

Summary
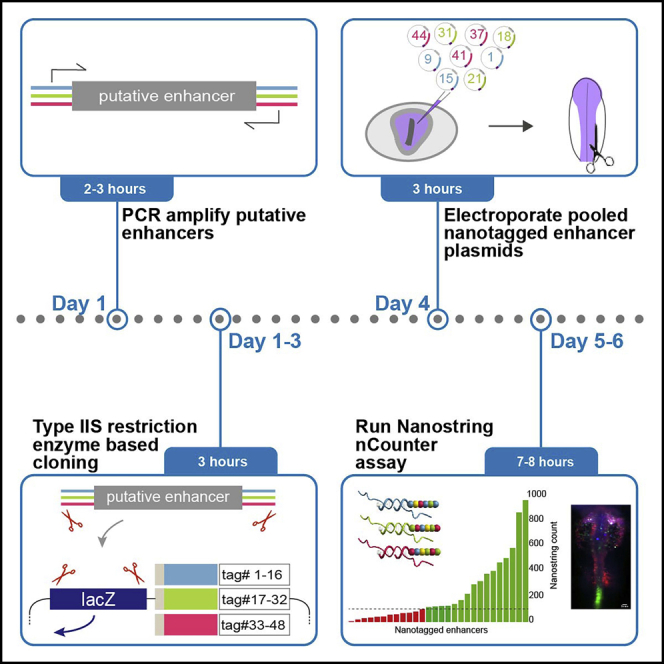
Here, we describe a highly efficient, medium-throughput strategy for cloning and in vivo screening of putative enhancers using the chick embryo. By incorporating 48 unique nanotags for use in NanoString nCounter® across three different fluorescent reporters and developing a rapid and efficient digestion/ligation type IIs restriction enzyme-based cloning protocol, we develop a multiplexed approach for rapidly identifying enhancer activity.
For complete details on the use and execution of this protocol, please see Williams et al. (2019).
Subject areas: Model Organisms, Molecular Biology, Gene Expression, CRISPR
Graphical abstract
Highlights
-
•
Highly efficient, medium-throughput protocol for cloning putative enhancers
-
•
Multiplexed, rapid screening strategy for detecting in vivo enhancer activity
-
•
Reproducible, non-mosaic validation of spatial and temporal enhancer activity in vivo
-
•
Functional conservation allows cross-species enhancer screening
Here, we describe a highly efficient, medium-throughput strategy for cloning and in vivo screening of putative enhancers using the chick embryo. By incorporating 48 unique nanotags for use in NanoString nCounter® across three different fluorescent reporters and developing a rapid and efficient digestion/ligation type IIs restriction enzyme-based cloning protocol, we develop a multiplexed approach for rapidly identifying enhancer activity.
Before you begin
Selecting putative enhancer regions
-
1.
We have found that chromatin accessibility analysis of specific cell types performed across different developmental timepoints yielded the best input data from which active enhancers can be identified. In short, we have used ATAC-seq to generate cell-type specific maps of putative regulatory elements of chick neural crest (NC) cells at two distinct stages of development, corresponding control non-NC cells, prospective NC progenitors during gastrulation, and, as an outlier population, another specific embryonic cell type (somites, myocardium etc.) (Williams et al., 2019). This approach provided us with a range of global chromatin accessibility profiles that can be used in unbiased clustering methods to identify dynamic spatiotemporal patterns of accessibility, thus enabling identification of tissue- and stage-specific elements for each population. Histone modifications and cross-species conservation are also useful parameters to consider for prediction of putative enhancers (Betancur et al., 2010; Rada-Iglesias et al., 2012; Simoes-Costa et al., 2012).
-
2.
Once candidate ATAC peaks are identified, primers should be designed to amplify the entire region underlying the peak +100 bp each way, this is generally ∼1 kb but some cloned enhancer elements may be up to 4 kb. This ensures all essential transcription factor binding sites are included, but potential repressor sites outside of the open region are excluded. If enhancer activity is observed, the region can be further trimmed down to identify the core element.
-
3.
Due to the significant level of conservation of enhancer function across amniotes, the chick system can be used to test and validate enhancer function of human cis-regulatory elements active during embryonic development (Hay et al., 2016; Hellner et al., 2016), and also to screen putative elements from other vertebrate species including zebrafish (unpublished data) and lamprey (Hockman et al., 2019).
Primer design
Timing: 5–15 min per primer pair
-
4.
We used Primer3 to predict target specific primers of approximately 20bp, with no off-target sites and no hairpin formation or self-complementarity.
-
5.
Ensure the sequence to be amplified does not contain any BsmBI sites (if this is unavoidable In-Fusion cloning should be used, see troubleshooting section).
-
6.
Primer tail sequences are provided in the table below. These include a BsmBI recognition site and excess sequence to facilitate enzyme binding, as well as appropriate overhangs to incorporate PCR amplicons into the reporter vector using BsmBI mediated cloning.
 |
Nanotag reporter vectors
-
7.
The Nanotag enhancer reporter vectors, including positive and negative controls, generated in the associated study can be obtained from Addgene. https://www.addgene.org/Tatjana_Sauka-Spengler/
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Fertilized chicken eggs | Henry Stewart & Co Ltd | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Kapa long range polymerase | Kapa Biosystems | #KK3501 |
| Kapa HiFi polymerase | Kapa Biosystems | #KK2103 |
| BsmBI | New England Biolabs | #R0580L |
| T4 DNA ligase | New England Biolabs | #M0202S |
| PlasmidSafe™ | Cambio | #E3101K |
| In-Fusion® HD Cloning Kit | Takara Clontech | #639649 |
| Critical commercial assays | ||
| EndoFree Maxiprep Kit | QIAGEN | #12362 |
| E.Z.N.A. EndoFree mini prep kit II | VWR (Omego Bio-Tek) | #D6950-02 |
| Wizard SV Gel and PCR Clean-up System | Promega | #A9282 |
| RNAqueous™-Micro Total RNA Isolation Kit | Ambion | #AM1931 |
| TURBO™ DNase | Ambion | #AM1907 |
| Deposited data | ||
| Nanotagged reporter vectors | https://www.addgene.org/Tatjana_Sauka-Spengler/ | IDs 130514 - 130573 |
| Selected positive enhancers | https://www.addgene.org/Tatjana_Sauka-Spengler/ | IDs 130625 - 130574 |
| Oligonucleotides | ||
| Cerulean forward | TTTTTTCGTCTC ccatgg nn nnnnnnnnnnnnnnnnnn |
N/A |
| Cerulean reverse | TTTTTTCGTCTC ggtcct nn +nnnnnnnnnnnnnnnnnn |
N/A |
| Citrine forward | TTTTTTCGTCTC gccagg nnn nnnnnnnnnnnnnnnnn |
N/A |
| Citrine reverse | TTTTTTCGTCTC caacag nnn nnnnnnnnnnnnnnnnn |
N/A |
| Cherry forward | TTTTTTCGTCTC gtgcag nnn nnnnnnnnnnnnnnnnn |
N/A |
| Cherry reverse | TTTTTTCGTCTC caccgt nnn nnnnnnnnnnnnnnnnn |
N/A |
| Inf. Cerulean forward | AGCTCGAGTT ccatg nnnn nnnnnnnnnnnnnnnn |
N/A |
| Inf. Cerulean reverse | CCGGGCTAGC ggtcc nnnn nnnnnnnnnnnnnnnn |
N/A |
| Inf. Citrine forward | AGCTCGAGTT gccag nnn nnnnnnnnnnnnnnnnn |
N/A |
| Inf. Citrine reverse | CCGGGCTAGC caaca nnnn nnnnnnnnnnnnnnnn |
N/A |
| Inf. Cherry forward | AGCTCGAGTT gtgca nnnn nnnnnnnnnnnnnnnn |
N/A |
| Inf. Cherry reverse | CCGGGCTAGC caccg nnnnn nnnnnnnnnnnnnnn |
N/A |
| pTK forward | CTAGCAAAATAGGCTGTCCC | N/A |
| pTK reverse | ATATTTCTTCCGGGGACACC | N/A |
| Other | ||
| Nanostring plasticware and reagents | NanoString Technologies | NAA-PPCK-048 |
| NanoString custom probe set (see Table S1) | N/A | |
Step-by-step method details
Cloning putative enhancers
PCR amplification of putative enhancers
Digestion/ligation reaction
Plasmid safe treatment to remove linearized vector
Colony screening
This protocol has been developed and optimized to facilitate rapid cloning of large numbers of enhancers in a streamlined, efficient manner. The vectors accompanying this protocol carry either Citrine, Cerulean or mCherry fluorescent reporters as well as one of 48 unique Nanotag sequences allowing multiplex testing of enhancers using Nanostring technology. The cloning insertion site contains the lacZ expression cassette flanked by asymmetric recognition sites for type IIS restriction enzyme BsmBI. Putative enhancers are amplified using primers extended by tails that contain the asymmetric BsmBI recognition site and specific cleavage sequences that will produce the sticky-end overhangs corresponding precisely to the ones remaining in the vector following BsmBI digestion. As a type IIS enzyme BsmBI cleaves outside its recognition sequence, the binding site is lost in both the vector and the amplified enhancer fragment following their ligation, precluding the re-digestion of the ligated constructs containing the intended fragment (Figure 1). As such the digestion-ligation reaction can be performed simultaneously in the same tube, using a modified GoldenGate assembly cycling reaction. During the digestion-ligation steps, the lacZ cassette is excised and the putative enhancer is inserted in a highly efficient fashion. Self-annealing of the vector does not occur as the overhangs generated by the excision of the lacZ cassette are not compatible. Positive colonies can then be easily identified by blue/white selection.
-
1.PCR amplification of putative enhancers
-
a.Amplify putative enhancer sequences from genomic DNA (gDNA) using a high-fidelity polymerase of your choice. We recommend Kapa LongRange and describe the conditions below.
Volume (μL) Reagent 27.5 Water 2.0 gDNA (100 ng) 10.0 5× buffer Long Range (no MgCl2) 3.5 MgCl2 (25 mM) 1.5 dNTPs (10 mM) 0.5 KAPA LR enzyme 2.5 F primer (10 μM) 2.5 R primer (10 μM) Temperature (°C) Time (s) # cycles 94 180 94
55∗
6815
15
Allow 1 min/kb10 94
63∗
6815
15
Allow 1 min/kb25 72 10 min 4 hold ∗Adjust according to primer Tm -
b.Run PCR samples on 1% agarose gel and gel purify. We used Promega Wizard SV gel and PCR clean-up system (A9282). Quantify on Nanodrop.
-
a.
Pause point: purified PCR’s can be stored at −20°C.
-
2.Digestion/Ligation reaction
-
a.Set up digestion/ligation reaction as below in 0.2 mL PCR tubes.
Volume (μL) Reagent 75 ng PCR product 75 ng Nanotag reporter vector 2.0 T4 DNA ligase buffer 1.0 T4 DNA ligase (NEB, M0202) 1.0 BsmBI (NEB, R0508) Up to 20 μL Water -
b.Run the digestion/ligation reaction in a thermal cycler
Temperature (°C) Time (min) # cycles 37 5 25 16 10 55 5 80 5  CRITICAL: Be sure to select the correct fluorescent reporter vector corresponding to the primers used to generate PCR products. Use different Nanotag vectors for each enhancer to be tested by Nanostring.
CRITICAL: Be sure to select the correct fluorescent reporter vector corresponding to the primers used to generate PCR products. Use different Nanotag vectors for each enhancer to be tested by Nanostring.
-
a.
Optional: Plasmid safe treatment to remove linearized vector
-
a.Set up the plasmid safe reaction as below
Volume (μL) Reagent 10.25 Digestion/ligation reaction 1.25 PlasmidSafe™ buffer (Cambio E3101K) 0.5 ATP (25 mM) 0.5 PlasmidSafe™ enzyme Temperature (°C) Time (min) 37 60 70 30
-
3.Transformation.
-
a.PlasmidSafe™ or digestion/ligation reactions can be directly transformed into competent cells of your choice. We used DH10B.
Optional: Colony Screening. This approach is only suitable for enhancers >500 bp, owing to the limited resolution of plasmid lysates by gel electrophoresis. Alternatively, colonies can be screened by PCR or constructs can be directly mini-prepped and checked by sequencing without a screening step. -
a.
-
a.For optimal results grow colonies to approximately 1 mm in diameter. Screen ∼6 colonies per putative enhancer.
-
b.Prepare 1% agarose gel, do not cover with TAE buffer.
-
c.Add 10 μL of Qiagen buffer P1 to each PCR tube or well of 96 plate to be used.
-
d.Using a sterile tip, touch selected colony, streak onto a new plate (multiple colonies can be streaked on a single plate marked out into a grid) and place tip in tube/well containing P1.
-
e.Resuspend cells in P1 by gently agitating tips.
-
f.Remove tips.
-
g.Add 10 μL buffer P2 and seal tubes/plate and heat at 60°C for 15 min in a PCR cycler.
-
h.Add 5 μL of 6× gel loading buffer.
-
i.Load samples onto gel ‘dry’ i.e., add only enough TAE running buffer to reach the sides of the gel, buffer should not cover the gel surface as the reaction mix does not sink in the buffer. Run electrophoresis for 2–5 min until samples have entered the gel then more running buffer can be added.
-
j.Discriminate positive colonies by comparing to super-coiled ladder. Nanotag reporter vectors with lacZ are 5 kb.
-
a.
-
4.
Prepare Endotoxin free preps of positive colonies. We used Qiagen EndoFree Maxi kit (#12362) or Omega BioTek EZNA Endofree plasmid mini prep II (VWR #D6950-02) and Sanger sequence using pTK Forward and pTK Reverse primers (see key resources table).
Figure 1.

Modified reporter plasmids for highly efficient enhancer cloning
(A) Modified pTK enhancer reporter vector containing Nanotag barcode downstream of the fluorescent reporter, but upstream of the polyA tail. BsmBI recognition sites flanking lacZ cassette are also shown.
(B) Sequence details of BsmBI mediated cloning of amplified putative enhancer regions into the modified pTK vector. Figure adapted and reprinted with permission from (Williams et al., 2019).
Electroporation of enhancer constructs into early chick embryos
Nanotagged putative enhancer preps are now ready to be pooled and electroporated into early chick embryos. Electroporated embryos are allowed to grow to the desired stage such that in vivo enhancer reporter activity can develop and be subsequently quantified using Nanostring nCounter system®. These same steps are then followed to electroporate combinations of 1-3 positive enhancers from the Nanostring screen driving different fluorophores and imaged to record specific spatial and temporal enhancer activity.
-
5.Combine Nanotag plasmids containing putative enhancers.
-
a.Plasmids with unique Nanotags should be combined at equal concentration (∼0.5 μg/μL), to a final concentration of 6 μg/μL in water (this is the maximum concentration chick embryos can reasonably tolerate), this equates to 10–12 plasmids including controls.
-
b.We provide a number of Nanotag reporter plasmids containing the FoxD3 neural crest enhancer (NC1) (Simoes-Costa et al., 2012) as positive control (Addgene #130570 - #130573). If this is not suitable, we have also made other tested chick enhancers available (Addgene #130574 - #130625). Some of these are more broadly active across chick embryonic tissues and stages, see (Williams et al., 2019) for specific expression patterns. We also provide several Nanotag reporter plasmids containing a short non-specific sequence to be used as negative control (Addgene #130558 - #130568). All controls were generated in a variety of Nanotags to facilitate flexibility in the pooling.CRITICAL: Ensure pooled plasmids contain unique Nanotags.
-
c.Add vegetable dye to visualize solution during electroporation.
-
a.
-
8.Electroporate pooled plasmids into early chick embryos. We describe this process in detail in the ‘Ex ovo electroporation of early chicken embryos’ protocol associated with (Williams et al., 2019).
-
a.Inject ∼1 μL pooled plasmid solution into the entire epiblast of HH4 (Hamburger and Hamilton, 1951) chick embryos.Note: At HH4, cells of ectodermal origin will be successfully transfected, if mesoderm or endoderm derivatives are targeted, electroporation should be performed at HH3, however this does impede survival so higher number of embryos should be used.
-
b.Electroporate using platinum plate electrodes and settings 5V, 100 ms ON, 50 ms OFF for 5 pulses.
-
c.Grow embryos on thin albumin at 37°C until desired stage is reached.
-
a.
-
9.
Dissect the tissue of interest.
Note: Isolation of specific cells is not necessary at this screening stage, but using dissected tissue will enrich for cells likely to harbour enhancer activity and thus provide more robust readout. Using whole embryos may result in false negatives due to dilution of enhancer positive cells.
NanoString assay
RNA extraction = 30 min hands on plus 1 h Turbo DNase incubation
Nanostring sample preparation = 30 min
Nanostring nCounter Prep station = 2 h
Nanostring nCounter digital analyser = 4 h
In this step RNA is extracted from the embryonic tissue of interest and subsequently hybridized with Nanostring probes such that enhancer reporter activity can be quantified using Nanostring nCounter®.
-
10.Prepare dissected tissue for RNA extraction.
-
a.We recommend Ambion RNAqueous™-micro total RNA isolation kit (AM1931).
-
b.Place dissected tissue directly into 100 μL lysis buffer, in a low-bind 1.5 mL tube, minimizing the amount of media added with tissue.
-
c.Keep on ice for 15 min and vortex for 5 s intermittently.
-
d.Flash freeze in liquid nitrogen and store at −80°C.
-
a.
-
11.Extract RNA from dissected tissue
-
a.Follow RNA extraction protocol. Elute RNA with 2× 10 μL EB from the kit.
-
b.Add 2 μL Turbo DNase (AM1907) to 20 μL of eluted RNA.
-
c.Incubate at 37°C for 1 h.
-
d.Add 2 μL resuspended inactivation reagent (AM1931) to DNase reaction and flick to mix.
-
e.Incubate at 20°C–24°C for 2 min, flicking after one minute to maintain solution.
-
f.Centrifuge at 10,000 g for 1.5 min, remove supernatant into new DNase/RNase free low-binding tube, careful not to take any pelleted inactivation reagent.
-
g.Verify the integrity of RNA using Agilent Bioanalyzer or Tapestation. Use 100 ng high quality (RIN >7) RNA for Nanostring analysis.
-
a.
-
12.
Hybridize samples with Nanostring CodeSet according to the manufacturer’s protocol (MAN-10056-03_CodeSet_hybridization_Setup).
-
13.
Load samples into the nCounter® prep station and subsequently run the chip on the digital analysis system following the manufacturer’s instructions (MANC0035_nCounter_Analysis_System_MAX_FLEX). This manual also provides details for extracting and processing data files.
Note: Nanostring nCounter® output data provides a simple count of mRNA transcripts detected by the custom CodeSet (Figure 2). By incorporating positive and negative controls, active enhancer candidates can be resolved. Typically, positive controls recorded counts between 600–900 and negative control counts were below 10 (Figure 2). We empirically determined that enhancers with a count above 100 drove fluorescent reporter activity in vivo. We found minimal variance between different Nanotag reporters containing the same enhancer.
Figure 2.

Nanostring screening of putative enhancers
(A) Nanotag transcripts are detected by a customized Nanostring codeset (Table S1), counts above 100 (in green, above dashed line) were determined to depict in vivo enhancer activity.
(B) Negative control plasmids achieved Nanostring counts less than 100. In vivo background expression is observed in the posterior extra-embryonic region.
(C) Enhancers with Nanostring count >100 are re-electroporated into early chick embryos at higher concentration such that fluorescence can be observed. Multiple enhancers carrying different fluorophores can be imaged in parallel, as shown for neural crest enhancers; NC1, NC2 and Sox10E2 (Betancur et al., 2010; Simoes-Costa et al., 2012). Figure adapted and reprinted with permission from (Williams et al., 2019).
-
14.
Electroporate candidate enhancers individually, or in combination of different fluorophores, into early chick embryos as step 6, but at higher concentration (2 μg/μL per enhancer) record spatiotemporal enhancer activity by confocal microscopy.
Expected outcomes
Following enhancer cloning using our modified GoldenGate BsmBI strategy we generally find >90% of colonies yield accurate sequencing results.
Using ATAC-seq datasets from a range of cell-types and performing comparative analysis we found approximately 75% of selected putative enhancers were indeed active in vivo.
Limitations
Currently high-throughput screening for enhancer activity depends on Nanostring nCounter® technology. In the absence of such resources it is feasible to use qPCR to detect Nanotag barcodes, however this approach has not optimized.
While Nanostring technology can detect up to 800 transcripts in a single sample, we have only optimized 48 different Nanotags. As these can be multiplexed in the embryo and up to 12 samples can be run on a single Nanostring chip we found this more than sufficient to screen over 500 putative enhancers.
Troubleshooting
Problem 1
BsmBI restriction sites within putative enhancer regions
Potential solution
In this scenario In-Fusion® HD Cloning kit is recommended, (Takara Clontech #639649). Nanotag reporter vectors are digested with BsmBI and PCR amplicons are generated with the following tails.
 |
In-Fusion® cloning protocol:
Digest Nanotag reporter vector with BsmBI
| Volume (μL) | Reagent |
|---|---|
| 1 μg | Nanotag reporter vector |
| 10.0 | NEB buffer 3.1 |
| 1.0 (10U) | BsmBI |
| Up to 100 μL | Water |
| Temperature (°C) | Time (h) |
|---|---|
| 55 | 4 |
Gel purify digested vector
Amplify putative enhancer regions as described above
Set up the In-Fusion® reaction as follows
| Volume (μL) | Reagent |
|---|---|
| 100 ng | PCR product |
| 100 ng | Digested vector |
| 2.0 | 5× In-Fusion® mix |
| Up to 10 μL | water |
| Temperature (°C) | Time (min) |
|---|---|
| 50 | 15 |
Immediately place reaction on ice and proceed directly to transformation
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tatjana Sauka-Spengler tatjana.sauka-spengler@imm.ox.ac.uk
Materials availability
Plasmids generated in this study have been deposited to Addgene. https://www.addgene.org/Tatjana_Sauka-Spengler/
Custom Nanotag Nanostring codeset is available from Nanostring technologies, sequences are provided here in a Table S1.
Data and code availability
Original/source data for figures in the paper is available (Williams et al., 2019)
Acknowledgments
We thank Dr. Jongmin Nam (Davidson Lab, Caltech) for providing nanotag vectors (Nam and Davidson, 2012) and Dr. Upeka Senanayake for assistance with cloning nanotag sequences into pTK reporter vectors. This work was supported by MRC (G0902418), The Lister Institute of Preventive Medicine Research Prize, John Fell Fund (131/038), and Leverhulme Trust (grant RPG-2015-026) to T.S.-S.
Author contributions
R.M.W. and T.S.-S. devised experimental strategy. R.M.W. performed experiments. R.M.W. and T.S.-S. analyzed data; R.M.W. and T.S.-S. wrote the original draft; T.S.-S. provided resources.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100507.
Contributor Information
Ruth M. Williams, Email: ruth.williams@imm.ox.ac.uk.
Tatjana Sauka-Spengler, Email: tatjana.sauka-spengler@imm.ox.ac.uk.
Supplemental information
References
- Betancur P., Bronner-Fraser M., Sauka-Spengler T. Genomic code for Sox10 activation reveals a key regulatory enhancer for cranial neural crest. Proc. Natl. Acad. Sci. U S A. 2010;107:3570–3575. doi: 10.1073/pnas.0906596107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- Hay D., Hughes J.R., Babbs C., Davies J.O.J., Graham B.J., Hanssen L., Kassouf M.T., Marieke Oudelaar A.M., Sharpe J.A., Suciu M.C. Genetic dissection of the alpha-globin super-enhancer in vivo. Nat. Genet. 2016;48:895–903. doi: 10.1038/ng.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellner K., Miranda F., Fotso Chedom D., Herrero-Gonzalez S., Hayden D.M., Tearle R., Artibani M., KaramiNejadRanjbar M., Williams R., Gaitskell K. Premalignant SOX2 overexpression in the fallopian tubes of ovarian cancer patients: Discovery and validation studies. EBioMedicine. 2016;10:137–149. doi: 10.1016/j.ebiom.2016.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockman D., Chong-Morrison V., Green S.A., Gavriouchkina D., Candido-Ferreira I., Ling I.T.C., Williams R.M., Amemiya C.T., Smith J.J., Bronner M.E. A genome-wide assessment of the ancestral neural crest gene regulatory network. Nat. Commun. 2019;10:4689. doi: 10.1038/s41467-019-12687-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J., Davidson E.H. Barcoded DNA-tag reporters for multiplex cis-regulatory analysis. PLoS One. 2012;7:e35934. doi: 10.1371/journal.pone.0035934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A., Bajpai R., Prescott S., Brugmann S.A., Swigut T., Wysocka J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell. 2012;11:633–648. doi: 10.1016/j.stem.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes-Costa M.S., McKeown S.J., Tan-Cabugao J., Sauka-Spengler T., Bronner M.E. Dynamic and differential regulation of stem cell factor FoxD3 in the neural crest is Encrypted in the genome. PLoS Genet. 2012;8:e1003142. doi: 10.1371/journal.pgen.1003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams R.M., Candido-Ferreira I., Repapi E., Gavriouchkina D., Senanayake U., Ling I.T.C., Telenius J., Taylor S., Hughes J., Sauka-Spengler T. Reconstruction of the global neural crest gene regulatory network in vivo. Dev Cell. 2019;51:255–276.e7. doi: 10.1016/j.devcel.2019.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original/source data for figures in the paper is available (Williams et al., 2019)