

Abstract
The aim of this study was to develop optimal micelles loaded with calcipotriol (Cal) and paclitaxel (PTX) for the treatment of pancreatic ductal adenocarcinoma (PDAC) using a Design of Experiment (DOE) approach. The central composite design (CCD), a type of DOE was used to tune the size and drug release properties of the drug-loaded micelles. This approach yielded optimal Cal and PTX co-loaded micelles (M-Cal/PTX) with size of 40-100 nm, a polydispersity index (PDI) of 0.25 and a zeta potential (ζ) of − 6.2 ± 0.8 mV. When evaluated in vitro, drug release from the micelles showed a biphasic pattern. The initial release, defined as the cumulative 2-hr drug release was less than 25% in all relevant media. This phase was followed by a gradual release with less than 80% of drugs released after 5 days. In vivo, the micelles prolonged the apparent biological half-life of Cal by more than 3 times and a marginal increase for PTX in an orthotopic mouse model of PDAC. The micelle-encapsulated drugs showed extended tumor accumulation when compared to non-encapsulated Cal and PTX at equivalent dose levels. Future studies on the antitumor activity of this novel dual drug payload delivery system are warranted.
Keywords: Calcipotriol, Paclitaxel, Micelle, Central composite design
Graphical Abstract
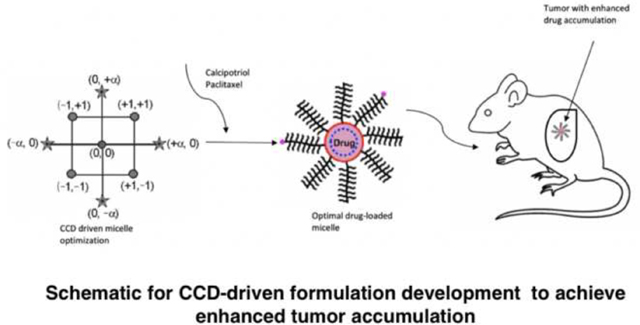
1. Introduction
Despite intense efforts, pancreatic ductal adenocarcinoma (PDAC) remains a treatment-refractory malignancy with a 5-year survival rate of 6% (Hall et al., 2018; Mcguigan et al., 2018). A literature search shows the survival rate is quite diverse, ranging from 2% to 9% (Ilic et al., 2016; Luo et al., 2013). Gemcitabine was shown to improve symptoms and extend survival in patients (Burris et al., 1997). However, since 1997, only two systemic therapies have shown marginal improvement in survival compared to gemcitabine monotherapy, but with serious side effects including febrile neutropenia, cardiac decompensation and sensory neuropathy which sometimes result in death (Vaccaro et al., 2011). The characteristic dense stromal cells in pancreatic tumors are known to impede drug delivery and contribute to chemoresistance.
A strategy that targets both tumor and stroma and improves drug delivery into these compartments has since been proposed as an alternative that could significantly improve survival outcomes in PDAC (Zhao et al., 2018a). Recently, it was shown that vitamin-D receptor-mediated stromal reprograming could improve pancreatic cancer therapy (Sherman et al., 2014). Calcipotriol (Cal), a synthetic vitamin-D analogue has shown promise in its ability to de-activate cancer associated fibroblasts within the stromal milieu, effectively “breaking” the stromal barrier to enhance the delivery of chemotherapy into the tumor (Sherman et al., 2014). Even as the least hypercalcemia-inducing compound within the vitamin-D derivatives class, Cal remains toxic at high doses, limiting its potential clinical use. Cal is an agonist of the vitamin D receptor (VDR), a ubiquitously expressed receptor found in a variety of tissues and controls the activity of several genes. Following activation, the VDR triggers a cascade of reactions that result in modifications in transcriptional output of genes (Pike and Meyer, 2012). Therefore, targeted activation of the VDR within the stroma is desirable to reduce off-target effects. Nanotechnology-based drug delivery systems have been proposed to offer the possibilities for efficient tumor targeting with diminished drug-related adverse events (Meng and Nel, 2018).
This study focused on developing a nano-polymeric micellar drug delivery system, loaded with Cal and PTX for the treatment of PDAC, using a Design of Experiment (DOE) approach. Two nanotechnology-based therapeutics, namely, Abraxane (albumin-bound PTX) and Onivyde (liposomal formulation of irinotecan), are currently available and prescribed for various types of cancer (Shi et al., 2017; Zhang et al., 2016). Conjugation of albumin to PTX improved its toxicity profile when compared to cremophor-based PTX formulation (Kim, 2017), and a combination regimen of albumin-bound PTX and gemcitabine out-performed gemcitabine monotherapy. Albumin-bound PTX is recognized to have some effects on stromal cells and modulates the tumor microenvironment. The improved therapeutic effect is largely attributed to this stromal-modulatory properties (Alvarez et al., 2013). However, these gains remain very modest. To this effect, our dual-payload drug delivery platform is designed to carry both a cytotoxic payload (PTX), and a bonafide stroma-modulating agent (Cal) for simultaneous targeting of stromal and tumor compartments.
We systematically optimized the delivery vehicle using the DOE approach to obtain a pre-specified micelle size and extend the blood circulation times of Cal and PTX. DOE is a mathematical modeling paradigm that involves developing relationships between process variables and response/output. Traditionally, at least in the area of drug development, this approach has been confined to optimizing process parameters (Gupta et al., 2015; Hejri et al., 2013; Zhang et al., 2013). Here, we extended its application to the selection of monomer units and degree of crosslinking in the synthesis of a polymeric drug delivery platform. Within the DOE domain, we used the central composite design (CCD) to establish a design space with a response surface, using fractional factorial designs with defined center and axial points to estimate the surface curvature. We characterized the synthesized polymers through the shape, size, storage stability and in vitro drug release profiles of Cal and PTX in the micelles. Finally, we studied the biodistribution of Cal and PTX from M-Cal/PTX in an aggressive orthotopic mouse model of PDAC with KrasG12D mutation.
2. Materials and Methods
2.1. Materials
Cal was purchased from Cayman Chemical Company (Ann Arbor, MI, USA) and PTX from LC laboratories (Woburn, MA. USA). The internal standards (IS) for Cal, calcipotriol-d4 (Cal-d4) and for PTX, paclitaxel-d5 (PTX-d5) were purchased from CRO laboratories Inc. (Dallas, TX, USA). Bead mill homogenizer (Model: Storm BBY24M) and bead lysis kits (Navy RINO screw 10 cap tubes) were purchased from Next Advantage (Troy, NY. USA). LC-MS grade water, methanol, hexane, isopropyl alcohol and dichloromethane were purchased from EMD Millipore Corporation (Billerica, MA. USA). All solutions were ultrasonically degassed before use. Blank C57BL6 whole blood (with Na EDTA) was purchased from Innovative Research (Novi, MI. USA). All other chemical reagents of ACS grade were purchased from Sigma-Aldrich (St. Louis, MO) or VWR (West Chester, PA).
2.2. Polymer synthesis
2.2.1. Polymer reaction initiator, 2-(benzyloxycarbonyl amino) ethyl 2-bromo-isobutyrate (Compound 1)
Cbz-N-ethanolamine 3 g (15.36 mmol) was dissolved in 20 mL of ethyl acetate in a round bottom flask and stirred vigorously with cooling in an ice bath for 1 hr. Triethylamine (2.35 mL) was directly added into the mixture in the round bottom flask. 2-Bromoisobutyryl bromide (3 mL, 165.1 mmol) in 5 mL of ethyl acetate was slowly added in dropwise under vigorous stirring. A cloudy product resulted. More ethyl acetate (up to 50 mL) was added when reaction mixture was too viscous. The ethyl acetate solution was subsequently washed with saturated NaHCO3, 5% HCl, and double-distilled water. Once separated, the aqueous phase (bottom layer) was discarded, and the product was dried with anhydrous MgSO4 and condensed in vacuo at 35°C. The resulting viscous oil was stored at 4°C (Fig 1., Compound 1).
Fig. 1.


(A) Polymer synthesis and formulation of Cal and PTX loaded micelles (M-Cal/PTX). TEA, trimethylamine; TEMED, tetramethylethylenediamine. The values a, b, c were variables used in the central composite design (CCD). (B) 1H-NMR spectrum of poly(OEG-MA)40-b-poly[HEMA-g-(ε-caprolactone)7]20 (polymer 2). Hydrogen atoms and their corresponding peak integrations are marked by a’ and b’.
2.2.2. Poly[oligo(monomethyl ether ethylene glycol) methacrylate)40-b-poly(2-hydroxyethyl methacrylate)20 (Polymer 1)
The synthetic schemes described in this manuscript are adaptations of the atomic radicalization polymerization reactions described by Matyjaszewski and Xia (Matyjaszewski and Xia, 2002).
Into a round-bottom flask was added Compound 1 (0.17 g, 70.2 μmol), oligo ethylene glycol methyl methacrylate (OEG20-MA, MW ~ 500, 10 g, 20 mmol), 2,2’-bipyridine, BPy (0.16 g, 1.02 mmol), and 5 mL of anhydrous methanol. The flask was flushed with anhydrous argon for 20 min and immersed in a 55°C oil bath. CuBr (0.072 g, 0.5 mmol) was quickly added under argon protection, and the reaction was allowed to proceed for 30 min. A separate mixture of 2-hydroxyethyl methacrylate (1.2 mL, 2.1 mmol) in 3.7 mL of de-oxygenized methanol was added into the reaction mixture. The polymerization continued for 18 h at 55°C and then stopped by exposure to open air for at least 4 h. After a dilution step with ethyl acetate, the reaction mixture was passed through a basic aluminum column to remove the CuBr catalyst and condensed in vacuo at 45°C to give Polymer 1 (Fig.1).
2.2.3. Poly(OEG-MA)40-b-poly[HEMA-g-(ε-caprolactone)7]20 (Polymer 2)
Polymer 1 (2.5 g, 0.11 mmol) was dried azeotropically by distillation with toluene at 140°C and allowed to cool for 1 hr. Anhydrous ε-caprolactone (CPL, 1.48 mL, 13.14 mmol) and tin (II) 2-ethylhexanoate [Sn(Oct)2, (0.015 g, 0.04 mmol)] were added. The flask was flushed with anhydrous argon and allowed to run overnight under N2 gas at 110°C. The polymerization mixture was precipitated in hexane and kept at −20°C for 4 h. Once fully precipitated, the product was filtered and dried under N2 gas for 1 h to obtain 3.8 g of Polymer 2 (Fig. 1A). The structure of Polymer 2 was confirmed by 1H NMR (Fig. 1B).
2.2.4. Poly(OEG-MA)40-b-poly[HEMA-g-(acryloyloxy ε-caprolactone)7]20-(Polymer 3)
To 1 g of Polymer 2, 8 mL of tetrahydrofuran (THF) was added, and polymer thoroughly dissolved on an ice bath. To this mixture, 70.5 μL of triethyl amine (TEA) was added while stirring. To the stirring mixture, 38.3 μL of acryloyl chloride (0.46 mmol) in 2 mL THF was added dropwise. The mixture reacted for 45 min, then was centrifuged at 6,000 g for 10 min after which the supernatant was collected (10 mL) to obtain Polymer 3 (Fig.1).
2.2.5. Preparation of micelles loaded with Cal and PTX (M-Cal/PTX) and stability assessments
To 5 mg of PTX and 1 mg of Cal, 2 mL of Polymer 3 in THF was added and vortexed to ensure the drugs were completely dissolved. The initial drug loading ratio of 5:1 PTX to Cal ratio was chosen after optimization with response surface methodology. Different ratios of PTX and Cal by weight were evaluated with encapsulation efficiency as the readout. We observed a 5:1 ratio of PTX to Cal yielded satisfactory encapsulation efficiencies (> 90% for PTX and > 65% for Cal). To the resulting mixture, 10 μL of tetramethylethylenediamine (TEMED) was added. While vortexing, 4 mL of distilled water was added and vigorously vortexed for 1 min. The organic solvent (THF) was removed in vacuo to leave concentrated micelles (4 mL). Potassium persulfate, K2S2O8 (10 mg) was added and then stirred for 1 hr. The loaded micelle (M-Cal/PTX) was dialyzed to remove unencapsulated drugs. Subsequently it was centrifuged at 10,000 g for 10 min, and supernatant collected to obtain micelles with a final PTX-to-Cal ratio of ~ 10:1. The freshly prepared micelles were stored at 4°C for short-term period of 6 weeks. For long-term storage (>3 months), the micelles were stored at −80 ° C using 5% sucrose as a cryoprotectant. Stability was evaluated by measuring the changes in micelle size and encapsulation efficiency.
2.3. Micelle optimization using central composite design (CCD)
To obtain micelles with size <100 nm and extended drug release properties, CCD was employed to tune formulation parameters, namely, the number of repeating units of methacrylate-grafted oligoethylene glycol (OEG-MA-500, a = 13.2 – 46.8 units), CPL (b = 4.3 – 7.6 units), and crosslinking density (XL, 10–90%; c = 2 – 18) (Fig. 1). To simplify the model and reduce the number of parameters to be optimized, the number of repeating units of 2-hydroxyethyl methacrylate was kept constant (20-HEMA) based on preliminary studies. A full factorial design was used to generate models using Design Expert Software v8 (StatEase, Minneapolis, MN) for statistical modeling and the generation of surface response plots. The independent factors of OEG-MA-500, CPL and XL, and their effects on the micelle size, and 2-hr accumulative drug release at pH 6 and pH 7.4 were coded at 5 levels as −α, −1, 0, +1, +α, with 0 as the central point (Table 1).
Table 1.
Formulation values of independent factors and measured response variables for M-PTX (N=3: Mean ± SD)
| a | b | c | Response 1 | Response 2 | Response 3 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Run | PEGMA-500 | CPL | XL | 2-hr release at pH 6 (%) | 2-hr release at pH 7.4 (%) | size (nm) | |||
| No. of units | No. of units | % | Mean (%) | SD | Mean (%) | SD | Mean (%) | SD | |
| 1 | 20 | 5 | 25 | 65.4 | 2.2 | 30.9 | 32.0 | 21.7 | 16.9 |
| 2 | 30 | 6 | 50 | 66.3 | 1.1 | 39.1 | 33.9 | 24.7 | 20.6 |
| 3 | 30 | 6 | 50 | 70.0 | 3.1 | 41.0 | 34.4 | 26.1 | 20.3 |
| 4 | 40 | 7 | 25 | 19.9 | 2.2 | 15.7 | 12.0 | 9.9 | 7.0 |
| 5 | 30 | 7.6 | 50 | 112.5 | 2.2 | 54.9 | 55.3 | 37.5 | 30.5 |
| 6 | 40 | 7 | 75 | 14.2 | 2.0 | 30.4 | 39.1 | 23.8 | 19.4 |
| 7 | 40 | 5 | 25 | 83.1 | 2.6 | 36.9 | 41.5 | 27.0 | 21.2 |
| 8 | 46.8 | 6 | 50 | 19.1 | 1.5 | 23.5 | 24.5 | 16.5 | 13.0 |
| 9 | 30 | 6 | 50 | 68.7 | 1.7 | 40.1 | 34.6 | 25.5 | 20.8 |
| 10 | 30 | 6 | 7.9 | 97.4 | 2.3 | 35.9 | 53.3 | 30.5 | 25.9 |
| 11 | 30 | 4.3 | 50 | 106.5 | 3.3 | 53.3 | 51.7 | 36.1 | 28.4 |
| 12 | 30 | 6 | 50 | 65.4 | 1.3 | 38.9 | 33.4 | 24.6 | 20.3 |
| 13 | 30 | 6 | 92 | 65.4 | 0.5 | 52.6 | 47.1 | 33.4 | 28.6 |
| 14 | 20 | 5 | 75 | 96.8 | 2.6 | 58.1 | 49.3 | 36.7 | 29.9 |
| 15 | 13.2 | 6 | 50 | 115.5 | 4.5 | 56.7 | 55.8 | 39.0 | 29.9 |
| 16 | 40 | 5 | 75 | 60.3 | 2.5 | 45.9 | 38.3 | 28.9 | 23.2 |
| 17 | 20 | 7 | 75 | 58.2 | 1.8 | 45.0 | 38.4 | 28.4 | 23.3 |
| 18 | 20 | 7 | 25 | 34.0 | 1.1 | 20.0 | 17.0 | 12.7 | 10.2 |
| 19 | 30 | 6 | 50 | 67.0 | 2.0 | 39.7 | 33.7 | 25.1 | 20.2 |
| 20 | 30 | 6 | 50 | 69.6 | 2.6 | 40.7 | 34.5 | 25.9 | 20.5 |
a, b, c corresponding to number of repeating units in Figure 1. XL = Crosslinking density was calculated as c/20*100, No. of units = Number of repeating units
2.4. Characterization of M-Cal/PTX
2.4.1. Cal and PTX encapsulation efficiency
Drug loading efficiency was evaluated by modifying a published protocol (Zhao et al., 2018a). Briefly, micelles were dissolved in methanol and vortexed at high speed, followed by a 5 min sonication to release encapsulated drugs. After centrifugation at 10,000 g for 30 min, the supernatant containing released drugs was collected and analyzed on Waters Acquity™ Ultra-high-pressure liquid chromatography (UPLC, Milford, MA. USA). Chromatographic separation was achieved using Kinetex C18 column (1.7 μm, 100 × 2.10 mm, Phenomenex. Torrance, CA, USA) under isocratic elution conditions with a total runtime of 3.5 min at a flow rate of 0.4 mL/min. The detection wavelengths for Cal and PTX were 210 nm and 227 nm respectively. The column and autosampler temperatures were 40°C and 10°C, respectively, and the injection volume was 10 μL. The composition of the mobile phase was ammonium acetate solution (5 mM)-methanol (15:85, v/v). Encapsulation efficiency (EE) was computed according to the formula
EE (%) = (Amounts of Cal and PTX recovered after dialysis) / (Amounts Cal and PTX added in micelles) × 100%
2.4.2. Morphology and size determination of M-Cal/PTX
Particle size, size distribution, and zeta potential were measured with a dynamic light scattering system on ZetaPlus particle sizer (Brookhaven Instruments Corp., Holtsville, NY). The dried state visualization of particle size and morphology of Cal and PTX-loaded micelles were examined on a transmission electron microscopy (JEOL USA, Inc., Peabody, MA) according to the method described in Zhao et al., 2018b, with digital images (Figure 4) collected on the AMT Imaging System (Advanced Microscopy Techniques Corp., Danvers, MA).
Fig.4.

TEM images of M-Cal/PTX at a scale of 500 nm and at 25,000X magnification (Images shown were from three sets of independently prepared M-Cal/PTX)
2.4.3. Drug release of polymeric micelles
To determine the drug release profiles, polymeric micelles containing 1.2 mg/mL PTX and 0.17 mg/mL Cal were diluted in the appropriate matrix (PBS at pH 7.4 or sodium acetate at pH 6, and mouse serum) and added to a microdialyzer (molecular weight cut-off ~ 3500, Thermo Scientific, Rockford, IL). The microdialyzers were incubated in PBS (pH 7.4), sodium acetate buffer (pH 6.0) or mouse plasma in a 37°C water bath with agitation. Aliquots (30 μL) were taken at predefined times from the microdialyzer. The samples were centrifuged at 6,000 g for 5 min and 10 μL of the supernatant was retrieved and analyzed for Cal and PTX concentrations using UPLC method in section 2.4.1.
2.5. UPLC-MS/MS Assay to measure concentrations of Cal and PTX in biological samples
Separation of Cal and PTX was achieved using the chromatographic conditions described in section 2.4.1. A triple quadrupole mass spectrometer with electrospray ionization source (ESI) (API 5500, Applied Biosystems/MDS SCIEX, Foster City, CA) was used for the detection using the positive ionization mode with multiple reaction monitoring. Analyte extraction from biological samples was achieved with a liquid-liquid extraction procedure. Whole blood samples were processed using a developed and validated UPLC-MS/MS assay for simultaneous quantifications of Cal and PTX (In press; Lincha et al., 2020).
Tissues samples were weighed and homogenized with bead mill homogenizer in 1 mL of water at 4°C. The tissue homogenate was transferred into a clean 1.5 mL Eppendorf tube from which 100 μL was extracted with 500 μL of water-MeOH (1:1, v/v) containing IS (10 ng/mL each of PTX-d5 and Cal-d4) by vortexing for 1 min. Additional processing and analyte measurement were as described in the referenced manuscript. Concentrations of analytes in tissues were normalized by the tissue weight. The UPLC-MS/MS assay was linear over a range of 0.5 – 500 ng/mL for both Cal and PTX with the lower limits of quantifications (LLOQs) of 0.5 ng/mL in whole blood, tumor, liver and spleen.
2.6. Biodistribution studies
All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) and were conducted in accordance with institutional guidelines. Kras* murine pancreatic cancer cells with a doxycycline-inducible mutation of KRASG12D were cultured and the Kras* murine PDAC model was developed according to a previously published protocol (Zhao et al., 2018b). Eight-weeks old C57BL/6 female mice (Taconic Biosciences, Rensselaer, NY) were injected with Kras* cells into the pancreas head under isoflurane anesthesia after a small opening was made in the abdomen to expose the pancreas. A 27-gauge needle was used for precise injection of 5 × 105 Kras* cells. Mice with palpable pancreatic tumors of diameter 5 mm were recruited and randomized into two study groups. The mice were each administered an intravenous bolus of M-Cal/PTX (N=3) at doses of 5 mg/kg PTX and 0.5 mg/kg Cal or the non-formulated drugs (N=3) at the equivalent doses. Blood samples were collected at pre-determined time points and the mice were euthanized 24 hours post dose. Tumor and liver tissues were collected for simultaneous quantifications of Cal and PTX concentrations using the developed and validated tandem UPLC-MS/MS assay referenced in section 2.5.
2.7. Statistical analysis
Data were evaluated using Student’s t-test or 1-way analysis of variance (ANOVA) followed by post hoc Tukey for multiple comparisons. The statistical significance was evaluated at p < 0.05.
3. Results and Discussion
3.1. Development of an optimal M-Cal/PTX using CCD
3.1.1. Selection of an optimal formulation with PTX as the model drug
The effects of the number of units of OEG-MA-500, CPL, and XL (%) on the size and 2-hr drug release kinetics of PTX were studied using 20 experimental runs (Table 1). PTX was chosen as the model drug for CCD formulation optimization, because PTX (MW= 853.9 g/mol, logP= 3.52, water solubility= 0.0056 mg/mL) and Cal (MW= 412.6 g/mol, logP=4.3, water solubility=0.0135 mg/mL) have similar physicochemical properties, making extrapolation from PTX to Cal feasible. Economic consideration was another factor for not using large amounts of Cal in the process of formulation optimization but validating with Cal in the optimized formulation. Micelle size from the experimental runs ranged between 16.6–100 nm, cumulative 2-hr PTX release at pH 6 and 7.4 were 17.7–115.4% and 6.6– 100 %, respectively, depending on the formulation compositions. The formulation variable-dependent drug release characteristics were best described by a linear model for pH 6 and, and a quadratic model for the release characteristics for pH 7.4. A log-transformed linear model best described the impacts of formulation variables on the micelle size, as summarized in Table 2. Positive values of regression were indicative of synergism between the dependent and independent variables, while negative coefficients signified antagonism (Hao et al., 2012). Surface response maps corroborated the mathematical models and showed that the extent of drug release decreased with increasing amount of PEGMA-500 at pH 6. However, at pH 7.4 the opposite trend was observed (Fig. 2A and 2B), while micelle size also decreased with higher PEGMA-500 amount (Fig. 2C). The decrease of nanoparticle size with increasing PEGMA-500, a capping agent, was anticipated, as it has long been recognized and discussed (Arulmozhi and Mythili, 2013). PEGMA-500, a long chain polymer with terminal hydroxy groups caps polymer growth by shielding and stabilizing the nanoparticle. As more PEGMA-500 is added, a greater amount of OEG groups are present to cap the end of particles, effectively reducing average size (Arulmozhi and Mythili, 2013). Since the first report of PEGylation for drug delivery (Abuchowski et al., 1977), this strategy has become a mainstay in drug formulation due to the unique stealth properties, resisting interaction with components of a biological matrix, usually blood that it confers on drugs (Suk et al., 2016). It is therefore no surprise that as the amount of PEGMA-500 increases, the micelle stabilization increases, and the cumulative PTX release decreases. This mechanism of extending circulation times of drugs underlines the reason why PEGylation is widely used to formulate drugs when longer circulation is desired (Arturson et al., 1983; Tan et al., 1993).
Table 2.
Equations for best-fit models and summary statistics for micelle size and 2-hr PTX release at pH 6 and 7.4
| Response variables | Model equations in terms of coded factors | R2 | p value |
|---|---|---|---|
| 2-hr PTX release at pH 6 | 47.49 − 19.77 × PEGMA-500 + 16.83 × CPL | 0.62 | 0.0035 |
| 2-hr PTX release at pH 7.4 | 48.34 − 19.70 ×PEGMA-500 + 11.19 × CPL + 2.97 × XL − 13.96 × PEGMA-500 × CPL + 11.26 × CPL2 − 8.26× XL2 | 0.83 | 0.0004 |
| Micelle size | 1.72 − 0.13 × PEGMA-500 − 0.11 × CPL + 0.10 × XL | 0.60 | 0.0262 |
Fig. 2.

Three-dimensional response surface plots showing the effects of polymer variables of PEGMA-500 and CPL units on A (PTX release at pH 6), B (PTX release at pH 7.4) and C (micelle size). For graphing purposes, the crosslinking density was set at 50%
Unlike PEGMA-500, as CPL increased, the cumulative release of PTX over 72 hr increased. The micelle size decreased with increasing CPL, with a negative correlation. CPL has widespread applications in drug delivery for its biocompatibility, biodegradability and being generally recognized as safe (Zelenková et al., 2014). Additionally, CPL undergoes slow degradation in the body (Karuppuswamy et al., 2015; Seremeta et al., 2013; Woodruff and Hutmacher, 2010). These properties make CPL suitable for drug delivery. It is widely expected to slow down drug release with increasing CPL amount, due to increased hydrophobic interaction between the polymeric matrix and the drug payloads. However, our observation was the opposite. We speculated that the interaction between CPL and other polymers might be the reason. CPL has excellent compatibility with other polymers in the formulation, including PEG, and therefore it is plausible the characteristics of this block of copolymer may change as it interacts with other ingredients. The compatibility of CPL with PEG makes it desirable when it is intended to control properties like degradation kinetics and hydrophilicity (Bilensoy et al., 2009; Payyappilly et al., 2015). The interaction between PEGMA-500 and CPL was indeed observed in our model to affect the PTX drug release at pH 7.4. This interaction was captured by the curvature for the surface response map from our CCD approach (Fig. 2B), and the mathematical model suggested the impact of the interaction was dominated by PEGMA-500.
To further improve stability of the polymeric micelles, we extended the end units of CPL with acryloyloxy to form crosslinks in the hydrophobic domain of the block copolymer by potassium persulfate mediated reaction. The goal was to obtain micelles that released drugs while in circulation but remained stable enough to control release. We therefore studied the extent of XL on the size and drug release characteristics. The XL did not significantly affect the drug release at pH 6, and thus was dropped and set at 50% for model simplicity and visualization efficiency (Table 2), in subsequent studies to generate the surface response maps. The optimal micelle consisted of 40 PEGMA-500 repeating units (a = 40), 7 repeating units of CPL (b = 7), 20 units of HEMA, and 75% crosslinking density (corresponding to c = 15). The structure of Polymer 2 from which polymer 3 and the optimal micelles were formed in one pot is confirmed by 1HNMR (Fig. 1B).
3.1.2. Selection of an optimal drug delivery system and validation of the CCD with Cal and PTX
Numerical optimization was used to obtain the optimal drug-loaded micelles. A desirability function was used to simultaneously optimize response by indicating a range for each independent variable within the experimental design. The limits for each response was assigned a minimum, maximum or target value. To allow flexibility in model predictions, a range for each response was indicated. The objectives of developing this drug delivery system were to minimize Cal toxicity by reducing free Cal in systemic circulation, extend the apparent biological half-life of Cal and PTX, and enhance their accumulation in the tumor. Based on preliminary experiments, we aimed at having no more than 25% of drugs released at 2 hr to control initial burst effect, and micelle size range of 40 −100 nm. The range for XL was set at 50–75%. Even though PEGMA-500 confers stealth properties on nanoparticles and increases circulation time, its hydrophilicity reduces cellular uptake (Ruiz et al., 2013; Wang and Thanou, 2010). However, nanoparticle size and shape also strongly impact cellular uptake (Albanese et al., 2012; Zhang et al., 2015). We desired micelles with a size of 40–100 nm, because this is the optimal size for cellular uptake (Jiang et al., 2008; Lu et al., 2009; Yuan et al., 2010; Zhang et al., 2009). Nanoparticles within such a size range have the appropriate entropic and enthalpic properties that influence adhesion strength between the particles and cellular receptors (Yuan et al., 2010; Zhang et al., 2009). The optimal micelle was thus selected with 40 repeating units of PEGMA-500 (a = 40), 20 repeating units of HEMA and 6 repeating units of CPL (b = 6) along with 75% XL (c= 15). The predicted average values of particle size, 2-hr drug release at pH 6 and 7.4 were 53 nm, 25% and 22%, respectively. The model was validated by performing confirmatory runs using the model-predicted optimal micelle composition, with co-encapsulation of Cal and PTX. The observed values for average micelle size was 51 nm. For PTX, 2-hour drug release at pH 6, 7.4 and in serum were 20%, 18% and 7%, respectively, and for Cal, 6%, 24% and 6% (Table 3). We postulated that the initial slow release of Cal and PTX in serum when compared to buffers tested could be due to binding of released drugs to serum. The observed responses correlated well with model-predicted response with biases between 3.7%−20% highlighting the reliability of using the CCD model for formulation development (Table 3). The full drug release profile (Fig. 3) showed that the cumulative percentage of Cal and PTX released at pH 6, 7.4 and in serum were 42%, 58%, 58% and 80%, 80%, 76%, respectively after 120 h, demonstrating the sustained drug release characteristics of the optimal micelles.
Table 3.
Model-predicted and observed outputs for PTX in optimal M-Cal/PTX
| Optimal M-Cal/PTX | Predicted | Observed N=3 Mean + SD) | %Bias |
|---|---|---|---|
| PTX size (nm) | 53 | 51 ± 2.7 | 3.7 |
| 2-hr Cumulative release (%) | |||
| pH 6 | 25 | 20 ± 4.5 | 20 |
| Ph 7.4 | 22 | 18 ± 3.3 | 18.2 |
Fig. 3.

Drug release profiles of Cal and PTX from optimal M-Cal/PTX (M-Cal, M-PTX) and from a 10% dimethyl acetamide cosolvent (Cal, PTX). Graph shows mean ± SD of 3 independent studies and an insert for 0–4 h.
3.2. Characterization of optimal M-Cal/PTX
3.2.1. TEM analysis
TEM analysis of the optimal M-Cal/PTX showed spherical particles with the size range of 40–100 nm (Fig. 4). The shape and size distribution were consistent among the three independently prepared micelles.
3.2.2. Encapsulation efficiency of optimal M-Cal/PTX
The initial ratio of PTX to Cal (w/w) affected the encapsulation efficiencies of both drugs. To obtain satisfactory encapsulation efficiencies, a DOE was used to evaluate different combinations of both drugs with encapsulation efficiency as the readout. A drug loading ratio of 5:1 PTX to Cal yielded a high PTX encapsulation efficiency of > 90% and an encapsulation efficiency of > 65% for Cal.
3.2.3. Polydispersity index and zeta potential of optimal M-Cal/PTX
The size distribution of the optimized formulation, reflected by PDI measured by dynamic light scattering (DLS), was 0.249 ± 0.004 and a zeta potential of 0.006 ± 0.001 (N=3 independent batches). In general, particles with a PDI < 0.3 are considered uniformly dispersed (Das and Chaudhury, 2011)
3.2.4. Storage stability at 4°C and −80 ° C
The stability of M-Cal/PTX in storage is shown in Figure 5. Particle size and EE (Fig. 5A), and PDI (Fig. 5B) were measured over a 6-week period from micelles stored at 4°C. The micelle size and encapsulation efficiencies of both drugs were not appreciably changed over the 6-week period and also after 3 months when stored at −80 °C in 5% sucrose. The PDI increased from 0.25 to 0.26 after 1 week to 0.27 after 6-week storage; nevertheless, but was still below the stable threshold PDI of 0.3. The optimized M-Cal/PTX formulation had a 75% crosslinking density and could be used within 6 weeks of storage at 4°C after preparation. For long term storage, micelle integrity is maintained by storing at −80°C in 5% sucrose up to 3 months. We attributed the stability of the micelles to the effective core crosslinking. The end hydroxy units of HEMA were conjugated with acryloyl chloride to enable crosslinking in the presence of potassium persulfate.
Fig. 5.

Storage stability of optimal M-Cal/PTX at 4°C (A, B) and −80°C (C, D). Data shows mean ± SD of measurements from 3 independently prepared batches. * p<0.05 (n = 3).
3.2.5. Pharmacokinetic and biodistribution of optimal M-Cal/PTX in healthy mice and an orthotopic KrasG12D mouse model of pancreatic cancer
The pharmacokinetic (PK) profiles of Cal (from dosing free Cal, dissolved in 10% dimethyl acetamide), PTX (from free PTX, dissolved in 10% dimethyl acetamide), M-Cal, and M-PTX (from dosing M-Cal/PTX) in healthy C57BL/6 mice were presented in Figure 6A. The PTX and M-PTX profiles appeared similar with no statistical difference at any time point. However, while M-Cal could be measured up to 12 hours post dose, Cal could only be measured up to 4 hours post dose. In addition, the elimination phase of free Cal appeared steeper than that of Cal in M-Cal. The biodistribution of Cal (Fig. 6B) and PTX (Fig. 6C) in tumor of PDAC Kras* tumor from M-Cal/PTX at 24 hr post dose were clearly higher than those from free Cal and free PTX. The Cal in liver from dosing of M-Cal/PTX was higher than that from free Cal (Fig. 6B), but those of PTX were similar between M-Cal/PTX and free PTX (Fig. 6C). The blood PK parameters were estimated using Phoenix ® version 8 software (Table 4). One of the main goals for developing the micellar formulation was to reduce systemic exposure of Cal. Predictably, the dose-normalized AUCinf, AUClast and Cmax of Cal was lower in mice administered the M-Cal/PTX, when compared to those that received the free Cal (Table 4). Specifically, the dose normalized AUCinf, AUClast and Cmax of micelle-treated group appeared to be more than 2 times lower than in free drug group, albeit not statistically significant. However, the corresponding parameters for PTX in both groups were relatively similar. We postulated that off-target activation of VDR is directly or indirectly responsible for some of the toxic side effects associated with Cal. Therefore, it was our expectation that reduced Cal exposure would correlate with the potential reduced toxicity of Cal (Table 4).
Fig.6.

Pooled whole blood PK profiles (A) and tissue biodistribution of optimal Cal (B) and PTX (C) from M-Cal/PTX and unformulated Cal/PTX at 24 h. Mice were administered a single IV bolus dose of 5 mg/kg PTX and 0.5 mg/kg Cal. Free Cal/PTX was prepared in 10% dimethylacetamide. Concentration time profiles were constructed from N=8 for M-Cal/PTX and N=7 for Cal/PTX groups. For the biodistribution study data show N=3 for M-Cal/PTX and free drugs groups. For mice received free Cal and free PTX, the drugs were measurable in 1 out of the 3 animals in the liver, and drug concentrations were below detection limit in the tumor in all animals. For Fig. 6A, free Cal and PTX were not measurable beyond 4 and 12 hours respectively. Data are presented as mean ± SD.
Table 4.
Whole blood PK parameters of Cal and PTX from M-Cal/PTX or unformulated Cal/PTX in healthy C57BL/6 mice after a single IV bolus dose of 5 mg/kg PTX and 0.5 mg/kg Cal, N=3 in each group
| M-Cal | Cal | M-PTX | PTX | ||||||
|---|---|---|---|---|---|---|---|---|---|
| t1/2 | h | 3.3# | (1.8) | 0.9 | (0.2) | 4.5 | (1.1) | 3.4 | (0.4) |
| Cmax/D | (ng/mL)/(mg/kg) | 124.5 | (29.3) | 315.5 | (178.6) | 70.3 | (29.4) | 60.2 | (26.7) |
| AUClast/D | h*(ng/mL)/(mg/kg) | 71.6 | (22.6) | 166.1 | (117.2) | 106.4 | (30.4) | 123.2 | (41.4) |
| AUCINF/D | h*(ng/mL)/(mg/kg) | 79.3 | (29.9) | 170.4 | (120.7) | 108.3 | (30.2) | 127.1 | (44.3) |
| CL | L/h/kg | 14.0 | (4.7) | 7.6 | (3.5) | 9.9 | (3.3) | 8.7 | (3.1) |
| MRTinf | h | 1.8 | (1.3) | 0.7 | (0.08) | 3.5 | (0.8) | 3.6 | (0.7) |
| Vss | L/kg | 22.2# | (10.5) | 5.5 | (2.30) | 33.2 | (8.8) | 36.4 | (7.2) |
Values shown are Mean (SD).
Parameter is significantly different from free Cal group, by Student’s t-test at p<0.05
With the dose of M-Cal/PTX, the apparent elimination half-life (t1/2) of Cal was 3.7 times longer than that with free Cal. The prolonged half-life resulted from the increased Vss, suggesting a greater particle uptake and longer residence time in tissues from M-Cal/PTX, and also consistent with the extended drug release characteristics of the micellar formulation. On the other hand, the t1/2 of PTX was not appreciably prolonged with M-Cal/PTX formulation.
We also measured Cal concentrations in liver and tumor from unformulated Cal or PTX and M-Cal/PTX. After a single IV bolus dose, only M-Cal/PTX yielded sustained levels of drugs in the tumors (Fig. 6B and 6C). These results confirmed the micellar drug delivery platform facilitated tumor accumulation. The shape and size are two physical characteristics of nanoparticles that play crucial roles in tumor uptake (Batist, 2007). Nanoparticles face several biobarriers while in circulation. A major barrier to tumor uptake is the body’s immune response which considers the formulation platform a foreign body. However, tuning the particle size and shape can provide stealth allowing the particles to circulate longer and accumulate in desired areas through the “Enhanced Permeability and Retention” (EPR) effect (Batist, 2007; Lasic and Papahadjopoulos, 1995). Nanoparticles of size greater than 200 nm do not generally extravasate into tumor (Nagayasu et al., 1999). Also, shapes of nanoparticles dictate their interactions with membranes and circular shapes are favored for tumor accumulation (Nagayasu et al., 1999). A combination of these factors possibly contributed to the enhanced uptake of Cal and PTX encapsulated in the M-Cal/PTX system.
4. Conclusion
Herein, we developed a micellar drug delivery platform to encapsulate and deliver Cal and PTX simultaneously for the treatment of pancreatic cancer. A sustained release delivery system was desired to reduce the systemic exposure of Cal in the circulation, a potential solution to Cal-associated toxicity. Additionally, micelles with a particle size < 100 nm were desired to improve tumor accumulation. We used the DOE approach to obtain the optimal delivery system with the size range of 40–100 nm which resulted in longer apparent biological half-life of Cal. The encapsulation efficiencies of Cal and PTX were sufficient to deliver therapeutically relevant doses in a mouse model of PDAC. Biodistribution studies using the micellar drug delivery system yielded Cal and PTX accumulation in tumors 24 hr after a single IV bolus, which was not replicated in mice that received non-encapsulated Cal and PTX. We postulated that the sustained release characteristics, size and shape of the optimal formulation permitted the sustained tumor accumulation. On-going studies will focus on demonstrating the proof-of-concept efficacy and safety of this micellar drug delivery system in relevant mouse models of PDAC.
Highlights.
The Design of Experiment approach enables faster drug delivery development with pre-defined performance parameters
The Design of Experiment approach enables the definition of a design space for potential modifications of a delivery system
Size-controlled micelles facilitate pancreatic tumor accumulation
Acknowledgement:
Special thanks to Dr. Mahua Sarkar for providing training for the use of Design Expert for statistical modeling
Funding
This work was supported in part by the Gillson-Longenbaugh Foundation, the John S. Dunn Foundation, and by a grant from The University of Texas MD Anderson Cancer Center Duncan Family Institute for Cancer Prevention and Risk Assessment [Grant number: G0501555]. The Research Animal Support Facility and Electron Microscopy Core Facility are supported by a Cancer Center Support Grant from the National Cancer Institute, National Institutes of Health [P30CA016672]. The listed funding sources had no role in the study design, collection, analysis and interpretation of data, nor in the writing of this report.
Footnotes
Editor’s comments
Can you please let me know whether you have NMR and MS data confirming the structure of polymer 3?
Response:
To address your inquiry about data for confirmation of polymers, we now provide NMR data for polymer 2 in Figure 1B.
Polymer 3 and micelle formation were done in one spot, so structural data for polymer 3 is not accessible.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF, 1977. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 252, 3582–3586. [PubMed] [Google Scholar]
- Albanese A, Tang PS, Chan WCW, 2012. The Effect of Nanoparticle Size, Shape, and Surface Chemistry on Biological Systems. Annu. Rev. Biomed. Eng. 14, 1–16. 10.1146/annurev-bioeng-071811-150124 [DOI] [PubMed] [Google Scholar]
- Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C, Muñoz M, Quijano Y, Cubillo A, Rodriguez-Pascual J, Plaza C, De Vicente E, Prados S, Tabernero S, Barbacid M, Lopez-Rios F, Hidalgo M, 2013. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 109, 926–933. 10.1038/bjc.2013.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arturson P, Laakso T, Edman P, 1983. Acrylic microspheres in vivo IX: Blood elimination kinetics and organ distribution of microparticles with different surface characteristics. J. Pharm. Sci. 72, 1415–1420. 10.1002/jps.2600721213 [DOI] [PubMed] [Google Scholar]
- Arulmozhi KT, Mythili N, 2013. Studies on the chemical synthesis and characterization of lead oxide nanoparticles with different organic capping agents. AIP Adv. 3. 10.1063/1.4858419 [DOI] [Google Scholar]
- Batist G, 2007. Cardiac safety of liposomal anthracyclines. Cardiovasc. Toxicol. 7, 72–74. 10.1007/s12012-007-0014-4 [DOI] [PubMed] [Google Scholar]
- Bilensoy E, Sarisozen C, Esendaǧli G, Doǧan AL, Aktaş Y, Şen M, Mungan NA, 2009. Intravesical cationic nanoparticles of chitosan and polycaprolactone for the delivery of Mitomycin C to bladder tumors. Int. J. Pharm. 371, 170–176. 10.1016/j.ijpharm.2008.12.015 [DOI] [PubMed] [Google Scholar]
- Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD, 1997. Improvements in survival and clinical benefit with gemcitabine as first- line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 15, 2403–2413. 10.1200/JCO.1997.15.6.2403 [DOI] [PubMed] [Google Scholar]
- Das S, Chaudhury A, 2011. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech 12, 62–76. 10.1208/s12249-010-9563-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta B, Poudel BK, Tran TH, Pradhan R, Cho HJ, Jeong JH, Shin BS, Choi HG, Yong CS, Kim JO, 2015. Modulation of Pharmacokinetic and Cytotoxicity Profile of Imatinib Base by Employing Optimized Nanostructured Lipid Carriers. Pharm. Res. 32, 2912–2927. 10.1007/s11095-015-1673-7 [DOI] [PubMed] [Google Scholar]
- Hall BR, Cannon A, Atri P, Wichman CS, Smith LM, Ganti AK, Are C, Sasson AR, Kumar S, Batra SK, 2018. Advanced pancreatic cancer: A meta-analysis of clinical trials over thirty years. Oncotarget 9, 19396–19405. 10.18632/oncotarget.25036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Wang F, Wang X, Zhang D, Bi Y, Gao Y, Zhao X, Zhang Q, 2012. Development and optimization of baicalin-loaded solid lipid nanoparticles prepared by coacervation method using central composite design. Eur. J. Pharm. Sci. 47, 497–505. 10.1016/j.ejps.2012.07.006 [DOI] [PubMed] [Google Scholar]
- Hejri A, Khosravi A, Gharanjig K, Hejazi M, 2013. Optimisation of the formulation of β-carotene loaded nanostructured lipid carriers prepared by solvent diffusion method. Food Chem. 141, 117–123. 10.1016/j.foodchem.2013.02.080 [DOI] [PubMed] [Google Scholar]
- Ilic M, & Ilic I. (2016). Epidemiology of pancreatic cancer. World Journal of Gastroenterology, 22(44), 9694. doi: 10.3748/wjg.v22.i44.9694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Kim BYS, Rutka JT, Chan WCW, 2008. Nanoparticle-mediated cellular response is size-dependent. Nat. Nanotechnol. 3, 145–150. 10.1038/nnano.2008.30 [DOI] [PubMed] [Google Scholar]
- Karuppuswamy P, Reddy Venugopal J, Navaneethan B, Luwang Laiva A, Ramakrishna S, 2015. Polycaprolactone nanofibers for the controlled release of tetracycline hydrochloride. Mater. Lett. 141, 180–186. 10.1016/j.matlet.2014.11.044 [DOI] [Google Scholar]
- Kim G, 2017. Nab-Paclitaxel for the treatment of pancreatic cancer. Cancer Manag. Res. 9, 85–96. 10.2147/CMAR.S127840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasic DD, Papahadjopoulos D, 1995. Liposomes revisited. Science (80-. ). 267, 1275–1276. 10.1126/science.7871422 [DOI] [PubMed] [Google Scholar]
- Lincha V, Hsiao C-H, Zhao J, Li C, Chow DSL, 2020. Sensitive and Rapid UHPLC-MS/MS assay for simultaneous quantifications of calcipotriol and paclitaxel in rat whole blood and plasma samples. Journal of Pharmaceutical and Biomedical Analysis (In press: JPBA-D-20–00406R1) [DOI] [PubMed] [Google Scholar]
- Lu F, Wu SH, Hung Y, Mou CY, 2009. Size effect on cell uptake in well-suspended, uniform mesoporous silica nanoparticles. Small 5, 1408–1413. 10.1002/smll.200900005 [DOI] [PubMed] [Google Scholar]
- Luo J, Xiao L, Wu C, Zheng Y, & Zhao N. (2013). The Incidence and Survival Rate of Population-Based Pancreatic Cancer Patients: Shanghai Cancer Registry 2004–2009. PLoS ONE, 8(10). doi: 10.1371/journal.pone.0076052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyjaszewski K, & Xia J. (2002). Fundamentals of Atom Transfer Radical Polymerization. Handbook of Radical Polymerization, 523–628. doi: 10.1002/0471220450.ch11 [DOI] [Google Scholar]
- Mcguigan A, Kelly P, Turkington RC, Jones C, Coleman HG, & Mccain RS (2018). Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World Journal of Gastroenterology, 24(43), 4846–4861. doi: 10.3748/wjg.v24.i43.4846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Nel AE, 2018. Use of nano engineered approaches to overcome the stromal barrier in pancreatic cancer. Adv. Drug Deliv. Rev. 130, 50–57. 10.1016/j.addr.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayasu A, Uchiyama K, Kiwada H, 1999. The size of liposomes: A factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv. Drug Deliv. Rev. 40, 75–87. 10.1016/S0169-409X(99)00041-1 [DOI] [PubMed] [Google Scholar]
- Payyappilly SS, Panja S, Mandal P, Dhara S, Chattopadhyay S, 2015. Organic solvent-free low temperature method of preparation for self assembled amphiphilic poly(ε-caprolactone)-poly(ethylene glycol) block copolymer based nanocarriers for protein delivery. Colloids Surfaces B Biointerfaces 135, 510–517. 10.1016/j.colsurfb.2015.07.075 [DOI] [PubMed] [Google Scholar]
- Pike JW, Meyer MB, 2012. The Vitamin D Receptor: New Paradigms for the Regulation of Gene Expression by 1,25-Dihydroxyvitamin D 3. Rheum. Dis. Clin. North Am. 38, 13–27. 10.1016/j.rdc.2012.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Salas G, Calero M, Hernández Y, Villanueva A, Herranz F, Veintemillas-Verdaguer S, Martínez E, Barber DF, Morales MP, 2013. Short-chain PEG molecules strongly bound to magnetic nanoparticle for MRI long circulating agents. Acta Biomater. 9, 6421–6430. 10.1016/j.actbio.2012.12.032 [DOI] [PubMed] [Google Scholar]
- Seremeta KP, Chiappetta DA, Sosnik A, 2013. Poly(e{open}-caprolactone), Eudragit® RS 100 and poly(e{open}-caprolactone)/Eudragit® RS 100 blend submicron particles for the sustained release of the antiretroviral efavirenz. Colloids Surfaces B Biointerfaces 102, 441–449. 10.1016/j.colsurfb.2012.06.038 [DOI] [PubMed] [Google Scholar]
- Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM, 2014. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93. 10.1016/j.cell.2014.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Kantoff PW, Wooster R, Farokhzad OC, 2017. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37. 10.1038/nrc.2016.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suk JS, Xu Q, Kim N, Hanes J, Ensign LM, 2016. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery HHS Public Access Graphical abstract. Adv Drug Deliv Rev 99, 28–51. 10.1016/j.addr.2015.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JS, Butterfield DE, Voycheck CL, Caldwell KD, Li JT, 1993. Surface modification of nanoparticles by PEO/PPO block copolymers to minimize interactions with blood components and prolong blood circulation in rats. Biomaterials 14, 823–833. 10.1016/0142-9612(93)90004-L [DOI] [PubMed] [Google Scholar]
- Vaccaro V, Sperduti I, Milella M, 2011. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 365, 768–769. 10.1056/NEJMc1107627 [DOI] [PubMed] [Google Scholar]
- Wang M, Thanou M, 2010. Targeting nanoparticles to cancer. Pharmacol. Res. 62, 90–99. 10.1016/j.phrs.2010.03.005 [DOI] [PubMed] [Google Scholar]
- Woodruff MA, Hutmacher DW, 2010. The return of a forgotten polymer - Polycaprolactone in the 21st century. Prog. Polym. Sci. 35, 1217–1256. 10.1016/j.progpolymsci.2010.04.002 [DOI] [Google Scholar]
- Yuan H, Li J, Bao G, Zhang S, 2010. Variable nanoparticle-cell adhesion strength regulates cellular uptake. Phys. Rev. Lett. 105, 1–4. 10.1103/PhysRevLett.105.138101 [DOI] [PubMed] [Google Scholar]
- Zelenková T, Fissore D, Marchisio DL, Barresi AA, 2014. Size control in production and freeze-drying of poly-ε-caprolactone nanoparticles. J. Pharm. Sci. 103, 1839–1850. 10.1002/jps.23960 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zeng L, Chen Y, Lian G, Qian C, Chen S, Li J, Huang K, 2016. Pancreatic Cancer Epidemiology, Detection, and Management. Gastroenterol. Res. Pract. 2016. 10.1155/2016/8962321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Gao H, Bao G, 2015. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 9, 8655–8671. 10.1021/acsnano.5b03184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S, 2009. Size-dependent endocytosis of nanoparticles. Adv. Mater. 21, 419–424. 10.1002/adma.200801393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Li X, Ye T, Chen F, Sun X, Kong J, Yang X, Pan W, Li S, 2013. Design, characterization, and in vitro cellular inhibition and uptake of optimized genistein-loaded NLC for the prevention of posterior capsular opacification using response surface methodology. Int. J. Pharm. 454, 354–366. 10.1016/j.ijpharm.2013.07.032 [DOI] [PubMed] [Google Scholar]
- Zhao J, Wang H, Hsiao CH, Chow DSL, Koay EJ, Kang Y, Wen X, Huang Q, Ma Y, Bankson JA, Ullrich SE, Overwijk W, Maitra A, Piwnica-Worms D, Fleming JB, Li C, 2018a. Simultaneous inhibition of hedgehog signaling and tumor proliferation remodels stroma and enhances pancreatic cancer therapy. Biomaterials 159, 215–228. 10.1016/j.biomaterials.2018.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Xiao Z, Li T, Chen H, Yuan Y, Wang YA, Hsiao CH, Chow DSL, Overwijk WW, Li C, 2018b. Stromal Modulation Reverses Primary Resistance to Immune Checkpoint Blockade in Pancreatic Cancer . ACS Nano 10.1021/acsnano.8b02481 [DOI] [PMC free article] [PubMed] [Google Scholar]