

Summary
Here, we present a revised protocol to derive neuroepithelial stem (NES) cells from human induced pluripotent stem cells. NES cells can be further differentiated into a culture of neurons (90%) and glia (10%). We describe how to derive and maintain NES cells in culture and how to differentiate them. In addition, we show the potential use of NES cells to study the role of reactive oxygen species in neuronal differentiation and a guideline for NES cell transfection.
For complete details on the use and execution of this protocol, please refer to Calvo-Garrido et al. (2019); Falk et al. (2012).
Subject areas: Neuroscience, Stem Cells, Cell Differentiation
Graphical abstract
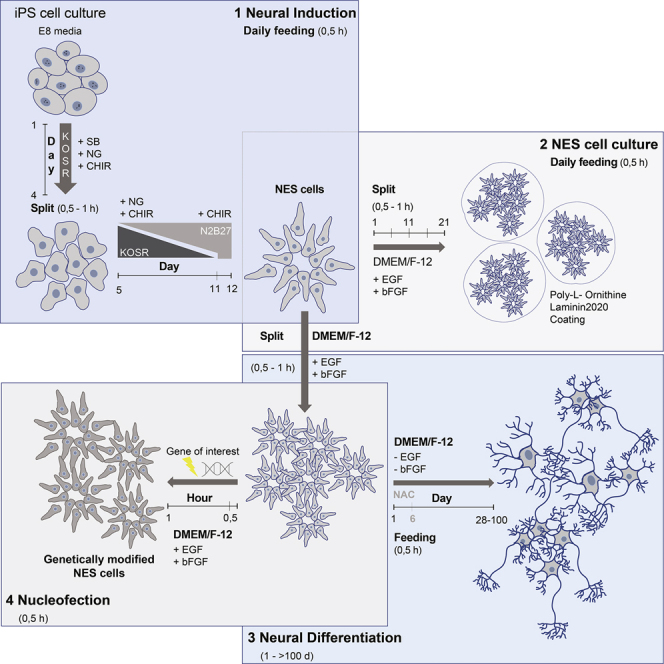
Highlights
-
•
NES cells, neurons, and glia generated through dual SMAD inhibition
-
•
Proliferate, maintain, and differentiate NES cells into neurons and glia in culture
-
•
Coating of the growth surface, cell density, and trypsinization need to be adjusted
-
•
Feasible model to study ROS effect in neuronal and glia differentiation
Here, we present a revised protocol to derive neuroepithelial stem (NES) cells from human induced pluripotent stem cells. NES cells can be further differentiated into a culture of neurons (90%) and glia (10%). We describe how to derive and maintain NES cells in culture and how to differentiate them. In addition, we show the potential use of NES cells to study the role of reactive oxygen species in neuronal differentiation and a guideline for NES cell transfection.
Before you begin
Reagent and factor preparation for iPS cell culture
Timing: 15 min
-
1.
Aliquot laminin521 and E8 supplement to avoid repeated freezing-thaw cycles.
Preparation of stock solutions for NES cell derivation, culture, and differentiation
Prepare aliquots of stock solutions to reduce freeze-thawing cycles. See Table 2 for storage and working concentrations.
-
2.
Resuspend and filter Poly-L-Ornithine in PBS to a final concentration of 100 μg/mL and aliquot.
-
3.
Resuspend Noggin in PBS at 500 μg/mL and aliquot 20 μL.
-
4.
Knock out serum replacement (KOSR) and non-essential amino acids, aliquot in volumes of 10 mL.
-
5.
Resuspend 1 mg of SB431542 in 260 μL of DMSO to a final concentration of 10 mM and aliquot.
-
6.
Resuspend 1 mg of CHIR99021 in 215 μL of DMSO to a final concentration of 3.3 μM and aliquot.
-
7.
Resuspend bFGF in PBS-BSA 0.15% and EGF in PBS to a final concentration of 100 ng/μL. Aliquot in a volume of 100 μL.
-
8.
Aliquot B27 in 500 μL aliquots and N2 and Penicillin/Streptomycin (P/S) in 5 mL aliquots.
-
9.
Optional! Prepare N-Acetyl-L-cysteine (NAC) stock solution at 612 mM. NAC is only necessary if differentiation under modified redox homeostasis is to be investigated. NAC is soluble in dH2O after heating at 37°C for five minutes with a concentration of 100 mg/mL or 612 mM (MW 163.19 g/mol). Dilute the stock in dH2O for working concentrations of 0.5 mM (low concentration) and 2.5 mM (high concentration).
Table 2.
Storage and working concentrations of reagents used to culture and differentiate NES cells
| Reagent | Storage Concentration | Working Concentration | Storing Conditions |
|---|---|---|---|
| bFGF | 100 ng/μL | 10 ng/mL | Long term storage at −80°C |
| EGF | 100 ng/μL | 10 ng/mL | |
| B27 | Directly aliquoted | 1:100 in induction medium 2. 1:1000 in growth medium. 1:100 in differentiation medium. |
Store at -20°C |
| N2 | Directly aliquoted | 1:200 in induction medium 2. 1:100 in both growth and differentiation media |
|
| hNoggin (NG) | 500 μg/mL | 500 ng/mL (1:1000) | |
| SB431542 | 10 mM | 10 μM (1:1000) | |
| CHIR 99021 | 10 mM | 3.3 μM (1:3000) | |
| mouse laminin | Directly aliquoted from a 1-2 mg/mL stock |
4 μg/mL | |
| laminin521 | Directly aliquoted from a 0.1 mg/mL stock |
5 μg/mL | |
| 2-mercaptoethanol | 50 mM | 90 μM | Store at 4°C |
| GlutaMAX | Directly aliquoted | 1:100 | |
| N-Acetyl-L-cysteine (NAC) | 612 mM | 0.5 mM/2.5 mM | |
| KnockOut Serum Replacement (KOSR) | Directly aliquoted | 1:5 | Store at −20°C (for long term storage) |
| Poly-L-Ornithine | 100 μg/mL | 100 μg/mL | |
| BSA | 200 mg/mL | 2 mg/mL | |
| Penicillin-Streptomycin (P/S) | 10,000 U/mL | 100 U/mL (1:100) | |
| ROCK inhibitor | 5 mM | 5 μM (1:1000) | |
| E8 supplement | Directly aliquoted | 1:50 |
Media for NES cell derivation, culture, and differentiation
-
10.Wash medium
-
a.Prepare a 200 mg/mL BSA stock solution in DMEM/F12 media.
-
b.Wash medium is prepared by adding 5 mL of BSA stock solution to 500 mL of DMEM/F12 to have a BSA working concentration of 2 mg/mL.
-
a.
- 11.
-
12.NES growth medium
-
a.Prepare NES growth medium according to Table 5.
-
a.
-
13.NES differentiation medium
-
a.Prepare NES differentiation medium according to Table 6.
-
b.NES differentiation medium consists of DMEM/F12 medium supplemented with N2 (1:100), B27 (1:100). Addition of P/S is optional (1:100). Follow NES cells establishment guidelines for correct storage of factors and medium.
-
c.See Table 2 for storage and working concentrations as well as storage conditions of all factors.
-
a.
Table 3.
Composition of neural induction medium-1
| KOSR-medium (medium 1) | Volume for 50 mL | Concentration |
|---|---|---|
| DMEM/F-12+GlutaMAX | 39.5 mL | - |
| KnockOut Serum Replacement (KOSR) | 10 mL | 1:5 |
| Non-essential Amino Acids | 500 μL | 1:100 |
| 2-mercaptoethanol | 91 μL | 1:550 |
| P/S | 500 μL | 1:100 |
Table 4.
Composition of neural induction medium-2
| N2B27-medium (medium 2) | Volume for 50 mL | Concentration |
|---|---|---|
| DMEM/F-12+GlutaMAX | 24 mL | - |
| Neurobasal | 24 mL | - |
| GlutaMAX | 250 μL | 1:100 (for Neurobasal) |
| 2-mercaptoethanol | 91 μL | 1:500 |
| N2 | 250 μL | 1:200 |
| B27 | 500 μL | 1:100 |
| P/S | 500 μL | 1:100 |
Table 5.
Composition of NES growth medium
| NES medium | Volume for 50 mL | Concentration |
|---|---|---|
| DMEM/F-12+GlutaMAX | 48.5 mL | - |
| N2 | 500 μL | 1:100 |
| B27 | 50 μL | 1:1000 |
| P/S | 500 μL | 1:100 |
| bFGF | 5 μL | 1:10 000 (10 ng/mL) |
| EGF | 5 μL | 1:10 000 (10 ng/mL) |
Table 6.
Composition of NES differentiation medium
| NES differentiation medium | Volume for 50 mL | Concentration |
|---|---|---|
| DMEM/F-12+GlutaMAX | 49 mL | - |
| N2 | 500 μL | 1:100 |
| B27 | 500 μL | 1:100 |
| P/S | 500 μL | 1:100 (optional) |
Preparation of stock solutions and media for NES cell nucleofection
-
14.
Aliquot buffer P3 from the Lonza nucleofection kit. Lonza offers several buffers to perform the nucleofection in P3 is the most efficient for NES cells.
-
15.
Measure plasmid DNA concentration and prepare the right amount in a small volume so buffer P3 composition is not altered. Try different concentrations of DNA to identify the one working the best for your experimental set-up.
-
16.NES growth medium
-
a.Follow NES cells growth procedure for reagents, factors preparation, and medium preparation.
-
a.
CRITICAL: Avoid repeated freeze-thaw cycles of frozen aliquots of EGF and bFGF. The volume of the aliquots should be determined according to the average volume used during a week.
Note: The reagents are not toxic or harmful to the user and do not require any special handling.
Coating of plastic dishes for iPS cell culture
-
17.Coating of cell culture plates with laminin521 for iPS cell culture
-
a.Dilute laminin521 to a working concentration of 5 μg/mL in PBS (we use PBS without Ca2+and Mg2+, but it should also be possible to use PBS with Ca2+and Mg2+) and add appropriate volume to each well as indicated in Table 1.
-
b.Incubate freshly coated plates at 4°C for at least 12 h. We recommend to seal plates and dishes with parafilm for extended storage (>24 h at +4°C).
-
a.
Table 1.
Volumes for different cell culture dishes to grow NES cells and coat culture dishes
| Culture dish | Growth area (cm2) | Volume of coating | Volume of medium |
|---|---|---|---|
| 24 well plate | 2 | 0.35 mL | 0.5 mL |
| 12 well plate | 3.5 | 0.5 mL | 0.6 mL |
| 6 well plate | 9.5 | 1.5 mL | 1.5–2 mL |
| T25 flask | 25 | 4 mL | 4–5 mL |
| T75 flask | 75 | 10 mL | 9–10 mL |
| T-150 flask | 150 | 20 mL | 18–20 mL |
Coating of plastic dishes for freshly established NES cells
-
18.Coat cell culture plates with Poly-L-Ornithine
-
a.Resuspend Poly-L-Ornithine in PBS for a working concentration of 100 μg/mL. Add Poly-L Ornithine-PBS solution to the culture plates and incubate for at least 2 h at 37°C (see Table 1 for volumes according to culture dish).
-
a.
-
19.Coat cell culture plates with mouse laminin and laminin521
-
a.Wash Poly-L-Ornithine coated plates three times with generous amounts of PBS (≥ 1 mL per 3 cm plate).
-
b.Dilute mouse laminin (4 mg/mL working concentration) and laminin521 (5 mg/mL working concentration) in PBS. Add appropriate volume of laminin dilution (see Table 1) into each well of the culture plate.
-
c.Incubate the plate for at least 12 h at +4°C or until they are needed. Alternatively, incubation at 37°C for at least 2 h is also possible, but coating will be less efficient and is not suitable for prolonged culturing (adequate volumes for different areas are indicated in the materials and equipment section).
-
a.
Coating of plastic dishes for established NES cell culture and NES cell differentiation
- 20.
-
21.Coat cell culture plates with mouse laminin
-
a.Wash Poly-L-Ornithine coated plates three times with generous amounts of PBS (≥ 1 mL per 3 cm plate).
-
b.Dilute mouse laminin to a working concentration of 4 μg/mL in PBS, add appropriate volume to each well as indicated in Table 1 and incubate the plates for at least 12 h at +4°C or until they are needed.
-
c.We recommend to seal plates and dishes with parafilm for extended storage (>24 h at +4°C).
-
a.
Coating of plastic dishes for nucleofection of NES cells
-
22.
Follow NES cell culture guidelines to coat plastic dishes and prepare the number of dishes required to perform the experiment.
-
23.
Prepare the appropriate number of coated culture dishes. When using strips for nucleofection it is recommended to seed each 16-strip well to a single well of a 24 multi-well plate. If cuvettes are used, we recommend using T25 flasks or equivalent plates per cuvette.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-β-tubulin III Working dilution, 1:1000 |
Merck | Cat T2200. RRID:AB_262133 |
| Rabbit monoclonal anti-SOX2 (D6D9) Working dilution, 1:1000 |
Cell Signaling | Cat #3579 RRID:AB_2195767 |
| Mouse monoclonal anti-Nestin Working dilution, 1:500 |
Millipore | MAB5326 RRID:AB_11211837 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM/F-12, GlutaMAXTM supplement | Thermo Fisher Scientific | 31331-028 |
| N-2 Supplement (100×) | Thermo Fisher Scientific | 17502-048 |
| B27TM Supplement (50×), Serum free | Thermo Fisher Scientific | 17504-044 |
| B27TM Supplement minus antioxidants (B27-MA ; 50×) Serum free | Thermo Fisher Scientific | 10889038 |
| Poly-L-ornithine hydrobromide | Merck | P3665. CAS Number: 27378-49-0 |
| Laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane | Merck | L2020. CAS Number: 114956-81-9 |
| Soybean Trypsin Inhibitor, powder | Thermo Fisher Scientific | 17075-029 |
| Human bFGF Recombinant Protein | Thermo Fisher Scientific | 13256-029 |
| Animal-Free Recombinant Human EGF | PeproTech | AF-100-15 Accession number: P01133 |
| N-Acetyl-L-cysteine | Merck | A9165 CAS Number: 616-91-1 |
| Essential 8 Medium Kit (E8) (Basal medium and supplement; 50×) | Thermo Fisher Scientific | A1517001 |
| ROCK Inhibitor (Y-27632) | Merck | SCM075 |
| Human recombinant laminin 521 | BioLamina | LN521-02 |
| KnockOut™ Serum Replacement | Thermo Fisher Scientific | 10828028 |
| Non-essential Amino Acids 100× | Thermo Fisher Scientific | 11140035 |
| 2-Mercaptoethanol 50 mM | Thermo Fisher Scientific | 31350010 |
| Penicillin-Streptomycin 10,000 U/mL | Thermo Fisher Scientific | 15140122 |
| Neurobasal™ Medium | Thermo Fisher Scientific | 21103049 |
| TrypLE™ Select Enzyme (1×) | Thermo Fisher Scientific | 12563011 |
| Recombinant Human Noggin | PeproTech | 120-10C |
| SB431542 | Merck | S4317-5MG |
| CHIR99021 | Merck | SML1046-5MG |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | 14190169 |
| BSA | Merck | A7030 |
| GlutaMAX™ Supplement | Thermo Fisher Scientific | 35050061 |
| Experimental models: cell lines | ||
| Human neuroepithelial stem cells | Falk Lab | N/A |
| Induced pluripotent stem cells | Falk lab | N/A |
| Critical commercial assays | ||
| Lonza P3 Primary Cell 4D-Nucleofector Kit S or L | Lonza (Fisher Scientific) | V4XP-3024 or -3032 (Lonza) 13429329 or 13439329 (Fisher Scientific) |
| Other | ||
| Plastic dishes | Sarstedt | N/A |
| Cryo vials | VWR | 479-1261P |
| 12-Well plates | VWR | 734-2324 |
| 4D-Nucleofector | Lonza | AAF-1002B |
Materials and equipment
-
•
All reagents mentioned in Table 2 can be stored in the conditions above for at least 1 year or as long as indicated by the manufacturer.
-
•
All buffers and reagents for the nucleofection can be stored in −20°C.
Step-by-step method details
Neural induction – derivation of NES cells from iPS cells
The following section describes how to culture iPSCs using laminin521 coating and E8 media, which is our preferential procedure.
Note: Other culture methods for stable culture of iPSCs are available. Please refer to the appropriate protocols if you wish to pursue them.
Culturing iPSCs
-
1.Culture iPS cells until a sufficient cell number for the intended experiment is reached.
-
a.Seed 17 500 – 25 000 cells/cm2 iPS cells (see Table 8) in a plate coated with laminin521.
-
b.Culture iPS cells in complete E8 media containing the E8 supplement (1:50) and P/S (1:100). Split the cells once they reached 80% confluency (after 3–4 days).
-
c.Change the media every day.
-
d.Add 1:1000 ROCK inhibitor to the media after every splitting and thawing.
-
a.
Table 8.
Suggested cell numbers for different cell culture dishes to grow iPS cells at day 0 (D0) and day 5 (D5).
| Culture dish | Cell numbers for seeding D0 and for stable NES cells (∗106) | Cell numbers for splitting D5 (∗106) |
|---|---|---|
| 24 well plate | 0.075–0.080 | 0.380–0.542 |
| 12 well plate | 0.150–0.200 | 0.700–1.000 |
| 6 well plate | 0.375–0.390 | 1.920–2.736 |
| T25 flask | 0.970–0.990 | 5.000–7.125 |
Neural induction
Day 0
-
3.
Split the iPS cells and seed according to the number of cells in 2-3 wells of your culture dish of choice.
-
4.
Plate the cells in an appropriate volume of complete Essential 8 media (E8 incl. P/S and E8 supplement).
-
5.
Add ROCK inhibitor 1:1000 to the media.
Day 1
Start of neural induction
-
6.
Warm an appropriate volume of KOSR media to 37°C.
-
7.
Prepare the complete KOSR media by adding the factors according to Table 3.
-
8.
Change the media in all wells to KOSR media, according to Table 9.
Table 9.
Media composition for each day of neural induction
| Day | KOSR Media | N2B27 Media | Factors |
|---|---|---|---|
| 1–4 | 100% | 0% | NG 1:1000 SB 1:1000 CHIR 1:3000 |
| 5–6 | 75% | 25% | NG 1:1000 CHIR 1:3000 |
| 7–8 | 50% | 50% | NG 1:1000 CHIR 1:3000 |
| 9–10 | 25% | 75% | NG 1:1000 CHIR 1:3000 |
| 11–12 | 0% | 100% | CHIR 1:3000 |
Day 2–4
-
9.
Change the media daily as stated above (see Table 9).
Day 4
-
10.
Coat double the number of wells used before in a fresh culture dish of the same size with laminin521. Keep refrigerated for ≥ 12 h at 4°C.
Day 5
Split cells to a fresh plate
-
11.
Prepare an appropriate volume (see Table 1) of KOSR/N2B27 media (see Tables 3, 4, and 9). Prepare 3 mL extra.
-
12.
Wash the cells once with PBS. Ensure that all PBS is removed after washing.
-
13.
Add 500 μL of TrypLE Express (500 μL are sufficient for culture dishes up to T25) to each well.
-
14.
Incubate at 37°C for maximum 3 min. Rounding and detachment of the cells from the culture surface can be confirmed under a standard light microscope.
-
15.
Meanwhile, prepare a sterile 15 mL tube with an equal volume of KOSR media to TrypLE used.
-
16.
Transfer the detached cells from the culture dish to the 15 mL tube with KOSR media.
-
17.
Centrifuge for 3 min at 300 g.
-
18.
Carefully aspirate the media without touching the pellet.
-
19.
Resuspend the cell pellet in 1–2 mL KOSR/N2B27 media and count the cells.
-
20.
Seed the cells with a density of 2–2.85 × 105 cells/cm2 and according to Table 8.
-
21.
Add ROCK inhibitor 1:1000 to the media.
Day 6-Day 11
-
22.
Change the media every day according to media composition in Table 9.
Day 11
Coat the necessary plates following NES cell culture section + additional laminin521 for splitting at day 12. Prepare the same type of plate as currently used for the cells. i.e., 12-well plate.
-
23.
Dilute an appropriate volume of poly-L-Ornithine 1:500 in PBS, according to Table 1.
-
24.
Incubate for at least 2 h at 37°C.
-
25.
Remove the Poly-L-Ornithine and wash each well with the volume of PBS stated in Table 1.
-
26.
Dilute mouse laminin for a final concentration of 4 μg/mL and laminin521 for 5 μg/mL in PBS (see Table 2 for working concentrations).
-
27.
Add an appropriate volume (see Table 1) of the diluted laminin solution to each well.
-
28.
Wrap the edges in parafilm and incubate for ≥ 12 h at +4°C.
Day 12
-
29.
Remove the media and wash the cells with PBS.
-
30.
Remove the PBS and add 500 μL of TrypLE to each well.
-
31.
Incubate at 37°C for 2–3 min.
-
32.
Transfer the detached cells from the wells into a tube with the same volume of NES media as the volume of TrypLE.
-
33.
Spin for 3 min at 300 g.
-
34.
Aspirate the supernatant carefully and without touching the cell pellet.
-
35.
Resuspend the cells in 1 mL of NES growth medium (Table 5) and count the cells.
-
36.
Plate 1 ∗106 cells per well (12 well plate) in a Poly-L-Ornithine- and laminin (mouse laminin and laminin521) coated 12 well plate.
-
37.
Add ROCK inhibitor 1:1000.
Establishment of NES cells
See “before you begin” for required stock solutions and media
Day 1–3
-
38.
Wash the newly established NES cells with PBS before changing the media for the first three days after neural induction.
-
39.
Exchange the media daily with fresh NES growth media (see Table 5). See Table 1 for details about the suggested medium volumes.
Day 4–5
-
40.
Change the media every day to fresh NES growth media (see Table 5 for media composition and Table 1 for medium volumes).
Day 6 and after the first passage
-
41.
Split the cells when they reached a confluency of 100%.
-
42.
Add ROCK inhibitor 1:1000 for this passage.
-
43.
Seed the cells onto normal NES cell plates (Poly-L-Ornithine and mouse laminin) (see section “Before You Begin” above.)
-
44.
Passage the cells 1:2 for 5 passages. After this the cells should be seeded with a density of 4 × 104 cells/cm2 and without adding ROCK inhibitor to the media.
-
45.
Exchange the media daily with fresh NES growth media (see Table 5 for medium composition and Table 1 for suggested volumes).
-
46.
Monitor the newly established cell lines for the formation of typical NES cell clusters in order to confirm the establishment of NES cells. NES cells will form small rosettes and express NES cell markers, such as NESTIN and SOX2 (Figure 1).
-
47.
Freeze several aliquots of the newly established NES cell line. For cryopreservation see the section below.
Figure 1.

NES cell characteristics
(A). Representative bright-field image of a healthy control NES cell line after 7 passages growing in the typical rosette shapes. Scale bar: 200 μm.
(B). Immunofluorescence analysis of an established healthy control NES cell line expressing the neural stem cell makers SOX2 (green) and NESTIN (red). Cell nuclei are shown in blue. Scale bar: 100 μm.
NES cell culture
Thawing of NES cells
-
48.
Frozen aliquots of NES cells should be rapidly thawed in a 37°C water bath for not longer than 30 seconds.
-
49.
Add 0.5 mL of warm wash medium dropwise to the vial the cells are in.
-
50.
Transfer the thawed NES cells to a fresh 15 mL sterile tube and add wash medium 5× the volume (see Table 1).
-
51.
Centrifuge the cell suspension for 3 min at 300 g and carefully remove the supernatant by pipetting or aspiration.
-
52.
Carefully resuspend the cells in 1 mL of wash media and count them.
-
53.
Calculate how many cells to seed according to the used cell culture plate (see Table 10). Increase the cell number slightly for this step.
-
54.
Add an appropriate volume for the planned culture plate of NES growth medium according to Table 1.
-
55.
Remove the laminin solution from the warm tissue culture plates by aspiration and rapidly seed the cells with an appropriate volume of NES growth medium.
-
56.
Replace the NES cell growth medium daily to ensure successful culturing and proliferation.
Table 10.
Required number of NES cells to be seeded after splitting or thawing
| Culture plate | Growth area (cm2) | Seeding number (∗106) |
|---|---|---|
| 24-MW | 2 | 0.1 |
| 12-MW | 3.8 | 0.15 |
| 6-MW | 9.5 | 0.5 |
| T-25 | 25 | 1 |
| T-75 | 75 | 3 |
| T-150 | 150 | 6 |
Maintenance of NES cells
-
57.
Replace the NES growth medium daily.
-
58.
Split the cells once they reached 100% confluency. This can take between 3–5 days, depending on the seeding density and the cell line.
-
59.
The stated cell numbers in Table 10 are suitable for normal NES cell expansion. For experimental set-ups like immunocytochemistry or electrophysiological recordings cell numbers must be adapted according to the needed cell density. The recommended cell density for NES cell maintenance is 4 × 104 cells/cm2.
Splitting of NES cells
-
60.
Remove all NES growth medium from the culture dish by slightly tilting the plate to one side. Add an appropriate volume of pre-warmed TrypLE select for 2–3 min to the cells.
-
61.
After incubation gently pipette the cell suspension to detach the remaining cells. If cells remain attached, a slight tapping to the side of the plates or an additional incubation for 1 min at 37°C can be performed.
-
62.
Transfer the cell suspension to a new 15 mL tube containing 5× the volume of wash media and centrifuge for 3 min at 300 g.
-
63.
Carefully remove the supernatant by pipetting, aspiration, or decanting, and resuspend the cell pellet in 5 volumes of wash medium (see Table 11).
-
64.
Centrifuge the cells for 3 min at 300 g.
-
65.
Count the cells.
-
66.
Remove the wash medium as above and carefully resuspend the cells in NES growth medium before seeding the cells at an appropriate concentration on coated plates (Table 10).
-
67.
Aspirate the mouse laminin dilution from the wells before adding the cells.
Table 11.
Volumes for different cell culture dishes to grow NES cells
| Culture dish | Volume of TrypLE select | Volume of wash medium |
|---|---|---|
| 24 well plate | 0.25 mL | 1.25 mL |
| 12 well plate | 0.5 mL | 2.5 mL |
| 6 well plate | 0.5 mL | 2.5 mL |
| T25 flask | 1 mL | 5 mL |
| T75 flask | 2.5 mL | 12.5 mL |
| T-150 flask | 5 mL | 25 mL |
Cryopreservation of NES cells
NES cells can be cryopreserved in NES growth medium, supplemented with 10% DMSO (NES freezing medium). Avoid using serum from animal origin. NES cells should be frozen slowly in appropriate freezing containers at −80°C and transferred to the vaporous phase of a liquid nitrogen tank or equivalent 24 h after freezing.
-
68.
Follow the guidelines for splitting of NES cells, but instead of a final resuspension in NES cell growth medium, resuspend the cell pellet in NES freezing medium in a concentration ranging from 2–5 x106 cells/mL and transfer to a cryotube.
Neural differentiation of NES cells
Change the medium for at least 14 days for a completely developed neuronal morphology. At least 45 days for a mixed culture of neurons and glia. Differentiated cells can be cultures for at least 2 months.
In order to induce neuronal differentiation NES growth medium is modified by removing growth factors bFGF and EGF and increasing B27 concentration 10 times (Falk et al., 2012; Koch et al., 2009). We assigned the name of NES differentiation medium to this medium (see Table 6). As it was indicated above for NES cell culture section, it is also necessary to pre-warm NES differentiation medium.
Day 0
-
69.
Culture NES cells according to the above guidelines.
-
70.
Seed NES cells in NES growth media with a density of 40 000 cells/cm2.
Day 1
-
71.
Remove NES growth medium by slightly tilting the culture plate and replace it with an equal volume of NES differentiation medium (see Table 1).
Day 2–7
-
72.
Replace half of NES differentiation medium every other day without tilting the plate.
Day 7 until intended day of differentiation
-
73.
Carefully replace half of NES differentiation medium every other day.
-
74.
Add mouse laminin (1:500) to the media for every feeding
Modulation of neuronal differentiation through redox homeostasis
The growth conditions mentioned above are sufficient for NES cell culturing and differentiation. One should be aware that redox homeostasis is tightly coupled to stem cell differentiation and function (Li et al., 2019 and Nugud et al., 2018). We here present consequences on NES cell differentiation, when antioxidant levels differ from those recommended in the standard NES differentiation medium mentioned above.
The following protocol compares differentiation potential using standard B27 (as above) or B27 minus antioxidants (B27-MA) in the NES cell differentiation medium in the presence or absence of the antioxidant N-Acetyl-L-cysteine (NAC). B27-MA is lacking antioxidant power: vitamin E, vitamin E acetate, superoxide dismutase, catalase, and glutathione. Thus, we can study how the lack of antioxidant supplementation in the medium is affecting neuronal differentiation.
-
75.
Follow NES cells establishment guidelines for correct storage of factors and medium. B27-MA is stored and aliquoted as B27.
-
76.
Prepare NES differentiation medium with B27 or B27-MA (see Table 7).
-
77.Prepare 6 wells to evaluate the effect of antioxidants on neuronal differentiation:
-
a.B27 no NAC
-
b.B27 + 0.5 mM NAC
-
c.B27 + 2.5 mM NAC
-
d.B27-MA no NAC
-
e.B27-MA + 0.5 mM NAC
-
f.B27-MA + 2.5 mM NAC
-
a.
Table 7.
Volume and working concentrations of reagents used to differentiate NES cells and modulate redox homeostasis
| NES differentiation medium+ antioxidant | Volume for 50 mL | Concentration |
|---|---|---|
| DMEM/F-12+GlutaMAX | 49 mL | - |
| N2 | 500 μL | 1:100 |
| B27/B27-MA | 500 μL | 1:100 |
| P/S | 500 μL | 1:100 (optional) |
| NAC | 40.85 μL (0.5 mM) or 204.25 μL (2.5 mM) | 1:1224 (0.5 mM) or 1:244.8 (2.5 mM) of a 0.612 M stock |
Neural induction medium 1 and 2 should be prepared fresh every day. NES growth medium, NES differentiation medium and NES differentiation medium + antioxidants can be stored at 4°C for 1 week.
Culture for at least 14 days for a completely developed neuronal morphology.
Day 1
-
78.
Plate the cells as mentioned in the NES cell differentiation part. Remove NES growth medium 24 h after seeding of the cells by slightly tilting the culture plate and replace it with an equal volume of the 6 different NES differentiation mediums mentioned above.
Day 2–14
-
79.
Replace half of NES differentiation medium every day.
-
80.
Add mouse laminin to the media every time the media gets changed.
Nucleofection of NES cells
We noticed extensive NES cell death after transfecting NES cells with Lipofectamine 3000 (Thermo Fisher Scientific) and therefore explored nucleofection as transfection method.
-
81.
Collect 2.5 × 105 NES cells for one well in a 16-strip and 2 × 106 for one cuvette according to NES cell culture protocol above but pellet cells by slow centrifugation at 80 g for 10 min.
-
82.
Resuspend cells in 20 μL of pre-warmed P3 buffer for one strip well and 100 μL for one cuvette.
-
83.
Add 0.6 μg and 5 μg of the desired DNA for the strips and cuvettes, respectively.
-
84.
Transfer the NES cells-DNA mix into the strip or the cuvette.
-
85.
Place the strips or cuvette into the nucleofector and run program DC100 (this is the most efficient program to nucleofect NES cells).
-
86.
After nucleofection let the cells recover for 10 min at 21°C to reduce cell death. Gently transfer the transformed cells to NES growth medium and seed them in a coated 24 multi-well plate (strip) or T25 flask (cuvette). Add ROCK inhibitor (1:1000) for better survival of the cells.
-
87.
After 24 h, change medium according to NES cell culture conditions (see Table 1 (volume) and Table 5 (media composition)).
-
88.
For stable line generation, antibiotics can be incorporated into the medium after 48 h. For transient experiments the peak of expression is reached at 48–72 h.
Expected outcomes
Albeit sensitive to culture conditions, NES cells are highly proliferative and can provide an almost unlimited source of cells of a neuronal cell lineage.
To verify the cell lineage, check for expression of the NES cell markers SOX2 and NESTIN by immunohistochemistry, FACS or qPCR. Additionally, the newly established NES cell lines should present the typical rosette shape (Figure 1).
During the first 4 days NES cells will arrest, after which (day 5–6) characteristic projections of neuronal morphology will appear. Cells will continue to differentiate and cluster in groups of cell bodies with bundled projections connecting the different clusters. After approximately 45 days of differentiation glial cells are formed. Representative images of neural differentiation are shown in Figure 2A.
Figure 2.

NES cells can efficiently differentiate into neural cells
(A) Representative bright-field image of a healthy control NES cell line differentiated for 51 days. After onset of differentiation, NES cells progressively assume a more branched morphology with neurites growing out from the cell body. The more differentiation progresses the more the cell bodies cluster together.
(B) To monitor neuronal differentiation in a healthy control cell line, cells can be immune-stained with B-III-Tubulin (green).
(C) Example of a differentiation deficient mutant NES cell line (1). Scale bar: 200 μm.
The outcome of differentiation can be tested by immunohistochemistry e.g., with the neuronal marker β-III tubulin (Figure 2B). Other neuronal markers might also be of use, but we recommend using β-III tubulin for a quick determination of the quality of differentiation. We also recommend monitoring the decline of stem cell markers, such as SOX2, during differentiation, since mature neurons should present very low levels in comparison to NES cells. After approximately 45 days of differentiation astrocytes expressing typical glial markers can be detected in the differentiating cells.
A failed differentiation attempt can be seen in Figure 2C.
Neuronal differentiation is blocked by the absence of antioxidants in NES differentiation medium with B27-MA at day 11 followed by neuronal cell death at day 12. NES differentiation medium with B27-MA supplemented with low NAC concentrations (0.5 mM) rescues this block allowing neurons to completely develop. High concentration NAC (2.5 mM) supplemented NES differentiation medium speeds up neuronal differentiation but failed to keep neurons alive inducing neuronal cell death at day 7 (Figure 3).
Figure 3.

Neuronal differentiation potential in standard NES cell differentiation media or media with high or low antioxidant capacity
Representative B-III-Tubulin staining (green) of NES cells at different points of differentiation (day 5 and day 12). Differentiation was started with either standard B27 (B27), or with B27 deprived of its antioxidant components (B27-MA). Moreover, differentiation medium was supplemented with either 0.5 mM or 2.5 mM N-Acetyl Cysteine (NAC), a commonly used antioxidant molecule. The complete removal of antioxidant power from the B27 results in non-viable differentiated cells. Addition of 0.5 mM NAC efficiently rescues cell death, but an increase of NAC concentration to 2.5 mM eventually leads to complete loss of the culture, suggesting that a precise balance between oxidant and antioxidant actions is required for normal differentiation. Scale bar: 100 μm.
Limitations
NES cells present a few limitations that are intrinsic to the model. Their stemness properties result in a broad spectrum of differentiation outcomes, eventually leading to a mixed culture of different subtypes of neurons and glia. It is possible to direct differentiation to a neuronal subtype of interest using alternative differentiation protocols, but we have not presented this here.
Moreover, it is important to use standardized culturing conditions, such as 5% CO2 and 21% O2, in order to have comparable cell models. If NES cells are not cultured under such conditions variability among cell lines can be introduced. The use of multiple lines, for both control and patient cell lines, is recommended to ensure reproducibility.
It should be considered that the isolation and culturing of NES cells might lead to clonal selection during the process due to the culturing conditions used, like cell density. It is therefore recommended to use the culture conditions discussed in this protocol. Always check the quality of your NES cell cultures before they are used in experiments.
The NES cell system is reductionistic, thus, it might be difficult to study interactions with other cell types similar to the in vivo situation. NES cells are generated from iPS cells and the reprogramming process to establish the iPS cells includes erasing the epigenetic landscape. The derived NES cells might not display the same epigenetic markers present in the individual donated cells for reprogramming.
It is difficult to study complex cell-cell interaction with other cell types as well as the impact of complex environmental factors, since these cells are kept in a monolayer culture.
This protocol describes 2D culture of NES cells. If the interactions of different neural cell types should be studied in 3D we recommend using protocols for brain organoids.
Using NES cells and differentiated cells in a 2D culture comes with the limitation of not knowing how and if a specific genotype translates into a specific behavior. For investigations regarding research like this a combined in vitro in vivo research approach is necessary.
Troubleshooting
Problem 1
NES cells detach from the dish (step 41–44)
Potential solution
Residual Poly-L-Ornithine or the absence of mouse laminin will prevent NES cells from attaching to the growth surface. We recommend at least three washes after Poly-L-Ornithine coating with PBS. A mouse laminin concentration of 2 μg/mL was only partially sufficient to maintain NES cell growth. We therefore suggest a 4 μg/mL concentration, which significantly improved NES cell growth. In addition, while preparing culture plates for NES cell culture, 12 h incubation at +4°C with mouse laminin showed better results in comparison to 2 h at 37°C. We also recommend adding fresh laminin (4 μg/mL concentration) for every feeding staring from day 7 of NES cell differentiation. Make sure that an appropriate culture dish is used (see key resource table).
Problem 2
Excessive cell death after trypsinization or during splitting (step 41–44)
Potential solution
Pre-warm TrypLE select, wash medium and NES growth medium and avoid excessive pipetting of NES cells. Never trypsinize the cells for more than 3 min and proceed with the splitting procedure as quick as possible so the cells don't have to stay outside of a culture plate for longer period of times. Aspirate laminin coating right before seeding the cells, and do not let the dish dry out. Double check time and speed of centrifugation.
Problem 3
NES cells remain quiescent (step 71–74)
Potential solution
Double check factors concentration and the dilution used to prepare the aliquots. Do not store the factors at +4°C. Use fresh bFGF, EGF and B27 to complete NES growth medium to improve factors activity. Avoid constant opening and closing of the incubator so the temperature and CO2 concentrations remain constant. It might also help to change only half of the media for every media change until the cells recovered. Check for the right seeding density. Seeding at a too low density can influence the cells proliferation rate.
Problem 4
Excessive cell death during neuronal differentiation (step 71–74)
Potential solution
Low cellular density can induce a quiescent state and cell death in NES cells. To achieve a successful neural differentiation, the confluences stated in this protocol should be used. Aberrant differentiation and high rates of cell death are expected below that percentage. In the beginning of the differentiation process cell death to some extend is expected.
Problem 5
Excessive cell death during nucleofection (step 81–88)
Potential solution
Increasing the number of cells and/or decreasing the amount of DNA used can reduce cell death. Avoid excessive pipetting and pre-warm NES growth medium. Adding ROCK inhibitor to the media before and after the nucleofection can help the survival rate as well.
Problem 6
Pelleting of NES cells after harvesting with a scraper (step 60–63)
Potential solution
Alternative to TrypLE trypsinization, cells can be harvested using a cell scraper. We recommend taking extra care as cells will readily detach. Preferably, harvest in NES growth medium followed by a PBS wash and a new centrifugation at 300 g for 3 min.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anna Wredenberg MD, PhD, anna.wredenberg@ki.se
Material availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze [datasets/code].
Acknowledgments
This study was supported by the Swedish Research Council (A.Wr. [VR2016-02179] and A.F. [VR2019-01498]). We thank the iPS Core at Karolinska Institutet.
Author contributions
J.C.-G., D.W., and C.M. conceived, planned, and carried out the experiments and wrote the manuscript. A.We., C.F., A.F., and A.Wr. conceived, planned, contributed to the definitive version of the manuscript, and provided financial support for the project leading to this publication.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Javier Calvo-Garrido, Email: javier.calvo.garrido@ki.se.
Anna Falk, Email: anna.falk@ki.se.
Anna Wredenberg, Email: anna.wredenberg@ki.se.
References
- Calvo-Garrido J., Maffezzini C., Schober F.A., Clemente P., Uhlin E., Kele M., Stranneheim H., Lesko N., Bruhn H., Svenningsson P. SQSTM1/p62-directed metabolic reprogramming is essential for normal neurodifferentiation. Stem Cell Rep. 2019;12:696–711. doi: 10.1016/j.stemcr.2019.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A., Koch P., Kesavan J., Takashima Y., Ladewig J., Alexander M., Wiskow O., Tailor J., Trotter M., Pollard S. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One. 2012;7 doi: 10.1371/journal.pone.0029597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P., Opitz T., Steinbeck J.A., Ladewig J., Brüstle O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. U S A. 2009;106:3225–3230. doi: 10.1073/PNAS.0808387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Choi E.H., Han I. Regulation of redox homeostasis by nonthermal biocompatible plasma discharge in stem cell differentiation. Oxid. Med. Cell. Longev. 2019;2019:2318680. doi: 10.1155/2019/2318680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugud A., Sandeep D., El-Serafi A.T. Two faces of the coin: minireview for dissecting the role of reactive oxygen species in stem cell potency and lineage commitment. J. Adv. Res. 2018 doi: 10.1016/j.jare.2018.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze [datasets/code].