

Abstract
Objective:
The apolipoprotein E (APOE) E4 isoform is the strongest genetic risk factor for sporadic Alzheimer disease (AD). Although APOE is predominantly expressed by astrocytes in the central nervous system, neuronal expression of APOE is of increasing interest in age-related cognitive impairment, neurological injury, and neurodegeneration. Here, we show that endogenous expression of E4 in stem-cell–derived neurons predisposes them to injury and promotes the release of phosphorylated tau.
Methods:
Induced pluripotent stem cells from 2 unrelated AD patients carrying the E4 allele were corrected to the E3/E3 genotype with the CRISPR/Cas9 system and differentiated into pure cultures of forebrain excitatory neurons without contamination from other cells types.
Results:
Compared to unedited E4 neurons, E3 neurons were less susceptible to ionomycin-induced cytotoxicity. Biochemically, E4 cells exhibited increased tau phosphorylation and ERK1/2 phosphoactivation. Moreover, E4 neurons released increased amounts of phosphorylated tau extracellularly in an isoform-dependent manner by a heparin sulfate proteoglycan–dependent mechanism.
Interpretation:
Our results demonstrate that endogenous expression of E4 by stem-cell–derived forebrain excitatory neurons predisposes neurons to calcium dysregulation and ultimately cell death. This change is associated with increased cellular tau phosphorylation and markedly enhanced release of phosphorylated tau. Importantly, these effects are independent of glial APOE. These findings suggest that E4 accelerates spreading of tau pathology and neuron death in part by neuron-specific, glia-independent mechanisms.
The apolipoprotein E (APOE) E4 allele, the strongest genetic risk factor for sporadic Alzheimer disease (AD), differs from the risk-neutral E3 allele by a single nucleotide polymorphism (SNP).1,2 E4 patients exhibit greater brain atrophy, accumulation of hyperphosphorylated tau protein, and deposition of amyloid, albeit by unclear mechanisms.3–6 Although most APOE is expressed by astrocytes in the brain,7–9 neuronal APOE is of increasing interest in age-related cognitive impairment, neurological injury, and neurodegeneration.10–12 Disease modeling using isogenic stem cells has demonstrated that, compared to E3, expression of E4 leads to distinct transcriptomic differences in multiple neural cell types and increases tau phosphorylation in neurons.13,14 However, the effects of APOE genotype on neuronal viability and tau release are unknown. Using genome editing and a reductionist human stem cell culture approach, we show that endogenous expression of E4 predisposes pure cultures of forebrain excitatory neurons to injury and promotes release of phosphorylated tau (p-tau). These findings suggest that neuronal APOE can accelerate brain atrophy and the spreading of tau pathology in E4-carrying AD patients independently of glia.
Subjects and Methods
Reprogramming and Culture of Stem Cells
Fibroblast lines from patients diagnosed with AD (AD1, AG11414; AD2, AG05810) were described previously15 and obtained from Coriell Institute for Medical Research. Cells were reprogrammed through Sendai-virus–mediated overexpression of Oct4, Sox2, Klf4, and c-myc (Thermo Fisher Scientific, Waltham, MA) per the manufacturer’s instructions. Reprogrammed cells were expanded on gamma-irradiated mouse embryonic fibroblasts (mEFs) in human induced pluripotent stem cell (hiPSC) medium (Dulbecco modified Eagle medium [DMEM]/F-12, 20% Knockout Serum Replacement [Thermo Fisher Scientific], 1% MEM nonessential amino acids [Thermo Fisher Scientific], 1% Glutamax [Thermo Fisher Scientific], and 0.1% 2-mercaptoethanol [Thermo Fisher Scientific]). Cells were visually inspected, and areas of differentiation were removed by manual dissection. Individual clones were manually picked, expanded in feeder-free conditions on Matrigel (Corning Incorporated, Corning, NY) in mTeSR1 (STEMCELL Technologies, Vancouver, BC, Canada), and passaged with ReLeSR (STEMCELL Technologies). Karyotyping (WiCell Research Institute, Madison, WI) was performed, and live cells were stained with Tra 1–81 and SSEA4 (R&D Systems, Minneapolis, MN) to confirm pluripotency prior to cryopreservation in mFrESR (STEMCELL Technologies). Cells were used from passages 5 to 8 after reprogramming for gene editing and passages 8 to 20 for differentiation.
Generation of Isogenic Cell Lines
Genome editing was performed as previously described16 with modifications. Guide RNA (gRNA) sequence was designed using an online Web tool (crispr.mit.edu), and was cloned into px458 plasmid (Addgene plasmid # 48138, gift of F. Zhang). Prior to editing, cells were dissociated to single-cell suspension with Accutase (Millipore, Billerica, MA), plated in 1:1 mixture of mTeSR1 (STEMCELL Technologies) and mEF-conditioned hiPSC medium supplemented with 10 ng/ml FGF-2 (R&D Systems) and 10 μM ROCK Inhibitor Y-27632 (Stemgent, Cambridge, MA). The next day, cells were cultured in medium without ROCK Inhibitor and grown for 4 to 5 days. Cells were washed with DPBS−/− (Sigma, St Louis, MO), dissociated to single-cell suspension with Accutase, and pelleted. Two million cells were resuspended in 95 μl of Human Nucleofector Solution 1 (Lonza, Basel, Switzerland), 2 μg of px458 containing APOE gRNA, and 100 μM of ssODN template (IDT, Coralville, IA), then transfected with Nucleofector IIb (Lonza) using program B-016. Cells were immediately treated with prewarmed, prebalanced mTeSR1 and transferred to a 10 cm Matrigel-coated dish in media plus 10 μM ROCK Inhibitor Y-27632. After 48 hours, green fluorescent protein–positive cells were sorted by flow cytometry (Aria; BD Bioscicences, Franklin Lakes, NJ) and plated at clonal density in media. Individual clones were picked and screened for editing by polymerase chain reaction (PCR) amplification (New England BioLabs, Ipswich, MA) and direct Sanger sequencing (ACGT, Wheeling, IL). Correctly edited clones were expanded in mTeSR1. Karyotyping was performed, and live cells were stained with anti-Tra-1–81 and anti-SSEA4 (R&D Systems) to confirm maintained pluripotency prior to cryopreservation in mFrESR (STEMCELL Technologies). In silico predicted off-target sites17 were directly sequenced.
Preparation of Lentivirus
Lentiviruses were generated in HEK293T cells using psPAX2 and pMD2.G, as described previously.18 rtTA plasmid was a gift of E. Kiskinis, and pTetO-Ngn2-puro was a gift of M. Wernig (Addgene plasmid # 52047).
Neuronal Differentiation
Cells were differentiated to forebrain excitatory neurons via lentiviral-mediated overexpression of neurogenin-2, as previously described19 with modifications to remove non-neuronal sources of APOE. hiPSCs was dissociated to single cells using Accutase (Millipore), and resuspended in mTeSR1 with 10 μM ROCK Inhibitor Y-27632 with lentiviruses encoding rtTA and pTetO-Ngn2-puro. The next day, medium was changed to KO-DMEM (Thermo Fisher Scientific), 1× MEM nonessential amino acids, 1× Glutamax, and 0.1% 2-mercaptoethanol, supplemented with 10 μM SB431542 (Stemgent), 100 nM LDN193189 (Stemgent), 2 μM XAV939 (Tocris Bioscience, Bristol, UK), and 3 μg/ml doxycycline (Sigma). Medium was gradually changed over 2 days to neural induction medium (DMEM/F-12, MEM nonessential amino acids, Glutamax, D-glucose [Sigma], N-2 [Thermo Fisher Scientific], 2 μg/ml heparin sulfate (Sigma), supplemented with 3 μg/ml doxycycline and 2 μg/ml puromycin [Sigma]). Induced neurons were replated onto tissue culture plates precoated with poly-L-ornithine (Sigma) and laminin (Roche, Basel, Switzerland) and cultured in neuronal maturation medium (BrainPhys Basal Medium [STEMCELL Technologies], B-27 and N-2 Supplements [Thermo Fisher Scientific], MEM nonessential amino acids, and Glutamax, supplemented with 3 μg/ml doxycycline and 10 ng/ml BDNF [R&D Systems]). After replating to eliminate proliferating cells, 2.75 μM Ara-C was used for 2 days. A 50% exchange with fresh neuronal maturation medium was performed every 2 to 3 days thereafter.
Neuronal Morphometry
Live neuronal cultures at 38 days postinduction were imaged by automated phase contrast microscopy with the IncuCyte S3 Live Cell Analysis System (Sartorius Corporation, Edgewood, NY). Somata were manually counted. Neurite length and neurite branch points were determined using automated morphometric analysis on phase contrast images with the IncuCyte NeuroTrack software module (Sartorius Corporation) as previously described20 using the following parameters: segmentation mode = brightness; segmentation adjustment = 0.2; hole fill = 0; adjust size = 0; minimum cell width = 7 μm; neurite filtering = best; neurite sensitivity = 0.6; neurite width = 2 μm. Neurite length and neurite branches were normalized to the number of somata per field, and the value for each well of a culture plate was determined by the average of at least 3 fields per well. To measure cell survival, live neuronal cultures at 38 days postinduction were treated with propidium iodide (Thermo Fisher Scientific), and cells were again visualized by epifluorescence microscopy using the IncuCyte S3 Live Analysis System. Positive cells were counted using automated morphometric analysis on epifluorescence images with the IncuCyte Basic Analyzer software module using the following parameters: segmentation adjustment = 0; minimum area = 300 μm2, maximum eccentricity = 0.97. The value for each well of a culture plate was determined by the average of at least 3 fields per well.
Immunoblotting
Cellular proteins were solubilized in M-PER (Bio-Rad Laboratories, Hercules, CA), and protein concentrations were determined by Pierce Rapid Gold BCA protein assay kit (Thermo Fisher Scientific), subjected to electrophoresis on 4 to 20% Tris-Glycine gels (Bio-Rad Laboratories), and transferred to polyvinylidene difluoride (Millipore). For APOE immunoblotting, recombinant human APOE3 protein and negative control lysate (RayBiotech, Norcross, GA) were used to generate a standard curve. Human adult brain lysate (Novus Biologicals, Littleton, CO) was used as a positive control. Antibodies used were goat anti-APOE (Millipore), rabbit anti–ß3-tubulin (Abcam, Cambridge, MA), donkey antigoat Alexa Fluor 680 (Thermo Fisher Scientific), and donkey antirabbit IRDye 800CW (LI-COR Biosciences, Lincoln, NE). Two exposures are shown for select lanes. Membranes were blocked in 5% normal donkey serum (Jackson ImmunoResearch, West Grove, PA), and signal was detected with an Odyssey CLx Imaging System (LI-COR Biosciences). For all other immunoblots, membranes were blocked with Blotto (Thermo Fisher Scientific) in Tris-buffered saline (Bio-Rad Laboratories). Primary antibodies were mouse anti–total tau (HT7; Thermo Fisher Scientific), mouse anti–p-tau (AT8; Thermo Fisher Scientific), rabbit anti-ERK1/2 (Cell Signaling Technology, Danvers, MA), rabbit anti–phospho-ERK1/2 (Thr202/Tyr204, Cell Signaling Technology), rabbit anti-GSK3ß (Cell Signaling Technology), rabbit anti–phospho-GSK3ß (Ser9, Cell Signaling Technology), anti–amyloid precursor protein (APP; Karen; a gift of V. Lee), and mouse anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore). Secondary antibodies were donkey antimouse horseradish peroxidase (HRP), donkey antirabbit HRP, or donkey antigoat HRP (Cell Signaling Technology). Membranes were incubated with enhanced chemiluminescent substrate (Thermo Fisher Scientific) and developed to film (GE Healthcare Life Sciences, Logan, UT). Quantitation of all blots was performed with ImageJ (NIH, Bethesda, MD).
Immunocytochemistry
Cells were grown on precoated glass coverslips (Neuvitro Corporation, Vancouver, WA) and fixed in 4% paraformaldehyde solution in phosphate-buffered saline. Cells were blocked with bovine serum albumin and normal serum from the species specific to the primary antibodies. Primary antibodies used were mouse IgG2b anti-TuJ1 (Sigma), chicken anti–glial fibrillary acidic protein (GFAP; Sigma), rabbit anti-S100ß (Dako Corporation, Carpinteria, CA), guinea pig anti-vGluT1 (Synaptic Systems, Göttingen, Germany), mouse anti–Tra-1–60 (Thermo Fisher Scientific), goat anti-Oct3/4 (Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit anti-Sox2 (Millipore). Fluorophore-conjugated donkey secondary antibodies against the primary antibody host species IgG were from Thermo Fisher Scientific. Nuclei were counterstained with DAPI (Thermo Fisher Scientific), and cells were mounted using Prolong Gold media (Thermo Fisher Scientific). Cell counts were performed in ImageJ, and values for each genotype and patient background were determined by the average of 3 fields per coverslip and 3 coverslips.
Enzyme-Linked Immunosorbent Assay
Cells were washed with basal media and then incubated for 4 days with a precise volume of fresh complete media. An aliquot of the cell culture supernatant was collected, mixed with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific), and centrifuged to remove cellular debris. Where indicated, cells were pretreated with 50 mM sodium chlorate (NaClO3, Sigma). Enzyme-linked immunosorbent assays were performed using kits from Thermo Fisher Scientific for Aß40, Aß42, p-tau Thr181, p-tau Ser199, p-tau Thr231, p-tau Ser396, and total tau per the manufacturer’s instructions.
Ionomycin Toxicity Assay
Cells were treated with dimethylsulfoxide (DMSO) or 7.5 μM ionomycin (Tocris Bioscience) in complete media, unless otherwise indicated. Cells were visualized by live phase-contrast microscopy using the IncuCyte S3 system as described above while in culture for 48 hours. At the end of the time course, cells were treated with media containing propidium iodide (Thermo Fisher Scientific), and cells were again visualized by epifluorescence microscopy using IncuCyte S3. Morphometric analysis was performed in Neurotrack and Basic Analyzer software (Sartorius Corporation) as described above.
Statistics
All values are reported as mean ± standard error of the mean. All statistical comparisons were between edited E3/E3 neurons of a single patient background and their respective unedited, isogenic, E4/E3 neurons, unless otherwise indicated for drug treatment experiments. The number of patient backgrounds, gene edited stem cell lines, and independent experimental replicates was chosen based on previously published studies.21–25 A 2-tailed unpaired Student t test was used to assess significance between 2 groups. Where multiple analytes were measured from the same sample, the level of significance was corrected for multiple comparisons by the Holm–Sidak method. A 2- or 3-way analysis of variance followed by Sidak multiple comparisons test was used to assess interactions between multiple variables. All statistical calculations were performed with Prism 8 (GraphPad Software, San Diego, CA) software. A probability level of p < 0.05 is considered significant for all tests.
Results
Human induced pluripotent stem cells (hiPSCs) were generated from fibroblasts from 2 unrelated individuals (AD1, AD2) with an APOE E4/E3 genotype, the most frequent genotype in the AD population.26 Neurons derived from these hiPSCs were previously shown to exhibit increased amyloid processing, tau phosphorylation, and vulnerability to calcium dysregulation compared to neurons derived from control patient hiPSCs.15,27 To determine whether the E4 allele influences this cellular phenotype, both E4/E3 hiPSC lines were corrected to E3/E3 genotype by overexpression of Cas9 nuclease, a single guide RNA sequence (sgRNA) targeted to the terminal exon of the gene, and a homology-directed repair template encoding a segment of the E3 allele (Fig 1). Genotypes of the untargeted and edited cell lines were confirmed by PCR amplification and direct Sanger sequencing. Editing did not alter the expression of markers of embryonic stem cells, including Tra-1–60, Oct3/4, or Sox2, and edited hiPSCs exhibited a normal karyotype. The top five in silico predicted off-target binding sites17 of the sgRNA were PCR-amplified and sequenced, and both edited cell lines were isogenic to their parent lines except for the intended SNP correction in the APOE locus.
FIGURE 1:
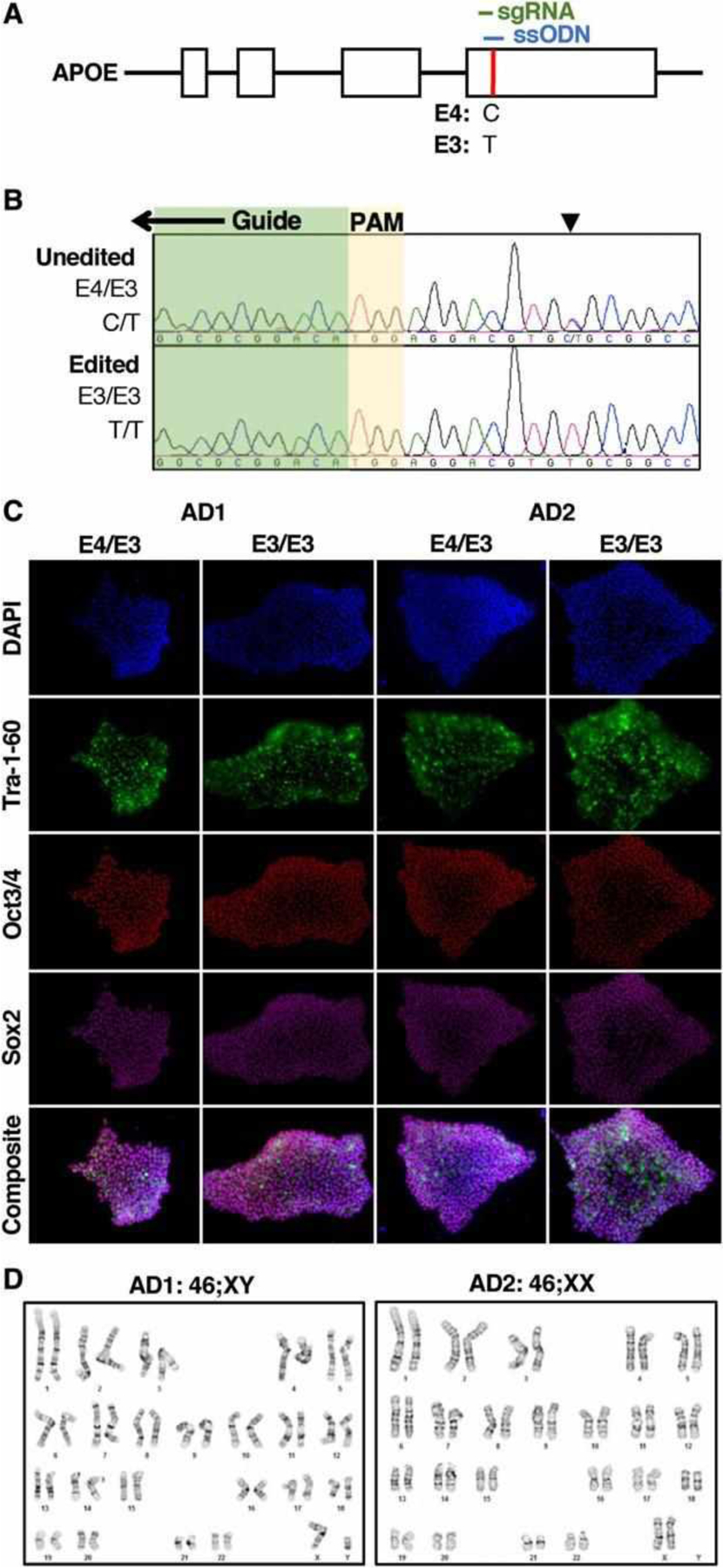
Genome editing of E4/E3 human induced pluripotent stem cells (hIPSCs). (A) The APOE gene’s E3 and E4 alleles are defined by a single nucleotide polymorphism (red) in its terminal exon. The nucleotide code for the E4 and E3 alleles are depicted below. sgRNA (green) = single guide RNA; ssODN (blue), single-stranded oligodeoxynucleotide template for homology-directed repair. (B) Representative chromatograms depicting the targeted editing of a cell line with the E4/E3 genotype (C/T) to E3/E3 (T/T) at the indicated single nucleotide polymorphism (arrowhead). A portion of the guide sequence (green) and the positionally adjacent motif (PAM) are indicated. (C) hiPSCs derived from Alzheimer disease patient fibroblasts with E4/E3 genotype expressed characteristic markers of embryonic stem cells. Editing hiPSCs to the E3/E3 genotype did not alter expression of pluripotency markers. (D) Editing iPSCs to the E3/E3 genotype did not alter their karyotype.
To assess whether human stem-cell–derived neurons express APOE endogenously, hiPSCs were differentiated to forebrain excitatory neurons via induced overexpression of neurogenin-2,19 with strict modifications to eliminate non-neuronal sources of APOE including glia and fetal bovine serum. Under these stringent conditions, cells expressed ß3-tubulin (TuJ1) and vesicular glutamate transporter 1 (vGluT1), while lacking detectable expression of GFAP or S100ß, demonstrating that hiPSCs differentiated into pure glutamatergic neurons without glial contamination (Fig 2). Isogenic E4/E3 and E3/E3 neurons differentiated with comparable efficiency and survival. There were no differences in soma density or average neurite length per cell in isogenic E4/E3 and E3/E3 neurons. However, E3/E3 neurons exhibited an increased number of neurite branch points per cell compared to unedited isogenic E4/E3 neurons, consistent with previous reports of murine neurons in vivo.28,29
FIGURE 2:
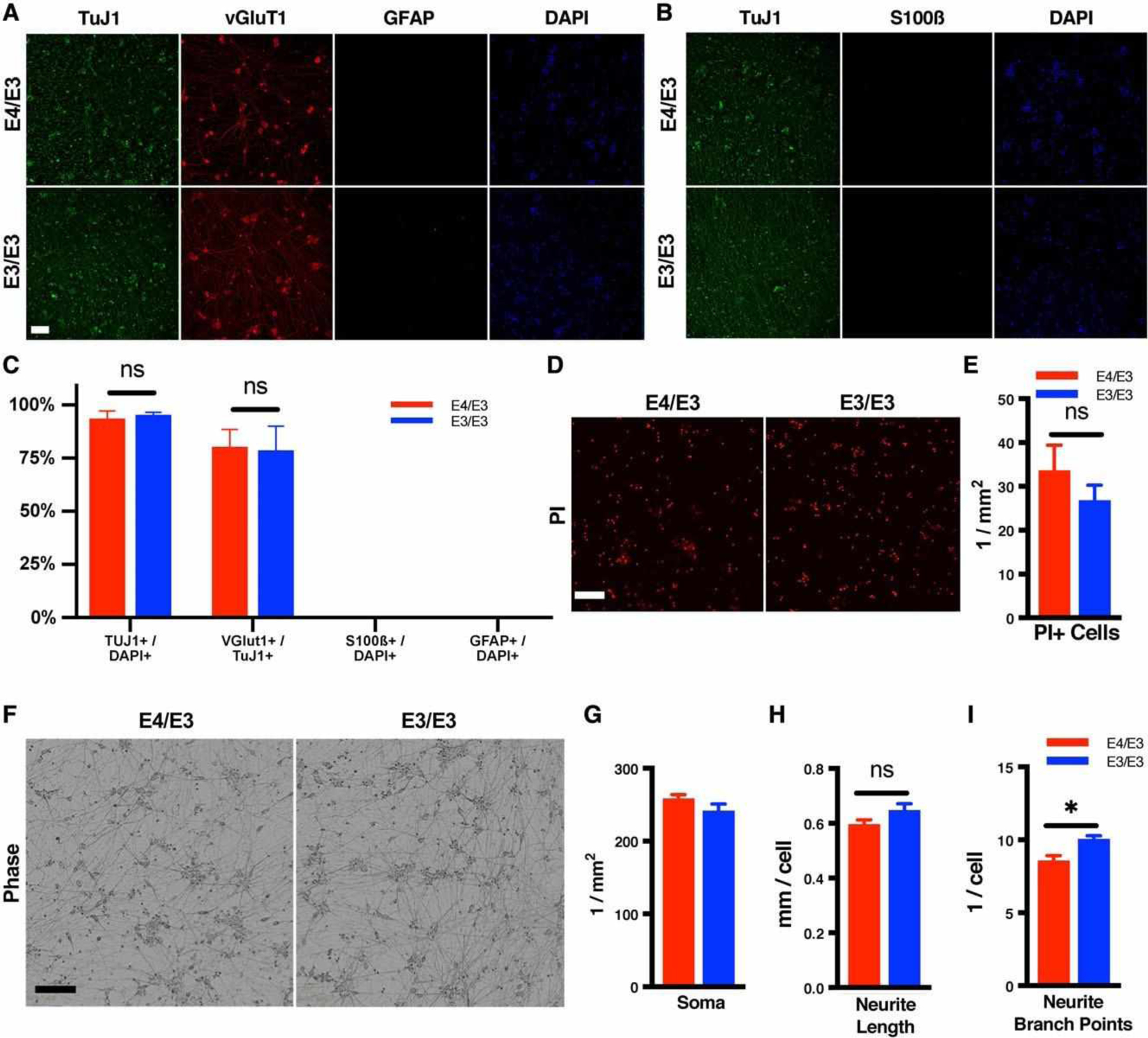
Isogenic E4/E3 and E3/E3 human induced pluripotent stem cells (hiPSCs) differentiate to forebrain excitatory neurons with comparable efficiency, survival, and morphology. (A, B) Representative photomicrographs of immunostained neuronal cultures with no glial contamination. Cultures were fixed at 38 days postinduction. Nuclei were counterstained with DAPI. (C) Editing iPSCs to the E3/E3 genotype did not alter the differentiation efficiency. Differentiation of isogenic E4/E3 and E3/E3 iPSCs yielded comparable proportions of vGlut1+TuJ1+ neurons. There were n = 3 independent differentiations per genotype. Each data point is the average of 3 fields per coverslip and 3 coverslips per differentiation. (D) Representative photomicrographs of propidium iodide (PI) labeling of dead cells in isogenic E4/E3 and E3/E3 neurons at 38 days postinduction (E) Quantitation of PI+ dead cells from D. There were n = 4 independent differentiations per genotype. Each data point is the average of 3 fields per differentiation. (F) Representative phase contrast photomicrographs of isogenic E4/E3 and E3/E3 neurons at 38 days postinduction. (G–I) Morphometric analysis of images in F. There were n = 4 independent differentiations per genotype. Each data point is the average of 3 fields per differentiation. For all measures, the statistical test was a 2-tailed unpaired Student t test, with Holm–Sidak correction for multiple comparisons: *p < 0.05; ns = not significant. Scale bar = 200 μM..
Physiologic phosphorylation of cellular tau protein (p-tau) and secretion of 40- and 42-amino acid peptides of amyloid ß (Aß40, Aß42) both steadily increased in our cultures (Fig 3). GAPDH expression increased on the same time course as expected for maturing neurons,30 and all comparative protein expression analyses were performed on lysates from synchronously differentiated cultures. APOE was detected in the cellular lysate of neurons. The doublet pattern of immunoreactivity mirrored that of human adult brain homogenate and likely represents sialylation, as has been reported previously.31,32 Recombinant human APOE3 was used to identify full-length unmodified APOE and generate a standard curve. Isogenic E4/E3 and E3/E3 neurons exhibited comparable full-length APOE protein.
FIGURE 3:
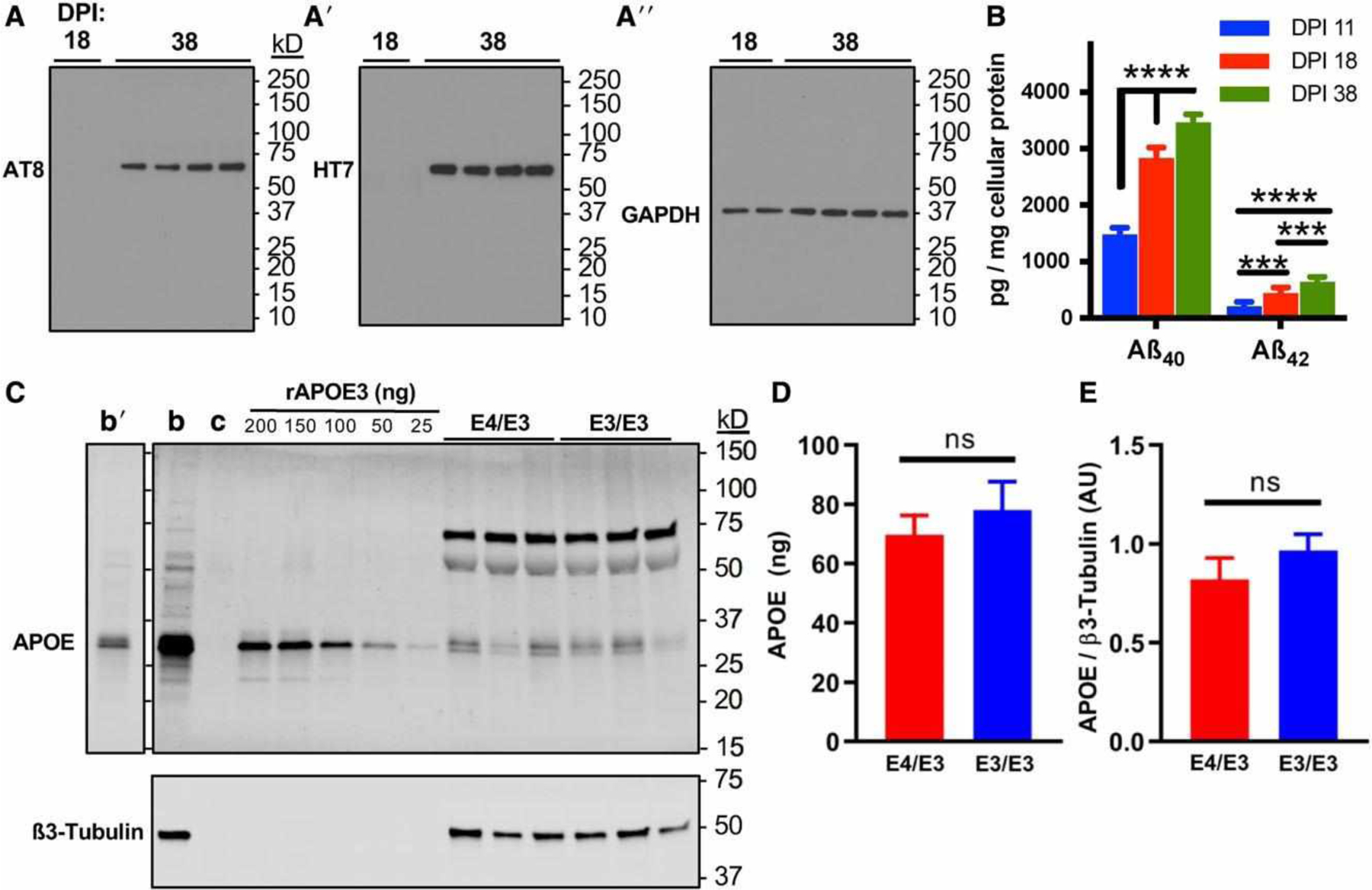
Forebrain excitatory neurons express p-tau, Aß, and apolipoprotein E (APOE). (A) Immunoblotting of p-tau detected by AT8, total tau detected by HT7, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) during the maturation of neuronal cultures at 18 and 38 days postinduction (DPI). Blots were stripped and probed sequentially. (B) Aß40 and Aß42 peptides were detected by peptide-specific immunoassay in the cell culture supernatant of developing and mature neurons by 11 DPI, and secretion increased over time until plateauing by 38 DPI. There were n = 3 independent differentiations. Each data point is the average of 3 measurements per differentiation. The statistical test was a 2-way repeated measures analysis of variance followed by Tukey range test: ***p < 0.001, ****p < 0.0001. (C) Isogenic E4/E3 and E3/E3 neurons at 38 DPI express APOE. Representative immunoblot depicts APOE with ß3-tubulin to control for neuronal protein loading. b = positive control human adult brain lysate; c = negative control lysate of HEK293 cells transfected with an empty vector. Recombinant APOE3 (rAPOE3) purified from HEK293 cells was serially diluted to generate a standard curve to quantify the expression of APOE in lysates from E4/E3 and E3/E3 neurons. Human brain lysates are also depicted at a shorter exposure time (b0). (D, E) Absolute (D) and relative (E) quantitation of immunoblotting experiments in C. There were n = 6 independent differentiations. The statistical test was 2-tailed unpaired Student t test: ns = not significant.
We previously reported that AD neurons exhibit an exaggerated glutamate-induced intracellular calcium response and increased susceptibility to calcium dysregulation compared to neurons of cognitively unimpaired patients.15 In view of the effects of neuronal APOE in aging,10,11 we used glia-free cultures of forebrain excitatory neurons to examine possible effects of neuronal APOE on cell viability. Cells were treated with DMSO vehicle or ionomycin, a potent calcium ionophore, for 48 hours, and neuronal morphology was observed by live phase-contrast microscopy. In both AD patient backgrounds, ionomycin reduced neurite length, whereas DMSO had no significant effect; however, neurite retraction was significantly reduced in E3/E3 neurons compared to similarly treated isogenic E4/E3 neurons (Fig 4A, C). To confirm this glia-independent effect of APOE isoform, neurons were treated with various doses of ionomycin for 48 hours, and dead cells then were labeled with propidium iodide. Concordant with changes in neuritic morphology, ionomycin was significantly less cytotoxic to E3/E3 neurons than to isogenic E4/E3 neurons at every treatment dose (see Fig 4B, D). These findings demonstrate that endogenous E4 sensitizes human neurons to calcium dysregulation and neurotoxic insult, suggesting that neuronal APOE, independent of glial APOE, is a modifier of genetic risk for and progression of AD.
FIGURE 4:
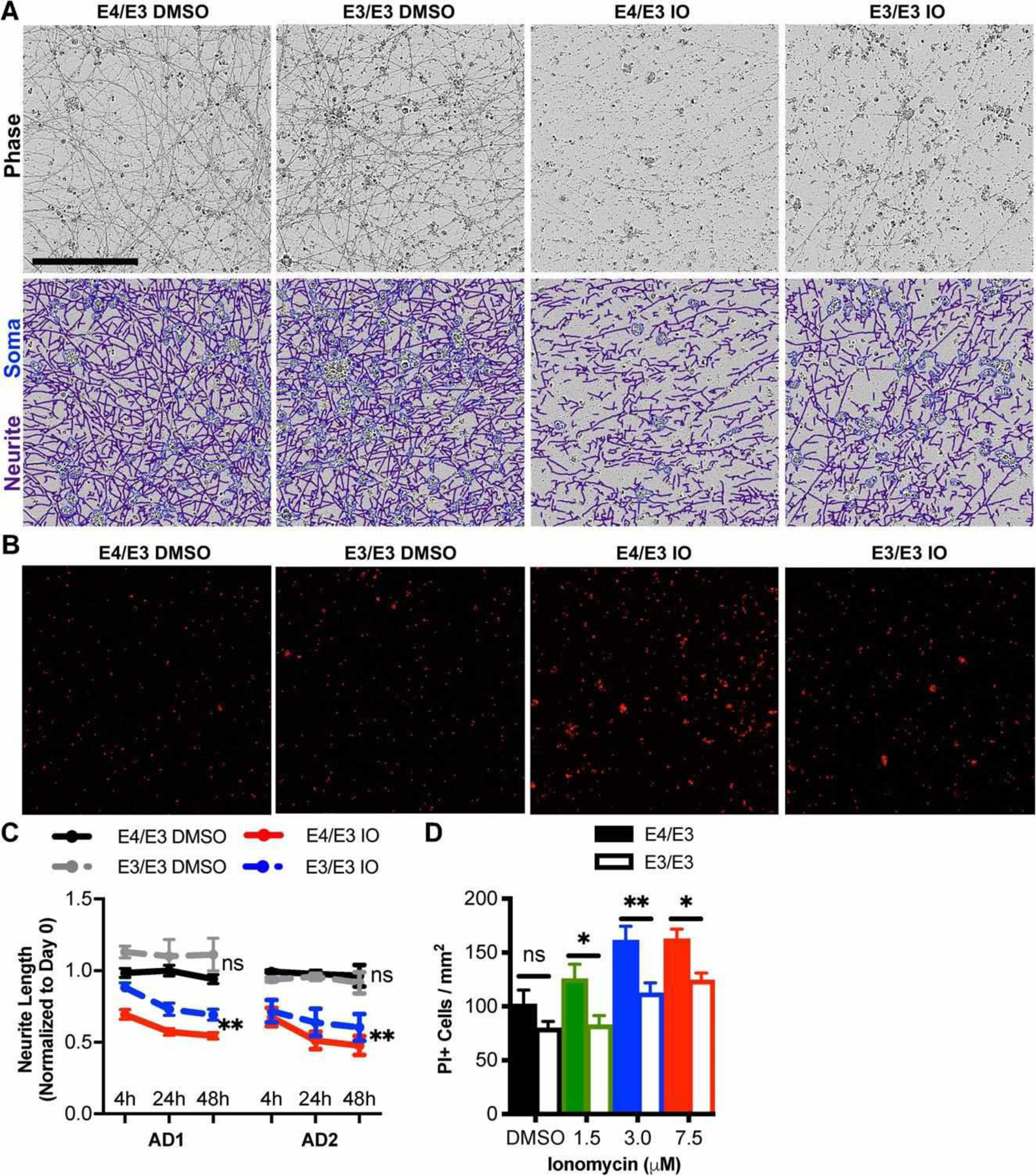
Genetic editing of E4 to E3 protects neurons from ionomycin-induced toxicity. (A) Phase contrast images of apolipoprotein E (APOE)-isogenic neurons were captured 0, 4, 24, and 48 hours after treatment with ionomycin (IO) or dimethylsulfoxide (DMSO) vehicle. Depicted are representative phase contrast images at 24 hours and corresponding neurite (purple) and cell body (blue) masks beneath. Scale bar = 200 μm. (B) Propidium iodide (PI) epifluorescence labeling dead cells were counted after cultures were treated for 48 hours with 1.5, 3.0, or 7.5 μM IO or DMSO vehicle. Depicted are representative images treated with 7.5 μM IO or DMSO vehicle. (C) Quantitation of neurite lengths from A. There were n = 4 independent treatments per genotype and patient background. Each data point is the average of 3 fields per treatment. The statistical test was a 2-way repeated measures analysis of variance for interaction between APOE genotype and time for each drug treatment: **p < 0.01; ns = not significant. Not all statistical comparisons are shown for clarity. (D) Quantitation of dead cells from B. There were n = 12 independent treatments per concentration and genotype. The statistical test was 2-way analysis of variance followed by Sidak multiple comparison test for each concentration: *p < 0.05, **p < 0.01.
Alterations in tau and Aß are both central to AD pathogenesis and are separately associated with calcium dysregulation in multiple model systems.33 Therefore, we next assessed whether neuronal APOE genotype altered tau phosphorylation or amyloid processing. There were no differences in total tau expression between genotypes, but in both patient backgrounds, the cellular p-tau/tau ratio was significant reduced in E3/E3 neurons compared to isogenic E4/E3 neurons (Fig 5). Contrastingly, APOE genotype did not alter the ratio of secreted Aß42/Aß40 or cellular expression of APP. Tau phosphorylation can result from multiple kinase signaling cascades, including calcium-dependent activation of ERK1/2 and Aß-induced activation of GSK3ß.34 Compared to isogenic E4/E3 neurons, E3/E3 neurons exhibited a significant reduction in the ratio of phosphoactivated to total ERK1/2 in both patient backgrounds but no change in the phosphoactivation of GSK3ß (Fig 6). There was no difference in the total level of expression of either kinase. This finding demonstrates that independent of injury, there is an isoform-dependent and neuron-specific effect of endogenous E4 on kinase activation and tau phosphorylation.
FIGURE 5:
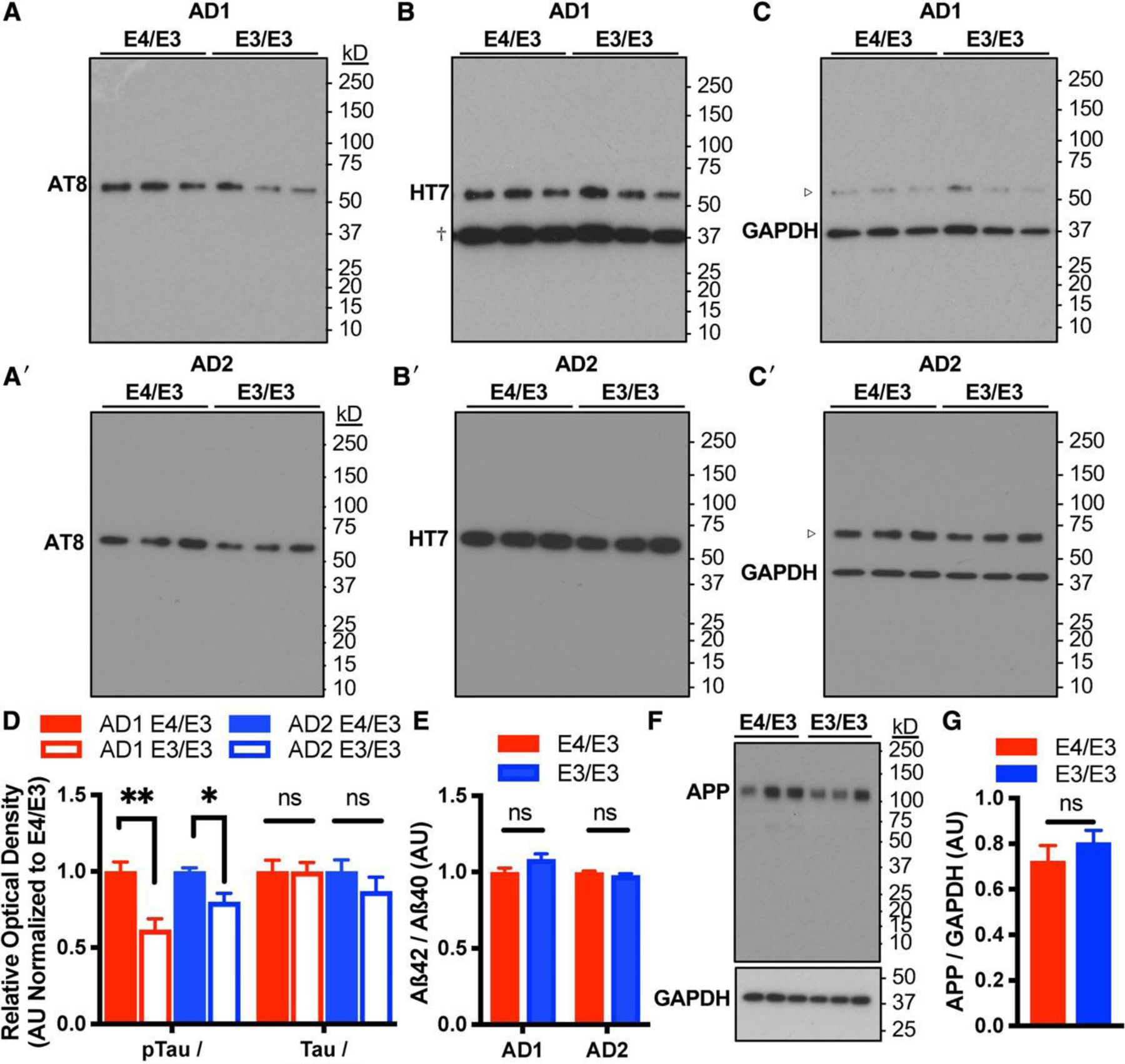
Genetic editing of E4 to E3 reduces tau phosphorylation but not amyloid processing. (A–C) Representative immunoblotting of lysates of isogenic E4/E3 and E3/E3 neurons from AD1 and AD2 for p-tau (AT8) and total tau (HT7). Blots were stripped and probed sequentially to distinguish phosphorylated and total forms of tau. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to control for protein loading. HT7 and GAPDH were detected simultaneously. Dagger indicates GAPDH immunoreactivity depicted at a longer exposure time not used for quantitation. Open arrowhead indicates HT7 immunoreactivity depicted at a shorter exposure time not used for quantitation. (D) Quantitation of p-tau/tau ratio and total tau expression. Measures are normalized to the E4/E3 genotype within each patient background. There were n = 6 independent differentiations per genotype and patient background. The statistical test was Student t test between isogenic E4/E3 and E3/E3 neuronal lysates with Sidak correction for multiple comparisons: *p < 0.05, **p < 0.01; ns = not significant. (E) Calculation of the secreted Aß42/Aß40 ratio in the conditioned media of isogenic E4/E3 and E3/E3 neurons detected by enzyme-linked immunoassay. There were n = 5 independent differentiations per genotype and patient background. Each data point is the average of 3 measurements per differentiation. (F) Representative immunoblotting of lysates of isogenic E4/E3 and E3/E3 neurons for amyloid precursor protein (APP; detected with Karen antibody). GAPDH was used to control for protein loading. (G) Quantitation of APP immunoblotting in F. There were n = 6 independent differentiations per genotype. The statistical test was 2-tailed unpaired Student t test.
FIGURE 6:
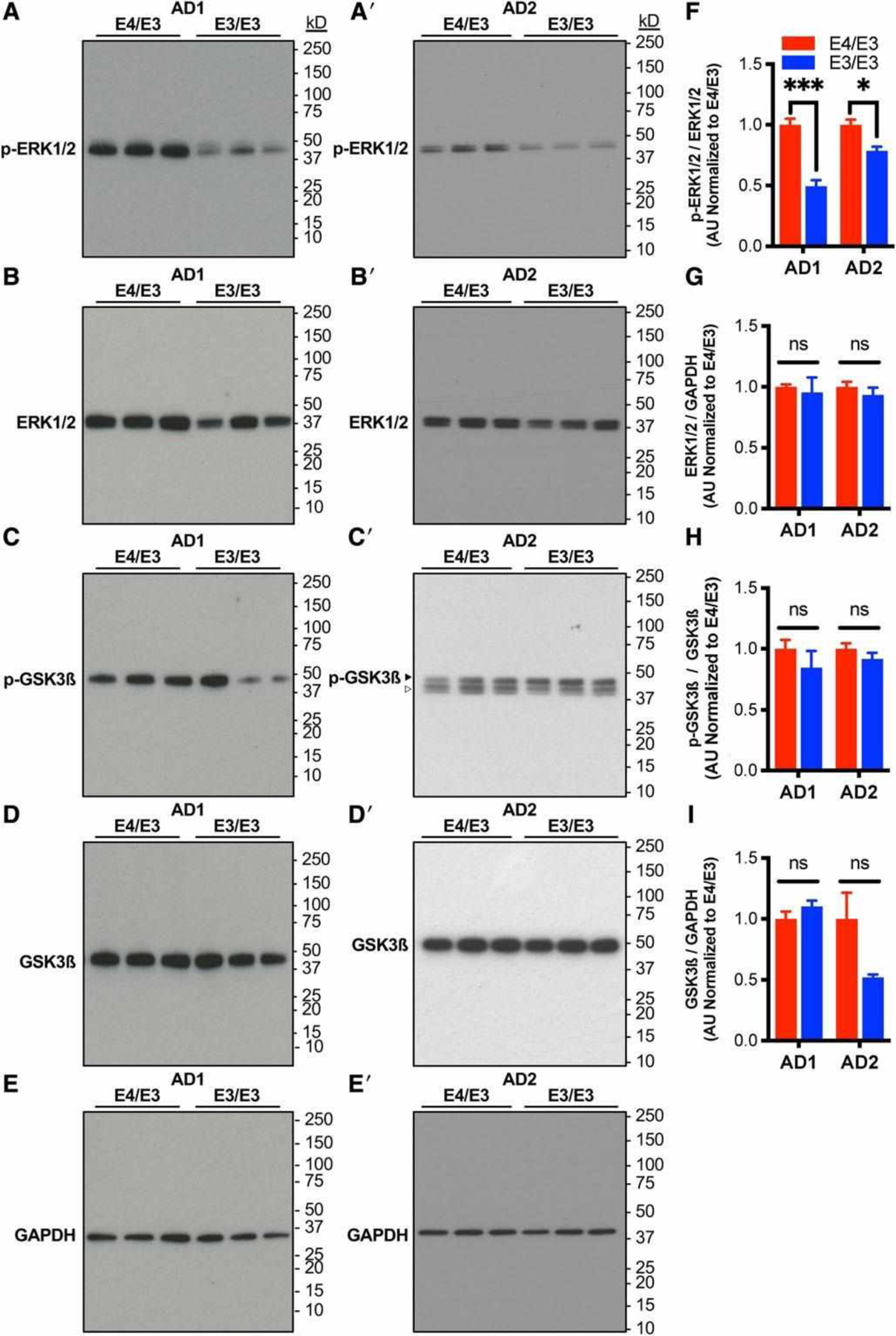
Neurons expressing E4 exhibit increased phosphoactivation of ERK1/2 but not GSK-3ß. (A–E) Representative immunoblotting of lysates of isogenic E4/E3 and E3/E3 neurons from AD1 and AD2 with antibodies directed against phosphorylated or total ERK1/2 and GSK3ß. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control for protein loading. Blots were stripped and probed sequentially. Open arrowhead indicates residual ERK1/2 immunoreactivity not used for quantitation. (F–I) Quantitation of phosphokinase/total kinase ratio and total kinase expression from A–E. There were n = 6 independent differentiations per genotype and patient background. Measures are normalized to the E4/E3 genotype within each patient background. The statistical test was Student t test between isogenic E4/E3 and E3/E3 neuronal lysates with Sidak correction for multiple comparisons: *p < 0.05, ***p < 0.001; ns = not significant.
The anatomically stereotyped and progressive spread of p-tau pathology is a fundamental feature of AD and is accelerated in E4-carriers.35,36 P-tau released by neurons into the cerebrospinal fluid (CSF) is a predictor of progressive cognitive impairment,37 and presymptomatic patients with the E4-allele exhibit an accelerated p-tau biomarker trajectory compared with E4-noncarriers.38 Given that E4 increased cellular tau phosphorylation at baseline in our glia-free neuronal cultures, we next examined whether tau release is altered similarly. Strikingly, in both patient backgrounds, the p-tau/tau ratio in the culture media was reduced in E3/E3 neurons compared to isogenic E4/E3 neurons for the Thr181, Thr231, and Ser396 phosphoepitopes (Fig 7A). In CSF, the presence of these epitopes distinguishes between AD and non-AD pathology.39–41 There was no difference in the level of secretion of total tau protein between the genotypes. We were not able to detect the Ser199 epitope in the cell culture supernatant of either E4/E3 or E3/E3 neurons. There were no differences in cell number or cell death (see Fig 2D–G).
FIGURE 7:
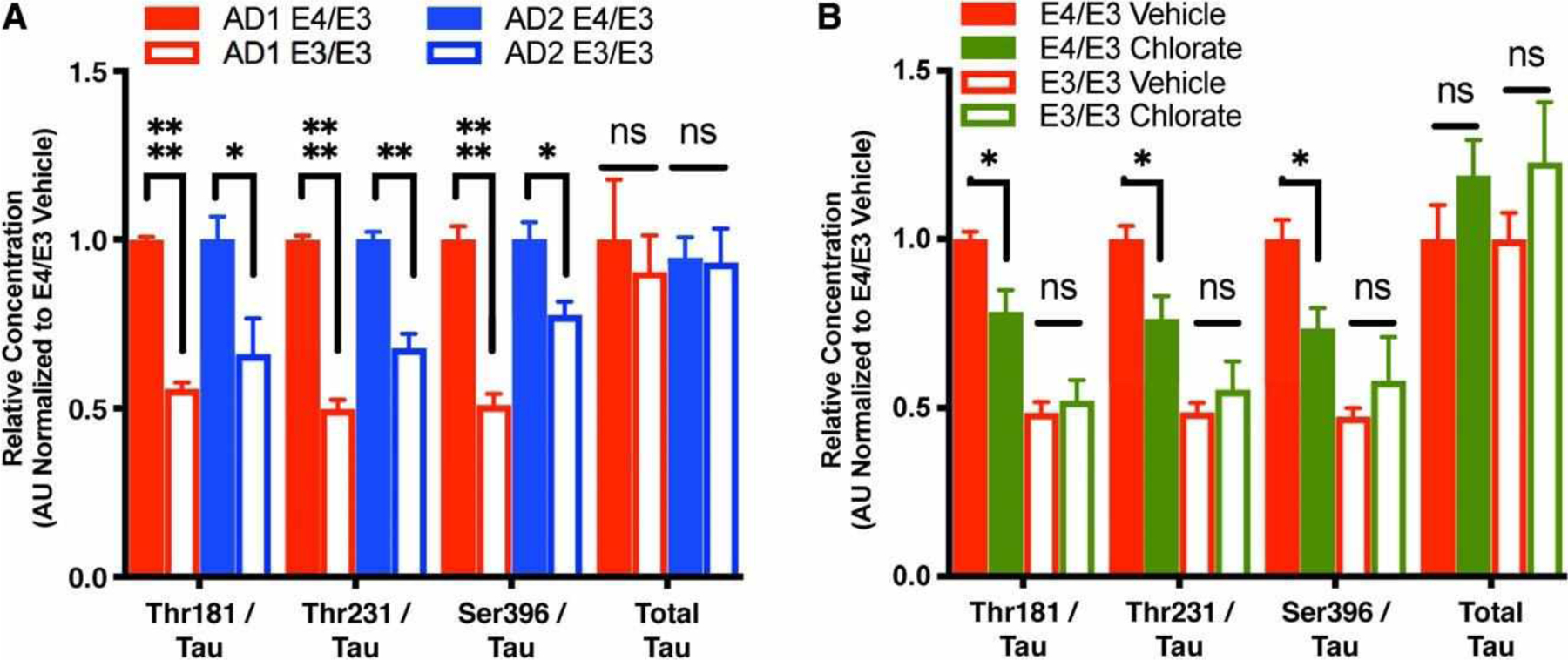
Neuronal E4 increases p-tau release by a heparin sulfate proteoglycan–dependent mechanism. (A) Calculation of p-tau/tau ratio for each phosphoepitope and total secreted tau in cell culture supernatant of isogenic E4/E3 and E3/E3 neurons from AD1 and AD2. Measures are normalized to the E4/E3 genotype within each patient background. There were n = 4 biologically independent differentiations per genotype and patient background. Each data point is the average of 3 measurements per differentiation. A 3-way interaction between genotype, analyte, and patient background by 3-way analysis of variance was not significant (F3, 56 = 0.3128, p = 0.8160); however, there was a main effect of apolipoprotein E (APOE) genotype (F1, 56 = 66.39, p < 0.0001). The analysis was therefore followed up with 2-tailed unpaired Student t tests between isogenic E4/E3 and E3/E3 neurons for each analyte, with Holm–Sidak correction for multiple comparisons: *p < 0.05, **p < 0.01, ****p < 0.0001; ns = not significant. (B) Calculation of p-tau/tau ratio for each phosphoepitope and total secreted tau in cell culture supernatant of isogenic APOE neurons after 3-day treatment with 50 mM sodium chlorate or saline vehicle. There were n = 3 independent treatments per genotype. Each data point is the average of 3 measurements per treatment. A 3-way interaction between genotype, chlorate treatment, and analyte by 3-way analysis of variance was not significant (F3, 32 = 0.7656, p = 0.5217); however, there was a significant interaction between APOE genotype and chlorate treatment (F1, 32 = 8.666, p = 0.006). The analysis was therefore followed up with 2-way analyses of variance for interaction between APOE genotype and treatment for each analyte, followed by Sidak multiple comparison test within each genotype. *p < 0.05.
We sought to determine whether this finding simply reflected the overall E4-associated increase of cellular p-tau, or whether E4 also facilitated tau release. In a heterologous expression system, p-tau is released extracellularly by type I unconventional protein secretion, an exosome-independent process dependent on cellular production of heparin sulfate proteoglycans (HSPGs).42 Pretreatment of E4/E3 neurons with sodium chlorate, which inhibits HSPG biosynthesis,43 reduced the p-tau/tau ratio for all 3 phosphoepitopes Fig. 7B. Intriguingly, similarly treating isogenic E3/E3 neurons had no effect suggesting that APOE participates in HSPG-dependent p-tau release in an isoform-dependent manner. Chlorate treatment had no effect on total tau protein secretion in either genotype, suggesting that this mechanism of release is specific to p-tau. Taken together, these data suggest that neuronal E4 may contribute to increased tau pathology in AD patients by 2 distinct mechanisms: increased cellular kinase activity and more rapid neuronal release of p-tau.
Discussion
Human stem-cell–based models of AD recapitulate pathophysiologic hallmarks of the disease, including amyloid alterations, tau hyperphosphorylation, and calcium dysregulation, even in the absence of overexpressed mutant alleles that precipitate amyloidosis or tauopathy.15,44–48 Given this feature, we leveraged the abilities to precisely edit the genome with the CRISPR/Cas9 system and to differentiate hiPSCs into a specific neural subtype to assess whether neuronal expression of APOE contributes to the AD cellular phenotype.
Most APOE in the central nervous system is synthesized and secreted by astrocytes. Neuronalization of a human neural cancer cell line was associated with a decrease of APOE expression suggesting that neurons do not express APOE.49 However, APOE also has been detected by in situ hybridization and immunofluorescence in primary human neurons in a manner indicative of de novo protein synthesis.12,50 Corroborating this, we find that APOE is expressed by human stem-cell–derived forebrain excitatory neurons. Furthermore, genetic correction of E4 to E3 did not alter the level of APOE expression in neurons, so the genotype-associated differences in cytotoxicity and tau phenotypes observed in the isogenic lines are attributable to APOE isoform-specific effects. Of note, when the human APOE alleles are expressed in both neuronal and non-neuronal cells under the control of the endogenous murine promoter, E4 protein expression is reduced compared to E3.51 The difference between the 2 systems may be due to a cell-type–specific effect at the post-transcriptional or post-translational level, as has been described in other model systems.47,52 Further experimentation is required to determine whether the differential effects of the E4 and E3 alleles involve gene regulatory elements, post-translational modifications, APOE secretion, or receptor-mediated signaling.
At baseline, E4 neurons exhibited decreased neurite branching per soma compared to isogenic E3 neurons, with no difference in other morphological characteristics. This finding is consistent with a report in murine cortical neurons in which expression of human E4 was associated with reduced dendritic complexity and arborization compared to E3-expressing littermates.28 The mouse study also noted that E4 reduced apical but not basal dendrite length. In our 2-dimensional culture system, there is no obvious cellular polarity, and we did not detect a difference in average neurite length per soma between APOE genotypes. Separately, it was shown that the effect of APOE genotype on neurite branching requires neuronal but not astrocytic expression.29 Taken together, these data suggest that APOE regulates neurite complexity in a cell-intrinsic and isoform-dependent manner, and in future studies, it may be possible to assess whether the gene exerts a similar effect on the length of subsets of neurites of human neurons cultured in a 3-dimensional system.
Although there were no differences between cultures in neurite length at baseline, when cells were subjected to ionomycin-induced calcium dysregulation, E4 neurons exhibited a marked increase in neurite retraction and cell death compared to genetically corrected E3 neurons. Effects of APOE isoform on calcium homeostasis have heretofore been attributed to differential binding of glial APOE on neuronal APOE receptors.53 Our observations indicate that endogenous expression of E4 in neurons sensitizes them to calcium dysregulation, independently of glial APOE. This finding offers an interesting human correlate to murine models that suggest a toxic gain-of-function role of E4 in neuronal injury and aging.10,11,54
Genetic editing of APOE also led to multiple distinct effects on tau. We observed an E4-associated increase in tau phosphorylation in pure cultures of forebrain excitatory neurons in both patient backgrounds. Our findings are consistent with previous reports differentiating hiPSCs into mixed cultures of glutamatergic and GABAergic neurons with a minority population of astrocytes.14,55 Of the many kinases for which tau is a substrate, we found that E4 was associated with increased phosphoactivation of ERK1/2. ERK1/2 interactions are strongly modulated by intracellular calcium,56 and increased activity of the signaling cascade is consistent with our hypothesis that E4 cells are more sensitive to calcium dysregulation.
In addition, E4 is associated with a remarkable increase in release of p-tau from neurons compared to isogenic E3 neurons, and we show for multiple epitopes a reduction in p-tau after treatment with sodium chlorate to inhibit HSPG biosynthesis. Because p-tau is more readily able to spread trans-synaptically,57,58 the increase in cellular p-tau may itself increase p-tau release. However, because sodium chlorate treatment had no effect on p-tau release in E3 neurons, our findings raise the interesting possibility that E4 specifically recruits this additional mechanism. Once in the extracellular milieu, p-tau would be accessible to other neurons, either by direct uptake or via other intermediates such as microglia.59,60 In AD, it has been argued that the highly stereotyped anatomical progression of tau pathology may be attributable to a prion-like, trans-synaptic spreading of tau assemblies,61 and phosphomimetic tau enhances this process in a transgenic mouse model.62 The more rapid spread of tau pathology observed in AD patients with the E4 allele36,37 may therefore be attributable in part to an enhanced release of p-tau by neuronal APOE.
Clinically, E4-carriers exhibit greater amyloid burden. Recombinant E4 has been reported to increase Aß production by receptor-mediated effects,63 and in transgenic mouse models, E4 promotes the seeding of amyloid plaques.4,5,63 Aß can in turn induce tau phosphorylation via GSK3ß activation.64 In our neuronal cultures, genetic correction of E4 did not increase APP expression, Aß42/Aß40 ratio, or GSK3ß phosphoactivation. Therefore, these findings suggest that neuronal APOE genotype does not influence amyloid processing or secretion, and furthermore, that the observed effect of APOE on tau in our cultures is likely to be amyloid-independent. The vast majority of Aß and APOE in the central nervous system is released into the extracellular milieu by neurons and astrocytes, respectively, where they interact in an isoform-dependent manner.65 The effect of APOE on amyloid processing, clearance, and plaque formation is therefore more likely to be a distinct function of glial APOE. Resolving the effects of APOE isoforms in different cell types in vitro will ultimately require a combinatorial coculture model with human neuronal and glial subtypes. The APOE-associated cellular tau phenotype of forebrain excitatory neurons is consistent with reports using alternative hiPSC differentiation strategies yielding mixed populations of neural cells.13,14 Taken together, these findings suggest that E4 increases tau phosphorylation by a cell-intrinsic mechanism. In contrast, APOE’s effect on neuronal Aß depends highly on cellular context; genetic correction of E4 to E3 had no effect in our pure excitatory neuronal cultures but reduced Aß secretion in mixed excitatory and inhibitory neuronal cultures.13 This suggests that an effect of E4 on Aß release may be activity-dependent.
Discrepancies in these stem-cell–based models stress the importance of cross-validation with intact neural systems. Our morphometric observations both at baseline and in response to calcium dysregulation are consistent with a murine model in which E4 is overexpressed in neurons. However, there is currently no murine AD model in which neuronal APOE’s effect on human tau can be modelled in vivowithout covarying glial APOE. Conditional expression models, in which either human E3 or E4 expression is restricted to a single neural cell type,10 may prove useful for this purpose when crossed with a knockin line expressing human wild-type tau or a tau variant associated with familial tauopathy.66 Importantly, our findings only demonstrate a role for neuronal APOE in AD pathophysiology, and editing of hiPSCs from nondemented control patients will be required to determine whether these effects are generalizable to aging or other disorders.
In summary, although it is widely believed that astrocytic and microglial APOE mediates the gene’s effect on neurodegeneration in AD,52,67 we demonstrate that endogenous expression of E4 in neurons is sufficient to alter multiple AD-relevant cellular pathways. Notably, E4-expressing human neurons are more susceptible to neurotoxic insult. Moreover, neuronal E4 increases both tau phosphorylation and its release, possibly explaining the more rapid progression of tau pathology and cognitive impairment experienced by E4-carying patients. Our observations indicate that APOE predisposes neurons to injury and accelerates neurodegeneration, at least in part, by a glia-independent mechanism. Almost certainly, APOE from non-neuronal sources has combinatorial but distinct effects, for instance on amyloid clearance and neuroinflammation. APOE’s multiple cell-type–specific effects may explain why the gene has been an elusive target for drug discovery despite its strong clinical correlates. Targeting neuron-specific effects of APOE, for example by reducing ERK1/2 activity, may slow tau spreading and offer a more successful neuroprotective strategy in E4-carriers.
Acknowledgment
This project was supported by NIH-NIA grant R01 AG054429 to J.A.K. A.R.W. was supported by NIH-NIA training grants F30AG051327 and T32AG20506, and NIH-NIGMS training grant T32GM008152.
We thank Drs L. Lyass and C.-Y. Peng for technical assistance.
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.Corder EH, Saunders AM, Strittmatter J, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 2.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993;90:1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi Y, Yamada K, Liddelow SA, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 2017;549:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huynh T-PV, Liao F, Francis CM, et al. Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron 2017;96:1013–1023.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu C-C, Zhao N, Fu Y, et al. ApoE4 accelerates early seeding of amyloid pathology. Neuron 2017;96:1024–1032.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasuda M, Mori E, Kitagaki H, et al. Apolipoprotein E epsilon 4 allele and whole brain atrophy in late-onset Alzheimer’s disease. Am J Psychiatry 1998;155:779–784. [DOI] [PubMed] [Google Scholar]
- 7.Raber J, Wong D, Buttini M, et al. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci U S A 1998; 95:10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell 2012;148:1204–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knoferle J, Yoon SY, Walker D, et al. Apolipoprotein e4 produced in GABAergic interneurons causes learning and memory deficits in mice. J Neurosci 2014;34:14069–14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buttini M, Masliah E, Yu G-Q, et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am J Pathol 2010;177:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Q, Bernardo A, Walker D, et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 2006; 26:4985–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y-T, Seo J, Gao F, et al. APOE4 Causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 2018;98:1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Najm R, Xu Q, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med 2018;24:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan L, Bhattacharyya BJ, Belmadani A, et al. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol Neurodegener 2014;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 2013;31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zufferey R, Dull T, Mandel RJ, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol 1998;72:9873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Pak C, Han Y, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013;78: 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mengel D, Hong W, Corbett GT, et al. PrP-grafted antibodies bind certain amyloid β-protein aggregates, but do not prevent toxicity. Brain Res 2018;pii:S0006–8993(18)30664–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Tay Y, Sim B, et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Reports 2017;8:619–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imamura K, Sahara N, Kanaan NM, et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci Rep 2016;6:34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reinhardt P, Schmid B, Burbulla LF, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Stem Cell 2013;12:354–367. [DOI] [PubMed] [Google Scholar]
- 24.Murakami N, Imamura K, Izumi Y, et al. Proteasome impairment in neural cells derived from HMSN-P patient iPSCs. Mol Brain 2017; 10:771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paquet D, Kwart D, Chen A, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016;533:125–129. [DOI] [PubMed] [Google Scholar]
- 26.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and alzheimer disease meta analysis consortium. JAMA 1997;278:1349–1356. [PubMed] [Google Scholar]
- 27.Kim H, Yoo J, Shin J, et al. Modelling APOE ε3/4 allele-associated sporadic Alzheimer’s disease in an induced neuron. Brain 2017;140: 2193–2209. [DOI] [PubMed] [Google Scholar]
- 28.Dumanis SB, Tesoriero JA, Babus LW, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci 2009;29:15317–15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jain S, Yoon SY, Leung L, et al. Cellular source-specific effects of apolipoprotein (Apo) E4 on dendrite arborization and dendritic spine development. PLoS One 2013;8:e59478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou L, Lim Q-E, Wan G, Too H-P. Normalization with genes encoding ribosomal proteins but not GAPDH provides an accurate quantification of gene expressions in neuronal differentiation of PC12 cells. BMC Genomics 2010;11:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brecht WJ, Harris FM, Chang S, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci 2004;24:2527–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pitas RE, Boyles JK, Lee SH, et al. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta 1987;917:148–161. [DOI] [PubMed] [Google Scholar]
- 33.Alzheimer’s Association Calcium Hypothesis Workgroup. Calcium hypothesis of Alzheimer’s disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 2017;13:178–182.e17. [DOI] [PubMed] [Google Scholar]
- 34.Brunden KR, Trojanowski JQ, Lee VMY. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat Rev Drug Discov 2009;8:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 36.Ohm TG, Kirca M, Bohl J, et al. Apolipoprotein E polymorphism influences not only cerebral senile plaque load but also Alzheimer-type neurofibrillary tangle formation. Neuroscience 1995;66: 583–587. [DOI] [PubMed] [Google Scholar]
- 37.Olsson B, Lautner R, Andreasson U, et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 2016;15:673–684. [DOI] [PubMed] [Google Scholar]
- 38.Sutphen CL, Jasielec MS, Shah AR, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol 2015;72:1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buerger K, Zinkowski R, Teipel SJ, et al. Differential diagnosis of Alzheimer disease with cerebrospinal fluid levels of tau protein phosphorylated at threonine 231. Arch Neurol 2002;59:1267–1272. [DOI] [PubMed] [Google Scholar]
- 40.Hampel H, Buerger K, Zinkowski R, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry 2004; 61:95–102. [DOI] [PubMed] [Google Scholar]
- 41.Buerger K, Ewers M, Pirttilä T, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 2006;129:3035–3041. [DOI] [PubMed] [Google Scholar]
- 42.Katsinelos T, Zeitler M, Dimou E, et al. Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep 2018;23:2039–2055. [DOI] [PubMed] [Google Scholar]
- 43.Safaiyan F, Kolset SO, Prydz K, et al. Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J Biol Chem 1999;274:36267–36273. [DOI] [PubMed] [Google Scholar]
- 44.Muratore CR, Rice HC, Srikanth P, et al. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet 2014;23:3523–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yagi T, Ito D, Okada Y, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet 2011;20:4530–4539. [DOI] [PubMed] [Google Scholar]
- 46.Sproul AA, Jacob S, Pre D, et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One 2014;9:e84547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondo T, Asai M, Tsukita K, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013;12: 487–496. [DOI] [PubMed] [Google Scholar]
- 48.Israel MA, Yuan SH, Bardy C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012;482:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferreira S, Dupire M, Delacourte A, et al. Synthesis and regulation of apolipoprotein E during the differentiation of human neuronal precursor NT2/D1 cells into postmitotic neurons. Exp Neurol 2000;166: 415–421. [DOI] [PubMed] [Google Scholar]
- 50.Dekroon RM, Armati PJ. Synthesis and processing of apolipoprotein E in human brain cultures. Glia 2001;33:298–305. [DOI] [PubMed] [Google Scholar]
- 51.Sullivan PM, Han B, Liu F, et al. Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging 2011;32:791–801. [DOI] [PubMed] [Google Scholar]
- 52.Xu Q, Walker D, Bernardo A, et al. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J Neurosci 2008;28:1452–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lane-Donovan C, Herz J. ApoE, ApoE receptors, and the synapse in Alzheimer’s disease. Trends Endocrinol Metab 2017;28:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tesseur I, Van Dorpe J, Bruynseels K, et al. Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am J Pathol 2000;157:1495–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chambers SM, Fasano CA, Papapetrou EP, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 2009;27:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agell N, Bachs O, Rocamora N, Villalonga P. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal 2002;14:649–654. [DOI] [PubMed] [Google Scholar]
- 57.Strittmatter WJ, Saunders AM, Goedert M, et al. Isoform-specific interactions of apolipoprotein-E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 1994;91:11183–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Futamura M, Dhanasekaran P, Handa T, et al. Two-step mechanism of binding of apolipoprotein E to heparin: implications for the kinetics of apolipoprotein E-heparan sulfate proteoglycan complex formation on cell surfaces. J Biol Chem 2005;280:5414–5422. [DOI] [PubMed] [Google Scholar]
- 59.Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 2015; 18:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015;138:1738–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2014;501: 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iba M, Guo JL, McBride JD, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 2013;33:1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Y-WA, Zhou B, Wernig M, Südhof TC. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Aβ secretion. Cell 2017;168:427–441.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Takashima A, Noguchi K, Michel G, et al. Exposure of rat hippocampal neurons to amyloid β peptide (25–35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3β. Neurosci Lett 1996; 203:33–36. [DOI] [PubMed] [Google Scholar]
- 65.Ladu MJ, Falduto MT, Manelli AM, et al. Isoform-specific binding of apolipoprotein-E to beta-amyloid. J Biol Chem 1994;269:23403–23406. [PubMed] [Google Scholar]
- 66.Frank S, Clavaguera F, Tolnay M. Tauopathy models and human neuropathology: similarities and differences. Acta Neuropathol 2007; 115:39–53. [DOI] [PubMed] [Google Scholar]
- 67.González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, et al. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci 2017;10:50. [DOI] [PMC free article] [PubMed] [Google Scholar]