

Abstract
Burkitt's lymphoma (BL) is a highly aggressive form of B-cell non-Hodgkin's lymphoma. The clinical outcome in children with BL has improved over the last years but the prognosis for adults is still poor, highlighting the need for novel treatment strategies. Here, we report that the combinational treatment with the Smac mimetic BV6 and TRAIL triggers necroptosis in BL when caspases are blocked by zVAD.fmk (TBZ treatment). The sensitivity of BL cells to TBZ correlates with MLKL expression. We demonstrate that necroptotic signaling critically depends on MLKL, since siRNA-induced knockdown and CRISPR/Cas9-mediated knockout of MLKL profoundly protect BL cells from TBZ-induced necroptosis. Conversely, MLKL overexpression in cell lines expressing low levels of MLKL leads to necroptosis induction, which can be rescued by pharmacological inhibitors, highlighting the important role of MLKL for necroptosis execution. Importantly, the methylation status analysis of the MLKL promoter reveals a correlation between methylation and MLKL expression. Thus, MLKL is epigenetically regulated in BL and might serve as a prognostic marker for treatment success of necroptosis-based therapies. These findings have crucial implications for the development of new treatment options for BL.
Keywords: Smac mimetics, TRAIL, Burkitt's lymphoma, Necroptosis, MLKL
Abbreviations: BL, Burkitt's lymphoma; Blast, Blasticidin; cIAP, cellular inhibitor of apoptosis; CRISPR, clustered regularly interspaced short palindromic repeats; DNMT, DNA-methyltransferase; DOX, doxycycline hydrochloride; EV, empty vector; FASL, FAS ligand; FASR, FAS receptor; FCS, fetal calf serum; IAP, inhibitor of apoptosis; KD, knockdown; KO, knockout; MLKL, mixed lineage kinase domain-like protein; Nec-1s, Necrostatin-1s; NSA, Necrosulfonamide; n.h.t, non-human target; OE, overexpression; PBMC, peripheral blood mononuclear cells; PI, propidium iodide; Puro, Puromycin; RIP, receptor interacting protein; RIPK1/3, receptor interacting serine/threonine kinase 1/3; SEM, standard error of the mean; TBZ, TRAIL/BV6/zVAD.fmk; TNFα, tumor necrosis factor alpha; TNFR1, tumor necrosis factor receptor 1; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TRAIL-R1, TRAIL receptor 1; TRAIL-R2, TRAIL receptor 2; zVAD.fmk, N-benzyloxycarbonyl-Val-Ala-Asp(O–Me) fluoromethylketone
Graphical Abstract
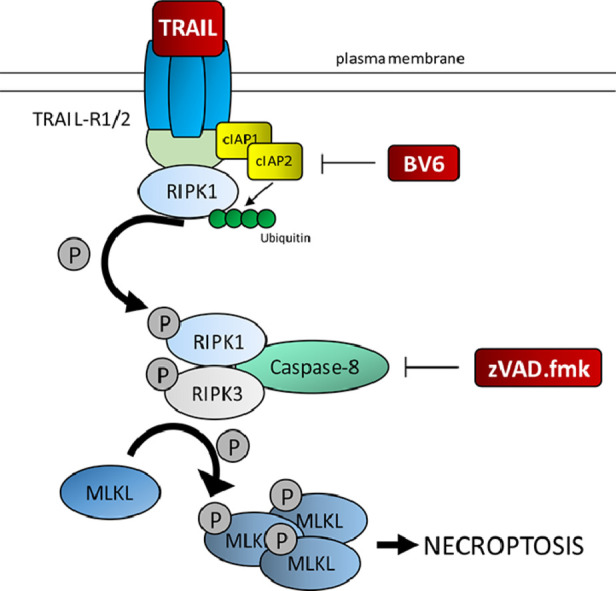
Introduction
Burkitt's lymphoma (BL) is a highly aggressive form of B-cell non-Hodgkin's lymphoma and one of the fastest growing human tumors [1]. Treatment strategies of BL for children and adults share multiagent chemotherapy but consensus of ideal first-line treatment is still lacking [2]. Nearly 90% of children can be cured due to their high tolerance to intensive chemotherapy [3]. However, treatment options for adults are limited, which highlights the need for advanced strategies.
Since evasion of apoptosis is a hallmark of cancer [4], the activation of alternative programmed cell death pathways might be an attractive strategy to induce cell death. Targeting necroptosis, a regulated form of necrosis, is considered as a possible therapeutic option to induce cell death especially for apoptosis-resistant cancers [5,6]. The necroptotic pathway can be activated by various stimuli including death receptor stimulation. Binding of tumor necrosis factor alpha (TNFα) to tumor necrosis factor receptor (TNFR) is considered to be the best described in vitro model of the necroptotic pathway [7]. Besides, other stimuli including FAS ligand (FASL) [8,9] or TNF-related apoptosis-inducing ligand (TRAIL) are known to initiate necroptotic cell death in various cancer cells [8,10]. Necroptosis execution depends on the formation of the necrosome, consisting of the kinases MLKL (mixed lineage kinase domain-like protein), RIPK1 and RIPK3 (receptor-interacting serine/threonine-protein kinase 1 and 3) [11]. Upon activation of necroptosis, RIPK1 and RIPK3 interact with their RIP homotypic interaction motifs (RHIM) to form heterodimeric filamentous structures [12] leading to phosphorylation and activation of RIPK3 [13]. MLKL is a direct substrate of RIPK3 and a central executor of necroptosis. RIPK3-dependent MLKL phosphorylation results in MLKL oligomerization and translocation to the plasma membrane [14]. Association of MLKL with the membrane then leads to its disruption and cell death [15].
A common strategy to overcome drug resistance is the combination of multiple drugs to induce cell death in cancer cells [16]. Several studies discovered that combining TRAIL with other anti-cancer drugs enhances the anti-cancer capability [17,18]. One approach to sensitize cancer cells is the neutralization of inhibitor of apoptosis proteins (IAPs), which are often overexpressed in cancer cells prohibiting cell death induction [19,20]. In our study, we concentrated on the cellular IAPs, cellular IAP (cIAP)1 and cIAP2, which both function as E3 ubiquitin ligases maintaining constitutive ubiquitylation of RIPK1 [21] thereby suppressing necrosome formation. cIAP1/2 can be antagonized by Smac mimetics, leading to their autoubiquitylation and subsequent proteasomal degradation [22], resulting in the induction of cell death [23]. Smac mimetics have previously been reported to display antitumor activity in BL [24]. The key regulators of necroptosis, i.e. RIPK3 and MLKL, are often downregulated in cancer cells [25,26]. However, in BL necroptotic cell death has not yet been studied. Since BL cell lines express the death receptors TRAIL-R1/2 [27], we hypothesized that the combination of Smac mimetics with TRAIL could induce necroptosis in BL, when caspases are inhibited.
Materials and methods
Cell culture and chemicals
Human BL cell lines RAMOS, DG-75, RAJI, BJAB and DAUDI were obtained from the German Collection of Microorganisms and Cell cultures (DSMZ, Braunschweig, Germany) and BL-2, BL-30, Seraphine, BL-60, BL-70 and Salina cells were kindly provided by T. Oellerich, Department of Medicine II, Hematology/Oncology, University Hospital Frankfurt, Germany. All cell lines were authenticated by STR profiling and continuously monitored for mycoplasma contamination. Cells were cultured in RPMI 1640 (Life Technologies), supplemented with 10% or 20% fetal calf serum (FCS) and 1% penicillin/streptomycin (Invitrogen). The bivalent Smac mimetic BV6 was kindly provided by Genentech. The broad-range caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethylketone (zVAD.fmk) was purchased from Bachem, Necrosulfonamide (NSA) from Toronto Research Chemicals Inc., GSK’872 and Necrostatin-1s (Nec-1s) from Merck and Dabrafenib from Selleck Chemicals. Recombinant human TRAIL was obtained from R&D Systems, human recombinant TNFα from PeproTech and human multimeric FASL from AdipoGen. Doxycycline hydrochloride (DOX) was purchased from Sigma-Aldrich. LCL-161 was purchased from Novartis and AT-406, Birinapant and SGI-110 (Guadecitabine) were obtained from Selleck Chemicals. All other chemicals were purchased from Sigma-Aldrich or Carl Roth unless indicated otherwise.
Cell death assay
All cells were seeded in a density of 2 × 105 cells/mL in a 96-well plate and instantly treated, except for BL-30 cells, which were seeded at a density of 1 × 105 cells/mL 24 h prior to treatment. Pretreatment with zVAD.fmk, Nec-1s, NSA, GSK’872 and Dabrafenib for 30 min was followed by the addition of Smac mimetics. After 1 h, TRAIL was applied. RAJI- and DAUDI MLKL- and/or RIPK3-overexpressing cells were pretreated with Nec-1s, GSK’872, NSA and Dabrafenib and zVAD.fmk overnight, then BV6 and DOX were added for 1 h followed by TRAIL. Cell death was assessed by measuring the uptake of propidium iodide (PI) using PI/Hoechst double staining diluted in PBS for 10 min using ImageXpress® Micro XLS system (Molecular devices, LLC, Biberach and der Riss, Germany), according to the manufacturer's instructions.
Stable overexpression of MLKL and RIPK3
For stable MLKL or RIPK3 overexpression constructs for human MLKL, RIPK3 and DOX-inducible RIPK3 were cloned into pSBbi-Blasticidin (Addgene plasmid #60526) and pSBtet-Puromycin (Addgene plasmid #60507) [28]. Cells were then transfected with 1800 ng pSBbi-RIPK3-blast or pSBbi-MLKL-blast and 200 ng pCMV(CAT)T7-SB100 (Addgene plasmid #34879) using the Neon Transfection System (Invitrogen) following the manufacturer's protocol. For stable co-overexpression of both MLKL and RIPK3, 1350 ng of pSBbi-MLKL-blast and 1350 ng of pSBtet-RIPK3-puro were used in combination with 300 ng of pCMV(CAT)T7-SB100. As a control, cells were transfected with 2700 ng of pSBbi-blast and 300 ng of pCMV(CAT)T7-SB100 for 72 h. Transfection was followed by at least two weeks of selection with puromycin (10 µg/mL) and/or blasticidin (20 µg/mL).
Methylation assay
Total DNA was isolated using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer's instructions. Bisulfite conversion of non-methylated cytosines to uracils was achieved using the EZ DNA Methylation-Lightning™Kit (Zymo Research), according to the manufacturer's protocol using 200 ng DNA. Amplification of bisulfite converted DNA by PCR (forward primer: ATTAGAGTTGAGGAAATGAAATTA, reverse primer: AAATAAACATAAAATCTTCCCAAAAA) was performed (recognizing uracil as thymine) to enrich parts of the CpG island in the promoter region of MLKL, followed by Sanger sequencing (forward primer: ATTAGAGTTGAGGAAATGAAATTA).
Western blot analysis
Western blot analysis was performed as described previously [29], using the following antibodies: rabbit anti-MLKL, rabbit anti-RIPK3, rabbit anti-phospho MLKL, rabbit anti-phospho RIKP1, rabbit anti-phospho RIPK3, rabbit anti-TNFR1, rabbit anti-cIAP2 (all from Cell Signaling), mouse anti-RIPK1 (BD Bioscience), mouse anti-GAPDH (BioTrend), rabbit anti-TRAIL-R1 (Abcam), rabbit anti-TRAIL-R2 (Millipore), rabbit anti-FasR, mouse anti-cIAP1 (all from Santa Cruz), mouse anti-Caspase-8 (Enzo). For detection, secondary antibodies goat anti-mouse IgG and goat anti-rabbit IgG (Abcam) conjugated to horseradish peroxidase and enhanced chemiluminescence were used (Amersham Bioscience).
RNA interference
Transient silencing of MLKL was performed by transfecting cells with Silencer® Select siRNAs (Life Technologies, Inc.): human siMLKL (s47087, s47088, s47089) using Neon Transfection System following the manufacturer's protocol. Cells were transfected with 200 nM of siRNA, followed by a second transfection after 24 h. 24 h after the second transfection cells were treated. Knockdown efficiency was confirmed by Western blot analysis.
Quantitative real-time PCR
PeqGOLD Total RNA kit (Peqlab) was used to isolate total RNA, followed by cDNA synthesis using RevertAid H Minus First Strand cDNA Synthesis Kit (MBI Fermentas GmbH) according to the manufacturer's instructions. mRNA levels were analyzed by SYBR green-based qRT-PCR using the QuantStudio 7 Flex real-time PCR system (Applied Biosystems). The samples were normalized to 28s-rRNA. Relative expression levels were calculated using ΔΔct-method. The following primers purchased from Eurofins were used for amplification: TRAIL-R1 (forward primer: GGGTCCACAAG ACCTTCAAGT, reverse primer: TGCAGCTGAGCTAGGTACGA); TRAIL-R2 (forward primer: AGACCCTGTGCTCGTTGTC, reverse primer: TTGTTGGGTGATCAGAGCAG), 28s (forward primer: TTGAAAATCCGGGGGAGAG, reverse primer: ACATTGTTCCAACATGCCAG).
Generation of CRISPR/Cas9-derived MLKL knockout cell lines
To generate CRISPR/Cas9-mediated knockout cells, sgRNAs against MLKL (CACCGATGACAATGGAGAATTGAGG,CACCGCCTGTTTCACCCATAAGCCA,CACCGTTCCCTTAGCAGAATCCACG) or GFP (as a non-human target (n.h.t)) (GGAGCGCACCATCTTCTTCA, GGCCACAAGTTCAGCGTGTC, GGGCGAGGAGCTGTTCACCG) were cloned into pLentiCRISPRv2 (Addgene #52961). Lentiviral particle production was then performed in HEK293T cells, by co-transfection of pLenti-CRISPRv2 with pPAX2 (Addgene #12260) and pMD2.G (Addgene #12259), using FuGENE® HD according to the manufacturer's protocol. BL-2, RAMOS, Seraphine and BL-30 cells were transduced with the collected supernatant containing viral particles by spin transduction, followed by selection with puromycin and single clone cultivation.
Statistical analysis
Statistical significance was assessed using Student's t-test (two-tailed distribution, equal variance) calculated with Excel and two-way ANOVA (Bonferroni test) calculated with GraphPad Prism (Version 5.0). Data were expressed as mean and SEM of at least three independent experiments performed in triplicates. P values were assigned as follows: *P< 0.5; **P< 0.01;***P< 0.001.
Data availability
Primary data are available on request from the authors.
Results
Smac mimetic BV6 induces cell death in BL cell lines
Smac mimetics are known to trigger cell death and sensitize cancer cells to various anticancer therapies [22]. To evaluate the therapeutic potential of Smac mimetics alone or in combination with other compounds in BL, we initially treated a panel of BL cell lines with increasing concentrations of the Smac mimetic BV6. While all cell lines showed a dose-dependent induction of cell death upon BV6 treatment, sensitivity varied among the cell lines (Fig. 1A). To investigate whether BV6 treatment can induce non-apoptotic cell death we applied the pan-caspase inhibitor zVAD.fmk to block apoptosis. BV6-induced cell death was caspase-independent and even increased compared to BV6 alone in RAMOS, BL-2, BL-30, Seraphine and DG-75 cells, while the other cell lines showed no increase of cell death (Fig. 1B). To further potentiate cell death induction, we additionally treated BL cells with a combination of TRAIL, BV6 and zVAD.fmk (TBZ). The TBZ combination resulted in a further increase of cell death in the BV6/zVAD.fmk-sensitive cell lines (Fig. 1B). To evaluate the potential of BL cells to undergo necroptosis, we monitored basal expression levels of key components of necroptosis signaling pathways (cIAP1, cIAP2, Caspase-8, RIPK1, RIPK3 and MLKL) in a panel of BL cell lines by Western blot analysis. Expression levels of Caspase-8 and RIPK1 did not differ much between the cell lines, while DG-75, BL-60 and BJAB cells displayed lower cIAP2 levels. RAJI, BJAB and DAUDI cells exhibited lower cIAP1 levels and BJAB and DAUDI cells lacked RIPK3 expression. Interestingly, MLKL was differentially expressed among the BL cell lines (Fig. 1C). Cells that expressed high MLKL levels including RAMOS, BL-2, BL-30, Seraphine and DG-75 cells were sensitive to TBZ-induced cell death. In contrast, BL-60, BL-70, Salina, RAJI, BJAB and DAUDI cells express low levels of MLKL, which is in line with their resistance to TBZ-induced cell death (Fig. 1B), underlining the importance of MLKL expression for necroptosis induction. Taken together, these data indicate that cell death induced by TBZ is caspase-independent and occurs in BL cells expressing high levels of MLKL.
Fig. 1.

TBZ combination treatment induces caspase-independent cell death in BL. (A) Cells were treated for 24 h with indicated concentrations of BV6. (B) Cells were treated for 24 h (BL-2: 48 h) with BV6 (DG-75, DAUDI: 2 µM, BL-2, BL-60, Salina, RAJI: 4 µM, BJAB: 5 µM, BL-30, BL-70: 6 µM, Seraphine: 7 µM, RAMOS: 18 µM), TRAIL (BL-30: 5 ng/mL, RAMOS/Seraphine: 10 ng/mL, BL-2, DG-75, BL-60, BL-70, Salina, RAJI, BJAB, DAUDI: 15 ng/mL) and zVAD.fmk (20 µM). Cell death was determined by analysis of PI/Hoechst staining and ImageXpress Micro XLS system. Mean and SEM of 3 independent experiments are shown. (C) Protein expression of RIPK1, RIPK3, MLKL, Caspase-8, cIAP1 and cIAP2 was analyzed by Western blotting. GAPDH served as loading control. The asterisk (*) indicates an unspecific band.
TBZ combination treatment triggers necroptosis
To investigate in detail necroptosis induction in BL cells upon TBZ treatment, we initially monitored cell death kinetics. Kinetic analysis revealed that BL-2, RAMOS, Seraphine and BL-30 cells undergo cell death in a time-dependent manner upon TBZ combination treatment in comparison to single agent treatment of either BV6 or TRAIL alone or BV6 in combination with zVAD.fmk (Fig. 2A).
Fig. 2.

TBZ treatment induces necroptotic cell death. Cells were treated for the indicated time points with BV6 (BL-2: 4 µM, BL-30: 6 µM, Seraphine: 7 µM, RAMOS: 18 µM) and TRAIL (BL-30: 5 ng/mL, RAMOS/ Seraphine: 10 ng/mL, BL-2: 15 ng/mL) and zVAD.fmk (20 µM). (A) Cell death was determined by analysis of PI/Hoechst staining and ImageXpress Micro XLS system. Mean and SEM of 3 independent experiments are shown. *P < 0.05; ***P < 0.001. (B) Protein levels of p-RIPK1, RIPK1, p-MLKL and MLKL were assessed by Western blotting, GAPDH served as loading control. (C) Cells were treated with TBZ in the presence and absence of Dabrafenib (BL-30: 5 µM, BL-2, RAMOS: 10 µM) or GSK’872 (10 µM), Nec-1s (20 µM) and NSA (1,5 µM) for 24 h (BL-2: 48 h). Cell death was determined by analysis of PI/Hoechst staining and ImageXpress Micro XLS system. Mean and SEM of 3 independent experiments are shown. ***P < 0.001.
Since we have demonstrated that upon TBZ treatment cell death occurs in a caspase-independent manner, we investigated whether TBZ treatment triggers necroptosis in BL cells. To this end, we assessed phosphorylation of MLKL and RIPK1, as critical steps of necroptosis execution by Western blot analysis. Time course analyses revealed that phosphorylation of RIPK1 and MLKL upon TBZ treatment occurred prior to cell death induction. Indeed, RIPK1 is phosphorylated already after 3 h of combination treatment followed by MLKL phosphorylation after 6 h (Fig. 2B). To further confirm the induction of necroptosis, pharmacological inhibitors of key necroptotic proteins were added prior to cell death induction by TBZ. Consistently, RIPK3 inhibitors Dabrafenib or GSK’872, RIPK1 inhibitor Nec-1s and MLKL inhibitor NSA significantly abrogated cell death induction upon TBZ in all cell lines, whereas cell death induced by only TRAIL and BV6 was not blocked by these inhibitors (Fig. 2C). Together, these findings demonstrate that the combination of TRAIL, BV6 and zVAD.fmk induces necroptosis in BL cell lines.
Smac mimetics induce necroptosis in BL
To illustrate the clinical relevance and translational potential of our findings we extended our investigation to other Smac mimetics. The monovalent Smac mimetics AT-406 and LCL-161 and the bivalent Smac mimetic Birinapant are currently being tested in clinical trials revealing promising anticancer activity [30], [31], [32]. Similar to TBZ combination treatment, application of AT-406, LCL-161 and Birinapant also induced cell death in BL cells in combination with TRAIL, when caspases were blocked by zVAD.fmk. Application of the pharmacological inhibitors Dabrafenib, Nec-1s and NSA significantly rescued TRAIL/Smac mimetic/zVAD.fmk-induced cell death, confirming necroptotic cell death induction in BL cell lines (Fig. 3A).
Fig. 3.

Smac mimetics induce necroptosis in BL. (A) Cells were treated with BV6 (BL-2: 4 µM, BL-30: 6 µM), AT-406, LCL-161 and Birinapant (2,5 µM), TRAIL (BL-30: 5 ng/mL, BL-2: 15 ng/mL) and zVAD.fmk (20 µM) in the presence and absence of Dabrafenib (BL-30: 5 µM, BL-2: 10 µM), Nec-1s (20 µM) and NSA (1,5 µM) for 24 h (BL-30) or 48 h (BL-2). Cell death was determined by analysis of PI/Hoechst staining and ImageXpress Micro XLS system. (B) Cells were treated for 24 h (BL-30, RAJI, RAMOS, Seraphine, DAUDI) or 48 h (BL-2) with BV6 (BL-2: 4 µM, DAUDI: 5 µM, BL-30: 6 µM, Seraphine: 7 µM, RAJI: 8 µM, RAMOS: 18 µM), TRAIL (BL-30: 5 ng/mL, RAMOS/Seraphine: 10 ng/mL, BL-2, RAJI, DAUDI: 15 ng/mL) and zVAD.fmk (20 µM). mRNA expression of TRAIL-R1 and TRAIL-R2 was analyzed by qRT-PCR and fold change normalized to 28s mRNA is shown. Mean and SEM of 3 independent experiments are shown; *P < 0.05; **P < 0.01; ***P < 0.001.
Importantly, TBZ treatment resulted in a higher percentage of cell death in cancer cells compared to normal blood cells obtained from healthy donors, underlining some tumor selectivity of the treatment and highlighting the potential clinical relevance of our findings (Supplementary Fig. S1). Interestingly, necroptosis can also be induced by combining BV6 and zVAD.fmk with FASL, since all cell lines express the FAS receptor (FASR) (Supplementary Fig. S2), while combination with TNFα failed to induce necroptosis in most of the cell lines (Supplementary Fig. S3).
Cell death can be promoted by the upregulation of death receptors [33,34]. Since all cell lines express TRAIL-R1 and TRAIL-R2 (Supplementary Fig. S4) and TRAIL is known to induce cell death in combination with Smac mimetics in multiple cancer cell lines [10], we analyzed if the levels of these receptors were further increased upon TBZ treatment. qRT-PCR revealed that the expression of TRAIL-R1 and TRAIL-R2 was upregulated in 5 out of 6 cell lines upon TBZ treatment (Fig. 3B), indicating a positive feedback loop to further sensitize the cells to TRAIL and potentiate cell death. Taken together, these data reveal that TRAIL receptors play an important role in the induction of necroptosis in BL cell lines and a broad range of Smac mimetics contribute to this cell death induction, further highlighting the potential clinical relevance of this combination treatment.
TBZ-mediated cell death depends on MLKL
Since MLKL is one of the key mediators in necroptotic signaling and essential for necroptosis induction [15], we hypothesized that the expression of MLKL determines responsiveness to necroptotic stimuli in BL cells. Therefore, we studied the relevance of MLKL in TBZ-induced necroptosis in BL cells using siRNA-mediated gene silencing of MLKL and confirmed knockdown efficiency via Western blotting (Fig. 4A). MLKL silencing showed a significant, although partial, inhibition of cell death upon TBZ treatment in BL-2 and RAMOS cells (Fig. 4B). To determine whether complete depletion of MLKL is sufficient to protect against TBZ-induced cell death, we performed CRISPR/Cas9 knockout of MLKL (Fig. 4C). Similar to our findings using siRNA, CRISPR/Cas9 knockout of MLKL completely blocked TBZ-induced cell death in BL-2, RAMOS, Seraphine and BL-30 cells (Fig. 4D), suggesting that MLKL depletion alone protects against TBZ-induced necroptosis in BL cells. To exclude possible effects on upstream signaling we performed Western blot analyses, which revealed that phosphorylation of RIPK1 still occurs upon treatment with TBZ when MLKL is downregulated by CRISPR/Cas9-mediated knockout (Fig. 4E). Taken together, we conclude that MLKL is required for the induction of TBZ-induced necroptosis in BL cell lines.
Fig. 4.

TBZ-induced cell death depends on MLKL. (A) BL-2 and RAMOS cells were transfected twice with 200 nM non-silencing control siRNA (siCtrl) or siRNAs against MLKL. Expression of MLKL was analyzed by Western blotting, GAPDH served as loading control. (B) MLKL knockdown cells were treated with BV6 (BL-2: 4 µM, RAMOS: 18 µM) and TRAIL (RAMOS: 10 ng/mL, BL-2: 15 ng/mL) in the presence of zVAD.fmk (20 µM) for 24 h (RAMOS) and 48 h (BL-2) and cell death was determined by PI/Hoechst staining. Mean and SEM of at least 3 experiments performed in triplicates are shown; **P < 0.01; ***P < 0.001. (C) MLKL knockout (KO) was performed by CRISPR/Cas9. MLKL levels were analyzed by Western blotting, GAPDH served as loading control. (D) MLKL KO cells and non-human target (n.h.t) control cells were treated for 24 h (RAMOS, Seraphine, BL-30) and 48 h (BL-2) with BV6 (BL-2: 4 µM, BL-30: 6 µM, Seraphine: 7 µM, RAMOS: 18 µM) and TRAIL (BL-30: 5 ng/mL, RAMOS/Seraphine: 10 ng/mL, BL-2: 15 ng/mL) in the presence of zVAD.fmk (20 µM). Cell death was measured by PI/Hoechst staining. Mean and SEM of at least 3 experiments performed in triplicates are shown; ***P < 0.001. (E) Cells were treated with BV6 (BL-2: 4 µM, BL-30: 6 µM, Seraphine: 7 µM, RAMOS: 18 µM) and TRAIL (BL-30: 5 ng/mL, RAMOS/Seraphine: 10 ng/mL, BL-2: 15 ng/mL) in the presence of zVAD.fmk (20 µM) for 8 h. Protein levels of p-RIPK1, RIPK1, p-MLKL, and MLKL were assessed by Western blotting, GAPDH served as a loading control.
High expression of MLKL and RIPK3 is essential for necroptosis induction
Since MLKL was shown to be required for TBZ-induced necroptosis, we further determined its biological relevance. To this end, we stably overexpressed MLKL in 2 exemplary cell lines (RAJI and DAUDI) that display low levels of endogenous MLKL expression (Fig. 1C). Although overexpression of MLKL resulted in increased expression of MLKL, only little MLKL phosphorylation was induced by TBZ combination treatment in RAJI cells, and none in DAUDI cells (Fig. 5A). This minor phosphorylation of MLKL alone was not sufficient to induce cell death (Fig. 5B). Therefore, we concluded that a lack of RIPK3 activity in RAJI and DAUDI cells (Fig. 1C) resulted in a lack of MLKL phosphorylation despite MLKL overexpression. To further investigate this interpretation, we next overexpressed RIPK3 in RAJI and DAUDI cells. Overexpression of RIPK3 alone led to a phosphorylation of RIPK3 upon TBZ treatment (Fig. 5C), but no cell death induction could be observed (Fig. 5D). Therefore, we hypothesized that BL cells might need high levels of both MLKL and RIPK3 to effectively execute necroptosis. To test this hypothesis, we performed stable overexpression of both MLKL and RIPK3 proteins in RAJI and DAUDI cells. RIPK3 expression was controlled by doxycycline (DOX)-induced transcriptional activation and could be detected already after 2 h of DOX addition (Supplementary Fig. S5). Of note, overexpression of MLKL and RIPK3 resulted in a pronounced phosphorylation of MLKL and RIPK3 upon TBZ treatment in RAJI (Fig. 5E) and DAUDI cells (Fig. 5H), as compared to the empty vector control. Ectopic RIPK3 and MLKL co-overexpression led to the induction of necroptotic cell death (Fig. 5F,I), whereas the empty vector control cells did not undergo cell death (Fig. 5G,J). Necroptotic cell death was confirmed using pharmacological inhibitors, showing a significant rescue of TBZ-induced cell death in the presence of GSK’872, Nec-1s and NSA (Fig. 5F, I). Intriguingly, siRNA-mediated knockdown of MLKL in RAJI and DAUDI cells co-overexpressing MLKL and RIPK3 resulted in a rescue of cell death (Supplementary Fig. S6). Taken together, these data highlight the importance of high levels of MLKL and RIPK3 expression for potential necroptosis-based therapies in BL.
Fig. 5.

TBZ treatment in MLKL and RIPK3 co-overexpressing cells leads to phosphorylation of RIPK3 and MLKL and the induction of necroptosis. (A) MLKL-overexpressing cells and (C) RIPK3-overexpressing cells were treated with BV6 (DAUDI: 5 µM, RAJI: 8 µM) and TRAIL (15 ng/mL) in the presence of zVAD.fmk (20 µM) for 2 h. Protein levels of p-RIPK3, RIPK3, p-MLKL, and MLKL were assessed by Western blotting, GAPDH served as loading control. (B) MLKL-overexpressing cells and (D) RIPK3-overexpressing cells were treated with BV6 (DAUDI: 5 µM, RAJI: 8 µM), TRAIL (15 ng/mL) and zVAD.fmk (20 µM) in the presence and absence of GSK’872 (20 µM), Nec-1s (40 µM) and NSA (3 µM) for 8 h. Cell death was determined by analysis of PI/Hoechst staining and ImageXpress Micro XLS system. Mean and SEM of 3 independent experiments are shown. (E, H) MLKL/RIPK3-overexpressing cells were treated with DOX (100 ng/mL), BV6 (DAUDI: 5 µM, RAJI: 8 µM), TRAIL (15 ng/mL) and zVAD.fmk (20 µM) for 2 h. Protein levels of p-RIPK3, RIPK3, p-MLKL, and MLKL were assessed by Western blotting, GAPDH served as loading control. (F, I) RAJI and DAUDI MLKL/RIPK3-overexpressing cells and (G,J) RAJI and DAUDI control cells (empty vector (EV)) were treated with DOX (100 ng/mL), BV6 (DAUDI: 5 µM, RAJI: 8 µM), TRAIL (15 ng/mL) and zVAD.fmk (20 µM) in the presence and absence of Nec-1s (40 µM), GSK’872 (20 µM) and NSA (3 µM) for 8 h. Cell death was determined by analysis of PI/Hoechst staining and ImageXpress Micro XLS system. Mean and SEM of 3 independent experiments are shown; *P < 0.05; **P < 0.01; ***P < 0.001.
MLKL expression is epigenetically regulated in BL
Gene silencing or loss of key necroptotic proteins in cancer cells is a major challenge for necroptosis induction used as a therapeutic option [26,35]. RIPK3 has been described to be epigenetically regulated in cancer [36,37], however, little is yet known about MLKL regulation. Therefore, we next analyzed the methylation status of the CpG island in the promoter region of MLKL and observed a higher methylation of cytosines in RAJI, DAUDI and Salina cells, expressing low levels of MLKL, while MLKL-expressing cells, i.e. BL-2, RAMOS, Seraphine and BL-30, exhibited no methylation of the MLKL promoter (Fig. 6A, Supplementary Fig. S7). To gain further insights into the regulation of MLKL in BL we applied the DNA methyltransferase (DNMT) inhibitor SGI-110 in cells lacking MLKL expression and observed an upregulation of MLKL in RAJI, DAUDI, and Salina cells after 72 h of SGI-110 treatment (Fig. 6B), indicating an enhanced methylation in the regulatory region of MLKL. Upregulation of total MLKL levels upon SGI-110 addition followed by treatment with TBZ in RAJI and DAUDI RIPK3-overexpressing cells and Salina cells resulted in phosphorylation of MLKL (Supplementary Fig. S8A). However, cell death assays revealed that a significant rescue with pharmacological inhibitors of RIPK3 (Dabrafenib and GSK’872) and MLKL (NSA) upon SGI-110/TBZ treatment could only be observed in the RAJI RIPK3-overexpressing cell line (Supplementary Fig. S8B), indicating that the effect of SGI-110 might be cell line-dependent. Together, these data indicate an epigenetic regulation of MLKL expression by methylation of the promoter region of MLKL in BL cells that are resistant to TBZ treatment.
Fig. 6.

MLKL expression is epigenetically regulated by methylation. (A) Non-methylated cytosines of extracted DNA were converted into uracils using sodium bisulfite. PCR of bisulfite converted DNA (recognizing uracils as thymines) was performed to enrich parts of the CpG island in the promoter of MLKL, followed by Sanger sequencing. (B) Cells were treated for 72 h with 500 nM SGI-110. Protein levels of MLKL were assessed by Western blotting, GAPDH served as a loading control.
Discussion
BL is a highly aggressive B-cell non-Hodgkin's lymphoma with adverse clinical outcomes in particular in adult patients due to the lack of effective therapy. Apoptosis resistance is another common therapeutic problem in cancer therapy including BL [38], [39], [40]. Therefore, targeting necroptosis (programmed necrosis) represents an alternative strategy to induce programmed cell death in cancer cells [6,41], however, its role in BL is unclear. Here, we report for the first time that treatment with both the Smac mimetic BV6 and TRAIL induced MLKL-dependent necroptosis in BL when caspases were inhibited. This conclusion is supported by several independent pieces of experimental evidence.
First, we discovered, that most BL cell lines express the key proteins of the necroptosis pathway RIPK1, RIPK3, and MLKL, which are reported to be lost or silenced in many types of cancer, and this is associated with poor overall survival [6,42]. Interestingly, cell death induction upon TBZ treatment correlated with the expression of MLKL, since we demonstrated that MLKL-expressing BL cells are highly sensitive to TBZ treatment. BL cells expressing low levels or lacking MLKL were resistant to necroptotic cell death.
Second, in our kinetic studies we show that phosphorylation of the kinases RIPK1 and MLKL occurs prior to cell death induction upon TBZ stimulation. Phosphorylation of RIPK1, RIPK3, and MLKL is known to be essential for necrosome formation and required for necroptosis execution [14,[43], [44], [45].
Third, pharmacological inhibition of RIPK1, RIPK3, and MLKL protects BL cells from TBZ-induced cell death, indicating susceptibility of BL cells to necroptosis and absence of alternative types of cell death.
Our findings have important implications for the development of necroptosis-inducing therapies based on Smac mimetic combinational treatments. Both Smac mimetics and recombinant TRAIL have been tested in several cancer models [10,[46], [47], [48], [49], [50], [51], but very little data are available on their therapeutic potential in BL. Besides BV6 combinations, in the present study, several other Smac mimetics that have already entered clinical trials [30,31,52] were tested in combination with TRAIL. Interestingly, all Smac mimetics tested (i.e., AT-406, LCL-161 and Birinapant) were capable of necroptosis induction in BL cells, confirming the contribution of IAP antagonism to necroptotic signaling. This is in line with previous studies, showing that IAP antagonism by Smac mimetics induces necroptosis in acute myeloid leukemia cells and pancreatic carcinoma cells, when caspases were blocked [53,54]. Additionally, we demonstrate that BL cells were more susceptible to TBZ treatment than normal blood cells, indicating some cancer selectivity of the combination. In the present study, we observed that all BL cell lines express TRAIL receptors and its expression can be further upregulated upon TBZ treatment. This leads to the speculation that this upregulation of TRAIL receptors might further sensitize cells to TRAIL, as it has previously been reported that treatment with TRAIL in combination with chemotherapeutic agents results in an upregulated expression of TRAIL-R1 and TRAIL-R2 [33]. These results highlight the impact of treatments targeting TRAIL signaling as promising therapeutic options for BL. Besides TRAIL, BL cell lines are also sensitive to FASL, as FASL/BV6/zVAD.fmk treatment induces necroptosis. Of note, prototypical necroptosis induction by TNFα occurred only in two cell lines, which is in line with our results showing that most BL cell lines lack expression of TNFR1. Together these findings underline the clinical relevance of Smac mimetics- and recombinant TRAIL-based treatments in BL in particular when apoptosis induction is compromised.
Not much is known about the regulation of necroptotic signaling in BL. Here, we provide compelling evidence showing that the pseudokinase MLKL is an essential mediator of TBZ-driven necroptosis in BL, since knockdown of MLKL partially and knockout completely abrogates cell death. These results are in line with previous studies demonstrating that MLKL knockout rescues TRAIL/Smac mimetic/zVAD.fmk-induced cell death in HeLa cells [55]. This highlights that TRAIL, in combination with Smac mimetics, induces MLKL-dependent necroptotic signaling when caspases are inhibited. MLKL is the key executioner of necroptotic signaling and required for cell death induction in BL. No other caspase-independent types of cell death are induced upon TBZ, when MLKL is not expressed or blocked by pharmacological inhibitors. We demonstrate that Smac mimetics in combination with TRAIL trigger caspase-independent, necroptotic cell death in a MLKL-dependent manner. This is confirmed by the fact that overexpression of MLKL, and in some cases RIPK3, is critical for necroptosis induction upon TBZ. Thus, high expression levels of both these kinases are required to induce necroptosis in cholangiocarcinoma cells [35], suggesting that analysis of expression patterns of MLKL and RIPK3 in patients might serve as an important tool to predict sensitivity to TBZ-induced necroptosis and the success of a potential therapy.
RIPK3 expression is known to be epigenetically regulated in cancer cells and can be reduced by overexpression of the DNA methyltransferase 1 (DNMT1) [36], or stimulation of DNMT1 [56], but the epigenetic regulation of MLKL has not yet been investigated. Here, we provide new insights into the epigenetic regulation of MLKL in BL cell lines. Interestingly, we could show that MLKL expression levels correlated with the methylation status of cytosines in the promoter region of MLKL. Consistently, application of the DNA methyltransferase inhibitor SGI-110 restores MLKL expression, indicating an epigenetic silencing of MLKL in cell lines exhibiting low levels of MLKL. Combined treatment with SGI-110 and TBZ led to phosphorylation of MLKL in all tested cell lines, indicating that hypomethylating agents facilitate the activation of necroptotic signaling. However, necroptotic cell death induction upon SGI-110/TBZ treatment might be cell line-specific, as SGI-110 not only impedes the methylation of the MLKL promoter but also regulates the expression of numerous other genes [57]. Therefore, our study reveals that, although hypomethylating agents facilitate the activation of necroptotic signaling upon TBZ treatment in MLKL-deficient cells, further studies are essentially required to investigate whether BL patients can benefit from receiving these agents.
In summary, our study is the first report demonstrating the susceptibility of BL cells to caspase-independent necroptotic cell death induction upon TBZ that depends on high expression levels of MLKL and RIPK3. In addition, the expression of MLKL is epigenetically regulated and therefore expression levels of MLKL and RIPK3 might serve as prognostic markers for the induction of necroptosis by TBZ and the following treatment success. Thus, the combination of Smac mimetics and death receptor ligands may offer a new approach to initiate necroptosis as an alternative form of programmed cell death in BL.
Declaration of competing interests
All authors declare that they have no competing financial or nonfinancial interests that might have influenced the performance or presentation of the work described in this manuscript.
Funding
This work was supported by grants from the Deutsche Krebshilfe (70113680 to SF and SJLvW) and BMBF (to AK).
Author contributions
AK performed the experiments; JR, BJ and SJLvW provided vectors for transduction experiments; AK, ND analyzed data; AK, ND and SF drafted study design and wrote the manuscript. All authors reviewed the manuscript.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neo.2021.03.003.
Appendix. Supplementary materials
References
- 1.Burkitt D. A sarcoma involving the jaws in African children. Br J Surg. 1958;46:218–223. doi: 10.1002/bjs.18004619704. [DOI] [PubMed] [Google Scholar]
- 2.Kalisz K., Alessandrino F., Beck R., Smith D., Kikano E., Ramaiya N.H., Tirumani S.H. An update on Burkitt lymphoma: a review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging. 2019;10:56. doi: 10.1186/s13244-019-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patte C., Auperin A., Gerrard M., Michon J., Pinkerton R., Sposto R., Weston C., Raphael M., Perkins S.L., McCarthy K., Cairo M.S., Committee F.L.I.S. Results of the randomized international FAB/LMB96 trial for intermediate risk B-cell non-Hodgkin lymphoma in children and adolescents: it is possible to reduce treatment for the early responding patients. Blood. 2007;109:2773–2780. doi: 10.1182/blood-2006-07-036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 6.Gong Y., Fan Z., Luo G., Yang C., Huang Q., Fan K., Cheng H., Jin K., Ni Q., Yu X., Liu C. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18:100. doi: 10.1186/s12943-019-1029-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fulda S. The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther. 2013;14:999–1004. doi: 10.4161/cbt.26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holler N., Zaru R., Micheau O., Thome M., Attinger A., Valitutti S., Bodmer J.L., Schneider P., Seed B., Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 9.Vercammen D., Brouckaert G., Denecker G., Van de Craen M., Declercq W., Fiers W., Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188:919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voigt S., Philipp S., Davarnia P., Winoto-Morbach S., Roder C., Arenz C., Trauzold A., Kabelitz D., Schutze S., Kalthoff H., Adam D. TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC Cancer. 2014;14:74. doi: 10.1186/1471-2407-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasparakis M., Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 12.Li J., McQuade T., Siemer A.B., Napetschnig J., Moriwaki K., Hsiao Y.S., Damko E., Moquin D., Walz T., McDermott A., Chan F.K., Wu H. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moriwaki K., Chan F.K. The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis. Int Rev Cell Mol Biol. 2017;328:253–275. doi: 10.1016/bs.ircmb.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 15.Wang H., Sun L., Su L., Rizo J., Liu L., Wang L.F., Wang F.S., Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 16.Leary M., Heerboth S., Lapinska K., Sarkar S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers. 2018;10 doi: 10.3390/cancers10120483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jo E.B., Lee Y.S., Lee H., Park J.B., Park H., Choi Y.L., Hong D., Kim S.J. Combination therapy with c-met inhibitor and TRAIL enhances apoptosis in dedifferentiated liposarcoma patient-derived cells. BMC Cancer. 2019;19:496. doi: 10.1186/s12885-019-5713-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hellwig C.T., Rehm M. TRAIL signaling and synergy mechanisms used in TRAIL-based combination therapies. Mol Cancer Ther. 2012;11:3–13. doi: 10.1158/1535-7163.MCT-11-0434. [DOI] [PubMed] [Google Scholar]
- 19.Lincoln F.A., Imig D., Boccellato C., Juric V., Noonan J., Kontermann R.E., Allgower F., Murphy B.M., Rehm M. Sensitization of glioblastoma cells to TRAIL-induced apoptosis by IAP- and Bcl-2 antagonism. Cell Death Dis. 2018;9:1112. doi: 10.1038/s41419-018-1160-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fulda S., Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 21.Bertrand M.J., Milutinovic S., Dickson K.M., Ho W.C., Boudreault A., Durkin J., Gillard J.W., Jaquith J.B., Morris S.J., Barker P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 22.Fulda S. Smac mimetics as IAP antagonists. Semin Cell Dev Biol. 2015;39:132–138. doi: 10.1016/j.semcdb.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Fulda S. Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin Cancer Res. 2015;21:5030–5036. doi: 10.1158/1078-0432.CCR-15-0365. [DOI] [PubMed] [Google Scholar]
- 24.Runckel K., Barth M.J., Mavis C., Gu J.J., Hernandez-Ilizaliturri F.J. The SMAC mimetic LCL-161 displays antitumor activity in preclinical models of rituximab-resistant B-cell lymphoma. Blood Adv. 2018;2:3516–3525. doi: 10.1182/bloodadvances.2018018168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nugues A.L., El Bouazzati H., Hetuin D., Berthon C., Loyens A., Bertrand E., Jouy N., Idziorek T., Quesnel B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014;5:e1384. doi: 10.1038/cddis.2014.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geserick P., Wang J., Schilling R., Horn S., Harris P.A., Bertin J., Gough P.J., Feoktistova M., Leverkus M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015;6:e1884. doi: 10.1038/cddis.2015.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tafuku S., Matsuda T., Kawakami H., Tomita M., Yagita H., Mori N. Potential mechanism of resistance to TRAIL-induced apoptosis in Burkitt's lymphoma. Eur J Haematol. 2006;76:64–74. doi: 10.1111/j.0902-4441.0000.t01-1-EJH2345.x. [DOI] [PubMed] [Google Scholar]
- 28.Kowarz E., Loscher D., Marschalek R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J. 2015;10:647–653. doi: 10.1002/biot.201400821. [DOI] [PubMed] [Google Scholar]
- 29.Fulda S., Sieverts H., Friesen C., Herr I., Debatin K.M. The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 1997;57:3823–3829. [PubMed] [Google Scholar]
- 30.Infante J.R., Dees E.C., Olszanski A.J., Dhuria S.V., Sen S., Cameron S., Cohen R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2014;32:3103–3110. doi: 10.1200/JCO.2013.52.3993. [DOI] [PubMed] [Google Scholar]
- 31.Hurwitz H.I., Smith D.C., Pitot H.C., Brill J.M., Chugh R., Rouits E., Rubin J., Strickler J., Vuagniaux G., Sorensen J.M., Zanna C. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT-406) in patients with advanced cancer: results of a first-in-man study. Cancer Chemother Pharmacol. 2015;75:851–859. doi: 10.1007/s00280-015-2709-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amaravadi R.K., Senzer N.N., Martin L.P., Schilder R.J., LoRusso P., Papadopoulos K.P., Weng D.E., Graham M., Adjei A.A. A phase I study of birinapant (TL32711) combined with multiple chemotherapies evaluating tolerability and clinical activity for solid tumor patients. Journal of Clinical Oncology. 2013;31 [Google Scholar]
- 33.Kim E.Y., Ryu J.H., Kim A.K. CAPE promotes TRAIL-induced apoptosis through the upregulation of TRAIL receptors via activation of p38 and suppression of JNK in SK-Hep1 hepatocellular carcinoma cells. Int J Oncol. 2013;43:1291–1300. doi: 10.3892/ijo.2013.2018. [DOI] [PubMed] [Google Scholar]
- 34.Hu R., Yang Y., Liu Z., Jiang H., Zhu K., Li J., Xu W. The XIAP inhibitor Embelin enhances TRAIL-induced apoptosis in human leukemia cells by DR4 and DR5 upregulation. Tumour Biol. 2015;36:769–777. doi: 10.1007/s13277-014-2702-6. [DOI] [PubMed] [Google Scholar]
- 35.Akara-Amornthum P., Lomphithak T., Choksi S., Tohtong R., Jitkaew S. Key necroptotic proteins are required for Smac mimetic-mediated sensitization of cholangiocarcinoma cells to TNF-alpha and chemotherapeutic gemcitabine-induced necroptosis. PLoS One. 2020;15 doi: 10.1371/journal.pone.0227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Q., Wang P., Zhang L., Tessema M., Bai L., Xu X., Li Q., Zheng X., Saxton B., Chen W., Willink R., Li Z., Zhang L., Belinsky S.A., Wang X., Zhou B., Lin Y. Epigenetic Regulation of RIP3 Suppresses Necroptosis and Increases Resistance to Chemotherapy in NonSmall Cell Lung Cancer. Transl Oncol. 2020;13:372–382. doi: 10.1016/j.tranon.2019.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koo G.B., Morgan M.J., Lee D.G., Kim W.J., Yoon J.H., Koo J.S., Kim S.I., Kim S.J., Son M.K., Hong S.S., Levy J.M., Pollyea D.A., Jordan C.T., Yan P., Frankhouser D., Nicolet D., Maharry K., Marcucci G., Choi K.S., Cho H., Thorburn A., Kim Y.S. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25:707–725. doi: 10.1038/cr.2015.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chumduri C., Gillissen B., Richter A., Richter A., Milojkovic A., Overkamp T., Muller A., Pott C., Daniel P.T. Apoptosis resistance, mitotic catastrophe, and loss of ploidy control in Burkitt lymphoma. J Mol Med (Berl) 2015;93:559–572. doi: 10.1007/s00109-014-1242-2. [DOI] [PubMed] [Google Scholar]
- 39.Farhat M., Poissonnier A., Hamze A., Ouk-Martin C., Brion J.D., Alami M., Feuillard J., Jayat-Vignoles C. Reversion of apoptotic resistance of TP53-mutated Burkitt lymphoma B-cells to spindle poisons by exogenous activation of JNK and p38 MAP kinases. Cell Death Dis. 2014;5:e1201. doi: 10.1038/cddis.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michael J.M., Lavin M.F., Watters D.J. Resistance to radiation-induced apoptosis in Burkitt's lymphoma cells is associated with defective ceramide signaling. Cancer Res. 1997;57:3600–3605. [PubMed] [Google Scholar]
- 41.Su Z., Yang Z., Xie L., DeWitt J.P., Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016;23:748–756. doi: 10.1038/cdd.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu B., Shi D., Lv X., Wu F., Chen S., Shao Z. Prognostic and clinicopathological significance of DEPTOR expression in cancer patients: a meta-analysis. Onco Targets Ther. 2018;11:5083–5092. doi: 10.2147/OTT.S167355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho Y.S., Challa S., Moquin D., Genga R., Ray T.D., Guildford M., Chan F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 45.Zhang D.W., Shao J., Lin J., Zhang N., Lu B.J., Lin S.C., Dong M.Q., Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 46.Dietz A., Dalda N., Zielke S., Dittmann J., van Wijk S.J.L., Vogler M., Fulda S. Proteasome inhibitors and Smac mimetics cooperate to induce cell death in diffuse large B-cell lymphoma by stabilizing NOXA and triggering mitochondrial apoptosis. Int J Cancer. 2020;147:1485–1498. doi: 10.1002/ijc.32976. [DOI] [PubMed] [Google Scholar]
- 47.Lecis D., Drago C., Manzoni L., Seneci P., Scolastico C., Mastrangelo E., Bolognesi M., Anichini A., Kashkar H., Walczak H., Delia D. Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. Br J Cancer. 2010;102:1707–1716. doi: 10.1038/sj.bjc.6605687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai Y., Liu M., Tang W., Li Y., Lian J., Lawrence T.S., Xu L. A Smac-mimetic sensitizes prostate cancer cells to TRAIL-induced apoptosis via modulating both IAPs and NF-kappaB. BMC Cancer. 2009;9:392. doi: 10.1186/1471-2407-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen Z., Chen J., Liu H., Dong W., Huang X., Yang D., Hou J., Zhang X. The SMAC Mimetic APG-1387 Sensitizes Immune-Mediated Cell Apoptosis in Hepatocellular Carcinoma. Front Pharmacol. 2018;9:1298. doi: 10.3389/fphar.2018.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.El-Mesery M., Shaker M.E., Elgaml A. The SMAC mimetic BV6 induces cell death and sensitizes different cell lines to TNF-alpha and TRAIL-induced apoptosis. Exp Biol Med (Maywood) 2016;241:2015–2022. doi: 10.1177/1535370216661779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allensworth J.L., Sauer S.J., Lyerly H.K., Morse M.A., Devi G.R. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Res Treat. 2013;137:359–371. doi: 10.1007/s10549-012-2352-6. [DOI] [PubMed] [Google Scholar]
- 52.Amaravadi R.K., Schilder R.J., Martin L.P., Levin M., Graham M.A., Weng D.E., Adjei A.A. A Phase I Study of the SMAC-Mimetic Birinapant in Adults with Refractory Solid Tumors or Lymphoma. Mol Cancer Ther. 2015;14:2569–2575. doi: 10.1158/1535-7163.MCT-15-0475. [DOI] [PubMed] [Google Scholar]
- 53.Safferthal C., Rohde K., Fulda S. Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene. 2017;36:1487–1502. doi: 10.1038/onc.2016.310. [DOI] [PubMed] [Google Scholar]
- 54.Hannes S., Abhari B.A., Fulda S. Smac mimetic triggers necroptosis in pancreatic carcinoma cells when caspase activation is blocked. Cancer Lett. 2016;380:31–38. doi: 10.1016/j.canlet.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 55.Park S.Y., Park H.H., Park S.Y., Hong S.M., Yoon S., Morgan M.J., Kim Y.S. Reduction in MLKL-mediated endosomal trafficking enhances the TRAIL-DR4/5 signal to increase cancer cell death. Cell Death Dis. 2020;11:744. doi: 10.1038/s41419-020-02941-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Z., Jiang B., Wang Y., Ni H., Zhang J., Xia J., Shi M., Hung L.M., Ruan J., Mak T.W., Li Q., Han J. 2-HG Inhibits Necroptosis by Stimulating DNMT1-Dependent Hypermethylation of the RIP3 Promoter. Cell Rep. 2017;19:1846–1857. doi: 10.1016/j.celrep.2017.05.012. [DOI] [PubMed] [Google Scholar]
- 57.Tellez C.S., Grimes M.J., Picchi M.A., Liu Y., March T.H., Reed M.D., Oganesian A., Taverna P., Belinsky S.A. SGI-110 and entinostat therapy reduces lung tumor burden and reprograms the epigenome. Int J Cancer. 2014;135:2223–2231. doi: 10.1002/ijc.28865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Primary data are available on request from the authors.