

Summary
EAST/SeSAME syndrome is a rare disease affecting the Central Nervous System (CNS), inner ear, and kidney. The syndrome is due to loss-of-function mutations in the KCNJ10 gene encoding the inward-rectifying potassium channel Kir4.1. EAST/SeSAME syndrome is mainly diagnosed during childhood with a tonic-clonic seizure being the usual first symptom. Due to a limited number of patients and recent identification of the disease, few data are available on the clinical progress of this disease in adulthood. In particular, neurologic and nephrological outcomes have not been reported. We present a case series of 4 adult patients harbouring homozygous missense mutation p.Ala167Val and homozygous frameshift mutations p.Asn232Glnfs*14 and p.Gly275Valfs*7. Effects of these mutations were predicted by in silico modelling and bioinformatic tools. Patients with truncating mutations were associated with more severe outcomes, both in tubulopathy severity and neurological symptomatology. Conversely, either missense or truncating mutations were correlated with similar severity of epilepsy, with a long free-of-event period up to 20 years old. No eGFR decline was documented. Modelling predicted that truncating mutations lead to complete Kir4.1 dysfunction. Finally, all patients had a mild increase in urinary protein excretion. Our study indicates that the prognosis of patients suffering from EAST/SeSAME syndrome is related to the severity of the mutation causing the disease. As predicted by in silico modelling, truncating mutations of KCNJ10 are associated with more severe disease, with recurrence of symptomatic hypokalemia and more severe neurological phenotype. The type of mutation should be considered for the therapy tailored to patients' phenotype.
Keywords: Kir4.1, potassium channel, tubulopathy
1. Introduction
EAST/SeSAME syndrome is a rare inherited disorder affecting the Central Nervous System (CNS), inner ear and kidney. The first description of the syndrome was in 2009, in two independent studies (1,2), where the syndrome was called EAST syndrome (Epilepsy, Ataxia, Sensorineural deafness and Tubulopathy) or SeSAME syndrome (Seizures, Sensorineural deafness, Ataxia, Mental retardation and Electrolyte imbalance). In both reports, loss-of-function mutations in KCNJ10 were described.
KCNJ10 gene encodes Kir4.1, a potassium channel mainly expressed in oligodendrocytes and basolateral membrane of the distal nephron including the cortical thick ascending limb of Henle's loop (TAL), distal convoluted tubule (DCT) 1/2, connecting tubule (CNT) and cortical collecting duct (cCD). Kir4.1 forms a hetero tetramer with another potassium channel, Kir5.1, to conduct inwardly-rectifying potassium currents (3). KCNJ16 gene encoding Kir5.1 is mainly expressed in the kidney and loss-of-function mutations cause a Gitelman like tubulopathy (3).
EAST/SeSAME syndrome is an ultra-rare disease affecting 1:1,000,000 (4). Tonic-clonic seizures responsive to common anticonvulsants is a usual presenting symptom and the syndrome is difficult to distinguish from primary epilepsy. Later on, ataxia and deafness manifest in the majority of patients. Other cerebellar symptoms and mental retardation may variably be present. Children can have neurodevelopmental delay, especially in the acquisition of motor and language skills (4). Whether this is related to the frequency and severity of seizures, or due to the disease itself is unclear. Impaired function of astrocytes and oligodendrocytes is likely responsible for the development of neurological symptoms (5). Since the Kir4.1 channel plays a crucial role in the production of endolymph and endochoclear potential (4), Sensorineural Hearing Loss (SHL) can be variably present and often requires the implantation of an acoustic device. Renal involvement is characterized by salt-losing nephropathy resembling Gitelman syndrome, with hypokalemic metabolic alkalosis, normo-hypomagnesemia and hypocalciuria.
EAST/SeSAME syndrome is usually diagnosed during infancy and consequently the data on this disease are mainly from childhood. Indeed, among the 54 cases reported on PubMed database identified as "EAST syndrome" or "EAST/SeSAME syndrome", none was centered on adult patients prognosis and clinical progress (2,6-8). Thus, several questions on the prognosis and evolution of renal and neurologic phenotype are still open. Here we present a case series of 4 adult patients with EAST/SeSAME syndrome and the correlation between their symptomatology and genetic background.
2. Patients and Methods
2.1. Patient data
We report retrospective data from a cohort of patients affected by genetically confirmed diagnosis of EAST/ SeSAME syndrome. Studies on patients were conducted according to the ethical standards of the Institutional Committee of the University of Campania and University of Catania, under the 1964 Helsinki Declaration and later amendments or comparable ethical standards. Informed consent was obtained from all participants and/or their legal guardians for the anonymous publication of the data.
2.2. In silico analysis
Homology model of human Kir4.1 (sequence 25 - 349) was generated by the Swiss-Model protein structure homology-modelling server (https://swissmodel.expasy.org), using the crystal structure of Kir3.1-prokaryotic Kir channel chimera (9) (PDB code : 2QKS) as a template. Sequence alignment between Kir4.1 and the template using PROMALS3D server (http://prodata.swmed.edu/promals3d/promals3d.php) showed 35.6% sequence identity. The model was refined by the 3Drefine server (http://sysbio.rnet.missouri.edu/3Drefine). Modelling of the Kir4.1-Ala167Val variant was performed similarly using refined model as a template. Molecular visualization was performed with PyMol software. Effects of the Ala167Val mutation were predicted by online bioinformatic tools PolyPhen-2, Mutation taster, PROVEAN and SIFT using default settings.
3. Results and Discussion
3.1. Patient presentation
We report four clinical cases of adult patients affected by EAST/SeSAME. Patient 1 and 2 are siblings and their parents share a common ancestor (Figure 1). Patient 1 is a male younger sibling of patient 2. He was born at full-term. Like his sister, epilepsy was the first sign of the disease, which occurred when he was 9 months old (m/ o). His head magnetic resonance imaging (MRI) was normal and he was misdiagnosed as idiopathic epilepsy responsive to valproate. Tonic-clonic seizures occurred again when he was 5 and 10 years old (y/o), although he was under anti-epileptic drug treatment. When he started primary school, slight cognitive disabilities with oral and written comprehension were diagnosed. Since no clear symptoms of psychiatric disorder or tubulopathy developed during adolescence, he never received potassium and/or magnesium salt replacement therapy until the age of 16, when he was screened for tubulopathy as part of a clinical work-up of his sister. At 14 y/o he was diagnosed with SHL, requiring an acoustic device. His last blood and urine electrolyte profile is shown in Table 1.
Figure 1.
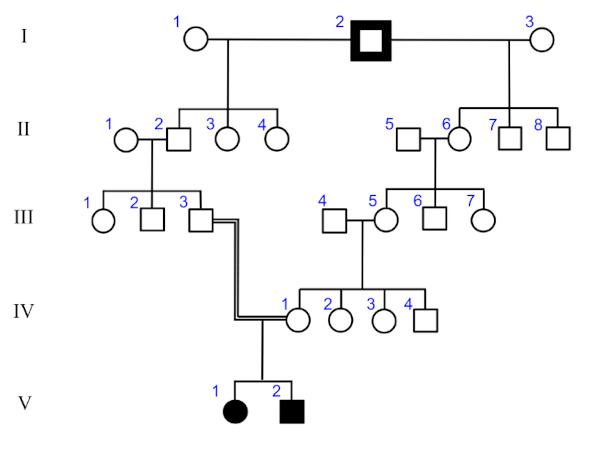
Pedigree of patient 1 and 2. Two siblings affected by Ala167Val mutation are indicated by filled symbols.
Table 1. Blood and 24h Urine parameters.
| Patients | 1 | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| Age | 16 | 20 | 32 | 33 | |
| Blood | |||||
| Creatinine | μM | 44 | 44 | 57.2 | 70.4 |
| eGFR | mL/min/1.73m2 | 161 | 140 | 129 | 118 |
| Na+ | mM | 141 | 137 | 132 | 140 |
| K+ | mM | 3.1 | 2.7 | 3.1 | 3.5 |
| Cl- | mM | 99 | 101 | 94 | 99 |
| Ca2+ | mg/dL | 8.8 | 10.6 | 9.9 | 9.4 |
| Mg2+ | mM | 0.66 | 0.66 | 0.66 | 0.58 |
| pH | 7.41 | 7.41 | 7.42 | 7.38 | |
| HCO3- | mM | 27.5 | 26.2 | 31.9 | 32.5 |
| pCO2 | mmHg | 46.5 | 45.9 | 50.3 | 53.9 |
| Urine | |||||
| Creatinine | mM | 4.5 | 3.53 | 3.71 | 8.3 |
| Na+ | mM | 120 | 82 | 78 | 94 |
| K+ | mM | 63 | 60 | 74 | 100 |
| Cl- | mM | 168 | 120 | 121 | 129 |
| Ca2+ | mg/dL | 2 | 5.8 | 3.3 | 11 |
| Phosphate | mg/dL | 3.9 | 2.7 | 3.9 | 2.6 |
| Mg2+ | mM | 3.55 | 3.05 | 4.2 | 11 |
| FE Na+ | % | 0.83 | 0.75 | 0.91 | 0.57 |
| FE K+ | % | 19.92 | 27.78 | 30.95 | 24.32 |
| FE Cl- | % | 1.66 | 1.48 | 2.00 | 1.11 |
| FE Mg2+ | % | 5.27 | 5.78 | 9.85 | 16.14 |
| FE Ca2+ | % | 0.22 | 0.68 | 0.52 | 1.04 |
| Ca2+/Creatinine | mg/dL/mg/dL | 0.04 | 0.15 | 0.08 | 0.12 |
| PCR | mg/mmol | 17.87 | 24.48 | 24.26 | 20.48 |
Patient 2 is a female, born by full-term delivery. Her psychomotor development was considered normal, being able to walk at around 15 m/o. At 9 m/o, she experienced her first seizure. Epilepsy was effectively controlled by valproate. This treatment was withdrawn at age 4 due to a paucity of events. Brain MRI ruled out morphological alterations causing epilepsy. She was free from seizures and off treatment until age 9 when she experienced a new tonic-clonic seizure. At 10 y/o, laboratory examinations revealed hypomagnesaemic and hypokalaemic metabolic alkalosis associated with salt-losing tubulopathy (low blood pressure and secondary aldosteronism). These electrolyte abnormalities suggested a syndromic disease and genetic testing was thus performed when she was 11 y/o. At this time, as part of a clinical work up for EAST/ SeSAME syndrome, she was found to be affected by SHL and moderate intellectual disability. However, she also had psychiatric symptomatology that is not usually part of the disease. Indeed, from 11 y/o and throughout adolescence she progressively manifested psychomotor agitation, panic attacks and outbursts of anger. This clinical scenario did not prevent her from attending school. Currently at 20 y/o, she has never experienced symptomatic hypokalaemia requiring hospital admission and her epilepsy is under control with no events for about 10 years. No clear signs of ataxia have been diagnosed so far.
Patient 3 is a male, born at term from consanguineous parents (first cousins). At 3 m/o he was admitted for tonic-clonic seizures and treated with carbamazepine and phenobarbital. At this age he was also affected by normo-kalaemic hypomagnesaemia and mild hypocalcaemia. At 6 m/o, when he was hospitalized for another seizure and obstinate constipation, hypokalemia was detected. He was started on a potassium and magnesium salt supplement alongside carbamazepine and phenobarbital. At 20 m/o, during an episode of dehydration and hypokalemia, spironolactone was introduced. However, this therapy did not prevent a hospital admission at 2 y/o for symptomatic hypokalaemia, after an episode of fever. Developmental delay was diagnosed at 1 y/ o when he was unable to maintain upright position and progressively worsened until 6 y/o when ataxia was diagnosed. Diffuse muscle hypotrophy became evident with adolescence. At 3 y/o he was diagnosed with SHL. Genetic testing was negative for most common hypokalemic tubulopathies during infancy. EAST/ SeSAME syndrome was confirmed only at 28 y/o when genetic analysis was extended to the KCNJ10 gene. During his childhood and adolescence he presented with salt craving symptoms and received ad libitum salt intake. Epilepsy was increasingly better controlled with no major events registered. The last severe episode of seizure was at 8 y/o with development of coma after discontinuation of carbamazepine. Currently, he is 32 y/ o and his last hospitalization for hypokalaemia-related symptoms occurred at 27 y/o. Therapy with potassium and magnesium supplements coupled with spironolactone was reinforced accordingly. His last clinical blood and urine electrolyte profile is shown in Table 1.
Patient 4 is a male, born prematurely from consanguineous parents (first grade cousins). Hypomagnesemia (Mg2+ 1.4 mg/dL) and hypokalaemia (K+ 2.8 mM) were diagnosed at birth and replacement therapy was started. Pregnancy was not affected by polyhydramnios. A detailed report of his neurologic findings was previously described (8). Briefly, he presented with tonic-clonic seizures at 3 m/o that recurred almost weekly, despite therapy with valproate and phenobarbital. An audiogram revealed normal hearing during childhood. His psychomotor development was delayed. At 4 y/o he was admitted to an intensive care unit for 14 days because of severe status epilepticus unresponsive to normal therapy. It is difficult to assess the impact of this event on the progression of his neurologic condition. However, he experienced progressive loss of motor ability and cerebellar symptoms together with myoclonus and dystonia of cervical muscles and the trunk up to 23 y/o, when he became wheel-chair bound. Currently he is 33 y/o and his epilepsy is well-controlled with valproate and phenobarbital with no major events reported in the last 10 years. His potassium level was well-controlled during childhood, but he experienced symptomatic hypokalemia requiring hospital admission 3 times in the last 9 years. As evident from the last clinical check (Table 1), no alteration in the estimated glomerular filtration rate (eGFR) was reported thus far.
3.2. Gene mutation modelling
Patients 1 and 2 carried a homozygous KCNJ10 c.500C>T variant, resulting in p.Ala167Val amino acid change. Patients 3 and 4 have rare homozygous variants c.693dup and c.822delG, producing truncated Kir4.1 p.Asn232Glnfs*14 and p.Gly275Valfs*7, respectively (Table 2). Homology modelling of human Kir4.1 revealed that Ala167Val at the junction between transmembrane domain 2 (TM2) and the C-terminus is predicted not to interrupt the sequence of the hydrogen bonds, thus not altering TM2 alpha-helix structure (Figure 2). Furthermore, effects of Ala167Val mutation were evaluated as "probably damaging" by PolyPhen-2 (score : 0.999) and "disease causing" by the Mutation taster tool with 0.999 probability, while PROVEAN and SIFT predicted Ala167Val as neutral (score : -2.45, cut-off = -2.5) and tolerated (score : 0.240, cut-off = 0.05), respectively. The frameshift mutations carried by patients 3 and 4, instead, confer a considerable loss of the C-terminal region of Kir4.1 resulting in a truncated protein which is highly likely nonfunctional.
Table 2. Clinical hallmarks over time.
| Patients | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Age (years) | 16 | 20 | 32 | 33 |
| Mutation | Missense | Missense | Frame-shift | Frame-shift |
| Protein variant | p.Ala167Val | p.Ala167Val | p.Asn232Glnfs*14 | p.Gly275Valfs*7 |
| Genetic variant | NM_002241.5: c.500C>T | NM_002241.5: c.500C>T | NM_002241.5: c.693dup. | NM_002241.5: c.822delG |
| Human (GRCh37/h19) | Chr1:160.011.823 | Chr1:160.011.823 | Chr1:160.011.630 | Chr1:160.011.500 |
| Disease Onset (months) | 9 | 9 | 3 | at birth |
| Symptomatic Hypokalemia | NO | NO | YES | YES |
| Ataxia | NO | NO | YES | YES |
| Seizures (frequency) | Low | Low | High | High |
| Speaking ability | YES | YES | NO | NO |
| Motor ability | YES | YES | Limited | NO |
| SHL | YES | YES | YES | NO |
| Psycosis | NO | YES | NO | NO |
| KCl dose (mmol/kg/day) | 0.17 | 1.1 | 2.72 | 1.44 |
| MgCl dose (mmol/kg/day) | 0.17 | 1.1 | -- | 1.43 |
Figure 2.
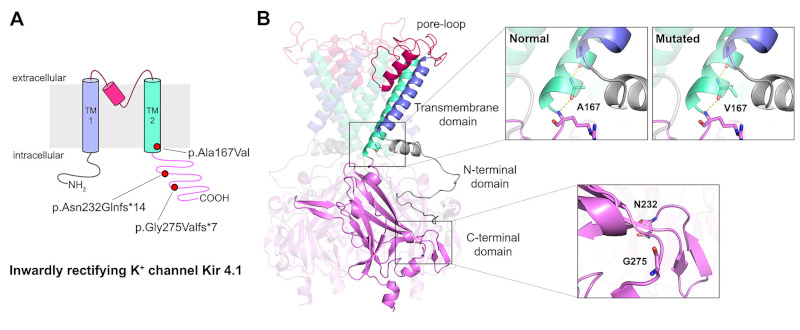
Mutations of the inwardly rectifying potassium channel Kir4.1 reported in this study. (A) Topology diagram of Kir 4.1 including the location of the sites of the three mutations. (B) Three-dimensional homology model of human Kir 4.1 homotetramer generated through Swiss- Model protein structure homology-modelling server (http://swissmodel.expasy.org). Molecular visualization was performed by PyMol software.
3.3. Symptoms severity
A complete overview of the last serum and urine electrolytes profile (Table 1) revealed hypokalemic metabolic alkalosis associated to hypomagnesemia and hypocalciuria are still present in adulthood. Only patient 3 currently takes potassium-sparing diuretics on top of electrolyte replacement therapy, due to frequent episodes of symptomatic hypokalemia. None of the patients have eGFR below 100 mL/min/1.73 m2, suggesting that even at the latest observed time point (33 y/o) no advanced signs of renal insufficiency can be detected. However, patients 3 and 4 present with severe muscle wasting and the use of Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula could be overestimating GFR. Singularly, all patients presented with a mild increase in urinary protein to creatinine ratio (PCR) independent of age, since similar values were observed during childhood. Whether this is an unfavourable prognostic factor for renal function, as well as a direct measure of GFR remains to be determined.
All patients reported here had seizures as a symptom of disease onset. Although Patient 4 had hypokalemia and hypomagnesemia at birth, no further diagnostic or therapeutic evaluation was completed before 3 m/o when the first seizure was diagnosed and a therapeutic program started. However, patients 1 and 2 had their first seizure later than patients 3 and 4 (Table 2, 9 vs. 3 m/ o). Furthermore, during childhood and adolescence, the neurologic involvement was also milder for patient 1 and 2 based on both frequency of epileptic events and appearance of other symptoms including ataxia, mental retardation and maintenance of language and motor abilities (Table 2). Indeed, patients 3 and 4 progressively lost their speaking and motor abilities. Preserved speaking and ambulatory ability of patients 1 and 2, at similar ages, suggests a potential influence of the type of mutation on the severity of phenotype. For all the patients reported here, it is of note that tonic-clonic seizures were very well-controlled by anti-epileptics during adulthood, with patients free of events for up to 10 years (Table 2). Finally, the severity of electrolyte imbalance secondary to salt-losing tubulopathy mainly manifest with the symptoms of hypokalemia. The patients carrying truncating mutations experienced more severe disease, as evaluated by the number of hypokalemic episodes requiring hospitalization and the dose of the supplement received (Table 2).
Neurologic phenotype has a major impact on the quality of life in patients affected by EAST/SeSAME syndrome. However, electrolyte disorders are potentially life-threatening throughout the patients' lives. Recently several studies have reported on the physiological role of Kir4.1 in the distal nephron that helps to explain mechanisms underlying the tubulopathy and suggest alternative treatments.
The distal nephron is heterogeneous in cellular composition (10), allowing fine control of sodium reabsorption and potassium secretion (11,12) by different cell types sharing common precursors (13) and a high degree of plasticity (14,15). Kir4.1 is expressed all along the distal nephron (from TAL to CD) (16,17) and is thus potentially crucial for a large number of cells. However, the renal clinical phenotype of EAST/SeSAME patients is consistent with defective DCT and activated CD function resembling Gitelman syndrome. Indeed, heteromer Kir4.1/Kir5.1 is essential for the activity of DCT and principal cells since it is the predominant potassium channel in the basolateral membrane and the main determinant of membrane potential. In DCT, Kir4.1 works as a sensor of circulating potassium levels. In response to hyperkalemia, inhibition of Kir4.1 leads to membrane depolarization and increased intracellular chloride (18). This in turn mediates a With No lysine (K) serine-threonine kinase (WNK)-dependent inhibition of sodium chloride co-transporter (NCC) on the apical side of the DCT (19), allowing increased delivery of sodium and chloride to the CNT and CD, and promotes potassium secretion into the urine secondary to epithelial sodium channel (ENaC) dependent sodium reabsorption. This ultimately restores the serum potassium concentration back to a normal level. Genetically encoded Kir4.1 dysfunction, as in EAST/SeSAME syndrome, results in a condition in which DCT wrongly senses hyperkalemia causing a sodium and potassium losing phenotype, despite hypokalemia and metabolic alkalosis. Defective activity of Kir4.1 in principal cells increases activity of the ENaC and renal outer medullary potassium channel (ROMK) further stimulating urinary potassium loss (20).
Homozygous Ala167Val and compound heterozygous with Arg297Cys were previously described as mild to moderate EAST/SeSAME syndrome (2,21). Patients 1 and 2 presented here belong to the same kindred showing a milder phenotype compared to patients 3 and 4 harboring truncating mutations. Ala167Val variation was evaluated as likely pathogenic by only two out of four bioinformatic prediction tools. This is mainly due to the type of substitution, which does not alter the TM2 alpha-helix structure (Figure 2), since alanine and valine share similar physical and chemical properties. Although Ala167 in Kir4.1 is highly conserved across various species (2), some members of Kir channels possess other hydrophobic, non-polar amino acids including valine in Kir3.4 at this position (22). Ala167Val retained approximately 60% of channel activity compared to wild-type (23), thus reduced channel function is not the sole cause of pathologies associated with Ala167Val. Indeed, mis-trafficking of Kir4.1-Ala167Val/Kir5.1 prevents the interaction between Kir4.1 and anchor protein MAGI-1 on the basolateral membrane, resulting in reduced basolateral potassium channels in the DCT leading to salt-wasting (24). Furthermore, Ala167 is located at the vicinity of the Kir4.1 channel gate, according to the crystal structure of Kir3.1 (9). Thus, reduced permeability to K+ flux cannot be excluded in Ala167Val mutants.
Other missense mutations including Ala201Thr and/ or Ile209Thr and Ile60Thr, despite an earlier time of presentation than patient 1 and 2 (before 7m/o), were associated with milder neurologic phenotypes compared to our truncating mutations (25,26). Finally, no signs of tubulopathy and hearing loss were diagnosed up to 3 y/o in patients with the Ala201Thr and/or Ile209Thr mutations and up to 20 y/o in patients carrying the Ile60Thr mutation. This is in line with patients bearing Thr57Ile showing signs of tubulopathy between 5 and 8 y/o (27). In our cohort of patients, tubulopathy was identified together with the first seizure (3 m/o) or even earlier for truncating mutations, but much later in two siblings bearing Ala167Val. Since patient 1 had hypokalemic and hypomagnesemic metabolic alkalosis diagnosed accidentally at 16 y/o, one can infer that not only the onset is delayed but also the severity of tubular defect is less.
Two frameshift mutations Asn232Glnfs*14 and Gly275Valfs*7 lack large parts of cytoplasmic C-terminus thus the channel is likely nonfunctional. Deletion of 47 amino acids from the C-terminus was sufficient to abolish channel activity of Kir4.1 (28), and Val259Ter and Arg199Ter with deletions of similar or larger parts of C-terminus compared to our truncating mutations result in nonfunctional channel as well (29). This could explain the severe neurological and renal phenotype found in our patients and more generally, an increase in patient phenotype severity when present as compound heterozygosity. Indeed, patients with compound heterozygosity for Arg199Ter and Arg65Pro, namely a nonsense mutation associated with a missense mutation that was biochemically characterized by residual channel function (30), present with a phenotype more severe than patients carrying homozygous Arg65Pro, at least as observed for serum potassium level, 2.9 vs. > 3 mM (1,2). Further follow-up studies on adult patients will be required for comprehensive prognostic evaluation of the disease.
Our study has certain limitations. First, this is a retrospective analysis on a small cohort of four Italian patients. Second, as often occurs with ultra-rare diseases, there are few reports of adult patients for a comprehensive comparison. A registry based multi-center prospective study will be necessary for compelling clinical evolution and prognostic evaluation of renal and neurologic phenotype.
4. Conclusion
We report here one of the largest cohorts of adult patients affected by EAST/SeSAME syndrome. The evolution of clinical picture and prognosis fit with the severity of mutations causing the disease. As predicted by in silico modelling, truncating mutations of KCNJ10 are associated with more severe adult prognosis in terms of recurrence of symptomatic hypokalemia and neurological phenotype, suggesting that the type of mutations should be taken into consideration in tailoring the electrolytes replacement and anticonvulsants therapy.
Acknowledgements
We are grateful to the patients and families for their contributions to this work.
Funding:
None.
Conflict of Interest
The authors have no conflicts of interest to disclose.
References
- 1. Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009; 360:1960-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009; 106:5842-5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paulais M, Bloch-Faure M, Picard N, Jacques T, Ramakrishnan SK, Keck M, Sohet F, Eladari D, Houillier P, Lourdel S, Teulon J, Tucker SJ. Renal phenotype in mice lacking the Kir5.1 (Kcnj16) K+ channel subunit contrasts with that observed in SeSAME/EAST syndrome. Proc Natl Acad Sci U S A. 2011; 108:10361-10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abdelhadi O, Iancu D, Stanescu H, Kleta R, Bockenhauer D. EAST syndrome: Clinical, pathophysiological, and genetic aspects of mutations in KCNJ10. Rare Dis. 2016; 4:e1195043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Su XT, Zhang C, Wang L, Gu R, Lin DH, Wang WH. Disruption of KCNJ10 (Kir4.1) stimulates the expression of ENaC in the collecting duct. Am J Physiol Renal Physiol. 2016; 310:F985-F993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Morin M, Forst A-L, Pérez-Torre P, Jiménez-Escrig A, Barca-Tierno V, García-Galloway E, Warth R, Lopez-Sendón Moreno JL, Moreno-Pelayo MA. Novel mutations in the KCNJ10 gene associated to a distinctive ataxia, sensorineural hearing loss and spasticity clinical phenotype. Neurogenetics. 2020; 21:135-143. [DOI] [PubMed] [Google Scholar]
- 7. Nicita F, Tasca G, Nardella M, Bellacchio E, Camponeschi I, Vasco G, Schirinzi T, Bertini E, Zanni G. Novel homozygous KCNJ10 mutation in a patient with non-syndromic early-onset cerebellar ataxia. Cerebellum. 2018; 17:499-503. [DOI] [PubMed] [Google Scholar]
- 8. Severino M, Lualdi S, Fiorillo C, Striano P, De Toni T, Peluso S, De Michele G, Rossi A, Filocamo M, Bruno C. Unusual white matter involvement in EAST syndrome associated with novel KCNJ10 mutations. J Neurol. 2018; 265:1419-1425. [DOI] [PubMed] [Google Scholar]
- 9. Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007; 26:4005-4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chambrey R, Trepiccione F. Relative Roles of Principal and Intercalated Cells in the Regulation of Sodium Balance and Blood Pressure. Curr Hypertens Rep. 2015; 17:538. [DOI] [PubMed] [Google Scholar]
- 11. Petrazzuolo O, Trepiccione F, Zacchia M, Capasso G. Hypertension and renal calcium transport. J Nephrol. 2010; 23:S112-S117. [PubMed] [Google Scholar]
- 12. Trepiccione F, Zacchia M, Capasso G. The role of the kidney in salt-sensitive hypertension. Clin Exp Nephrol. 2012; 16:68-72. [DOI] [PubMed] [Google Scholar]
- 13. Trepiccione F, Soukaseum C, Iervolino A, Petrillo F, Zacchia M, Schutz G, Eladari D, Capasso G, Hadchouel J. A fate-mapping approach reveals the composite origin of the connecting tubule and alerts on "single-cell"-specific KO model of the distal nephron. Am J Physiol Renal Physiol. 2016; 311:F901-F906. [DOI] [PubMed] [Google Scholar]
- 14. Iervolino A, Prosperi F, De La Motte LR, Petrillo F, Spagnuolo M, D'Acierno M, Siccardi S, Perna AF, Christensen BM, Frische S, Capasso G, Trepiccione F. Potassium depletion induces cellular conversion in the outer medullary collecting duct altering Notch signaling pathway. Sci Rep. 2020; 10:5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trepiccione F, Capasso G, Nielsen S, Christensen BM. Evaluation of cellular plasticity in the collecting duct during recovery from lithium-induced nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2013; 305:F919-F929. [DOI] [PubMed] [Google Scholar]
- 16. Zhang C, Wang L, Su XT, Lin DH, Wang WH. KCNJ10 (Kir4.1) is expressed in the basolateral membrane of the cortical thick ascending limb. Am J Physiol Renal Physiol. 2015; 308:F1288-F1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lachheb S, Cluzeaud F, Bens M, Genete M, Hibino H, Lourdel S, Kurachi Y, Vandewalle A, Teulon J, Paulais M. Kir4.1/Kir5.1 channel forms the major K+ channel in the basolateral membrane of mouse renal collecting duct principal cells. Am J Physiol Renal Physiol. 2008; 294:F1398-F1407. [DOI] [PubMed] [Google Scholar]
- 18. Su XT, Klett NJ, Sharma A, Allen CN, Wang WH, Yang CL, Ellison DH. Distal convoluted tubule Cl-concentration is modulated via K+ channels and transporters. Am J Physiol Renal Physiol. Am J Physiol Physiol. 2020; 319:F534-F540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int. 2018; 93:893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Penton D, Vohra T, Banki E, Wengi A, Weigert M, Forst AL, Bandulik S, Warth R, Loffing J. Collecting system-specific deletion of Kcnj10 predisposes for thiazide- and low-potassium diet-induced hypokalemia. Kidney Int. 2020; 97:1208-1218. [DOI] [PubMed] [Google Scholar]
- 21. Parrock S, Hussain S, Issler N, et al. KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol. 2013; 123:7-14. [DOI] [PubMed] [Google Scholar]
- 22. D'Avanzo N, Cheng WWL, Wang S, Enkvetchakul D, Nichols CG. Lipids driving protein structure? Evolutionary adaptations in Kir channels. Channels (Austin). 2010; 4:139-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Williams DM, Lopes CMB, Rosenhouse-Dantsker A, Connelly HL, Matavel A, O-Uchi J, McBeath E, Gray DA. Molecular basis of decreased Kir4.1 function in SeSAME/ EAST syndrome. J Am Soc Nephrol. 2010; 21:2117-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanemoto M, Abe T, Uchida S, Kawahara K. Mislocalization of K+ channels causes the renal salt wasting in EAST/SeSAME syndrome. FEBS Lett. 2014; 588:899-905. [DOI] [PubMed] [Google Scholar]
- 25. Zhang H, Zhu L, Wang F, Wang R, Hong Y, Chen Y, Zhu B, Gao Y, Luo H, Zhang X, Sun H, Zhou Y, Yao Y, Wang X. Novel KCNJ10 compound heterozygous mutations causing EAST/SeSAME-like syndrome compromise potassium channel function. Front Genet. 2019; 10:912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Al Dhaibani MA, El-Hattab AW, Holroyd KB, Orthmann- Murphy J, Larson VA, Siddiqui KA, Szolics M, Schiess N. Novel mutation in the KCNJ10 gene in three siblings with seizures, ataxia and no electrolyte abnormalities. J Neurogenet. 2018; 33:21-26. [DOI] [PubMed] [Google Scholar]
- 27. Scholl UI, Dave HB, Lu M, Farhi A, Nelson-Williams C, Listman JA, Lifton RP. SeSAME/EAST syndrome-phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol. 2012; 27:2081-2090. [DOI] [PubMed] [Google Scholar]
- 28. Tanemoto M, Abe T, Ito S. PDZ-binding and di-hydrophobic motifs regulate distribution of Kir4.1 channels in renal cells. J Am Soc Nephrol. 2005; 16:2608-2614. [DOI] [PubMed] [Google Scholar]
- 29. Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, Ognjanovic M, Dorison N, Reichold M, Bockenhauer D, Kleta R, Zdebik AA. KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron Physiol. 2011; 119:40-48. [DOI] [PubMed] [Google Scholar]
- 30. Tang X, Hang D, Sand A, Kofuji P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem Biophys Res Commun. 2010; 399:537-541. [DOI] [PMC free article] [PubMed] [Google Scholar]