearrangement in the presence of HNMe2 99 (Scheme 45).

Scheme 45
Ben Jannet et al.100 obtained the corresponding ethoxymethyleneamino derivative 136 by the reaction of 5-aminopyrazole-4-carbonitrile 135 with TEOF101 and demonstrated that imidate 136 reacted at its two electrophilic centers with aliphatic amines 116 to form pyrazolopyrimidines 138a–c in two steps via intermediates 137a–c. In the first step, the condensation of imidate 136 with amines 116 in EtOH in the presence of a catalytic amount of AcOH leads to intermediate compounds 137a–c due to the nucleophilic attack of the amino group at the imide carbon atom. In the second step, the isolated amidines 137a–c undergo intramolecular cyclization with the in situ formation of intermediates I which are isomerized to thermodynamically more stable pyrazolopyrimidine derivatives 138a–c via tandem basecatalyzed opening and closure of the pyrimidine ring (Scheme 46). This rearrangement corresponds to those discussed in earlier studies.102–104 The reaction of compounds 136 with aromatic amines 90 leads to N-aryl-3-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amines 139a–d via the Dimroth rearrangement of the intermediate compounds J100 (Scheme 46).

Scheme 46
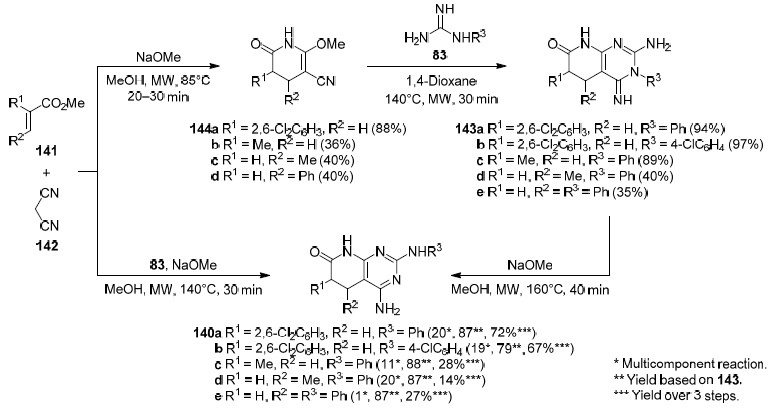
Borrell's group105 developed two methods for the synthesis of 2-arylamino-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-ones 140a–e. One of them relies on a multicomponent reaction between α,β-unsaturated ester 141, malononitrile (142), and arylguanidine 83 (obtained preliminary from the carbonate salt) in the presence of NaOMe in MeOH, whereas the other is based on the Dimroth rearrangement of 3-aryl-substituted pyridopyrimidines 143a–e, formed during the treatment of pyridones 144a–d with arylguanidines 83 in 1,4-dioxane, into 2-arylaminopyridopyrimidines 140a–e upon heating in MeOH in the presence of NaOMe (Scheme 47). For comparison, the yields (for each step and the combined yield) of a series of 2-arylamino-substituted pyridopyrimidines 140a–e using both methods are shown in Scheme 47.
Scheme 47
Scheme 47 demonstrates that a) the total yields of 4-amino-5,6-dihydropyrido[2,3-d]pyrimidin-7(8H)-ones 140a–e formed via 3-aryl-substituted pyridopyrimidines 143a–e, as a rule, are higher than those obtained as a result of the multicomponent reaction; b) when the α,β-unsaturated ester 141 has a substituent at the β-position (R2), the yields are generally lower than when it is present at the α-position (R1); and c) although the multicomponent reaction gives lower yields than the three-step procedure, in some cases it can be a good alternative as it allows the desired pyridopyrimidine 140 to be obtained in one step. The proposed mechanism for the formation of compounds 143a and 140a105 is given in Scheme 48.

Scheme 48
Rearrangement of condensed heterocyclic systems containing six-membered rings with nitrogen and sulfur atoms
Wu's group106 developed a highly selective and efficient temperature-dependent chemodivergent method for the synthesis of 4H-benzo[d][1,3]thiazin-4-ones and 2-thioxo-2,3-dihydroquinazolin-4(1H)-ones from isothiocyanates 145 and isatins 146. The method incorporates a cascade of oxidation and decarboxylation processes followed by cyclization; carrying out the reaction at room temperature gives rise to 2-amino-4H-benzo[d][1,3]thiazin-4-one derivatives 147a–l (Scheme 49), while derivatives of 2-thioxo-4(3H)-quinozalinones 148a–l are formed at 80°С (Scheme 50).

Scheme 49

Scheme 50
As shown in Scheme 49, the use of halogenated (3-F, 3-Cl, 4-Cl, and 4-Br) isothiocyanatobenzenes 145 in the synthesis of 4H-benzo[d][1,3]thiazin-4-ones 147a–l leads to target products 147b–e in good yields (71–86%). The electron-withdrawing group (4-NO2) had a positive effect on the reaction and the desired product 147f was obtained in good yield (85%). Electron-donating groups (4-Me, 3-MeO) also led to the corresponding compounds 147g,h in moderate yields (48 and 58%, respectively). Moreover, 3-isothiocyanopyridine underwent the reaction to form product 147i in good yield (86%). As for the substituents in the isatin fragment, electron-neutral and electron-donating groups do not affect the course of the reaction since products 147j–l were obtained in good yields (80–89%) in all of the variations of substituents.106
In the case of the synthesis of 2-thioxo-2,3-dihydroquinozalin-4(1H)-one derivatives 148a–l, the reactions of isothiocyanatobenzenes containing halogen atoms and electron-withdrawing groups in the benzene ring proceeded with the formation of the target products 148b–e in high yields (65–78%). Electron-neutral and electron-donating groups in different positions had a minor effect on the yields of products 148b–e (51–65%). The sterically hindered 2-isothiocyanatonaphthalene also reacted with the formation of the target product 148i in 70% yield. In addition, isatin derivatives with electron-neutral and electron-donor groups showed good reactivity (compounds 148j–l, 72–81% yields)106 (Scheme 50).
A mechanism was proposed for the reaction of phenyl isothiocyanates with isatins based on the results of the following reactions of phenyl isothiocyanate (145a) with anhydride 149 at different temperature conditions with the formation of compounds 147a and 148a and treatment of 2-(phenylamino)-4H-benzo[d][1,3]thiazin-4-one (147а) with Na2CO3 in DMSO at 100°С resulting in the Dimroth rearrangement with the formation of compound 148a (Scheme 51).106

Scheme 51
In the initial step of the process initiated by the nucleophilic attack of tert-butylperoxy anion on isatin 146a (R2 = H), intermediate A is formed which is then converted into anhydride 149 by a mechanism similar to the Baeyer–Villiger oxidation. Then, cyclization of phenyl isothiocyanate (145а) with anhydride 149 with simultaneous decarboxylation leads to 2-(phenylamino)-4H-benzo[d]-[1,3]thiazin-4-one 147а which in the presence of Na2CO3 at high temperature undergoes the Dimroth rearrangement producing stable 2-thioxo-2,3-dihydroquinazolin-4(1H)- one 148a106 (Scheme 52).

Scheme 52
In conclusion, a highly selective, base-catalyzed, temperature-controlled method has been developed for the synthesis of 4H-benzo[d][1,3]thiazin-4-ones and 2-thioxo-2,3-dihydroquinazolin-4(1H)-ones. The method involves a kinetically controlled tandem oxidation–cyclization process with decarboxylation in the reactions of isothiocyanates with isatins. Carrying out the reaction at room temperature yields access to 4H-benzo[d][1,3]thiazin-4-ones, whereas when the temperature is risen to 80°C, 2-thioxo-2,3-dihydroquinazolin-4(1H)-ones can be obtained.106
Wu et al.107 employed 2-haloaryl isothiocyanates instead of isothiocyanobenzenes in the reaction with commercially available isatins resulting in the development of an efficient method for the synthesis of 12H-benzo[4,5]thiazolo[2,3-b]-quinazolin-12-one derivatives without the use of transition metals. A key step toward the synthesis of these compounds is the Dimroth rearrangement.
As shown in Scheme 53, the electronic properties and the position of the substituent on the phenyl ring of the 2-haloaryl isothiocyanate derivative 150 practically do not affect the course of the reaction. Compounds with fluorine atoms in positions 3, 4, and 6 react well forming target products 151a–c in good yields (64–81%). The substituents at position 6 of the phenyl ring lead to a slight decrease in yield which can probably be attributed to steric hindrance.

Scheme 53
The presence of electron donor groups (4-MeO, 4-Me, and 5-Me) decreases the yields of products 151d–f (61–72%). The use of other halogens as substituents in the starting compounds 150 does not affect the course of the reaction and does not decrease the yields of products 151g–i (66-80%).107
Based on the above results and literature data, a possible reaction mechanism was proposed using isatin 146a (R2 = H) and 1-fluoro-2-isothiocyanatobenzene (150a) as models. Initially, isatin 146a is transformed into intermediate A as a result of a nucleophilic attack of the tert-butylperoxy anion followed by intramolecular rearrangement with the formation of anhydride 149, an oxidation product of the Baeyer–Villiger-type oxidation. Cyclization of anhydride 149 with 1-fluoro-2-isothiocyanatobenzene (150а) with decarboxylation gives intermediate benzothiazinone 152. Then, the Dimroth rearrangement of compound 152 occurs which is interrupted by intramolecular aromatic nucleophilic substitution to form intermediate C. Finally, intramolecular amidation of compound D leads to product 151a107 (Scheme 54).

Scheme 54
To conclude, an analysis of the literature data made it possible to draw conclusions on the importance of pyrimidines and their condensed analogs and, in this regard, the need to develop new methods for their preparation, as well as the prospects of research of the directions of their practical use. The Dimroth rearrangement is a simple and efficient way of constructing condensed pyrimidines, often in a one-pot manner, from available starting reagents. An important positive aspect of the Dimroth rearrangement is the variability of the starting reagents which makes it possible to obtain various condensed systems with the pyrimidine ring with various substituents. At the same time, the dependence of the regional orientation and selectivity of reactions on many factors makes research in this direction interesting and unpredictable. The potential of condensed pyrimidine analogs as compounds with practically important properties, which has not yet been fully disclosed, guarantees in the future the constant interest of synthetic chemists both in this class of compounds in general and in methods of constructing the pyrimidine ring in combination with other carbo- and heterocyclic rings using also the Dimroth rearrangement. We hope that the systematization of literature data on the synthesis of various condensed pyrimidine analogs can serve as a foundation for the development of approaches to the synthesis of both natural compounds and their modified analogs with predictable biological activity.
Footnotes
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2021, 57(4), 342–368
References
- 1.Rathke B. Ber. Dtsch. Chem. Ges. 1888;21:867. doi: 10.1002/cber.188802101154. [DOI] [Google Scholar]
- 2.Dimroth O. Justus Liebigs Ann. Chem. 1909;364:183. doi: 10.1002/jlac.19093640204. [DOI] [Google Scholar]
- 3.Brown, D. J.; Hoerger, E.; Mason, S. F. J. Chem. Soc.1955, 4035.
- 4.Carrington, H. C.; Curd, F. H. S.; Richardson, D. N. J. Chem. Soc.1955, 1858.
- 5.El Ashry, E. S. H.; El Kilany, Y.; Rashed, N.; Assafir, H. In Advances in Heterocyclic Chemistry; Katritzky, A. R., Ed.; Elsevier: Amsterdam, 1999, Vol. 75, p. 79.
- 6.Brown, D. J.; Harper, J. S. J. Chem. Soc.1963, 1276.
- 7.L'abbé G, Vanderstede E. J. Heterocycl. Chem. 1989;26:1811. doi: 10.1002/jhet.5570260652. [DOI] [Google Scholar]
- 8.Nagamatsu T, Fujita T. Heterocycles. 2002;57:631. doi: 10.3987/COM-02-9450. [DOI] [Google Scholar]
- 9.Loakes D, Brown DM, Salisbury SA. Tetrahedron Lett. 1998;39:3865. doi: 10.1016/S0040-4039(98)00634-0. [DOI] [Google Scholar]
- 10.Loakes, D.; Brown, D. M.; Salisbury, S. A. J. Chem. Soc., Perkin Trans. 11999, 1333.
- 11.Ogata Y, Takagi K, Hayashi E. Bull. Chem. Soc. Jpn. 1977;50:2505. doi: 10.1246/bcsj.50.2505. [DOI] [Google Scholar]
- 12.Fanghaenel E, Kordts B, Richter AM, Dutschmann K. J. Prakt. Chem. 1990;332:387. doi: 10.1002/prac.19903320317. [DOI] [Google Scholar]
- 13.Guerret P, Jacquier R, Maury G. J. Heterocycl. Chem. 1971;8:643. doi: 10.1002/jhet.5570080419. [DOI] [Google Scholar]
- 14.Brown DJ, Nagamatsu T. Aust. J. Chem. 1977;30:2515. doi: 10.1071/CH9772515. [DOI] [Google Scholar]
- 15.El Ashry, E. S. H.; Nadeem, S.; Shah, M. R.; El Kilany, Y. In Advances in Heterocyclic Chemistry; Katritzky, A. R., Ed.; Elsevier: Amsterdam, 2010, Vol. 101, p. 161.
- 16.Liu KC, Shih BJ, Chern JW. J. Heterocycl. Chem. 1990;27:391. doi: 10.1002/jhet.5570270253. [DOI] [Google Scholar]
- 17.Vaughan K, LaFrance RJ, Tang Y, Hooper DL. J. Heterocycl. Chem. 1991;28:1709. doi: 10.1002/jhet.5570280711. [DOI] [Google Scholar]
- 18.Stevens MFG, Chui WK, Castro MA. J. Heterocycl. Chem. 1993;30:849. doi: 10.1002/jhet.5570300401. [DOI] [Google Scholar]
- 19.Wang, Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons: Hoboken, 2010, Vol. 1, p. 905.
- 20.Kurti L, Czako B. Strategic Applicatons of Named Reactions in Organic Synthesis. Boston: Academic Press; 2005. [Google Scholar]
- 21.Hassner A, Namboothiri I. Organic Syntheses Based on Name Reactions. Amsterdam: Elsevier; 2012. [Google Scholar]
- 22.Fujii T, Itaya T. Heterocycles. 1998;48:359. doi: 10.3987/REV-98-500. [DOI] [Google Scholar]
- 23.Brown, D. J.; Harper, J. S. In Pteridine Chemistry; Pfleiderer, W.; Taylor, E. C., Eds.; Macmillan: New York, 1964, p. 219.
- 24.Brown, D. J. In Mechanism of Molecular Migrations; Thyagarajan, B. S., Ed.; Interscience Publishers: New York, 1968, Vol. 1, p. 209.
- 25.Brown DJ. The Chemistry of Heterocyclic Compounds, The Pyrimidines. Supplement 1. Hoboken: John Wiley & Sons; 1970. [Google Scholar]
- 26.L'abbé G. Ind. Chim. Belge. 1971;36:3. [Google Scholar]
- 27.Fujii, T.; Itaya, T.; Saito, T. Symp. Heterocycl.1977, 129.
- 28.L'abbé G. J. Heterocycl. Chem. 1984;21:627. doi: 10.1002/jhet.5570210301. [DOI] [Google Scholar]
- 29.Maiboroda, D. А.; Babaev, Е. V. Chem. Heterocycl. Compd.1995, 31, 1251. [Khim. Geterotsikl. Soedin.1995, 1445.]
- 30.Krajczyk A, Boryski J. Curr. Org. Chem. 2017;21:2515. doi: 10.2174/1385272821666170427125720. [DOI] [Google Scholar]
- 31.Steenackers H, Ermolat'ev D, Trang TTT, Savalia B, Sharma UK, De Weerdt A, Shah A, Vanderleyden J, Van der Eycken EV. Org. Biomol. Chem. 2014;12:3671. doi: 10.1039/C3OB42282H. [DOI] [PubMed] [Google Scholar]
- 32.Koltun DO, Parkhill EQ, Elzein E, Kobayashi T, Notte GT, Kalla R, Jiang RH, Li X, Perry TD, Avila B, Wang W-Q, Smith-Maxwell C, Dhalla AK, Rajamani S, Stafford B, Tang J, Mollova N, Belardinelli L, Zablocki JA. Bioorg. Med. Chem. Lett. 2016;26:3202. doi: 10.1016/j.bmcl.2016.03.101. [DOI] [PubMed] [Google Scholar]
- 33.Sirakanyan, S. N.; Avetisyan, N. G.; Naravyan, A. S. Chem. Heterocycl. Compd.2012, 48, 470. [Khim. Geterotsikl. Soedin.2012, 500.]
- 34.Krinochkin AP, Kopchuk DS, Giri K, Shtaitz YK, Starnovskaya ES, Khalymbadzha IA, Drokin RA, Ulomsky EN, Santra S, Zyryanov GV, Rusinov VL, Chupakhin ON. ChemistrySelect. 2018;3:8202. doi: 10.1002/slct.201801244. [DOI] [Google Scholar]
- 35.Li Z, Chen J, Wu L, Ren A, Lu P, Wang Y. Org. Lett. 2020;22:26. doi: 10.1021/acs.orglett.9b03708. [DOI] [PubMed] [Google Scholar]
- 36.Ali TE, Assiri MA, Abdel-Kariem SM, Yahia IS. J. Sulfur Chem. 2018;39:472. doi: 10.1080/17415993.2018.1455837. [DOI] [Google Scholar]
- 37.Khalladi K, Touil S. J. Sulfur Chem. 2012;33:27. doi: 10.1080/17415993.2011.639021. [DOI] [Google Scholar]
- 38.Wan Z, Hu D, Li P, Xie D, Gan X. Molecules. 2015;20:11861. doi: 10.3390/molecules200711861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang M, Zhang G, Wang Y, Wang J, Zhu M, Cen S, Wang Y. Bioorg. Med. Chem. Lett. 2020;30:127143. doi: 10.1016/j.bmcl.2020.127143. [DOI] [PubMed] [Google Scholar]
- 40.Capuzzi SJ, Sun W, Muratov EN, Martínez-Romero C, He S, Zhu W, Li H, Tawa G, Fisher EG, Xu M, Shinn P, Qiu X, García-Sastre A, Zheng W, Tropsha A. J. Med. Chem. 2018;61:3582. doi: 10.1021/acs.jmedchem.8b00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sroor FM, Basyouni WM, Tohamy WM, Abdelhafez TH, El-awady MK. Tetrahedron. 2019;75:130749. doi: 10.1016/j.tet.2019.130749. [DOI] [Google Scholar]
- 42.Mohamed MS, Sayed AI, Khedr MA, Nofal S, Soror SH. Eur. J. Pharm. Sci. 2019;127:102. doi: 10.1016/j.ejps.2018.10.022. [DOI] [PubMed] [Google Scholar]
- 43.Balaraman S, Nayak N, Subbiah M, Elango KP. Med. Chem. Res. 2018;27:2538. doi: 10.1007/s00044-018-2256-z. [DOI] [Google Scholar]
- 44.Bassetto M, Leyssen P, Neyts J, Yerukhimovich MM, Frick DN, Brancale A. Eur. J. Med. Chem. 2016;123:31. doi: 10.1016/j.ejmech.2016.07.035. [DOI] [PubMed] [Google Scholar]
- 45.Venkatesham A, Saudi M, Kaptein S, Neyts J, Rozenski J, Froeyen M, Van Aerschot A. Eur. J. Med. Chem. 2017;126:101. doi: 10.1016/j.ejmech.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 46.Kelley JL, Linn JA, Davis RG, Selway JWT. Eur. J. Med. Chem. 1990;25:623. doi: 10.1016/0223-5234(90)90188-9. [DOI] [Google Scholar]
- 47.Shin YS, Jarhad DB, Jang MH, Kovacikova K, Kim G, Yoon, J.-s.; Kim, H.-R.; Hyun, Y. E.; Tipnis, A. S.; Chang, T.-S.; van Hemert, M. J.; Jeong, L. S. Eur. J. Med. Chem. 2020;187:111956. doi: 10.1016/j.ejmech.2019.111956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoon, J.-s.; Kim, G.; Jarhad, D. B.; Kim, H.-R.; Shin, Y.-S.; Qu, S.; Sahu, P. K.; Kim, H. O.; Lee, H. W.; Wang, S. B.; Kong, Y. J.; Chang, T.-S.; Ogando, N. S.; Kovacikova, K.; Snijder, E. J.; Posthuma, C. C.; van Hemert, M. J.; Jeong, L. S. J. Med. Chem.2019, 62, 6346. [DOI] [PMC free article] [PubMed]
- 49.Khodak LA, Mushenko LV, Rzhevskaya OA. Mezhdunarodn. med. zh. 2005;2:124. [Google Scholar]
- 50.Yu W, Goddard C, Clearfield E, Mills C, Xiao T, Guo H, Morrey JD, Motter NE, Zhao K, Block TM, Cuconati A, Xu X. J. Med. Chem. 2011;54:5660. doi: 10.1021/jm200696v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rashad AE, Hegab MI, Abdel-Megeid RE, Micky JA, Abdel-Megeid FME. Bioorg. Med. Chem. 2008;16:7102. doi: 10.1016/j.bmc.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 52.Chern J-H, Shia K-S, Hsu T-A, Tai C-L, Lee C-C, Lee Y-C, Chang C-S, Tseng S-N, Shih S-R. Bioorg. Med. Chem. Lett. 2004;14:2519. doi: 10.1016/j.bmcl.2004.02.092. [DOI] [PubMed] [Google Scholar]
- 53.Chen T-C, Chang H-Y, Lin P-F, Chern J-H, Hsu JT-A, Chang C-Y, Shih S-R. Antimicrob. Agents Chemother. 2009;53:2740. doi: 10.1128/AAC.00101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gueiffier A, Lhassani M, Elhakmaoui A, Snoeck R, Andrei G, Chavignon O, Teulade J-C, Kerbal A, Essassi EM, Debouzy J-C, Witvrouw M, Blache Y, Balzarini J, De Clercq E, Chapat J-P. J. Med. Chem. 1996;39:2856. doi: 10.1021/jm9507901. [DOI] [PubMed] [Google Scholar]
- 55.Micewicz ED, Khachatoorian R, French SW, Ruchala P. Bioorg. Med. Chem. Lett. 2018;28:452. doi: 10.1016/j.bmcl.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 56.Jacquier R, Lopez H, Maury G. J. Heterocycl. Chem. 1973;10:755. doi: 10.1002/jhet.5570100513. [DOI] [Google Scholar]
- 57.Chavignon O, Teulade JC, Madesclaire M, Gueiffier A, Blache Y, Viols H, Chapat JP, Dauphin G. J. Heterocycl. Chem. 1992;29:691. doi: 10.1002/jhet.5570290403. [DOI] [Google Scholar]
- 58.Abignente E, Sacchi A, Laneri S, Rossi F, D'Amico M, Berrino L, Calderaro V, Parrillo C. Eur. J. Med. Chem. 1994;29:279. doi: 10.1016/0223-5234(94)90097-3. [DOI] [Google Scholar]
- 59.Anaflous A, Benchat N, Mimouni M, Abouricha S, Ben-Hadda T, El-Bali B, Hakkou A, Hacht B. Lett. Drug Des. Discovery. 2004;1:224. doi: 10.2174/1570180043398885. [DOI] [Google Scholar]
- 60.Borisov, A. V.; Tolmachev, A. A.; Zavada, O. A.; Zhuravel, I. A.; Kovalenko, S. N. Chem. Heterocycl. Compd.2013, 49, 704. [Khimiya geterotsikl. soedinenii2013, 754.]
- 61.Chatzopoulou M, Martinize RF, Willis NJ, Claridge TDW, Wilson FX, Wynne GM, Davies SG, Russell A. J. Tetrahedron. 2018;74:5280. doi: 10.1016/j.tet.2018.06.033. [DOI] [Google Scholar]
- 62.Rozentsveig, I. B.; Serykh, V. Yu.; Chernysheva, G. N.; Kondrashov, E. V.; Fedotova, A. I.; Ushakov, I. A.; Tretyakov, E. V.; Romanenko, G. V. Eur. J. Org. Chem.2014, 6547.
- 63.Carballares S, Cifuentes MM, Stephenson GA. Tetrahedron Lett. 2007;48:2041. doi: 10.1016/j.tetlet.2006.11.181. [DOI] [Google Scholar]
- 64.Tang C, Li Z, Wang Q, Beilstein J. Org. Chem. 2013;9:2629. doi: 10.3762/bjoc.9.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Astakhov, A. V.; Chernyshev, V. M. Chem. Heterocycl. Compd.2012, 48, 1417. [Khim. Geterotsikl. Soedin.2012, 1519.]
- 66.Son HY, Song Y-H. J. Korean Chem. Soc. 2010;54:350. doi: 10.5012/jkcs.2010.54.3.350. [DOI] [Google Scholar]
- 67.Okamuru T, Kurogi Y, Hashimoto K, Nishikawa H, Nagao Y. Bioorg. Med. Chem. Lett. 2004;14:2443. doi: 10.1016/j.bmcl.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 68.Lovelette CA, Geagan K. J. Heterocycl. Chem. 1982;19:1345. doi: 10.1002/jhet.5570190618. [DOI] [Google Scholar]
- 69.Nagamatsu T, Ahmed S, Hossion AML, Ohno S. Heterocycles. 2007;73:777. doi: 10.3987/COM-07-S(U)58. [DOI] [Google Scholar]
- 70.Shawali AS, Hassaneen HM, Shurrab NK. Tetrahedron. 2008;64:10339. doi: 10.1016/j.tet.2008.08.082. [DOI] [Google Scholar]
- 71.Shawali AS, Hassaneen HM, Shurrab NK. Heterocycles. 2008;75:1479. doi: 10.3987/COM-07-11311. [DOI] [Google Scholar]
- 72.Lauria A, Patella C, Abbate I, Martorana A, Almerico AM. Eur. J. Med. Chem. 2013;65:381. doi: 10.1016/j.ejmech.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 73.Lauria A, Abbate I, Patella C, Martorana A, Dattolo G, Almerico AM. Eur. J. Med. Chem. 2013;62:416. doi: 10.1016/j.ejmech.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 74.Lauria A, Diana P, Barraja P, Montalbano A, Cirrincione G, Dattolo G, Almerico AM. Tetrahedron. 2002;58:9723. doi: 10.1016/S0040-4020(02)01245-0. [DOI] [Google Scholar]
- 75.Lauria A, Patella C, Diana P, Barraja P, Montalbano A, Cirrincione G, Dattolo G, Almerico AM. Heterocycles. 2003;60:2669. doi: 10.3987/COM-03-9877. [DOI] [Google Scholar]
- 76.Tang J-H, Shi D-X, Zhang L-J, Zhang Q, Li J-R. Synth. Commun. 2010;40:632. doi: 10.1080/00397910902908822. [DOI] [Google Scholar]
- 77.Roger R, Neilson D. Chem. Rev. 1961;61:179. doi: 10.1021/cr60210a003. [DOI] [Google Scholar]
- 78.Ghashang M, Mansoor SS, Aswin K. J. Adv. Res. 2014;5:209. doi: 10.1016/j.jare.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Foucourt A, Dubouilh-Benard C, Chosson E, Corbière C, Buquet C, Iannelli M, Leblond B, Marsais F, Besson T. Tetrahedron. 2010;66:4495. doi: 10.1016/j.tet.2010.04.066. [DOI] [Google Scholar]
- 80.Karimi N, Davoodnia A, Pordel M. Heterocycl. Commun. 2018;24:31. doi: 10.1515/hc-2017-0228. [DOI] [Google Scholar]
- 81.Ebrahimi Z, Davoodnia A, Motavalizadehkakhky A, Mehrzad J. Org. Prep. Proced. Int. 2019;51:357. doi: 10.1080/00304948.2019.1596472. [DOI] [Google Scholar]
- 82.Zhen B, Jiao Q, Zhang Y, Wu Q, Li H, Shi D, Li J. Catal. Commun. 2013;32:1. doi: 10.1016/j.catcom.2012.11.029. [DOI] [Google Scholar]
- 83.Liu M, Li J, Chen S, Huang D, Chai H, Zhang Q, Shi D. RSC Adv. 2014;4:35629. doi: 10.1039/C4RA05346J. [DOI] [Google Scholar]
- 84.Hosseininasab N, Davoodnia A, Rostami-Charati F, Tavakoli-Hoseini N, Khojastehnezhad A. J. Heterocycl. Chem. 2018;55:161. doi: 10.1002/jhet.3019. [DOI] [Google Scholar]
- 85.Chandregowda V, Rao GV, Reddy GC. Org. Process Res. Dev. 2007;11:813. doi: 10.1021/op700054p. [DOI] [Google Scholar]
- 86.Besson T, Chosson E. Comb. Chem. High Throughput Screen. 2007;10:903. doi: 10.2174/138620707783220356. [DOI] [PubMed] [Google Scholar]
- 87.Sirisoma N, Pervin A, Zhang H, Jiang S, Willardsen JA, Anderson MB, Mather G, Pleiman CM, Kasibhatla S, Tseng B, Drewe J, Cai SX. J. Med.Chem. 2009;52:2341. doi: 10.1021/jm801315b. [DOI] [PubMed] [Google Scholar]
- 88.Kasibhatla S, Baichwal V, Cai SX, Roth B, Skvortsova I, Skvortsov S, Lukas P, English NM, Sirisoma N, Drewe J, Pervin A, Tseng B, Carlson RO, Pleiman CM. Cancer Res. 2007;67:5865. doi: 10.1158/0008-5472.CAN-07-0127. [DOI] [PubMed] [Google Scholar]
- 89.Brocklesby KL, Waby JS, Cawthorne C, Smith G. Tetrahedron Lett. 2017;58:1467. doi: 10.1016/j.tetlet.2017.02.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chau NG, Haddad R. J. Clin. Cancer Res. 2013;19:524. doi: 10.1158/1078-0432.CCR-12-2353. [DOI] [PubMed] [Google Scholar]
- 91.Hennequin LF, Stokes ESE, Thomas AP, Johnstone C, Plé PA, Ogilvie DJ, Dukes M, Wedge SR, Kendrew J, Curwen JO. J. Med. Chem. 2002;45:1300. doi: 10.1021/jm011022e. [DOI] [PubMed] [Google Scholar]
- 92.Hennequin LF, Thomas AP, Johnstone C, Stokes ESE, Plé PA, Lohmann J-JM, Ogilvie DJ, Dukes M, Wedge SR, Curwen JO, Kendrew J, Lambert-van der Brempt C. J. Med. Chem. 1999;42:5369. doi: 10.1021/jm990345w. [DOI] [PubMed] [Google Scholar]
- 93.Gold H. Angew. Chem. 1960;72:956. doi: 10.1002/ange.19600722406. [DOI] [Google Scholar]
- 94.Marinho E, Araujo R, Proença F. Tetrahedron. 2010;66:8681. doi: 10.1016/j.tet.2010.09.013. [DOI] [Google Scholar]
- 95.Han Y, Ebinger K, Vandevier LE, Maloney JW, Nirschl DS, Weller HN. Tetrahedron Lett. 2010;51:629. doi: 10.1016/j.tetlet.2009.11.071. [DOI] [Google Scholar]
- 96.Loidreau Y, Marchand P, Dubouilh-Benard C, Nourrisson M-R, Duflos M, Besson T. Tetrahedron Lett. 2012;53:944. doi: 10.1016/j.tetlet.2011.12.042. [DOI] [Google Scholar]
- 97.Loidreau Y, Marchand P, Dubouilh-Benard C, Nourrisson M-R, Duflos M, Lozach O, Loaec N, Meijer L, Besson T. Eur. J. Med. Chem. 2012;58:171. doi: 10.1016/j.ejmech.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 98.Kosheleva, E. A.; Shikhaliev, Kh. S.; Ponomareva, L. F. Proceedings of Voronezh State University. Series: Chemistry. Biology. Pharmacy2016, 7.
- 99.Senhorães N, Costa AL, Silva DI, Proença MF, Dias AM. Tetrahedron. 2013;69:10014. doi: 10.1016/j.tet.2013.09.063. [DOI] [Google Scholar]
- 100.Rahmouni A, Romdhane A, Ben Said A, Majouli K, Ben Jannet H. Turk. J. Chem. 2014;38:210. doi: 10.3906/kim-1303-20. [DOI] [Google Scholar]
- 101.Eljazi IA, Samar AA. Molecules. 2001;6:621. doi: 10.3390/60700621. [DOI] [Google Scholar]
- 102.Ducray R, Ballard P, Barlaam BC, Hickinson MD, Kettle JG, Ogilvie DJ, Trigwell CB. Bioorg. Med. Chem. Lett. 2008;18:959. doi: 10.1016/j.bmcl.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 103.Shawali AS, Hassaneen HM, Shurrab NK. Tetrahedron. 2008;64:10339. doi: 10.1016/j.tet.2008.08.082. [DOI] [Google Scholar]
- 104.Oliveira-Campos, A. M. F.; Salaheldin, A. M.; Rodrigues, L. M. ARKIVOC2007, (xvi), 92.
- 105.Galve I, Puig de la Bellacasa R, Sánchez-García D, Batllori X, Teixidó J, Borrell J, Mol I. Diversity. 2012;16:639. doi: 10.1007/s11030-012-9398-6. [DOI] [PubMed] [Google Scholar]
- 106.Zhou Z-W, Jia F-C, Xu C, Jiang S-F, Wu Y-D, Wu A-X, Asian J. Org. Chem. 2017;6:1773. [Google Scholar]
- 107.Zhou Z-W, Jia F-C, Xu C, Jiang S-F, Wu Y-D, Wu A-X. Chem. Commun. 2017;53:1056. doi: 10.1039/C6CC09376K. [DOI] [PubMed] [Google Scholar]