

Abstract
Bile acids (BAs) play important functions in the development of alcohol‐associated liver disease (ALD). In the current study, urine BA concentrations in 38 patients with well‐described alcohol‐associated hepatitis (AH) as characterized by Model for End‐Stage Liver Disease (MELD), 8 patients with alcohol‐use disorder (AUD), and 19 healthy controls (HCs) were analyzed using liquid chromatography–mass spectrometry. Forty‐three BAs were identified, and 22 BAs had significant changes in their abundance levels in patients with AH. The potential associations of clinical data were compared to candidate BAs in this pilot proof‐of‐concept study. MELD score showed positive correlations with several conjugated BAs and negative correlations with certain unconjugated BAs; taurine‐conjugated chenodeoxycholic acid (CDCA) and MELD score showed the highest association. Cholic acid, CDCA, and apocholic acid had nonsignificant abundance changes in patients with nonsevere ALD compared to HCs but were significantly increased in those with severe AH. Receiver operating characteristic analysis showed that the differences in these three compounds were sufficiently large to distinguish severe AH from nonsevere ALD. Notably, the abundance levels of primary BAs were significantly increased while most of the secondary BAs were markedly decreased in AH compared to AUD. Most importantly, the amount of total BAs and the ratio of primary to secondary BAs increased while the ratio of unconjugated to conjugated BAs decreased as disease severity increased. Conclusion: Abundance changes of specific BAs are closely correlated with the severity of AH in this pilot study. Urine BAs (individually or as a group) could be potential noninvasive laboratory biomarkers for detecting early stage ALD and may have prognostic value in AH morbidity.
The abundance changes of bile acids (BAs) in urine are closely correlated with the severity of alcohol‐associated hepatitis (AH). BAs (individually or as a group) could be potential noninvasive laboratory biomarkers for detecting early stage of alcohol‐associated liver disease and may have prognostic value in AH morbidity.

Abbreviations
- AC
alcohol‐associated cirrhosis
- AH
alcohol‐associated hepatitis
- ALD
alcohol‐associated liver disease
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- AST
aspartate aminotransferase
- AUC
area under the curve
- AUD
alcohol use disorder
- AUDIT
Alcohol Use Disorders Identification Test
- BA
bile acid
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- CI
confidence interval
- DCA
deoxycholic acid
- GCA
glycocholic acid
- GCDCA
glycine‐conjugated chenodeoxycholic acid
- GHCA
glycohyocholic acid
- GUDCA
glycoursodeoxycholic acid
- HC
healthy control
- LC
liquid chromatography
- LCA
lithocholic acid
- MCA
muricholic acid
- MDF
Maddrey’s discriminant function
- MELD
Model for End‐Stage Liver Disease
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PLS‐DA
partial least squares–discriminant analysis
- ROC
receiver operator characteristic
- TCA
taurine‐conjugated cholic acid
- TCDCA
taurine‐conjugated chenodeoxycholic acid
- THCA
taurine‐conjugated hyocholic acid
- TαMCA
taurine‐conjugated α‐muricholic acid
Alcohol‐associated liver disease (ALD) is a leading form of liver disease. Harmful use of alcohol caused more than 3 million deaths worldwide in 2016,( 1 ) and this accounts for almost 1% of all global deaths and 50% of all liver disease‐attributable deaths.( 2 ) Liver damage can be reversible at early stages, and it is generally asymptomatic; however, it can progress undetected and can easily proceed to permanent liver damage. Identification of ALD in the early stages could reduce mortality and the health care burden. Alcohol‐associated hepatitis (AH; also commonly termed acute alcoholic hepatitis) is a severe form of ALD that carries an important morbidity and mortality rate. Without any U.S. Food and Drug Administration‐approved therapy( 3 ) and limited effective diagnostic and/or prognostic determinants, gaps in the overall diagnosis and medical management of AH remain a challenge. Thus, there is an urgent need for an accurate, noninvasive, diagnostic marker that can distinguish subjects at various stages of ALD and/or predict mortality in advanced ALD.
Chronic alcohol consumption can be associated with changes in bile acid (BA) profiles( 4 ) in patients with AH.( 5 ) AH can be accompanied by cholestasis( 6 ) and high levels of BAs in the liver and serum.( 7 ) Under normal conditions, 95% of primary and secondary BAs are recirculated in blood, usually in their conjugated forms, and only 5% of BAs are excreted from the intestine through the colon.( 8 ) Dysregulation of BA metabolism is a common mechanistic factor in liver injury in ALD.( 9 , 10 )
Early studies identified the up‐regulated primary BAs in AH as cholic acid (CA) and chenodeoxycholic acid (CDCA).( 7 , 11 ) In addition to primary BAs, increases in taurine‐ and glycine‐conjugated BAs, including the sulfated BAs, and notable decreases in deoxycholic acid (DCA) and glycine‐conjugated CDCA (GCDCA) in AH were reported.( 12 ) Recent studies confirmed the increase of conjugated BAs (e.g., taurine‐conjugated CA [TCA], taurine‐conjugated CDCA [TCDCA], GCDCA) in the serum of active drinkers with alcohol‐associated cirrhosis (AC) or AH( 10 ) compared to controls. However, different results were observed for the unconjugated BAs, which were either increased in AC or decreased in AH.( 5 , 10 ) Importantly, some BAs were positively correlated with disease severity as measured by the Model for End‐Stage Liver Disease (MELD) score( 5 ) or AH.( 7 ) These existing studies demonstrate the potential of using BAs as diagnostic markers for AH.
In AH, BA excretion into intestine can be reduced due to the decreased expression of BA‐related transporters,( 10 , 11 ) thereby increasing BA levels in liver and blood. There is some excretion of BAs from blood into urine,( 13 ) and early studies suggested that detectable changes in the levels of urinary BAs in ALD were helpful in characterizing liver pathology.( 14 , 15 , 16 ) Additionally, urinary BAs have the potential to serve as valuable biomarkers due to the high stability observed in liver diseases( 17 ) and their easy accessibility. Therefore, urine BA levels could be well‐suited to be noninvasive diagnostic markers for ALD. Thus, we investigated whether the concentrations of candidate BAs in human urine could serve as potential diagnostic and/or prognostic determinants of ALD.
Participants and Methods
Patient Recruitment and Sample Collection
This study was approved by the University of Louisville Institutional Review Board, and information on patient definitions and recruitment are detailed in another publication.( 18 ) All study participants provided informed consent before participation in the study, including appropriate authorization for data and sample collection. A total of 19 healthy controls (HCs), 8 patients with alcohol‐use disorder (AUD) with mild liver injury who were evaluated for suspected AH but did not meet National Institute on Alcohol Abuse and Alcoholism (NIAAA) consortium criteria,( 3 ) and 38 patients with AH (both moderate and severe stages) were enrolled in this study. All study participants had a complete history, physical examination, and laboratory evaluation on study enrollment.
Healthy participants did not have any clinically diagnosed liver or organ system (or inflammation) disease that could cause altered laboratory values for comparison analyses. Inclusion criteria comprised individuals who were 21 years or older and enrolled in the University of Louisville Hospital System. Individuals who were unwilling or unable to provide informed consent, had significant comorbid conditions (liver, heart, kidney, lung, neurologic or psychiatric illnesses, sepsis) and active drug abuse, pregnant and lactating women, and prisoners or other vulnerable subjects were excluded from the study.
All patients with AH met the diagnosis for AH based on the clinical and laboratory guidelines published by the NIAAA consortium on AH.( 3 ) Detailed eligibility for patients with AH from this trial can be reviewed in our publications.( 18 , 19 ) Patients who did not meet the criteria for MELD score ≥12 were used as a disease control group, meaning they had AUD with only minimal biochemical evidence of liver inflammation/injury in this study.
All participants’ specimens (urine and blood) were collected in the morning after overnight fasting, and their clinical data were analyzed at the University of Louisville. All de‐identified data from participants who provided urine and blood samples were collected at baseline as well as information on subsequent death, if available. Clinical data included participant demographics (age, sex), drinking history (Alcohol Use Disorders Identification Test [AUDIT]), medical assessments at admission, and medical history. Confirmatory tests for AH (laboratory and imaging) and markers of liver severity (MELD score and Maddrey’s discriminant function [MDF]) were collected and analyzed. A laboratory panel specific for this study was composed of a comprehensive metabolic panel (including liver injury panel), coagulation measures, and complete blood count tests.
BA Extraction
Urine samples were collected in 10‐mL tubes and were stored at −80°C until analyzed.( 20 ) BAs in urine samples were extracted by solid phase extraction (SPE), as described( 21 ) with some modifications, and the analysis was optimized using a mixture of 46 BAs. In brief, the pH of urine samples was adjusted to pH ≥8.5 by ammonium hydroxide and centrifuged at 14,000g for 20 minutes at 4°C. Two hundred µL of supernatant was transferred to a fresh tube and lyophilized. Each dried sample was reconstituted in 200 µL water (pH ≥8.5) and then loaded onto an OASIS HLB cartridge (Waters Corporation, Milford, MA) that had been activated and equilibrated with methanol and water (pH ≥8.5) following the manufacturer’s instructions. The cartridge was washed with 1 mL of H2O (pH ≥8.5) 3 times and then eluted 3 times with 100 µL of 70% acetonitrile in water (pH ≥8.5). The eluate was combined and lyophilized overnight. The residue was then reconstructed in 50 µL solvent that had the same chemical composition as the start point of the liquid chromatography (LC) gradient described below. The sample was then centrifuged at 14,000g for 20 minutes at 4°C. The upper clear solution was transferred to an LC vial for LC‐mass spectrometry (MS) analysis. Group‐based pooled samples were also prepared by mixing a small portion of the supernatant of samples in the same group.
To extract BAs from serum samples, 50 µL serum was first mixed with 200 µL acetonitrile. After vigorous vortex, the mixture was centrifuged at 14,000g for 20 minutes at 4°C. Then, 200 µL supernatant was transferred to a fresh tube and lyophilized overnight. The remaining steps of SPE are identical to those of processing the urine samples, as described above.
LC‐Tandem MS Analysis
A Thermo Q Exactive HF Hybrid Quadrupole‐Orbitrap Mass Spectrometer coupled with a Thermo DIONEX UltiMate 3000 Ultra Performance Liquid Chromatography (UPLC) system (Thermo Fisher Scientific, Waltham, MA) was used in this study. The UPLC system was equipped with a Cortecs T3 column (100 × 2.1 mm; inner diameter, 1.8 µm; Waters Corporation). The temperatures of the column and autosampler were set to 60°C and 12°C, respectively. Sample injection volume was 2 µL. Mobile phase A was 1 mM ammonia acetate (pH 4.15), and mobile phase B was 1 mM ammonia acetate (pH 4.15) in 96% acetonitrile. The LC gradient was as follows: 0 to 0.1 minute, 20% mobile phase B; 0.1 to 12 minutes, mobile phase B increased linearly from 20% to 60%; 12 to 16 minutes, mobile phase B increased linearly from 60% to 76%; and 16 to 17 minutes, mobile phase B increased to 100% and kept constant at 100% for 2 minutes. The flow rate was 0.4 mL/minute. The operating parameters for MS were the same as in our previous study,( 21 ) with the exception that the full scan range was changed to 150‐800 m/z.
All BA samples were analyzed by LC‐MS in random order under negative mode to obtain full MS data for their quantification. The group‐based pooled samples were analyzed by LC‐tandem MS (MS/MS) in negative mode to acquire MS/MS spectra at three collision energies (20, 40, and 60 eV) for BA identification. Forty‐six BA standards purchased from Steraloids Inc. (Newport, RI) and Cayman Chemical (Ann Arbor, MI) were also analyzed by LC‐MS/MS under negative mode in different collision energies (20, 40, and 60 eV), and the results were recorded in our in‐house database.
Data Analysis
XCMS software was used for spectrum deconvolution,( 22 ) and MetSign software was used for metabolite identification, cross‐sample peak list alignment, and normalization, as described.( 23 ) To identify BAs, the LC‐MS/MS data of the pooled samples were matched to the MS/MS spectra of 46 BA standards recorded in our in‐house database that contains parent ion m/z, MS/MS spectra, and retention time. The threshold for the MS/MS spectrum similarity was set as ≥0.4, and the thresholds of the retention time difference and m/z variation window were set as ≤0.15 minutes and ≤5 ppm, respectively.
Absolute quantification of each BA was achieved by external calibration, where the calibration curve of each BA was constructed using the LC‐MS data of the BA standard with different concentrations. Statistical analyses were performed using SPSS version 25 (IBM Corporation, Armonk, NY) and MetSign software. Partial least squares–discriminant analysis (PLS‐DA), a supervised technique that uses the PLS algorithm to explain and predict the membership of samples to groups, was performed using MetSign to give an overview of the metabolic profile difference among groups. Distributional assumptions of continuous outcomes were checked and, if needed, a data transformation (e.g., log transformation) was applied to meet the normality assumption. Univariate analysis of BA abundance among groups was conducted using one‐way analysis of variance (ANOVA) with Bonferroni post hoc analysis. The group cross linear‐by‐linear association test was used for trend analysis; Mann‐Whitney U test was used for clinical measures; receiver operating characteristic (ROC) analysis was used to classify patients based on the abundances of BAs; and Spearman’s rank correlation was used to measure the association of BAs with the clinical parameters. The thresholds of statistical significance tests were set as P ≤ 0.05, area under the ROC curve (AUC) >0.7 or <0.3, and Spearman’s rank correlation test coefficient ≥ 0.5.
Results
Patient Characterization by Clinical Measures
A total of 65 individuals were assessed, including 19 patients who were HCs, 8 patients with AUD, and 38 patients with moderate or severe AH. Using the MELD score to categorize these patients, the 8 patients with AUD (disease controls) had MELD <12 (mild liver injury), the 13 patients with moderate AH had 12≤MELD≤19, and the 25 patients with severe AH had MELD ≥20. The 8 patients with AUD and 13 patients with moderate AH were grouped together for some analyses as “nonsevere ALD.” The detailed demographic and clinical characteristics of the study cohort are shown in Table 1. Patients with AH were older (mean, 49 years old) compared to the age of the HC group (mean, 36 years old). As expected, patients with AH had significantly lower mean levels of serum albumin (2.7 g/dL in AH vs. 4.2 g/dL in HCs; P < 0.001) and high mean levels of serum bilirubin (6.7 mg/dL in AH vs. 0.7 mg/dL in HCs; P < 0.001). Liver aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were significantly elevated in patients with AH relative to controls (P < 0.05) (Table 1). The mean MELD score of all patients was 18.
Table 1.
Demographic, Drinking, and Liver Indicators of Study Participants by Group
| Variables | HCs (n = 19) | ALD | P Value* | |||
|---|---|---|---|---|---|---|
| Nonsevere ALD (n = 21) | Severe AH (n = 25) | Total Patients (n = 46) | ||||
| AUD (n = 8) | Moderate AH (n = 13) | |||||
| Age (years) | 36 (24‐60) | 51 (39‐67) | 50 (34‐65) | 47 (27‐66) | 49 (27‐67) | ‐ |
| Total bilirubin (mg/dL) | 0.7 (0.4‐1.3) | 1.5 (0.8‐2.6) | 4.2 (1.2‐18.2) | 12.9 (3.7‐34.2) | 6.7 (0.8‐34.2) | <0.001 |
| Male (female) | 13 (6) | 4 (4) | 7 (6) | 20 (5) | 31 (15) | ‐ |
| INR | N/A | 1.2 (1.1‐1.7) | 1.5 (1.2‐2.8) | 2.0 (1.0‐3.2) | 1.7 (1.0‐3.2) | ‐ |
| AST (U/L) | 27 (19‐66) | 59 (21‐120) | 119 (53‐347) | 88 (16‐190) | 90 (16‐347) | <0.001 |
| ALT (U/L) | 25 (16‐109) | 36.8 (14.0‐60.0) | 48 (18‐194) | 35 (16‐66) | 39 (14‐194) | 0.015 |
| Alkaline phosphatase (IU/L) | 52 (37‐62) | 124 (89‐232) | 173 (80‐518) | 144 (71‐336) | 148 (71‐518) | <0.001 |
| Albumin (g/dL) | 4.2 (3.8‐4.3) | 3.9 (2.6‐4.9) | 2.8 (1.9‐4.5) | 2.4 (1.4‐4.3) | 2.7 (1.4‐4.9) | <0.001 |
| Creatinine (mg/dL) | 0.88 (0.69‐1.07) | 0.69 (0.36‐1.40) | 0.68 (0.32‐1.30) | 0.89 (0.39‐5.68) | 0.79 (0.32‐5.68) | 0.082 |
| MELD score | N/A | 9.2 (6.0‐11.0) | 16 (12‐19) | 24 (20‐39) | 18 (6‐39) | ‐ |
| AUDIT | 2 (1‐5) | 18 (9‐36) | 21 (11‐32) | 22 (10‐39) | 21 (9‐39) | <0.001 |
| MDF | N/A | N/A | 20.42 (3.24‐82.86) | 48.41 (17.52‐121.68) | 34.14 (−8.80 to 121.68) | ‐ |
Values are presented as mean with ranges.
Mann‐Whitney U test between HCs and patients abusing alcohol.
Abbreviations: INR, international normalized ratio; N/A, not available.
BA Abundance Changes in Urine Samples
A total of 43 BAs were identified and quantified from the urine samples. To study BA metabolic profile differences, we first categorized the 46 patient samples into two groups by MELD score as nonsevere ALD (MELD ≤ 19, n = 21) and severe AH (MELD ≥20, n = 25). Therefore, we had three groups including HCs (n = 19). Sample group information categorized by MDF, AST:ALT, and AUDIT is listed in Supporting Table S1.
After samples were grouped, the BA profile differences among the groups were analyzed by PLS‐DA. BA profile differences among the groups, as categorized by MELD score, are depicted in Fig. 1. It is clear that the BA profile of AH is distinct from HCs and that a slight overlap exists between nonsevere ALD and severe AH. Overall, the large values of R 2 = 0.63 and Q 2 = 0.53 indicate that the PLS‐DA model fitted the data well with excellent predictability. The PLS‐DA model also showed good performance in differentiating the BA profile when samples were categorized by MDF (R 2 = 0.52), but the predictive value was relatively poor (Q 2 = 0.39; Supporting Fig. S1A). The performance of PLS‐DA and its predictive value were worse when the samples were categorized by AST:ALT (Supporting Fig. S1B) and AUDIT (Supporting Fig. S1C). These results suggest that MELD score is the best clinical parameter to categorize urine samples for analyzing the BA profile in AH using PLS‐DA.
FIG. 1.
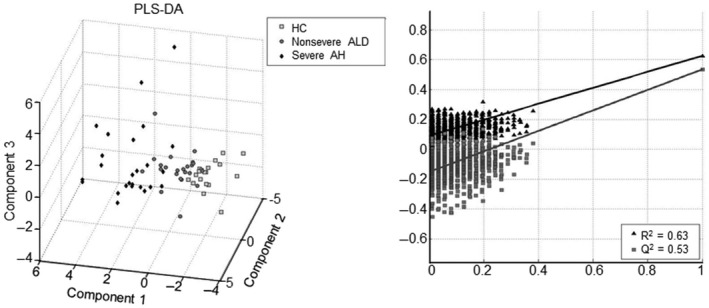
BA profiling in urine samples using PLS‐DA. All 65 samples were categorized into one of three groups: HCs, nonsevere ALD, or severe AH. The left panel is a PLS‐DA three‐dimensional score plot, and the right panel is the performance plot of the PLS‐DA model.
To assess the concentration changes of BAs, 11 solutions of BA standards with different concentrations were analyzed to construct calibration curves. By categorizing samples using MELD score, total BAs were significantly elevated in severe AH and nonsevere ALD compared with HCs (Fig. 2A). The ratio of unconjugated and conjugated BAs markedly decreased (Fig. 2B). In contrast, the ratio of primary and secondary BAs was significantly increased (Fig. 2C). These changes were induced by the increased concentrations of conjugated and primary BAs and the decreased concentrations of unconjugated and secondary BAs. A similar trend was also observed when the samples were categorized by MDF (Supporting Fig. S2A), AST:ALT (Supporting Fig. S2B), and AUDIT (Supporting Fig. S2C). Results of serum when the samples were categorized by MELD score show that a similar result was also obtained in serum samples of those patients (Supporting Fig. S2D).
FIG. 2.
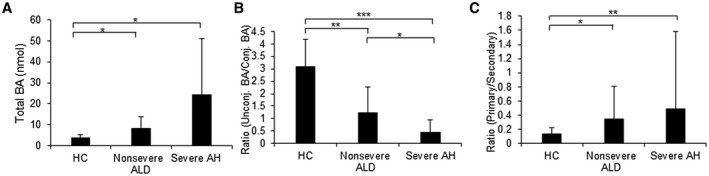
Changes in the amount of BAs in urine samples. All samples were grouped into HCs, nonsevere ALD, and severe AH, using MELD score. (A) Patients with AH have significantly higher urine levels of total BAs than HCs. (B) The ratio of unconjugated to conjugated BAs was significantly decreased in patients with AH in a disease severity‐dependent manner. (C) The ratio of primary to secondary BAs was significantly increased in patients with AH compared to the HC group. Student t test, *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: conj., conjugated; unconj., unconjugated.
To identify the relative abundance changes of each BA, the abundance of 43 identified BAs was analyzed by one‐way ANOVA (Table 2). Of these, 22 BAs had statistically significant changes in their abundance levels among the three groups (HCs, nonsevere ALD, and severe AH) when the samples were categorized by MELD score. TCDCA had the largest abundance increase in severe AH compared with its abundance level in HCs. TCDCA was increased 108.2‐fold in severe AH and 4.3‐fold in nonsevere ALD (Table 2). Furthermore, taurine‐conjugated hyocholic acid (THCA) was increased 75.2‐fold and 13.2‐fold, respectively, and GCDCA was increased 51‐fold and 5.7‐fold, respectively. These three BAs also had the largest fold change when the patients were categorized by MDF, AST:ALT, and AUDIT (Supporting Table S2), suggesting that urine BAs were dramatically changed with AH severity.
Table 2.
BAs With Significant Abundance Changes Among Groups
| Name | tR (minutes) | P Value* | P Value † | AUC ‡ | 95% CI ‡ | Fold Change § | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 2‐1 | P Value* | 3‐1 | P Value* | 3‐2 | P Value* | ||||||
| GCA | 5.13 | <0.001 | 0.001 | 0.791 | 0.663‐0.919 | 3.82 | <0.001 | 13.44 | <0.001 | 3.52 | <0.001 |
| GCDCA | 6.71 | <0.001 | <0.001 | 0.903 | 0.819‐0.986 | 5.71 | <0.001 | 50.96 | <0.001 | 8.93 | <0.001 |
| TCDCA | 5.76 | <0.001 | <0.001 | 0.944 | 0.883‐1.000 | 4.29 | <0.001 | 108.19 | <0.001 | 25.21 | <0.001 |
| GHCA | 4.46 | <0.001 | <0.001 | 0.915 | 0.837‐0.993 | 5.42 | <0.001 | 45.47 | <0.001 | 8.39 | <0.001 |
| Apocholic acid | 9.37 | <0.001 | <0.001 | 0.918 | 0.826‐1.000 | 0.69 | 0.134 | 2.25 | <0.001 | 3.27 | <0.001 |
| DCA | 10.58 | <0.001 | <0.001 | 0.090 | 0.000‐0.188 | 0.57 | 0.065 | 0.16 | <0.001 | 0.28 | <0.001 |
| 23‐Nordeoxycholic acid | 9.19 | <0.001 | <0.001 | 0.150 | 0.027‐0.273 | 0.74 | <0.001 | 0.54 | <0.001 | 0.74 | <0.001 |
| 6,7‐Diketolithocholic acid | 9.05 | <0.001 | <0.001 | 0.906 | 0.811‐1.000 | 3.36 | <0.001 | 11.28 | <0.001 | 3.36 | <0.001 |
| CDCA | 10.23 | <0.001 | 0.008 | 0.731 | 0.581‐0.881 | 1.21 | 0.518 | 1.86 | <0.001 | 1.54 | 0.006 |
| 23‐Norcholic acid | 6.50 | <0.001 | 0.005 | 0.737 | 0.598‐0.877 | 2.26 | 0.002 | 4.67 | <0.001 | 2.07 | 0.003 |
| GUDCA | 4.96 | <0.001 | <0.001 | 0.705 | 0.542‐0.868 | 7.64 | <0.001 | 17.85 | <0.001 | 2.34 | 0.022 |
| ω‐MCA | 6.24 | 0.002 | ‐ || | ‐ | ‐ | 0.38 | 0.098 | 0.21 | 0.002 | 0.55 | 0.326 |
| Nutriacholic acid | 8.99 | <0.001 | ‐ | ‐ | ‐ | 2.73 | 0.009 | 4.56 | <0.001 | 1.67 | 0.265 |
| 12‐Ketolithocholic acid | 9.24 | <0.001 | ‐ | ‐ | ‐ | 0.30 | <0.001 | 0.25 | <0.001 | 0.82 | 0.400 |
| TαMCA | 3.00 | <0.001 | 0.006 | 0.735 | 0.588‐0.881 | 6.95 | 0.007 | 40.16 | <0.001 | 5.78 | 0.005 |
| CA | 7.93 | 0.002 | 0.004 | 0.757 | 0.605‐0.908 | 1.23 | 1.000 | 3.01 | 0.002 | 2.44 | 0.007 |
| TCA | 4.43 | 0.001 | 0.033 | 0.680 | 0.527‐0.833 | 2.38 | 0.099 | 5.73 | <0.001 | 2.41 | 0.053 |
| THCA | 3.74 | <0.001 | 0.005 | 0.739 | 0.595‐0.883 | 13.20 | <0.001 | 75.22 | <0.001 | 5.70 | 0.003 |
| LCA | 13.42 | 0.014 | ‐ | ‐ | ‐ | 0.52 | 0.014 | 0.55 | 0.027 | 1.05 | 1.000 |
| α‐MCA | 6.45 | 0.002 | 0.030 | 0.699 | 0.530‐0.867 | 0.31 | 0.002 | 0.60 | 1.000 | 1.91 | 0.075 |
| TLCA | 7.60 | 0.005 | 0.003 | 0.767 | 0.624‐0.910 | 0.87 | 0.683 | 1.24 | 0.149 | 1.43 | 0.005 |
| TDCA | 6.08 | 0.035 | 0.011 | 0.265 | 0.103‐0.428 | 1.37 | 0.090 | 0.99 | 1.000 | 0.73 | 0.035 |
| 3‐Oxocholic acid | 0.018 | 0.719 | 0.549‐0.889 | 0.64 | 0.92 | 1.44 | |||||
One‐way ANOVA with Bonferroni post hoc test.
ROC analysis.
AUC and asymptotic 95% confidence interval of ROC.
Fold change that was calculated by the mean of each group, where 2‐1 means nonsevere ALD/HC, 3‐1 means severe AH/HC, and 3‐2 means severe AH/nonsevere ALD.
Data not collected.
Abbreviations: TDCA, taurodeoxycholic acid; TLCA, taurolithocholic acid; tR, retention time in minutes.
To evaluate the diagnostic performance of the changes in BAs, ROC analysis was performed for severe AH in comparison with nonsevere ALD. When the samples were categorized by MELD score, 19 BAs had statistically significant abundance changes. Seventeen of them had AUC larger than 0.7 or smaller than 0.3 (Table 2), suggesting the potential of using these BAs as a diagnostic marker, especially TCDCA (AUC, 0.94; 95% confidence interval [CI], 0.883‐1.000). When the samples were categorized respectively by MDF, AST:ALT, and AUDIT, 11, one, and four BAs were detected with statistically significant abundance changes between severe AH and nonsevere ALD (Supporting Table S2). However, no other BAs were detected with substantial abundance changes using all four sample grouping methods, showing that each of the four clinical parameters had limited accuracy in describing AH.
When the samples were grouped by MELD score, the abundance levels of four BAs had a disease severity stepwise increase from HCs to nonsevere ALD and ultimately the highest in severe AH, including unconjugated BAs: CA, nutriacholic acid, 23‐norcholic acid, and 6,7‐dicketolithocholic acid (Fig. 3A). Moreover, the levels of four other BAs, i.e., DCA, lithocholic acid (LCA), 23‐nordeoxycholic acid, and 12‐ketolithocholic acid, were significantly decreased in a stepwise manner (Fig. 3B). The linear‐by‐linear association test showed a statistically significant trend for all these eight BAs (Fig. 3), suggesting that the levels of these BAs changed in a disease severity stepwise manner.
FIG. 3.
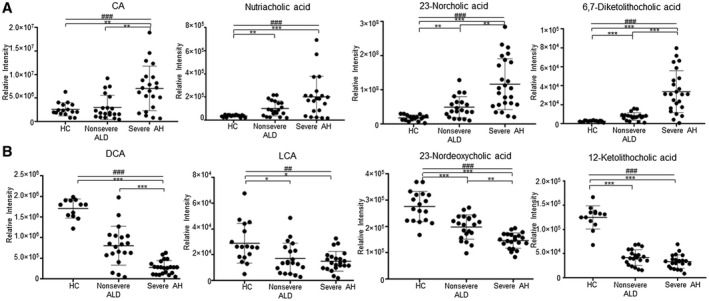
BA changes in urine among HCs, nonsevere ALD, and severe AH. (A) BAs that had increased levels with the increase in AH severity. (B) BAs that had decreased levels with the increase in AH severity. One‐way ANOVA with Bonferroni post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001. P values of linear‐by‐linear association test, ##P < 0.01, ###P < 0.001. Data points represent individual patients; horizontal lines in plots show mean ± SD.
The abundance levels of CA, CDCA, and apocholic acid did not have significant changes between HCs and nonsevere ALD. However, they were markedly increased in severe AH compared to HCs and nonsevere ALD (Fig. 4A). ROC analysis showed that the AUC of these three BAs were 0.76 (95% CI, 0.605‐0.908), 0.73 (95% CI, 0.581‐0.881), and 0.92 (95% CI, 0.826‐1.000), respectively, with P < 0.01 (Fig. 4B), suggesting that these three BAs could be used as biomarkers for severe AH.
FIG. 4.
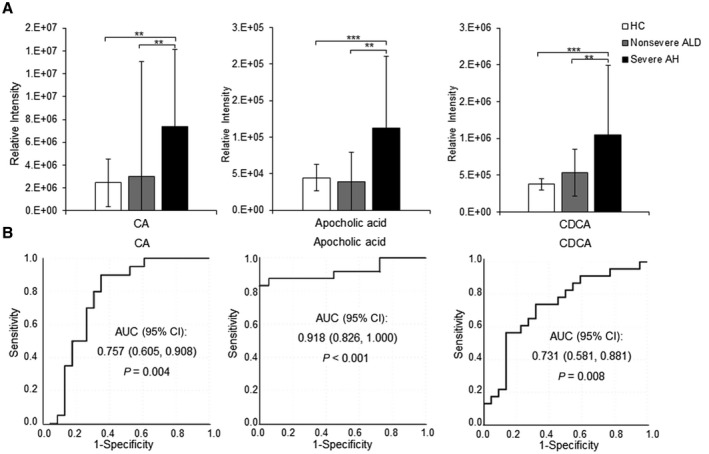
Using CA, CDCA, and apocholic acid, as potential biomarkers to discern AH disease severity. (A) Histogram of the three BAs showing a dramatic increase in patients with severe AH. Data show mean ± SD. (B) ROC curves of each BA for differentiating patients with severe AH from patients with nonsevere ALD (moderate AH, including AUD with mild liver injury). One‐way ANOVA with Bonferroni post hoc test, **P < 0.01, ***P < 0.001.
Correlation of BA Abundance and Clinical Parameters
Spearman’s rank correlation coefficients between the clinical parameters (MELD, MDF, AST:ALT, and AUDIT) of patients and the LC‐MS instrument response (i.e., peak area) of a specific BA are listed in Table 3. Eight BAs had at least one correlation coefficient ≥ 0.5 with these clinical parameters. When the samples were categorized by MELD score, seven BAs had large correlation coefficients: TCDCA (ρ = 0.75), GCDCA (ρ = 0.68), glycohyocholic acid (GHCA) (ρ = 0.67), apocholic acid (ρ = 0.58), 6,7‐diketolithocholic acid (ρ = 0.63), 23‐nordeoxycholic acid (ρ = −0.58), and DCA (ρ = −0.65). TCDCA had the largest correlation coefficient with the MELD score, which is consistent with reported information.( 5 ) To further evaluate the diagnostic performance of these seven BAs in differentiating severe AH from nonsevere ALD, their peak areas were used for ROC analyses. The AUC was 1.000 (95% CI, 0.899‐1.000) with P < 0.001 (Fig. 5), indicating excellent and highly reproducible true positivity.
Table 3.
Spearman’s Rank Correlation of Urine BAs With Clinical Parameters
| Name | Spearman's Rank Coefficient | |||||||
|---|---|---|---|---|---|---|---|---|
| MELD | P Value | MDF | P Value | AST:ALT | P Value | AUDIT | P Value | |
| DCA | −0.65 | <0.001 | −0.50 | <0.001 | ‐ | ‐ | −0.52 | 0.001 |
| 23‐Nordeoxycholic acid | −0.58 | <0.001 | −0.50 | 0.001 | ‐ | ‐ | ‐ | ‐ |
| GβMCA | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | −0.54 | <0.001 |
| 6,7‐Diketolithocholic acid | 0.63 | <0.001 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Apocholic acid | 0.58 | <0.001 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| GHCA | 0.67 | <0.001 | 0.50 | <0.001 | ‐ | ‐ | ‐ | ‐ |
| GCDCA | 0.68 | <0.001 | 0.58 | <0.001 | ‐ | ‐ | ‐ | ‐ |
| TCDCA | 0.75 | <0.001 | 0.53 | <0.001 | ‐ | ‐ | ‐ | ‐ |
Abbreviation: GβMCA, glycine‐β‐muricholic acid.
FIG. 5.
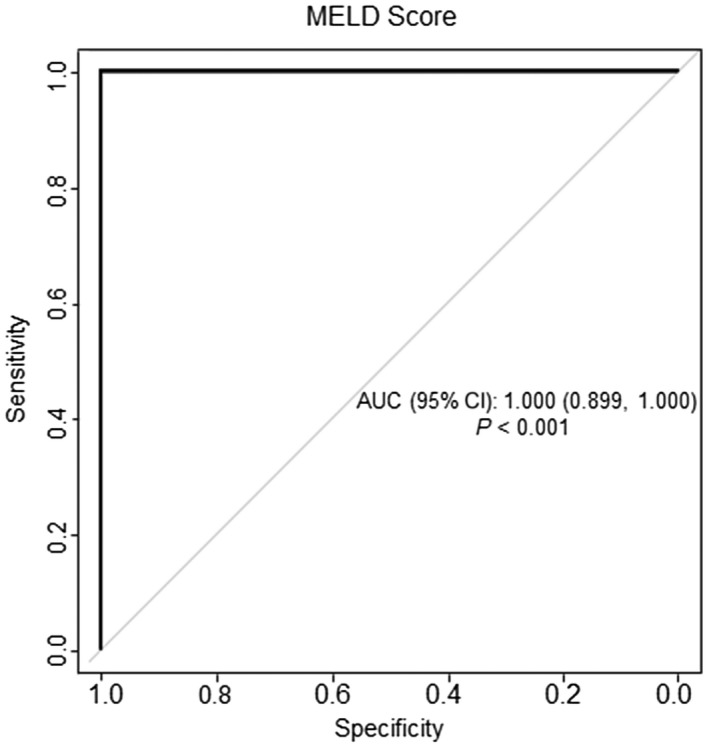
ROC curves constructed to differentiate severe AH and nonsevere ALD, using the levels of the seven BAs that have the best correlation with MELD score, i.e., TCDCA, GCDCA, GHCA, apocholic acid, 6,7‐diketolithocholic acid, 23‐nordeoxycholic acid, and DCA. ROC is the bold solid curve. The light grey line is a guideline line for comparison.
By categorizing the samples using MDF, AST:ALT, and AUDIT, GCDCA had the largest correlation coefficient with MDF (ρ = 0.58) and glycine‐β‐muricholic acid (GβMCA) had the largest correlation coefficient with AUDIT (ρ = −0.54). All BAs had a correlation coefficient <0.5 with AST:ALT (Table 3). ROC curves constructed using the abundances of BAs that had ≥ 0.5 for differentiating severe AH from nonsevere ALD, where samples were categorized by MDF and AUDIT, respectively, are shown in Supporting Fig. S3. The large AUC values of the ROC curves indicate that the MELD score and MDF perform much better than AUDIT and AST:ALT.
BAs Continuously Change in Patients With AH
Samples from patients with MELD score <12 were grouped as AUD. All samples from patients with moderate and severe AH were grouped into AH. The abundance levels of 11 BAs (glycocholic acid [GCA], GCDCA, TCDCA, TCA, GHCA, THCA, 23‐norcholic acid, 6,7‐diketolithocholic acid, nutriacholic acid, glycoursodeoxycholic acid [GUDCA], and taurine‐conjugated α‐muricholic acid [TαMCA]) were significantly increased from HCs to AUD and ultimately to AH in a stepwise manner (Fig. 6A). On the other hand, the levels of five unconjugated BAs (12‐ketolithocholic acid, LCA, DCA, 23‐nordeoxycholic acid, and ω‐MCA) were decreased from HCs to AUD and AH in a stepwise fashion (Fig. 6B). These observations suggest that BAs can be investigated for their role as diagnostic markers for liver injury at early and advanced stages due to heavy alcohol consumption.
FIG. 6.
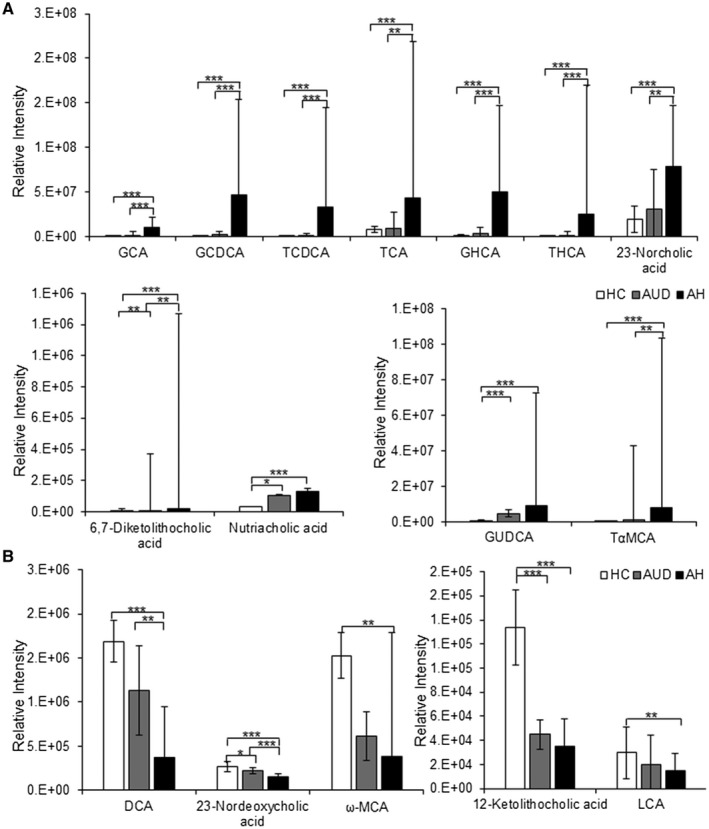
BA changes in urine collected from HCs, patients with AUD, and patients with moderate and severe AH. (A) The three panels display significantly increased levels of BAs in AH. (B) The two panels show the levels of secondary BAs were significantly decreased from HCs to AUD and to AH. Data show mean ± SD. One‐way ANOVA with Bonferroni post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001.
When categorizing samples by AST:ALT, most of the above‐mentioned BAs had a similar trend as that when samples were categorized by MELD score (Supporting Fig. S4).
Discussion
A novel aspect of this study is the use of urine as our biomarker specimen source. Studies have shown that urine BAs are affected to a lesser extent by food intake( 17 ) and nutritional interaction,( 24 ) providing higher stability. Urine samples are convenient to collect and store( 25 ) and are easier to handle because the extraction of serum or plasma from blood samples is an initial step of laboratory processing before metabolomic analyses.( 26 ) Urine samples can also be easily collected in great volume, and thus sample availability is an advantage compared to blood samples for many clinical studies. This is especially true when study patients cannot go to clinical laboratories in situations such as the corona virus disease 2019 pandemic.
We selected 46 BAs for this pilot proof‐of‐concept study based on their importance in BA metabolic pathways. BA quantification showed that the levels of most of the detected primary BAs, i.e., GCA, TCDCA, GCDCA, and GHCA, were markedly increased as the severity of AH increased (Table 2), suggesting that the synthesis of BAs increased. This hypothesis is supported by findings from our study’s urine and serum data (Supporting Fig. S5) as well as literature reports, i.e., elevated BAs in ALD.( 5 , 10 ) This evidence suggests that urine can be a source for ALD biomarker discovery.
Both sulfated and nonsulfated BAs are present in urine in liver diseases,( 27 ) and we observed both forms in the urine and serum samples of patients with AH as well. Sulfation of BAs is a mechanism involved in their elimination and systemic detoxification. Around 70% of BAs are sulfated, and this can reduce the toxic effects of BAs,( 28 ) increase their solubility,( 29 ) decrease their intestinal absorption,( 30 ) and enhance their fecal and urinary excretion.( 31 )
MELD score is a better clinical parameter than MDF, AST:ALT, and AUDIT to categorize urine samples for analysis of BA abundance changes in AH. When the samples were categorized by MELD score, large R 2 and Q 2 values showed that PLS‐DA had the best fitting and predictability for BA profile analysis. ROC analysis showed that more BAs have statistically significant abundance changes with very large AUC (>0.9). Furthermore, the changes of total BAs, the ratio of unconjugated and conjugated BAs, as well as the ratio of primary and secondary BAs showed the best stepwise trend from HCs to nonsevere ALD and ultimately to severe AH. All these results suggest that MELD score is the best parameter to categorize urine samples for BA biomarker analysis.
BAs are derivatives of cholesterol.( 32 ) Primary BAs are synthesized by the liver, and secondary ones arise from bacterial action in the colon. BAs are transported into the intestine to facilitate the absorption of lipids, steroids, and other small molecules. In the intestine, primary BAs (e.g., CA, CDCA) are converted into secondary BAs (e.g., DCA, LCA, ursodeoxycholic acid) by bacterial action. BAs play a critical role in maintaining metabolic homeostasis. As important nutrient sensors, BAs work as emulsifiers in the gut to facilitate the absorption of dietary fats, steroids, and lipid‐soluble vitamins.( 32 ) BAs also serve as signaling molecules and regulate hepatic glucose, lipid, and energy metabolism by binding with nuclear farnesoid X receptor and membrane Takeda G protein‐coupled receptor 5 (also called G protein‐coupled BA receptor 1).( 33 , 34 ) In addition to their roles in the regulation of metabolism, BAs promote hepatic inflammation during cholestasis by activating a signaling network in hepatocytes( 35 , 36 , 37 , 38 ) and thus can promote the development of liver diseases.
Primary BAs play important roles in ALD progression. It has been reported that GCDCA promotes liver fibrosis in mice with hepatocellular cholestasis by increasing hepatic messenger RNA (mRNA) expression of alpha‐smooth muscle actin and collagen 1a,( 39 ) and it directly contributes to liver disease development. GCDCA can also induce oxidative stress and cell death by reducing the transcription of endothelial nitric oxide synthase( 40 ) and thus can promote the development of ALD. CA, CDCA, and TCA have also been reported to have roles in liver disease progression. Mice fed with a CA‐containing diet for 5 days showed increased liver weight, cell proliferation index, and oxidative stress,( 41 ) all of which resulted in enhanced visceral adiposity, atherosclerosis, and fatty liver disease.( 42 ) TCA is an active promoter of the progression of liver cirrhosis through activating hepatic stellate cells through up‐regulating toll‐like receptor 4 expression.( 43 ) High levels of TCA and CDCA in the liver directly activate a signaling network in hepatocytes that induces the mRNA expression of numerous proinflammatory mediators and also promote hepatic inflammation( 35 ) and thus contribute to liver disease development. In the present study, most of the detected primary BAs in urine were increased in a stepwise manner from HCs to nonsevere ALD and to severe AH, suggesting that urine BAs may accurately reflect liver disease severity.
In contrast to primary BAs, secondary BAs, such as DCA, LCA and 23‐nordeoxycholic acid, were significantly decreased in a stepwise manner (Fig. 3B). Secondary BAs arise from primary BAs synthesized in the liver and secreted into the gut where they are transformed by intestinal microbiota. This decrease may be due to a reduction of BA secretion into the intestine and/or alterations of gut microbiota( 5 , 10 ) and renal reabsorption. Studies have shown that alcohol consumption changed the composition of gut microbiota,( 44 , 45 ) and the abundance of ileal Bacteriodetes, ( 46 ) which is a major microbiota phyla responsible for BA metabolism,( 47 ) was decreased in subjects with ALD.
MELD score correlated closely with most of the 43 BAs identified, particularly with GCDCA, GHCA, TCDCA, and DCA (Table 3), and this is consistent with earlier studies. It has been reported that LCA and DCA were negatively correlated with the MELD score in AC( 48 ) and that TCDCA had the highest correlation with the MELD score in AH.( 5 ) Our data are consistent with these reports with the exception that the negative correlation between LCA and MELD score is not significant in the current study (ρ = −0.16). The continuum of increased/decreased abundance levels of BAs in urine from patients provided us with new insights into AH diagnosis (Figs. 3 and 6). The ROC curve built by the abundances of seven BAs (TCDCA, GCDCA, GHCA, apocholic acid, 23‐nordeoxycholic acid, 6,7‐diketolithocholic acid, and DCA) has a very large AUC (1.000; 95% CI, 0.899‐1.000; P < 0.001) (Fig. 5), suggesting that these BAs could be used to determine the progress of AH severity with high accuracy.
This pilot proof‐of‐principle study has limitations. The sample size is relatively small, therefore identifying the roles of metabolites with minor effect sizes was not possible and we were unable to identify any underlying sex effects. Secondary BAs, such as LCA and DCA, are more toxic and easier to be sulfated and excreted into urine; and primary BAs, such as CA and CDCA, are less toxic and less sulfated. We do not have sulfation data on some BAs, which limits our data interpretation on those primary BAs. HCs were younger than patients, and we do not yet know what, if any, impact that had on the study. Moreover, more extensive studies are needed to precisely elucidate the role of various demographic measures. This study was not designed as a treatment‐based investigation; thus, identifying any role of BAs in therapeutic–mechanistic pathways was also not within the scope of this study. However, given the potential prognostic value of the candidate BAs, this study supports the need for longitudinal AH studies where efficacy of treatment and change in liver function can be measured in relation to the candidate metabolites identified in this study. Lastly, we did no fecal analysis to define the role of the microbiome in BA changes.
In summary, our data showed that certain urinary BA concentrations were altered in patients with AH/AUD depending on disease severity. Primary BAs were increased, while secondary BAs were decreased in the patients with severe AH compared to the patients with nonsevere ALD and HCs. Three BAs, CA, CDCA and apocholic acid, had increased levels in patients with severe AH and might be diagnostic of severe liver disease. The large ROC AUC between the patients with nonsevere ALD and severe AH as well as a high correlation between the levels of BAs and MELD score indicate that levels of BAs are closely correlated with severity of liver injury. BAs could, therefore, serve as potential laboratory markers to assist in the diagnosis/prognosis of ALD/AH. Lastly, urine proved to be an excellent biomarker specimen source.
Supporting information
Supplementary Material
Acknowledgment
We thank Ms. Marion McClain for review of this manuscript.
Supported by the University of Louisville Alcohol Research Center (grant 1P50AA024337 to C.J.M.), the National Institutes of Health (S10OD020106 to X.Z.; 1P20GM113226, 1U01AA026934, 1U01AA026936, 1U01AA026980, and 1R01AA023681 to C.J.M.), and the Department of Veterans Affairs (1I01BX002996‐01A2 to C.J.M.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ClinicalTrials.gov identifier numbers NCT01922895 and NCT01809132.
Potential conflict of interest: Nothing to report.
Contributor Information
Craig J. McClain, Email: craig.mcclain@louisville.edu.
Xiang Zhang, Email: xiang.zhang@louisville.edu.
References
Author names in bold designate shared co‐first authorship.
- 1. World Health Organization . Global status report on alcohol and health 2018. https://apps.who.int/iris/bitstream/handle/10665/274603/9789241565639‐eng.pdf?ua=9789241565631. Accessed January 7, 2021. [Google Scholar]
- 2. Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res 2015;39:763‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crabb DW, Bataller R, Chalasani NP, Kamath PS, Lucey M, Mathurin P, et al.;NIAAA Alcoholic Hepatitis Consortia . Standard definitions and common data elements for clinical trials in patients with alcoholic hepatitis: recommendation from the NIAAA Alcoholic Hepatitis Consortia. Gastroenterology 2016;150:785‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nestel P, Simons L, Homma Y. Effects of ethanol on bile acid and cholesterol metabolism. Am J Clin Nutr 1976;29:1007‐1015. [DOI] [PubMed] [Google Scholar]
- 5. Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura‐Cots M, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte‐Rojo A, Louvet A, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 2014;146:1231‐1239.e1231‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trinchet JC, Gerhardt MF, Balkau B, Munz C, Poupon RE. Serum bile acids and cholestasis in alcoholic hepatitis. Relationship with usual liver tests and histological features. J Hepatol 1994;21:235‐240. [DOI] [PubMed] [Google Scholar]
- 8. Chambers KF, Day PE, Aboufarrag HT, Kroon PA. Polyphenol effects on cholesterol metabolism via bile acid biosynthesis, CYP7A1: a review. Nutrients 2019;11:2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciocan D, Voican CS, Wrzosek L, Hugot C, Rainteau D, Humbert L, et al. Bile acid homeostasis and intestinal dysbiosis in alcoholic hepatitis. Aliment Pharmacol Ther 2018;48:961‐974. [DOI] [PubMed] [Google Scholar]
- 10. Yang Z, Kusumanchi P, Ross RA, Heathers L, Chandler K, Oshodi A, et al. Serum metabolomic profiling identifies key metabolic signatures associated with pathogenesis of alcoholic liver disease in humans. Hepatol Commun 2019;3:542‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Habior A, Ostrowski J, Trzeciak L, Wojciechowski K. Comparison of the value of serum caffeine and bile acid concentrations as indicators of liver injury. [in Polish] Pol Arch Med Wewn 1990;84:284‐291. [PubMed] [Google Scholar]
- 12. Rachakonda V, Gabbert C, Raina A, Bell LN, Cooper S, Malik S, et al. Serum metabolomic profiling in acute alcoholic hepatitis identifies multiple dysregulated pathways. PLoS One 2014;9:e113860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hofmann A, Klein P. Characterisation of bile acid metabolism in man using bile acids labelled with stable isotopes. In: Baillie TA, ed. Stable Isotopes. London, United Kingdom: Palgrave Macmillan; 1978:189‐204. [Google Scholar]
- 14. Bremmelgaard A, Sjovall J. Bile acid profiles in urine of patients with liver diseases. Eur J Clin Invest 1979;9:341‐348. [DOI] [PubMed] [Google Scholar]
- 15. Jonsson G, Hedenborg G, Wisen O, Norman A. Presence of bile acid metabolites in serum, urine, and faeces in cirrhosis. Scand J Clin Lab Invest 1992;52:555‐564. [DOI] [PubMed] [Google Scholar]
- 16. Shinozaki K, Nakagawa S. Sulfated bile acid in urine of patients with hepatobiliary diseases. Lipids 1973;8:47‐49. [DOI] [PubMed] [Google Scholar]
- 17. Bathena SPR, Thakare R, Gautam N, Mukherjee S, Olivera M, Meza J, et al. Urinary bile acids as biomarkers for liver diseases I. Stability of the baseline profile in healthy subjects. Toxicol Sci 2015;143:296‐307. [DOI] [PubMed] [Google Scholar]
- 18. Vatsalya V, Cave MC, Kong M, Gobejishvili L, Falkner KC, Craycroft J, et al. Keratin 18 is a diagnostic and prognostic factor for acute alcoholic hepatitis. Clin Gastroenterol Hepatol 2020;18:2046‐2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Y, Vatsalya V, Gobejishvili L, Lamont RJ, McClain CJ, Feng W. Porphyromonas gingivalis as a possible risk factor in the development/severity of acute alcoholic hepatitis. Hepatol Commun 2018;3:293‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X, Yin P, Shao Y, Wang Z, Wang B, Lehmann R, et al. Which is the urine sample material of choice for metabolomics‐driven biomarker studies? Anal Chim Acta 2020;1105:120‐127. [DOI] [PubMed] [Google Scholar]
- 21. He L, Wei X, Ma X, Yin X, Song M, Donninger H, et al. Simultaneous quantification of nucleosides and nucleotides from biological samples. J Am Soc Mass Spectrom 2019;30:987‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web‐based platform to process untargeted metabolomic data. Anal Chem 2012;84:5035‐5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He L, Li F, Yin X, Bohman P, Kim S, McClain CJ, et al. Profiling of polar metabolites in mouse feces using four analytical platforms to study the effects of cathelicidin‐related antimicrobial peptide in alcoholic liver disease. J Proteome Res 2019;18:2875‐2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glicksman C, Pournaras DJ, Wright M, Roberts R, Mahon D, Welbourn R, et al. Postprandial plasma bile acid responses in normal weight and obese subjects. Ann Clin Biochem 2010;47:482‐484. [DOI] [PubMed] [Google Scholar]
- 25. Cornelis R, Heinzow B, Herber R, Molin Christensen J, Poulsen OM, Sabbioni E, et al. Sample collection guidelines for trace elements in blood and urine. IUPAC Commission of Toxicology. J Trace Elem Med Biol 1996;10:103‐127. [DOI] [PubMed] [Google Scholar]
- 26. A J, Trygg J, Gullberg J, Johansson AI, Jonsson P, Antti H, et al. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal Chem 2005;77:8086‐8094. [DOI] [PubMed] [Google Scholar]
- 27. Makino I, Hashimoto H, Shinozaki K, Yoshino K, Nakagawa S. Sulfated and nonsulfated bile acids in urine, serum, and bile of patients with hepatobiliary diseases. Gastroenterology 1975;68:545‐553. [PubMed] [Google Scholar]
- 28. Hofmann AF. Detoxification of lithocholic acid, a toxic bile acid: relevance to drug hepatotoxicity. Drug Metab Rev 2004;36:703‐722. [DOI] [PubMed] [Google Scholar]
- 29. Gu JJ, Hofmann AF, Ton‐Nu HT, Schteingart CD, Mysels KJ. Solubility of calcium salts of unconjugated and conjugated natural bile acids. J Lipid Res 1992;33:635‐646. [PubMed] [Google Scholar]
- 30. Rodrigues CM, Kren BT, Steer CJ, Setchell KD. The site‐specific delivery of ursodeoxycholic acid to the rat colon by sulfate conjugation. Gastroenterology 1995;109:1835‐1844. [DOI] [PubMed] [Google Scholar]
- 31. Alnouti Y. Bile acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci 2009;108:225‐246. [DOI] [PubMed] [Google Scholar]
- 32. Chiang JYL. Bile acids: regulation of synthesis. J Lipid Res 2009;50:1955‐1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res 2017;1:3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shapiro H, Kolodziejczyk AA, Halstuch D, Elinav E. Bile acids in glucose metabolism in health and disease. J Exp Med 2018;215:383‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 2011;178:175‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Anwer MS. Intracellular signaling by bile acids. J Biosci (Rajshari) 2012;20:1‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luedde T, Schwabe RF. NF‐kappaB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011;8:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Halilbasic E, Baghdasaryan A, Trauner M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin Liver Dis 2013;17:161‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hohenester S, Kanitz V, Kremer AE, Paulusma CC, Wimmer R, Kuehn H, et al. Glycochenodeoxycholate promotes liver fibrosis in mice with hepatocellular cholestasis. Cells 2020;9:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. González‐Rubio S, López‐Sánchez L, Muñoz‐Castañeda J, Linares CI, Aguilar‐Melero P, Rodríguez‐Perálvarez M, et al. GCDCA down‐regulates gene expression by increasing Sp1 binding to the NOS‐3 promoter in an oxidative stress dependent manner. Biochem Pharmacol 2015;96:39‐51. [DOI] [PubMed] [Google Scholar]
- 41. Miura T, Tachikawa M, Ohtsuka H, Fukase K, Nakayama S, Sakata N, et al. Application of quantitative targeted absolute proteomics to profile protein expression changes of hepatic transporters and metabolizing enzymes during cholic acid‐promoted liver regeneration. J Pharm Sci 2017;106:2499‐2508. [DOI] [PubMed] [Google Scholar]
- 42. Yamada S, Kawaguchi H, Yamada T, Guo X, Matsuo K, Hamada T, et al. Cholic acid enhances visceral adiposity, atherosclerosis and nonalcoholic fatty liver disease in microminipigs. J Atheroscler Thromb 2017;24:1150‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Z, Zhang Z, Huang M, Sun X, Liu B, Guo Q, et al. Taurocholic acid is an active promoting factor, not just a biomarker of progression of liver cirrhosis: evidence from a human metabolomic study and in vitro experiments. BMC Gastroenterol 2018;18:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bajaj JS. Alcohol, liver disease and the gut microbiota. Nat Rev Gastroenterol Hepatol 2019;16:235‐246. [DOI] [PubMed] [Google Scholar]
- 45. Engen PA, Green SJ, Voigt RM, Forsyth CB, Keshavarzian A. The gastrointestinal microbiome: alcohol effects on the composition of intestinal microbiota. Alcohol Res 2015;37:223‐236. [PMC free article] [PubMed] [Google Scholar]
- 46. Bull‐Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, et al. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS One 2013;8:e53028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Staley C, Weingarden AR, Khoruts A, Sadowsky MJ. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl Microbiol Biotechnol 2017;101:47‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol 2014;306:G929‐G937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material