

Abstract
Single-cell transcriptomic analysis is widely used to study human tumors. However it remains challenging to distinguish normal cell types in the tumor microenvironment from malignant cells and to resolve clonal substructure within the tumor. To address these challenges, we developed an integrative Bayesian segmentation approach called CopyKAT (Copynumber Karyotyping of Aneuploid Tumors) to estimate genomic copy number profiles at an average genomic resolution of 5Mb from read depth in high-throughput scRNA-seq data. We applied CopyKAT to analyze 46,501 single cells from 21 tumors, including triple-negative breast cancer, pancreatic ductal adenocarcinomas, anaplastic thyroid cancer, invasive ductal carcinoma and glioblastoma to accurately (98%) distinguish cancer cells from normal cell types. In three breast tumors, CopyKAT resolved clonal subpopulations that differed in the expression of cancer genes such as KRAS and signatures including EMT, DNA repair, apoptosis and hypoxia. These data show that CopyKAT can aid the analysis of scRNA-seq data in a variety of solid human tumors.
Single-cell RNA sequencing (scRNA-seq) methods have emerged as powerful tools to delineate normal cell types in the tumor microenvironment (TME) and understand the expression programs of tumor cells in a variety of human cancers1–3. The development of high-throughput sequencing technologies including microdroplet systems (Drop-Seq4, Indrop5, 10X Chromium6) and nanowells (Wafergen iCELL87, SeqWell8, CelSee7) make it possible to sequence thousands of single cells in parallel for less than $1 US per cell. However, a major challenge in the analysis of large-scale datasets is in distinguishing tumor cells from the stromal and immune cells in the TME, so that they can be studied independently. An effective approach to distinguish tumor from normal cells involves the identification of aneuploid copy number profiles, which are common (88%) in most human tumors9 and are not found in stromal cell types that have diploid genomes. Previous methods such as inferCNV3 and HoneyBadger10 have shown that it is possible to estimate genomic copy number profiles from RNA read counts at sufficiently large genomic regions. However, these methods were designed for the analysis of first-generation of scRNA-seq technologies with lower cell throughput and higher coverage depth. These methods are not suitable for the analysis of newly developed high-throughput scRNA-seq platforms (microdroplet and nanowell platforms) that perform whole transcriptome amplification (WTA) and sequence only the 3’ or 5’ end of mRNA at very sparse coverage depth. Furthermore, previous methods could not accurately resolve the genomic locations of specific chromosome breakpoints or classify tumor and normal cells from their aneuploid copy number profiles. To address these challenges, we developed CopyKAT and applied it to a variety of human tumors to identify aneuploid tumor cells and delineate the clonal substructure of different subpopulations that co-exist within the tumor mass.
Results
Overview of CopyKAT workflow
The statistical workflow of CopyKAT combines a Bayesian approach with hierarchical clustering to calculate genomic copy number profiles of single cells and define clonal substructure from high-throughput 3’ scRNA-seq data (Fig. 1, Online Methods). The workflow takes the gene expression matrix of unique molecular identifiers (UMIs) counts as input for the calculations. The analysis begins with the annotation of genes in rows to order them by their genomic coordinates. Freeman-Tukey Transformation (FTT)11 is performed to stabilize variance, followed by polynomial dynamic linear modeling (DLM)12 to smooth the outliers in the single cell UMI counts (Fig. 1a). The next step is to detect a subset of diploid cells with high confidence to infer the copy number baseline values of the normal 2N cells. To do this, we pool single cells into several small hierarchical clusters and estimate the variance of each cluster using a Gaussian Mixture Model (GMM) (Fig. 1b). The cluster with minimal estimated variance is defined as the ‘confident diploid cells’ by following a strict classification criterion. Potential misclassifications may occur when the data has only a few normal cells, or when the tumor cells have near-diploid genomes with limited CNA events. In this case CopyKAT provides a ‘GMM definition’ mode to identify the diploid normal cells one-by-one, where a mixture of three Gaussian models of gene expression in single cells are assumed to represent genomic gains, losses and neutral states. A single cell is then defined as ‘confident diploid cell’ when genes in neutral states account for at least 99% of the expressed genes.
Figure 1 – Overview of the CopyKAT analysis workflow.

a, The CopyKAT workflow begins with a UMI count matrix to order genes by their genomic positions and uses the raw count matrix to perform log-Freeman Turkey Transformation to stabilize variance and smooth outliers using a polynomial dynamic linear model. b, A subset of normal cells is defined using integrative clustering and GMM method to infer the copy number baseline. c, Relative gene expression values in single cells are used for MCMC segmentation and segments are merged by KS testing. d, Aneuploid tumor and normal cell clusters are classified using a normal cell enrichment and GMM distribution tests. e, Clonal substructure of tumor cells are delineated by clustering and subclones are used for differential expression analysis.
To detect chromosome breakpoints, we integrate a PoissonGamma model and Markov Chain Monte Carlo (MCMC) iterations to generate posterior means per gene-window and then apply Kolmogorov-Smirnov (KS) tests to join adjacent windows that do not have significant differences in their means (Fig. 1c). To speed up the calculations, we split thousands of single cells into clusters, find consensus chromosome breakpoints and merge them together to form a union of genomic breakpoints for the whole population of cells in the sample. The final copy number values for each window are then calculated as the posterior averages of all genes spanning across the adjacent chromosome breakpoints in each cell. We further convert resulting copy number values from gene space to genomic positions by rearranging genes into 220Kb variable genomic bins13 to obtain a genome-wide copy number profile for the single cells at an approximate resolution of 5Mb. The genomic resolution was estimated based on the median neighboring gene distance (~20Kb) across the genome multiplied by the gene window (25 genes) size (Online Methods, Supplementary Fig. 1a–c). We then perform hierarchical clustering of the single cell copy number data to identify the largest distance between the aneuploid tumor cells and the diploid stromal cells, however if the genomic distance is not significant we switch to the “GMM definition” model to predict single tumor cells one-by-one (Fig. 1d). Finally, we cluster the single cell copy number data to identify clonal subpopulations and calculate consensus profiles representing the subclonal genotypes for further analysis of their gene expression differences (Fig. 1e).
Evaluation of technical performance
To evaluate the performance of CopyKAT we sequenced 1,480 single tumor cells from a premalignant breast tumor (DCIS1) by high-throughput 3’ scRNA-seq (10X Genomics) (Fig. 2, Online Methods, Supplementary Table 1). We calculated the genome-wide copy number profiles from scRNA-seq data using CopyKAT (Fig. 2a–b), and compared the results to an analysis performed on the same data using a previously published method called inferCNV3 (Fig. 2c–d). To generate a ground-truth reference of the DNA copy number profile, we flow-sorted millions of aneuploid tumor cells from DCIS1 for whole genome bulk DNA sequencing (Fig. 2e–f). Our results showed that CopyKAT achieved high concordance (Pearson’s correlation = 0.82) with the bulk reference DNA copy number profile at 220Kb genomic resolution. Most of the major copy number aberrations (CNAs) detected in the bulk DNA-seq data were identified in the scRNA-seq data, including chromosomal gains in chr4, 6, 8, 12, 17, 20, X and losses in chr2, 3, 9, 10, 15 (Fig. 2a–b). We ran inferCNV3 on the same dataset and manually identified stromal cells based on the fibroblast marker genes ACTA2 and FN1, which were used to provide an internal baseline reference that is required.
Figure 2 – Comparison of bulk DNA and single cell RNA copy number profiles.

Copy number profiles estimated from scRNA-seq data for DCIS1 using CopyKAT and inferCNV. a, Clustered heatmap of 1,100 scRNA-seq copy number profiles estimated by CopyKAT. b, Line plot of the consensus of scRNA-seq copy number profiles estimated by CopyKAT where values are the median segments of all cells in the population. c, Clustered heatmap of 1,100 single tumor cell RNAseq copy number profiles estimated by inferCNV. d, Line plot of the consensus copy number profiles estimated by inferCNV. e, Heatmap of DNA copy number profile calculated from bulk DNA sequencing data from DCIS1, representing the ground truth reference profile. f, Line plot of bulk DNA-seq copy number profile from DCIS1. g, Boxplot comparing the relative distances of inferred copy numbers for all gene windows to the ground truth DNA copy number values for CopyKAT and inferCNV. h, Boxplot comparing the stability of gene interval sizes, showing the variation in averaged copy number values across different gene intervals. In g and h, ***, p-value < 0.001 of pair-wise two side t-tests comparing n= 12,167 gene windows between CopyKAT and inferCNV results. In both boxplots, the boxes are centered at median values, where the range of boxes are the inter quartile range (IQR) bounded by first quartile (Q1) and third quartile (Q3). The upper whiskers are located at the smaller of the data maximum and Q3 + 1.5 IQR, whereas the lower whiskers are located at the larger value of the data minimum and Q1 – 1.5 IQR.
We further compared the data by converting the results of inferCNV3 to the same genomic resolution as CopyKAT using 220Kb variable bins13. Although the signal of inferCNV3 was lower, these data also achieved a high concordance with the bulk DNA-seq data reference by correlation analysis (Pearson correlation = 0.79) (Fig. 2c–d). However, a major limitation of inferCNV3 was that it can only report smoothed averages of gene windows, and does not detect specific coordinates of chromosome breakpoints or copy number segments, which was achieved by CopyKAT. We further calculated the relative distance of the inferred copy number states from the two methods to the reference bulk DNA-seq copy number profile by repetitively sampling adjacent local regions of different gene size intervals (Online Methods). This analysis showed that CopyKAT segmentation results were significantly (p-value < 0.001, T-test) closer to the reference DNA copy number states, compared to the sliding window averages reported by inferCNV3 (Fig. 2g). Furthermore, we found that CopyKAT exhibited more stable performance across different sizes of gene intervals ranging from 5 to 500 genes (Fig. 2h).
We next evaluated the sensitivity and efficiency of CopyKAT in detecting chromosome breakpoints from the DCIS1 data by comparing the scRNA-seq results to a “ground truth” standard - the bulk DNA-seq copy number profile (Online Methods, Supplementary Fig. 1d). Bootstrapping was performed to resample single cells and estimate the variation in detection sensitivity. On average we estimated that 19% of CNAs were detected at 1Mb resolution, 56% at 5Mb resolution and 88% at 20Mb resolution respectively, which agrees with our theoretical calculation of a 5Mb average genomic resolution. We also determined that CopyKAT is sensitive to the segmentation parameter (KS.cut), which is set as an ad hoc pruning cutoff parameter to join adjacent chromosome segments. The breakpoint detection becomes more stringent as the KS.cut value increases, resulting in fewer breakpoints detected (Supplementary Fig.1e). We observed a significant drop in the accuracy of segmentation values when KS.cut exceeds a value of 0.3 (range: 0 to 1) (Supplementary Fig. 1f). These data suggested that CopyKAT can accurately infer DNA copy number profiles at a moderate genomic resolution (5Mb) from high throughput 3’ scRNA-seq data.
Classification of tumor and normal cells in solid tumors
We applied CopyKAT to previously published 3’ scRNA-seq data from 5 pancreatic adenocarcinoma (PDAC) patients1, as well as 3’ scRNA-seq data that we generated from 5 triple-negative breast cancer (TNBC) patients and 5 anaplastic thyroid cancer (ATC) patients to distinguish tumor and normal cells based on their copy number differences (Fig. 3). We analyzed 9,717 single cell transcriptomes from 5 PDAC patients with CopyKAT and successfully identified aneuploid tumor cell subpopulations in all of the patients (Fig. 3a and Supplementary Fig. 2a). The predicted tumor cells had genome-wide copy number aberrations (CNAs) including frequent amplifications of 1q, 3q, 7p, 8q, 17, 19, 20 and losses of 3p, 6, 8p that are commonly reported in PDAC14–16 tumors, whereas normal cells with diploid profiles had no recurrent CNAs. The UMAP projection of the aneuploid tumor cells that we classified co-localized to the expression clusters that showed high epithelial gene scores detected using a panel of four established tumor epithelial markers (EPCAM, KRT19, KRT18 and KRT8). Notably, in all 5 PDAC patients the genome-wide CNAs were only detected in one (c1) of the two epithelial clusters, suggesting the other cluster was likely normal diploid epithelial cells (c2), which could not be resolved by gene expression alone. We designated the c1 cluster as the tumor cells since they corresponded to the aneuploid copy number profiles and contained higher expression levels of KRT19, which is a widely used marker to identify cancer cells in PDAC tumors17. We estimated that CopyKAT achieved high accuracy (98.5% concordance) in the identification of tumor cells by calculating their co-localization to the c1 expression cluster (Online Methods, Supplementary Table 2). From these data we estimated the tumor purity, which ranged from 6–18% (Fig. 3d) and was consistent with previous histopathological data showing that PDAC patients generally have low tumor purity due to high stromal cell populations18–20.
Figure 3 – Classification of cancer and normal cells in human tumors.

Classification of tumor and normal cells by aneuploidy estimation with CopyKAT and mapping of the inferred profiles to scRNA-seq expression data from PDAC, ATC and TNBC tumors. a, UMAPs of scRNA-seq data from 5 PDAC tumors, with upper panels mapping the aneuploid clusters to the gene expression data, and the lower panels showing epithelial scores (average expression of four epithelial markers). Circles indicate expression clusters with high epithelial scores and include both tumor and normal epithelial cells. b, UMAPs of scRNA-seq data from 5 ATC tumors, with upper panels mapping the aneuploid clusters to the scRNA-seq gene expression data, and lower panels showing epithelial scores. c, UMAPs of 5 TNBC tumors, with upper panels mapping the aneuploid clusters to the scRNA-seq gene expression data, and the lower panels show epithelial scores. d-f, Stacked bar graph showing percentages of predicted aneuploid tumor cell and normal diploid cell purities of the d, PDAC tumors e, ATC tumors and f, TNBC tumors.
We also performed 3’ high-throughput scRNA-seq of 19,568 cells from 5 ATC tumors using the 10X Genomics platform (Online Methods, Supplementary Table 1). Analysis of the scRNA-seq data using CopyKAT resulted in the identification of aneuploid tumor cells in all of the 5 ATC tumors (Fig. 3b). Common CNAs that were frequently reported in ATC tumors 21, 22 included amplifications of 1p, 2p, 5p, 7, 8q, 11p, 12, 18p, 20 and losses of 1q, 6p, 13, 17, 22 that were identified in inferred copy number data (Supplementary Fig. 2b). In the UMAP expression data, the predicted aneuploid cells corresponded to 1–2 clusters in all patients, which showed high epithelial panel scores, including KRT8 that has previously been used to identify ATC by histopathology23. Based on the co-localization of the predicted aneuploid tumor cells to c1 (c1 and c2 in ATC1), we estimated the mean prediction accuracy for identifying tumor cells to be 97% (Online Methods, Supplementary Table 2). We observed a wide range of tumor purities (2–80%) among the ATC patients (Fig. 3e), in which two tumors (ATC2, ATC3) had low purities (2, 12%) and three patients (ATC1, ATC4, ATC5) had high tumor purities (42–80%), consistent with ranges reported in pathological data for ATC tumors24–26.
We further performed 3’ scRNA-seq of 8,944 single cells from 5 untreated TNBC patients (Online Methods, Supplementary Table 1). In all 5 TNBC samples, CopyKAT identified distinct groups of single cells with aneuploid and diploid copy number profiles. From the clustered heatmaps of the estimated single cell copy number profiles we identified frequent CNAs that have been previously reported in TNBC patients27–29, including gains of 1q, 6p, 8, 10p,18p, Xq and losses of 1p, 5q, 17p, Xp (Supplementary Fig. 2c). Dimensionality reduction showed that the predicted aneuploid tumor cells corresponded to gene expression clusters that were positive for the epithelial gene panel score, which included EPCAM that is often used to identify tumor cells in TNBC patients (Fig. 3c). In 4 tumors (TNBC1, TNBC2, TNBC4, TNBC5) the clusters (c1-c2) with positive epithelial scores corresponded directly to the predicted aneuploid clusters by CopyKAT, however in one tumor (TNBC3) only 1 out of 3 clusters that were positive for the epithelial gene panel (c1) showed genome-wide CNAs. We compared the predicted aneuploid tumor cells to the epithelial cell expression clusters (c1 in TNBC3) and estimated a high prediction accuracy across the TNBC tumors (98%). Notably, in three cases (TNBC1, TNBC2 and TNBC5) the inferred copy number profiles localized to two tumor-specific expression clusters that were both positive for epithelial gene panel expression, suggesting that there were multiple aneuploid clones present in the tumor mass. In contrast to the PDAC and ATC tumors, the TNBC samples all had high tumor purities (34–83%) across the patients (Fig. 3f and Supplementary Table 2). Collectively, these data suggest that CopyKAT can accurately (98% ± 3% S.D.) distinguish tumor and normal cells in a variety of solid tumors based on the aneuploid copy number profiles inferred from the scRNA-seq data alone, without the need of specific gene expression markers.
Application to other scRNA-seq technologies
While our data suggest that CopyKAT can accurately estimate copy number data from 3’ scRNA-seq data, we further investigated whether this approach can be applied broadly to first-generation scRNA-seq data (SMART-Seq2) as well as 5’ scRNA-seq data (10X Genomics). We performed 5’ scRNA-seq on two estrogen-receptor positive invasive ductal carcinoma (ER+ IDC) tumors (IDC1, IDC2) and analyzed SMART-Seq2 data from a previously published study30 involving 2 glioblastoma multiforme (GBM) patients (GBM1, GBM2) (Supplementary Table 1). In total 7,780 single cells were sequenced from IDC1 and IDC2 using the 5’ scRNA-seq (10X Genomics). CopyKAT analysis identified two cluster in each tumor representing normal cells (N) and tumor cells (T), in which both tumors showed a large amplification of chromosome 8p (MYC) (Fig. 4a, c). Clustering of the scRNA-seq expression data showed that the inferred aneuploid tumor cells in both tumors co-localized with the cluster that displayed high epithelial scores (Fig. 4b, d), validated the accuracy of the CopyKAT prediction. Similar to the TNBC samples, both ER+ IDC tumors showed high tumor cellularity (97% and 87% respectively).
Figure 4 – Classification of tumor and normal cells sequenced by different scRNA-seq technologies.

Clustered heatmaps of single cell copy number profiles estimated by CopyKAT from 5’ scRNA-seq data for invasive breast cancer samples (a) IDC1 and (c) IDC2, and full-length SMART-seq2 scRNA-seq data for GBM sample (e) GBM1 and (g) GBM2. CopyKAT classification of diploid normal cells (N) and aneuploid cells tumor cells (T) are indicated on the left side annotation bars. High-dimensional UMAP embedding of scRNA-seq data with annotation of the inferred CopyKAT diploid and aneuploid copy number profiles for (b) IDC1, (d) IDC2, (f) GBM1 and (h) GBM2.
Next, we analyzed a previously published scRNA-seq dataset sequenced using a first generation full-length scRNA-seq (SMART-seq2) approach30 from two GBM patients: GBM1 (MGH125) and GBM2 (MGH128) (GSE131928). In contrast to the high-throughput 3’ or 5’ scRNA-seq methods (10X Genomics) that have only partial gene body coverage, the full-length SMART-seq2 data has sequence reads that cover the whole gene transcripts. However, SMART-seq2 has much lower cell throughput (332 and 184 cells per patient respectively) and do not have UMI barcodes that can mitigate amplification bias. To perform CopyKAT analysis, we used the TPM/10 matrix that represent normalized gene expression count data. In both samples, we observed distinct separation between the clusters of aneuploid tumor cells and diploid normal cells (Fig. 4e, g). The aneuploid tumor cell cluster inferred by CopyKAT expressed high levels of EGFR (Fig. 4f, h), which is an established tumor cell marker in GBM patients31. Collectively, these data suggest that CopyKAT is compatible with a wide range of scRNA-seq technologies.
Inferring the clonal substructure of breast tumors
To delineate clonal substructure and link cancer genotypes to phenotypes, we applied CopyKAT to scRNA-seq data generated from three TNBC patients (Fig. 5). The inferred copy number profiles were clustered to identify subpopulations based on copy number differences and a consensus copy number profile was computed from the clusters of single cell profiles to identify genomic regions with copy number differences. From the consensus profiles of the subclones, we performed differential expression (DE) and gene signature analysis to identify phenotypic differences between the subclones (Online Methods).
Figure 5 – Clonal substructure of three triple-negative breast tumors.
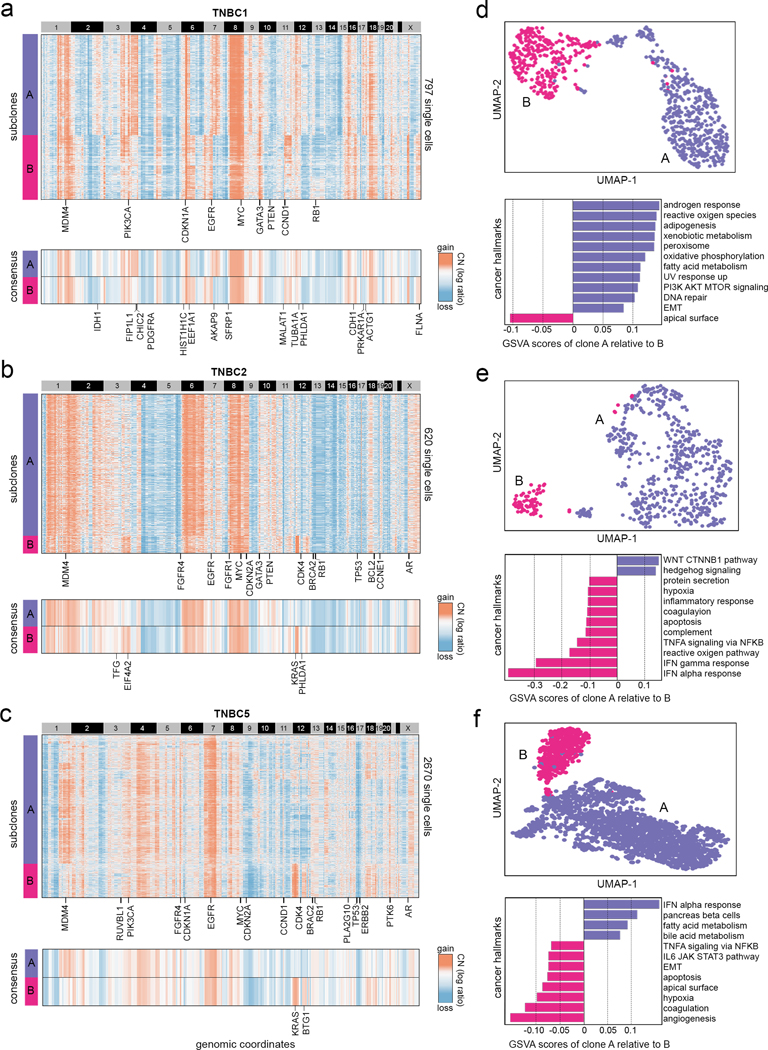
Clonal substructure of TNBC1, TNBC2, TNBC5 delineated by clustering single cell copy number profiles inferred from scRNA-seq data by CopyKAT. (a-c) The upper panels show the clustered heatmap of single cells of two major subclones in TNBC1, TNBC2 and TNBC5 with cancer genes annotated in clonal events, while the lower panels show consensus copy number profiles of the two major clones with subclonal cancer genes annotated. (d, e, f) Upper panels show UMAP projections of the scRNA-seq expression data of the two major clones in TNBC1, TNBC2 and TNBC5 with inferred aneuploid copy number profiles marked, while lower panels show GSVA analysis of the top 12 cancer hallmark signatures between the two major subclones.
In one TNBC sample (TNBC1) the clustering of 797 aneuploid copy number profiles identified two major subclones (A, B) that comprised 44% and 28% of the tumor mass (Fig. 5a upper panel) and were separate by two distinct lineages in their neighbor-joining (NJ) trees (Supplementary Fig. 3a). Clustered heatmaps identified clonal amplifications (1q, 6p, 8q, 10p, 16p 18p) and clonal deletions (1p, 4q, 5q, 8p, 10q, 13, 14) that were shared across all of the tumor cells. These genomic regions included many known breast cancer genes in TCGA32 including MDM4, PIK3CA, EGFR, MYC, GATA3, PTEN, CCND1, RB1 and other genes (Fig. 5a upper panel). The clustered heatmaps of the consensus copy number profiles revealed subclonal CNA events, including subclonal amplifications in clone A (4p, 7q, 9p13.2-q22.2, 17q) and subclonal amplifications in clone B (3p26.3-p25.1, 6q, 7p, 11q, Xp11.23, Xq) that varied in the tumor mass (Fig. 5a lower panel). DE analysis identified 329 differentially expressed genes between the two subclones (FDRadj p-value < 0.01, | log2(Fold Change) | ≥ 0.5), of which 47% were located in the subclonal CNA regions (Supplementary Fig. 4a) and included known cancer genes33 such as IDH1 on Chr2q that was overexpressed in subclone A, and CDH1 on chr16q that was overexpressed in subclone B (Fig. 5a lower panel). The two aneuploid subclones corresponded to distinct expression clusters in high-dimensional space (Fig. 5d upper panel). Single cell Gene Set Variation Analysis (GSVA)34 identified several cancer hallmark signatures that were enriched in subclone A relative to subclone B, including androgen response, Epithelial-to-Mesenchymal Transition (EMT) as well as other cancer signatures (Fig. 5d lower panel).
In another TNBC patient (TNBC2), the clustering of 620 single cell aneuploid copy number profiles inferred from scRNA-seq data resolved two subclones, including a major subclone (A) that comprised most of the tumor (53%) and a minor subclone (B) that represented a small fraction of the tumor mass (7%). NJ trees constructed from this data showed that the two subclones represented distinct clonal lineages that emerged from a common ancestor and corresponded to the clustering results (Supplementary Fig. 3b). The clustered heatmap identified many clonally amplified regions (1, 2p, 6, 8, 9p, Xq) and clonal deletions (4q, 5, 9q, 13, 14, 15, 16q, 20, Xp) that were shared among all tumor cells and encompassed known breast cancer genes including MDM4, EGFR, MYC, CDKN2A, GATA3, PTEN, BRCA2, RB1, TP53 and other genes (Fig. 5b upper panel). The comparison of the consensus profiles further revealed subclonal CNAs that were specific to subclone A (gains of 16p13.3-p13.2) or specific to subclone B (gains of 3q, 12p13.1-q12, 12q21.33–24.12). DE analysis identified 158 genes (FDRadj p-value < 0.01, | log2(Fold Change) | ≥ 0.5) that were differentially expressed between the two subclones, of which 42% were located in subclonal CNA regions (Supplementary Fig. 4b). A major subclonal event that emerged in subclone B was the focal amplification of chr12p13.1-q12 that resulted in the overexpression of KRAS (Fig. 5b lower panel and Supplementary Fig. 4d). The copy number profiles of the minor subclone B mapped to a distinct region in the high-dimensional analysis of the scRNA-seq data (Fig. 5e upper panel). Single cell GSVA analysis showed that the major subclone A had increased WNT and Hedgehog signaling, whereas the minor subclone B had multiple cancer hallmarks upregulated, including interferon responses, TNF-alpha signaling, hypoxia and other signatures (Fig. 5e lower panel).
In the third TNBC patient (TNBC5), the clustering of 2,670 aneuploid single copy number profiles inferred from scRNA-seq data identified two subclones, including a major subclone (A) that constituted the majority of the tumor (65%) and a minor subclone (B) that comprised a smaller fraction of the tumor mass (18%). The NJ tress constructed from the CNA data identified two distinct lineages that shared a common ancestor and corresponded to the clustering results (Supplementary Fig. 3c). Clonal CNAs detected across all of the tumor cells included gains of 4q, 7p, 8q and losses of 1p, 2q, 9, 10, 11, 17p that intersected several breast cancer genes including MDM4, PIK3CA, FGFR4, EGFR, MYC, TP53, CDKN2A and others (Fig. 5c upper panel). Comparison of the consensus copy number profiles of the subclones revealed a number of subclonal CNAs, such as amplification of chr1p and 14p that were specific to subclone A and amplification of chromosomes 12p and 12q that were specific to subclone B (Fig. 5c lower panel). The aneuploid copy number profiles of the two subpopulations mapped to two different regions in the high-dimensional expression space, suggesting that they had different transcriptional programs (Fig. 5f upper panel). DE analysis identified 89 genes (FDRadj p-value < 0.01, | log2(Fold Change) | ≥ 0.5) that were differentially expressed between the two subclones, of which 66% were located in subclonal CNA regions and included cancer genes such as KRAS and SMARCA4 (Supplementary Fig. 4c). Notably, the minor subclone B harbored major amplification of chr12p13.33-q12 that led to increased expression of KRAS, consistent with the minor subclone (B) detected in the previous TNBC patient (TNBC2) (Supplementary Fig. 4e) Single cell GSVA analysis showed differences in cancer hallmarks in subclone A including the upregulation of interferon alpha response and fatty acid metabolism, while subclone B showed increases in angiogenesis, hypoxia, EMT among other signatures (Fig. 5f lower panel). Taken together, these results suggest that CopyKAT can resolve clonal copy number substructure in tumors from scRNA-seq data and identify subclonal differences in breast cancer genes and cancer phenotypes that exist within the tumor mass.
Discussion
Here, we report the development of an integrative Bayesian segmentation approach to quantify genomic copy number profiles from high-throughput scRNA-seq data. A major application of CopyKAT is in the identification of tumor cells in unbiased scRNA-seq data, which often consists of not only tumor cells, but also many different stromal and immune cell types in the TME. Normal epithelial cells are often the most difficult to distinguish from malignant tumor cells by expression profiles alone, since they can express many of the same epithelial markers as the cancer cells. Using CopyKAT, we exploit a unique property of cancer cells in solid tumors, namely that they often harbor aneuploid copy number events in their genomes, while most stromal and immune cells have diploid copy number profiles. We show that the classification of aneuploid genomes from scRNA-seq data is feasible in several different solid cancer types, including PDAC, ATC, DCIS, TNBC, IDC and GBM - even in cases where the tumor purity is very low (<15%) or very high (>90%). Thus, we expect that CopyKAT will be a valuable tool for identifying tumor cells in scRNA-seq experiments that are comprised of mixtures of many different TME cell types.
Another application of CopyKAT is the delineation of clonal substructure in solid tumors based on differences in copy number alterations. We applied CopyKAT to resolve clonal substructure in three TNBC tumors, which identified two major subpopulations in each tumor that differed by distinct CNA events. Further, we show that from these data we can link the genotypes of the subclones to their phenotypes (transcriptional programs) to understand how the genomic alterations influenced different cancerous properties. Our analysis in TNBC showed differences in gene signatures and signaling pathways among the subclones within the tumor mass, including variation in EMT, DNA repair, hypoxia, apoptosis and angiogenesis. Interestingly, in two of the TNBC tumors we identified rare subclones (7%, 18%) with amplifications of the KRAS oncogene on chromosome 12p that upregulated gene expression. These rare KRAS subclones may be of interest for diagnostics or therapeutic targeting, if they are found to be common subpopulations in larger cohorts of TNBC patients in future studies.
Two previous methods have also been developed to estimate copy number alterations3,10. InferCNV3 utilizes an average moving window of gene expression, after excluding high and low expressed genes, however has limited ability to accurately resolve chromosome breakpoints. Another method, HoneyBadger10 was designed to predict CNVs from scRNA-seq data by jointly analyzing allelic imbalance of many variant sites in pooled clusters of single cells, but is highly dependent on obtaining full coverage data of the gene body. Thus a limitation of these previous methods is that they are not compatible with 3’ and 5’ scRNA-seq methods (10X Genomics6, Drop-Seq4, InDrop5) that are now widely used in the field of single cell genomics, but were instead developed for first-generation scRNA-seq methods such as Fluidigm35 and SMART-seq236. In contrast CopyKAT is compatible with high-throughput scRNA-seq methods that generate sparse data (e.g. 100K reads per cell) on thousands of cells that are sequenced in parallel, and is also compatible with data from first generation scRNA-seq methods.
A notable limitation of CopyKAT (and other methods) is that not all cancer types have aneuploid copy number events that can be used to distinguish normal and tumor cells. In particular, pediatric cancers and hematopoietic cancers (e.g. AML, CLL) are known to have few copy number alterations and therefore may not be suitable for CopyKAT analysis. Another limitation is that CopyKAT is mainly limited to the detection of CNA events based on changes in read depth across the genome and cannot be used to detect other genomic events that contribute to genomic diversity including chromosomal structural rearrangements, indels and somatic mutations. Furthermore, CopyKAT cannot provide reliable copy number information on the genomes of individual cells with unique genotypes, due to the technical variability of 3’ scRNA-seq data. This makes CopyKAT more suitable for the analysis of subclones in tumors where many cells have expanded and share similar genotypes, rather than the analysis of replicating cells or extremely rare subpopulations. A potential issue that we noted using CopyKAT is that when scRNA-seq datasets are without any tumor cells, CopyKAT may attempt to incorrectly detect CNA events in clusters with the highest gene expression levels. However in such cases the inferred CNA events will be inconsistent with known cytogenetic events in these cancers and can therefore be dismissed.
In summary, CopyKAT provides a powerful automated tool to classify tumor/normal cells and delineate clonal substructure in solid tumors analyzed by high-throughput scRNA-seq methods. We anticipate that this tool will be applied widely to many types of solid tumors in addition to the cancer types analyzed in this study. These studies will greatly improve our understanding of the malignant expression programs of tumor cells by providing a pure signal of the tumor cells, whereas previous bulk RNA-seq methods have been challenged by the intermixing of stromal and immune cells with the tumor cells, resulting in the incorrect assessment of cancer phenotypes. Moreover, these studies will lead to new insights into how chromosome alterations lead to gene dosage effects that reprogram cancer phenotypes in human tumors during disease progression.
Methods
Tumor tissue samples
Fresh tumor tissues samples from DCIS and invasive triple-negative breast cancers were obtained from the MD Anderson Cancer Center under IRB approved protocols in which patients were fully consented. The cancers were classified by pathological evaluation of H&E stained tissue sections and by staining for by immunohistochemistry for estrogen receptor (<1%) and progesterone receptor (<1%), and fluorescence in situ hybridization analysis of HER2 amplification using the CEP-17 centromere control probe (ratio of HER2/CEP-17 < 2.2). The invasive breast cancer (IDC) and anaplastic thyroid cancer (ATC) samples were also obtained from patients at the MD Anderson cancer center under an IRB approved protocol in which the patients were consented. The classification of cancer types was determined by histopathological evaluation of the H&E stained tissue sections.
Single cell RNA sequencing of fresh tumor tissues
To prepare viable single cell suspensions, 1–3cm3 fresh tumors were placed in a 10cm dish with 5ml dissociation solution, minced with scalpels into 1mm3 pieces and transferred to a 50ml conical tube with 30ml dissociation solution to dissociate the tissue suspension at 37°C in a rotating hybridization oven for 15 minutes to 1 hour. For trypsinization, the tissue suspension was centrifuged at 450g for 5 minutes to remove the supernatant, and the pellet was resuspended into 5ml trypsin (Corning #25053CI) and incubated at 37°C in a rotating hybridization oven for 5 minutes. Trypsin was neutralized by 10ml DMEM (Sigma #D5796) containing 10% fetal bovine serum (FBS) (Sigma #F0926). The tissue suspension was filtered through a 70μm strainer by using a syringe plunger flange to grind the leftover unfiltered tissue. The strainer was rinsed and grinded with DMEM to ensure any remaining single cells were filtered. The flow-through was centrifuged at 450g for 5 minutes and the supernatant was removed. If red blood cells (RBCs) were presented in the pellet, 10–20ml 1x MACS RBC lysis buffer [1:10 dilution of 10x MACS RBC lysis (MACS #130–094-183) into miliQ H2O] was applied by nutating at room temperature for 10 minutes. To stop RBC lysis, 20ml DMEM was added and the mixture was centrifuged at 450g for 5min. The supernatant was discarded and cell pellet was washed by 10ml 4°C DMEM. After centrifuging at 450g for 5 minutes, supernatant was discarded and cells were resuspended into cold PBS (Sigma #D8537) +0.04%BSA solution (Ambion #AM2616) and passed through 40um flowmi (Bel-Art #h13680–0040). To make dissociation solution, collagenase A (Sigma #11088793001) was dissolved in 75% v/v DMEM F12/HEPES media (Gibco #113300) and 25% v/v BSA fraction V (Gibco# 15260037) to prepare a concentration of 1mg/ml.
Single cell capture, barcoding and library preparation was performed by following the 10X Genomics Single Cell Chromium 3’ protocol (PN-120237) or 5’ protocol (PN-1000006) using V3 or V2 chemistry reagents (10X Genomics). The final libraries containing barcoded single cell transcriptomes were sequenced at 100 cycles on an S2 flowcell on the Novoseq 6000 system (Illumina). Data were processed using the CASAVA 1.8.1 pipeline (Illumina Inc.), and sequence reads were converted to FASTQ files and UMI read counts using the CellRanger software (10X Genomics).
Preprocessing and transformation of scRNA-seq data
The analysis starts with the UMI count matrix that has genes in rows and cell IDs in columns. To remove ambient and low viability cells, we filter out single cells that have less than 200 genes detected. To mitigate gene dropout effects, we require at least 7000 genes that are detected in at least 5% of cells in the population. Genes that are detected in less than 5% of cells in the population are excluded from our analysis. Further, we require that each chromosome should have at least 5 genes detected to represent its copy number status. All row IDs are annotated by gene symbols and ordered by their genomic coordinates. To mitigate the segmentation artifacts caused by HLAs genes in chr6p in immune cells, we removed HLA genes from the copy number calculation pipeline. Similarly, we removed a set of cellular cycling genes (c5.all.v6.2.cyclegene)37 to reduce artificial segments associated with cells that are actively dividing.
Supposed be the raw UMI count matrix, we first transform using log-Freeman-Tukey Transformation (FTT)11 to stabilize variance as below:
And then apply ‘dlmModPoly’ model in R package ‘dlm’ to model gene expression by polynomial dynamic linear model (DLM)12 and the ‘dlmSmooth’ function to smooth outliers in single cell UMI counts.
Estimating copy number baseline values in diploid cells
To estimate the ground state copy number baseline, we predefine a subset of ‘confident normal’ cells using a combined approach. First, the normalized and smoothed scRNA-seq data are clustered using ward linkage for hierarchical clustering. The average silhouette width Wd2 in the 2-cluster separation with the ‘cutree’ function is then calculated. Next, single cells are pooled into 6 clusters and the consensus gene expression profiles are calculated for each of the 6 clusters as the medians of all single cells within the cluster. The variance of consensus profile is calculated using Gaussian Mixture Model (GMM). The minimal variance is compared to the maximal variance using Fishers’ F test:
The cluster with minimal variance is predefined as ‘confident normal’ cell cluster if the stringent criteria is met: p-value < 0.05, Wd2 ≥ 0.15 and at least 5 ‘confident normal’ cells to serve as copy number baseline.
In the alternative “GMM definition” approach, we evaluate the diploid status of single cells one-at-a-time by calculating the fraction of neutral copy number events in each cell. The normalized and smoothed expression values of all genes in a single cell are assumed to be a mixture of three Gaussian distributions to represent three copy number states: gain, neutral and loss. To fit the GMM model, we initiate the modeling with equal variances in the three distributions, i.e. half of the sample standard deviation, with initial mean values of 0.2, 0, −0.2 respectively. Next, we use the modeled means and relative frequencies of the three distributions to determine the fractions of neutral events in each cell as:
A single cell is predefined as normal cells if more than 99% of genes fall into the neutral distribution ( Nfrac ≥ 0.99 ). Finally, the baseline copy number values are calculated as the median of all predefined ‘confident normal’ cells, and the relative gene expression values are obtained by subtraction of these baseline values from each individual gene.
Copy number segmentation
To perform segmentation, we first transform the cluster consensus to find chromosome breakpoints for the within-cluster of cells using Monte Carlo Simulation based on a Poisson Likelihood with a Gamma prior by applying the ‘MCpoissongamma’ function in R package ‘MCMCpack’38. We assumed a Poisson distribution of segment mean and a conjugated Gamma distribution of the Poisson parameter as follows:
where is the back-transformed UMI counts; are shape parameters of gamma distribution. We simulated 1,000 posterior means for each window and then use Kolmogorov–Smirnov (K-S) tests to determine if adjacent windows should be joined together. We calculate the KS test statistic D as the largest vertical distance between the accumulative distributions of posterior means of the two adjacent windows:
where are posterior means of adjacent windows i and j. If the KS test statistic D is greater than a cutoff value, then a breakpoint is defined. If less than 25 breakpoints are defined in the first round with the initial cutoff, we decrease the cutoff by 50%. We repeat this process twice at most for each cluster. We unify all breakpoints from each cluster to form the consensus breakpoints for the whole population. We then calculate the posterior means of each window between adjacent breakpoints as its segment means by applying ‘MCpoissongamma’ function to all consensus windows of each single cell. We then log-transform the means and center the data as the final relative copy number ratios of each gene. Finally, we convert individual gene copy numbers to genomic bins by calculating averages of the genes that fall into 220kb genomic bins28 across the human genome to estimate the genome-wide copy number profile for each cell from the scRNA-seq data.
Theoretical estimation of genomic copy number resolution
To estimate the expected resolution of the copy number profiles inferred from the single cell RNA data, we downloaded the BED file of all gene entries in GRCh38 (v28) from UCSC. Since chromosome Y was not included in our copy number calculation, we only considered genes located in chr1:22 and chrX, which harbor a total of 56,051 genes entries. We estimated the genomic center position of individual genes by taking the averages of the gene start position and gene end position. Next we ordered all genes by their genomic positions and estimated the distance between two adjacent gene centers by calculating the distance between two gene centers. In total, we defined 56,028 gene intervals across the genome. From chr1 to 22 and X, the number of gene intervals are as follows: 5127, 3872, 2925, 2430, 2779, 2802, 2292, 2189, 2137, 3189, 2857, 1279, 2152, 2081, 2440, 2911, 1133, 2917, 1350, 795, 1300, 2281. The first quartile, median, mean, third quartile and maximum of gene interval across the whole genome are as follows: 9430bp, 24532 bp, 52806bp, 58485bp and 21765992bp. Since the size distribution of gene intervals is heavily skewed to the right, we calculated the median value to estimate the copy number resolution. Since we require at least 7000 genes to be detected across the whole single cell population in our pipeline, this number represents an equivalent to a median of 7000/56051 ≈ 12.5% gene detection rate. Finally, we calculated the minimum gene interval in our analysis as 24,532 bp ÷ 12.5% ≈ 200 Kb per gene interval. Last, we initiated our copy number analysis with 25-gene window, therefore we estimated the minimum size of a segment is 200Kb × 25 = 5 Mb genomic resolution for the detection of copy number events across the genome of each cell.
Estimation of bulk DNA copy number from single cell DNA data
The bulk DNA copy numbers of DCIS1 tumor cells were obtained from a single cell DNA sequencing data in which the median copy number profiles were computed. Briefly, we flow-sorted aneuploid single cells from the single nuclei suspensions and performed multiplexed single nucleus sparse whole genome sequencing as previously described39. In total we sequenced 539 single cells. Single cell copy number profiles were calculated using a 220kb variable binning method13 to quantify sequence reads in genomic bins and the CBS method40 to segment genomic bins, followed by MergeLevels41 to join adjacent segments with non-significant differences in segment ratios. The final bulk DNA copy number profile of this tumor was calculated by taking the median values of the all 539 single cell copy numbers of each genomic bin to generate a pseudo-bulk profile as a reference for the single cell RNA sequencing data.
Empirical estimation of CopyKat sensitivity from a breast tumor sample
We quantified the breakpoint detection efficiency using the breast tumor sample DCIS1 that has both scRNA-seq and bulk DNA-seq data describes above. The breakpoints detected in the bulk DNA-seq data were taken as the ‘ground truth’. The estimation was performed by comparing the breakpoints detected by CopyKAT in the scRNA-seq data to the ‘ground truth’ in bulk DNA-seq data. To evaluate the variation in genomic resolution detection, we performed bootstrapping 1000 times to resample 200 single cells with replacement and sent them for segmentation in CopyKAT. For each ‘ground truth’ breakpoint, we located a closet CopyKAT breakpoint that had the smallest genomic distance to the ‘ground truth’. If the distance fell within a given range, e.x. 200Kb, we defined that this breakpoint was successfully detected at 200Kb resolution; similarly breakpoints that fell into 5Mb range were determined as successfully detected at 5Mb resolution, and so forth. Lastly, we calculated the resolution at a given resolution as the percentages of breakpoints that were successfully detected with the given range of genomic distances, i.e. the total number of breakpoints detected divided by the total ‘ground truth’ breakpoints. We repeated this process 1,000 times to calculate the averaged sensitivity at the given resolution by bootstrapping resampling.
Classification of tumor and normal cells
Classification of tumor and normal cells was performed in two steps. We assumed that the major genetic distance among the cell populations is the difference between diploid and aneuploid genomes and therefore forced the single cells into two major clusters using hierarchical clustering with Ward linkage and Euclidean distance. To determine the identities of each clusters, we integrated the clustering results with the predefinition of the ‘confident normal cells’ that are defined by a very stringent criteria (see Online Methods section on Estimating Copy Number Baseline Values in Diploid Cells).The cluster that has significantly higher enrichment of predefined normal cells is defined as the normal diploid cell cluster. In cases where there is no significant difference in the enrichment test, we switch to the ‘GMM definition’ approach to determine if the consensus profiles of each cluster pass the ‘normal cell criteria’, where at least 95% of the regions fall into the neutral distribution. In some challenging samples that have aneuploidy too close to 2N, we use an alternative slower approach by predicting the cells one-by-one using the ‘GMM definition’ approach and ‘normal cell criteria’.
To evaluate the accuracy of this copy number-based classification of tumor and normal cells, we applied an empirical approach to decide tumor and normal cells based on clustering and expression of cancer-specific marker genes. We first clustered all single cells within a tumor using ‘SNN’ method in R package ‘Seurat’42. Next we obtained the expression levels of a panel of four epithelial markers (EPCAM, KRT19, KRT18, and KRT8). We calculated the average expression values of this epithelial markers panel as a consolidated epithelial score in each cell. Single cell gene expression clusters with high epithelial scores (kernel density center is above 0) were labeled as putative tumor cell clusters. In tumors that have both normal epithelial and tumor epithelial cell clusters, we further applied evaluated cancer type specific markers, including KRT19 for PDAC tumor epithelial cells, KRT8 for ATC, EPCAM for TNBC and IBC, and EGFR for GBM cancer cells. Furthermore, expression clusters that expressed immune cells markers (CD45, CD3, CD4, CD8) or fibroblast markers (ACTA2, FN1) were classified as normal cells. Single cells that had consistent aneuploid prediction results in both CopyKAT and by gene expression clusters with high epithelial score were considered to be tumor cells. The prediction accuracy of CopyKAT using aneuploid copy number profiles alone was then calculated as the number of cells with the correct prediction divided by the total number of single cells in the analysis.
Differential gene expression of subclones
We compared the single cell RNA gene expression data of the two major subclones within each tumor using a bimodal algorithm, MAST43, to mitigate gene detection rates across cells. The significant differentially expressed genes (DEG) were defined as having FDR adjusted p-value < 0.01 and | log2(Fold Change) | ≥ 0.5. The list of DEG was intersected with COSMIC33 human cancer gene list and a TCGA defined breast cancer gene list32 to identify known cancer genes. The genomic positions of genes were annotated by using R package ‘biomaRt’44.
Single cell gene set enrichment analysis
To identify enrichment of cancer hallmark signatures between the subclones, we applied single-sample GSVA (GSVA)34 to calculate enrichment scores for each gene set of the single cells using log2(UMI + 1) data. We first obtained GSVA scores for the 50 cancer hallmark gene signatures45 for each cell, and then compared the enrichment scores between two clones by using R package ‘limma’46. Differentially enriched signatures were defined as having FDR adjusted p-values < 0.05 and | mean score difference | ≥ 0.1 as described previously47.
Construction of neighbor-joining trees from copy number profiles
To construct the neighbor-joining (NJ) trees of breast tumors, the pearson’s correlation distances were first calculated from the single tumor cell copy number matrix generated by by CopyKAT. The NJ trees were built from the distance matrix using the ‘nj’ function and re-rooted to an artificial normal diploid cell (2N copy number across the entire genome) using the interactive ‘root.phylo’ function in R package ‘ape’48. The final trees were plotted as a downward phylogram plots in R.
Supplementary Material
Acknowledgements
This work was supported by grants to N.E.N. from the American Cancer Society (129098-RSG-16–092-01-TBG), the National Cancer Institute (RO1CA240526, RO1CA236864), the Emerson Collective Cancer Research Fund and the CPRIT Single Cell Genomics Center (RP180684). N.E.N. is a AAAS Wachtel Scholar, AAAS Fellow, Andrew Sabin Family Fellow, and Jack & Beverly Randall Innovator. This study was supported by the MD Anderson Breast Cancer Moonshot Program. This study was supported by the MD Anderson Sequencing Core Facility Grant (CA016672). This project was also supported by Susan Komen Postdoctoral Fellowship to R.G (PDF17487910). Other grant supports include Anaplastic Thyroid Cancer Research Fund (S.Y.L. and J.R.W.) and an institutional Multi-investigator Research Program grant to S.Y.L..
Footnotes
Competing Interests Statement
The authors have no competing interests to declare.
Ethical Compliance
This study has complied with all relevant ethical regulations for the human subjects.
Data availability
Single cell RNA-seq data from this study was deposited to the Gene Expression Omnibus (GEO): GSE148673.
Software availability
Software is available at GitHub (https://github.com/navinlabcode/copykat).
References (for main text only)
- 1.Peng J. et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29, 725–738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma L. et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 36, 418–430 e416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng GX et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao R. et al. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nat Commun 8, 228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gierahn TM et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods 14, 395–398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor AM et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 33, 676–689 e673 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan J. et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res 28, 1217–1227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freeman MF & Tukey JW Transformations Related to the Angular and the Square Root. Ann Math Stat 21, 607–611 (1950). [Google Scholar]
- 12.Petris G. An R Package for Dynamic Linear Models. J Stat Softw 36, 1–16 (2010). [Google Scholar]
- 13.Baslan T. et al. Genome-wide copy number analysis of single cells. Nat Protoc 7, 1024–1041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harada T. et al. Genome-wide DNA copy number analysis in pancreatic cancer using high-density single nucleotide polymorphism arrays. Oncogene 27, 1951–1960 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samuel N. et al. Integrated genomic, transcriptomic, and RNA-interference analysis of genes in somatic copy number gains in pancreatic ductal adenocarcinoma. Pancreas 42, 1016–1026 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e. & Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 32, 185–203 e113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao H. et al. Glypican-3 and KRT19 are markers associating with metastasis and poor prognosis of pancreatic ductal adenocarcinoma. Cancer Biomark 17, 397–404 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Girgis AH, Bui A, White NM & Yousef GM Integrated genomic characterization of the kallikrein gene locus in cancer. Anticancer Res 32, 957–963 (2012). [PubMed] [Google Scholar]
- 19.Dijk F. et al. Unsupervised class discovery in pancreatic ductal adenocarcinoma reveals cell-intrinsic mesenchymal features and high concordance between existing classification systems. Sci Rep 10, 337 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heid I. et al. Co-clinical Assessment of Tumor Cellularity in Pancreatic Cancer. Clin Cancer Res 23, 1461–1470 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Ravi N. et al. Identification of Targetable Lesions in Anaplastic Thyroid Cancer by Genome Profiling. Cancers (Basel) 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribeiro FR, Meireles AM, Rocha AS & Teixeira MR Conventional and molecular cytogenetics of human non-medullary thyroid carcinoma: characterization of eight cell line models and review of the literature on clinical samples. BMC Cancer 8, 371 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo D. et al. Cytokeratin-8 in Anaplastic Thyroid Carcinoma: More Than a Simple Structural Cytoskeletal Protein. Int J Mol Sci 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunt JL Molecular pathology of endocrine diseases. (Springer, New York; 2010). [Google Scholar]
- 25.Barletta JA Endocrine Pathology: Advances, Updates, and Diagnostic Pearls. Surg Pathol Clin 12, xi-xii (2019). [DOI] [PubMed] [Google Scholar]
- 26.Asa SL & LiVolsi VA New diagnostic and management approaches in endocrine pathology. Arch Pathol Lab Med 132, 1228–1230 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Turner N. et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 29, 2013–2023 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao R. et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet 48, 1119–1130 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andre F. et al. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res 15, 441–451 (2009). [DOI] [PubMed] [Google Scholar]
- 30.Neftel C. et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849 e821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brennan CW et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forbes SA et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45, D777–D783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanzelmann S, Castelo R. & Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xin Y. et al. Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proc Natl Acad Sci U S A 113, 3293–3298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picelli S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098 (2013). [DOI] [PubMed] [Google Scholar]
References (for Methods only)
- 37.Liberzon A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin AD, Quinn KM & Park JH MCMCpack: Markov Chain Monte Carlo in R.” Journal of Statistical Software. J Stat Softw 42, 22 (2011). [Google Scholar]
- 39.Kim C. et al. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 173, 879–893 e813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olshen AB, Venkatraman ES, Lucito R. & Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Willenbrock H. & Fridlyand J. A comparison study: applying segmentation to array CGH data for downstream analyses. Bioinformatics 21, 4084–4091 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Stuart T. et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finak G. et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome biology 16, 278 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durinck S, Spellman PT, Birney E. & Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liberzon A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao R. et al. Nanogrid single-nucleus RNA sequencing reveals phenotypic diversity in breast cancer. Nature communications 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paradis E. & Schliep K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019). [DOI] [PubMed] [Google Scholar]
- 49.do Valle IF et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinformatics 17, 341 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roth A. et al. in Nat. Methods, Vol. 11 396–398 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malikic S, McPherson AW, Donmez N. & Sahinalp CS Clonality inference in multiple tumor samples using phylogeny. Bioinformatics 31, 1349–1356 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Gerstung M. et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nature communications 3, 811 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Navin N. et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nilsen G, Liestol K. & Lingjaerde OC copynumber: Segmentation of single- and multi-track copy number data by penalized least squares regression. R package version 1.12.0 (2013). [Google Scholar]
- 55.Hennig C. fpc: Flexible Procedures for Clustering. R package version 2.1–10 (2015). [Google Scholar]
- 56.Paradis E, Claude J. & Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Gendoo DM et al. Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32, 1097–1099 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breiman L. Manual On Setting Up, Using, And Understanding Random Forests V3.1. (2002). [Google Scholar]
- 59.Curtis C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira B. et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nature communications 7, 11479 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.