

Abstract
Estrogen receptor alpha (ER)-positive breast cancer is commonly treated with endocrine therapies, including anti-estrogens that bind and inhibit ER activity, and aromatase inhibitors that suppress estrogen biosynthesis to inhibit estrogen-dependent ER activity. Paradoxically, treatment with estrogens such as 17b-estradiol can also be effective against ER+ breast cancer. Despite the known efficacy of estrogen therapy, the lack of a predictive biomarker of response and understanding of the mechanism of action have contributed to its limited clinical use. Herein, we demonstrate that ER overexpression confers resistance to estrogen deprivation through ER activation in human ER+ breast cancer cells and xenografts grown in mice. However, ER overexpression and the associated high levels of ER transcriptional activation converted 17b-estradiol from a growth-promoter to a growth-suppressor, offering a targetable therapeutic vulnerability and a potential means of identifying patients likely to benefit from estrogen therapy. Since ER+ breast cancer cells and tumors ultimately developed resistance to continuous estrogen deprivation or continuous 17b-estradiol treatment, we tested schedules of alternating treatments. Oscillation of ER activity through cycling of 17b-estradiol and estrogen deprivation provided long-term control of patient-derived xenografts, offering a novel endocrine-only strategy to manage ER+ breast cancer.
INTRODUCTION
Breast cancer is the most commonly diagnosed cancer in women, and the majority of these cases are estrogen receptor alpha (ER)-positive (1, 2). Breast tumors that express ER are typically dependent upon estrogen-induced ER transcriptional activation for growth, and the main therapeutic strategies for ER+ breast cancer target this dependence. Drugs from three classes of endocrine therapy are routinely used for ER+ breast cancer: aromatase inhibitors (AIs; e.g., letrozole, anastrozole, exemestane) that inhibit estrogen biosynthesis; selective estrogen receptor modulators (SERMs; e.g., tamoxifen) that antagonize ER but can have agonistic effects; and selective estrogen receptor downregulators (SERDs; e.g., fulvestrant) that inhibit ER and promote its degradation. Although patients with early-stage ER+ breast cancer benefit overall from adjuvant endocrine therapy with tamoxifen or an AI, approximately one-third of these patients eventually experience disease recurrence (3). Despite the development of therapies for endocrine-resistant disease (e.g., inhibitors of CDK4/6, mTOR, or PI3K), advanced ER+ breast cancer is rarely cured and there exists a need for improved therapeutic options for these patients.
Although the anti-cancer effects of estrogens have been known since the 1940s (4), clinical use of estrogen therapy has been limited since the introduction of anti-estrogens that have improved adverse events profile (5, 6). More recent clinical studies have demonstrated that treatment with estrogens [i.e., 17β-estradiol (E2), diethylstilbestrol, or ethinylestradiol] elicits anti-tumor effects in approximately 30% of patients with advanced ER+ breast cancer previously treated with anti-estrogens and/or AIs (7–11). Despite the proven efficacy of estrogen therapy, its clinical use has been hindered by a lack of mechanistic understanding and a predictive biomarker to identify patients likely to benefit from this treatment.
Clinical evidence indicates that the patients most likely to benefit from estrogen therapy are post-menopausal (12), suggesting that a period of adaption to low levels of endogenous estrogens increases tumor sensitivity to estrogen therapy. Preclinical studies using MCF-7 cells and the WHIM16 patient-derived xenograft model, as well as a clinical case report, suggest that amplification of the gene encoding ER (ESR1) is associated with therapeutic response to estrogen (13–15). Herein, we demonstrate that high ER levels confer resistance to estrogen deprivation and increase estrogen-independent ER activity. These high levels of ER elicit therapeutic responses to E2. We leverage this mechanistic understanding to develop novel treatment paradigms to improve long-term endocrine management of breast tumors.
RESULTS
Breast cancer cells with acquired resistance to estrogen deprivation and sensitivity to estrogen therapy are dependent upon ER activity when estrogen-deprived
MCF-7 and HCC-1428 ER+ breast cancer cells are growth-inhibited by estrogen deprivation and growth-induced by E2. Long-term estrogen-deprived (LTED) counterparts of these cell lines have acquired resistance to estrogen deprivation and are growth-inhibited by treatment with 1 nM E2 (Fig. 1A). MCF-7/LTED cells are ESR1-amplified [Fig. S1 and refs. (14, 15)], and both MCF-7/LTED and HCC-1428/LTED cells express higher levels of endogenous ER than parental controls (Fig. 1B). LTED cells also express increased levels of ESR1 mRNA and ER target genes compared to parental cells (Fig. 1C), suggesting that LTED cells exhibit increased estrogen-independent ER activity. Treatment with the SERD fulvestrant suppressed estrogen-independent LTED cell growth (Figs. 1D and S2) and expression of ER target genes (Fig. 1E), indicating that LTED cells are reliant upon estrogen-independent ER activity for growth.
Figure 1. Breast cancer cells that therapeutic respond to E2 are ER-dependent despite estrogen deprivation.
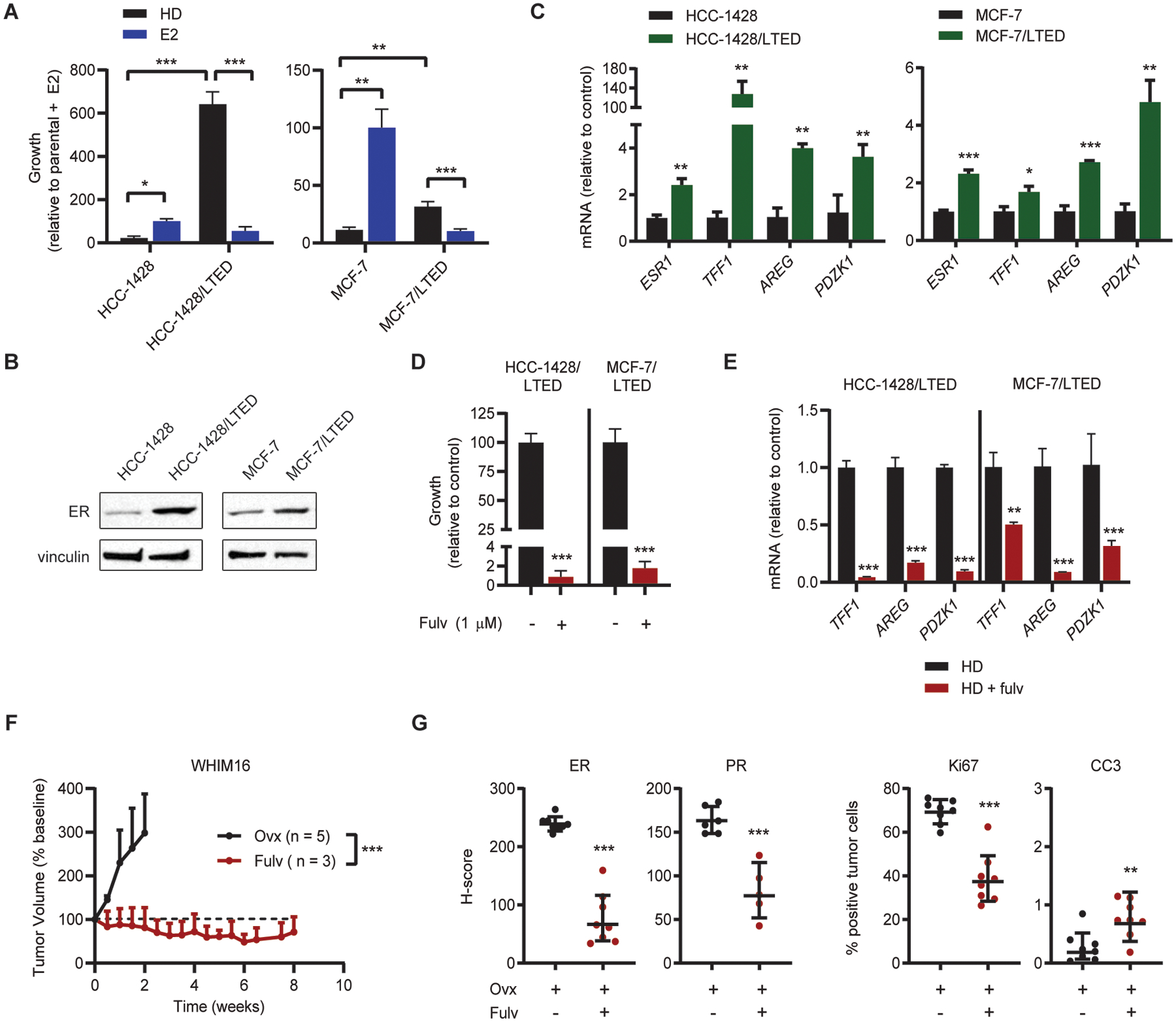
(A) Cells were seeded at low density and treated in triplicate as indicated for 4 wk. Colonies were fixed and stained with crystal violet, and colony area was quantified. Parental cells were hormone-deprived (HD) for 3 d prior to seeding. (B) Lysates from parental cells (hormone-deprived for 3 d) and LTED cells were analyzed by immunoblot. (C) RNA isolated from parental cells (hormone-deprived for 3 d) and LTED cells was analyzed by RT-qPCR. Values for indicated transcripts were normalized to ACTB expression. (D) LTED cells were seeded at low density and treated in triplicate ± fulvestrant (fulv) as indicated for 4 wk. Colony area was analyzed as in (A). (E) LTED cells were treated ± 1 μM fulv for 3 d. RNA was isolated and analyzed by RT-qPCR as in (B). (F) Ovx mice bearing WHIM16 tumors were randomized to treatment as indicated. (G) Tumors were harvested from mice treated as in (F) after 8 d of treatment. Expression of ER, Ki67, cleaved caspase 3 (CC3) and progesterone receptor (PR) were analyzed by IHC. *p≤0.05, **p≤0.005, ***p≤0.0005 by two-tailed t-test (D, G), Bonferroni multiple comparison-adjusted post-hoc test (C, E), or linear mixed modeling (F). Data are shown as mean ± SD.
The WHIM16 patient-derived xenograft model is ESR1-amplified (14) and grows in ovariectomized (ovx) mice, mimicking resistance to low levels of estrogen that occur in post-menopausal patients and AI-induced estrogen depletion. We and others showed that E2 treatment of mice induces regression of WHIM16 tumors (14, 15). Fulvestrant treatment inhibited WHIM16 tumor growth (Figs. 1F and S3), decreased tumor levels of ER, Ki67 (marker of cell proliferation), and PR (encoded by the ER-inducible gene PGR), and modestly increased cleaved caspase-3 levels (marker of apoptosis) (Figs. 1G and S4). These data suggest that WHIM16 tumors, which are therapeutically sensitive to E2, are dependent upon ER activity for growth in estrogen-depleted conditions.
Forced ER overexpression induces estrogen-independent ER transcriptional activity and resistance to estrogen deprivation
To determine whether high levels of ER expression are sufficient to induce resistance to estrogen deprivation, we stably transfected MCF-7 and HCC-1428 cells with FLAG-tagged ESR1 (Fig. 2A). Constitutive FLAG-ESR1 overexpression increased estrogen-independent growth and expression of ER target genes under estrogen deprivation compared to vector controls (Fig. 2B/C). Vector control cells did not survive estrogen deprivation for the duration of selection, therefore parental cells hormone-deprived for 3 d were used as a control for experiments with FLAG-ESR1 cells. We also stably transfected T47D ER+ breast cancer cells with the pInducer20 system (16) encoding ESR1 or empty vector under the control of a doxycycline (dox)-inducible promoter (Fig. 2D). Dox-induced ER overexpression increased estrogen-independent growth and expression of ER target genes (Fig. 2E/F). Treatment with fulvestrant suppresses estrogen-independent increases in ER transcriptional activity and growth in ER-overexpressing models, indicating that these effects are directly dependent upon elevated levels of ER expression (Fig. S5). Together, these data suggest that high levels of ER expression drive estrogen-independent ER transcriptional activity and growth.
Figure 2. Overexpression of ER induces estrogen-independent ER signaling and resistance to estrogen deprivation.
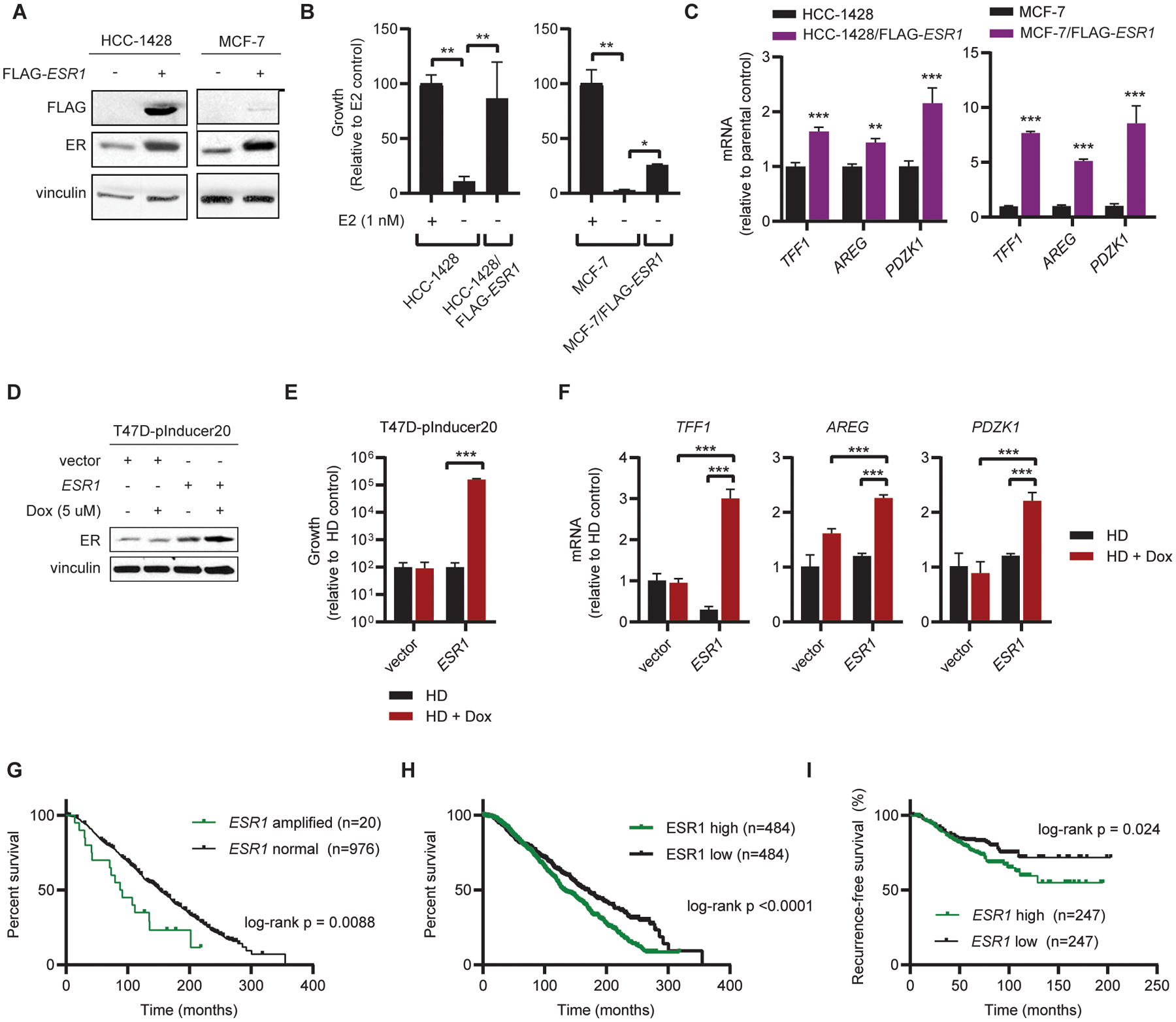
(A) MCF-7 and HCC-1428 cells stably expressing FLAG-ESR1 were generated, and transgene expression was confirmed by immunoblot. Parental cells were hormone-deprived for 3 d prior to collection of lysates. (B) Parental and FLAG-ESR1 cells were seeded at low density and treated as indicated for 4 wk. Parental cells were hormone-deprived for 3 d prior to seeding. Colonies were fixed and stained, and relative colony area was quantified. (C) RNA was isolated from ER-overexpressing cells and parental controls. Expression of indicated transcripts was analyzed by RT-qPCR, normalized to expression of ACTB. Parental cells were hormone-deprived for 3 d prior to RNA isolation. (D) Protein was harvested from T47D cells stably expressing the pInducer20 vector with either doxycycline-inducible ESR1 or empty vector control. Cells were hormone-deprived for 3 d and treated with doxycycline (dox) as indicated for 2 d. ER expression was analyzed by immunoblot. (E) T47D/pInducer20 cells were hormone-deprived for 3 d and seeded at low density. Cells were treated as indicated in triplicate for 4 wk, and colony area was quantified as in (B). (F) T47D/pInducer20 cells were hormone-deprived for 5 d and treated ± 5 μM doxycycline on Day 3 of hormone deprivation. RNA was harvested and analyzed as in (C). (G) Overall survival analysis of patients with ESR1-amplified (n=20) and -non-amplified (n=976) ER+/HER2- breast cancer. Analysis is limited to patients with ER+ status confirmed by IHC, HER2- status measured by SNP6 array, and receipt of endocrine therapy in ref. (17). (H) Overall survival analysis of patients with high (n=484) vs. low (n=484) ESR1 mRNA expression from dataset in (G). (I) Recurrence-free survival analysis of 494 patients with ESR1-high (n= 247) vs. ESR1-low (n=247) ER+/HER2- breast cancer described in ref. (19). Data were stratified by median ESR1 mRNA expression. Data in (B-F) are shown as mean ± SD. *p≤0.05, **p≤0.005, ***p≤0.0005 by two-tailed t-test (C) or Bonferroni multiple comparison-adjusted post-hoc test (B, E, F).
We then evaluated whether ESR1 amplification is associated with resistance to conventional anti-estrogen and AI therapies in patients. An analysis of overall survival among a cohort of 996 patients with early-stage ER+/HER2- breast cancer treated with adjuvant endocrine therapy (17) revealed that ESR1 amplification in primary tumors is associated with poor outcome compared to ESR1-non-amplified tumors (Fig. 2G). Of note, the 20-patient subset with ESR1-amplified tumors contained a higher proportion of tumors associated with the Luminal B subtype (n=12) compared to Luminal A (n=2), while the ESR1-non-amplified subset contained a higher proportion of Luminal A vs. B (n=474 vs. n=337). Luminal B tumors confer worse prognosis compared to Luminal A (18). However, this ESR1-amplified subset was too small to statistically test for differences in molecular subtype distribution between subsets. Analysis of ESR1 mRNA expression in the same cohort revealed that high ESR1 mRNA levels are similarly associated with poor outcome compared to tumors expressing low ESR1 (Fig. 2H). Molecular subtype distributions were significantly different between these subsets (Chi-Square p<0.0001) with an increased proportion of ESR1-high tumors assigned to the Luminal B subtype compared to ESR1-low tumors (45.0% vs. 25.4%; Table S1). Additionally, an analysis of recurrence-free survival in a second cohort of 490 patients with ER+/HER2- breast cancer treated with adjuvant endocrine therapy, chemotherapy, or both (19) demonstrated that high tumor ESR1 mRNA expression is associated with poor outcome compared to low ESR1 (Fig. 2I).
Forced overexpression of ER sensitizes cells to the anti-cancer effects of 17β-estradiol
Since ESR1-amplified tumors and cells are growth-inhibited by E2, we tested the effects of partial ER knockdown on response to E2. We generated LTED cell lines expressing dox-inducible shRNA targeting ESR1 or a non-silencing control. Partial knockdown of ER partly rescued LTED cells from the growth-inhibitory effects of E2 (Figs. 3A and S6). This suggests that high levels of endogenous ER are necessary for the growth-inhibitory effects of E2.
Figure 3. Overexpression of ER converts E2 from a growth promoter to a growth suppressor.
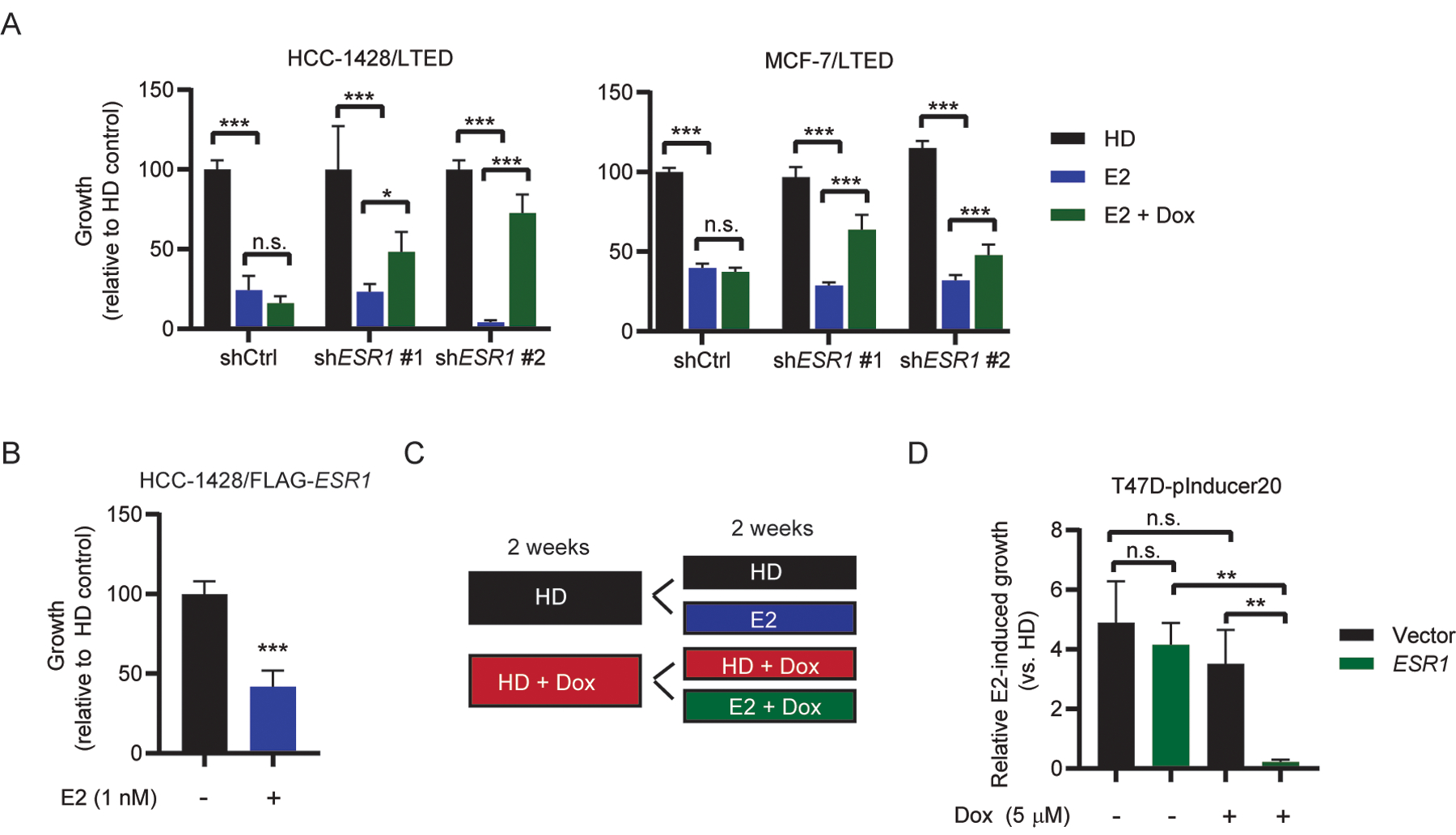
(A) LTED cells stably expressing doxycycline-inducible shRNA targeting ESR1 or non-coding control were seeded at low density and treated as indicated with doxycycline (dox; HCC-1428/LTED, 100 nm; MCF-7/LTED, 1 μM) and E2 (1 nM) for 4 wk. Colonies were fixed and stained with crystal violet, and colony area was quantified. (B) HCC-1428 cells stably overexpressing FLAG-ESR1 and maintained in hormone-depleted medium were seeded at low density and treated ± E2 as indicated. Colonies were fixed, stained, and quantified as in (A). (C) Experimental design to test E2 sensitivity in T47D-pInducer20 cell lines. (D) Cells were seeded at low density and treated ± 1 nM E2 and 5 μM doxycycline as indicated. Data shown are mean of triplicates ± SD. *p≤0.05, **p≤0.005, ***p≤0.0005 by Bonferroni multiple comparison-adjusted post-hoc test (A, D) or two-tailed t-test (B).
Conversely, we tested the effects of overexpression of exogenous FLAG-ER on response to E2. HCC-1428/FLAG-ESR1 cells were growth-inhibited by E2 (Fig. 3B). T47D/pInducer20-ESR1 cells treated with dox for 2 wk to induce ER overexpression were growth-inhibited by E2, while non-dox-treated cells were growth-stimulated by E2 (Fig 3C/D). T47D/pInducer20-vector cells were growth-stimulated by E2 regardless of the presence or absence of dox. Thus, E2 can be converted from a growth-promoter to a growth-suppressor by increasing ER levels.
ER transcriptional hyperactivation is associated with growth-inhibitory effects of 17β-estradiol
We next sought to determine whether high levels of both ER expression and ligand stimulation were necessary for the anti-cancer effects of E2. In LTED cells, low-picomolar doses of E2 stimulated growth, while cell growth was inhibited by higher doses (0.1–1 nM) within the physiological range in sera of pre-menopausal women (Fig. 4A). Growth-stimulating low-picomolar doses of E2 were associated with small increases in ER transcriptional activity in LTED cells compared to estrogen-deprived controls, while high levels of ER transcriptional activity co-occurred with growth inhibition (Fig. 4A). In ovx mice bearing WHIM16 tumors, tumor regression was dose-dependent: high doses of orally administered E2 induced complete regression, while lower doses only slowed tumor growth (Fig. 4B and S7A–E). The lack of growth stimulation with low doses of exogenous E2 in vivo may be due to the low levels of endogenous E2 in ovx mice. However, treatment with the AI letrozole did not decrease serum E2 levels in ovx mice, so we were unable to test this hypothesis (Fig. S7F). The dose-dependent therapeutic responses to E2 observed in mice and cultured cells, and the stimulation of growth with low levels of E2 in vitro, suggest that ER activation is not intrinsically growth-inhibitory in these models, but rather that high levels of ER activation must occur to elicit anti-cancer effects.
Figure 4. Therapeutic response to E2 is accompanied by hyperactivation of ER transcriptional activity.
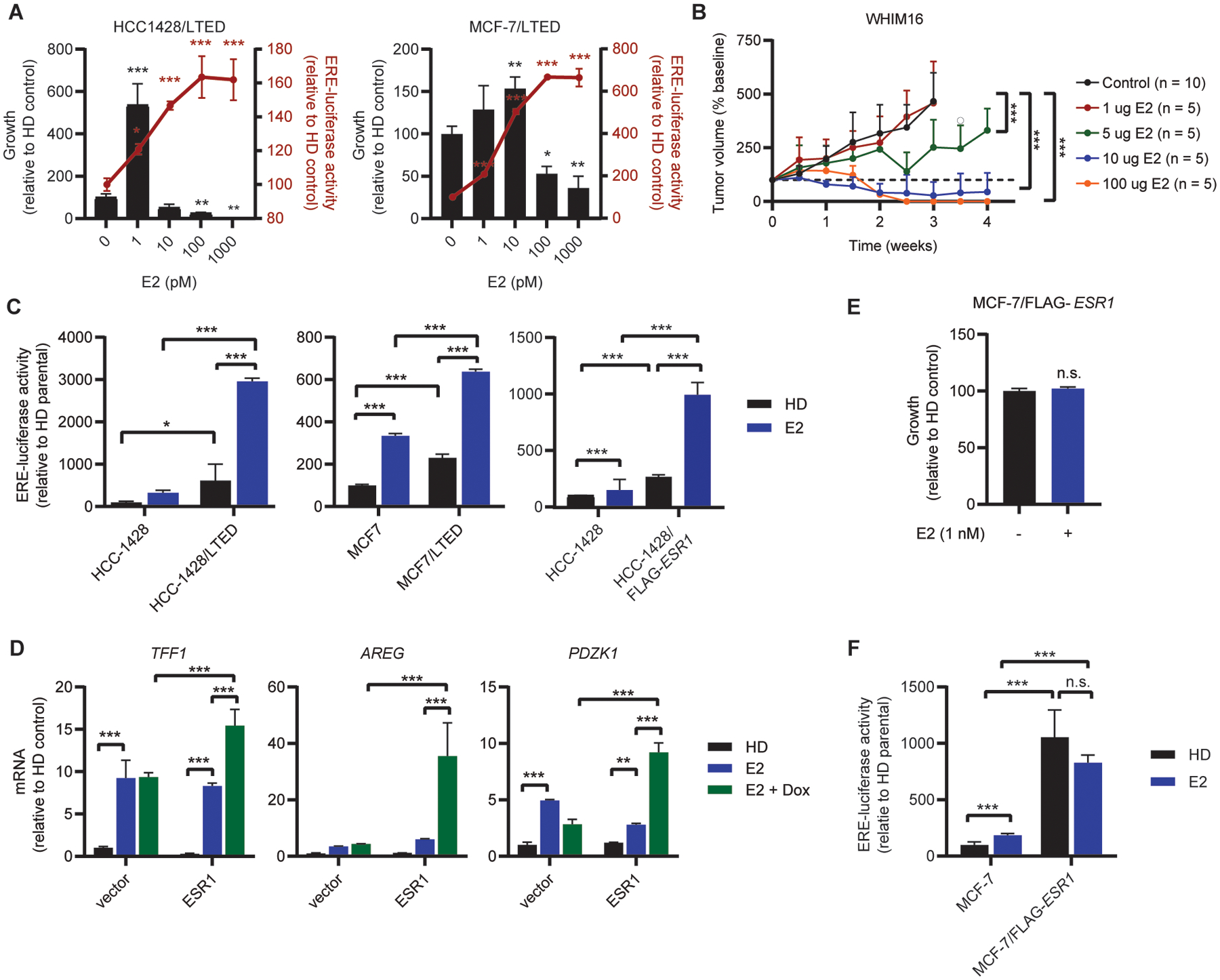
(A) Cells were seeded at low density and treated with E2 as indicated for 4 wk, then fixed and stained with crystal violet. Colony area was quantified using ImageJ (left y-axis). To measure ER activity, cells were transfected with ERE-driven firefly luciferase and CMV-Renilla. One day following transfection, cells were treated with E2 as indicated for 24 h, and luciferase activities were measured (right y-axis). (B) Ovx mice bearing WHIM16 tumors were randomized to treatment with E2 (p.o., BID) at the indicated doses. Tumor volumes were measured twice weekly. Data are shown as mean + SD. (C) HCC-1428 and MCF-7 parental, LTED, and FLAG-ESR1 cells were transfected with luciferase vectors and analyzed as in (A). Parental cells were hormone-deprived for 3 d prior to transfection. (D) RNA was isolated from T47D-pInducer20 cells treated ± 5 μM doxycycline for 2 wk, and ± 1 nM E2 for 24 h. Expression of indicated transcripts was analyzed by RT-qPCR and normalized to ACTB. (E) MCF-7/FLAG-ESR1 cells were seeded at low density, treated with E2 as indicated for 4 wk, and colony area analyzed as in (A). (F) MCF-7/FLAG-ESR1 and MCF-7 parental cells were transfected with luciferase vectors and analyzed as in (A). Parental cells were hormone-deprived for 3 d prior to transfection. Data are shown as mean of 3 (A/C/D/E) or 6 (F) replicates ± SD. *p≤0.05, **p≤0.005, ***p≤0.0005 by Bonferroni multiple comparison-adjusted post-hoc test (A/C/D/F), two-tailed t-test (E) or linear mixed modeling (B).
To further test this concept, we treated parental and LTED cells with 1 nM E2, a dose that induces parental cell growth but inhibits LTED cell growth (Fig. 1A). E2 treatment induced relative hyperactivation of ER in LTED cells compared to parental cells (Fig. 4C). Similarly, hyperactivation of ER was observed in HCC-1428/FLAG-ESR1 and dox-induced T47D-pInducer20-ESR1 cells treated with E2 compared to vector controls (Fig. 4C/D). Notably, MCF-7 cells constitutively overexpressing exogenous ESR1 (MCF-7/FLAG-ESR1) cells were not growth-inhibited by 1 nM E2 (Fig. 4E), which is likely due to a lack of ER hyperactivation upon E2 treatment compared to control-treated cells (Fig. 4F). These findings suggest that there is a window of optimal ER transcriptional activity for inducing cell/tumor growth, and a combination of high ER expression and E2 stimulation converge to provide ER hyperactivation that elicits anti-cancer effects.
Resistance to 17β-estradiol therapy is associated with ER downregulation
E2 treatment of mice induced complete regression of WHIM16 tumors. However, all tumors ultimately recurred during E2 treatment (Fig. 5A), indicating resistance to E2 therapy. Withdrawal of E2 from mice bearing such recurrent tumors stunted tumor growth (Fig. 5A), demonstrating that tumors that resumed growth during E2 treatment were partly growth-stimulated by E2. Tumors were collected at baseline (Day 0), after short-term (Day 3) and long-term (Day 56) E2 treatments, after recurrence on E2 (Day 126), and after E2 withdrawal (Day 129) for molecular analyses (outlined in Fig. 5B). E2 treatment initially decreased tumor cell proliferation (Day 0 vs. Day 3). In recurrent tumors, E2 withdrawal reduced proliferation (Day 126 vs. Day 129; Figs. 5C and S8). Apoptosis was apparent following 3 d of E2 treatment, but not at time points thereafter; this may explain in part why recurrent tumors were growth-inhibited but did not regress when treated with E2 withdrawal (Fig. 5A/C). E2 did not significantly increase PR expression in these tumors, which are PR+ at baseline in ovx mice (Day 0 vs. Day 3). However, in tumors that recurred on E2, withdrawal of E2 decreased PR levels (Day 126 vs. Day 129; Fig. 5C). These observations suggest that ER transcriptional activity is partly estrogen-dependent in tumors that recur on E2.
Figure 5. Resistance to E2 therapy is associated with downregulation of ER levels and activity.
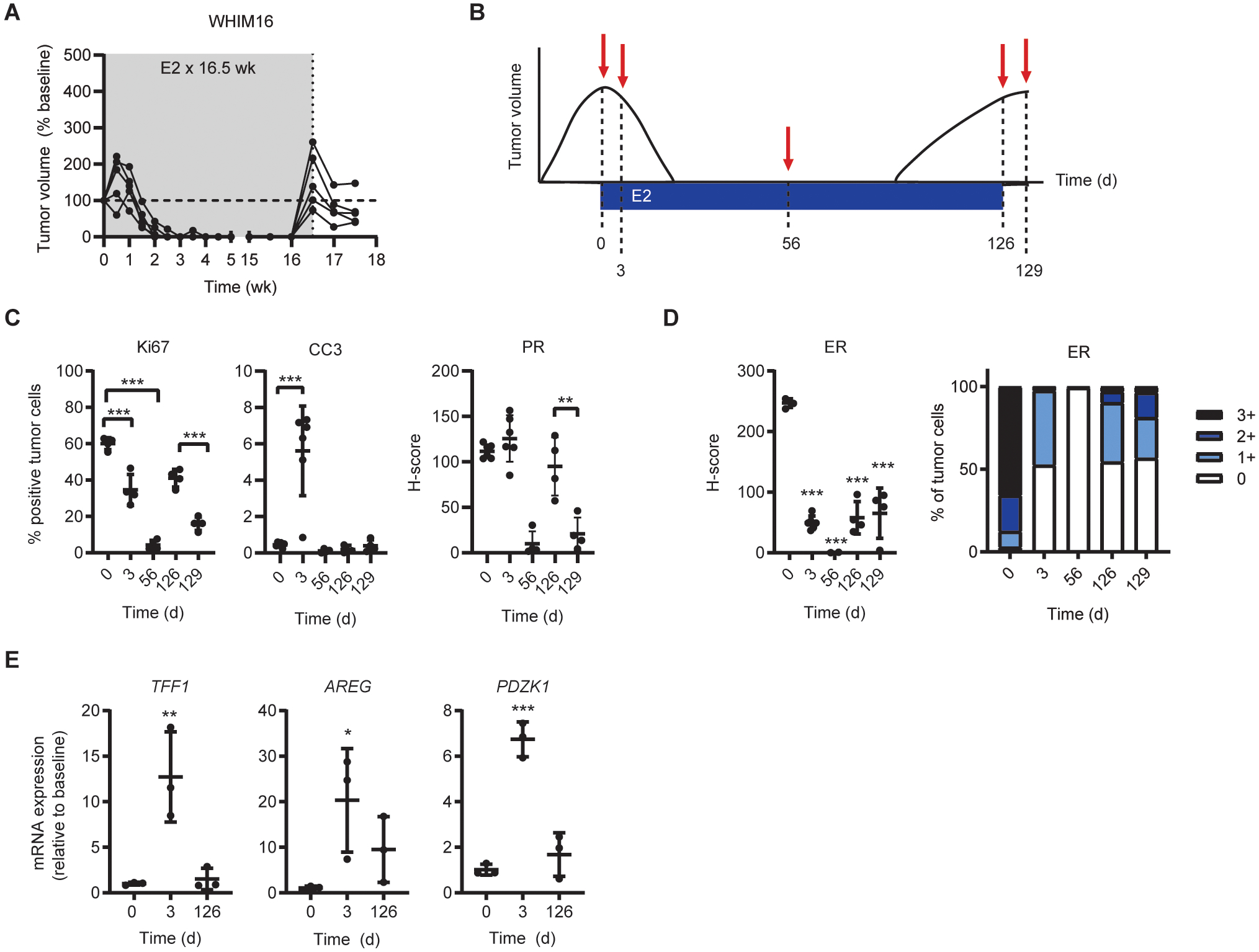
(A) Ovx mice bearing WHIM16 tumors were treated with E2 by s.c. pellet, replaced every 30 d (gray shading). When tumors re-grew to baseline volume, E2 was withdrawn. Tumor volumes were measured twice weekly. Each line represents one tumor. (B) Experimental design for comparison of tumors differentially sensitive to E2 and estrogen deprivation. Tumors were harvested for molecular analysis at indicated timepoints. (C/D) Expression of indicated proteins was analyzed by IHC, and expression was quantified. (E) RNA was isolated from tumor samples and analyzed by RT-qPCR. Expression of indicated transcripts was normalized to ACTB. Data are shown as mean ± SD. *p≤0.05, **p≤0.005, ***p≤0.0005 by Bonferroni multiple comparison-adjusted post-hoc test.
Consistent with the hypothesis that high levels of ER activity promote the anti-cancer effects of estrogen, tumors that recurred on E2 exhibited lower ER levels than at baseline, even after E2 withdrawal (compare Day 129 vs. Day 0 in Fig. 5D). Downregulation of ESR1 mRNA also occurred at Day 129 without loss of ESR1 gene amplification (Fig. S9A/B). Analysis of ER target gene expression indicated that E2 initially stimulated ER transcriptional activity (by Day 3) in tumors that regressed in response to E2, while expression of these genes return to near-baseline levels in tumors that acquired resistance to E2 (Fig. 5E). Interestingly, analysis of ER expression in surviving tumor cells at Day 56 suggested complete loss of ER at this time point. Breast cancer stem cells typically do not express ER (20, 21), which is consistent with the hypothesis that cells surviving E2 treatment are stem-like and later give rise to E2-resistant ER+ cells as observed at Day 126.
Cycling 17β-estradiol and estrogen deprivation provides long-term control of ER+ breast tumors
Since recurrent WHIM16 tumors are partially growth-stimulated by E2 (Fig. 5A/C), we hypothesized that residual tumor cells surviving E2 therapy could be targeted by switching to estrogen deprivation prior to the emergence of E2-resistant tumors. WHIM16 tumors in ovx mice treated with E2 for 74 d or 35 d recurred at similar time points (Fig. S10A–C), suggesting that after initial tumor regression, E2 treatment no longer elicited anti-tumor effects. Additionally, tumors that recurred following A) 35 d of E2 plus B) subsequent estrogen deprivation were growth-inhibited by a second round of E2 treatment (Fig. S10B), suggesting that oscillating between estrogen therapy and estrogen deprivation may be an effective strategy for controlling tumor growth long-term.
In order to refine the optimal treatment schedule for oscillating between E2 and estrogen deprivation therapies, ovx mice bearing WHIM16 tumors were treated with E2 for either 1 wk or 4 wk followed by estrogen deprivation, or treated continuously with E2. Tumors in all treatment groups regressed completely (Figs. 6A and S11A–D). Treatment with E2 for 1 wk (followed by estrogen deprivation) or with continuous E2 resulted in similar times to recurrence. In contrast, 4 wk of E2 treatment (followed by estrogen deprivation) significantly delayed recurrence compared to the other treatment schedules (Fig. 6B). These observations suggest that i) there is an optimal duration of E2 therapy to elicit maximal anti-cancer effects that extends beyond the time of maximum measurable response (i.e., change in volume), and ii) E2 may elicit anti-cancer effects at early time points and pro-cancer effects at later time points.
Figure 6. Cycling E2 and estrogen deprivation is an effective strategy for maintaining long-term tumor control.
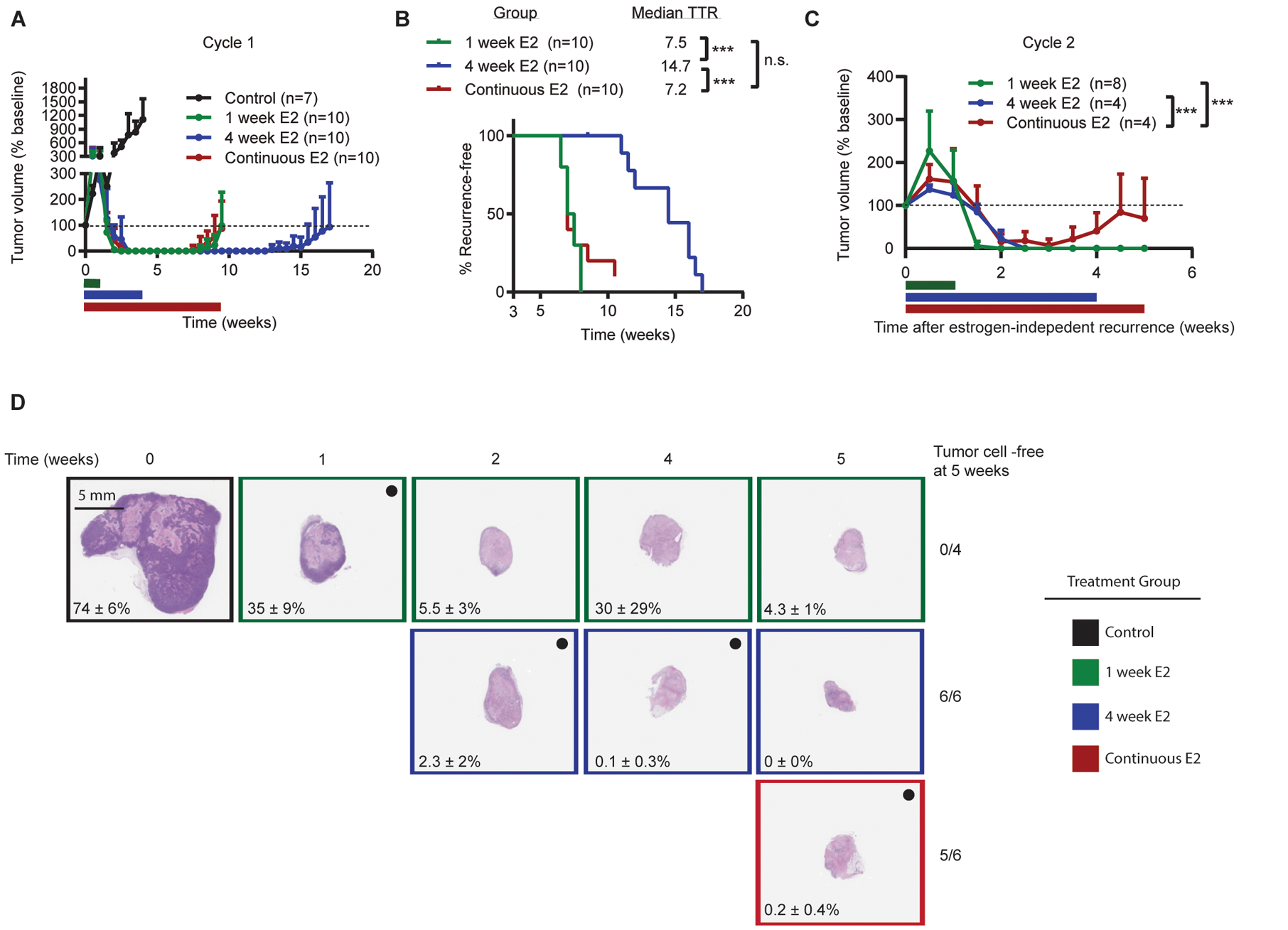
(A) Ovx mice bearing WHIM16 tumors were randomized to treatment ± E2 by s.c. pellet for 1 wk, 4 wk, or continuous treatment until tumors re-grew to baseline volume. Colored bars represent duration of E2 treatment. Horizontal bars indicate duration of E2 treatment. (B) Time to recurrence was measured as time from treatment start until tumors re-grew to baseline volume. (C) When tumors treated with E2 for 1 wk or 4 wk (followed by estrogen deprivation) re-grew to baseline, mice were re-treated with 1 wk or 4 wk of E2. When tumors treated with continuous E2 re-grew to baseline volume, mice were switched to estrogen deprivation; when these tumors resumed growth on estrogen deprivation, mice were re-treated with E2. (D) Tumors were harvested from mice treated ± E2 by s.c. pellet for 1 wk, 4 wk, or continuous treatment at the indicated timepoints. Tumor sections were H&E-stained, and the percentage of viable tumor cells in each section was quantified by a pathologist. Black dot indicates tumors receiving E2 treatment at time of analysis. Percentages indicate mean % viable tumor cells ± SD for each timepoint. Data are shown as mean + SD. *p≤0.05, ***p≤0.0005 by log-rank test (B) or nonlinear mixed modeling (C).
Nearly all WHIM16 tumors in mice treated with E2 on all schedules recurred (Figs. 6A/B and S11B–D). Mice bearing tumors that recurred on continuous E2 were then treated with estrogen deprivation; once tumors resumed estrogen-independent growth (defined as an increase in tumor volume at two consecutive timepoints), mice were re-challenged with E2. Similarly, mice with tumors that recurred after 1 wk or 4 wk of E2 followed by estrogen deprivation were re-challenged with E2 using the same schedule as in the prior cycle (Fig. 6C). In mice exposed to first-line short-term (1 or 4 wk) E2, tumors completely regressed during the second cycle of E2 treatment. However, mice exposed to first-line continuous E2 showed incomplete tumor regression when re-challenged with E2 (Figs. 6C and S11B–D). These results indicate that pre-emptively switching between E2 and estrogen deprivation therapies before resistant tumors emerge is an effective strategy for long-term control of tumor burden.
To better understand tumor cell responses to different treatment schedules, tumors were acquired from mice treated with E2 using the above-described schedules. Tumors from mice treated with E2 for ≥2 wk contained few viable malignant cells (Fig. 6D). Tumors from mice treated with 1 wk of E2 followed by estrogen deprivation contained more tumor cells than those treated with E2 for 2–5 wk, suggesting that 1 wk of E2 is less effective at eliminating tumor cells. By Week 4 of E2 treatment, there were no detectable tumor cells in resected tumor beds. At week 5 of treatment, all mice on the 4 wk treatment schedule remained free of detectable tumor cells, while only 5/6 tumor beds from mice on continuous E2 treatment remained tumor cell-free (Fig. 6D). These observations may explain in part the improved long-term outcome of the 4-week E2 treatment schedule compared to the 1-week and continuous schedules.
DISCUSSION
Several previous studies have suggested that high levels of ER are associated with resistance to anti-estrogen therapies. A study by Li et al. demonstrated that T47D cells overexpressing wild-type ER exhibited increased growth under estrogen deprivation compared to vector controls (14). Amplification of ESR1 was also associated with resistance to adjuvant tamoxifen in patients with early stage ER+ breast cancer (22). Prior reports also hinted at a connection between ESR1 amplification and response to estrogen therapy. MCF-7/LTED cells and WHIM16 PDX tumors exhibit amplification of ESR1 (14, 15), and both are growth-inhibited by E2. A patient with ESR1-amplified metastatic breast cancer was also noted to experience a partial response upon treatment with E2 (13). However, a direct relationship between ER expression levels and the anti-cancer effects of estrogen therapy has not been previously demonstrated.
Numerous clinical studies have demonstrated the therapeutic efficacy of estrogen therapy in patients with ER+ breast cancer (5–11, 23–25). One of the major clinical hurdles to more widespread use of estrogen therapy for breast cancer is the lack of a predictive marker to select patients likely to benefit from treatment. A study by Carter et al. demonstrated that higher doses of the estrogen diethylstilbestrol increased response rates in post-menopausal breast cancer patients, but the duration of response did not differ between dose cohorts (23). This suggests that a threshold of ER activation exists for therapeutic response to estrogen. We surmise that tumors expressing higher levels of ER will require less exogenous E2 to surpass this threshold, while tumors expressing lower levels of ER are unlikely to achieve sufficient ER activation to trigger a therapeutic response with doses of E2 safely achievable in humans. Taken together with our results, this suggests that patients with very high levels of ER expression are most likely to benefit from estrogen therapy.
Our work indicates that amplification of ESR1 is likely predictive of response to estrogen therapy. Although uncommon in primary tumors (26), ESR1 copy number gain is detected in a subset (13–25%) of metastatic ER+ breast tumors, and increases in ESR1 copy number are correlated with increased ESR1 mRNA expression (26, 27). However, identifying patients as candidates for estrogen therapy based on tumor ESR1 amplification would likely miss patients with high tumor ER expression in the absence of ESR1 copy number gain (as in HCC-1428/LTED cells) who could still benefit from treatment. Further work is necessary to determine what constitutes ‘high’ ER expression in tumors, and what other tumor features contribute to sensitivity to estrogen therapy. Our data suggest that high levels of ER transcriptional activation are required to elicit anti-cancer effects upon estrogen treatment; why some but not all ER-overexpressing models further activate ER upon treatment with E2 remains to be elucidated.
Some patients with acquired resistance to AI therapy are re-sensitized to treatment with AIs after experiencing clinical benefit from E2 therapy (8). Our results suggest that this re-sensitization is due to the downregulation of ER upon acquisition of resistance to E2 therapy. Additionally, our data suggest that cycling between estrogen therapy and estrogen deprivation therapy prior to the emergence of resistance is more effective at controlling tumor growth than either treatment strategy alone. Although unconventional, alternation of treatments pre-emptively before recurrence may result in improved long-term disease management. Short-term E2 treatment may also reduce adverse events, thereby increasing the therapeutic index of E2. However, one limitation of our study is the lack of available in vivo models to test the generalizability of the finding that short-term E2 is superior to continuous E2 treatment. We were unable to establish LTED cell line xenografts in ovx mice, likely due to the fact that the measured levels of serum E2 in ovx mice is approximately 10 pg/mL (equivalent to ~30 pM), which is a growth-inhibiting dose for LTED cells in vitro (Fig. 4A and S7F).
Although we tested alternation between estrogen treatment and estrogen deprivation, oscillation between activation and inhibition of ER activity may also be successful with other anti-estrogen therapies. Direct comparison of treatment with the SERD fulvestrant vs. the AI anastrozole in patients with advanced ER+ breast cancer demonstrated that fulvestrant provided longer time-to progression (28). In addition, approximately 30% of patients with tumors exhibiting acquired resistance to AI therapy respond to treatment with fulvestrant (29, 30). This raises the possibility that cycling between estrogen and fulvestrant therapies may be beneficial in patients with acquired resistance to AIs. However, the long half-life of fulvestrant (31) may prevent rapid changes in ER activity upon cycling treatments, limiting the efficacy of this treatment strategy. Further work is necessary to determine whether the observed benefit of alternating estrogen and estrogen deprivation can be replicated with other types of anti-estrogen therapies.
Similar to estrogen therapy in breast cancer, treatment with androgens elicits anti-cancer effects in a subset of patients with castration-resistant prostate cancer (32, 33), and treatment with intermittent high doses of androgens (i.e., bipolar androgen therapy) has been used as a strategy to delay the onset of drug resistance (34, 35). In accordance with our results, high levels of androgen receptor (AR) expression are associated with therapeutic response to androgens (36, 37).
Our findings raise the question of why high levels of ER activation are toxic to breast cancer cells. Previous work, conducted primarily in LTED derivatives of MCF-7 cells, demonstrated that E2 induces an unfolded protein response that drives apoptosis (15, 38–41); the mechanism underlying the initiation of this unfolded protein response has not been reported. It is reasonable to infer that high levels of ER activation could flood cells with new transcripts to be translated, inducing a sudden influx of newly translated polypeptides and resulting in an unfolded protein response. ER also regulates its own expression in a negative feedback loop, with high levels of ER activation resulting in downregulation of ESR1 transcription (42–44). It is therefore also possible that following the initial high levels of ER activation stimulated with estrogen therapy, ER expression is downregulated enough to elicit anti-estrogen effects.
In conclusion, the effects of high ER expression are context-dependent, providing a growth advantage under estrogen deprivation but a disadvantage in E2-replete conditions. High expression of ER is therefore targetable with estrogen therapy, and modulating ER activation by cycling estrogen and estrogen deprivation provides long-term control of tumor growth. Further work is necessary to determine how to best apply this knowledge clinically to benefit patients.
MATERIALS AND METHODS
Cell culture
Parental cancer cell lines (MCF-7, HCC-1428, T47D) were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA), and LentiX cells were obtained from Clontech (Mountain View, CA, USA). MCF-7, HCC-1428, T47D, and LentiX cells were cultured in DMEM with 10% FBS (HyClone Laboratories; Logan, UT, USA). MCF-7/LTED cells were a gift from Matthew Ellis (Baylor College of Medicine, Houston, TX, USA) (45). HCC-1428/LTED cells were generated through long-term (>1 yr) culture in phenol-red-free DMEM with 10% dextran/charcoal-treated FBS (DCC-FBS; HyClone Laboratories) as previously described (46). LTED cells were maintained in phenol-red-free DMEM + 10% DCC-FBS supplemented with 2 mM GlutaMAX (ThermoFisher Scientific, Waltham, MA, USA), and were passaged using phenol-red-free 0.25% trypsin plus 2.21 mM EDTA (Corning, Tewksbury, MA, USA). Cell lines were verified by STR genotyping at the University of Vermont Cancer Center DNA Analysis Facility and confirmed to be free of mycoplasma (Universal Mycoplasma Detection Kit; ATCC). Assays were performed using cells cultured for <3 months thereafter.
Generation of stably transfected cell lines
Plasmids encoding SmartVector doxycycline-inducible shRNA targeting ESR1 or non-coding control were obtained from Dharmacon (cat. # V3SH11252–225116931 and V3SH11252–225648231; Lafayette, CO, USA; Table S2). The pInducer20 lentiviral genome plasmid (16) was a gift from Thomas Westbrook (Baylor College of Medicine). The Gateway PLUS Shuttle Clone encoding ESR1 cDNA was obtained from GeneCopoeia (cat. # GC-A0322-B; Rockville, MD, USA), and the ESR1 sequence was inserted into pInducer20 by Gateway Cloning (GenScript, Piscataway, NJ, USA). LentiX cells were cultured in phenol red-free DMEM with 10% DCC-FBS for 3 d prior to transfection. Lentivirus was produced in LentiX cells using a standard three-plasmid system consisting of a genome plasmid (SmartVector/shControl, SmartVector/shESR1, or pInducer-ESR1), pMD2.G (#12259), and psPAX2 (#12260) (the latter 2 plasmids were gifts from Didier Trono obtained from Addgene; Watertown, MA, USA). Plasmids were transfected into LentiX cells using polyethylenimine. Media containing lentivirus was used to transduce MCF7/LTED, HCC1428/LTED, HCC-1428, MCF-7, and T47D cells, and stably transfected cells were selected with G418 (2 μg/mL) or puromycin (1 μg/mL) for 1 wk. Parental cell lines were hormone-deprived for 3 d prior to lentiviral transduction. Cells were selected with puromycin (1 μg/mL) for 1 wk, and stably expressing cell lines were maintained in hormone-depleted medium.
Luciferase Reporter Assays
Plasmids encoding firefly-luciferase under an estrogen-response element (ERE)-driven promoter (gift from Dorraya El-Ashry, University of Minnesota) and CMV-Renilla luciferase (Promega, Madison, WI, USA) were co-transfected into cells. The day following transfection, cells were treated with E2 as indicated for 24 h. Luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) as per manufacturers’ instructions. Firefly luciferase signal was normalized to Renilla transfection control signal.
Mouse Studies
Studies were approved by the Dartmouth College IACUC. Ovariectomized (ovx) female NOD/SCID/Il2γ−/−(NSG) mice were obtained from the Norris Cotton Cancer Center Mouse Modeling Shared Resource. The WHIM16 tumor model was obtained from the Washington University HAMLET Core (St. Louis, MO, USA). Tumor fragments (~8 mm3) were implanted subcutaneously (s.c.). into ovx mice aged 4–6 weeks. Tumor dimensions were measured twice weekly with calipers, and volumes were calculated as [length × width2/2]. When tumor volume reached ≥200 mm3, mice were randomized to treatment groups. No specific randomization or blinding techniques were used for these studies. E2 was administered by s.c. beeswax pellet containing 1 mg E2 (47). For long-term E2 treatment, pellets were replaced every 30 d. Fulvestrant (Ontario Chemicals, Inc., Guelph, ON, Canada) was dissolved in ethanol at 500 mg/mL, and diluted 10-fold with castor oil to yield a 50-mg/mL solution. Fulvestrant (5 mg/mouse) or vehicle was administered weekly by s.c. injection. Letrozole (Selleck Chemicals, Houston, TX, USA) was dissolved in 0.5% methylcellulose + 0.2% Tween-20, and 5 mg/kg was administered daily by intraperitoneal injection. Tumors harvested for molecular analyses were cut in half and either formalin-fixed and paraffin-embedded (FFPE), or frozen in liquid nitrogen.
Immunoblotting
All chemicals were purchased from Sigma (St. Louis, MO, USA) unless noted otherwise. Cells were lysed in RIPA buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 5 mM NaPPi, 50 mM NaF, 10 mM Na β-glycerophosphate) plus fresh HALT protease inhibitor cocktail (Pierce; Rockford, IL, USA) and 1 mM Na3VO4 (New England Biolabs; Ipswich, MA, USA). Lysates were sonicated for 15 s and centrifuged at 17 000 × g for 10 min at 4°C. Protein content in supernatant was quantified by BCA assay (Pierce). Protein extracts were reduced and denatured using NuPAGE (ThermoFisher Scientific) plus 1.25% β-mercaptoethanol. SDS-PAGE-separated proteins were transferred to nitrocellulose, and membranes were stained with Ponceau S to visually confirm even protein loading. Blots were probed with antibodies against ER (Santa Cruz Biotechnology, Dallas, TX, USA; cat.# sc-8002), vinculin, β-actin (Cell Signaling Technology; Danvers, MA, USA; cat.# 13901), and FLAG (Millipore Sigma; Burlington, MA, USA; cat.# F3165). Signal was detected using horseradish-peroxidase labeled secondary antibodies (GE Healthcare; Waukesha, WI, USA) and ECL substrates (Pierce).
Histology and immunohistochemistry (IHC)
Slide-mounted 5-micron sections of FFPE tissue were deparaffinized in xylene and rehydrated in a graded ethanol series. For histology, sections were stained with Eosin-Y (Richard-Allan Scientific, San Diego, CA, USA) and counterstained with hematoxylin (Richard-Allan Scientific). Viable tumor cells in hematoxylin and eosin (H&E) stained slides were quantified by a pathologist. For IHC, heat-induced antigen retrieval was performed in either Tris-EDTA buffer, pH 9 or citrate buffer, pH 6 (VWR; Radnor, PA, USA). Sections were permeabilized in 0.2% Triton X-100 in PBS, and blocked in 5% goat serum in PBS for 30 min. Sections were incubated overnight at 4°C in primary antibody against ER (Dako; Santa Clara, CA, USA; cat.# IR084), Ki67 (BioCare Medical; Pacheco, CA, USA; cat.# 325), cleaved caspase-3 (Cell Signaling Technology; cat.# 9664), or progesterone receptor A/B (Cell Signaling Technology; cat.# 3153). Sections were treated with in 0.3% hydrogen peroxide, and signal was developed using VectaStain Elite ABC-HRP kit and DAB substrate (Vector Laboratories; Burlingame, CA, USA). Sections were counterstained with hematoxylin. IHC staining in each section was quantified in 3 representative microscopic fields at 200x magnification using HALO software (Indica Labs; Alburquerque, NM, USA).
qPCR and RT-qPCR
RNA was isolated from cells and frozen tumors using RNeasy Universal Plus Mini Kit (Qiagen; Germantown, MD, USA). RNA was reverse-transcribed using the iScript cDNA Synthesis kit (Bio-Rad; Hercules, CA, USA). Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen). Real-time qPCR was performed using iQ SYBR Green SuperMix (Bio-Rad) using the Bio-Rad CFX96 thermocycler. Data was analyzed using the ΔΔCT method. Primer sequences are listed in Table S1.
Growth Assays
Cells were seeded in triplicate at 2500 cells/well in 6-well plates, and treated as indicated for 4 wk. Parental cells were hormone-deprived for 3 d prior to seeding. Cells were fixed and stained with 0.5% crystal violet in 20% methanol for 10 min, then rinsed with water. Plates were scanned, and area fraction of staining was determined for each well using ImageJ software.
Statistical analysis
Data from IHC, long-term growth assays, RT-qPCR, and luciferase assays were analyzed by two-tailed t-test (two-sample experiments) or ANOVA with Bonferroni-adjusted post-hoc testing (experiments with 3 or more samples). Cell culture experiments were repeated on 3 independent occasions, and the data met the assumptions of all tests. Tumor growth data was analyzed using the following linear mixed model : Log10(tumor volumeit) = ai + b * t + eit, where i represents the ith mouse, t represents time of tumor volume measurement, ai represents the mouse-specific log tumor volume at t = 0, b represents the rate of tumor volume growth, and eit represents deviation of measurements from the model over time (48, 49). Mouse heterogeneity (the baseline tumor volume) is represented by variance of ai, and b*loge(10) * 100 indicates tumor volume increase (%) per week. Treatment groups were compared using a z-test for slopes with standard error derived from the output of the function lme from the library nlme in R. Mouse studies were performed once. Sample size for initial tumor growth experiments were based on an untreated tumor growth rate of 8.7% per day. Power analysis indicated that with n=10 mice per group a minimum difference of 1.6% per day between two treatment groups would be detectable with Type I error of 5% and 80% power. Sample size was modified in subsequent experiments to account for larger anticipated treatment effects.
For overall survival (OS) analyses, a cohort of patients with hormone-therapy treated ER+/HER2- breast tumors were selected using cBioPortal (50, 51); patients were grouped by ESR1 copy number (amplified vs. non-amplified) or by ESR1 expression Z-score, stratified on the median. For recurrence-free survival analysis, a cohort of patients with ER+/HER2- breast cancer were selected for analysis of recurrence-free survival using Kaplan-Meier Plotter (19). Statistical analysis of survival curves was performed by log-rank test, with post-hoc Bonferroni correction for multiple comparisons.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by Susan G. Komen (CCR1533084 to TWM) and NIH (R01CA200994 and R01CA211869 to TWM, F31CA243409 to NAT, Dartmouth College Norris Cotton Cancer Center Support Grant P30CA023108). We thank the following Norris Cotton Cancer Center Shared Resources for their support: Mouse Modeling; Pathology; Biostatistics; Microscopy; Genomics.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–51. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. [DOI] [PubMed] [Google Scholar]
- 3.Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M, et al. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11(12):1135–41. [DOI] [PubMed] [Google Scholar]
- 4.Haddow A, Watkinson JM, Paterson E, Koller PC. Influence of Synthetic Oestrogens on Advanced Malignant Disease. Br Med J. 1944;2(4368):393–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingle JN, Ahmann DL, Green SJ, Edmonson JH, Bisel HF, Kvols LK, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304(1):16–21. [DOI] [PubMed] [Google Scholar]
- 6.Beex L, Pieters G, Smals A, Koenders A, Benraad T, Kloppenborg P. Tamoxifen versus ethinyl estradiol in the treatment of postmenopausal women with advanced breast cancer. Cancer Treat Rep. 1981;65(3–4):179–85. [PubMed] [Google Scholar]
- 7.Lonning PE, Taylor PD, Anker G, Iddon J, Wie L, Jorgensen LM, et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67(2):111–6. [DOI] [PubMed] [Google Scholar]
- 8.Zucchini G, Armstrong AC, Wardley AM, Wilson G, Misra V, Seif M, et al. A phase II trial of low-dose estradiol in postmenopausal women with advanced breast cancer and acquired resistance to aromatase inhibition. Eur J Cancer. 2015;51(18):2725–31. [DOI] [PubMed] [Google Scholar]
- 9.Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302(7):774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrawal A, Robertson JF, Cheung KL. Efficacy and tolerability of high dose “ethinylestradiol” in post-menopausal advanced breast cancer patients heavily pre-treated with endocrine agents. World J Surg Oncol. 2006;4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, et al. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: a prospective study. Br J Cancer. 2013;109(6):1537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Council on Drugs. Androgens and estrogens in the treatment of disseminated mamary carcinoma : Retrospective study of 1944 patients. JAMA. 1960;172:1271–83. [Google Scholar]
- 13.Kota K, Brufsky A, Oesterreich S, Lee A. Estradiol as a Targeted, Late-Line Therapy in Metastatic Breast Cancer with Estrogen Receptor Amplification. Cureus. 2017;9(7):e1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hosford SR, Shee K, Wells JD, Traphagen NA, Fields JL, Hampsch RA, et al. Estrogen therapy induces an unfolded protein response to drive cell death in ER+ breast cancer. Mol Oncol. 2019;13(8):1778–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A. 2011;108(9):3665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu NY, Iftimi A, Yau C, Tobin NP, van ‘t Veer L, Hoadley KA, et al. Assessment of Long-term Distant Recurrence-Free Survival Associated With Tamoxifen Therapy in Postmenopausal Patients With Luminal A or Luminal B Breast Cancer. JAMA Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li QY, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Tr. 2010;123(3):725–31. [DOI] [PubMed] [Google Scholar]
- 20.Simoes BM, O’Brien CS, Eyre R, Silva A, Yu L, Sarmiento-Castro A, et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015;12(12):1968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma R, Karthik GM, Lovrot J, Haglund F, Rosin G, Katchy A, et al. Estrogen Receptor beta as a Therapeutic Target in Breast Cancer Stem Cells. J Natl Cancer Inst. 2017;109(3):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen KV, Ejlertsen B, Muller S, Moller S, Rasmussen BB, Balslev E, et al. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res Treat. 2011;127(2):345–55. [DOI] [PubMed] [Google Scholar]
- 23.Carter AC, Sedransk N, Kelley RM, Ansfield FJ, Ravdin RG, Talley RW, et al. Diethylstilbestrol: recommended dosages for different categories of breast cancer patients. Report of the Cooperative Breast Cancer Group. JAMA. 1977;237(19):2079–8. [PubMed] [Google Scholar]
- 24.Peethambaram PP, Ingle JN, Suman VJ, Hartmann LC, Loprinzi CL. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res Treat. 1999;54(2):117–22. [DOI] [PubMed] [Google Scholar]
- 25.Matelski H, Greene R, Huberman M, Lokich J, Zipoli T. Randomized trial of estrogen vs. tamoxifen therapy for advanced breast cancer. Am J Clin Oncol. 1985;8(2):128–33. [DOI] [PubMed] [Google Scholar]
- 26.Basudan A, Priedigkeit N, Hartmaier RJ, Sokol ES, Bahreini A, Watters RJ, et al. Frequent ESR1 and CDK Pathway Copy-Number Alterations in Metastatic Breast Cancer. Molecular Cancer Research. 2019;17(2):457–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. Journal of Clinical Oncology. 2016;34(16):1872–+. [DOI] [PubMed] [Google Scholar]
- 28.Robertson JF, Llombart-Cussac A, Rolski J, Feltl D, Dewar J, Macpherson E, et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: results from the FIRST study. J Clin Oncol. 2009;27(27):4530–5. [DOI] [PubMed] [Google Scholar]
- 29.Perey L, Paridaens R, Hawle H, Zaman K, Nole F, Wildiers H, et al. Clinical benefit of fulvestrant in postmenopausal women with advanced breast cancer and primary or acquired resistance to aromatase inhibitors: final results of phase II Swiss Group for Clinical Cancer Research Trial (SAKK 21/00). Ann Oncol. 2007;18(1):64–9. [DOI] [PubMed] [Google Scholar]
- 30.Ingle JN, Suman VJ, Rowland KM, Mirchandani D, Bernath AM, Camoriano JK, et al. Fulvestrant in women with advanced breast cancer after progression on prior aromatase inhibitor therapy: North Central Cancer Treatment Group Trial N0032. J Clin Oncol. 2006;24(7):1052–6. [DOI] [PubMed] [Google Scholar]
- 31.Robertson JF, Harrison M. Fulvestrant: pharmacokinetics and pharmacology. Br J Cancer. 2004;90 Suppl 1:S7–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mathew P Prolonged control of progressive castration-resistant metastatic prostate cancer with testosterone replacement therapy: the case for a prospective trial. Ann Oncol. 2008;19(2):395–6. [DOI] [PubMed] [Google Scholar]
- 33.Szmulewitz R, Mohile S, Posadas E, Kunnavakkam R, Karrison T, Manchen E, et al. A randomized phase 1 study of testosterone replacement for patients with low-risk castration-resistant prostate cancer. Eur Urol. 2009;56(1):97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schweizer MT, Wang H, Luber B, Nadal R, Spitz A, Rosen DM, et al. Bipolar Androgen Therapy for Men With Androgen Ablation Naive Prostate Cancer: Results From the Phase II BATMAN Study. Prostate. 2016;76(13):1218–26. [DOI] [PubMed] [Google Scholar]
- 35.Teply BA, Wang H, Luber B, Sullivan R, Rifkind I, Bruns A, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol. 2018;19(1):76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: results from a pilot clinical study. Sci Transl Med. 2015;7(269):269ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chatterjee P, Schweizer MT, Lucas JM, Coleman I, Nyquist MD, Frank SB, et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J Clin Invest. 2019;130:4245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, Ramos P, et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A. 2011;108(47):18879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan P, Cunliffe HE, Maximov PY, Agboke FA, McDaniel RE, Zou X, et al. Integration of Downstream Signals of Insulin-like Growth Factor-1 Receptor by Endoplasmic Reticulum Stress for Estrogen-Induced Growth or Apoptosis in Breast Cancer Cells. Mol Cancer Res. 2015;13(10):1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan P, Griffith OL, Agboke FA, Anur P, Zou X, McDaniel RE, et al. c-Src modulates estrogen-induced stress and apoptosis in estrogen-deprived breast cancer cells. Cancer Res. 2013;73(14):4510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan P, Tyagi AK, Agboke FA, Mathur R, Pokharel N, Jordan VC. Modulation of nuclear factor-kappa B activation by the endoplasmic reticulum stress sensor PERK to mediate estrogen-induced apoptosis in breast cancer cells. Cell Death Discov. 2018;4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berkenstam A, Glaumann H, Martin M, Gustafsson JA, Norstedt G. Hormonal regulation of estrogen receptor messenger ribonucleic acid in T47Dco and MCF-7 breast cancer cells. Mol Endocrinol. 1989;3(1):22–8. [DOI] [PubMed] [Google Scholar]
- 43.Saceda M, Lippman ME, Chambon P, Lindsey RL, Ponglikitmongkol M, Puente M, et al. Regulation of the estrogen receptor in MCF-7 cells by estradiol. Mol Endocrinol. 1988;2(12):1157–62. [DOI] [PubMed] [Google Scholar]
- 44.Ellison-Zelski SJ, Solodin NM, Alarid ET. Repression of ESR1 through actions of estrogen receptor alpha and Sin3A at the proximal promoter. Mol Cell Biol. 2009;29(18):4949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, Gao F, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13(2):R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120(7):2406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeRose YS, Gligorich KM, Wang G, Georgelas A, Bowman P, Courdy SJ, et al. Patient-derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol. 2013;Chapter 14:Unit14 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demidenko E Mixed models : theory and applications with R. Second edition. ed. Hoboken, New Jersey: Wiley; 2013. xxvii, 717 pages p. [Google Scholar]
- 49.Demidenko E Advanced statistics with applications in R. Hoboken, NJ: Wiley,; 2020. [Google Scholar]
- 50.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.