

Summary
Late-stage functionalization (LSF) has over the past years emerged as a powerful approach in the drug discovery process. At its best, it allows for rapid access to new analogues from a single drug-like molecule, bypassing the need for de novo synthesis. To be successful, methods able to tolerate the diverse functional groups present in drug-like molecules that perform under mild conditions are required. C−H methylation is of particular interest due to the magic methyl effect in medicinal chemistry. Herein we report an iridium-catalyzed carboxylate-directed ortho C−H methylation and d3-methylation of benzoic acids. The method uses commercially available reagents and precatalyst and requires no inert atmosphere or exclusion of moisture. Substrates bearing electron-rich and electron-poor groups were successfully methylated, including compounds with competing directing/coordinating groups. The method was also applied to the LSF of several marketed drugs, forming analogues with increased metabolic stability compared with the parent drug.
Subject areas: Chemistry, Organic chemistry, Green chemistry, Applied chemistry
Graphical abstract

Highlights
-
•
Direct access to high-value methylated and d3-methylated analogues from benzoic acids
-
•
Process utilizing bench-stable precatalyst and reagents, as well as a recyclable solvent
-
•
Applications to late-stage functionalization (LSF) of structurally complex drugs
-
•
Positive effect on biological and physical properties of pharmaceuticals demonstrated
Chemistry; Organic chemistry; Green chemistry; Applied chemistry
Introduction
C−H bonds are ubiquitous in the vast majority of pharmaceuticals; in 2018, 199 of the top 200 small molecule pharmaceuticals by retail sales contained C−H bonds (Njardarson 2018). The direct transformation of such a frequently appearing motif is challenging, but highly advantageous, as it allows rapid access to molecular diversity in a single step. In the late-stage functionalization (LSF) context, this would allow to surpass the need of de novo synthesis of new analogues (Blakemore et al., 2018; Cernak et al., 2016; Börgel and Ritter 2020). This is beneficial with respect to time, overall step count, and atom economy and thus highly advantageous for the drug discovery process. In an ideal situation, a wide array of lead compound analogues could be obtained from a single common intermediate, enabling fast structure-activity relationships studies, or even leading to new candidate drugs. To fully unleash the potential of this approach and to access as much chemical space as possible, a variety of synthetic tools with high regioselectivity are needed (Hartwig 2017). Methodologies allowing the selective introduction of methyl-, fluoro- or other small groups on each position of a lead molecule should be widely utilized in the drug discovery process (Blakemore et al., 2018). The field of directed C−H activation has been reviewed (Sambiagio et al., 2018; Rej et al., 2020), presenting an attractive strategy to functionalize otherwise unreactive C−H bonds with high regioselectivity. This approach has been applied to LSF in the context of medicinal chemistry (Moir et al., 2019), and the field is still developing. Given the frequency that methyl groups bound to a carbon atom appear in marketed drugs (Njardarson 2018), combined with the overwhelming presence of C−H bonds in drug molecules, the C−H to C−Me transformation presents an appealing strategy. The potential impact of the C−H methylation has been highlighted by Cernak (Schönherr and Cernak 2013), and the effect of methylation in medicinal chemistry has been extensively reviewed by Barreiro et al. (2011). Owing to the significance of the so-called magic methyl effect, a call for new C−H methylation reactions was made (Schönherr and Cernak 2013). Methylation is used to tune biological and physical properties of drug candidates, such as metabolic stability (Gomtsyan et al., 2007), solubility (Jones et al., 2008), off-target selectivity (Shamovsky et al., 2009), binding mode (Zimmerman et al., 1993), and affinity (for more examples of the aforementioned effects see Schönherr and Cernak, [2013]). The substitutions of single C−H for C−Me functionality resulting in more than a 100-fold increase in potency have been reported (Angell et al., 2008; Coleman et al., 2012).
Although some progress in late-stage C(sp3)−H methylation has been made in recent years, exemplified by the excellent work of the White group (Feng et al., 2020), we only mention this transformation briefly as the focus of this article is on C(sp2)−H methylations.
In late-stage functionalization methods, C(sp2)−Me bonds from C−H bonds can in principle be obtained by two distinct approaches: introduction of a synthetic handle followed by methylation (2 steps, Figure 1A, left) and directed C−H methylation (1 step, Figure 1A, right). Although the latter approach presents benefits from a step and atom economy perspective, the two approaches are complementary. In the first approach, the regioselectivity of the synthetic handle introduction is typically governed by the electronic and steric properties of the substrate. The “classic” tactics for this approach are halogenations via electrophilic aromatic substitution (SEAr). The halogenation occurs on the most electron-rich position of the aromatic system; however, steric effects can influence the selectivity. Applications of this approach to LSF of complex substrates can be complicated by the formation of undesired polyhalogenated species and/or mixtures of regioisomers. A recent approach to this method utilizing electrochemical generation of the halogenating agent tackles some of these issues and affords monohalogenated analogues of a selection of complex molecules with high regioselectivity (Tan et al., 2017). The arylhalides obtained can in turn be converted to C−Me via transition-metal catalysis. A different, well-established approach to install synthetic handles in the form of boronic acid pinacol esters (BPin) is the Hartwig-Miyaura C−H borylation (Ishiyama et al., 2002; Larsen and Hartwig 2014). The method utilizes an Ir(I) catalyst to introduce the synthetic handle, typically on the most sterically accessible position, thus complimenting the SEAr regioselectivity. The BPin analogues can in turn be methylated by transition-metal catalysis (He et al., 2018; Haydl and Hartwig 2019). The Ritter group developed a charge-transfer-directed para-selective radical amination to introduce the TEDA (N-(methyl)triethylenediamine) synthetic handle, which can be subsequently methylated via nickel catalysis (Serpier et al., 2018). This methodology showcases high para-selectivity in complex molecules and was successfully applied to LSF of small molecule pharmaceuticals. More recently the group has reported similar transformation utilizing a thiantrenation protocol followed by a Negishi coupling (Berger et al., 2019). Another example of radical functionalization in heterocycles was reported by the Baran group (Gui et al., 2014). This report also demonstrates the preparation of d3-methylated analogues, as well as difluoromethylated compounds. The alternative approach, direct C−H methylation, presents a more step-economical solution. A Minisci-type photoredox methylation published was used to convert a wide array of heterocyclic compounds, including small molecule drugs in an LSF fashion (DiRocco et al., 2014).
Figure 1.

Approaches and opportunities in C(sp2)−H functionalization
(A) Possible approaches to C(sp2)−H methylations and their respective regioselectivity. Left: undirected approaches. Right: ortho-directed approach.
(B) C−H map of repaglinide, highlighting possible reaction sites. The ortho-directed site highlighted in green. The functionalization descriptors are based on the review article published by Cernak et al. (2016). Descriptors: C--H = Innate insertion or H-abstraction; H+ = Deprotonation; δ- = Addition-elimination at nucleophilic C(sp2); DG = Guided by directing group; )( = Guided by sterics.
Alternatively, the presence of a Lewis basic coordinating group (directing group) can allow for transition-metal-catalyzed selective activation of C−H bonds in its vicinity (Figure 1A, right). Methods relying on designer directing groups can enable access to challenging structural motifs, as demonstrated in the excellent diversification protocol from the Yu lab (Dai et al., 2011); however, such reactions are not considered true LSF based on the recent perspective from Ritter (Börgel and Ritter 2020). A more straightforward approach is to utilize directing groups already present in the molecule. Although a multitude of directed C−H methylation methodologies have been developed (Evano and Theunissen 2019), successful LSF applications are scarce. Recently a methodology for cobalt-catalyzed C−H methylation was reported by Ackermann, Johansson and coworkers, enabling access to 22 drug analogues (Friis et al., 2020). The major strength of this method is the ability to utilize a large variety of inherent directing groups in a predictable manner. However, one important class of functional groups incompatible with the aforementioned method is carboxylic acids. Carboxylic acids, and particularly benzoic acids, are not only an important class of building blocks frequently utilized in synthesis but also represent a structural motif present in numerous drugs and natural products (Lamberth and Dinges 2016). Martín-Matute and coworkers have previously reported C−H iodinations of benzoic acids (Erbing et al., 2018; Weis et al., 2020). Directed C−H methylations of benzoic acids are known (Shang et al., 2016; Lv et al., 2019; Giri et al., 2007), whereas examples of LSF applications are limited. The pioneering applications came from the Yu group, which demonstrated palladium-catalyzed LSF with two compounds, a medicinally relevant compound BMS-98947-055-01 (Thuy-Boun et al., 2013), and in the synthesis of natural product Hongoquercin A, in collaboration with the Baran group (Rosen et al., 2013). Although both aforementioned examples are important to the field, LSF via C−H methylations of drug-like benzoic acids with high functional group diversity remains relatively unexplored.
A general and accessible methodology for the late-stage methylation of benzoic acids containing multiple potential C−H activation sites and a variety of Lewis basic groups (e.g., repaglinide, Figure 1B) is desirable. High levels of regioselectivity and selectivity toward the monomethylation are also crucial, as the separation of mixtures can be difficult. Furthermore, air- and moisture-tolerant reactions utilizing bench-stable, easy to dispense, commercially available reagents are highly desired for automated synthesis and high-throughput experimentation (HTE) (Mennen et al., 2019) and for broader applications.
With this work we aim to develop a methodology for ortho-C−H methylation of benzoic acids, which fulfills the aforementioned criteria, tolerates a broad array of functional groups, and allows for functionalization of building blocks, advanced intermediates, as well as marketed drugs with high regioselectivity in a single step.
Results and discussion
Optimization of reaction conditions
At the onset of the project we set several desirable criteria, such as the use of commercially available starting materials, reagents, and catalyst, and that the reaction tolerates air and moisture, to facilitate HTE experimentation. The initial hit identification and subsequent optimization was conducted in several rounds of plate screening, and it is described in detail in the supplemental information (Tables S1–S11). A summary of the optimization with meta-toluic acid is shown in Table 1. Further in Table 1, deviations from the optimized conditions are presented to highlight the characteristics and requirements of the final methodology. A series of transition-metal precatalysts were tested for the envisioned reaction; however, from the Co, Rh, Ru, and Ir series that were tested (Table 1, entries 1 to 4), only [Cp∗IrCl2]2 showed conversion to the desired product. By simply increasing the loading of the methyl source the reaction proceeded with full conversion (Table 1, entry 5, standard conditions). Under these standard conditions, K2HPO4 is used as base, MeBF3K as methyl source, Ag2CO3 as the stoichiometric oxidant, and HFIP as solvent. The reaction is performed under air atmosphere at 60°C over 18 h. Under these conditions, complete consumption of starting material was observed, with clean conversion to the desired product, which is obtained in 90% isolated yield after purification.
Table 1.
Optimization and effect of deviations from optimized conditions.
 | ||
|---|---|---|
| Entry | Deviations from optimized conditions | Conversion (%) (SFC-MS, UV-trace) |
| 1 | Cp∗Co(CO)I2 (6 mol%) | NR |
| 2 | [Cp∗RhCl₂]₂ (3 mol%) | NR |
| 3 | [(p-cymene)RuCl2]2 (3 mol%) | NR |
| 4 | [Cp∗IrCl₂]₂ (3 mol%), MeBF₃K (1 equiv) | 21 |
| 5 | No change (standard conditions) | >99a |
| 6 | [Cp∗Ir(H2O)3]SO4 (6 mol%) | 11 |
| 7 | IrCl3 (6 mol%) | NR |
| 8 | Inert atmosphere (N₂) | NR |
| 9 | Solvent TFE | 41 |
| 10 | Solvent DCE | NR |
| 11 | Solvent acetone | NR |
| 12 | 40 °C | 60 |
| 13 | 23 °C | 13 |
| 14 | [Cp∗IrCl₂]₂ (2 mol%) | 61 |
| 15 | [Cp∗IrCl₂]₂ (1 mol%) | 11 |
| 16 | No base | 2 |
| 17 | MeB(OH)₂ (2.5 equiv) | 4 |
| 18 | Methylboronic acid MIDA ester (2.5 equiv) | NR |
| 19 | K₂CO₃ (2.0 equiv) as base | 97 |
| 20 | 0.2 M reaction concentration | >99b |
| 21 | AgF (2.5 equiv) as oxidant | 6 |
| 22 | AgOAc (2.5 equiv) as oxidant | 25 |
| 23 | RBF3K, R: nBu, Et, cyclopropyl, vinyl, Ph, CF3 | NR |
NR = no reaction; SFC-MS = supercritical fluid chromatography mass spectrometry; TFE = 2,2,2-trifluoroethanol; DCE = 1,2-dichloroethane; MIDA = N-methyliminodiacetic acid.
90% isolated yield.
82% yield determined by 1H NMR spectroscopy. With the exception of entries 5 and 20, by-products were not detected. Thus, the conversions corresponded well with the yields.
The use of the [Cp∗IrCl2]2 precatalyst was once again proved to be crucial when other iridium catalysts were tested. [Cp∗Ir(H2O)3]SO4 provided only 11% conversion, whereas the use of IrCl3 resulted in no conversion. The importance of the air atmosphere was showcased when the reaction was set up under an atmosphere of N2, resulting in no conversion (Table 1, entry 8). This indicates a crucial role of O2 in either precatalyst activation or the catalytic cycle itself. The use of alternative solvents proved detrimental to the reaction outcome (Table 1, entries 9 to 11), with only TFE providing conversion (Table 1, entry 9). The reaction successfully progressed at decreased temperatures, albeit with significantly lower conversions (Table 1, entries 12 and 13). Similarly, lowering of catalyst loading led to decreased conversions (Table 1, entries 14 and 15). In the absence of base only trace amounts of product were formed (Table 1, entry 16), while pressure generation due to the formation of a large amount of gas was observed. One of the major components of the gas was identified as methane by 1H NMR spectroscopy (see the supplemental information). Alternative boron-based methyl sources (Table 1, entries 17 and 18) showed little or no conversion. When K2CO3 was used as base instead of K2HPO4, only a slightly lower conversion was observed (Table 1, entry 19). Increasing the reaction concentration to 0.2 M led to less clean reaction profile and slightly decreased conversion to the desired product (82% by qNMR, entry 20). Use of alternative Ag(I) oxidants led to lower conversions (Table 1, entries 21 and 22). Finally, other alkylations, vinylations, and arylations were unsuccessful (Table 1, entry 23, and supplemental information, Table S12).
Scope and limitations: building blocks
With the optimized conditions in hand (Table 1, entry 5), we investigated the C−H methylation of a variety of ortho- and meta-substituted benzoic acid derivatives, bearing electron-donating and electron-withdrawing groups (Scheme 1). Compounds 2a, 2b, and 2c, containing ortho substituents, were obtained in good yields. Regarding the character of the ortho substituent, whereas the substituent electronics had little effect on the reaction outcome, the apparent limitation was the steric bulk of the substituent. The presence of the phenyl substituent in 1d resulted in a significant decrease in yield, although complete regioselectivity for methylation at the 6 position was observed. This selectivity is complementary to related copper and palladium chemistry, where lactonization on the 2′ position in the neighboring ring is observed instead (Gallardo-Donaire and Martin 2013; Li et al., 2013). The scope of meta-substituted compounds is significantly broader in this respect, as substituent sterics played no significant role in the reaction outcome. Compounds 1e and 1f gave the expected methylation product with the activation of the less sterically hindered C−H bond.
Scheme 1.

Substrate scope of C−H methylations of benzoic acids
Isolated yields are shown. Potential competing directing groups highlighted in blue. aAg2CO3 (2.5 equiv), MeBF3K (4.0 equiv).
An important, yet seldom explored (Lu et al., 2019), aspect of C−H activation chemistry is the selective activation of a single C−H bond in compounds bearing multiple directing groups (Tomberg et al., 2019). Here a series of compounds with two coordinating groups were methylated with complete regioselectivity, i.e., exclusively ortho to the carboxylic acid moiety. The acetanilide group, a well-established directing group for C−H activations (Yang et al., 2007; Li et al., 2011; Jiang and Wang 2012), was well tolerated and provided the desired compound 2g with complete regioselectivity, resulting from the functionalization of the C−H group ortho to the carboxylic acid moiety exclusively. While aryl esters can also serve as a directing group in C−H activation chemistry (Wang et al., 2018; Zhao and Snieckus 2016), the methylation of 2h occurred once again with the anticipated regioselectivity. Ketones, another well-established directing group (Bettadapur et al., 2015; Huang et al., 2015), were also tolerated, as shown in the reaction of 2i, with an enolizable ketone, and of 2j, bearing an aryl ketone, yielding the desired products with complete regioselectivity, ortho to the carboxylic functional group. The final directing group tested was the amide moiety (Wencel-Delord et al., 2012; Schröder et al., 2012), giving the methylated benzoic acid derivative 2k, once again, as a single regioisomer. The observed selectivity is of particular importance for further applications in LSF. The current state of the art in palladium-catalyzed C−H methylation of benzoic acids shows no known tolerance to these directing groups (Giri et al., 2007; Lv et al., 2019), with the exception of a single tolerated ester from the Yu group (Thuy-Boun et al., 2013). Although the utilization of directing groups other than carboxylates was shown in an excellent contribution by Nakamura and coworkers (Shang et al., 2016), compatibility and/or competition with directing groups contained within a single substrate was only demonstrated with one example.
The reaction outcome was also unaffected by the presence of electron-withdrawing groups at the meta position: trifluoromethyl-substituted 1L and nitro-substituted 1m formed the corresponding products, 2L and 2m, in good yields. Interestingly, the methylation of benzoic acid 1n, having an alkyl carboxylic acid substituent, resulted in the formation of not only the anticipated product 2n but also the corresponding methyl ester 2o. The selectivity observed is once again complementary to current palladium chemistry, which frequently utilizes the acetyl carboxylic acid moieties of phenylacetic acids as directing groups (Thuy-Boun et al., 2013; Giri et al., 2007). Regarding the esterification observed in 2o, this occurred with complete regioselectivity for the benzylic carboxylate over the aryl carboxylate. However, control experiments conducted on phenylacetic acid showed no conversion to the ester (Table S13). The naphthalene 2p was obtained with complete regioselectivity for methylation at the 2 position, suggesting the preferential formation of a 5-membered iridacycle over a 6-membered one in the C−H activation step. This transformation of 1-napththoic acid presents an improvement in both yield and selectivity when compared with the current state of the art using palladium chemistry (Lv et al., 2019). Naphthalene 1q was also selectively methylated at the more sterically accessible position, yielding 2q.
Examples of more highly substituted benzoic acids, such as 2r and 2s containing a 1,2,3,4-substitution pattern, were successfully prepared, as well as the 1,2,4,6-substituted 2t and the 1,2,4,5-substituted 2u. All the presented examples so far underwent methylation at a sterically accessible ortho-position in the absence of a neighboring meta substituent. The formation of 1,2,3-substitution patterns with the newly introduced group at the 2-position is seldom reported. With this chemistry, the methylation of such systems was possible under two specific sets of conditions. In 2v, with one of the ortho positions blocked, the methyl group was installed between the methoxy group and the directing group. In 1w, which has both ortho positions unsubstituted, and both meta positions occupied, the methylation product 2w was obtained with complete regioselectivity ortho to the fluoride and ortho to the carboxylic acid. Although the application of the current method to heterocyclic functionalization is limited, two successful examples are presented: the N-acylated indole 1x was methylated with complete regioselectivity at the C5 position over the C3 position, as well as over the potential acyl-directed product at positions C2 and C7. This presents a rare example of indole C5 C−H activation. Thiophene 2y was methylated at the 2-position, once again with complete regioselectivity. Finally, although methylation of para-substituted substrates could not be achieved with useful selectivity toward ortho monosubstituion, the reaction could be pushed toward dimethylation by simply increasing the equivalents of Ag2CO3 and MeBF3K. This afforded dimethylated 2z and 2aa in good yield, of 62% and 73%, respectively.
Solvent recovery and recycling
With the pressing challenges of sustainability and environmental concern over the use of solvents in chemistry (Welton 2015), an investigation of recovery and recyclability of the reaction solvent HFIP was undertaken.
After the first round under standard conditions, 95% of the reaction solvent was successfully recovered by distillation, and the reaction product was isolated from the leftover residue with an 88% isolated yield (Scheme 2). In the recyclability study the recovered solvent was shown to have satisfactory purity by 1H NMR spectroscopy and was successfully reused for the same reaction, yielding 87% of the desired product.
Scheme 2.

Solvent recovery and recycling study
Isolated yields are shown. After the first round of reaction 95% of HFIP was successfully recovered.
Scope and limitations: LSF
As applications for LSF were our ultimate goal for this methodology, a series of compounds currently in clinical use were subjected to the standard methylation conditions (Scheme 3). Repaglinide, a drug used for blood glucose reduction in type 2 diabetes mellitus (Culy and Jarvis 2001), was selectively methylated at the ortho position with respect to the carboxylic acid-directing group. The single-step synthesis of 2ab, with a 42% isolated yield directly from the commercially available compound, presents a big advantage over de novo synthesis, which would take 16 steps based on reported procedures (Vardanyan and Hruby 2016; Grell et al., 1998), including the synthesis of a novel methylated core (Scheme S2). Bexarotene, an antineoplastic agent used for the treatment of cutaneous T cell lymphoma (Duvic et al., 2001; Gniadecki et al., 2007), afforded the monomethylation product 2ac as the major product, as well as the dimethylated product 2ad as a minor product. The terminal alkene functionality was unaffected by the reaction. Although both methylated cores contained in the analogues are commercially available, the de novo synthesis of the compounds would still take four steps each (Boehm et al., 1994). Finally, tranilast, an anti-allergic drug, also used in treatments of a variety of other diseases (Darakhshan and Pour 2015), was successfully methylated ortho to the carboxylate, yielding compound 2ae. No functionalization directed by the anilide moiety was observed. Owing to the relatively low yield in this particular case, the unreacted starting material was also isolated (61%).
Scheme 3.

Late-stage methylation of selected pharmaceuticals under standard conditions
For specific conditions see Scheme 1. Isolated yields are shown. Complete regioselectivity was observed in all cases. rsm, recovered starting material.
We then investigated the possibility of using this methodology for selective introduction of deuterium by means of d3-methylation. Similar to methylation, d3-methylation can improve key physicochemical, pharmacokinetic, or metabolic properties of a drug candidate. The difference between the isotope-labeled analogue and its unlabeled counterpart is a potentially increased metabolic stability of the newly acquired methyl group and/or redirected metabolic pathways as a result of a primary kinetic isotope effect (Russak and Bednarczyk 2019). The potential benefits have been demonstrated in several studies (Stringer et al., 2014; Parcella et al., 2017; Gant 2014), and in 2017 the first deuterated drug, Austedo, was approved by the Food and Drug Administration (FDA) (DeWitt and Maryanoff 2018). However, only a limited number of articles concerning C−H d3-methylations have been published (Sun and Yoshikai 2018; Han et al., 2019; Gao et al., 2019), and to the best of our knowledge, no such method is known in the context of benzoic acid functionalization and/or LSF. To access this transformation, we prepared the deuterated methyl source MeBF3K-d3 according to a published procedure (Falb et al., 2017). We were pleased to see that when this reagent was used under otherwise standard conditions, d3-methylation was observed with very similar yields compared with the non-isotope-labeled material. Thus, the isotope-labeled compounds 3a, 3b, and 3c were obtained in similar yields to the standard methylation procedure (Scheme 4). Importantly, no D−H exchange was observed under the reaction conditions (determined by 1H NMR spectroscopy), and thus the products were fully deuterated at the methyl groups.
Scheme 4.

Late-stage d3-methylation of benzoic acids and selected pharmaceuticals
MeBF3K-d3 used under otherwise standard conditions. Isolated yields are shown. No D−H exchange was observed. rsm, recovered starting material.
The reaction using MeBF3K-d3 was also applied in an LSF fashion, affording yields and regioselectivities nearly identical to those obtained when using the non-deuterated trifluoroborate salt. The d3-methylation of repaglinide yielded 40% of the desired analogue 3d in a single step. The de novo synthesis of this compound would take up to 17 steps (Scheme S3). Both mono- and di-d3-methylated analogues of bexarotene were obtained (Scheme 3, 3e and 3f) with similar yields to their hydrogen-containing counterparts 2ac and 2ad (Scheme 1). However, as the deuterated benzoic acid cores required for the synthesis of the analogues are not commercially available, the de novo synthesis would be significantly longer. We have estimated that this would be achieved in 7 steps in the case of 3e (Scheme S4) and 15 steps in the case of 3f (Scheme S5). Finally, the tranilast analogue 3g was also successfully obtained in a single step, improving the proposed de novo synthesis by three steps (Scheme S6).
Studies of drug-like properties
With the set of compounds derived from pharmaceuticals in hand, we became interested in the effect of methylation on their drug-like properties. For the metabolic stability studies six compounds were chosen (Table 2, see Table S14 for the complete study results), consisting of the parent compounds repaglinide and tranilast, and both their methylated and d3-methylated analogues.
Table 2.
LogD and metabolic stability data in HLM and human and rat hepatocytes for Repaglinide, Tranilast, and their analogues
| Entry | Structure | Name | logD | Rat Heps Clint (μL/min/106 cells) |
HLM Clint (μL/min/mg) |
Hheps Clint (μL/min/106 cells) |
|---|---|---|---|---|---|---|
| 1 |  |
Repaglinide | 2.25 | 26.1 | 47.3 | 35.1 |
| 2 | 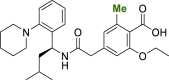 |
2ab | 1.65 | 14.4 | 18.8 | 10.1 |
| 3 | 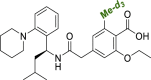 |
3d | 1.70 | 15.2 | 21.3 | – |
| 4 | 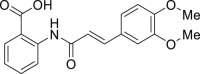 |
Tranilast | 0.15 | 14.6 | <3.0 | 5.17 |
| 5 | 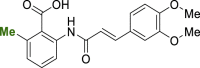 |
2ae | −0.85 | <1.0 | <3.0 | 2.37 |
| 6 | 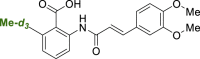 |
3g | −0.70 | <1.0 | <3.0 | – |
Rat Heps Clint = Intrinsic clearance in rat hepatocytes, HLM Clint = Intrinsic clearance in human liver microsomes, Hheps Clint = Intrinsic clearance in human hepatocytes. For additional data see Table S14.
Crucial parameters for all drug molecules, regardless of their therapeutic indication, are their physicochemical properties, such as lipophilicity, and metabolic stability. In this respect, the introduction of a non-polar group such as methyl is expected to cost a lipophilic penalty by increasing the logD value (Schönherr and Cernak 2013). Interestingly, with all the analogues prepared (Table 2) a small decrease in logD was observed. With a 1,2,3-substitution pattern in all cases (Entries 2, 3, 5 and 6), we speculate that the decrease in lipophilicity is the result of changes in dihedral angles between the carboxylic acid moiety and the aryl system. The same trend for methylations ortho to a polar group was recently reported (Friis et al., 2020).
When the metabolic stability of repaglinide and its analogues 2ab and 3d was studied, a significant decrease in intrinsic clearance (Clint), and thus increase in metabolic stability in both rat hepatocyte (Rat Heps) and human liver microsome (HLM) assays was observed. Somewhat surprising was the fact that no significant difference between the values obtained for 2ab and 3d were observed, as benzylic methyl groups are often prone to cytochrome P450 oxidation (Zhang and Tang 2018). The function of the newly introduced benzylic methyl groups as a metabolic hotspot is thus unlikely. For this reason, only repaglinide and its analogue 2ab were taken to the human hepatocytes (Hheps) Clint study, which again showed a significant increase of metabolic stability for the methylated analogue.
Similar results were obtained when tranilast and its analogues 2ae and 3g were compared, with an order of magnitude decrease of Clint in rat hepatocytes for both analogues. This study also served as the basis for a metabolite identity (MetID) study, in which suppression of glucuronidation was observed with tranilast analogue 2ae (see supplemental information). As the Clint in human microsomes for all compounds was under the detection threshold no conclusions can be made from this part of the study. Finally, when Clint in human hepatocytes between tranilast and 2ae were compared the clearance of the parent compound was double of the methylated analogue.
Conclusion
A catalytic method for ortho C−H methylation and d3-methylation of benzoic acids was developed. The method tolerates air and moisture, and apart from MeBF3K-d3, utilizes commercially available reagents and precatalyst. Substrates bearing electron-donating and electron-withdrawing groups, as well as a number of compounds containing other known directing groups were functionalized in a predictable manner, with complete regioselectivity ortho to the carboxylic acid moiety. An efficient late-stage methylation of selected pharmaceuticals was performed in useful yields. In total four methylated and four d3-methylated analogues of three clinically relevant drugs were prepared with this single-step procedure, saving time, resources, and over 60 synthetic steps in total. Our results clearly demonstrate the effect of methylation on lipophilicity and metabolic stability of the described marketed drugs. Given the potential for a significant improvement of drug-like compound properties with a single methylation, this method represents a valuable addition in the late-stage functionalization toolbox. Both methylated and d3-methylated analogues can be obtained, facilitating labeling studies, particularly studies of deuteration on metabolic stability. Finally, recycling of the HFIP solvent was demonstrated, an important point in respect of halogenated solvent use. It is our belief that the combined characteristics of this method, together with the importance of the benzoic acid structural motif in both building blocks and pharmaceuticals, will render it valuable for the synthetic and medicinal chemistry communities.
Limitations of the study
A number of functional groups, including nitrogen-containing heterocycles, nitriles, and aniline nitrogens, were not tolerated under the reaction conditions, which is important to consider when applying the methodology to LSF of drug-like molecules. The introduction of sterically demanding ortho substituents was shown to decrease conversion. All the problematic compounds tested are summarized in the supplemental information.
Resource availability
Lead contact
Further information and requests for resources should be directed to the lead contact, Prof. Dr Belén Martín-Matute (belen.martin.matute@su.se).
Materials availability
All the reagents used, except CD3BF3K and 1-acetyl-1H-indole-4-carboxylic acid (1x), were commercially available and used without further purification. CD3BF3K was prepared according to published procedure (Falb et al., 2017). CD3B(OH)2 was purchased from Cambridge Isotope Laboratories. The synthesis of 1x is described in the supplemental information. [Cp∗IrCl2]2 was purchased from STREM, HFIP from Chem-Impex International, MeBF3K from Combi-Blocks, and Et3N and Ag2CO3 from Aldrich. Benzoic acids were purchased from Combi-Blocks, Aldrich, Enamine, Fluorochem, Lancaster, Acros, Ubichem, WuXi AppTec, and Fluka.
Data and code availability
Not applicable.
Methods
All methods can be found in the accompanying transparent methods supplemental file.
Acknowledgments
We would like to thank Dr. Stig D. Fris for scientific discussions. E.W. is supported through a grant from the Swedish Foundation for Strategic Research (SSF). The authors are grateful for support from the Swedish Research Council through Vetenskapsrådet and Formas, and from the Swedish Foundation for Strategic Environmental Research (Mistra SafeChem, project number 2018/11).
Author contributions
E.W., B.M.-M., and M.J.J. conceived and designed the project. E.W. conducted the methodology development, synthesis, and characterization of compounds. M.A.H. analyzed the data from biological studies. B.M.-M. and M.J.J. supervised the project and secured funding. All authors contributed to the discussion of experimental results. All authors contributed to writing the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102467.
Contributor Information
Magnus J. Johansson, Email: magnus.j.johansson2@astrazeneca.com.
Belén Martín-Matute, Email: belen.martin.matute@su.se.
Supplemental information
References
- Angell R., Aston N.M., Bamborough P., Buckton J.B., Cockerill S., deBoeck S.J., Edwards C.D., Holmes D.S., Jones K.L., Laine D.I. Biphenyl amide p38 kinase inhibitors 3: improvement of cellular and in vivo activity. Bioorg. Med. Chem. Lett. 2008;18:4428–4432. doi: 10.1016/j.bmcl.2008.06.048. [DOI] [PubMed] [Google Scholar]
- Barreiro E.J., Kümmerle A.E., Fraga C.A.M. The methylation effect in medicinal chemistry. Chem. Rev. 2011;111:5215–5246. doi: 10.1021/cr200060g. [DOI] [PubMed] [Google Scholar]
- Berger F., Plutschack M.B., Riegger J., Yu W., Speicher S., Ho M., Frank N., Ritter T. Site-selective and versatile aromatic C−H functionalization by thianthrenation. Nature. 2019;567:223–228. doi: 10.1038/s41586-019-0982-0. [DOI] [PubMed] [Google Scholar]
- Bettadapur K.R., Lanke V., Prabhu K.R. Ru (II)-Catalyzed C–H activation: ketone-directed novel 1,4-addition of ortho C–H bond to maleimides. Org. Lett. 2015;17:4658–4661. doi: 10.1021/acs.orglett.5b01810. [DOI] [PubMed] [Google Scholar]
- Blakemore D.C., Castro L., Churcher I., Rees D.C., Thomas A.W., Wilson D.M., Wood A. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 2018;10:383–394. doi: 10.1038/s41557-018-0021-z. [DOI] [PubMed] [Google Scholar]
- Boehm M.F., Zhang L., Badea B.A., White S.K., Mais D.E., Berger E., Suto C.M., Goldman M.E., Heyman R.A. Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J. Med. Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- Börgel J., Ritter T. Late-stage functionalization. Chemistry. 2020;6:1877–1887. [Google Scholar]
- Cernak T., Dykstra K.D., Tyagarajan S., Vachal P., Krska S.W. The medicinal chemist's toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016;45:546–576. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]
- Coleman P.J., Schreier J.D., Cox C.D., Breslin M.J., Whitman D.B., Bogusky M.J., McGaughey G.B., Bednar R.A., Lemaire W., Doran S.M. Discovery of [(2R,5R)-5-{[(5-Fluoropyridin-2-yl)oxy]methyl}-2-methylpiperidin-1-yl][5-methyl-2-(pyrimidin-2-yl)phenyl]methanone (MK-6096): a dual orexin receptor antagonist with potent sleep-promoting properties. ChemMedChem. 2012;7:415–424. doi: 10.1002/cmdc.201200025. [DOI] [PubMed] [Google Scholar]
- Culy C.R., Jarvis B. Repaglinide: a review of its therapeutic use in type 2 diabetes mellitus. Drugs. 2001;61:1625–1660. doi: 10.2165/00003495-200161110-00008. [DOI] [PubMed] [Google Scholar]
- Dai H.-X., Stepan A.F., Plummer M.S., Zhang Y.-H., Yu J.-Q. Divergent C–H functionalizations directed by sulfonamide pharmacophores: late-stage diversification as a tool for drug discovery. J. Am. Chem. Soc. 2011;133:7222–7228. doi: 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- Darakhshan S., Pour A.B. Tranilast: a review of its therapeutic applications. Pharmacol. Res. 2015;91:15–28. doi: 10.1016/j.phrs.2014.10.009. [DOI] [PubMed] [Google Scholar]
- DeWitt S.H., Maryanoff B.E. Deuterated drug molecules: focus on FDA-approved deutetrabenazine. Biochemistry. 2018;57:472–473. doi: 10.1021/acs.biochem.7b00765. [DOI] [PubMed] [Google Scholar]
- DiRocco D.A., Dykstra K., Krska S., Vachal P., Conway D.V., Tudge M. Late-stage functionalization of biologically active heterocycles through photoredox catalysis. Angew. Chem. Int. Ed. 2014;53:4802–4806. doi: 10.1002/anie.201402023. [DOI] [PubMed] [Google Scholar]
- Duvic M., Hymes K., Heald P., Breneman D., Martin A.G., Myskowski P., Crowley C., Yocum R.C. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J. Clin. Oncol. 2001;19:2456–2471. doi: 10.1200/JCO.2001.19.9.2456. [DOI] [PubMed] [Google Scholar]
- Erbing E., Sanz-Marco A., Vázquez-Romero A., Malmberg J., Johansson M.J., Gómez-Bengoa E., Martín-Matute B. Base- and additive-free Ir-catalyzed ortho-iodination of benzoic acids: scope and mechanistic investigations'. ACS Catal. 2018;8:920–925. [Google Scholar]
- Evano G., Theunissen C. Beyond friedel and crafts: directed alkylation of C−H bonds in arenes. Angew. Chem. Int. Ed. Engl. 2019;58:7202–7236. doi: 10.1002/anie.201806629. [DOI] [PubMed] [Google Scholar]
- Falb E., Ulanenko K., Tor A., Gottesfeld R., Weitman M., Afri M., Gottlieb H., Hassner A. A highly efficient Suzuki–Miyaura methylation of pyridines leading to the drug pirfenidone and its CD3 version (SD-560) Green. Chem. 2017;19:5046–5053. [Google Scholar]
- Feng K., Quevedo R.E., Kohrt J.T., Oderinde M.S., Reilly U., White M.C. Late-stage oxidative C(sp3)–H methylation. Nature. 2020;580:621–627. doi: 10.1038/s41586-020-2137-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis S.D., Johansson M.J., Ackermann L. Cobalt-catalysed C–H methylation for late-stage drug diversification. Nat. Chem. 2020;12:511–519. doi: 10.1038/s41557-020-0475-7. [DOI] [PubMed] [Google Scholar]
- Gallardo-Donaire J., Martin R. Cu-Catalyzed Mild C(sp2)–H Functionalization Assisted by Carboxylic Acids en Route to Hydroxylated Arenes. J. Am. Chem. Soc. 2013;135:9350–9353. doi: 10.1021/ja4047894. [DOI] [PubMed] [Google Scholar]
- Gant T.G. Using deuterium in drug discovery: leaving the label in the drug. J. Med. Chem. 2014;57:3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- Gao Q., Shang Y., Song F., Ye J., Liu Z.-S., Li L., Cheng H.-G., Zhou Q. Modular dual-tasked C–H methylation via the catellani strategy. J. Am. Chem. Soc. 2019;141:15986–15993. doi: 10.1021/jacs.9b07857. [DOI] [PubMed] [Google Scholar]
- Giri R., Maugel N., Li J.-J., Wang D.-H., Breazzano S.P., Saunders L.B., Yu J.-Q. Palladium-catalyzed methylation and arylation of sp2 and sp3 C−H bonds in simple carboxylic acids. J. Am. Chem. Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- Gniadecki R., Assaf C., Bagot M., Dummer R., Duvic M., Knobler R., Ranki A., Schwandt P., Whittaker S. The optimal use of bexarotene in cutaneous T-cell lymphoma. Br. J. Dermatol. 2007;157:433–440. doi: 10.1111/j.1365-2133.2007.07975.x. [DOI] [PubMed] [Google Scholar]
- Gomtsyan A., Bayburt E.K., Keddy R., Turner S.C., Jinkerson T.K., Didomenico S., Perner R.J., Koenig J.R., Drizin I., McDonald H.A. α-Methylation at benzylic fragment of N-aryl-N′-benzyl ureas provides TRPV1 antagonists with better pharmacokinetic properties and higher efficacy in inflammatory pain model. Bioorg. Med. Chem. Lett. 2007;17:3894–3899. doi: 10.1016/j.bmcl.2007.04.105. [DOI] [PubMed] [Google Scholar]
- Grell W., Hurnaus R., Griss G., Sauter R., Rupprecht E., Mark M., Luger P., Nar H., Wittneben H., Müller P. Repaglinide and related hypoglycemic benzoic acid derivatives. J. Med. Chem. 1998;41:5219–5246. doi: 10.1021/jm9810349. [DOI] [PubMed] [Google Scholar]
- Gui J., Zhou Q., Pan C.-M., Yabe Y., Burns A.C., Collins M.R., Ornelas M.A., Ishihara Y., Baran P.S. C–H methylation of heteroarenes inspired by radical SAM methyl transferase. J. Am. Chem. Soc. 2014;136:4853–4856. doi: 10.1021/ja5007838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Yuan Y., Shi Z. Rhodium-catalyzed selective C–H trideuteromethylation of indole at C7 position using acetic-d6 anhydride. J. Org. Chem. 2019;84:12764–12772. doi: 10.1021/acs.joc.9b01114. [DOI] [PubMed] [Google Scholar]
- Hartwig J.F. Catalyst-controlled site-selective bond activation. Acc. Chem. Res. 2017;50:549–555. doi: 10.1021/acs.accounts.6b00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydl A.M., Hartwig J.F. Palladium-catalyzed methylation of aryl, heteroaryl, and vinyl boronate esters. Org. Lett. 2019;21:1337–1341. doi: 10.1021/acs.orglett.9b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z.-T., Li H., Haydl A.M., Whiteker G.T., Hartwig J.F. Trimethylphosphate as a methylating agent for cross coupling: a slow-release mechanism for the methylation of arylboronic esters. J. Am. Chem. Soc. 2018;140:17197–17202. doi: 10.1021/jacs.8b10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z., Lim H.N., Mo F., Young M.C., Dong G. Transition metal-catalyzed ketone-directed or mediated C–H functionalization. Chem. Soc. Rev. 2015;44:7764–7786. doi: 10.1039/c5cs00272a. [DOI] [PubMed] [Google Scholar]
- Ishiyama T., Takagi J., Hartwig J.F., Miyaura N. A stoichiometric aromatic C H borylation catalyzed by iridium(I)/2,2′-Bipyridine complexes at room temperature. Angew. Chem. Int. Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Jiang T.-S., Wang G.-W. Palladium-catalyzed ortho-alkoxylation of anilides via C–H activation. J. Org. Chem. 2012;77:9504–9509. doi: 10.1021/jo301964m. [DOI] [PubMed] [Google Scholar]
- Jones C.D., Andrews D.M., Barker A.J., Blades K., Byth K.F., Finlay M.R.V., Geh C., Green C.P., Johannsen M., Walker M., Weir H.M. Imidazole pyrimidine amides as potent, orally bioavailable cyclin-dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2008;18:6486–6489. doi: 10.1016/j.bmcl.2008.10.075. [DOI] [PubMed] [Google Scholar]
- Lamberth C., Dinges J. Wiley-VCH; 2016. Bioactive Carboxylic Compound Classes: Pharmaceuticals and Agrochemicals. [Google Scholar]
- Larsen M.A., Hartwig J.F. Iridium-catalyzed C–H borylation of heteroarenes: scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc. 2014;136:4287–4299. doi: 10.1021/ja412563e. [DOI] [PubMed] [Google Scholar]
- Li C., Wang L., Li P., Zhou W. Palladium-catalyzed ortho-acylation of acetanilides with aldehydes through direct C-H bond activation. Chem. Eur. J. 2011;17:10208–10212. doi: 10.1002/chem.201101192. [DOI] [PubMed] [Google Scholar]
- Li Y., Ding Y.-J., Wang J.-Y., Su Y.-M., Wang X.-S. Pd-catalyzed C–H lactonization for expedient synthesis of biaryl lactones and total synthesis of cannabinol. Org. Lett. 2013;15:2574–2577. doi: 10.1021/ol400877q. [DOI] [PubMed] [Google Scholar]
- Lu Q., Mondal S., Cembellín S., Greßies S., Glorius F. Site-selective C–H activation and regiospecific annulation using propargylic carbonates. Chem. Sci. 2019;10:6560–6564. doi: 10.1039/c9sc01703h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv W., Wen S., Liu J., Cheng G. Palladium-catalyzed ortho-C–H methylation of benzoic acids. J. Org. Chem. 2019;84:9786–9791. doi: 10.1021/acs.joc.9b01204. [DOI] [PubMed] [Google Scholar]
- Mennen S.M., Alhambra C., Allen C.L., Barberis M., Berritt S., Brandt T.A., Campbell A.D., Castañón J., Cherney A.H., Christensen M. The evolution of high-throughput experimentation in pharmaceutical development and perspectives on the future. Org. Process. Res. Dev. 2019;23:1213–1242. [Google Scholar]
- Moir M., Danon J.J., Reekie T.A., Kassiou M. An overview of late-stage functionalization in today’s drug discovery. Expert Opin. Drug Discov. 2019;14:1137–1149. doi: 10.1080/17460441.2019.1653850. [DOI] [PubMed] [Google Scholar]
- Njardarson Top 200 SMALL Molecule Drugs by Sales in 2018. 2018. https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/Top%20200%20Small%20Molecule%20Pharmaceuticals%202018V4.pdf
- Parcella K., Eastman K., Yeung K.-S., Grant-Young K.A., Zhu J., Wang T., Zhang Z., Yin Z., Parker D., Mosure K. Improving metabolic stability with deuterium: the discovery of BMT-052, a pan-genotypic HCV NS5B polymerase inhibitor. ACS Med. Chem. Lett. 2017;8:771–774. doi: 10.1021/acsmedchemlett.7b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rej S., Das A., Chatani N. Strategic evolution in transition metal-catalyzed directed C–H bond activation and future directions. Coord. Chem. Rev. 2020:213683. [Google Scholar]
- Rosen B.R., Simke L.R., Thuy-Boun P.S., Dixon D.D., Yu J.-Q., Baran P.S. C-H functionalization logic enables synthesis of (+)-Hongoquercin A and related compounds. Angew. Chem. Int. Ed. 2013;52:7317–7320. doi: 10.1002/anie.201303838. [DOI] [PubMed] [Google Scholar]
- Russak E.M., Bednarczyk E.M. Impact of deuterium substitution on the pharmacokinetics of pharmaceuticals. Ann. Pharmacother. 2019;53:211–216. doi: 10.1177/1060028018797110. [DOI] [PubMed] [Google Scholar]
- Sambiagio C., Schönbauer D., Blieck R., Dao-Huy T., Pototschnig G., Schaaf P., Wiesinger T., Zia M.F., Wencel-Delord J., Besset T. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018;47:6603–6743. doi: 10.1039/c8cs00201k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr H., Cernak T. Profound methyl effects in drug discovery and a call for new C-H methylation reactions. Angew. Chem. Int. Ed. Engl. 2013;52:12256–12267. doi: 10.1002/anie.201303207. [DOI] [PubMed] [Google Scholar]
- Schröder N., Wencel-Delord J., Glorius F. High-yielding, versatile, and practical [Rh(III)Cp∗]-Catalyzed ortho bromination and iodination of arenes. J. Am. Chem. Soc. 2012;134:8298–8301. [Google Scholar]
- Serpier F., Pan F., Ham W.S., Jacq J., Genicot C., Ritter T. Selective methylation of arenes: a radical C−H functionalization/cross-coupling sequence'. Angew. Chem. Int. Ed. 2018;57:10697–10701. doi: 10.1002/anie.201804628. [DOI] [PubMed] [Google Scholar]
- Shamovsky I., de Graaf C., Alderin L., Bengtsson M., Bladh H., Börjesson L., Connolly S., Dyke H.J., van den Heuvel M., Johansson H. Increasing selectivity of CC chemokine receptor 8 antagonists by engineering nondesolvation related interactions with the intended and off-target binding sites. J. Med. Chem. 2009;52:7706–7723. doi: 10.1021/jm900713y. [DOI] [PubMed] [Google Scholar]
- Shang R., Ilies L., Nakamura E. Iron-catalyzed ortho C–H methylation of aromatics bearing a simple carbonyl group with methylaluminum and tridentate phosphine ligand. J. Am. Chem. Soc. 2016;138:10132–10135. doi: 10.1021/jacs.6b06908. [DOI] [PubMed] [Google Scholar]
- Stringer R.A., Williams G., Picard F., Sohal B., Kretz O., McKenna J., Krauser J.A. Application of a deuterium replacement strategy to modulate the pharmacokinetics of 7-(3,5-dimethyl-1H-1,2,4-triazol-1-yl)-3-(4-methoxy-2-methylphenyl)-2,6-dimethylpyrazolo[5,1-b]oxazole, a novel CRF1 antagonist. Drug Metab. Dispos. 2014;42:954. doi: 10.1124/dmd.114.057265. [DOI] [PubMed] [Google Scholar]
- Sun Q., Yoshikai N. Cobalt-catalyzed directed ortho-methylation of arenes with methyl tosylate. Org. Chem. Front. 2018;5:2214–2218. [Google Scholar]
- Tan Z., Liu Y., Helmy R., Rivera N.R., Hesk D., Tyagarajan S., Yang L., Su J. Electrochemical bromination of late stage intermediates and drug molecules. Tetrahedron Lett. 2017;58:3014–3018. [Google Scholar]
- Thuy-Boun P.S., Villa G., Dang D., Richardson P., Su S., Yu J.-Q. Ligand-accelerated ortho-C–H alkylation of arylcarboxylic acids using alkyl boron reagents. J. Am. Chem. Soc. 2013;135:17508–17513. doi: 10.1021/ja409014v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomberg A., Muratore M.É., Johansson M.J., Terstiege I., Sköld C., Norrby P.-O. Relative strength of common directing groups in palladium-catalyzed aromatic C−H activation. iScience. 2019;20:373–391. doi: 10.1016/j.isci.2019.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardanyan R., Hruby V. Chapter 26 - hyperglycemic and hypoglycemic drugs. In: Vardanyan R., Hruby V., editors. Synthesis of Best-Seller Drugs. Academic Press; 2016. pp. 419–458. [Google Scholar]
- Wang S.-M., Moku B., Leng J., Qin H.-L. Rh-catalyzed carboxylates directed C–H activation for the synthesis of ortho-carboxylic 2-arylethenesulfonyl fluorides: access to unique electrophiles for SuFEx click chemistry. Eur. J. Org. Chem. 2018;32:4407–4410. [Google Scholar]
- Weis E., Johansson M.J., Martín-Matute B. IrIII-catalyzed selective ortho-monoiodination of benzoic acids with unbiased C−H bonds. Chem. Eur. J. 2020;26:10185–10190. doi: 10.1002/chem.202002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welton T. Solvents and sustainable chemistry. Proc. R. Soc. A. 2015;471:20150502. doi: 10.1098/rspa.2015.0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wencel-Delord J., Nimphius C., Wang H., Glorius F. Rhodium(III) and hexabromobenzene—a catalyst system for the cross-dehydrogenative coupling of simple arenes and heterocycles with arenes bearing directing groups. Angew. Chem. Int. Ed. Engl. 2012;51:13001–13005. doi: 10.1002/anie.201205734. [DOI] [PubMed] [Google Scholar]
- Yang S., Li B., Wan X., Shi Z. Ortho arylation of acetanilides via Pd(II)-Catalyzed C−H functionalization'. J. Am. Chem. Soc. 2007;129:6066–6067. doi: 10.1021/ja070767s. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Tang W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B. 2018;8:721–732. doi: 10.1016/j.apsb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Snieckus V. Ester-directed Ru-catalyzed C–O activation/C–C coupling reaction of ortho-methoxy naphthoates with organoboroneopentylates. Chem. Comm. 2016;52:1681–1684. doi: 10.1039/c5cc09121g. [DOI] [PubMed] [Google Scholar]
- Zimmerman D.M., Leander J.D., Cantrell B.E., Reel J.K., Snoddy J., Mendelsohn L.G., Johnson B.G., Mitch C.H. Structure-activity relationships of trans-3,4-dimethyl-4-(3-hydroxyphenyl)piperidine antagonists for .mu.- and .kappa.-opioid receptors. J. Med. Chem. 1993;36:2833–2841. doi: 10.1021/jm00072a001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.