

Summary
Small molecular probes designed for photopharmacology and opto-chemogenetics are rapidly gaining widespread recognition for investigations of transient receptor potential canonical (TRPC) channels. This protocol describes the use of three photoswitchable diacylglycerol analogs—PhoDAG-1, PhoDAG-3, and OptoDArG—for ultrarapid activation and deactivation of native TRPC2 channels in mouse vomeronasal sensory neurons and olfactory type B cells, as well as heterologously expressed human TRPC6 channels. Photoconversion can be achieved in mammalian tissue slices and enables all-optical stimulation and shutoff of TRPC channels.
For complete details on the use and execution of this protocol, please refer to Leinders-Zufall et al. (2018).
Subject areas: Cell Biology, Microscopy, Neuroscience, Molecular/Chemical Probes
Graphical abstract
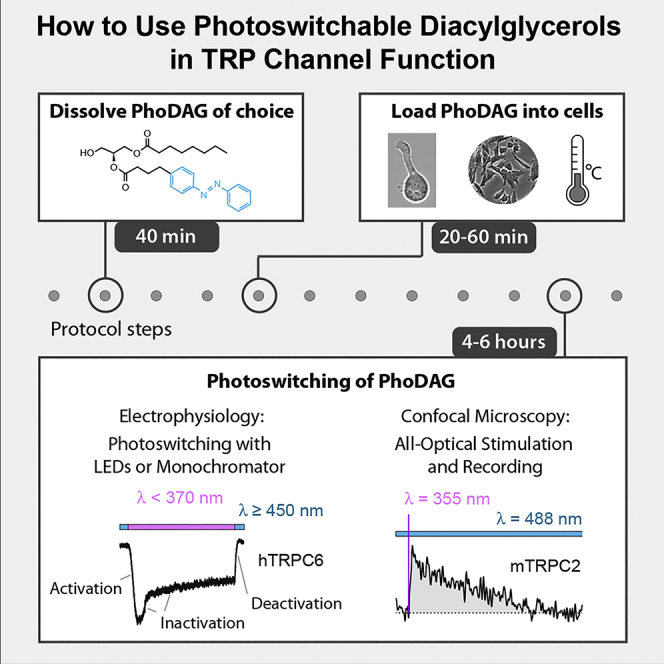
Highlights
-
•
DAG photoswitching enables ultrarapid activation and deactivation of TRPC channels
-
•
Multiple photoswitchable DAG analogs are now available
-
•
DAG photoconversion is sufficient for the gating of TRPC2, TRPC3, and TRPC6
-
•
Photoswitching combined with Ca2+ imaging enables all-optical stimulation and recording
Small molecular probes designed for photopharmacology and opto-chemogenetics are rapidly gaining widespread recognition for investigations of transient receptor potential canonical (TRPC) channels. This protocol describes the use of three photoswitchable diacylglycerol analogs—PhoDAG-1, PhoDAG-3, and OptoDArG—for ultrarapid activation and deactivation of native TRPC2 channels in mouse vomeronasal sensory neurons and olfactory-type B cells, as well as heterologously expressed human TRPC6 channels. Photoconversion can be achieved in mammalian tissue slices and enables all-optical stimulation and shutoff of TRPC channels.
Before you begin
Preparation of photoswitchable diacylglycerols
Timing: 30–60 min
For TRP channel activation and deactivation, we used three distinct photoswitchable diacylglycerol (DAG) analogs (PhoDAGs) that can be transformed from the inactive to the active form and back again by exposure to light of different wavelengths (Figure 1):
Figure 1.

Chemical structures of three distinct photoswitchable diacylglycerols: PhoDAG-3 and PhoDAG-1 (new commercial name, 18:0-PhoDAG) (Frank et al., 2016; Leinders-Zufall et al., 2018), and OptoDArG (Lichtenegger et al., 2018)
These DAG analogs isomerize between their trans- and cis configurations in response to UV-A (λ < 370 nm) and blue (λ > 460 nm) illumination, respectively. The cis forms of these DAG analogs have been shown to activate mouse TRPC2, human TRPC3, and human TRPC6 (Leinders-Zufall et al., 2018; Lichtenegger et al., 2018). Compare also Figures 3 and 4 shown below.
(1) PhoDAG-3 (FW 468.59) (Leinders-Zufall et al., 2018); (2) PhoDAG-1 (FW 664.96) (Leinders-Zufall et al., 2018) which is now commercially available under the name 18:0-PhoDAG (CAS #: 1985595-31-0); and (3) OptoDArG (FW 704.39) (Lichtenegger et al., 2018) which is now also commercially available (CAS #: 2230617-93-1). The two commercially available compounds can be obtained at amounts of 5 or 100 mg, respectively. The initial design and chemical synthesis of PhoDAG-1 and PhoDAG-3 have been described by Frank et al. (2016).
Note that several recent reviews have appeared on this topic (Chen et al., 2020; Curcic et al., 2019; Hüll et al., 2018; Iseppon and Arcangeletti, 2020).
-
1.
Prepare stock solutions of 10 mM or 100 mM PhoDAG or of 50 mM OptoDArG in anhydrous DMSO. These compounds are weakly soluble in aqueous solutions.
-
2.
Aliquot the stock solutions in portions of 3 to 100 μL and dilute with extracellular solution to achieve a final concentration of 10 μM PhoDAG with 0.1% DMSO, 100 μM PhoDAG with 0.1% DMSO or 30 μM OptoDArG with 0.06% DMSO.
-
3.
If PhoDAG does not dissolve or precipitates from the aqueous solution, warm it up to 40°C for 5–10 min. This will usually solve the problem. As a second step, the solution can be sonicated briefly.
-
4.
Store the various stock solutions in the dark and in a desiccator at either −20°C for long-term use (1–6 months) or at 20°C–24°C for immediate use. In the case of the 50 mM OptoDArG stock solution, aliquots should be warmed up to 40°C for 5–10 min before preparing the final concentration.
Note: The concentration of the stock solution should be carefully considered since it influences the amount of DMSO in the final concentration of PhoDAG or OptoDArG delivered to the cells. DMSO is known to influence the integrity of the cell membrane and could thus change the physiological response to be studied (Gurtovenko and Anwar, 2007; Notman et al., 2006). A DMSO concentration of ≤0.1% should be sufficient to keep the compound in solution and not damage the cells. Various cell lines seem to be less prone to DMSO damage. Control experiments should be performed if the DMSO concentration is >0.1%. The preparation of photoswitchable DAGs does not need to be performed in the dark. Eliminating any light will convert these molecules back into the trans form (Frank et al., 2016; Leinders-Zufall et al., 2018).
Preparation of cells or tissue slices
This step provides a short description of the procedures for obtaining (I) mouse chemosensory neurons expressing TRPC2 channels from (a) the vomeronasal organ (VNO) or (b) the main olfactory epithelium (MOE), as well as (II) HEK293T cells expressing human TRPC6 channels. Make sure that all animal experimental procedures are performed in accordance with the guidelines established by the animal welfare committees of the respective institutions.
Note: The protocols in this section are optimizations of previous protocols (Bleymehl et al., 2016; Leinders-Zufall et al., 2000; Leinders-Zufall et al., 2004; Leinders-Zufall et al., 2009; Leinders-Zufall et al., 2018; Lucas et al., 2003; Spehr et al., 2009; Ukhanov et al., 2007). Other practical details of the VNO preparation and the preparation of acute VNO tissue slices including several instructive videos have been published (Brechbühl et al., 2011; Leinders-Zufall and Zufall, 2013; Ma et al., 2011).
-
5.Preparation of chemosensory neurons that express native TRPC2 cation channels: Vomeronasal sensory neurons (VSNs) obtained from the mouse VNO
-
a.Euthanize a mouse with a CO2 overdose (or alternative procedure) followed by decapitation.
-
b.Take the mouse head and eliminate all skin and fur to expose the skull and nasal bones. Start with a caudal to rostral cut along the sagittal suture to the tip of the nose using sharp Metzenbaum scissors. Remove the skin sideways and cut it off.
-
c.Detach the underjaw (mandible) from the head bone by cutting on both sides through the temporomandibular joint with sharp blunt scissors.
-
d.Remove the soft palate using blunt curved forceps by taking hold of the palate near the incisors and pulling the forceps with soft palate caudally. The two vomer bones, the septum, and the nasal cavity will now be visible.
-
e.Using curved spring scissors, cut through the bony structure connecting the left and right incisive bone to the interincisive foramen.
-
f.Grab the septum at the caudal end of the two vomer bones using blunt forceps. In a gentle motion break the septum and move the whole bone structure (septum and vomer bones) up and to the tip of the nose to ensure that the rostral septum connections are broken. Then place the whole bone structure in a 35 mm culture dish which is placed on an ice block underneath a binocular dissection microscope.
-
g.The vomer bone is very brittle. Carefully remove it from the VNO tissue with high-grade forceps (Biologie #5 Dumont forceps; 0.05 x 0.02 mm). The most difficult part will be the dorsal part of the vomer bone through which VNO axon bundles protrude. The VNO tissue is easily damaged at this point. This step requires patience and practice.
-
h.Place the VNO as soon as possible in a dish containing oxygenated S1 solution to prevent dehydration and oxygen deprivation.
-
a.
Note: If the VNO is used for acutely dissociated VSNs, a little damage will not be problematic. Please follow step 7 (Preparation of freshly dissociated VSNs) for directions.
Note: If the VNO is used for tissue slices, damage to the tissue could prevent the production of slices or undermine the health of VSNs in the tissue. Please follow the next steps to produce tissue slices.
-
6.Preparation of VNO tissue slices
-
a.Check the VNO carefully for any leftover bone pieces that might damage the tissue when using the tissue slicer (step 6f below).
-
b.Prepare 3% agarose solution (3 g in 100 mL S6 solution; kept in a warm water bath at 40°C) and pour into a 35 mm petri dish which is placed onto the ice block underneath the binocular dissection microscope.
-
c.Carefully place the VNO into the agarose solution and move the VNO around, so that the S1 solution around the VNO is gently replaced by the agarose solution and all air bubbles are removed.
-
d.Align and position the two VNOs vertically in the dish so that they stand on their anterior or posterior ends. Keep a little distance between the VNOs, so that they can be cut simultaneously, but prevent ripping them out of the settled agarose during the slicing procedure. Let the agarose cool down.
-
e.When the agarose is completely solidified, cut a truncated square pyramid shaped block through the agarose using a scalpel knife (blade #11), with the VNOs positioned in the middle. The truncated pyramid shape will help during the slicing procedure to place the most pressure on the base of the pyramid.
-
f.Depending on the tissue slicer used in the laboratory (i.e., vibratome or microtome), position the truncated pyramid block containing the VNOs onto the stage and cut coronal VNO slices of 250 μm minimum thickness on ice-cold oxygenated S1 solution (see materials and equipment).
-
g.For loading the slices with both a calcium indicator and photoswitchable DAG, proceed to step 12.
-
a.
-
7.Preparation of freshly dissociated VSNs
-
a.Carefully dissect the two VNOs out of their bone shells and place them in a dish containing 50 μL ice-cold S1 solution (see: Materials and Equipment) supplemented with 0.25 mg/mL papain and 1 U/mL DNase.
-
b.Mince the tissue using a scalpel knife with blade #10, transfer to an Eppendorf tube containing 150 μL S1 solution supplemented with papain and DNAse and incubate for 15 min at 37°C in an O2/CO2 incubator (21.7% O2, 5% CO2) to prevent hypoxia.
-
c.Transfer the VNO tissue, which is at the bottom of the Eppendorf tube, to a 15 mL glass tube containing 2 mL enzyme-free oxygenated S1 solution. Wash the tissue by gently resuspending it in the enzyme-free oxygenated S1 solution using a fire-polished glass Pasteur pipette having a big opening and without producing air bubbles. To fire-polish the glass Pasteur pipette, take the glass pipette and hold its tip into the pilot flame of a Bunsen burner (Leinders-Zufall and Zufall, 2013) for a few seconds while rotating. Upon inspection, the tip will look smoother and the opening will be narrower.
-
d.Centrifuge for 10 min at 40 rcf (g force) at 7°C.
-
e.Remove the supernatant and resuspend the cells in 1.5 mL oxygenated S1 solution (see: Materials and Equipment) through gentle trituration to avoid damage to the microvilli using a fire-polished glass Pasteur pipette having half of its normal opening to achieve a uniform suspension. Be careful not to produce air bubbles.
-
f.Plate the dissociated cells on glass coverslips previously coated with poly-L-lysine (0.01%) and laminin (0.1%) dissolved in distilled water (Fischer et al., 2008).
-
g.Wait 10–20 min to let the cells settle and attach to the coverslip at 20°C–24°C, then proceed to step 10.
-
a.
-
8.Preparation of chemosensory neurons that express native TRPC2 cation channels: TRPC2-expressing olfactory neurons obtained from the main olfactory epithelium (MOE)
-
a.Euthanize a mouse (e.g., Trpc2-IRES-taumCherry mouse) with a CO2 overdose (or alternative procedure) followed by decapitation.
-
b.Take the mouse head and eliminate all skin and fur to expose the skull and nasal bones. Start with a caudal to rostral cut along the sagittal suture to the tip of the nose using sharp Metzenbaum scissors. Remove the skin sideways and cut it off.
-
c.Detach the underjaw (mandible) from the head bone by cutting on both sides through the temporomandibular joint with sharp blunt scissors.
-
d.Cut off the top of the mouse incisors (front teeth) to ensure a stable position of the ventral part of the head on the surface of a cutting board.
-
e.Using a single edge razor blade, make a cut through the left nasal cavity and the whole mouse head. More precisely, when placing the head on the cutting board, the razor should be positioned with one of its sharp points next to the tip of the nose. To cut through the nasal cavity, the sharp surface of the razor blade is carefully placed at the same angle as the dorsal nose/head bones, parallel to the internasal suture (midline) and approximately 1/3 the distance of the nasal bone from the suture. Put pressure onto the angled top part of the razor blade and cut through the bones and brain tissue in one stroke.
-
f.Carefully take the turbinates out of the nasal cavity and place them in a dish containing 400 μL low Ca2+-buffered, oxygenated (95% O2 / 5% CO2) S2 solution (see: Materials and Equipment).
-
g.Take out the septum and remove the turbinates from the contralateral nasal cavity and place them onto the dish.
-
h.Mince the tissue and incubate for 15 min at 37°C in an O2/CO2 incubator (21.7% O2, 5% CO2) to prevent hypoxia.
-
i.Transfer the MOE tissue to a 15 mL glass tube containing 2.4 mL extracellular oxygenated (95% O2 / 5% CO2) S1 solution. Gently passage the tissue using a fire-polished glass Pasteur pipette.
-
j.Plate the dissociated cells on glass coverslips previously coated with poly-L-lysine (0.01%) and laminin (0.1%) dissolved in distilled water (Fischer et al, 2008).
-
k.Wait 10–20 min to let the cells settle and attach to the coverslip, then proceed to step 10 or 12.
-
a.
Note: To discriminate olfactory type B cells from type A cells (both of which are located in the MOE and express TRPC2) using Trpc2-IRES-taumCherry mice, post hoc immunostaining for adenylyl cyclase type 3 (Adcy3) is performed directly in the recording chamber after physiological recording, in order to distinguish Trpc2+ Adcy3- (type B) cells from Trpc2+ Adcy3+ (type A) cells (Bleymehl et al., 2016; Leinders-Zufall et al., 2018). We found that acute dissociation of type A or type B cells from the MOE does not require enzymatic treatment (Bleymehl et al., 2016; Leinders-Zufall et al., 2018).
Note: Be meticulous in preventing the ciliary or microvillous surfaces of the cavernous tissue and the sensory epithelium to touch each other. Cilia from both sensory and nonsensory epithelium could be lost due to their entanglement (velcro hook-and-loop closure effect) and therefore cause the absence of chemosensory responses.
CRITICAL: The cilia and microvilli of olfactory and vomeronasal sensory neurons are easily damaged, therefore take utmost care in the trituration of the tissue (air bubbles) and the centrifugation step.
-
9.Preparation of HEK293T cells expressing human TRPC6 channels
-
a.Maintain HEK293T cells (from Leibniz-Institute DSMZ, ACC 635) in culture medium containing Earl’s minimal essential medium with 2 mM glutamine (EMEM, M4655, Sigma-Aldrich) supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (P4333, Sigma-Aldrich) and with 10% fetal calf serum (FCS, 10270106, Gibco). Keep the cells at 37°C in a humified atmosphere with 5% CO2.
Reagent Final concentration Amount EMEM - 1 L Penicillin and streptomycin 100 U mL−1 (penicillin), 100 μg mL−1 (streptomycin) 10 mL FCS 10% 100 mL -
b.Four days prior to the experiment, seed 3 x 105 HEK293T cells per well in a 6-well plate. After 48 h, the cells have reached a confluence of approximately 80 to 90% and are ready for transfection.
-
c.Transiently transfect the cells with 1 to 2 μg μL−1 cDNA coding for hTRPC6 (NP_004612) in pIRES2-EGFP expression vector (Clontech) using lipofection with GeneJuice® (Merck Millipore) according to the manufacturer’s instructions.
-
d.24 to 48 h after transfection, and 1–2 h prior to the loading with PhoDAG or OptoDArG, split the transfected HEK293T cells from one well plate onto 3 to 5 glass-bottom dishes (WPI, FluoroDish FD35-100, diameter of glass bottom 23 mm). For equal distribution and separation of the transfected HEK293T cells, wash the cells once with 1 mL Dulbecco’s phosphate buffered saline modified without calcium chloride and magnesium chloride (DPBS, D8537, Sigma-Aldrich) and detach the cells by applying 300 μL Trypsin-EDTA solution (T3924, Sigma-Aldrich) for 20–30 s followed by application of 1.5 mL culture medium. Gently triturate the cell suspension 10 to 15 times using a 1000 μL pipette tip to separate the cells before seeding.
-
e.Proceed to step 11 for loading with PhoDAG or OptoDArG.
-
a.
Loading of photoswitchable DAGs
The time required for loading of PhoDAGs into living cells is variable in our hands and depends on the particular cell type studied and on the chemical structure of a given PhoDAG (Figure 1).
-
10.
Incubate dissociated olfactory and vomeronasal neurons with 10 μM PhoDAG in S1 solution for 20–30 min at 20–24°C in the dark. During a stable physiological recording (electrophysiology) it is possible to measure the responses before loading, during loading, and after sufficient loading to determine the optimal time for loading of the photoswitchable compound. For calcium imaging experiments, proceed to step 12.
-
11.
Incubate HEK293T cells for about 45–60 min at 20°C–24°C in the dark with S3 solution supplemented with 100 μM PhoDAG or with 30 μM OptoDArG prior to measurements.
Note: As mentioned above in ‘Preparation of Photoswitchable Diacylglycerols’, an increased temperature helps to dissolve high concentrations of these compounds. Similarly, a temperature of 32°C–37°C will improve the loading of PhoDAGs into cells. This effect was most obvious for the loading of OptoDArG into olfactory cells. The loading of HEK293T cells with OptoDArG was done at 20°C–24°C.
Loading of photoswitchable DAGs together with calcium indicator dye for use in all-optical stimulation and recording experiments
-
12.
Loading of the calcium indicator
To prevent repeated exposure to moisture, use calcium indicator packaged in sets of 20 separate vials, each containing 50 μg for reconstitution in DMSO as required.-
a.Freshly prepare 20% Pluronic F127 in anhydrous DMSO (= 0.02 g in 100 μL DMSO).
-
b.Take a tube of 50 μg fluo4/AM (FW 1096.95) out of the freezer (−20°C) and warm it in your hands to 20–24°C.
-
c.Add 5 μL anhydrous DMSO and 5 μL 20% Pluronic F127 to the tube containing 50 μg fluo4/AM to make 4.56 mM fluo4/AM (100% DMSO, 20% Pluronic F127).
-
d.Mix solution using a p200 tip on your pipettor.
-
e.Add 90 μL of S1 solution (see: Materials and Equipment) to the tube containing 10 μL 4.56 mM fluo4/AM stock solution to make 456 μM fluo4/AM (10% DMSO, 2% Pluronic F127). Mix using a vortex for 30 s. This solution can be used for up to 1 week at 4°C.
-
f.For cells that respond to low oxygen levels, such as olfactory type B cells (Bleymehl et al., 2016), prepare the calcium indicator solution in oxygenated S1 solution. This is obtained by using an O2/CO2 incubator (21.7% O2, 5% CO2) at 32°C or, if the loading is done at 20°C–24°C, a solution oxygenated with carbogen (95% O2, 5% CO2).
-
g.For the preparation of 1 mL calcium indicator loading solution having a final concentration of 9.1 μM (0.2% DMSO, 0.04% Pluronic F127): add 20 μl of the 456 μM fluo4/AM solution to 280 μL S1 solution (see: Materials and Equipment) and sonicate for 5 min; then add 700 μL S1 solution oxygenated for 15 min at 20°C–24°C with carbogen (95% O2, 5% CO2).
-
h.Incubate cells or tissue slices for 30 min (dissociated cells) to 1.5 h (250–300 μm tissue slices) in an O2/CO2 incubator (21.7% O2, 5% CO2) at 32°C, or at the bench at 20°C–24°C.
-
a.
-
13.Loading of photoswitchable DAGs
-
a.For dissociated cells that require only 30 min loading with the calcium indicator, PhoDAG-1, PhoDAG-3, or OptoDArG are added directly at the same time to the cells to produce a final concentration of 10 μM.
-
a.
In case of tissue slices, add PhoDAG to the tissue slice for the last 30 min of calcium indicator incubation.
Note: If the tube containing 50 μg fluo4/AM is not yet at 20°C–24°C, the DMSO will solidify preventing the mixing of the solution. Solution: Warm the tube to 20°C–24°C and mix.
Note: Dilute aqueous solutions of the calcium indicator for cell loading should be used within 1 week after they have been prepared. The final 9.1 μM solution should be used on the day it was made.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Adcy3 polyclonal antibody (1:1000) | Santa Cruz | RRID:AB_630839 |
| Alexa Fluor 633 rabbit secondary antibody (1:800) | Thermo Fisher Scientific | RRID:AB_2535731 |
| Hoechst 33342 nuclear counterstain (1:10000) | Thermo Fisher Scientific | Cat #: H3570 |
| Chemicals, peptides, and recombinant proteins | ||
| PhoDAG-1 | Frank et al., 2016 | PubChem CID: 315492946; CAS #: 1985595-31-0 |
| 18:0-PhoDAG (= PhoDAG-1) | Merck or Avanti | PubChem CID: 315492946; CAS #: 1985595-31-0 |
| PhoDAG-3 | Frank et al., 2016 | PubChem CID: 315492951 |
| OptoDArG | AOBIOUS or Glixx Laboratories | CAS # 2230617-93-1 |
| Fluo-4 AM | Thermo Fisher Scientific | CAS# 273221-67-3 |
| DMSO, anhydrous | Sigma-Aldrich | CAS# 67-68-5 |
| Pluronic F-127 | Sigma-Aldrich | CAS# 9003-11-6 |
| Agarose, low gelling temperature type XI | Sigma-Aldrich | CAS# 9012-36-6 |
| Papain (15 units/mg) | Worthington Biochemical Company | CAS# 9001-73-4 |
| RNase-free DNase (1 U/μL) | Promega | M610A |
| Poly-L-lysine (FW 70,000–150,000) | Sigma-Aldrich | CAS# 25988-63-0 |
| Laminin, A chain | Sigma-Aldrich | CAS# 123063-31-0 |
| Earle’s Minimum Essential Medium (EMEM) | Sigma-Aldrich | M4655 |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 |
| Fetal calf serum | Gibco | 10270106 |
| Dulbecco’s phosphate buffered saline (DPBS) | Sigma-Aldrich | D8537 |
| Trypsin-EDTA solution | Sigma-Aldrich | T3924 |
| BES (2-hydroxyethyl)-2-aminoenthanesulfonic acid) | Sigma-Aldrich | B9879, CAS# 10191-18-1 |
| HEPES (2-[4-(2-hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid) | Sigma-Aldrich | H0887, CAS# 7365-45-9 |
| EGTA (ethyleneglycol-bis(β-aminoethyl)-N,N,Nʹ,Nʹ-tetraacetic acid) | Sigma-Aldrich | 03777, CAS# 67-42-5 |
| BAPTA (1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) | Sigma-Aldrich | A4926, CAS# 85233-19-8 |
| CsCl | Sigma-Aldrich | 203025, CAS# 7647-17-8 |
| Na3-GTP | Sigma-Aldrich | G8877, CAS# 36051-31-7 |
| CsOH | Sigma-Aldrich | C8518, CAS# 12260-45-6 |
| Tetrodotoxin | Sigma-Aldrich | T5651, CAS# 4368-28-9 |
| Experimental models: cell lines | ||
| HEK293T cells (T293; passage 2 –24) | Leibniz Institute DSMZ | ACC 635 |
| Critical commercial assays | ||
| GeneJuice transfection reagent | Merck | 70967 |
| Experimental models: organisms/strains | ||
| Mouse: 129/Sv (6–15 weeks old, either sex) | Charles River | N/A |
| Mouse: C57BL/6N (6–15 weeks old, either sex) | Charles River | N/A |
| Mouse: B6;129P2-Trpc2<tm2Mom>/MomJ (6–15 weeks old, either sex) | The Jackson Laboratory | RRID:IMSR_JAX:006733 |
| Recombinant DNA | ||
| Human TRPC6 | Hofmann et al., 1999 | NP_004612 |
| Software and algorithms | ||
| Patchmaster | Heka | N/A |
| Origin | OriginLab | N/A |
| Zen | Zeiss | N/A |
| ImageJ | NIH | N/A |
| DC4104 LED controller | Thorlabs | N/A |
| Igor Pro | WaveMetrics | N/A |
| SPSS | IBM | N/A |
| Other | ||
| Leica vibratome | Leica | VT1200S |
| O2/CO2 incubator | Binder (Germany) | N/A |
| Upright fixed-stage microscope | Olympus | BX50WI |
| Inverted microscope | Olympus | IX 70 |
| Monochromator | TILL Photonics | Polychrome V |
| Recording chamber | Warner Instruments | N/A |
| Amplifier | Heka | EPC9 and EPC10 |
| Gravity perfusion and suction device | Warner Instruments | N/A |
| Micromanipulator | Narishige | N/A |
| Micromanipulator | Luigs & Neumann | N/A |
| Ground electrode, silver wire | Harvard Apparatus | W3 64-1319 |
| LED driver | Thorlabs | DC4104 |
| LED connector hub | Thorlabs | DC4100-HUB |
| Fiber-coupled LED, 365 nm | Thorlabs | N/A |
| Fiber-coupled LED, 470 nm | Thorlabs | N/A |
| Bifurcated fiber bundle | Thorlabs | BFY400HS02 |
| Optic mating sleeve | Thorlabs | ADAFCSMA1 |
| Ferrule patch cable | Thorlabs | M86L01 |
| LSM880 INDIMO confocal microscope | Zeiss (Germany) | N/A |
| 355 nm Laser (60 mW) | Coherent, Inc. | N/A |
| FluoroDish FD35-100 | World Precision Instruments | FD35-100 |
| 20 x 1.0 NA Plan-Apochromat water immersion objective | Zeiss (Germany) | N/A |
| 40 x 0.8 NA Plan-Apochromat water immersion objective | Olympus | N/A |
| 20 x 0.85 UPlanS-Apochromat oil immersion objective | Olympus | N/A |
| 0.2 μm Membrane filter | Merck | GNWP02500 |
Materials and equipment
For preparation of all physiological solutions, we use ultrapure water (>18.2 MΩ-cm resistivity at 25°C, low ppt in divalent cations). Various laboratory systems are available to filter pretreated water (reverse osmosis or demineralized water) and to produce ultrapure water with < 5 ppb total organic carbon (TOC) to reduce organic contaminants.
Extracellular S1 solution
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 120 mM | 7.0128 g |
| NaHCO3 | 25 mM | 2.100 g |
| KCl | 5 mM | 0.3728 g |
| BES (2-hydroxyethyl)-2-aminoenthansulfonic acid) | 5 mM | 1.066 g |
| MgSO4 (anhydrous) | 1 mM | 0.1203 g |
| CaCl2 (anhydrous) | 1 mM | 0.1110 g |
| Glucose | 10 mM | 1.8016 g |
| Total | n/a | 1 L |
Note: The pH will be ∼7.3 after 10 min aeration with carbogen (95% O2 / 5% CO2), with an osmolarity of 300 mOsm kg−1. If a higher osmolarity is required, it can be adjusted by adding more glucose (1 mM equals 1 mOsm kg−1). Filter the solution twice using a 0.2 μm membrane filter to eliminate dust particles and possible bacterial contaminations. Store the solution at 4°C up to 1 month. Make sure that the solution is at 20°C–24°C before used in an experiment and aerated for 10 min with carbogen (95% O2 / 5% CO2) before use.
Low Ca2+-buffered, oxygenated extracellular S2 solution
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 120 mM | 7.0128 g |
| NaHCO3 | 25 mM | 2.100 g |
| KCl | 5 mM | 0.3728 g |
| BES (2-hydroxyethyl)-2-aminoenthansulfonic acid) | 5 mM | 1.066 g |
| MgSO4 (anhydrous) | 1 mM | 0.1203 g |
| CaCl2 (anhydrous) | 4.8 mM | 0.5327 g |
| EGTA (Ethyleneglycol- bis(β-aminoethyl)-N,N,Nʹ,Nʹ-tetraacetic Acid) | 5 mM | 1.9017 g |
| NaOH | 10 mM | 0.3999 g |
| Glucose | 10 mM | 1.8016 g |
| Total | n/a | 1 L |
Note: The pH will be ∼7.3 after 10 min aeration with carbogen (95% O2 / 5% CO2), with an osmolarity of 300 mOsm kg−1. If a higher osmolarity is required, it can be adjusted by adding more glucose (1 mM equals 1 mOsm kg−1). Filter the solution twice using a 0.2 μm membrane filter to eliminate dust particles and possible bacterial contaminations and store the solution at 4°C up to 1 month. Make sure that the solution is at 20°C–24°C before used in an experiment and aerated for 10 min with carbogen (95% O2 / 5% CO2) before use.
Extracellular S3 solution
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 140 mM | 8.1816 g |
| CsCl | 5 mM | 0.8418 g |
| HEPES (2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid) | 10 mM | 2.3830 g |
| MgCl2 (anhydrous) | 1 mM | 0.0952 g |
| CaCl2 (anhydrous) | 2 mM | 0.2220 g |
| Glucose | 10 mM | 1.8016 g |
| Total | n/a | 1 L |
Note: Adjust the pH to 7.4 with NaOH. The osmolarity is 295–302 mOsm kg−1. If a higher osmolarity is required, it can be adjusted by adding more glucose (1 mM equals 1 mOsm kg−1). Filter the solution twice using a 0.2 μm membrane filter to eliminate dust particles and possible bacterial contaminations and store the solution at 4°C up to 1 month.
Intracellular S4 solution (pipette/recording electrode solution)
| Reagent | Final concentration | Amount |
|---|---|---|
| CsCl | 120 mM | 20.2032 g |
| NaCl | 9.4 mM | 0.5493 g |
| HEPES (2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid) | 10 mM | 2.3830 g |
| Na3-GTP | 0.2 | 0.1046 g |
| MgCl2 (anhydrous) | 1 mM | 0.0952 g |
| CaCl2 (anhydrous) | 3.949 mM | 0.4383 g |
| BAPTA (1,2-Bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) | 10 mM | 4.7643 g |
| Total | n/a | 1 L |
Note: Adjust the pH to 7.1 with CsOH. The osmolarity is 295–302 mOsm kg−1. If a higher osmolarity is required, it can be adjusted by adding more glucose (1 mM equals 1 mOsm kg−1). Filter the solution twice using a 0.2 μm membrane filter to eliminate dust particles and possible bacterial contaminations and store the solution at 4°C up to 1 week.
Intracellular S5 solution (pipette/recording electrode solution)
| Reagent | Final concentration | Amount |
|---|---|---|
| CsCl | 140 mM | 23.5704 g |
| HEPES (2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid) | 10 mM | 2.3830 g |
| EGTA (Ethyleneglycol- bis(β-aminoethyl)-N,N,Nʹ,Nʹ-tetraacetic Acid) | 1 mM | 0.3804 g |
| CsOH | 2 mM | 1.4991 g |
| Total | n/a | 1 L |
Note: Adjust the pH to 7.1 with CsOH. The osmolarity is adjusted to 290 mOsm kg−1 with glucose. If a higher osmolarity is required, it can be adjusted by adding more glucose (1 mM equals 1 mOsm kg−1). Filter the solution twice using a 0.2 μm membrane filter to eliminate dust particles and possible bacterial contaminations and store the solution at 4°C up to 1 month.
Extracellular S6 solution
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 145 mM | 8.4738 g |
| KCl | 5 mM | 0.3728 g |
| HEPES (2-[4-(2-Hydroxyethyl)-1-piperazinyl]-ethanesulfonic acid) | 10 mM | 2.3830 g |
| MgCl2 (anhydrous) | 1 mM | 0.0952 g |
| CaCl2 (anhydrous) | 1 mM | 0.1110 g |
| Total | n/a | 1 L |
Note: Adjust the pH to 7.4 with NaOH. The osmolarity is 295–302 mOsm kg−1. If a higher osmolarity is required, it can be adjusted by adding more glucose (1 mM equals 1 mOsm kg−1). Filter the solution twice using a 0.2 μm membrane filter to eliminate dust particles and possible bacterial contaminations and store the solution at 4°C up to 1 month.
Equipment
-
•
Photoswitching using fiber-coupled light-emitting diodes (LEDs)
We use an upright fixed-stage microscope (Olympus BX50WI) equipped for fluorescence microscopy. The stage contains a recording chamber (Warner Instruments), ground electrode, gravity perfusion, and suction device, as well as two micromanipulators (Figures 2A and 2B). One micromanipulator holds the headstage of the Heka EPC9 amplifier with the electrode holder for the recording electrode pipette, the other micromanipulator holds the glass fiber for photomanipulation via the fiber-coupled LEDs.
Figure 2.

DAG photoswitching using fiber-coupled LEDs
(A) Image showing the stage of an upright fixed-stage microscope to indicate the recording chamber located underneath a 40x objective, the location of the electrode holder with its glass recording electrode pipette (P), and the holder with the glass fiber for photomanipulation.
(B) Enlargement of the recording chamber showing the 40x objective, the recording pipette (P), and the ferrule glass fiber cable. Light (λ = 470 nm) from the fiber-coupled LED can be seen due to its reflection from the glass bottom of the recording chamber that contains dissociated VSNs.
(C) Schematic of the LED configuration used for DAG photoswitching. A TTL signal from the amplifier (Heka EPC9) triggers the LED driver which, in turn, activates the LED connector hub. The connector hub drives either of two fiber-coupled LEDs (λ = 470 nm or 365 nm). The LED light is sent through a bifurcated fiber bundle to an optic mating sleeve which aligns the 400-μm thick bifurcated fiber with a thinner optical fiber. This fiber is placed in the bath solution close to the cells.
(D) UV exposure (365 nm) of a freshly dissociated VSN preincubated with PhoDAG-3 at 5 μM causes rapid activation of sustained channel activity that terminates upon exposure to 470 nm light. Whole-cell voltage-clamp recording; holding potential, −70 mV. No such currents could be induced in VSNs not preincubated with PhoDAG-3 (lower trace).
(E) Steady-state current-voltage relationships of photoactivated currents after digital subtraction of the currents obtained before PhoDAG-3 treatment (no PhoDAG-3, dark) from an individual VSN isolated from either a C57BL6 mouse or a TRPC2 knockout (–/–) mouse. D and E are adopted with permission from Figure 1B, 1H, and 1J in Leinders-Zufall et al., 2018. See also Leinders-Zufall et al. (2018) for detailed information regarding voltage step and clamp protocols.
For photoswitching, a TTL signal from the amplifier (Heka EPC9; see the manufacturer’s manual of Patchmaster) triggers the LED driver (Thorlabs) that, in turn, controls the brightness of two fiber-coupled LEDs via the LED connector hub (Figure 2C). The two LEDs produce light at a wavelength of 470 nm or 365 nm. Depending on which LED is activated, the light is sent through a bifurcated fiber bundle to an optic mating sleeve which aligns the 400-μm thick bifurcated fiber with a thinner optical fiber that is placed directly in the bath solution in front of the cells (Figure 2B).
-
•
Photoswitching using confocal laser scanning microscopy
We use an upright confocal laser scanning microscope (Zeiss LSM 880 INDIMO) with individualized modifications performed by the Zeiss Company to enable the use of a UV 355 nm laser (Coherent, Inc.) for photoswitching combined with an Argon laser (488 nm) for excitation at a wavelength of 488 nm (fluo-4 excitation). Preferably, the microscope should be equipped with a 20 x 1.0 NA Plan-Apochromat water immersion objective. The correct dichroic mirrors have to be chosen for the photomanipulation during the 488 nm-dependent calcium fluorescence measurements. A critical advantage of laser scanning technology is that it enables precise spatial and temporal control of the photoswitch. For example, spatially localized photoswitching can be obtained within small subcellular compartments such as cilia, microvilli, and dendritic endings (Leinders-Zufall et al., 2018). With this set-up, laser scanning illumination can also be used to perform large-scale mapping of cellular activity across a population of cells in a tissue slice or an entire organ (Leinders-Zufall et al., 2018). Furthermore, we have used spatially controlled UV laser scanning for photolysis of inositol 1,4,5-trisphosphate (InsP3) in subcellular compartments (Chamero et al., 2017).
-
•
Photoswitching using a monochromator
We use an inverse microscope (Olympus IX-70) equipped for fluorescence microscopy to deliver light produced by the monochromator Polychrome V (Till Photonics, Planegg, Germany). The stage contains the recording chamber, a ground electrode, and a micromanipulator holding the headstage of the Heka EPC10 amplifier with the electrode holder for the recording electrode pipette. For photoswitching, the monochromator is controlled using PolyCon 3.2 software (Till Photonics). The microscope is equipped with a 20 x 0.85 UPlanS-Apochromat oil immersion objective (FN26.5), and with a filter cube containing a laser-dichroic mirror (H 488 LPXR superflat (T=1 mm), F48-487, AHF analysentechnik, Tübingen, Germany) that allowed photomanipulation at the wavelengths λ = 350 nm for PhoDAG-1, λ = 360 nm for OptoDArG, and λ = 450 nm for PhoDAG-1 and OptoDArG. The setup is schematically displayed in Figure 4A.
Figure 4.

Photoswitching of OptoDArG activates human TRPC6 channels expressed in HEK293T cells
(A) Schematic depiction of the setup for whole-cell measurements of HEK293T cells using an inverse microscope. Photoswitching was performed with light produced by the monochromator Polychrome V that was controlled by the PolyCon software. The stage of the microscope contained the recording chamber. The Heka EPC10 amplifier with Patchmaster software and the recording electrode are displayed. The microscope was equipped with a 20 x oil immersion objective and with a filter cube containing a dichroic mirror. Pipette solution (S4); bath solution (S3).
(B) Voltage protocols and resulting currents of HEK293T cells expressing hTRPC6 and preincubated with 30 μM OptoDArG. Currents were measured over time and after stimulation with λ = 450 nm (blue) or λ = 360 nm light (pink). The gray line illustrates the ramp voltage protocols. Ramp currents were examined using 50 ms voltage ramps from −100 to 100 mV (4 V/s). The ramp current with 450 nm exposure is indicated in blue and with a blue data point at 100 mV (1). The ramp current with 360 nm exposure is indicated in pink and with a pink data point at 100 mV (2).
(C) Single data points obtained from ramp currents of the experiment shown in B are plotted at 100 and −100 mV vs. time. Exposure to 360 nm UV light caused the switching of OptoDArG from the trans to the cis form and the activation of hTRPC6 channels. The data points within the gray box are from the current ramps shown in B, whereby the colored data points depict the value at 100 mV. Switching back to 450 nm light exposure returned the ramp currents at 100 and −100 mV back to their original value. The colored data points (1–3) are plotted as I-V curves in D.
(D) I-V curves obtained from the ramp protocols shown in B. The numbers (1, 2, and 3) indicate the time points at which the I-V curves are extracted from C.
(E and F) Data points obtained from ramp currents of an untransfected HEK297T cell preincubated with 30 μM OptoDArG are plotted at 100 and −100 mV vs. time using the same ramp and analysis protocols as in B and C. The colored data points (4–6) are plotted as I-V curves in F. Exposure to 360 nm UV light failed to induce a current under these conditions.
(G) Representative time course of a light-evoked hTRPC6 current recorded at a holding potential (VH) of −60 mV. The activation phase, inactivation with fast and slow phases, and deactivation phase of the hTRPC6 current are indicated. (B, C, E and G) Illumination protocol using light of the wavelengths λ = 360 nm or λ = 450 nm is depicted above each trace.
Step-by-step method details
We provide step-by-step method details for performing either electrophysiology in combination with LED-evoked photoswitching or monochromator-based illumination; or an all-optical method combining laser scanning-controlled photoswitching with intracellular calcium imaging. All experimental procedures should be performed in agreement with animal protocols approved by the institutional animal care and use committees.
Optical control of TRPC channel activity recorded by electrophysiology
-
1.
Produce patch pipettes made of borosilicate glass with resistances of 2.0–3.5 MΩ (HEK293T cells) or 5–8 MΩ (olfactory neurons) for whole-cell measurements.
-
2.
Place the slice in a perfusion chamber of the electrophysiology microscope and perfuse the cells/slice with bath solution S1 (aerated with 95% O2 / 5% CO2) or S3 (HEK293T cells) at an appropriate flow rate (e.g., 100 μL/s) to ensure the health of the cells. Control the flow of the bath perfusion solution, e.g., with a pump or gravity feed. The solution speed in a gravity feed system should be strictly controlled using a combination of solution volume (container) and gravity flow controllers. Keep track of the perfusion solution volume to maintain relatively equal pressure on the tubing lines. For measurements of TRPC6 channel activity in transfected HEK293T cells, mount the glass bottom dishes with the loaded cells onto the stage of the inverse microscope. Perfusion is not necessary in this case.
-
3.
Fill the recording electrode with the artificial intracellular solution (depending on the experiment: S4 or S5; see: Materials and Equipment).
-
4.
Place the recording electrode in the pipette holder of the amplifier headstage. Apply positive pressure using a 10 or 50 mL syringe to prevent the formation of air bubbles when transferring the electrode into the bath solution and attachment of debris onto the glass electrode.
-
5.
Set the amplifier according to the manufacturer’s manual, and apply a test pulse to monitor the pipette/seal resistance.
-
6.
Bring the recording electrode into the bath solution, monitor, and correct your junction potential between the solutions and slowly approach the cell of interest.
-
7.
Using the standard whole-cell patch-clamp configuration technique, begin with the formation of a gigaseal between the cell membrane and recording pipette electrode by releasing the positive pressure and applying a little suction (cell-attached configuration). After gigaseal formation, change the voltage to the predicted resting potential of the cell (−60 mV for most neurons) and correct for fast capacitance. Access to the cell interior is made by rupturing the cell membrane under the electrode by introducing a sudden burst of low pressure (suction). This step requires training and practice.
-
8.Data are usually acquired using the acquisition software bundled with the patch-clamp amplifier. The following recording voltage step protocols were used in Leinders-Zufall et al. (2018):
-
a.VSN recording protocols
-
i.Obtain families of whole-cell currents to a series of depolarizing and hyperpolarizing voltage steps in complete darkness and during activating and inactivating photoswitching illuminations.
-
ii.Keep VSNs at a holding potential of −70 mV before initiating a cycle of step protocols.
-
iii.A prepulse to +80 mV eliminates the activity of most voltage-activated sodium and calcium channels due to their inactivation kinetics (see Lucas et al., 2003). The prepulse is followed by (1) a short step to 0 mV, then to (2) a specific test voltage, (3) back to 0 mV and (4) ending again at the holding potential −70 mV. The test voltages range between −80 and +80 mV in incremental steps of 10 mV.
-
iv.Perform experiments in the presence of 1 μM tetrodotoxin to additionally block voltage-gated sodium channels. Voltage-activated potassium channels are blocked by using the Cs+-based S5 solution.
-
i.
-
b.HEK293T cell recording protocols
-
i.Obtain current-voltage relations from triangular voltage ramps ( –100 to +60 mV with a slope of 0.4 V s−1) applied at a frequency of 1 Hz.
-
ii.Extract the current amplitudes at minimal or maximal currents at ±60 mV, respectively.
-
iii.To estimate the fast activation and deactivation kinetics of TRPC6 currents, use the following protocols: Either apply fast up-ramps (−100 to +100 mV with a slope of 4 V s−1) at a frequency of 12.5 Hz or continuously apply a constant holding potential of −60 mV.
-
iv.Data are usually acquired at a frequency of 5 kHz after filtering at 1.67 kHz. In case of measurements at continuous constant holding potential, acquire data at a frequency of 200 kHz after filtering at 2.5 kHz.
-
i.
-
a.
-
9.Photoswitching using (a) a monochromator or (b) LEDs:
-
a.Settings for the monochromator: Fluorescence light of λ = 350 or 360 nm, and λ = 450 nm at maximal intensity (100%) should be applied using a monochromator (Polychrome V, Till Photonics) controlled by the PolyCon 3.2 software.
-
b.Settings for the use of LEDs:
-
a.
For photoswitching in patch-clamp experiments using VSNs (Figure 2), light of appropriate wavelengths through two fiber-coupled LEDs with nominal wavelengths of λ = 365 nm (4.1 mW) or λ = 470 nm (10.1 mW) is delivered to switch PhoDAG between the active and inactive state, respectively. A bifurcated fiber bundle with a 400 μm core (0.39 NA) was directly coupled to the LEDs, and a fiber optic mating sleeve (Thorlabs) aligned the 400-μm thick optical fiber with a thinner optical fiber (200 μm, 0.22 NA) which is placed directly in the bath solution in front of the cell preparations. Light switching can be controlled by a DC4104 LED controller (Thorlabs) using a 1-ms switching time, triggered by a TTL signal controlled by the patch-clamp amplifier.
-
10.
Analyze the data using Igor Pro (WaveMetrics), Origin (OriginLab), or similar software packages. Current-voltage (I-V) curves are acquired by (a) a series of depolarizing and hyperpolarizing voltage steps or (b) by voltage-ramp protocols (see voltage-clamp protocols in Figures 3 and 4). Important parameters can be derived from these curves such as reversal potential, ionic properties and selectivity, voltage-dependence (rectification of I-V curves), activation threshold, and overall conductance properties. By using fitting routines obtained in Igor Pro, the activation, inactivation, and deactivation time constants can be determined. A commonly used fitting routine for these procedures is the Levenberg-Marquardt algorithm to minimize least squared errors.
Note: In some instances, it could be an advantage to (additionally) infuse photoswitchable DAGs through the patch pipette if there appears to be a loading problem. In this case, use the 100 mM PhoDAG-1 stock solution and dilute it with pipette solution S4 to a final concentration of 100 μM PhoDAG-1. Proceed with “Optical control of TRPC channel activity recorded by electrophysiology”.
Figure 3.

Combined DAG photoswitching and intracellular calcium recording using an all-optical confocal laser scanning approach
(A) Sketch and stimulus protocol for an upright Zeiss LSM880 laser scanning confocal microscope that contains modifications to enable the combined use of a UV laser (355 nm) for photomanipulation and an Argon laser (488 nm) for excitation of the calcium indicator dye. The Argon laser exciting the calcium fluorophore will scan the preset image size of 512 x 512 pixels at a set cycle time with only a short gap between the scans. The photomanipulation with the UV laser switching the PhoDAG to the active cis-form will interrupt the collection of images from the calcium fluorophore. This pause will depend on the duration of the scanning time in one or multiple regions of interest. In the example shown, one ROI (cell # 1) is excited by the UV laser. Due to the gap in collecting the fluorophore signals of all ROIs, the ΔF/F analysis will thus display a pause in the recording.
(B) Example of the Ca2+ response of a TRPC2-expressing olfactory type B cell loaded with PhoDAG-1 (10 μM). Photoswitching to 355 nm (10 mW) for 61 ms (thin magenta line) evoked a transient elevation in intracellular Ca2+. The photostimulus consisted of 40 individual scans with a pixel dwell time of 2.05 μs, resulting in a total UV exposure time of 61 ms. Decay time constant of the Ca2+ transient is indicated. Figure reprinted with permission from Figure 4D in Leinders-Zufall et al., 2018.
(C) Example of the Ca2+ response of an olfactory TRPC2-expressing cell loaded with OptoDArG (30 μM). Photoswitching to 355 nm (30 mW) for 54 ms (thin magenta line) evoked a transient elevation in intracellular Ca2+. The photostimulus consisted of 100 individual scans with a pixel dwell time of 2.05 μs, resulting in a total UV exposure time of 54 ms.
Combined laser scanning-controlled photoswitching and calcium imaging
-
11.
Dissociated cells or tissue slices co-loaded with a calcium indicator and PhoDAG are placed in a laminar flow recording chamber and continuously perfused with extracellular S1 solution (aerated with 95% O2 / 5% CO2) at an appropriate flow rate (e.g., 100 μL/s) to ensure the health of the cells. Control the flow of the bath perfusion solution using a pump or gravity feed. The solution speed in a gravity feed system should be strictly controlled using a combination of solution volume (container) and gravity flow controllers. Keep track of the perfusion solution volume to maintain relatively equal pressure on the tubing lines.
-
12.
Visualize the cells within a scanning field of 512 x 512 pixels and a depth of ∼9 μm.
-
13.
Adjust the pixel dwell time of the laser to a total image scanning of ∼1 s.
-
14.
To measure the basic activity of the cells and to ensure there is no drift in the recording, perform calcium fluorescence imaging of the cells/tissue using the Argon laser (excitation wavelength of 488 nm) for 4 min. The Zen software (Zeiss) will capture the emission fluorescence (510–560 nm). All scanning head settings need to be kept constant during the entire experiment to ensure comparison of the image data. At 488 nm wavelength, PhoDAGs are known to exist in the inactive trans configuration (Frank et al., 2016).
-
15.
For photoswitching of PhoDAG to the active cis form, UV light of 355 nm is used to scan preselected regions-of-interests (ROIs) via the Zen software tool ‘Bleaching’. To prevent damage to the highly sensitive gallium-arsenide-phosphide (GaAsP) detectors, a pre-installed shutter should be activated via the Zen software protocol which will block UV light reaching the detector. Unfortunately, the collection of the calcium indicator fluorescence is halted during photoswitching of the PhoDAG compounds, but resumes at the end of the UV stimulation. An example of the imaging acquisition and stimulus protocol is shown in Figure 3A.
-
16.
Analyze images using a combination of Zen (Zeiss), ImageJ (NIH), and Igor (WaveMetrics) software. Raw fluorescence images can be acquired via the manufacturer’s software Zen (Zeiss) and, by using Zen or ImageJ (NIH), regions-of-interests (ROIs) are marked for analysis. The raw fluorescence intensity of the ROIs versus time are exported to an analysis program, e.g., Igor Pro (WaveMetrics), which enables to determine the average baseline fluorescence values (Fo). Cytosolic changes in fluorescence intensity are then calculated as (F–Fo)/Fo. Onset time of Ca2+ transients is defined as the difference between the time point at which 488 nm excitation started after UV exposure and the time at which Ca2+ fluorescence exceeded twice the standard deviation of the mean of the baseline noise. Other parameters (such as Fpeak, and activation or deactivation time courses) can then be determined and fitted using the standard tools provided by Igor Pro.
Note that a variety of state-of-the-art methods for calcium imaging data analysis and various software tools have been summarized recently (Pnevmatikakis, 2019).
Note: UV laser power will have to be adjusted depending on the DAG analog being used and the final concentration loaded into the cells. PhoDAG-1 and PhoDAG-3 could be photoswitched to the active cis form using a UV laser power of 10 mW (Figure 3B). However, OptoDArG required 30 mW laser power to obtain a similar Ca2+ response (Figure 3C).
Note: The depth of focus can be increased from 9 μm to 16 μm to ensure, together with the region of interest (ROI) diameter, illumination of individual cells, localized areas of a given cell, or a subcellular region containing the microvilli or dendritic knobs of VSNs in tissue slices.
Note: Prior to photoswitching, UV laser light can be optimally focused in 18-μm thick tissue sections by loading them with Hoechst 33342 (1:10000) and using the semi-automated correction tool of the Zen software (Zeiss).
Expected outcomes
With these protocols, it has been possible to establish DAG photoswitching as an effective optical tool for investigating the functions of DAG-sensitive TRPC channels such as mouse TRPC2 and human TRPC6 (Leinders-Zufall et al. 2018). Other studies have used DAG photoswitching for experiments investigating the gating mechanisms of human TRPC3 (Lichtenegger et al., 2018). Photoswitchable DAGs are minimally invasive tools that enable channel activation and deactivation with unprecedented speed and temporal precision. In combination with confocal laser scanning microscopy to locally deliver spatially and temporally controlled light pulses, photoswitching can be used to activate TRPC channels in small subcellular compartments such as dendritic spines or cilia and microvilli. Furthermore, DAG photoswitching can be combined with Ca2+ imaging and various genetic manipulations (Leinders-Zufall et al. 2018).
While we have used PhoDAG-1 and PhoDAG-3 for our initial studies (Leinders-Zufall et al. 2018), we now have also included in the current protocols the use of OptoDArG, an optimized photochromic DAG featuring two arachidonyl-mimetic photoswitchable azobenzene chains characterized as a highly effective ligand for the control of TRPC3 channel activity (Lichtenegger et al. (2018). In freshly dissociated olfactory cells that natively express TRPC2 and that respond reliably to PhoDAG-1 stimulation, we also achieved photostimulated Ca2+ responses with OptoDArG (Figures 3B and 3C). However, these responses required a somewhat higher concentration of OptoDArG, enhanced laser intensities, and optimized loading conditions (Figure 3C). Using PhoDAG-1 or PhoDAG-3 stimulation, we were able to activate TRPC2-expressing olfactory cells in 100% of the cases (23/23, see Leinders-Zufall et al., 2018). Using OptoDArG, we could activate a Ca2+ response in 42% (5/12) of TRPC2-expressing olfactory neurons (Figure 3C). Negative control experiments are discussed below (see “Troubleshooting” section). We conclude that PhoDAG-1 or PhoDAG-3 remain the preferred DAG analogs for this type of experiment. By contrast, OptoDArG worked very well with heterologously expressed TRPC6 channels (see below and Figure 4). Therefore, the results summarized in these protocols indicate that there are considerable differences in the effects of the three photoswitchable DAG analogs on mTRPC2, hTRPC3, and hTRPC6 within the various cellular preparations (Figure 1).
By analyzing PhoDAG-1 stimulated hTRPC6 channels in a heterologous overexpression system, we found that PhoDAG-1 is suitable to evoke fast photoswitchable TRPC6 channel activation and deactivation (Leinders-Zufall et al. 2018). However, the initial success rate of these measurements was relatively low (1 out of 10 measurements). Therefore, we have tested here whether OptoDArG might be advantageous for the analysis of TRPC6 current properties. Indeed, we observed that the success rate was considerably higher by using OptoDArG (1 out of 2 measurements were successful). In addition, OptoDArG was effective at a lower concentration than PhoDAG-1 (30 μM vs. 100 μM). Cells loaded with OptoDArG responded to switches of the fluorescence light to 360 nm or 450 nm with rapidly increasing or decreasing outward and inward currents, respectively, at holding potentials of ±100 mV (Figure 4B). These currents exhibited the characteristic current-voltage (I-V) relationships of TRPC6 channels (Figure 4D). As a control for the target specificity of OptoDArG, untransfected OptoDArG-loaded HEK293T cells were exposed to light switching from 360 to 450 nm. In this case, there was no current activation, thus ruling out a significant contribution of endogenously expressed ion channels to the responses (Figures 4E and 4F).
To precisely define the fast activation and deactivation kinetics of light-evoked TRPC6 currents, we additionally applied a constant holding potential of -60 mV and monitored the light-induced changes of TRPC6 inward currents (Figure 4G). We observed that TRPC6 inward currents possess fast activation and deactivation phases at the beginning and end of the 360 nm light stimulus, respectively. During the 360 nm stimulation, current inactivation comprises two distinct phases: a fast inactivation that is followed by a slower inactivation phase (Figure 4G). Half-life time constants τ1/2 were estimated as follows: ∼150 ms for current activation, ∼150 ms for fast inactivation and ∼20 s for slow inactivation, and ∼50 ms for deactivation. Thus, current deactivation was three times faster than current activation. These parameters were consistent with the kinetic parameters estimated with the current-time course at -100 mV (see Figure 4B). Here, the values for τ1/2 were estimated as ∼160 ms for activation, ∼200 ms for fast inactivation, ∼11 s for slow inactivation, and ∼80 ms for deactivation. However, using the fast up-ramp protocol (Figures 4B–4D) detailed kinetic information becomes lost since the recording frequency is not sufficient. Nevertheless, one advantage of this protocol is the sampling of I-V relationships that allow for estimation of leak currents that might occur during measurements. Specifically for the analysis of fast activation and deactivation kinetics and for the fast inactivation phase of TRPC6 currents, a stimulation protocol with a constant holding potential is beneficial and allows for estimation of exact, reliable, and reproducible kinetic parameters. However, one should keep in mind that leak currents might influence the kinetics.
In summary, we find that these photoswitchable DAG-analogs are well-suited tools to study the biophysical properties of TRPC channels and allow for a detailed and reliable analysis of kinetic parameters of TRPC6 whole-cell currents. We note that photoswitchable DAG-analogs enable for the first time a detailed and valid analysis of fast deactivation parameters.
With these new DAG photoswitches, it should now also be possible to resolve the long-standing question whether DAG signaling is directly involved in TRP channel activation of Drosophila photoreceptors (e.g., see Delgado et al., 2019) to better define the second messenger mechanisms underlying phototransduction in insects and other invertebrates.
Similarly, DAG photoswitching – perhaps in combination with photolysis of InsP3 or Ca2+ – should resolve the long-standing question whether and how DAG-signaling interacts with inositol trisphosphate- and Ca2+-signaling networks in a given cell or its subcellular compartments (e.g., see Chamero et al., 2017; Eckstein et al., 2020).
Future experiments should also use DAG photoswitching to assess the functions of additional TRPC channel subtypes whose second messenger-driven signaling mechanisms still remain unclear, such as TRPC5 (Blum et al., 2019; Storch et al., 2017). For example, DAG photoswitching can now be applied in brain slices where TRPC5 is known to contribute to complex oscillatory activity and hormone-activated long-lasting plateau potentials in dopamine neurons of the hypothalamus (Blum et al., 2019).
Outlook and future visions: we expect that DAG photoswitching should be a valuable tool in further studies aimed at understanding the gating mechanisms of DAG-sensitive TRP channels, perhaps even in combination with structural methods such as single-particle electron cryomicroscopy. DAG photoswitching provides an important advance to explore the pathophysiological relevance of DAG-sensitive TRP channels in the maintenance of body homeostasis (Leinders-Zufall et al., 2018).
Quantification and statistical analysis
Statistical analyses are generally performed using specialized statistics software packages, e.g., SPSS (IBM Corporation, New York, U.S.A.) or Origin Pro (OriginLab Corporation, Northampton, MA, USA). Assumptions of normality and homogeneity of variance should be tested before conducting the following statistical approaches. Student’s t-test is used to measure the significance of the differences between two distributions. Multiple groups are compared using one- or two-way analysis of variance (ANOVA) with Tukey or Fisher’s least significant difference (LSD) test as a post hoc comparison. In case the results fail the test of normality, the Kruskal-Wallis ANOVA and Mann-Whitney U test can be used. The probability of error level (alpha) is chosen to be 0.05.
The inclusion criteria identify the study population in a consistent, reliable, uniform, and objective manner. The exclusion criteria include factors or characteristics that make the recruited population ineligible for the study. For calcium imaging experiments, cells that did not take up the calcium indicator dye are excluded from the study. For electrophysiology experiments, parameters determined from the current-voltage steps should be appropriate for the cells. In both types of experiments, the stability of the experiment is determined by the stability of the baseline. Rapid fluctuation baselines indicate either problems with movements, solution exchange, and loss of calcium indicator dye during the calcium imaging, or problems with poor seal resistance in the electrophysiology experiments. Data from such experiments should be discarded.
Limitations
During the time when the image acquisition protocol switches back to using the 488 nm laser to resume monitoring of calcium fluorescence, PhoDAG will be switched back into the inactive trans form. Depending on the speed of this transition and the duration of the gap in collecting calcium fluorescence data due to the UV light scan, it may happen that no change in the calcium signal is detected for a short while. Therefore, care has to be taken regarding the simultaneous exposure of multiple regions of interest (ROIs) that are widely distributed over a given scanning field (in our case 512 x 512 pixels). To ensure the fasted time of collecting the change in calcium fluorescence and activation of PhoDAG, only single cells or even subcellular compartments (resulting in smaller regions of interest) should be scanned by the UV laser. A UV exposure time of only 1.7 ms can be achieved with this approach by scanning subcellular regions.
To improve temporal resolution in some of the single-cell scanning experiments, we minimized the total number of scanned pixels per image, reducing the total scanning time to 900 ms. Under these conditions, UV stimulation of a cell with a typical diameter of 10 μm consisted of 20 individual scans with a pixel dwell time of 1.54 μs, resulting in a total UV exposure time of 23 ms. UV stimulation for an olfactory type B cell typically consisted of 40 individual scans with a pixel dwell time of 2.05 μs, resulting in a total UV exposure time of 61 ms. With these procedures, it has been possible to demonstrate that millisecond DAG pulses are sufficient to activate native TRPC2 channels expressed in mouse chemosensory neurons, and that rapid channel deactivation occurs after returning the photoswitch back into its inactive form (Leinders-Zufall et al., 2018).
In future experiments, the optical switching time should be reduced even further with the aim to provide a minimal DAG photoactivation time that is sufficient to achieve activation (gating) of TRPC channels. Such experiments, in combination with specific channel mutations such as those obtained for hTRPC3 (Lichtenegger et al., 2018), should be highly useful to further explore the gating mechanisms of DAG-sensitive TRP channels.
Troubleshooting
Problem 1
Control experiments to rule out potential off-target effects caused by DAG photoswitching (step 9 of ‘Optical control of TRPC channel activity recorded by electrophysiology’; step 15 of ‘Combined laser scanning-controlled photoswitching and calcium imaging’).
Potential solution
For experiments with native cells, the gold standard to rule out off-target effects is the use of knockout mice. We have used TRPC2 knockout mice as controls for photoswitching experiments in VSNs and found no current activation under these conditions, thus ruling out any off-target effects (Leinders-Zufall et al., 2018).
For experiments using olfactory type B cells, we have compared the effects of photoswitching with two other cell types: OSNs that lack TRPC2 expression and olfactory type A cells that do express TRPC2 (Leinders-Zufall et al., 2018). As expected, OSNs did not show any currents whereas type A cells exhibited similar currents as type B cells. Together these experiments have provided clear evidence for target specificity of the DAG photoactivation under our conditions.
Since all TRPC channels are sensitive to DAG (Storch et al., 2017; Hofmann et al., 1999), it should be ruled out that endogenously expressed channels in HEK293T cells are activated by photoswitchable DAGs. Use non-transfected HEK293T cells as a negative control. We found that non-transfected HEK293T cells loaded with photoswitchable DAGs showed no current increases upon light stimulation (Figures 4E and 4F) (see also Leinders-Zufall et al., 2018). Notably, TRPC6 is directly activated by DAG in a membrane-delimited fashion, independently of protein kinases C (PKC) activated by DAG (Hofmann et al., 1999), suggesting that off-target effects resulting from PKC stimulation can be neglected.
Problem 2
Control experiments to ensure optimal activity of PhoDAG (step 9 of ‘Optical control of TRPC channel activity recorded by electrophysiology’; step 15 of ‘Combined laser scanning-controlled photoswitching and calcium imaging’).
Potential solution
To ensure optimal activity of PhoDAG, we have performed concentration-response measurements with PhoDAG-3 in VSNs (Leinders-Zufall et al., 2018). The size of the resulting currents can be compared to other experiments in which we used direct current injection to generate action potentials in VSNs (Ukhanov et al., 2007). We found that only 2 pA of inward current is sufficient to generate repetitive action potentials in these neurons, and saturation of action potential frequency occurs at approximately 15 pA. Such small currents can readily be generated by loading the cells with 5 μM of PhoDAG-3 and performing photostimulation, thus providing an estimate for a physiological concentration range and an optimal activity of PhoDAG-3. As a further control, we found that the concentration dependence of PhoDAG-3 in VSNs was closely similar to the effects obtained with application of the DAG analog 1-stearoyl-2-arachidonoyl-sn-glycerol (Lucas et al., 2003).
Problem 3
Few responses are detected after UV-illumination of the cells (step 9 of ‘Optical control of TRPC channel activity recorded by electrophysiology’; step 15 of ‘Combined laser scanning-controlled photoswitching and calcium imaging’).
Potential solution
Test whether the cells were injured or died during the preparation or imaging procedures. This can be verified by exposing OSNs and VSNs with known chemostimuli or, more generally, by depolarizing cells with high extracellular KCl solution (e.g., 20 or 60 mM instead of 5 mM KCl).
Try to load cells with a different photoswitchable DAG which may be better tolerated by a given cell type.
Increase the UV-light intensity to switch DAG analogs from the trans- to the cis form. OptoDArG with two azobenzene moieties requires a higher light intensity to switch to the cis form.
Check for phototoxicity (see: Problem 4).
Problem 4
Cellular responses after UV-illumination appear to be adversely affected by the UV-light intensity – phototoxicity (step 9 of ‘Optical control of TRPC channel activity recorded by electrophysiology’; step 15 of ‘Combined laser scanning-controlled photoswitching and calcium imaging’).
Potential solution
Check if cells are declining in health as a result of exposure to UV-light or to the excitation wavelength of the calcium indicator dye. Calcium indicator dyes are known to produce reactive oxygen species during their excited state. Reducing the amount of or using another calcium indicator dye could be beneficial.
Reduce the UV-light intensity. You may need to optimize the amount of uploaded PhoDAG versus the intensity of λ < 370 nm and λ ≥ 450 nm light.
Use another of the photoswitchable DAGs. For example, more light intensity is needed to switch the OptoDArG with two azobenzene moieties from the cis to the trans form and vice versa.
Reduce the illumination time (i.e., by changing exposure time or pixel dwell time).
For additional suggestions to minimize phototoxicity, see Kiepas et al. (2020).
Problem 5
The UV-stimulated cis form of a given photoswitchable DAG does not switch back to the inactive trans form, which could potentially damage the TRP channel-expressing cells by activation of a persistent inward current, followed by accumulation of intracellular calcium (step 9 of ‘Optical control of TRPC channel activity recorded by electrophysiology’; step 15 of ‘Combined laser scanning-controlled photoswitching and calcium imaging’).
Potential solution
Switch to complete darkness or increase the λ ≥ 450 nm light intensity.
Use another of the photoswitchable DAGs. In the calcium imaging experiments, decrease the Argon laser (488 nm) used for calcium monitoring to low light intensities (<2% of 10 mW) in order to prevent the production of damaging reactive oxygen species, and to enable the measurements of calcium changes. However, a low 488 nm laser intensity could limit the formation of the inactive trans form of the photoswitchable DAGs, keeping some of the molecules in their active cis form.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Frank Zufall (frank.zufall@uks.eu).
Material availability
This study did not generate new unique reagents.
Data and code availability
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author on request.
Acknowledgments
We thank Frank Filip Steinbauer for excellent technical assistance. This research was supported by Deutsche Forschungsgemeinschaft (DFG) Grant Sonderforschungsbereich-Transregio TR152, project no. 239283807 (to T.L.-Z., T.G., and F.Z.), DFG Grant project no. 406028471 (to U.S.), DFG Instrumentation Grant INST 256/427-1 FUGB (to T.L.-Z.), and the Volkswagen Foundation (to T.L.-Z.).
Author contributions
T.L.-Z., T.G., and F.Z. conceived the project. T.L.-Z., U.S., M.M.y.S., T.G., and F.Z. designed experiments. T.L.-Z., U.S., N.K.O., and K.K. performed experiments, and T.L.-Z., U.S., N.K.O., K.K., and M.M.y.S. performed analyses. T.L.-Z. wrote the first draft of the manuscript. F.Z., U.S., M.M.y.S., and T.G. contributed to the writing of the manuscript. All authors edited the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- Bleymehl K., Pérez-Gómez A., Omura M., Moreno-Pérez A., Macías D., Bai Z., Johnson R.S., Leinders-Zufall T., Zufall F., Mombaerts P. A sensor for low environmental oxygen in the mouse main olfactory epithelium. Neuron. 2016;92:1196–1203. doi: 10.1016/j.neuron.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum T., Moreno-Pérez A., Pyrski M., Bufe B., Arifovic A., Weissgerber P., Freichel M., Zufall F., Leinders-Zufall T. Trpc5 deficiency causes hypoprolactinemia and altered function of oscillatory dopamine neurons in the arcuate nucleus. Proc. Natl. Acad. Sci. U S A. 2019;116:15236–15243. doi: 10.1073/pnas.1905705116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brechbühl J., Gaëlle L., Moine F., Rodriguez I., Broillet M.-C. Imaging pheromone sensing in a mouse vomeronasal acute tissue slice preparation. J. Vis. Exp. 2011;58:e3311. doi: 10.3791/3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamero P., Weiss J., Alonso M.T., Rodríguez-Prados M., Hisatsune C., Mikoshiba K., Leinders-Zufall T., Zufall F. Type 3 inositol 1,4,5-trisphosphate receptor is dispensable for sensory activation of the mammalian vomeronasal organ. Sci. Rep. 2017;7:10260. doi: 10.1038/s41598-017-09638-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Sooch G., Demaree I.S., White F.A., Obukhov A.G. Transient receptor potential canonical (TRPC) channels: then and now. Cells. 2020;9:1983. doi: 10.3390/cells9091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcic S., Tiapko O., Groschner K. Photopharmacology and opto-chemogenetics of TRPC channels-some therapeutic visions. Pharmacol. Ther. 2019;200:13–26. doi: 10.1016/j.pharmthera.2019.04.003. [DOI] [PubMed] [Google Scholar]
- Delgado R., Delgado M.G., Bastin-Heline L., Glavic A., O'Day P.M., Bacigalupo J. Light-induced opening of the TRP channel in isolated membrane patches excised from photosensitive microvilli from Drosophila photoreceptors. Neuroscience. 2019;396:66–72. doi: 10.1016/j.neuroscience.2018.11.017. [DOI] [PubMed] [Google Scholar]
- Eckstein E., Pyrski M., Pinto S., Freichel M., Vennekens R., Zufall F. Cyclic regulation of Trpm4 expression in female vomeronasal neurons driven by ovarian sex hormones. Mol. Cell. Neurosci. 2020;105:103495. doi: 10.1016/j.mcn.2020.103495. [DOI] [PubMed] [Google Scholar]
- Fischer A.H., Jacobson K.A., Rose J., Zeller R. Preparation of slides and coverslips for microscopy. Cold Spring Harb. Protoc. 2008;3 doi: 10.1101/pdb.prot4988. [DOI] [PubMed] [Google Scholar]
- Frank J.A., Yushchenko D.A., Hodson D.J., Lipstein N., Nagpal J., Rutter G.A., Rhee J.S., Gottschalk A., Brose N., Schultz C. Photoswitchable diacylglycerols enable optical control of protein kinase C. Nat. Chem. Biol. 2016;12:755–762. doi: 10.1038/nchembio.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtovenko A.A., Anwar J. Modulating the structure and properties of cell membranes: the molecular mechanism of action of dimethyl sulfoxide. J. Phys. Chem. B. 2007;111:10453–10460. doi: 10.1021/jp073113e. [DOI] [PubMed] [Google Scholar]
- Hofmann T., Obukhov A.G., Schaefer M., Harteneck C., Gudermann T., Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hüll K., Morstein J., Trauner D. In vivo photopharmacology. Chem. Rev. 2018;118:10710–10747. doi: 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- Iseppon F., Arcangeletti M. Optogenetics and photopharmacology in pain research and therapeutics. STEMedicine. 2020;1:e43. [Google Scholar]
- Kiepas A., Voorand E., Mubaid F., Siegel P.M., Brown C.M. Optimizing live-cell fluorescence imaging conditions to minimize phototoxicity. J. Cell Sci. 2020;133:jcs242838. doi: 10.1242/jcs.242834. [DOI] [PubMed] [Google Scholar]
- Leinders-Zufall T., Lane A.P., Puche A.C., Ma W., Novotny M.V., Shipley M.T., Zufall F. Ultrasensitive pheromone detection by mammalian vomeronasal neurons. Nature. 2000;405:792–796. doi: 10.1038/35015572. [DOI] [PubMed] [Google Scholar]
- Leinders-Zufall T., Brennan P., Widmeyer P., Chandramani P.S., Maul-Pavicic A., Jäger M., Li X.-H., Breer H., Zufall F., Boehm T. MHC class I peptides as chemosensory signals in the vomeronasal organ. Science. 2004;306:1033–1037. doi: 10.1126/science.1102818. [DOI] [PubMed] [Google Scholar]
- Leinders-Zufall T., Ishii T., Mombaerts P., Zufall F., Boehm T. Structural requirements for the activation of vomeronasal sensory neurons by MHC peptides. Nat. Neurosci. 2009;12:1551–1558. doi: 10.1038/nn.2452. [DOI] [PubMed] [Google Scholar]
- Leinders-Zufall T., Zufall F. The electrovomeronasogram: field potential recordings in the mouse vomeronasal organ. Methods Mol. Biol. 2013;1068:221–236. doi: 10.1007/978-1-62703-619-1_16. [DOI] [PubMed] [Google Scholar]
- Leinders-Zufall T., Storch U., Bleymehl K., Mederos y Schnitzler M., Frank J.A., Konrad D.B., Trauner D., Gudermann T., Zufall F. PhoDAGs enable optical control of diacylglycerol-sensitive transient receptor potential channels. Cell Chem. Biol. 2018;25:215–223. doi: 10.1016/j.chembiol.2017.11.008. [DOI] [PubMed] [Google Scholar]
- Lichtenegger M., Tiapko O., Svobodova B., Stockner T., Glasnov T.N., Schreibmayer W., Platzer D., de la Cruz G.G., Krenn S., Schober R. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018;14:396–404. doi: 10.1038/s41589-018-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas P., Ukhanov K., Leinders-Zufall T., Zufall F. A diacylglycerol-gated cation channel in vomeronasal neuron dendrites is impaired in TRPC2 mutant mice: mechanism of pheromone transduction. Neuron. 2003;40:551–561. doi: 10.1016/s0896-6273(03)00675-5. [DOI] [PubMed] [Google Scholar]
- Ma L., Haga-Yamanaka S., Yu Q.E., Qiu Q., Kim S., Yu C.R. Imaging neuronal responses in slice preparations of vomeronasal organ expressing a genetically encoded calcium sensor. J. Vis. Exp. 2011;58:e3404. doi: 10.3791/3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notman R., Noro M., O'Malley B., Anwar J. Molecular basis for dimethylsulfoxide (DMSO) action on lipid membranes. J. Am. Chem. Soc. 2006;128:13982–13983. doi: 10.1021/ja063363t. [DOI] [PubMed] [Google Scholar]
- Pnevmatikakis E.A. Analysis pipelines for calcium imaging data. Curr. Opin. Neurobiol. 2019;55:15–21. doi: 10.1016/j.conb.2018.11.004. [DOI] [PubMed] [Google Scholar]
- Spehr J., Hagendorf S., Weiss J., Spehr M., Leinders-Zufall T., Zufall F. Ca2+-calmodulin feedback mediates sensory adaptation and inhibits pheromone-sensitive ion channels in the vomeronasal organ. J. Neurosci. 2009;29:2125–2135. doi: 10.1523/JNEUROSCI.5416-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch U., Forst A.L., Pardatscher F., Erdogmus S., Philipp M., Gregoritza M., Mederos y Schnitzler M., Gudermann T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. U S A. 2017;114:E37–E46. doi: 10.1073/pnas.1612263114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukhanov K., Leinders-Zufall T., Zufall F. Patch-clamp analysis of gene-targeted vomeronasal neurons expressing a defined V1r or V2r receptor: ionic mechanisms underlying persistent firing. J. Neurophysiol. 2007;98:2357–2369. doi: 10.1152/jn.00642.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author on request.