

Abstract
Hepcidin, a peptide hormone produced by hepatocytes, is the central regulator of systemic iron homeostasis through its interaction with ferroportin, the major cellular iron export protein. Hepcidin binding to ferroportin results in reduced iron export from macrophages and intestinal absorptive cells, leading to decreased serum iron levels. Hepcidin expression is influenced by several factors that include serum and liver iron stores, erythropoiesis, hypoxia, inflammation, and infection. Erythropoietic drive and hypoxia suppress hepcidin expression and promote red cell production. In contrast, inflammation and infection are associated with increased hepcidin production to sequester iron intracellularly as a means of depriving microorganisms of iron. Chronic inflammation may up‐regulate hepcidin expression through the interleukin‐6 (IL‐6)–Janus kinase 2 (JAK2)–signal transducer and activator of transcription 3 (STAT3) pathway. The bone morphogenetic protein (BMP)–mothers against decapentaplegic homolog (SMAD) pathway is a major positive driver of hepcidin expression in response to either increased circulating iron in the form of transferrin or iron loading in organs. Hereditary hemochromatosis (HH) consists of several inherited disorders that cause inappropriately reduced hepcidin expression in response to body iron stores, leading to increased iron absorption from a normal diet. The most common form of HH is due to a mutation in the HFE gene, which causes a failure in the hepatocyte iron–sensing mechanism, leading to reduced hepcidin expression; the clinical manifestations of HFE‐HH include increased serum transferrin–iron saturation and progressive iron loading in the liver and other tissues over time among patients who express the disease phenotype. In this article, we review the physiologic mechanisms and cellular pathways by which hepcidin expression is regulated, and the different forms of HH resulting from various mutations that cause hepcidin deficiency. We also review other drivers of hepcidin expression and the associated pathophysiologic consequences.
Abbreviations
- ALK
activin receptor‐like kinase
- BMP
bone morphogenetic protein
- BMPR
BMP receptor
- ERFE
erythroferrone
- HAMP
hepatic antimicrobial protein
- HH
hereditary hemochromatosis
- HIF
hypoxia inducible factor
- HJV
hemojuvelin
- IL‐6
interleukin‐6
- IRP
iron‐regulatory protein
- JAK
Janus kinase
- LPS
lipopolysaccharide
- mRNA
messenger RNA
- NASH
nonalcoholic steatohepatitis
- RES
reticuloendothelial system
- SMAD
mothers against decapentaplegic homolog
- STAT
signal transducer and activator of transcription
- TMPRSS6
matriptase‐2
- TFR
transferrin receptor
- TLR4
toll‐like receptor 4
Regulation of Systemic Iron Stores
Iron metabolism is tightly regulated to maintain a balance between availability of a sufficient quantity of this metal to maintain vital cellular processes and the risk of iron‐related tissue toxicity. This balance is maintained through control of iron absorption in the intestine, storage and release from intracellular compartments in hepatocytes and reticuloendothelial system (RES) cells, as well as iron utilization by mitochondria.( 1 , 2 ) Dietary iron is absorbed in the proximal duodenum (Fig. 1) and is exported from enterocytes and macrophages into the systemic circulation through ferroportin.( 1 , 2 ) Ferroportin is the only known transmembrane protein responsible for exporting iron, and is found in macrophages, enterocytes, and hepatocytes.( 3 ) Hepcidin binds to and reduces ferroportin activity either by promoting internalization and lysosomal degradation of ferroportin or directly blocking iron export by ferroportin, leading to a reduction in circulating iron stores by decreasing duodenal iron absorption and increasing iron sequestration in the RES.( 1 , 2 , 4 , 6 , 7 ) Hepcidin is predominantly produced in the liver as a prepropeptide containing 84 amino acids with an endoplasmic reticulum signal sequence at the N‐terminal and a C‐terminal furin consensus cleavage site.( 4 , 5 ) After cleavage, the now‐bioactive 25 amino acid peptide is secreted into circulation, where the protein exerts its effects on ferroportin degradation and subsequent iron storage and absorption.( 4 , 6 )
FIG. 1.
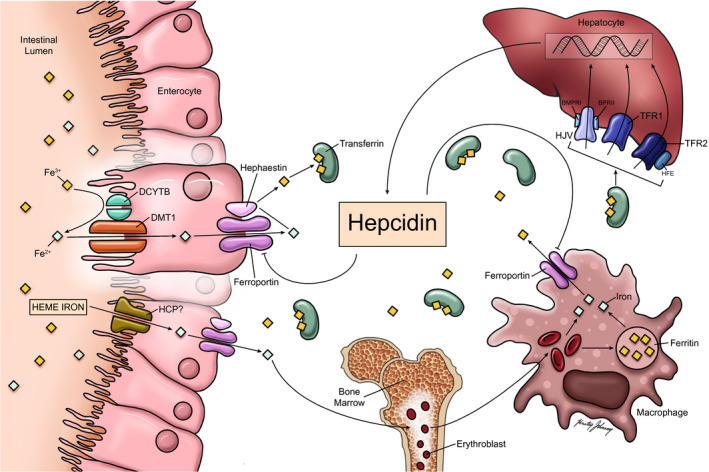
Hepcidin, the master regulator of iron absorption and secretion. Plasma iron levels are controlled primarily at the level of absorption by duodenal enterocytes and RES macrophages. Heme‐iron can also be directly absorbed from the gastrointestinal tract through a carrier protein, possibly heme carrier protein. Non‐heme (inorganic) iron is absorbed via divalent metallic transporter 1 after conversion from ferric (Fe 3+) to ferrous (Fe 2+) iron through duodenal cytochrome b‐related ferric reductase. RES macrophages acquire iron through erythrophagocytosis. Efflux of iron from both enterocytes and RES macrophages occurs through the iron export protein ferroportin. Fe 2+ is reduced back to ferric iron (Fe 3+) through hephaestin before being transported out of the enterocyte. Iron is bound to transferrin as it enters the plasma, which then travels through the circulation where it is taken up by various organs and used in the development of erythrocytes and other biological processes. The principal regulator of iron levels is the hormone hepcidin, which is produced by hepatocytes. As iron stores increase, hepcidin inhibits iron efflux from both duodenum enterocytes and RES macrophages through down‐regulation of ferroportin. Serum iron levels influence hepcidin expression through the interaction of the HFE protein with TFR1/TFR2 along with BMP6 and HJV. Abbreviations: DCYTB, duodenal cytochrome b‐related ferric reductase; DMT1, divalent metallic transporter 1; HCP, heme carrier protein.
Dietary iron is absorbed in two forms: heme, which is characterized as iron coordinated to a porphyrin, and nonheme.( 8 ) The absorption of nonheme iron begins with the conversion of the ferric (Fe 3+) to the ferrous (Fe 2+) form of iron, primarily through duodenal cytochrome b‐related ferric reductase, located on the luminal side of the duodenal enterocyte.( 8 ) Once in the ferrous form, iron can cross the apical membrane of the duodenal enterocyte through the divalent metal transporter 1.( 8 ) In contrast, although heme carrier protein and heme‐responsive gene 1 have been proposed as heme transporters, the mechanism for heme absorption remains unclear.( 1 , 9 ) Iron is released from heme by heme oxygenase and then enters a common pathway with inorganic iron within the enterocyte.( 8 )
Iron homeostasis is also regulated at the intracellular level by the iron‐responsive element/iron‐regulatory protein (IRP) system.( 10 , 11 ) This system affects both the influx of iron into the cell as well as sequestration of iron within the cell.( 10 , 11 ) Under conditions of intracellular iron shortage, IRP1 and 2 stabilize transferrin receptor 1 (TFR1) messenger RNA (mRNA) and suppress ferritin and ferroportin mRNA translation, resulting in increased cellular iron uptake and reduced iron export.( 2 , 11 ) In the setting of iron sufficiency or excess, IRP1 is converted to cytosolic aconitase, and IRP2 is proteosomally degraded.( 2 )
Bone Morphogenetic Protein–Mothers Against Decapentaplegic Homolog Pathway in Iron Regulation
The bone morphogenetic protein (BMP)–mothers against decapentaplegic homolog (SMAD) pathway occupies a key function in modulating iron homeostasis through regulation of hepcidin expression at the level of transcription.( 12 ) BMPs are part of the transforming growth factor β superfamily of molecules involved in various signaling pathways.( 12 ) BMP2 and BMP6, produced by sinusoidal endothelial cells in the liver, are an important component of hepcidin signaling in response to increased tissue iron stores.( 13 ) Expression of BMP6 and phosphorylation of SMAD 1/5/8 was regulated by iron status and correlated with hepcidin mRNA expression in mice.( 14 , 15 ) BMP6 was then shown to be a positive regulator of hepcidin expression in response to iron status, and that absence of BMP6 resulted in iron overload.( 16 ) Finally, BMP6 was shown to bind directly to hemojuvelin (HJV), which is expressed on the hepatocyte cell membrane and part of the iron sensing complex that includes HFE, HJV, and TFR2.( 12 , 16 , 17 ) The current evidence, albeit much of it from genetically modified murine models, proposes that BMP 2 and 6, produced in response to iron, bind to a receptor complex consisting of BMPR1 (activin receptor‐like kinase [ALK] 3 and ALK2) and BMPR2, possibly as heterodimers along with HJV leading to SMAD 1/5/8 phosphorylation, which then forms a complex with SMAD4.( 18 , 19 ) The complex translocates to the nucleus and then increases hepcidin mRNA expression through promoter binding (Fig. 2).( 12 , 18 ) This positive pathway for hepcidin is suppressed by a serine protease, encoded by matriptase‐2 (TMPRSS6); this enzyme renders HJV inactive by cleavage to its soluble form, thus decreasing hepcidin at the transcriptional level.( 12 , 20 ) It has been proposed that BMP2 and 6 are secreted by liver sinusoidal endothelial cells in response to tissue iron overload, whereas TFR1 and TFR2, along with HFE, are the sensors for circulating iron stores measured through transferrin‐iron saturation( 13 ); others have proposed that BMP2 contributes to sensing of circulating iron, while BMP6 serves as a sensor for storage iron.( 13 )
FIG. 2.
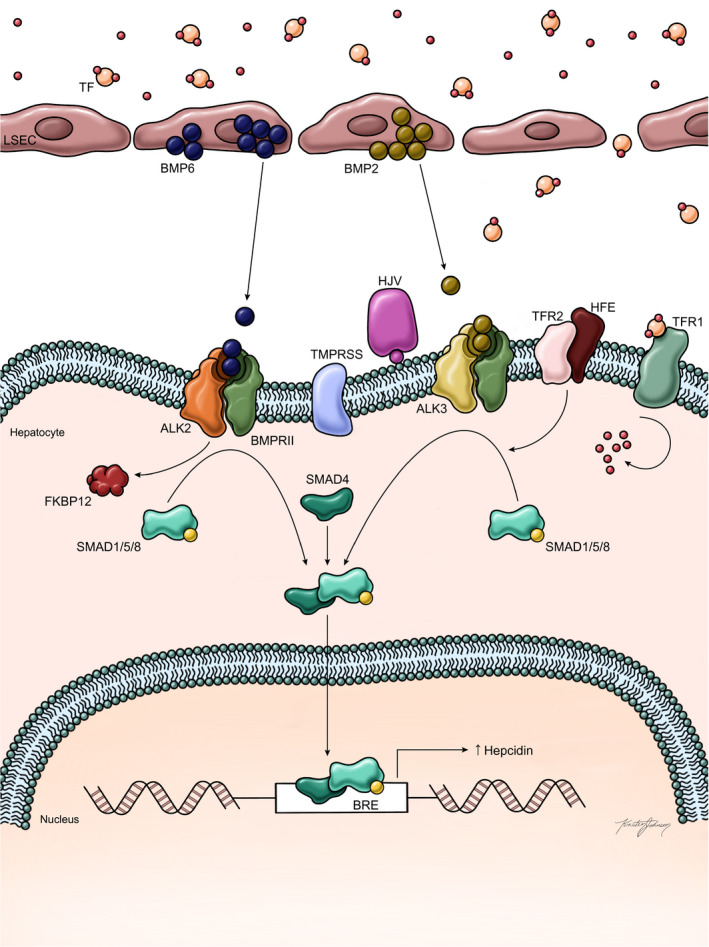
Regulation of hepcidin expression in the setting of iron overload (adapted from Camaschella et al.( 2 )). It is proposed that tissue iron stores are sensed by liver sinusoidal‐lining endothelial cells, which secrete BMP6 (and to a lesser extent BMP2) in response to increased storage iron. Circulating iron is sensed by a complex interaction among HFE, TFR1, and TFR2 in conjunction with HJV. BMP6 and BMP2 bind to the receptors ALK2 and ALK3, respectively, resulting in SMAD1/5/8 phosphorylation, forming a complex with SMAD4 that enters the nucleus and increases hepcidin transcription through promoter binding. HJV as a BMP co‐receptor and TFR2 also contribute to ALK3‐dependent SMAD phosphorylation. Similarly, the interaction among HFE, TFR1, TFR2, and HJV also contributes to SMAD1/5/8 phosphorylation and ultimately increased hepcidin expression. Thus, the response to increased circulating and tissue iron is increased hepcidin production, which then binds to ferroportin in both duodenal enterocytes and macrophages, leading to reduced iron export from cells and decreased iron absorption in the duodenum. In HFE hemochromatosis, the mutant HFE is trapped intracellularly and leads to a dampened hepcidin response to circulating iron, resulting in inappropriately high iron absorption in relation to body iron stores. Abbreviations: LSEC, liver sinusoidal‐lining endothelial cell; TF, transcription factor.
Alteration of the BMP/HJV/SMAD Pathway in Hereditary Hemochromatosis
Several important discoveries have led to our current understanding of the mechanisms by which perturbations in the BMP/SMAD/HJV pathway result in reduced hepcidin expression and lead to iron overload because of inappropriately increased iron absorption.( 12 , 13 ) Disruption of the iron‐sensing mechanism in hepatocytes resulting in deficient hepcidin production leads to several forms of hereditary hemochromatosis (HH), classified as type 1, type 2A and type 2B, type 3, and type 4 HH( 21 ) (Table 1).
TABLE 1.
Classification of HH
| Type | Gene | Gene Location |
|---|---|---|
| 1 (classical HH) | HFE | 6p21.3 |
| 2a | HJV | 1p21 |
| 2b | HAMP | 19q13 |
| 3 | TFR2 | 7q22 |
| Ferroportin disease (formerly type 4A) | SLC40A1 | 2q32.2 |
| 4 (formerly type 4B) | SLC40A1 | 2q32.2 |
Abbreviation: SLC40A1, solute carrier family 40 member 1.
In type 1 HH, mutations in HFE are believed to result in the reduced ability of hepatocytes to sense circulating iron bound to transferrin.( 22 ) Type 1 HH, the most common inherited form of iron overload, is caused by a C282Y or H63D mutation in the HFE gene located on chromosome 6 (6p21.3), responsible for encoding a major histocompatibility complex class I–like protein.( 23 , 24 ) The role of HFE in iron regulation remains incompletely understood; one hypothesis suggests that increased transferrin‐iron saturation displaces HFE from TFR1, and stabilizes TFR2, which may then allow HFE to increase hepcidin expression through BMP/SMAD signaling.( 1 , 2 ) As mentioned previously, the BMP‐SMAD 1/5/8 signaling pathway has been found to be an essential mechanism to the regulation of hepcidin synthesis. BMPR1A (also known as ALK3) has been studied as a down‐stream target of HFE, and BMPR1A deficiency in mice has been shown to prevent the increase in hepcidin expression that would be expected after HFE overexpression.( 25 ) HJV is known to act as a BMP co‐receptor for BMPR1A/1B.( 26 ) The ligands BMP2 and BMP6 bind as a heterodimer, and the resultant signaling complex phosphorylates and thereby activates SMAD proteins that bind to promoter elements on the HAMP (hepatic antimicrobial protein) gene, which encodes hepcidin and induces transcription.( 27 ) Although BMP receptors are present on hepatocytes, BMPs 2 and 6 are produced by hepatic endothelial cells and regulate the pathway through paracrine action.( 28 , 29 ) Recent studies in mice suggest that BMP2 and BMP6 may function in concert to regulate hepcidin, and that neither is able to influence expression of the protein in the absence of the other, although BMP2 may be less responsive to iron stores than BMP6 and may contribute to maintaining basal hepcidin levels.( 2 , 30 ) Furthermore, HFE may regulate the BMP‐SMAD pathway at least partly through a mechanism independent of BMP2, as double HFE/BMP2 knockout mice demonstrate a more severe iron overload phenotype than single BMP2 knockout.( 30 ) A previous study found that HFE may bind BMPR1A directly, stabilizing the receptor and increasing its expression at the cell surface.( 31 ) Consistent with these findings, altered BMP signaling has been found in patients with type 1 HH.( 32 , 33 ) There remains a need for further in vivo and patient data to provide meaningful insights into the mechanisms by which HFE, TFR1, and TFR2 regulate hepcidin expression.
The most common variants in the HFE gene are the C282Y and H63D mutations.( 21 ) The defects are inherited in an autosomal recessive pattern; however, the phenotypic expression is highly variable.( 21 ) Many patients with classical HH are asymptomatic, especially early in the disease process.( 34 ) When early symptoms are present, they are often nonspecific and include weakness, fatigue, lethargy, and weight loss.( 35 , 36 ) Specific symptoms are usually related to the affected organ and can include abdominal pain, arthralgias, diabetes, amenorrhea, erectile dysfunction, palpitations, swelling, and increased skin pigmentation.( 13 , 34 ) It is estimated that up to 10% of Caucasian Americans have at least one mutant allele, but the rate of occurrence of genetic mutations is much higher than the prevalence of the disease.( 21 ) The variability in presentation and progression of HH has been ascribed to both genetic and environmental risk factors. Alcohol intake of ≥ 14 g per day is significantly associated with advancement to cirrhosis.( 37 ) Those with classical HH who are also carriers of mutations that cause juvenile HH, such as in HAMP and HJV, are at increased risk for development of cirrhosis at <30 years of age; rarely, HFE mutations may be accompanied by TFR2 variants, resulting in complications of iron overload.( 38 , 39 , 40 , 41 ) Women are at decreased risk for cirrhosis due to iron loss through menstruation and pregnancy, and possible hormonal effects and modifier genes.( 33 , 34 ) However, ferritin concentrations do increase after menopause increases the likelihood of phenotypic expression.( 34 ) Most symptomatic patients with HH are homozygous for the C282Y mutation (80%‐90%), whereas C282Y/H63D compound heterozygotes make up 7%‐8% of affected patients.( 21 )
Other forms of hereditary hemochromatosis are now recognized to be caused by disruption of other steps involved in hepcidin production through the iron‐sensing function of HJV in hepatocytes, resulting in markedly reduced hepcidin production.( 42 , 43 ) Juvenile or type 2 hemochromatosis is subclassified into type 2a and type 2b. Mutations in HJV are associated with juvenile hemochromatosis, now classified as type 2a HH.( 33 ) HJV is located on chromosome 1 (1q21) and encodes the membrane protein HJV, a member of the response guidance molecule family.( 12 , 17 ) Mutations within the HJV gene ultimately lead to decreased hepcidin expression and subsequent iron overload.( 33 ) In a landmark study, HJV was found to be a BMP co‐receptor, enhancing endogenous BMP signaling by directly binding BMP receptors, specifically BMPR1A/1B.( 25 )
HJV binds to BMP ligands and promotes activation of BMP signaling, although the precise mechanism remains unclear.( 12 ) However, HJV mutations were found to be associated with a severe hemochromatosis phenotype.( 33 , 44 ) Although the precise mechanism has not been elucidated, the HJV mutations presumably impair SMAD1/5/8 signaling, thus preventing SMAD4 binding and resulting in reduced hepcidin transcription through promoter binding.( 45 ) Type 2A HH is inherited as an autosomal recessive disorder; the most common mutation is the p. Gly320Val (G320V) mutation, and accounts for slightly more than 50% of mutations worldwide.( 44 , 46 )
Type 2b HH is characterized by homozygous autosomal recessive mutations in the HAMP gene.( 47 , 48 ) HAMP, found on chromosome 19 (19q13), encodes hepcidin, and several homozygous mutations resulting in loss of function have been reported.( 48 ) The original description of HAMP mutations resulting in juvenile hemochromatosis described a deletion of a guanine in exon 2 at position 93 of HAMP complementary DNA (93delG).( 47 ) This frameshift mutation resulted in an elongated prohepcidin, resulting in a misformed protein lacking activity.( 47 ) The second R56X mutation produced a truncated and ineffective prohepcidin protein.( 47 ) Other HAMP variants have been recently described.( 48 )
The clinical phenotypes associated with juvenile hemochromatosis were recently published in a comprehensive review of the literature.( 49 ) Cardiac disease and hypogonadism are more common in juvenile HH. Other clinical features such as diabetes, skin pigment changes, and hepatic fibrosis are also more common in juvenile HH than in type 1 HH, but arthropathy was less prevalent.( 49 ) Patients present with severe end‐organ manifestations, particularly cardiac disease, may die before the age of 30 and frequently have advanced hepatic fibrosis at presentation; however, phenotypic expression in HJV‐related HH may vary, and cases with late‐onset presentation have been described.( 50 , 51 ) It has been suggested that early, and markedly increased, plasma iron is due to greatly reduced or absence of hepcidin production, leading to the severe iron‐loading and associated phenotype in type 2 HH.( 50 ) Finally, ferroportin gene mutations can lead to ferroportin disease (previously called type 4A HH), in which loss‐of‐function mutations result in reduced iron export and iron retention in macrophage compartment, with the spleen being a predominant site of iron overload.( 21 ) These patients may have normal or transferrin saturation, but elevated serum ferritin. Type 4 HH (also known as type 4B HH) is associated with a “gain‐of‐function” mutation in ferroportin and severe iron overload.( 2 , 20 , 21 )
Role of Transferrin Receptors in Iron Regulation
Transferrin receptors are a class of proteins responsible for binding of transferrin and internalization of transferrin‐bound iron.( 51 ) Iron absorbed by duodenal enterocytes is bound to transferrin as monoferric or diferric transferrin.( 1 ) Transferrin then binds to TFR1, a cell‐surface protein that is responsible for iron uptake through receptor‐mediated endocytosis.( ) There is recent evidence that TFR1 in hepatocytes may also influence hepcidin expression with iron loading.( 52 ) Hepatocyte‐specific deletion of TFR1 led to increased hepcidin relative to liver iron content.( 52 ) The model proposed that iron deficiency increased TFR1, which bound to HFE and reduced activation of the HFE‐TFR2 complex and subsequent induction of hepcidin expression.( 12 , 52 ) TFR2 is homologous to TFR1 but is liver‐specific and produced in hepatocytes.( 53 ) The interaction among TFR1, TFR2, and HFE is recognized to be important for hepcidin expression in response to circulating iron stores, although some evidence indicates that the effects on hepcidin expression by HFE may be independent of TFR2.( 54 , 55 , 56 ) As discussed previously, the HFE‐TFR2 complex then increases hepcidin production through activation of SMAD 1/5/8 via phosphorylation by HJV/BMP, to form a multiprotein complex (HFE‐TFR2‐HJV) (Fig. 2).( 15 , 16 , 57 , 58 )
Type 3 HH results from a mutation in TFR2 located on chromosome 7 (7q22), leading to a misfolded protein that cannot be expressed on the cell surface.( 45 , 59 , 60 ) TFR2 mutations also result in impaired SMAD 1/5/8 signaling and are accompanied by reduced hepcidin production and parenchymal iron loading, with a phenotype similar to type 1 HH, although earlier clinical presentation and more extreme phenotypic expression has been described.( 12 , 13 , 44 , 45 , 61 , 62 , 63 , 64 )
Regulators of Hepcidin Other Than Iron Status
Circulating and tissue iron stores are the most important regulators of hepcidin expression. In addition, hepcidin production is also influenced by erythropoiesis, inflammation, hypoxia, and other signals.( 12 , 18 ) Chronic diseases resulting in increased systemic inflammation may be associated with increased basal hepcidin production, leading to the “anemia of chronic disease.” These other signaling pathways for hepcidin are discussed in the following section.
Pathways Inducing Hepcidin Expression
Inflammation can result in up‐regulation of hepcidin through the interleukin‐6 (IL‐6)–induced Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway (Fig. 3).( 18 ) The induction of hepcidin by lipopolysaccharide (LPS) is consistent with a decades‐old hypothesis by Weinberg, proposing that restricting availability of iron to pathogens represents an antimicrobial response.( 65 , 66 ) In the setting of acute or chronic inflammation, IL‐6 binds IL‐6 receptor‐α and gp130, activating JAKs and leading to the phosphorylation of STAT3.( 13 , 59 ) Subsequently, phospho‐STAT3 translocates to the nucleus and induces hepcidin production by binding to the hepcidin. However, the BMP‐SMAD pathway remains important in inflammation‐related hepcidin response.( 12 , 18 )
FIG. 3.
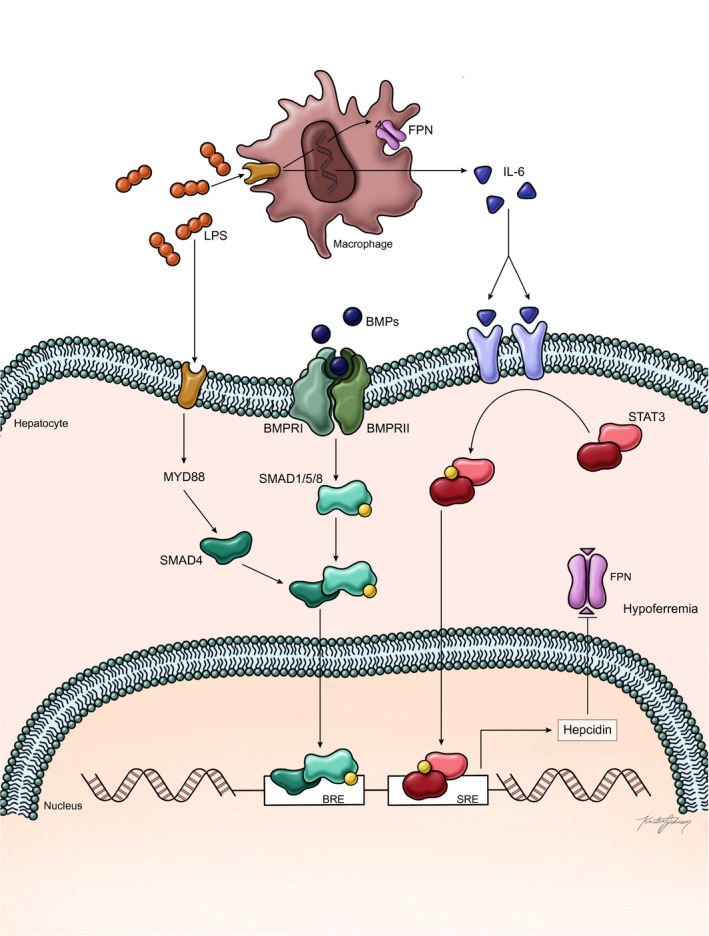
Increased hepcidin production in response to inflammation (adapted from Camaschella et al.( 2 )). The current evidence suggests that inflammation influences hepcidin expression primarily through the JAK‐STAT pathway. Increased LPS produced in the setting of infection or inflammation increases production of IL‐6 in macrophages through TLR4 binding. IL‐6 then binds to its receptor on hepatocytes and may bind directly to hepatocytes, resulting in activation of the JAK2‐STAT3 signaling pathway through phosphorylation of STAT3; phosphorylated STAT3 then enters the nucleus and increases hepcidin transcription by binding to the STAT3 responsive element on the hepcidin promoter. LPS‐activated myeloid differentiation protein 88 in hepatocytes may also increase SMAD signaling through SMAD4. In addition, it has been proposed from observations in animals that cross‐talk between BMP and STAT3 may also contribute to hepcidin expression. Abbreviations: BRE, BMP responsive element; FPN, ferroportin; MYD888, myeloid differentiation protein 88; SRE, STAT3 responsive element.
In the setting of endoplasmic reticulum (ER) stress and presence of misfolded or unfolded proteins in the ER, transcription of hepcidin is up‐regulated through promoter binding of CREBEL3 (cyclic adenosine monophosphate response element binding protein 3), alternatively known as CREBH (cyclic adenosine monophosphate response element binding protein H), although the BMP/SMAD pathway remains essential.( 67 , 68 , 69 ) Hepcidin transcription is also increased via nutrient and hormone gluconeogenic signals through the transcriptional cofactors CREBH and PPARGC1A (peroxisome proliferator‐activated receptor gamma coactivator 1 alpha).( 70 )
The tumor‐suppression gene, p53, also has a role in iron regulation.( 71 , 72 ) This occurs through the p53 response element (p53RE) found within the promoter region of the hepcidin gene HAMP.( 72 ) Activation of p53 leads to increased hepcidin mRNA levels in human hepatoma cells.( 72 ) It has been postulated that p53‐induced hepcidin expression may be an anti‐carcinogenic mechanism for this tumor‐suppressor gene.( 72 )
Pathways Associated With Reduced Hepcidin Expression
A major physiologic process that results in reduced hepcidin expression is erythropoiesis.( 12 , 18 ) The mechanism appears linked to a recently identified hormone erythroferrone (ERFE), which is produced by erythroblasts in response to erythropoietin (EPO) and acts directly in the liver to suppress hepcidin.( 73 ) Hepcidin suppression by blood loss, anemia, and erythropoiesis is mediated by ERFE, and the proposed mechanism is ERFE binding of BMP ligands.( 12 , 18 , 74 ) Increased ERFE levels were reported among patients with beta‐thalassemia and iron‐deficiency anemia.( 74 , 75 ) In summary, a substantial amount of data supports the action of ERFE as a key hormone acting directly on the liver to reduce hepcidin expression in response to erythropoietic stimuli, consistent with a logical physiological mechanism to increase iron absorption and release during times of a physiologic need to increase erythropoiesis. However, other factors are also likely involved, such as reduced iron stores as a consequence of increased erythropoietic demand in the bone marrow.
Hypoxia inducible factors (HIFs) are transcription factors that regulate the response to hypoxia.( 18 , 76 ) Under hypoxic conditions, posttranslational modulation of HIF results in translocation to the nucleus and binding to hypoxia‐responsive elements.( 76 ) In the liver, HIF‐2 activation suppresses hepcidin through EPO and possibly through direct binding of HIF‐1 to the hepcidin promoter, although it has also been proposed that the effect of hypoxia on hepcidin expression could be indirectly mediated by activation of ERFE and platelet‐derived growth factor–BB, due to erythropoiesis and by HIF‐1α binding to the TMPRSS‐6 promoter.( 76 , 77 , 78 ) An additional pathway by which hepcidin production is inhibited during hypoxic conditions occurs through furin, a processing enzyme usually found in the trans‐Golgi network.( 18 ) Furin acts by cleaving HJV to its soluble form, leading to the sequestration of BMP6 followed by suppression of hepcidin expression.( 79 )
Other Factors Affecting Hepcidin Expression
Several hormones and growth factors have been implicated in hepcidin regulation. It is well known that male patients with hemochromatosis have increased body iron stores than women.( 13 , 34 ) Although physiologic blood loss through menses is certainly likely to play a role, other factors have been implicated, such as testosterone‐mediated suppression of hepcidin production.( 80 ) There are conflicting data on the effect of estrogen on hepcidin expression; 17β‐estradiol was suggested to increase hepcidin expression in HepG2 cells in one study, while 17β‐estradiol was shown to decrease hepcidin production in human liver HuH7 and HepG2 cells.( 81 , 82 ) Progesterone was reported to increase hepcidin expression through PGRMC1 (progesterone receptor membrane component‐1).( 83 ) Other growth factors known to suppress hepcidin include hepatocyte growth factor and epidermal growth factor through the BMP‐SMAD pathway.( 84 ) Toll‐like receptor 4 (TLR4) activation in macrophages may be a source of hepcidin in response to infection.( 18 ) LPS‐driven TLR4 activation in hepatocytes results in hepcidin promoter binding through JNK.( 85 ) TLR4 may also activate nuclear factor kappa B (NF‐kB) signaling, which is also the putative mechanism by which alcohol suppresses hepcidin production and may be a mechanism explaining the increased hepatic iron stores frequently found in patients with alcohol‐associated liver disease.( 86 ) We recently showed that bone marrow–derived macrophages treated with iron demonstrated increased expression levels of macrophage M1 markers (CCL2 [chemokine (C‐C motif) ligand 2], CD14 [clusters of differentiation 14], iNOS [inducible nitric oxide synthase], IL‐1β, IL‐6, and TNF‐α [tumor necrosis factor α]) as well as increased protein levels, an effect that could be abrogated by desferrioxamine.( 87 ) Furthermore, iron loading of macrophages in the presence of IL‐4 led to down‐regulation of M2 markers (arginase‐1, Mgl‐1, and the M2‐specific transcriptional regulator, Kruppel‐like factor 4 [KLF4]) and reduced phosphorylation of STAT6, which also regulates M2 activation. In addition, patients with nonalcoholic steatohepatitis (NASH) with iron in RES cells showed increased hepatic gene expression of M1 markers and reduced gene expression of TGM2, an M2 marker, in comparison to patients with NASH with a hepatocellular iron pattern.( 87 ) RES iron deposition is observed in 10% of the NASH population and is associated with more severe disease.( 87 , 88 ) It is possible that TLR4‐related and NF‐kB‐related hepcidin production in response to repeated cycles of inflammation in NASH may explain the phenomenon of RES iron deposition in NASH.
Therapies Based on Hepcidin Biology
A greater understanding of HJV‐BMP‐SMAD pathways regulating hepcidin may provide therapeutic targets for drug development. Inhibitors of BMP‐SMAD signaling have been shown to improve anemia associated with inflammation and iron refractory iron deficiency anemia.( 12 ) Development of hepcidin mimetics for disorders of hepcidin deficiency such as HH are also being developed and in clinical trials.( 12 , 18 , 21 ) Due to the short half‐life of natural hepcidin, research is now focusing on methods to prolong its activity through hepcidin mimetics.( 89 , 90 ) Minihepcidins—small 7‐9 amino acid peptides designed to mimic hepcidin function—have been shown to prevent and reverse iron overload in a mouse model and hold potential for clinical trials.( 91 , 92 )
Potential additional future therapies include TMPRSS6 inhibitors.( 93 ) Because TMPRSS6 cleaves HJV, leading to reduced phosphorylation of the SMAD 1/5/8 complex and resulting in decreased hepcidin expression, inhibition of TMPRSS6 should therefore result in increased hepcidin production and decreased serum iron levels.
Conclusions
Investigation into the pathophysiology of iron signaling has identified hepcidin as the main regulator of iron metabolism. It is now clear that the liver is central to sensing the level of circulating iron stores and that hepcidin is the “master regulator” of iron status by controlling the amount of iron absorbed in enterocytes by its action on ferroportin. A complex interaction among HFE, TFR1, TFR2, and HJV expressed on the hepatocyte cell membrane is involved in sensing serum transferrin‐iron saturation, and increased iron levels induce hepcidin transcription through the SMAD 1/5/8 pathway. In addition, increased tissue iron stores are sensed by endothelial cells in the liver, which result in release of BMP2 and BMP6. BMPs form a complex on hepatocytes by binding to their receptors with HJV as a coreceptor. This complex results in phosphorylation of SMAD1/5/8, which then translocate to the nucleus and increase hepcidin transcription by promoter binding, resulting in blocking iron absorption and release.
Hepcidin levels are also influenced by infection, inflammation, erythropoiesis, and hypoxia. Infection and inflammation increase hepcidin levels, whereas erythropoiesis and hypoxia reduce hepcidin levels. “Anemia of chronic disease” is likely a phenomenon of unopposed hepcidin production due to ongoing chronic inflammation. Our understanding of the molecular biology and cell biology of hepcidin will allow for rational therapies using agonists or antagonists to hepcidin activity, and such compounds are already being studied in clinical trials.
Potential conflict of interest: Dr. Kowdley advises, is on the speakers’ bureau for, and received grants from Gilead and Intercept. He advises and received grants from HighTide. He consults for Altimmune, Roche, and Boeringer Ingelheim. He advises Assembly and Calliditas. He is on the speakers’ bureau for Abbvie. He received grants from Janssen, Allergan, Genfit, CymaBay, Novartis, Enanta, Protagonist, Pfizer, BMS, Celgene, Intercept, Madrigal, and Viking.
References
Author names in bold designate shared co‐first or second authorship.
- 1. Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr 2017;106(Suppl. 6):1559S‐1566S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020;105:260‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab 2015;22:777‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001;276:7806‐7810. [DOI] [PubMed] [Google Scholar]
- 5. Valore EV, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol Dis 2008;40:132‐138. 10.1016/j.bcmd.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090‐2093. [DOI] [PubMed] [Google Scholar]
- 7. Aschemeyer S, Qiao BO, Stefanova D, Valore EV, Sek AC, Ruwe TA, et al. Structure‐function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018;2018:899‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andrews NC. Disorders of iron metabolism [published correction appears in N Engl J Med. 2000;342(91):364]. N Engl J Med 1999;341:1986‐1995. [DOI] [PubMed] [Google Scholar]
- 9. Shayeghi M, Latunde‐Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, et al. Identification of an intestinal heme transporter. Cell 2005;122:789‐801. [DOI] [PubMed] [Google Scholar]
- 10. Muckenthaler MU, Galy B, Hentze MW. Systemic iron homeostasis and the iron‐responsive element/iron‐regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr 2008;28:197‐213. [DOI] [PubMed] [Google Scholar]
- 11. Hentze MW, Kuhn LC. Molecular control of vertebrate iron metabolism: mRNA‐based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci U S A 1996;93:8175‐8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiao X, Alfaro‐Magallanes VM, Babitt JL. Bone morphogenic proteins in iron homeostasis. Bone 2020;138:115495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parrow NL, Fleming RE. Liver sinusoidal endothelial cells as iron sensors. Blood 2017;129:397‐398. [DOI] [PubMed] [Google Scholar]
- 14. Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang R‐H, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad 7, Id1, and Atoh8 in the mouse liver. Blood 2008;15:1503‐1509. [DOI] [PubMed] [Google Scholar]
- 15. Meynard D, Kautz L, Darnaud V, Canonne‐Hergaux F, Coppin H, Roth MP. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet 2009;41:478‐481. [DOI] [PubMed] [Google Scholar]
- 16. Andriopoulos B Jr, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 2009;41:482‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Babitt JL, Zhang Y, Samad TA, Xia Y, Tang J, Campagna JA, et al. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co‐receptor. J Biol Chem 2005;19:29820‐29827. [DOI] [PubMed] [Google Scholar]
- 18. Rishi G, Subramaniam VN. Signaling pathways regulating hepcidin. Vitam Horm 2019;110:47‐70. [DOI] [PubMed] [Google Scholar]
- 19. Xu Y, Alfaro‐Magallanes VM, Babitt JL. Physiological and pathophysiological mechanisms of hepcidin regulation: clinical implications for iron disorders. Br J Haematol 2020. Dec 14. 10.1111/bjh.17252. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piperno A, Pelucchi S, Mariani R. Inherited iron overload disorders. Transl Gastroenterol Hepatol 2020;5:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kowdley KV, Brown KE, Ahn J, Sundaram V. ACG clinical guideline: hereditary hemochromatosis [published correction appears in Am J Gastroenterol 2019;114:1927]. Am J Gastroenterol 2019;2019:1202‐1218. [DOI] [PubMed] [Google Scholar]
- 22. Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates Hfe‐dependent regulation of hepcidin expression. Cell Metab 2008;7:205‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bennett MJ, Lebrón JA, Bjorkman PJ. Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor. Nature 2000;403:46‐53. [DOI] [PubMed] [Google Scholar]
- 24. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I‐like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996;13:399‐408. [DOI] [PubMed] [Google Scholar]
- 25. Traeger L, Enns CA, Krijt J, Steinbicker AU. The hemochromatosis protein HFE signals predominantly via the BMP type I receptor ALK3 in vivo. Commun Biol 2018;1:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet 2006;38:531‐539. [DOI] [PubMed] [Google Scholar]
- 27. Wang C‐Y, Babitt JL. Liver iron sensing and body iron homeostasis. Blood 2019;133:18‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Canali S, Zumbrennen‐Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017;129:405‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koch PS, Olsavszky V, Ulbrich F, Sticht C, Demory A, Leibing T, et al. Angiocrine Bmp2 signaling in murine liver controls normal iron homeostasis. Blood 2017;129:415‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiao X, Dev S, Canali S, Bayer A, Xu Y, Agarwal A, et al. Endothelial bone morphogenetic protein 2 (Bmp2) knockout exacerbates hemochromatosis in homeostatic iron regulator (Hfe) knockout mice but not Bmp6 knockout mice. Hepatology 2020;72:642‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu XG, Wang Y, Wu Q, Cheng WH, Liu W, Zhao Y, et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 2014;124:1335‐1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolondi G, Garuti C, Corradini E, Zoller H, Vogel W, Finkenstedt A, et al. Altered hepatic BMP signaling pathway in human HFE hemochromatosis. Blood Cells Mol Dis 2010;45:308‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ryan JD, Ryan E, Fabre A, Lawless MW, Crowe J. Defective bone morphogenic protein signaling underlies hepcidin deficiency in HFE hereditary hemochromatosis. Hepatology 2010;52:1266‐1273. [DOI] [PubMed] [Google Scholar]
- 34. Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet 2016;388:706‐716. [DOI] [PubMed] [Google Scholar]
- 35. Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G–> A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002;359:211‐218. [DOI] [PubMed] [Google Scholar]
- 36. Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, et al. Iron‐overload‐related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221‐230. [DOI] [PubMed] [Google Scholar]
- 37. Barton JC, McLaren CE, Chen WP, Ramm GA, Anderson GJ, Powell LW, et al. Cirrhosis in hemochromatosis: independent risk factors in 368 HFE p.C282Y homozygotes. Ann Hepatol 2018;17:871‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jacolot S, Le GG, Scotet V, Quere I, Mura C, Ferec C. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood 2004;103:2835‐2840. [DOI] [PubMed] [Google Scholar]
- 39. Le Gac G, Scotet V, Ka C, Gourlaouen I, Bryckaert L, Jacolot S, et al. The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Hum Mol Genet 2004;13:1913‐1918. [DOI] [PubMed] [Google Scholar]
- 40. Pietrangelo A, Caleffi A, Henrion J, Ferrara F, Corradini E, Kulaksiz H, et al. Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes. Gastroenterology 2005;128:470‐479. [DOI] [PubMed] [Google Scholar]
- 41. Pelucchi S, Mariani R, Trombini P, Coletti S, Pozzi M, Paolini V, et al. Expression of hepcidin and other iron‐related genes in type 3 hemochromatosis due to a novel mutation in transferrin receptor‐2. Haematologica 2009;94:276‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kent P, Wilkinson N, Constante M, Fillebeen C, Gkouvatsos K, Wagner J, et al. Hfe and Hjv exhibit overlapping functions for iron signaling to hepcidin. J Mol Med 2015;93:489‐498. [DOI] [PubMed] [Google Scholar]
- 43. Wu Q, Wang H, An P, Tao Y, Deng J, Zhang Z, et al. HJV and HFE play distinct roles in regulating hepcidin. Antioxid Redox Signal 2015;22:1325‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Papanikolaou G, Samuels ME, Ludwig EH, MacDonald MLE, Franchini PL, Dubé M‐P, et al. Mutations in HFE2 cause iron overload in chromosome 1q‐linked juvenile hemochromatosis. Nat Genet 2004;36:77‐82. [DOI] [PubMed] [Google Scholar]
- 45. Casanovas G, Mleczko‐Sanecka K, Altamura S, Hentze MW, Muckenthaler MU. Bone morphogenetic protein (BMP)‐responsive elements located in the proximal and distal hepcidin promoter are critical for its response to HJV/BMP/SMAD. J Mol Med 2009;87:471‐480. [DOI] [PubMed] [Google Scholar]
- 46. Kong X, Xie L, Zhu H, Song L, Xing X, Yang W, et al. Genotypic and phenotypic spectra of hemojuvelin mutations in primary hemochromatosis patients: a systematic review. Orphanet J Rare Dis 2019;14:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003;33:21‐22. [DOI] [PubMed] [Google Scholar]
- 48. Piperno A, Bertola F, Bentivegna A. Juvenile hemochromatosis. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., eds. GeneReviews®. Seattle, WA: University of Washington; 2005. Feb 17 [updated 2020 Jan 9]:1993‐2021. [PubMed] [Google Scholar]
- 49. Sandhu K, Flintoff K, Chatfield MD, Dixon JL, Ramm LE, Ramm GA, et al. Phenotypic analysis of hemochromatosis subtypes reveals variations in severity of iron overload and clinical disease. Blood 2018;132:101‐110. [DOI] [PubMed] [Google Scholar]
- 50. Corradini E, Buzzetti E, Pietrangelo A. Genetic iron overload disorders [published online ahead of print, 2020 Sep 7]. Mol Aspects Med 2020;75:100896. [DOI] [PubMed] [Google Scholar]
- 51. Hamdi‐Rozé H, Ben Ali Z, Ropert M, Detivaud L, Aggoune S, Simon D, et al. Variable expressivity of HJV related hemochromatosis: “Juvenile” hemochromatosis? Blood Cells Mol Dis 2019;74:30‐33. [DOI] [PubMed] [Google Scholar]
- 52. Gammella E, Buratti P, Cairo G, Recalcati S. The transferrin receptor: the cellular iron gate. Metallomics 2017;9:1367‐1375. [DOI] [PubMed] [Google Scholar]
- 53. Fillebeen C, Charlebois E, Wagner J, Katsarou A, Mui J, Vali H, et al. Transferrin receptor 1 controls systemic iron homeostasis by fine‐tuning hepcidin expression to hepatocellular iron load. Blood 2019;133:344‐355. [DOI] [PubMed] [Google Scholar]
- 54. Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor‐like family. J Biol Chem 1999;274:20826‐20832. [DOI] [PubMed] [Google Scholar]
- 55. Schmidt PJ, Fleming MD. Transgenic HFE‐dependent induction of hepcidin in mice does not require transferrin receptor‐2. Am J Hematol 2012;87:588‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rishi G, Crampton EM, Wallace DF, Subramaniam VN. In situ proximity ligation assays indicate that hemochromatosis proteins Hfe and transferrin receptor 2 (Tfr2) do not interact. PLoS One 2013;8:e77267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wallace DF, Summerville L, Crampton EM, Frazer DM, Anderson GJ, Subramaniam VN. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009;50:1992‐2000. [DOI] [PubMed] [Google Scholar]
- 58. Corradini E, Schmidt PJ, Meynard D, Garuti C, Montosi G, Chen S, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology 2010;139:1721‐1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. D’Alessio F, Hentze MW, Muckenthaler MU. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane‐associated protein complex for hepcidin regulation. J Hepatol 2012;57:1052‐1060. [DOI] [PubMed] [Google Scholar]
- 60. Wallace DF, Summerville L, Crampton EM, Subramaniam VN. Defective trafficking and localization of mutated transferrin receptor 2: implications for type 3 hereditary hemochromatosis. Am J Physiol Cell Physiol 2008;294:C383‐C390. [DOI] [PubMed] [Google Scholar]
- 61. Camaschella C, Roetto A, Calì A, De Gobbi M, Garozzo G, Carella M, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000;25:14‐15. [DOI] [PubMed] [Google Scholar]
- 62. Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood 2005;105:1803‐1806. [DOI] [PubMed] [Google Scholar]
- 63. Piperno A, Roetto A, Mariani R, Pelucchi S, Corengia C, Daraio F, et al. Homozygosity for transferrin receptor‐2 Y250X mutation induces early iron overload. Haematologica 2004;89:359‐360. [PubMed] [Google Scholar]
- 64. Ravasi G, Rausa M, Pelucchi S, Arosio C, Greni F, Mariani R, et al. Transferrin receptor 2 mutations in patients with juvenile hemochromatosis phenotype. Am J Hematol 2015;90:E226‐E227. [DOI] [PubMed] [Google Scholar]
- 65. Weinberg ED. Nutritional immunity. Host’s attempt to withhold iron frommicrobial invaders. JAMA 1975;231:39‐41. [DOI] [PubMed] [Google Scholar]
- 66. Weinberg ED. Infection and iron metabolism. Am J Clin Nutr 1977;1977:1485‐1490. [DOI] [PubMed] [Google Scholar]
- 67. Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, et al. ER stress controls iron metabolism through induction of hepcidin. Science 2009;325:877‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, et al. STAT3 is required for IL‐6‐gp130‐dependent activation of hepcidin in vivo. Gastroenterology 2007;132:294‐300.Erratum in: Gastroenterology 2007;132:1208. [DOI] [PubMed] [Google Scholar]
- 69. Canali S, Vecchi C, Garuti C, Montosi G, Babitt JL, Pietrangelo A. The SMAD pathway is required for hepcidin response during endoplasmic reticulum stress. Endocrinology 2016;157:3935‐3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vecchi C, Montosi G, Garuti C, Corradini E, Sabelli M, Canali S, et al. Gluconeogenic signals regulate iron homeostasis via hepcidin in mice. Gastroenterology 2014;146:1060‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gao J, Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: the mechanisms involved in inhibiting cell‐cycle progression. Blood 2001;98:842‐850. [DOI] [PubMed] [Google Scholar]
- 72. Weizer‐Stern O, Adamsky K, Margalit O, Ashur‐Fabian O, Givol D, Amariglio N, et al. Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br J Haematol 2007;138:253‐262. [DOI] [PubMed] [Google Scholar]
- 73. Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism [published correction appears in Nat Genet 2020;52:463]. Nat Genet 2020;2014:678‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ganz T, Jung G, Naeim A, Ginzburg Y, Pakbaz Z, Walter PB, et al. Immunoassay for human serum erythroferrone. Blood 2017;130:1243‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. El Gendy FM, El‐Hawy MA, Shehata AMF, Osheba HE. Erythroferrone and iron status parameters levels in pediatric patients with iron deficiency anemia. Eur J Haematol 2018;100:356‐360. [DOI] [PubMed] [Google Scholar]
- 76. Renassia C, Peyssonnaux C. New insights into the links between hypoxia and iron homeostasis. Curr Opin Hematol 2019;26:125‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ravasi G, Pelucchi S, Buoli Comani G, Greni F, Mariani R, Pelloni I, et al. Hepcidin regulation in a mouse model of acute hypoxia. Eur J Haematol. 2018;100:636‐643. [DOI] [PubMed] [Google Scholar]
- 78. Maurer E, Gütschow M, Stirnberg M. Matriptase‐2 (TMPRSS6) is directly up‐regulated by hypoxia inducible factor‐1: identification of a hypoxia‐responsive element in the TMPRSS6 promoter region. Biol Chem 2012;393:535‐540. [DOI] [PubMed] [Google Scholar]
- 79. Silvestri L, Pagani A, Camaschella C. Furin‐mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood 2008;111:924‐931. [DOI] [PubMed] [Google Scholar]
- 80. Latour C, Kautz L, Besson‐Fournier C, Island M‐L, Canonne‐Hergaux F, Loréal O, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology 2014;59:683‐694. [DOI] [PubMed] [Google Scholar]
- 81. Ikeda Y, Tajima S, Izawa‐Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, et al. Estrogen regulates hepcidin expression via GPR30‐BMP6‐dependent signaling in hepatocytes. PLoS One 2012;7:e40465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang Q, Jian J, Katz S, Abramson SB, Huang X. 17β‐Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half‐site. Endocrinology 2012;153:3170‐3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li X, Rhee DK, Malhotra R, Mayeur C, Hurst LA, Ager E, et al. Progesterone receptor membrane component‐1 regulates hepcidin biosynthesis. J Clin Invest 2016;126:389‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology 2012;56:291‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin‐1 and interleukin‐6. Proc Natl Acad Sci U S A 2005;102:1906‐1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zmijewski E, Lu S, Harrison‐Findik DD. TLR4 signaling and the inhibition of liver hepcidin expression by alcohol. World J Gastroenterol 2014;20:12161‐12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Handa P, Thomas S, Morgan‐Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol 2019;105:1015‐1026. [DOI] [PubMed] [Google Scholar]
- 88. Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp‐Arida A; Nonalcoholic Steatohepatitis Clinical Research Network . Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011;53:448‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu J, Sun B, Yin H, Liu S. Hepcidin: a promising therapeutic target for iron disorders: a systematic review. Medicine 2016;95:e3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Blanchette NL, Manz DH, Torti FM, Torti SV. Modulation of hepcidin to treat iron deregulation: potential clinical applications. Expert Rev Hematol 2016;9:169‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, et al. Minihepcidins prevent iron overload in a hepcidin‐deficient mouse model of severe hemochromatosis. Blood 2012;120:3829‐3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood 2018;131:1790‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, et al. Reducing TMPRSS6 ameliorates hemochromatosis and β‐thalassemia in mice. J Clin Invest 2013;123:1531‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]