

Abstract
Ubiquitylation and deubiquitylation are reversible protein post-translational modification (PTM) processes involving the regulation of protein degradation under physiological conditions. Loss of balance in this regulatory system can lead to a wide range of diseases, such as cancer and inflammation. As the main members of the deubiquitinases (DUBs) family, ubiquitin-specific peptidases (USPs) are closely related to biological processes through a variety of molecular signaling pathways, including DNA damage repair, p53 and transforming growth factor-β (TGF-β) pathways. Over the past decade, increasing attention has been drawn to USPs as potential targets for the development of therapeutics across diverse therapeutic areas. In this review, we summarize the crucial roles of USPs in different signaling pathways and focus on advances in the development of USP inhibitors, as well as the methods of screening and identifying USP inhibitors.
Keywords: ubiquitin-specific peptidases, signaling pathways, drug screening, USP inhibitors
1. Introduction
As a complex regulatory mechanism of biological functions, post-translational modification (PTM) is essential for cell growth and stress response. Generally, intracellular proteins will experience multiple types of modifications after translation, such as phosphorylation, acetylation, methylation, and ubiquitylation, each corresponding to one or more specific functions [1]. Among them, ubiquitylation is responsible for regulating protein–protein interactions, cellular localization, and enzymatic activities of its protein substrates, and it is also related to proteasome-mediated protein degradation. A large number of studies have identified the ubiquitin-driven degradation pathways as one of the most important ways to help maintain protein balance within eukaryotic cells [1,2]. Therefore, the ubiquitylation of proteins plays indispensable regulatory roles in various biological phenomena [2].
Eukaryotic cells are equipped to recognize and degrade proteins by the ubiquitin–proteasome system (UPS). Upon conjugated to chains of ubiquitin, proteins are then directed to the 26S proteasome, a macromolecular protease, and degraded [3]. Ubiquitin is a small peptide (8.5 kDa) consisting of 76 amino acids that is ubiquitous in eukaryotic cells. The peptide sequence is highly conserved and contains seven lysine sites (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63), a glycine site at the C-terminus, and a methionine at the N-terminus (Met1). In general, the poly-ubiquitin chain linked by Lys48 is a degradation marker for proteasome, while the Lys63-linked poly-ubiquitin chain usually works with non-proteasome pathways (such as DNA repair, DNA replication, and cell signal transduction) [4,5,6]. It has been reported that the ubiquitin chains connected to the target protein through Lys6, Lys11, Lys27, Lys29 or Lys33 are also related to proteasome-mediated degradation [7]. In addition, under certain circumstances Lys63-linked ubiquitin chains can also bind and target proteins that need to be degraded by the proteasome [8].
Similar to other PTMs, the ubiquitin modification of protein is a dynamic and reversible process. Ubiquitin modification can be removed by a series of ubiquitin-specific proteases, which is called deubiquitylation. These proteases are named deubiquitinases (DUBs). Deubiquitinases specifically recognize and excise the tumbling molecules on the target protein, and also participate in the editing of poly-ubiquitin, thus playing an important role in the cleavage of ubiquitin precursors and ubiquitin monomers [9,10]. DUBs also regulate gene expression, apoptosis, cell cycle, DNA repair, and cytokines [11,12,13,14,15].
There are nearly a hundred known DUBs including cysteine proteases (USPs, UCHs, MJDs and OTUs) and metalloproteinases (containing metal catalytic domains) according to different catalytic mechanisms. They are divided into the following superfamilies: ubiquitin-specific protease (USP), ubiquitin C-terminal hydrolase (UCH), ovarian tumor protease (OTU), Machado-Josephin domain superfamily (MJD), and zinc-containing metalloproteases [16,17].
In recent years, the vast majority of DUBs have been shown to be associated with a variety of diseases, including cancer, diabetes, neurodegenerative diseases, and infectious diseases [18,19,20,21]. As the largest superfamily with over 50 members, USPs have aroused increasing attention as potential therapeutic targets in recent years [22]. It is interesting to compare USPs with kinases as drug targets since they are both involved in protein posttranslational modifications. Discovery in protein phosphorylation was awarded the Nobel Prize in 1992, and in the past thirty years numerous efforts have been invested in kinase inhibitors which resulted in a good number of clinically approved drugs including Gleevec. However, the research in the ubiquitin system caught the attention of medicinal chemists much later, and only in the past ten years USPs inhibitors have started to gradually emerge. We envision that USPs represent a new reservoir of therapeutic targets, which will reach its prime time in the twenty years to come. To date, no USP inhibitor has yet been approved for clinical use. In this review, we focus on advances in the development of USP inhibitors within the past decade.
2. The Ubiquitylation System
2.1. The Ubiquitylation and Deubiquitylation Processes
The role of ubiquitin in protein degradation has been discovered and the main enzymatic reactions of this system have been elucidated by biochemical studies. In this system, proteins are targeted for degradation via covalent ligation to ubiquitin, a 76-amino-acid protein. The key biochemical steps in ubiquitylation and deubiquitylation are illustrated in Figure 1.
Figure 1.

Key events in the ubiquitylation and deubiquitylation process [1,2,3]. E1: ubiquitin-activating enzyme; E2: ubiquitin-conjugating enzyme; E3: ubiquitin-protein ligase; Ub: ubiquitin. Really interesting new gene (RING) E3 ligases which represent the vast majority of E3 ligases are depicted here as an example.
Ubiquitin-protein ligation requires the sequential action of three enzymes: ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin-protein ligases (E3s) [1,2,3]. Firstly, the E1 enzyme activates the C-terminal carboxyl group of ubiquitin in an ATP-dependent manner, resulting in a covalent high-energy thioester linkage between ubiquitin and the active-site cysteine of E1. Then, the activated ubiquitin is trans-thiolated to an E2, forming a new thioester bond with the E2 catalytic cysteine. Finally, an E3 ligase assists or directly catalyzes the transfer of ubiquitin from E2 to the substrate, generally via the ε-amino group of a substrate lysine. To be specific, E3 ligases from the really interesting new gene (RING) family and RING-related families such as the U-box family can mediate a direct transfer by catalyzing the formation of an isopeptide bond between ubiquitin and the substrate, while the E6AP carboxyl terminus (HECT) E3s and RING-between-RING (RBR) E3s go through a third thioester intermediate with another thioester bond between ubiquitin and the E3 cysteine [23].
Proteins ligated to poly-ubiquitin chains are usually degraded by the 26S proteasome complex that requires ATP hydrolysis. The 26S proteasome is composed of a 20S proteolytic core and 19S regulatory complexes, which is the main proteolytic machine in mammalian cells [24]. The 19S complexes are responsible for the recognition of ubiquitylated proteins, while the 20S core is mainly responsible for catalyzing the degradation of proteins [24].
As mentioned above, the post-translational modification of cellular proteins through ubiquitylation is a dynamic and reversible process coordinated by the action of ubiquitinating and deubiquitinating enzymes. DUBs have fundamental roles in the ubiquitin system through their ability to specifically deconjugate ubiquitin from substrate proteins. Extensive studies suggested that DUBs can act at many different stages throughout the ubiquitin–proteasome pathway [25]. Due to their capability of preventing proteasomal degradation pathways, DUBs regulate the level and/or activities of various proteins such as tumor suppressors, DNA repair proteins, epigenetic modulators, etc., emerging as a compelling target for the development of novel therapies [11,12,13,14,15].
2.2. Structural Characteristics of USPs
The USPs, with >50 members, constitute the largest DUBs family described to date. All of them have highly conserved USP domains formed by three sub-domains resembling the palm, thumb and fingers of a right hand. The catalytic site is located between the palm and thumb domains, while the finger domain is responsible for interactions with distal ubiquitin [22]. Furthermore, many USPs exhibit additional domains and terminal extensions, which have important roles in activity and specificity. However, despite their relative structural diversity, most USPs share the common feature of a typical conformational change upon ubiquitin binding, which drives the transition from an inactive form to a catalytically active state. As cysteine proteases, USPs’ catalytic capability mainly depends on the nucleophilic attack by a cysteine in the catalytic site.
Here, we will take USP7 as an example to demonstrate the catalytic process of USPs. As a typical representative of the USPs family, USP7 is also known as herpes virus-associated ubiquitin-specific protease (HAUSP) and is closely related to various diseases such as prostate cancer, colon cancer, lung cancer, and multiple myeloma [26].
USP7 has an N-terminal tumor necrosis factor-receptor associated factor (TRAF) region, a catalytic region, and a C-terminal region (Figure 2). The TRAF region directly binds to substrates such as p53, Mdm2, etc. The catalytic region consists of three domains, resembling the shape of an extended right hand, and forms a binding surface for incoming ubiquitin. The C-terminal region contains five consecutive ubiquitin-like (UBL) regions [27].
Figure 2.

Structure of USP7. (A) Schematic of the USP7 domain organization [27]. (B) Crystal structure of the dimer of the USP7CD-UBL4/5-ubiquitin complex (PDB ID: 5JTV). Ubiquitin: blue; USP7 catalytic domain: orange; UBL4/5: gray. In this structure, the USP7 N-terminal TRAF-like domain and ubiquitin-like (UBL) domains 1–3 were truncated. The catalytic triad of USP7 (Cys223-His464-Asp481) was shown as red sticks.
The catalytic domain contains the classic catalytic triad: Cys, His, and Asp. The ubiquitin-binding pocket is located in the catalytic region and consists of a papain-like structure and an unfolded finger-like region [28]. The cleavage of the isopeptide bond between the target protein and ubiquitin can be summarized in three steps: binding to the substrate, acylation, and deacylation. First, USP7 specifically binds to its substrate, followed by a conformational change to an activated state. Next, the catalytic cysteine is deprived of a proton by histidine, and the resulting sulfhydryl group undergoes a nucleophilic attack toward the carbonyl Gly76 of ubiquitin from the USP7-ubiquitin intermediate. Finally, the ubiquitin is released by hydrolysis of the thioester bond between USP7 and ubiquitin [29].
3. Roles of USPs in Cancers
Aberrant regulation of protein ubiquitylation is closely related to the occurrence and development of tumorigenesis and other pathologies such as neurodegenerative diseases, autoimmunity, inflammatory disorders, infection, muscle dystrophies, etc. [30]. Given that the target proteins for USPs contain a large number of cell homeostasis regulators, as well as products of known oncogenes or tumor suppressor genes, USPs might be attractive and promising targets for the development of novel cancer therapies.
Studies have shown the involvement of USPs in the regulation of multiple known cancer-related pathways, including p53, transforming growth factor-β (TGF-β), protein kinase B (Akt), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Janus kinase/signal transducers and activators of transcription (JAKs/STATs), and G protein-coupled receptor (GPCR). For example, the overexpression of USP2a stabilizes p53-murine double minute 2 (MDM2) through direct deubiquitylation, without reducing MDM2-mediated p53 ubiquitylation, and thus enhances p53 degradation [31]. Since p53 functions as a tumor suppressor and is vital for normal cellular process controlling, such downregulation of p53 can ultimately cause tumor progression [31]. USP7, however, deubiquitylates both MDM2 and p53, while its affinity to MDM2 is confirmed to be higher [32,33]. Another notable example is USP26. It has been reported to be a novel negative regulator of the TGF-β pathway and the loss of USP26 expression may be an important factor in glioblastoma pathogenesis and breast cancer [34]. Low levels of USP26 degrade drosophila mothers against decapentaplegic protein 7 (SMAD7) and stabilize TGFβ, while high levels of USP26 stabilize SMAD7 by deubiquitylation and form a complex with SMAD ubiquitylation regulatory factor 2 (SMURF2), which degrades the TGF-β receptor by ubiquitylation [34].
Here, in Table 1 we summarized the roles of USPs implicated in tumorigenesis according to the different signaling pathways on which they act.
Table 1.
Cancer-related pathways regulated by USPs.
| Pathway | USPs Involved | Refs |
|---|---|---|
| DNA damage repair | USP1, USP28 | [35,36] |
| TGF-β | USP2a, USP4, USP9X, USP15, USP26 | [34,37,38,39,40] |
| Wnt/β-catenin | USP4, USP5, USP9X, USP14 | [41,42,43,44,45] |
| p53 | USP2, USP4, USP5, USP7, USP10, USP15, USP24, USP42 | [31,32,33,46,47,48,49,50,51,52,53,54,55] |
| c-Myc | USP2, USP10, USP22, USP28, USP36, USP37 | [56,57,58,59,60,61] |
| Akt | USP4, USP12, USP14, USP22, USP46 | [62,63,64,65,66,67] |
| JAKs-STATs | USP7 | [68] |
| NF-κB | USP4, USP11, USP14, USP15, USP18, USP19, USP20, USP35, USP24, USP48 | [69,70,71,72,73,74,75,76,77,78,79,80,81,82] |
| GPCR | USP4, USP8, USP14, USP20, USP30 | [83,84,85,86] |
4. Methods in Screening and Identification of Inhibitors for USPs
In order to enable the continuous discovery and development of inhibitors for USPs, a number of biological testing methods have been developed to screen and identify small molecule inhibitors (Figure 3).
Figure 3.

USPs inhibitor screening methods. (A) Activity-based probe (ABP) structures; (B) Ub-7-amino-4-methylcoumarin (AMC) structure; (C) Ub-phospholipase A2 (PLA2) assay; (D) Time-resolved fluorescence resonance energy transfer (TR-FRET) assay; (E) UBA52 structure and the SDS-PAGE-Coomassie assay; (F) Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) method.
4.1. Activity-Based Probes
Activity-based probes (ABPs), in which an electrophilic warhead is introduced onto the C-terminal glycine of ubiquitin, provide a way to test compounds in a cellular environment. Currently reported probe molecules include ubiquitin-vinylmethyl sulfone (Ub-VS) [87], ubiquitin-vinylmethyl ester (Ub-VME) [88], and ubiquitin-propargylic acid (Ub-PA) [89]. They can covalently label the nucleophilic cysteine of DUBs, resulting in a band shift on SDS-PAGE.
The advantage of this method is that it is closely related to the physiological environment of cells. The disadvantage is that it is time-consuming and laborious, so it is not currently recommended for screening USP inhibitors.
Interestingly, in order to overcome the lack of target selectivity, a novel Ub-based activity probe (Rh-M20-PA) bearing specific mutations to achieve selectivity for USP16 was developed by combining structural modelling and computation. A number of USP16-specific inhibitors were successfully discovered using these USP16-selective ABPs [90].
4.2. Ub-AMC
Ub-AMC, which has the C-terminus of a ubiquitin molecule linked to 7-amino-4-methylcoumarin (AMC), is a rather simple method, and has been widely applied in the determination of deubiquitinating enzyme activities [91]. The advantages of the Ub-AMC method are low cost per test and commercial availability. However, Ub-AMC is an unnatural substrate, and light-emitting substances (auto-fluorescence or fluorescence quenchers) can interfere with the reading. Later, researchers improved the method by replacing AMC with rhodamine-110 (Rho110) or tetramethylrhodamine, so the wavelength was red-shifted and the interference was reduced [92,93].
4.3. Ub-PLA2
In the Ub-phospholipase A2 (PLA2) method, the PLA2 does not directly emit fluorescence after being cleaved from the ubiquitin chain, but it acts on a fluorescent substrate and causes it to emit fluorescence [94]. It is also called the Ub-CHOP method and makes the screening at lower enzyme concentrations possible by amplifying the activity of deubiquitinating enzymes [95]. Its signal intensity and duration are better than the Ub-AMC. Besides, the excitation wavelength is not in the ultraviolet region, and currently there are commercial kits. However, the Ub-PLA2 method is not sensitive to some deubiquitinating enzymes of the UCH family, and its price is higher than the Ub-AMC method. USP inhibitors identified by this method include shionone and P22077 [95].
4.4. TR-FRET
The time-resolved fluorescence resonance energy transfer (TR-FRET) method is based on a full-length ubiquitin substrate that is site-specifically labeled with a yellow fluorescent protein (YFP) at the N-terminus and a terbium donor at the C-terminus. This substrate has strong fluorescence resonance energy transfer (FRET) between the two groups, while the cleavage by USPs will decrease the extent of FRET [96,97,98].
The advantage of the TR-FRET method is that it is equally sensitive to the four deubiquitinating enzymes of the UCH family. However, there is no commercial kit available.
The expansion of this application is to use diubiquitin molecules (diUb) as substrates, and this diubiquitin molecule can be connected through different lysine sites to simulate different ubiquitin chain forms [99].
4.5. SDS-PAGE-Coomassie
The development of a highly reliable assay based on a readily available SDS-PAGE-Coomassie system using UBA52 as the substrate protein has been reported recently [100]. A number of effective USP2 inhibitors were identified using this assay. Natural substrate UBA52 was used and quantitative measurement was based on the infrared emission of Coomassie dye on SDS-PAGE.
This method uses readily available and inexpensive materials and has excellent reproducibility without the interference problem that is intrinsic to any fluorescence-based approaches. It also has the advantage of using a natural protein substrate, avoiding any artifacts that may be introduced by unnatural substrates. However, this assay was not amenable to high-throughput screening. It is useful for the accurate determination of IC50 values during fine-tuning of the structures during the structure-activity studies.
4.6. MADAL-TOF
A sensitive and fast assay to quantify in vitro DUBs enzyme activities using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry has been developed [101]. This method realized the high specificity of many members of the OTU and JAB/MPN/Mov34 metalloenzyme DUB families. It used unmodified substrates, such as di-ubiquitin topoisomers, and can be used to assess the potency and specificity of deubiquitylation inhibitors.
5. Recent Development of USP Inhibitors
5.1. USP Inhibitors in Clinical Trials
A novel chalcone compound b-AP15 (Figure 4) has been reported to induce the cathepsin-dependent apoptosis by inhibiting the UPS system [102]. Later, it was determined that b-AP15 targeted the proteasome-bound USP14 and UCHL5, which belong to the USP and UCH families, respectively [103]. b-AP15 specifically inhibits the deubiquitylation activity of USP14 and UCHL5, and its affinity to USP14 is slightly higher than that of UCHL5. It was shown that b-AP15 caused the apoptosis of Bcl-2-overexpressing and p53-deficient cells, thus it may serve as a potential treatment for bortezomib-resistant patients [104,105]. b-AP15 was also shown to reduce the viability and proliferation of multiple myeloma cells, which mainly correlates with the reduced levels of cell division cycle 25C (CDC25C), cyclin-dependent kinases 1 (CDK1), cyclin B1 and subsequent caspase-mediated apoptosis and activation of unfolded protein response (UPR) [106]. Besides, the rapid apoptotic response caused by b-AP15 is related to the enhancement of oxidative stress and the rapid activation of the cJun N-terminal kinase and activator protein-1 (JNK-AP1) signaling pathway [107,108]. It also showed efficacy in the multiple myeloma xenograft tumor model. However, the poor solubility and stability of b-AP15 in aqueous solution limited its drugability toward the clinic use.
Figure 4.

Structures of reported USP inhibitors in clinical trials.
After an attempted to improve the physicochemical properties of b-AP15, compound VX1570 was obtained in 2015 as the first DUB inhibitor to enter clinical trials [109]. Although there was no significant improvement in the aqueous solubility of VX1570, it required an excipient containing a lower percentage of Kolliphor EL for injection. Compared to b-AP15, it contains an azepane as the central ring instead of a piperidine, swaps the position of nitro groups, and has additional fluoro substitution (Figure 4). It is more inclined to inhibit proteasome DUB activity. VLX1570 binds to USP14 in vitro and inhibits its activity, while its inhibitory activity on UCHL5 is relatively low (Table 2).
Table 2.
List of reported USP inhibitors.
| Target | Compound ID | PDB | Refs |
|---|---|---|---|
| USP inhibitors in clinical trials | |||
| USP14/UCHL5 | b-AP15 | None | [103] |
| VLX1570 | None | [109] | |
| USP inhibitors of reported new chemical entities | |||
| USP1 | Pimozide | None | [113] |
| GW7647 | None | ||
| Trifluoperazine | None | ||
| Rottlerin | None | ||
| C527 | None | [114] | |
| SJB2-043 | None | ||
| SJB3-091A | None | ||
| ML323 | None | [115,116] | |
| USP2 | Q29 | None | [117] |
| ML364 | None | [118] | |
| LCAHA | None | [119] | |
| STD1T | None | [120] | |
| 6TG | 5XU8 | [121] | |
| USP2/4/5/7/8/15/20/28/47/UCHL1/UCHL3/UCHL5 | PR619 | None | [122,123] |
| USP2/5/8/UCHL1/UCHL3 | AM416 | None | [124] |
| RA9 | None | ||
| RA14 | None | ||
| USP2/7 | NSC632839 | None | [98] |
| Compound 14 | None | [125] | |
| USP4/5 | Vialinin A | None | [126] |
| USP5/7/8/13/14/15/22 | Curcusone D | None | [127] |
| USP5/9X/14/24/UCHL5 | WP1130 (Degrasny) | None | [128,129,130,131] |
| EOAI3402143 (G9) | None | [132] | |
| USP7 | HBX-41108 | None | [133] |
| HBX-19818 | None | [134] | |
| HBX-28258 | None | ||
| Spongiacidin A | None | [135] | |
| GEN-6640 | 5UQV | [136] | |
| GEN-6776 | 5UQX | ||
| XL188 | 5V6S | [137] | |
| ALM2 | 5N9R | [138] | |
| ALM45 | 6F5H | [139] | |
| FT671 | 5NGE | [140,141] | |
| FT827 | 5NGF | ||
| Compound 2 | 5WHC | [142] | |
| Compound 28 | None | ||
| Parthenolide (PTL) | None | [143] | |
| Costunolide | None | ||
| α-santonin | None | ||
| USP7/8 | HY50736 | None | [144] |
| HY50737A | None | ||
| USP7/10 | Compound 3 | None | [145] |
| Compound 9 | None | ||
| USP7/47 | P22077 | None | [122,146,147] |
| P5091 | None | [148,149,150] | |
| P50429 | None | [151] | |
| USP10/13 | Spautin-1 | None | [152] |
| USP11/15 | Mitoxantrone | 6GH9 | [153,154] |
| USP14 | IU1 | 6IIK | [155] |
| IU1-47 | 6IIL | [156,157] | |
| IU1-206 | 6IIM | [157] | |
| IU1-248 | 6IIN | [157] | |
| USP14/UCHL5 | Auranofin | None | [158,159] |
| CuPT | None | [160,161] | |
| PtPT | None | [162] | |
| NiPT | None | [163] | |
| USP25/28 | AZ1 | None | [164] |
| USP30 | MF-094 | None | [165] |
| 15-oxospiramilactone (S3) | None | [166] | |
In vivo studies on multiple myeloma revealed that VLX1570 was more effective than b-AP15 in inhibiting tumor progression in mice [110].
However, the phase 1 clinical trial of VLX1570 in combination with dexamethasone in patients with relapsed or refractory multiple myeloma was recently suspended due to pulmonary toxicity [111]. In this trial, two patients underwent severe, abrupt, and progressive respiratory insufficiency, which was associated with diffuse pulmonary infiltrates, similar to those rarely noted with bortezomib and other inhibitors of the 20S proteasome. Two possible mechanisms of toxicity have been proposed: the activation of NF-κB and lung inflammation upon drug withdrawal and damage to lung tissues due to the accumulation of active drug metabolites [112].
5.2. USP Inhibitors at Early Stage of Drug Discovery
5.2.1. USP1 Inhibitors
Given the fact that USP1 functions as a USP1/UAF1 complex, studies have been conducted to identify inhibitors against the USP1/UAF1 complex. In 2011, the first USP1/UAF1 inhibitors were reported using a Ub-Rho110-based high-throughput screening (HTS) [113]. Among them, pimozide and GW7647 (Figure 5) as the most potent compounds (IC50 = 2 and 5 μM) were shown to inhibit USP1/UAF1 by a noncompetitive and reversible mechanism with reasonable selectivity against other human USPs and UCH-family DUBs. Further studies demonstrated that both compounds showed efficacy in cisplatin-resistant non-small cell lung cancer (NSCLC) cells when used in combination with cisplatin, as well as enhanced proliferating cell nuclear antigen (PCNA) and FANCD2 monoubiquitylation in human embryonic kidney 293T (HEK293T) cells. However, pimozide and GW7647 were both known to bind proteins unrelated to DUBs, which has limited their use.
Figure 5.

Structures of representative USP1 inhibitors.
In 2013, C527 (IC50 = 0.88 μM) (Figure 5) was identified as a potent USP1/UAF1 inhibitor using a similar Ub-Rho110-based HTS [114]. Later, more potent derivatives SJB2-043 (IC50 = 0.544 μM) and SJB3-019A (IC50 = 0.0781 μM) were reported. However, their target selectivity remained limited.
To improve the selectivity for USP1/UAF1, following up the previously reported GW7647, ML323 was developed (Figure 5), which was a nanomolar inhibitor of USP1/UAF1 (IC50 = 76 nM) with remarkable selectivity over 18 DUBs, deSUMOylase, deneddylase, 70 unrelated proteases, and 451 kinases [115,116]. By inhibiting the deubiquitylation of PCNA and FANCD2, ML323 sensitized cisplatin-resistant NSCLC cells and impaired DNA repair. This study provided ML323 as a best-in-class chemical probe to investigate the function and regulation of the USP1/UAF1 complex.
5.2.2. USP2 Inhibitors
By screening a commercial library, a series of ortho quinones with considerable inhibition against USP2 at a concentration of 5 μM was identified [117]. Among them, Q29 (Figure 6) was at that time in advanced clinical trials for pancreatic cancer treatment. Q29 was shown to generate reactive oxygen species (ROS) in the presence of low concentration 1,4-dithiothreitol (DTT), and its activity was due to selective and irreversible oxidation of the catalytic cysteine of USP2.
Figure 6.

Structures of representative USP2 inhibitors.
Through a high-throughput screening followed by structural optimization, ML364 (Figure 6) was identified as a USP2 inhibitor with an IC50 of 1.1 μM for the Lys48-linked substrate and 1.7 μM for the Lys63-linked substrate. It was shown that ML364 led to elevated cellular cyclin D1 level and cell cycle arrest in G0/G1 phase, resulting in a downregulation of DNA repair [118].
A series of lithocholic acid (LCA) derivatives was investigated for their anticancer activity, and LCAHA (Figure 6) was found to induce G0/G1 arrest in human colon cancer 116 (HCT116) cells accompanied by decreased expression of cyclin D1, thus leading to an assumption that USP2a was involved [119]. Further studies confirmed that the most potent compound LCAE directly inhibited USP2a activity with an IC50 of 5.8 μM by an uncompetitive mode.
A nuclear magnetic resonance (NMR)-based fragment screening led to the discovery of compound STD1T as a selective USP2 inhibitor (IC50 = 3.3 μM) (Figure 6) [120].
Although its inhibitory activity is moderate (IC50 = 40 μM), 6-thioguanine (6TG) (Figure 6) was the first and the sole small molecule cocrystal with USP2 to date (USP2-Ub-6TG, PDB ID: 5XU8) [121]. As shown in Figure 7, 6TG has polar interactions with nearby residues Asn279, Gln283, Ser576 and Tyr558, and the sulfur atom of it shows a disulfide bond with the catalytic Cys276. Besides, a 3.2 Å shift of Asp575 toward His557 is also observed [121]. As Asp575 is a conserved residue in the USPs with a role in the protonation of His557 for catalytic competency, the shift of Asp575 may also contribute to the inhibition of USP2.
Figure 7.

Cocrystal structure of the USP2-Ub-6TG complex and a close-up on the 6TG-binding site (PDB ID: 5XU8). Ubiquitin: magenta; USP2 catalytic domain: gray; 6TG: cyan sticks. In the close-up figure, hydrogen bonds made by 6TG are indicated by yellow dotted lines. Key residues involved in the interactions and the catalytic triad of USP2 (Cys276-His557-Asp574) are shown as sticks, with Cys276 in orange.
5.2.3. USP2/4/5/7/8/15/20/28/47/UCHL1/UCHL3/UCHL5 Inhibitors
Activity-based chemical proteomics was used to screen a diversity-based library for modulators of USP7 activity, and PR619 (Figure 8) was identified as a broad-spectrum DUBs inhibitor (USP2, USP4, USP5, USP7, USP8, USP15, USP20, USP28, USP47, UCHL1, UCHL3, UCHL5), but with limited activity against other families of proteases [122]. In addition, this study demonstrated that DUBs inhibitors induced the accumulation of poly-ubiquitylated proteins in cells without directly affecting proteasome activity. It was also found that PR619 could regulate the microtubule network by inhibiting deubiquitinating enzymes in the improvement of neurodegenerative diseases [123].
Figure 8.
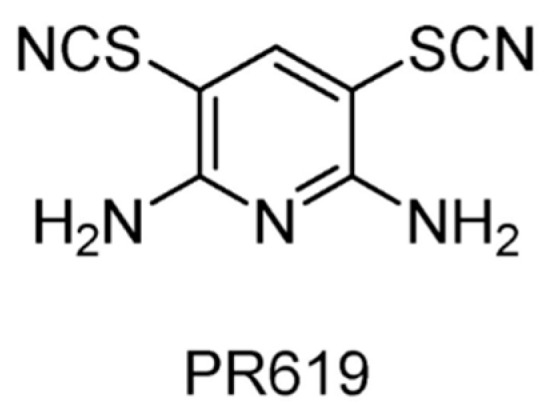
Structure of compound PR619.
5.2.4. USP2/5/8/UCHL1/UCHL3 Inhibitors
Three chalcone derivatives, AM146, RA-9 and RA-14 (Figure 9), were reported to show direct inhibition of USP2, USP5, USP8, UCHL1, and UCHL3, but with no significant effect on USP7 and USP14. They inhibited tumor cell proliferation by affecting protein ubiquitylation and upregulating p53, p27, and p16 levels [124].
Figure 9.

Structure of compound RA-9. The exact chemical structures of AM146 and RA-14 were not reported.
5.2.5. USP2/7 Inhibitors
A previously reported DUB inhibitor, NSC632839 (Figure 10), was identified as the inhibitor of USP2/7. It also exhibited the inhibition of deSUMOylase SUMO specific protease 2 (SENP2), suggesting that this compound is a relatively nonselective isopeptidase inhibitor [94]. Later, using a unique quenched pair assay, compound 14 was identified as a reversible inhibitor of USP2 with an uncompetitive mechanism and an IC50 of 250 nM. It is also worth mentioning that the introduction of a fluorine atom reversed the selectivity between USP2 and USP7.
Figure 10.

Structures of reported USP2/7 inhibitors.
5.2.6. USP4/USP5 Inhibitors
Vialinin A (Figure 11), which was isolated from the Chinese mushroom Thelephora vialis, was shown to act as an inhibitor of USP5 (IC50 = 5.9 μM) and USP4 (IC50 = 1.5 μM) and possess significant anti-inflammatory activity [126]. However, no significant inhibition was observed for USP2, USP8, and UCHL3, which suggested that vialinin A was a semi-selective inhibitor. It led to a reduction in the ubiquitylation level of the inhibitor of NF-κB (I-κB) and the release of tumor necrosis factor-α (TNF-α).
Figure 11.
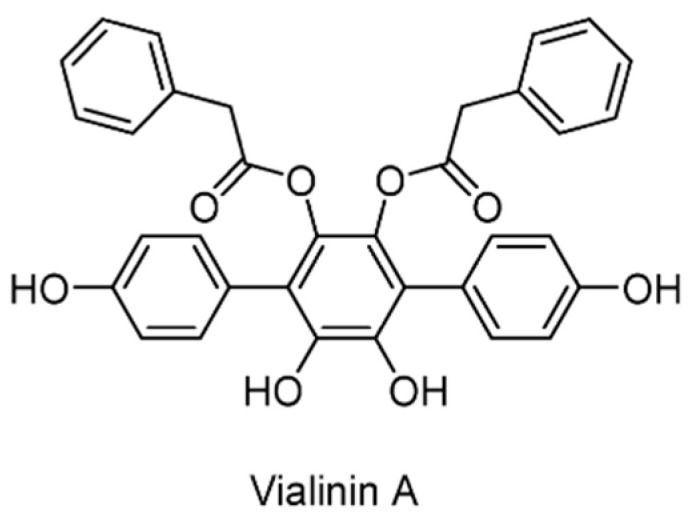
Structure of vialinin A.
5.2.7. USP5/7/8/13/14/15/22 Inhibitors
Inspired by the discovery that the α,β-unsaturated carbonyl in prostaglandins could react with the catalytic cysteine of DUBs to form covalent bonds and result in the inhibition of the DUBs, curcusone D (Figure 12) was reported as a novel USPs inhibitor [127]. Curcusone D is a diterpene isolated from Jatropha curcas (Barbados nut), a herbal plant that has been used in traditional folk medicine in many tropical countries. As a non-selective DUBs inhibitor, curcusone D was shown to inhibit USP5, USP7, USP8, USP13, USP14, USP15, and USP22, but had no effect on UCHL1, UCHL3, and UCHL5. It also showed efficacy in multiple myeloma when used in combination with bortezomib.
Figure 12.
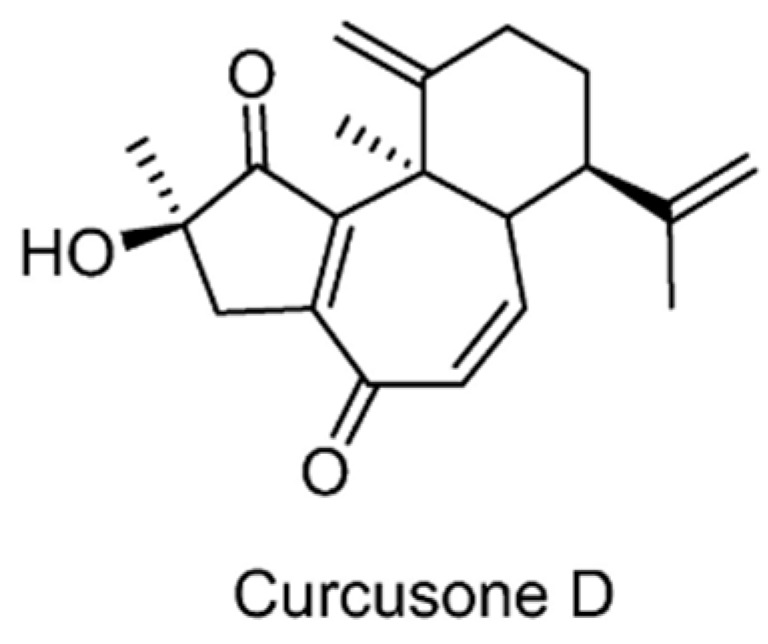
Structure of curcusone D.
5.2.8. USP5/9X/14/24/UCHL5 Inhibitors
WP1130 (Degrasny) (Figure 13), which was initially identified as a JAKs/STATs pathway inhibitor, was later shown to directly inhibit USP9X, USP5, USP14, and UCHL5 [128,129]. As a cell-permeable DUBs inhibitor, WP1130 caused rapid accumulation of Lys48/Lys63 poly-ubiquitylated proteins, which led to the formation of near-nuclear aggresomes and eventually apoptosis without affecting 20S proteasome activity. Another study demonstrated that combined treatment with WP1130 sensitized hepatocellular carcinoma (HCC) cells to doxorubicin via USP9X-depedent p53 degradation [130]. Furthermore, it was later shown that WP1130-mediated USP9X inhibition prevents the growth of ERG-positive prostate tumors in vitro and in a mouse xenograft model [131].
Figure 13.

Structures of reported USP5/9X/14/24/UCHL5 inhibitors.
EOAI3402143 (G9) (Figure 13) was identified in the study of WP1130 analogs, and showed improved drug-like properties and potency against USP9X [132]. Furthermore, G9 displayed USP24 inhibitory activity and potent apoptotic activity against myeloma and diffuse large B-cell lymphomas. A mechanism of action study indicated that G9 inhibits USP9X through a covalent and slow reversible conjugation with the cysteine residue.
5.2.9. USP7 Inhibitors
Through high-throughput screening of a proprietary library of 65,092 chemically diverse compounds, HBX-41108 (Figure 14) was identified as a USP7 inhibitor (IC50 = 0.424 μM) [133]. It showed an uncompetitive reversible inhibition according to kinetics assay. It was shown to affect USP7-mediated p53 deubiquitylation in vitro and in cells. Later, HBX-19818 and HBX-28258 were also identified as USP7 inhibitors [134]. They were shown to be covalent inhibitors binding to the active site, and exhibited no cross-reactivity on other USP members including USP2, USP5, USP8, and USP20.
Figure 14.

Structures of representative USP7 inhibitors.
Spongiacidin A, isolated from a marine sponge, was identified as the first USP7 inhibitor (IC50 = 3.8 μM) from a natural source [135]. It exhibited the inhibition of USP21c (IC50 = 16.6 μM) as well. However, it did not show any cytotoxic activity.
An extensive screening, including HTS and NMR fragment screening followed by counter-screening against USP5 and USP47, was carried out, and a series of fragments with favorable properties was identified [136]. The optimization of these hit fragments ultimately yielded GNE-6640 and GNE-6776 (Figure 14), which showed considerable inhibitory activity against both full length (IC50 = 0.75 and 1.34 μM) and the USP7 catalytic domain (IC50 = 0.43 and 0.61 μM) [136]. Surprisingly, co-crystal structures revealed that this scaffold binds to a hydrophobic pocket approximately 12 Å away from the catalytic triad (PDB ID: 5UQV and 5UQX), indicating a new mode of inhibition rather than competing with the ubiquitin C-terminus (Figure 15A). By comparing with the ubiquitin-bound structure, GNE-6640 and GNE-6776 appeared to exert their inhibitory activity by sterically hindering ubiquitin binding and preventing the transition of USP7 α5 helix to the active conformation. Moreover, it is notable that GNE-6776 is orally bioavailable and promotes on-target pathway modulation.
Figure 15.

Comparison of the structures of USP7-GNE-6776 (PDB ID: 5UQX) and USP7-ALM2 (PDB ID: 5N9R). A ubiquitin molecule is modeled into the complexes based on a superposition with a USP7CD-ubiquitin complex (PDB ID: 1NBF). Ubiquitin: magenta; USP7 catalytic domain: gray; GNE-6776: green sticks; ALM2: cyan sticks. Hydrogen bonds: yellow dotted lines. Key residues involved in binding: sticks. (A) Crystal structure of USP7-GNE-6776 and a close-up of the GNE-6776-binding site. In the close-up figure, the ubiquitin peptide AGKQLED is omitted for clarity. (B) Crystal structure of USP7-ALM2 and a close-up of the ALM2-binding site. In the close-up figure, the catalytic triad of USP7 (Cys223-His464-Asp481) is also shown as sticks, with Cys223 in orange, and the ubiquitin C-terminal peptide LRLRGG is omitted for clarity.
A potent and selective inhibitor of USP7, XL188, with an IC50 of 90 nM, was developed by structure-based design [137]. XL188 specifically binds to the S4-S5 pocket of USP7 (PDB ID: 5VS6), indicating that XL188 is a non-covalent active-site inhibitor. Further studies have shown that XL188 caused the elevation of tumor suppressor proteins p53 and p21, thereby inhibiting the occurrence and development of tumors.
Fragment-based screening using surface plasmon resonance (SPR) on 1946 fragments against the catalytic domain of USP7 was carried out. By combining the fragment hits with known features of published USP7 inhibitors, followed by crystallography and rational structural modification, the authors achieved a highly potent inhibitor ALM2. It consistently exhibited IC50 values in the single-digit nanomolar range (IC50 = 6 nM, fluorescence polarization (FP) assay; IC50 = 1.5 nM, Ub-Rho110 assay) and demonstrated high selectivity (>10,000-fold) against other members of the USP and DUB families [138]. It is a non-competitive inhibitor. Furthermore, its antiproliferative effects were explored, and cell lines hypersensitive to USP7 inhibition (EC50 < 30 nM), including both haematological (RS4; 11) and solid tumor cell lines (LNCaP), were identified. The crystal structure of the USP7-ALM2 complex (PDB ID: 5N9R) shows that ALM2 occupies the ubiquitin C-terminal tail-binding channel, thus creating a steric clash with the ubiquitin C-terminus (Figure 15B). Key interactions include three hydrogen bonds contributed by the core 4-hydroxypiperadine, four hydrogen bonds contributed by the 3H-pyrimidin-4-one group, and hydrophobic interactions with the side chain of Phe409. Notably, the stereochemistry of the methyl group in ALM2 is later confirmed to greatly affect the potency of the compounds, with the favored conformation of this scaffold being the (R)-stereoiomer.
By structure-guided design high-resolution crystallography, ALM45 was developed as a selective USP7 inhibitor with an IC50 of 0.1 μM (PDB ID: 6F5H) [139]. In particular, ALM45 demonstrated excellent biochemical and the absorption, distribution, metabolism, and excretion (ADME) profile, as well as promising pharmacokinetic profile.
A class of pyrazolo[3,4-d]pyrimidin-4-one-piperidine (PyrzPPip) compounds was identified through screening a diverse collection of approximately 500,000 compounds using a ubiquitin-Rho110 assay [140]. Further structural optimization of this series led to a non-covalent inhibitor, FT671 (IC50 = 52 nM), and a covalent inhibitor, FT827 (kincat/Ki= 66 M-1S-1) [141]. The cocrystal structures revealed that these two inhibitors target a dynamic pocket near the catalytic center of the auto-inhibited apo form of USP7, which differs from other USP deubiquitinases (PDB ID: 5NGE and 5NGF). Moreover, it was shown that FT671 destabilizes USP7 substrates including MDM2, upregulates p53 level, and results in the transcription of the target genes of p53. It also showed efficacy in the MM.1S xenograft mouse model.
Through integrated NMR and in silico techniques, two series of inhibitors were identified [142]. First, an oxadiazole series was identified by ligand-based virtual screening on an internal library and was represented by compound 2. Further biophysical characterization, including two-dimensional [1H-15N] TROSY spectrum and crystallization, revealed that compound 2 binds to a novel site within the “palm” regions (PDB ID: 5WHC). Second, an aminopyridine series was identified via an NMR-driven scaffold-hopping strategy. As a representative, compound 28 demonstrated submicromolar activity for USP7 (IC50 = 0.75 μM) and MDM2 expression (EC50 = 0.3 μM).
In a recent study in 2020, sesquiterpene lactone parthenolide (PTL) was reported as an inhibitor of USP7 (IC50 = 6.58 μM, Ub-AMC assay; IC50 = 15.42 μM, Ub-Rho110 assay) [143]. Treatment with PTL partially destabilized β-catenin, thereby inhibiting the activity of the Wnt pathway. Cytostatic experiments demonstrated that PTL prevented the proliferation of colorectal cancer cells and induced apoptosis. Two more sesquiterpene lactones (costunolide and α-santonin) were also identified to be USP7 inhibitors, indicating that the α-methylene-γ-butyrolactone can serve as a new scaffold for future development of USP7 inhibitors.
5.2.10. USP7/8 Inhibitors
On the basis of the USP7 inhibitor HBX-41108, USP7/8 activity was explored by structural modification [144]. In particular, the introduction of O-alkyloxime moieties at C-9 of the tricyclic scaffold gave the first known USP8-specific inhibitors with an IC50 below 1 μM, which was exemplified by HY50536 and HY50737A (IC50 = 0.28 and 0.24 μM) (Figure 16).
Figure 16.

Structures of reported USP7/8 inhibitors.
5.2.11. USP7/10 Inhibitors
A small series of HBX19818 analogs was evaluated for their inhibitory activity on USP10 [145]. Among them, compound 9 (Figure 17) inhibited USP10 similarly to HBX19818, but with no inhibition against USP7 (IC50 >> 100 μM). Compound 3 showed a lower anti-proliferation EC50 and induced FMS-like tyrosine kinase 3 (FLT3) degradation at lower concentrations.
Figure 17.

Structures of reported USP7/10 inhibitors.
5.2.12. USP7/47 Inhibitors
The thiophenyl compound P22077 (Figure 18) was found to be a selective inhibitor of USP7 (IC50 = 8.0 μM) and induce cell death in HCT116 and HEK293T cells [122]. It was later shown to play a role in the treatment of non-small cell lung cancer [146]. It was also revealed that P22077 mediated the increase in intracellular reactive oxygen species by enhancing intracellular oxidative stress response and endoplasmic reticulum stress response, thereby causing apoptosis [147].
Figure 18.

Structures of reported USP7/47 inhibitors.
Through a high throughput screening, the trisubstituted thiophenyl compound P5091 was discovered to be a USP7 inhibitor (IC50 = 4.2 μM) [148]. It showed selectivity toward USP7, with no effect on other DUBs such as USP2 and USP8. P5091 can induce the apoptosis of multiple myeloma (MM) cells resistant to traditional therapy or bortezomib by inhibiting the activity of USP7, and can also play a synergistic role when used in combination with dexamethasone or lenalidomide. Furthermore, it exhibited no cytotoxicity in the USP7 knockout HCT116 cell line, indicating that the cytotoxicity of P5091 depends on the intracellular USP7. It can induce cell death in ovarian cancers with different p53 status [149]. It also suppressed in vivo tumor growth in the HCT116 xenograft mouse model, which is consistently associated with reduced expression of β-catenin and Wnt target genes [150].
Progenra conducted the structural modification of P5091 and demonstrated that after the acetyl group in the C-2 position was changed to various amides, the derivative P50429 showed an IC50 of 0.42 μM and 1.0 μM against USP7 and USP47, respectively [151]. It has no inhibitory effect on apoptosis-related caspase 1/3 or 20S proteasome. In addition, the IC50 values of P50429 for other USP family members such as USP2, USP5, USP8, USP21, and USP28 are all greater than 31.6 μM.
5.2.13. USP10/13 Inhibitors
Through an imaging-based screening and subsequent structural modification, a potent autophagy inhibitor spautin-1 (Figure 19) was identified, which was demonstrated to inhibit USP10 and USP13 and thus promote the degradation of Vps34 PI3 kinase complexes [152].
Figure 19.
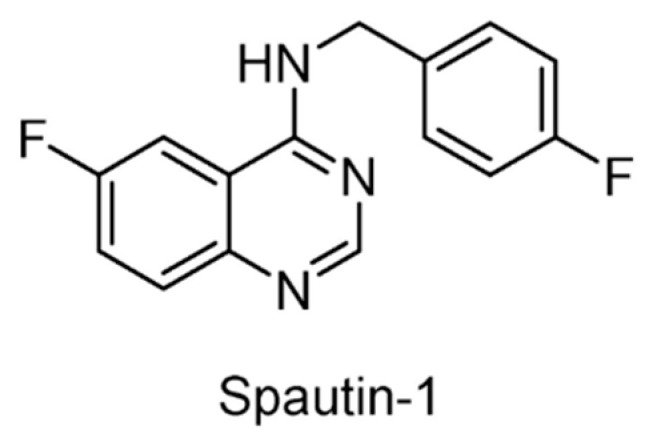
Structure of Spautin-1.
5.2.14. USP11/15 Inhibitors
Mitoxantrone (Figure 20), a clinical drug used to treat acute myeloid leukemia, hormone refractory prostate cancer, and multiple sclerosis treatment, was reported to inhibit USP11 (IC50 = 3.15 μM) and impact pancreatic ductal adenocarcinoma (PDA) cell survival [153]. In addition, it was found that mitoxantrone weakly inhibits the activity of USP15 with an IC50 of 33 µM. The crystal structure of the USP15-mitoxantrone complex (PDB ID: 6GH9) revealed predominantly hydrophobic interactions between mitoxantrone and USP15 residues Tyr855, Gly856, Gly860 and His862, which are located near the catalytic Cys269 (Figure 21) [154].
Figure 20.
Structure of mitoxantrone.
Figure 21.

Crystal structure of the USP15-mitoxantrone complex and a close-up of the binding site (PDB ID: 6GH9). A di-ubiquitin molecule is modeled into the complex based on a superposition with a USP30 C77A Lys6-linked di-ubiquitin structure (PDB ID: 5OHP), the closest available USP structure in complex with a substrate. Ubiquitin: magenta; USP15 catalytic domain: gray; mitoxantrone: cyan sticks. In the close-up figure, key residues involved in binding and the catalytic triad (Cys269-His862-Asp879) are shown as sticks, with Cys269 marked orange, and the di-ubiquitin molecule is omitted for clarity.
5.2.15. USP14 Inhibitors
By screening 63,052 compounds, IU1 (Figure 22) was identified as the first USP14 inhibitor (IC50 = 4 μM) [155]. It interacts with the active form of USP14, blocking its docking to proteasome. It had little or no inhibitory activity against eight other DUBs: isopeptidase T (IsoT/USP5), UCHL5, BAP1, UCHL1, UCHL3, USP15, USP2, and USP7. IU1 promoted the degradation of several proteasome substrates that have been implicated in neurodegenerative disease, indicating that IU1 may play an important role in the drug development for neurodegenerative disease.
Figure 22.

Structures of representative USP14 inhibitors.
Later, during the structural modification of IU1, IU1-47 was identified as a potent USP14 inhibitor with an IC50 of 0.6 μM, accompanied by a modest increase in selectivity over IsoT/USP5 to approximately 33-fold [156]. Moreover, using the microtubule-associated protein tau that has been implicated in many neurodegenerative diseases as a reporter, IU1-47 was shown to enhance protein degradation in cells.
The high-resolution co-crystal structures of USP14 (PDB ID: IU1, 6IIK; IU1-47, 6IIL) revealed that IU1 and its analogs bind competitively with the C-terminus of ubiquitin to the active site of USP14, thereby abrogating the catalytical activity of USP14 [157]. Subsequent structure-guided design led to the discovery of IU1-248 with an IC50 of 0.83 μM. Taking the USP14-IU1-248 cocrystal structure as an example, as shown in Figure 23, the phenyl ring of the inhibitor extends into the inner hydrophobic pocket constituting of Phe331, His246, and Tyr436, while the piperidine ring provides both hydrogen bond and hydrophobic interactions.
Figure 23.

Cocrystal structure of USP14-IU1-248 and a close-up of the binding site (PDB ID: 6IIN). A ubiquitin molecule is modeled into the complex based on the superposition with a USP14CD-ubiquitin complex (PDB ID: 2AYO). Ubiquitin: magenta; USP14 catalytic domain: gray; IU1-248: cyan sticks. Hydrogen bonds: yellow dotted lines. Key residues involved in binding and the catalytic triad (Cys114-His435-Asp451) are shown as sticks, with Cys114 in orange. The ubiquitin C-terminal peptide RLRGG is omitted for clarity.
5.2.16. USP14/UCLH5 Inhibitors
In recent years, several metal-based compounds have been found to target USP14 with promising therapeutic value [158]. One of the representative drugs is auranofin (Aur) (Figure 24), a gold-containing compound, which has been used clinically to treat rheumatic arthritis since 1985. Aur recently entered phase 2 clinical trials as a cancer therapy. Later, it was demonstrated that Aur targets both UCHL5 and USP14 [159]. Moreover, the in vivo efficacy of Aur was evaluated in mouse xenograft models, suggesting that Aur can accumulate proteasome substrates and inhibit tumor growth.
Figure 24.

Structures of representative USP14/UCLH5 inhibitors.
In addition, various pyrithione (PT)-metal chelates, including copper, zinc, nickel, and platinum, have been reported as USP14/UCHL5 inhibitors [160,161,162,163]. ZnPT, an FDA-approved drug, was reported to target USP14 and UCHL5, as well as efficiently inducing apoptosis in primary cancer cells from leukemia patients and suppressing tumor growth in mouse xenografts [161]. Platinum pyrithione (PtPT) and nickel pyrithione (NiPT) were also successfully explored as inhibitors inducing typical proteasome inhibition via targeting USP14/UCHL5 [162,163]. CuPT has also been reported as a novel class of USP14/UCHL5 inhibitors [160].
5.2.17. USP25/28 Inhibitors
By high-throughput screening on a directed library of approximately 40,000 compounds, AZ1 (Figure 25) was identified as the first known USP28 inhibitor exhibiting an IC50 of 0.7 μM [164]. Interestingly, subsequent selectivity profiling against USPs and DUBs demonstrated that AZ1 had strong inhibition against USP25 (IC50 = 0.62 μM), whilst no significant effect was observed against any of the other family members tested (<10% inhibition at 10 μM). Furthermore, it was shown that AZ1 was responsible for the modulation of both the levels and the half-life of c-Myc, and apoptosis and loss of cell viability in a range of cancer cell lines.
Figure 25.

Structure of compound AZ1.
5.2.18. USP30 Inhibitors
During a hit-to-lead optimization, MF-094 (Figure 26) was identified as a potent and selective USP30 inhibitor (IC50 = 0.12 μM) [165]. It was shown that MF-094 accelerated the disappearance of 5-bromo-2′-dexoyuridine (BrdU) from the mitochondrial DNA, indicating the role of MF-094 in the acceleration of mitophagy.
Figure 26.

Structures of reported USP30 inhibitors.
In a study to understand mitochondrial fusion and fission, a diterpenoid 15-oxospiramilactone (S3) was found to induce remarkable mitochondrial elongation in cells that lack mitofusin 1 (Mfn1). Later, a mechanism of action study determined that S3 directly interacted with USP30, suggesting that USP30 is a target of S3 [166].
6. Conclusions
Ubiquitylation and deubiquitylation control nearly all aspects of human cell biology and physiology, while any defects can cause diseases. Accordingly, ubiquitylation and related processes have drawn tremendous attention as potential therapeutic targets. USPs, as the most widely studied enzymes in the deubiquitinating enzyme family, have been proposed as promising targets for anticancer, antiviral, and anti-infective treatments. During the past decades, in-depth studies on USPs in human cancer progression have achieved significant advances such as in the identification of their pathological functions, mode of actions, intrinsic molecular mechanisms, and regulation in cancer. Several USPs are responsible for tumorigenesis by various cancer-related signaling pathways, including the DNA repair pathway, TGF-β pathway, p53 pathway, etc., whereas one USP can generally affect more than one pathway. All these findings contributed to the development of small molecule inhibitors against USPs with anticancer potency, which could, in turn, help to explore or/and confirm the precise functions of the targeted USPs. For example, USP7 has been discovered to stabilize both MDM2 and p53 by deubiquitylation, with a higher binding affinity for MDM2 according to structural and biochemical data [32,33]. In another work, HBX-41108, a USP7-specific inhibitor, increases p53 levels in HCT116 colon cancer cells by inhibiting MDM2 deubiquitylation [133].
Considering the fact that proteolysis-targeting chimeras (PROTACs), perhaps one of the most exciting technologies to arise in recent years, implement aimed protein degradation based on the UPS system, it is worthy to briefly compare USP inhibitors with PROTACs. These two strategies do differ greatly and have totally distinct advantages and disadvantages. The PROTACs technology employs E3 ligase ligands via a flexible chemical linker to the target protein to elicit ectopic ubiquitylation, thus promoting the UPS-mediated degradation of a specific target protein, whereas USP inhibitors induce targeted degradation through the inhibition of deubiquitylation process. Since PROTACs are still an emerging technology, though their small molecule nature makes PROTACs simple to use in experiments as inhibitors, the conversion of an E3 ligase ligand into a PROTAC can still be time-consuming, and, of course, not all proteins or subcellular locations are amenable yet. USP inhibitors, however, do not have these hindrances but generally suffer from a lack of selectivity because members of USPs share a high degree of homology.
The discovery of USP inhibitors reported before 2014 mainly relied on high-throughput screening. These studies rarely included any structure-activity relationship (SAR), lacking compound optimization and rigorous characterization. Recently, only after the co-crystal structures of USP-inhibitor complexes were reported, it became feasible to take advantage of structure-guided drug design and carry out SAR optimization. Interestingly, several inhibitors have been shown to bind to the vicinity of the catalytic site instead of the central region where the catalytic cysteine is located. Meanwhile, biophysical and biochemical assays including SPR, isothermal titration calorimetry (ITC), differential scanning fluorimetry (DSF) and hydrogen-deuterium exchange mass spectrometry (HDX-MS) started to be used for on-target binding validation. Current efforts are mainly focused on the improvement of the compound inhibitory selectivity against the entire USPs or DUBs family. With the continued improvements in selectivity profile, target engagement, and phenotypic discovery, we will expand our understanding of the exact action mode of USP inhibitors and their selectivity. More potent and selective USP inhibitors or even clinical candidates will be discovered and eventually developed into useful therapeutic agents.
Funding
We thank the National Key Research and Development Program of China (2017YFA0505200), National Science Foundation of China (81573264), and E-Institutes of Shanghai Universities (EISU) Chemical Biology Division for financial support of this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Millar A.H., Heazlewood J.L., Giglione C., Holdsworth M.J., Bachmair A., Schulze W.X. The scope, functions, and dynamics of posttranslational protein modifications. Annu. Rev. Plant Biol. 2019;70:119–151. doi: 10.1146/annurev-arplant-050718-100211. [DOI] [PubMed] [Google Scholar]
- 2.Swatek K.N., Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleiger G., Mayor T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014;24:352–359. doi: 10.1016/j.tcb.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chau V., Tobias J.W., Bachmair A., Marriott D., Ecker D.J., Gonda D.K., Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 5.Hershko A., Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 6.Haglund K., Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu P., Duong D.M., Seyfried N.T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saeki Y., Kudo T., Sone T., Kikuchi Y., Yokosawa H., Tohe A., Tanaka K. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 2009;28:359–371. doi: 10.1038/emboj.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickart C.M., Rose I.A. Ubiquitin carboxyl-terminal hydrolase acts on ubiquitin carboxyl-terminal amides. J. Biol. Chem. 1985;260:7903–7910. doi: 10.1016/S0021-9258(17)39538-8. [DOI] [PubMed] [Google Scholar]
- 10.Mevissen T.E.T., Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- 11.Frappier L., Verrijzer C.P. Gene expression control by protein deubiquitinases. Curr. Opin. Genet. Dev. 2011;21:207–213. doi: 10.1016/j.gde.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 12.He M., Zhou Z., Wu G., Chen Q., Wan Y. Emerging role of DUBs in tumor metastasis and apoptosis: Therapeutic implication. Pharmacol. Ther. 2017;177:96–107. doi: 10.1016/j.pharmthera.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darling S., Fielding A.B., Sabat-Pospiech D., Prior I.A., Coulson J.M. Regulation of the cell cycle and centrosome biology by deubiquitylases. Biochem. Soc. Trans. 2017;45:1125–1136. doi: 10.1042/BST20170087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kee Y., Huang T.T. Role of deubiquitinating enzymes in DNA repair. Mol. Cell. Biol. 2016;36:524–544. doi: 10.1128/MCB.00847-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo B., Baek K.H. Regulatory interplay between deubiquitinating enzymes and cytokines. Cytokine Growth Factor Rev. 2019;48:40–51. doi: 10.1016/j.cytogfr.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komander D., Clague M.J., Urbe S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 17.Clague M.J., Barsukov I., Coulson J.M., Liu H., Rigden D.J., Urbe S. Deubiquitylases from genes to organism. Physiol. Rev. 2013;93:1289–1315. doi: 10.1152/physrev.00002.2013. [DOI] [PubMed] [Google Scholar]
- 18.D’Arcy P., Wang X., Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015;147:32–54. doi: 10.1016/j.pharmthera.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Balaji V., Pokrzywa W., Hoppe T. Ubiquitylation pathways in insulin signaling and organismal homeostasis. Bioessays. 2018;40:e1700223. doi: 10.1002/bies.201700223. [DOI] [PubMed] [Google Scholar]
- 20.Ruan J., Schluter D., Wang X. Deubiquitinating enzymes (DUBs): DoUBle-edged swords in CNS autoimmunity. J. Neuroinflamm. 2020;17:102. doi: 10.1186/s12974-020-01783-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nanduri B., Suvarnapunya A.E., Venkatesan M., Edelmann M.J. Deubiquitinating enzymes as promising drug targets for infectious diseases. Curr. Pharm. Des. 2013;19:3234–3247. doi: 10.2174/1381612811319180008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young M.J., Hsu K.C., Lin T.E., Chang W.C., Hung J.J. The role of ubiquitin-specific peptidases in cancer progression. J. Biomed. Sci. 2019;26:42. doi: 10.1186/s12929-019-0522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metzger M.B., Hristova V.A., Weissman A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012;125:531–537. doi: 10.1242/jcs.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldberg A.L. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 25.Reyes-Turcu F.E., Ventii K.H., Wilkinson K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009;78:363–397. doi: 10.1146/annurev.biochem.78.082307.091526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J., Wang J., Chen C., Yuan H., Wen X., Sun H. USP7: Target validation and drug discovery for cancer therapy. Med. Chem. 2018;14:3–18. doi: 10.2174/1573406413666171020115539. [DOI] [PubMed] [Google Scholar]
- 27.Rouge L., Bainbridge T.W., Kwok M., Tong R., Di Lello P., Wertz I.E., Maurer T., Ernst J.A., Murray J. Molecular understanding of USP7 substrate recognition and C-terminal activation. Structure. 2016;24:1335–1345. doi: 10.1016/j.str.2016.05.020. [DOI] [PubMed] [Google Scholar]
- 28.Hu M., Li P., Li M., Li W., Yao T., Wu J.W., Gu W., Cohen R.E., Shi Y. Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. doi: 10.1016/S0092-8674(02)01199-6. [DOI] [PubMed] [Google Scholar]
- 29.Daviet L., Colland F. Targeting ubiquitin specific proteases for drug discovery. Biochimie. 2008;90:270–283. doi: 10.1016/j.biochi.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Popovic D., Vucic D., Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014;20:1242–1253. doi: 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 31.Stevenson L.F., Sparks A., Allende-Vega N., Xirodimas D.P., Lane D.P., Saville M.K. The deubiquitinating enzyme USP2a regulates the p53 pathway by targeting Mdm2. EMBO J. 2007;26:976–986. doi: 10.1038/sj.emboj.7601567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu M., Gu L., Li M., Jeffrey P.D., Gu W., Shi Y. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: Implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 2006;4:e27. doi: 10.1371/journal.pbio.0040027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meulmeester E., Pereg Y., Shiloh Y., Jochemsen A.G. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle. 2005;4:1166–1170. doi: 10.4161/cc.4.9.1981. [DOI] [PubMed] [Google Scholar]
- 34.Kit Leng Lui S., Iyengar P.V., Jaynes P., Isa Z., Pang B., Tan T.Z., Eichhorn P.J.A. USP26 regulates TGF-β signaling by deubiquitinating and stabilizing SMAD7. EMBO Rep. 2017;18:797–808. doi: 10.15252/embr.201643270. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Dianov G.L., Meisenberg C., Parsons J.L. Regulation of DNA repair by ubiquitylation. Biochemistry. 2011;76:69–79. doi: 10.1134/S0006297911010093. [DOI] [PubMed] [Google Scholar]
- 36.Zhang D., Zaugg K., Mak T.W., Elledge S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y., Wang X., Wang Q., Deng Y., Li K., Zhang M., Zhang Q., Zhou J., Wang H.Y., Bai P., et al. USP2a supports metastasis by tuning TGF-β signaling. Cell Rep. 2018;22:2442–2454. doi: 10.1016/j.celrep.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L., Zhou F., Drabsch Y., Gao R., Snaar-Jagalska B.E., Mickanin C., Huang H., Sheppard K.A., Porter J.A., Lu C.X., et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat. Cell Biol. 2012;14:717–726. doi: 10.1038/ncb2522. [DOI] [PubMed] [Google Scholar]
- 39.Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M.J., Soligo S., Morsut L., et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–135. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 40.Iyengar P.V., Jaynes P., Rodon L., Lama D., Law K.P., Lim Y.P., Verma C., Seoane J., Eichhorn P.J. USP15 regulates SMURF2 kinetics through C-lobe mediated deubiquitination. Sci. Rep. 2015;5:14733. doi: 10.1038/srep14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yun S.I., Kim H.H., Yoon J.H., Park W.S., Hahn M.J., Kim H.C., Chung C.H., Kim K.K. Ubiquitin specific protease 4 positively regulates the WNT/β-catenin signaling in colorectal cancer. Mol. Oncol. 2015;9:1834–1851. doi: 10.1016/j.molonc.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y., Li Y., Xue J., Gong A., Yu G., Zhou A., Lin K., Zhang S., Zhang N., Gottardi C.J., et al. Wnt-induced deubiquitination FoxM1 ensures nucleus β-catenin transactivation. EMBO J. 2016;35:668–684. doi: 10.15252/embj.201592810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Premarathne S., Murtaza M., Matigian N., Jolly L.A., Wood S.A. Loss of Usp9x disrupts cell adhesion, and components of the Wnt and Notch signaling pathways in neural progenitors. Sci. Rep. 2017;7:8109. doi: 10.1038/s41598-017-05451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang B., Zhang S., Wang Z., Yang C., Ouyang W., Zhou F., Zhou Y., Xie C. Deubiquitinase USP9X deubiquitinates β-catenin and promotes high grade glioma cell growth. Oncotarget. 2016;7:79515–79525. doi: 10.18632/oncotarget.12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang G., Li L., Zhou W. USP14 activation promotes tumor progression in hepatocellular carcinoma. Oncol. Rep. 2015;34:2917–2924. doi: 10.3892/or.2015.4296. [DOI] [PubMed] [Google Scholar]
- 46.Wang C.L., Wang J.Y., Liu Z.Y., Ma X.M., Wang X.W., Jin H., Zhang X.P., Fu D., Hou L.J., Lu Y.C. Ubiquitin-specific protease 2a stabilizes MDM4 and facilitates the p53-mediated intrinsic apoptotic pathway in glioblastoma. Carcinogenesis. 2014;35:1500–1509. doi: 10.1093/carcin/bgu015. [DOI] [PubMed] [Google Scholar]
- 47.Li Z., Hao Q., Luo J., Xiong J., Zhang S., Wang T., Bai L., Wang W., Chen M., Wang W., et al. USP4 inhibits p53 and NF-κB through deubiquitinating and stabilizing HDAC2. Oncogene. 2016;35:2902–2912. doi: 10.1038/onc.2015.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X.N., Berger F.G., Yang J.H., Lu X.B. USP4 inhibits p53 through deubiquitinating and stabilizing ARF-BP1. EMBO J. 2011;30:2177–2189. doi: 10.1038/emboj.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dayal S., Sparks A., Jacob J., Allende-Vega N., Lane D.P., Saville M.K. Suppression of the deubiquitinating enzyme USP5 causes the accumulation of unanchored polyubiquitin and the activation of p53. J. Biol. Chem. 2009;284:5030–5041. doi: 10.1074/jbc.M805871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Potu H., Peterson L.F., Pal A., Verhaegen M., Cao J., Talpaz M., Donato N.J. Usp5 links suppression of p53 and FAS levels in melanoma to the BRAF pathway. Oncotarget. 2014;5:5559–5569. doi: 10.18632/oncotarget.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tavana O., Sun H., Gu W. Targeting HAUSP in both p53 wildtype and p53-mutant tumors. Cell Cycle. 2018;17:823–828. doi: 10.1080/15384101.2018.1456293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takayama K.I., Suzuki T., Fujimura T., Takahashi S., Inoue S. Association of USP10 with G3BP2 inhibits p53 signaling and contributes to poor outcome in prostate cancer. Mol. Cancer Res. 2018;16:846–856. doi: 10.1158/1541-7786.MCR-17-0471. [DOI] [PubMed] [Google Scholar]
- 53.Zou Q., Jin J., Hu H., Li H.S., Romano S., Xiao Y., Nakaya M., Zhou X., Cheng X., Yang P., et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014;15:562–570. doi: 10.1038/ni.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L., Nemzow L., Chen H., Lubin A., Rong X., Sun Z.Y., Harris T.K., Gong F. The deubiquitinating enzyme USP24 is a regulator of the UV damage response. Cell Rep. 2015;10:140–147. doi: 10.1016/j.celrep.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hock A.K., Vigneron A.M., Carter S., Ludwig R.L., Vousden K.H. Regulation of p53 stability and function by the deubiquitinating enzyme USP42. EMBO J. 2011;30:4921–4930. doi: 10.1038/emboj.2011.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benassi B., Flavin R., Marchionni L., Zanata S., Pan Y., Chowdhury D., Marani M., Strano S., Muti P., Blandino G., et al. MYC is activated by USP2a-mediated modulation of microRNAs in prostate cancer. Cancer Discov. 2012;2:236–247. doi: 10.1158/2159-8290.CD-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Z., Yang H., Tan C., Li J., Liu Z., Quan Q., Kong S., Ye J., Gao B., Fang D. USP10 antagonizes c-Myc transcriptional activation through SIRT6 stabilization to suppress tumor formation. Cell Rep. 2013;5:1639–1649. doi: 10.1016/j.celrep.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D., Hong A., Park H.I., Shin W.H., Yoo L., Jeon S.J., Chung K.C. Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J. Cell. Physiol. 2017;232:3664–3676. doi: 10.1002/jcp.25841. [DOI] [PubMed] [Google Scholar]
- 59.Weili Z., Zhikun L., Jianmin W., Qingbao T. Knockdown of USP28 enhances the radiosensitivity of esophageal cancer cells via the c-Myc/hypoxia-inducible factor-1 α pathway. J. Cell. Biochem. 2019;120:201–212. doi: 10.1002/jcb.27305. [DOI] [PubMed] [Google Scholar]
- 60.Sun X.X., Sears R.C., Dai M.S. Deubiquitinating c-Myc: USP36 steps up in the nucleolus. Cell Cycle. 2015;14:3786–3793. doi: 10.1080/15384101.2015.1093713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pan J., Deng Q., Jiang C., Wang X., Niu T., Li H., Chen T., Jin J., Pan W., Cai X., et al. USP37 directly deubiquitinates and stabilizes c-Myc in lung cancer. Oncogene. 2015;34:3957–3967. doi: 10.1038/onc.2014.327. [DOI] [PubMed] [Google Scholar]
- 62.Xing C., Lu X.X., Guo P.D., Shen T., Zhang S., He X.S., Gan W.J., Li X.M., Wang J.R., Zhao Y.Y., et al. Ubiquitin-specific protease 4-mediated deubiquitination and stabilization of PRL-3 is required for potentiating colorectal oncogenesis. Cancer Res. 2016;76:83–95. doi: 10.1158/0008-5472.CAN-14-3595. [DOI] [PubMed] [Google Scholar]
- 63.McClurg U.L., Summerscales E.E., Harle V.J., Gaughan L., Robson C.N. Deubiquitinating enzyme Usp12 regulates the interaction between the androgen receptor and the Akt pathway. Oncotarget. 2014;5:7081–7092. doi: 10.18632/oncotarget.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu D., Shan B., Lee B.H., Zhu K., Zhang T., Sun H., Liu M., Shi L., Liang W., Qian L., et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. eLife. 2015;4:e10510. doi: 10.7554/eLife.10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhuang Y.J., Liao Z.W., Yu H.W., Song X.L., Liu Y., Shi X.Y., Lin X.D., Zhou T.C. ShRNA-mediated silencing of the ubiquitin-specific protease 22 gene restrained cell progression and affected the Akt pathway in nasopharyngeal carcinoma. Cancer Biol. Ther. 2015;16:88–96. doi: 10.4161/15384047.2014.987029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gui D., Peng W., Jiang W., Huang G., Liu G., Ye Z., Wang Y., Xu Z., Fu J., Luo S., et al. Ubiquitin-specific peptidase 46 (USP46) suppresses renal cell carcinoma tumorigenesis through AKT pathway inactivation. Biochem. Biophys. Res. Commun. 2019;519:689–696. doi: 10.1016/j.bbrc.2019.09.036. [DOI] [PubMed] [Google Scholar]
- 67.Li X., Stevens P.D., Yang H., Gulhati P., Wang W., Evers B.M., Gao T. The deubiquitination enzyme USP46 functions as a tumor suppressor by controlling PHLPP-dependent attenuation of Akt signaling in colon cancer. Oncogene. 2013;32:471–478. doi: 10.1038/onc.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Z., Huo S., Shan Y., Liu H., Xu Y., Yao K., Li X., Zhang X. STAT3 repressed USP7 expression is crucial for colon cancer development. FEBS Lett. 2012;586:3013–3017. doi: 10.1016/j.febslet.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 69.Mahul-Mellier A.L., Pazarentzos E., Datler C., Iwasawa R., AbuAli G., Lin B., Grimm S. De-ubiquitinating protease USP2a targets RIP1 and TRAF2 to mediate cell death by TNF. Cell Death Differ. 2012;19:891–899. doi: 10.1038/cdd.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiao N., Li H., Luo J., Wang R., Chen H., Chen J., Wang P. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFα-induced cancer cell migration. Biochem. J. 2012;441:979–986. doi: 10.1042/BJ20111358. [DOI] [PubMed] [Google Scholar]
- 71.Xu C., Peng Y., Zhang Q., Xu X.P., Kong X.M., Shi W.F. USP4 positively regulates RLR-induced NF-κB activation by targeting TRAF6 for K48-linked deubiquitination and inhibits enterovirus 71 replication. Sci. Rep. 2018;8:13418. doi: 10.1038/s41598-018-31734-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun W., Tan X., Shi Y., Xu G., Mao R., Gu X., Fan Y., Yu Y., Burlingame S., Zhang H., et al. USP11 negatively regulates TNFα-induced NF-κB activation by targeting on IκBα. Cell. Signal. 2010;22:386–394. doi: 10.1016/j.cellsig.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mialki R.K., Zhao J., Wei J., Mallampalli D.F., Zhao Y. Overexpression of USP14 protease reduces I-κB protein levels and increases cytokine release in lung epithelial cells. J. Biol. Chem. 2013;288:15437–15441. doi: 10.1074/jbc.C112.446682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meng Q., Cai C., Sun T., Wang Q., Xie W., Wang R., Cui J. Reversible ubiquitination shapes NLRC5 function and modulates NF-κB activation switch. J. Cell Biol. 2015;211:1025–1040. doi: 10.1083/jcb.201505091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Villeneuve N.F., Tian W., Wu T., Sun Z., Lau A., Chapman E., Fang D., Zhang D.D. USP15 negatively regulates Nrf2 through deubiquitination of Keap1. Mol. Cell. 2013;51:68–79. doi: 10.1016/j.molcel.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu X., Li H., Zhong B., Blonska M., Gorjestani S., Yan M., Tian Q., Zhang D.E., Lin X., Dong C. USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J. Exp. Med. 2013;210:1575–1590. doi: 10.1084/jem.20122327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang Z., Xian H., Hu J., Tian S., Qin Y., Wang R.F., Cui J. USP18 negatively regulates NF-κB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 2015;5:12738. doi: 10.1038/srep12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lei C.Q., Wu X., Zhong X., Jiang L., Zhong B., Shu H.B. USP19 inhibits TNF-α- and IL-1β-triggered NF-κB activation by deubiquitinating TAK1. J. Immunol. 2019;203:259–268. doi: 10.4049/jimmunol.1900083. [DOI] [PubMed] [Google Scholar]
- 79.Yasunaga J., Lin F.C., Lu X., Jeang K.T. Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-κB signaling. J. Virol. 2011;85:6212–6219. doi: 10.1128/JVI.00079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu C., Wang L., Chen W., Zhao S., Yin C., Lin Y., Jiang A., Zhang P. USP35 activated by miR let-7a inhibits cell proliferation and NF-κB activation through stabilization of ABIN-2. Oncotarget. 2015;6:27891–27906. doi: 10.18632/oncotarget.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Y.C., Wu Y.S., Hung C.Y., Wang S.A., Young M.J., Hsu T.I., Hung J.J. USP24 induces IL-6 in tumor-associated microenvironment by stabilizing p300 and β-TrCP and promotes cancer malignancy. Nat. Commun. 2018;9:3996. doi: 10.1038/s41467-018-06178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schweitzer K., Naumann M. CSN-associated USP48 confers stability to nuclear NF-κB/RelA by trimming K48-linked Ub-chains. Biochim. Biophys. Acta. 2015;1853:453–469. doi: 10.1016/j.bbamcr.2014.11.028. [DOI] [PubMed] [Google Scholar]
- 83.Milojevic T., Reiterer V., Stefan E., Korkhov V.M., Dorostkar M.M., Ducza E., Ogris E., Boehm S., Freissmuth M., Nanoff C. The ubiquitin-specific protease Usp4 regulates the cell surface level of the A2A receptor. Mol. Pharmacol. 2006;69:1083–1094. doi: 10.1124/mol.105.015818. [DOI] [PubMed] [Google Scholar]
- 84.Berlin I., Higginbotham K.M., Dise R.S., Sierra M.I., Nash P.D. The deubiquitinating enzyme USP8 promotes trafficking and degradation of the chemokine receptor 4 at the sorting endosome. J. Biol. Chem. 2010;285:37895–37908. doi: 10.1074/jbc.M110.129411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mines M.A., Goodwin J.S., Limbird L.E., Cui F.F., Fan G.H. Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK ativation. J. Biol. Chem. 2009;284:5742–5752. doi: 10.1074/jbc.M808507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berthouze M., Venkataramanan V., Li Y., Shenoy S.K. The deubiquitinases USP33 and USP20 coordinate β2 adrenergic receptor recycling and resensitization. EMBO J. 2009;28:1684–1696. doi: 10.1038/emboj.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borodovsky A., Kessler B.M., Casagrande R., Overkleeft H.S., Wilkinson K.D., Ploegh H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001;20:5187–5196. doi: 10.1093/emboj/20.18.5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borodovsky A., Ovaa H., Kolli N., Gan-Erdene T., Wilkinson K.D., Ploegh H.L., Kessler B.M. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem. Biol. 2002;9:1149–1159. doi: 10.1016/S1074-5521(02)00248-X. [DOI] [PubMed] [Google Scholar]
- 89.Sommer S., Weikart N.D., Linne U., Mootz H.D. Covalent inhibition of SUMO and ubiquitin-specific cysteine proteases by an in situ thiol-alkyne addition. Bioorg. Med. Chem. 2013;21:2511–2517. doi: 10.1016/j.bmc.2013.02.039. [DOI] [PubMed] [Google Scholar]
- 90.Gjonaj L., Sapmaz A., Flierman D., Janssen G.M.C., van Veelen P.A., Ovaa H. Development of a DUB-selective fluorogenic substrate. Chem. Sci. 2019;10:10290–10296. doi: 10.1039/C9SC02226K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dang L.C., Melandri F.D., Stein R.L. Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry. 1998;37:1868–1879. doi: 10.1021/bi9723360. [DOI] [PubMed] [Google Scholar]
- 92.Hassiepen U., Eidhoff U., Meder G., Bulber J.F., Hein A., Bodendorf U., Lorthiois E., Martoglio B. A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin-rhodamine110-glycine as substrate. Anal. Biochem. 2007;371:201–207. doi: 10.1016/j.ab.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 93.Tirat A., Schilb A., Riou V., Leder L., Gerhartz B., Zimmermann J., Worpenberg S., Eldhoff U., Freuler F., Stettler T., et al. Synthesis and characterization of fluorescent ubiquitin derivatives as highly sensitive substrates for the deubiquitinating enzymes UCH-L3 and USP-2. Anal. Biochem. 2005;343:244–255. doi: 10.1016/j.ab.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 94.Nicholson B., Leach C.A., Goldenberg S.J., Francis D.M., Kodrasov M.P., Tian X., Shanks J., Sterner D.E., Bernal A., Mattern M.R., et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008;17:1035–1043. doi: 10.1110/ps.083450408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goldenberg S.J., McDermott J.L., Butt T.R., Mattern M.R., Nicholson B. Strategies for the identification of novel inhibitors of deubiquitinating enzymes. Biochem. Soc. Trans. 2008;36:828–832. doi: 10.1042/BST0360828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Geurink P.P., El Oualid F., Jonker A., Hameed D.S., Ovaa H. A general chemical ligation approach towards isopeptide-linked ubiquitin and ubiquitin-like assay reagents. ChemBioChem. 2012;13:293–297. doi: 10.1002/cbic.201100706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ohayon S., Spasser L., Aharoni A., Brik A. Targeting deubiquitinases enabled by chemical synthesis of proteins. J. Am. Chem. Soc. 2012;134:3281–3289. doi: 10.1021/ja2116712. [DOI] [PubMed] [Google Scholar]
- 98.Horton R.A., Strachan E.A., Vogel K.W., Riddle S.M. A substrate for deubiquitinating enzymes based on time-resolved fluorescence resonance energy transfer between terbium and yellow fluorescent protein. Anal. Biochem. 2007;360:138–143. doi: 10.1016/j.ab.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 99.Faesen A.C., Luna-Vargas M.P., Geurink P.P., Clerici M., Merkx R., van Dijk W.J., Hameed D.S., El Oualid F., Ovaa H., Sixma T.K. The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem. Biol. 2011;18:1550–1561. doi: 10.1016/j.chembiol.2011.10.017. [DOI] [PubMed] [Google Scholar]
- 100.Wang Z., Xie W., Zhu M., Zhou H. Development of a highly reliable assay for ubiquitin-specific protease 2 inhibitors. Bioorg. Med. Chem. Lett. 2017;27:4015–4018. doi: 10.1016/j.bmcl.2017.07.059. [DOI] [PubMed] [Google Scholar]
- 101.Ritorto M.S., Ewan R., Perez-Oliva A.B., Knebel A., Buhrlage S.J., Wightman M., Kelly S.M., Wood N.T., Virdee S., Gray N.S., et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014;5:4763. doi: 10.1038/ncomms5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Berndtsson M., Beaujouin M., Rickardson L., Havelka A.M., Larsson R., Westman J., Liaudet-Coopman E., Linder S. Induction of the lysosomal apoptosis pathway by inhibitors of the ubiquitin-proteasome system. Int. J. Cancer. 2009;124:1463–1469. doi: 10.1002/ijc.24004. [DOI] [PubMed] [Google Scholar]
- 103.D’Arcy P., Brnjic S., Olofsson M.H., Fryknas M., Lindsten K., De Cesare M., Perego P., Sadeghi B., Hassan M., Larsson R., et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011;17:1636–1640. doi: 10.1038/nm.2536. [DOI] [PubMed] [Google Scholar]
- 104.D’Arcy P., Linder S. Proteasome deubiquitinases as novel targets for cancer therapy. Int. J. Biochem. Cell Biol. 2012;44:1729–1738. doi: 10.1016/j.biocel.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 105.Chitta K., Paulus A., Akhtar S., Blake M.K., Caulfield T.R., Novak A.J., Ansell S.M., Advani P., Ailawadhi S., Sher T., et al. Targeted inhibition of the deubiquitinating enzymes, USP14 and UCHL5, induces proteotoxic stress and apoptosis in Waldenstrom macroglobulinaemia tumour cells. Br. J. Haematol. 2015;169:377–390. doi: 10.1111/bjh.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tian Z., D’Arcy P., Wang X., Ray A., Tai Y.T., Hu Y., Carrasco R.D., Richardson P., Linder S., Chauhan D., et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123:706–716. doi: 10.1182/blood-2013-05-500033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brnjic S., Mazurkiewicz M., Fryknas M., Sun C., Zhang X., Larsson R., D’Arcy P., Linder S. Induction of tumor cell apoptosis by a proteasome deubiquitinase inhibitor is associated with oxidative stress. Antioxid. Redox Signal. 2014;21:2271–2285. doi: 10.1089/ars.2013.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang X., Espinosa B., Saei A.A., D’Arcy P., Zubarev R.A., Linder S. Oxidative stress induced by the deubiquitinase inhibitor b-AP15 is associated with mitochondrial impairment. Oxid. Med. Cell. Longev. 2019;2019:1659468. doi: 10.1155/2019/1659468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang X., D’Arcy P., Caulfield T.R., Paulus A., Chitta K., Mohanty C., Gullbo J., Chanan-Khan A., Linder S. Synthesis and evaluation of derivatives of the proteasome deubiquitinase inhibitor b-AP15. Chem. Biol. Drug Des. 2015;86:1036–1048. doi: 10.1111/cbdd.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Paulus A., Akhtar S., Caulfield T.R., Samuel K., Yousaf H., Bashir Y., Paulus S.M., Tran D., Hudec R., Cogen D., et al. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib- or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016;6:e492. doi: 10.1038/bcj.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rowinsky E.K., Paner A., Berdeja J.G., Paba-Prada C., Venugopal P., Porkka K., Gullbo J., Linder S., Loskog A., Richardson P.G., et al. Phase 1 study of the protein deubiquitinase inhibitor VLX1570 in patients with relapsed and/or refractory multiple myeloma. Investig. New Drugs. 2020;38:1448–1453. doi: 10.1007/s10637-020-00915-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kharel P., Uprety D., Chandra A.B., Hu Y., Belur A.A., Dhakal A. Bortezomib-induced pulmonary toxicity: A case report and review of literature. Case Rep. Med. 2018;2018:2913124. doi: 10.1155/2018/2913124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen J., Dexheimer T.S., Ai Y., Liang Q., Villamil M.A., Inglese J., Maloney D.J., Jadhav A., Simeonov A., Zhuang Z. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol. 2011;18:1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mistry H., Hsieh G., Buhrlage S.J., Huang M., Park E., Cuny G.D., Galinsky I., Stone R.M., Gray N.S., D’Andrea A.D., et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dexheimer T.S., Rosenthal A.S., Luci D.K., Liang Q., Villamil M.A., Chen J., Sun H., Kerns E.H., Simeonov A., Jadhav A., et al. Synthesis and structure-activity relationship studies of N-benzyl-2-phenylpyrimidin-4-amine derivatives as potent USP1/UAF1 deubiquitinase inhibitors with anticancer activity against nonsmall cell lung cancer. J. Med. Chem. 2014;57:8099–8110. doi: 10.1021/jm5010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liang Q., Dexheimer T.S., Zhang P., Rosenthal A.S., Villamil M.A., You C., Zhang Q., Chen J., Ott C.A., Sun H., et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014;10:298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ohayon S., Refua M., Hendler A., Aharoni A., Brik A. Harnessing the oxidation susceptibility of deubiquitinases for inhibition with small molecules. Angew. Chem. Int. Ed. Engl. 2015;54:599–603. doi: 10.1002/ange.201408411. [DOI] [PubMed] [Google Scholar]
- 118.Davis M.I., Pragani R., Fox J.T., Shen M., Parmar K., Gaudiano E.F., Liu L., Tanega C., McGee L., Hall M.D., et al. Small Molecule Inhibition of the Ubiquitin-specific Protease USP2 Accelerates cyclin D1 Degradation and Leads to Cell Cycle Arrest in Colorectal Cancer and Mantle Cell Lymphoma Models. J. Biol. Chem. 2016;291:24628–24640. doi: 10.1074/jbc.M116.738567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Magiera K., Tomala M., Kubica K., De Cesare V., Trost M., Zieba B.J., Kachamakova-Trojanowska N., Les M., Dubin G., Holak T.A., et al. Lithocholic Acid Hydroxyamide Destabilizes Cyclin D1 and Induces G0/G1 Arrest by Inhibiting Deubiquitinase USP2a. Cell Chem. Biol. 2017;24:458–470.e18. doi: 10.1016/j.chembiol.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tomala M.D., Magiera-Mularz K., Kubica K., Krzanik S., Zieba B., Musielak B., Pustula M., Popowicz G.M., Sattler M., Dubin G., et al. Identification of small-molecule inhibitors of USP2a. Eur. J. Med. Chem. 2018;150:261–267. doi: 10.1016/j.ejmech.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 121.Chuang S.J., Cheng S.C., Tang H.C., Sun C.Y., Chou C.Y. 6-Thioguanine is a noncompetitive and slow binding inhibitor of human deubiquitinating protease USP2. Sci. Rep. 2018;8:3102. doi: 10.1038/s41598-018-21476-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Altun M., Kramer H.B., Willems L.I., McDermott J.L., Leach C.A., Goldenberg S.J., Kumar K.G., Konietzny R., Fischer R., Kogan E., et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 123.Seiberlich V., Goldbaum O., Zhukareva V., Richter-Landsberg C. The small molecule inhibitor PR-619 of deubiquitinating enzymes affects the microtubule network and causes protein aggregate formation in neural cells: Implications for neurodegenerative diseases. Biochim. Biophys. Acta. 2012;1823:2057–2068. doi: 10.1016/j.bbamcr.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 124.Issaenko O.A., Amerik A.Y. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle. 2012;11:1804–1817. doi: 10.4161/cc.20174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vamisetti G.B., Meledin R., Gopinath P., Brik A. Halogen Substituents in the Isoquinoline Scaffold Switches the Selectivity of Inhibition between USP2 and USP7. ChemBioChem. 2019;20:282–286. doi: 10.1002/cbic.201800612. [DOI] [PubMed] [Google Scholar]
- 126.Okada K., Ye Y.Q., Taniguchi K., Yoshida A., Akiyama T., Yoshioka Y., Onose J., Koshino H., Takahashi S., Yajima A., et al. Vialinin A is a ubiquitin-specific peptidase inhibitor. Bioorg. Med. Chem. Lett. 2013;23:4328–4331. doi: 10.1016/j.bmcl.2013.05.093. [DOI] [PubMed] [Google Scholar]
- 127.Cao M.N., Zhou Y.B., Gao A.H., Cao J.Y., Gao L.X., Sheng L., Xu L., Su M.B., Cao X.C., Han M.M., et al. Curcusone D, a novel ubiquitin-proteasome pathway inhibitor via ROS-induced DUB inhibition, is synergistic with bortezomib against multiple myeloma cell growth. Biochim. Biophys. Acta. 2014;1840:2004–2013. doi: 10.1016/j.bbagen.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 128.Kapuria V., Peterson L.F., Fang D., Bornmann W.G., Talpaz M., Donato N.J. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265–9276. doi: 10.1158/0008-5472.CAN-10-1530. [DOI] [PubMed] [Google Scholar]
- 129.Luo H., Jing B., Xia Y., Zhang Y., Hu M., Cai H., Tong Y., Zhou L., Yang L., Yang J., et al. WP1130 reveals USP24 as a novel target in T-cell acute lymphoblastic leukemia. Cancer Cell Int. 2019;19:56. doi: 10.1186/s12935-019-0773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu H., Chen W., Liang C., Chen B.W., Zhi X., Zhang S., Zheng X., Bai X., Liang T. WP1130 increases doxorubicin sensitivity in hepatocellular carcinoma cells through usp9x-dependent p53 degradation. Cancer Lett. 2015;361:218–225. doi: 10.1016/j.canlet.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 131.Wang S., Kollipara R.K., Srivastava N., Li R., Ravindranathan P., Hernandez E., Freeman E., Humphries C.G., Kapur P., Lotan Y., et al. Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc. Natl. Acad. Sci. USA. 2014;111:4251–4256. doi: 10.1073/pnas.1322198111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Peterson L.F., Sun H., Liu Y., Potu H., Kandarpa M., Ermann M., Courtney S.M., Young M., Showalter H.D., Sun D., et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood. 2015;125:3588–3597. doi: 10.1182/blood-2014-10-605584. [DOI] [PubMed] [Google Scholar]
- 133.Colland F., Formstecher E., Jacq X., Reverdy C., Planquette C., Conrath S., Trouplin V., Bianchi J., Aushev V.N., Camonis J., et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009;8:2286–2295. doi: 10.1158/1535-7163.MCT-09-0097. [DOI] [PubMed] [Google Scholar]
- 134.Reverdy C., Conrath S., Lopez R., Planquette C., Atmanene C., Collura V., Harpon J., Battaglia V., Vivat V., Sippl W., et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 2012;19:467–477. doi: 10.1016/j.chembiol.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 135.Yamaguchi M., Miyazaki M., Kodrasov M.P., Rotinsulu H., Losung F., Mangindaan R.E., de Voogd N.J., Yokosawa H., Nicholson B., Tsukamoto S. Spongiacidin C, a pyrrole alkaloid from the marine sponge Stylissa massa, functions as a USP7 inhibitor. Bioorg. Med. Chem. Lett. 2013;23:3884–3886. doi: 10.1016/j.bmcl.2013.04.066. [DOI] [PubMed] [Google Scholar]
- 136.Kategaya L., Di Lello P., Rouge L., Pastor R., Clark K.R., Drummond J., Kleinheinz T., Lin E., Upton J.P., Prakash S., et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550:534–538. doi: 10.1038/nature24006. [DOI] [PubMed] [Google Scholar]
- 137.Lamberto I., Liu X., Seo H.S., Schauer N.J., Iacob R.E., Hu W., Das D., Mikhailova T., Weisberg E.L., Engen J.R., et al. Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem. Biol. 2017;24:1490–1500.e11. doi: 10.1016/j.chembiol.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gavory G., O’Dowd C.R., Helm M.D., Flasz J., Arkoudis E., Dossang A., Hughes C., Cassidy E., McClelland K., Odrzywol E., et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat. Chem. Biol. 2018;14:118–125. doi: 10.1038/nchembio.2528. [DOI] [PubMed] [Google Scholar]
- 139.O’Dowd C.R., Helm M.D., Rountree J.S.S., Flasz J.T., Arkoudis E., Miel H., Hewitt P.R., Jordan L., Barker O., Hughes C., et al. Identification and structure-guided development of pyrimidinone based USP7 inhibitors. ACS Med. Chem. Lett. 2018;9:238–243. doi: 10.1021/acsmedchemlett.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ioannidis S., Talbot A.C., Follows B., Buckmelter A.J., Wang M., Campbell A.-M., Schmidt D.R., Guerin D.J., Caravella J.A., Diebold R.B., et al. Pyrrolo and Pyrazolopyrimidines as Ubiquitin-Specific Protease 7 Inhibitors. 09902728. U.S. Patent. 2018 Feb 27;
- 141.Turnbull A.P., Ioannidis S., Krajewski W.W., Pinto-Fernandez A., Heride C., Martin A.C.L., Tonkin L.M., Townsend E.C., Buker S.M., Lancia D.R., et al. Molecular basis of USP7 inhibition by selective small-molecule inhibitors. Nature. 2017;550:481–486. doi: 10.1038/nature24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Di Lello P., Pastor R., Murray J.M., Blake R.A., Cohen F., Crawford T.D., Drobnick J., Drummond J., Kategaya L., Kleinheinz T., et al. Discovery of small-molecule inhibitors of ubiquitin specific protease 7 (USP7) using integrated NMR and in Silico techniques. J. Med. Chem. 2017;60:10056–10070. doi: 10.1021/acs.jmedchem.7b01293. [DOI] [PubMed] [Google Scholar]
- 143.Li X., Kong L., Yang Q., Duan A., Ju X., Cai B., Chen L., An T., Li Y. Parthenolide inhibits ubiquitin-specific peptidase 7 (USP7), Wnt signaling, and colorectal cancer cell growth. J. Biol. Chem. 2020;295:3576–3589. doi: 10.1074/jbc.RA119.011396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Colombo M., Vallese S., Peretto I., Jacq X., Rain J.C., Colland F., Guedat P. Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem. 2010;5:552–558. doi: 10.1002/cmdc.200900409. [DOI] [PubMed] [Google Scholar]
- 145.Weisberg E.L., Schauer N.J., Yang J., Lamberto I., Doherty L., Bhatt S., Nonami A., Meng C., Letai A., Wright R., et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat. Chem. Biol. 2017;13:1207–1215. doi: 10.1038/nchembio.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhang C., Lu J., Zhang Q.W., Zhao W., Guo J.H., Liu S.L., Wu Y.L., Jiang B., Gao F.H. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells. Int. J. Biochem. Cell Biol. 2016;79:209–221. doi: 10.1016/j.biocel.2016.08.025. [DOI] [PubMed] [Google Scholar]
- 147.Lee G., Oh T.I., Um K.B., Yoon H., Son J., Kim B.M., Kim H.I., Kim H., Kim Y.J., Lee C.S., et al. Small-molecule inhibitors of USP7 induce apoptosis through oxidative and endoplasmic reticulum stress in cancer cells. Biochem. Biophys. Res. Commun. 2016;470:181–186. doi: 10.1016/j.bbrc.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 148.Chauhan D., Tian Z., Nicholson B., Kumar K.G., Zhou B., Carrasco R., McDermott J.L., Leach C.A., Fulcinniti M., Kodrasov M.P., et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012;22:345–358. doi: 10.1016/j.ccr.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang M., Zhang Y., Wang T., Zhang J., Zhou Z., Sun Y., Wang S., Shi Y., Luan X., Zhang Y., et al. The USP7 inhibitor P5091 induces cell death in ovarian cancers with different P53 status. Cell. Physiol. Biochem. 2017;43:1755–1766. doi: 10.1159/000484062. [DOI] [PubMed] [Google Scholar]
- 150.An T., Gong Y., Li X., Kong L., Ma P., Gong L., Zhu H., Yu C., Liu J., Zhou H., et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem. Pharmacol. 2017;131:29–39. doi: 10.1016/j.bcp.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 151.Weinstock J., Wu J., Cao P., Kingsbury W.D., McDermott J.L., Kodrasov M.P., McKelvey D.M., Suresh Kumar K.G., Goldenberg S.J., Mattern M.R., et al. Selective dual inhibitors of the cancer-related deubiquitylating proteases USP7 and USP47. ACS Med. Chem. Lett. 2012;3:789–792. doi: 10.1021/ml200276j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu J., Xia H., Kim M., Xu L., Li Y., Zhang L., Cai Y., Norberg H.V., Zhang T., Furuya T., et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Burkhart R.A., Peng Y., Norris Z.A., Tholey R.M., Talbott V.A., Liang Q., Ai Y., Miller K., Lal S., Cozzitorto J.A., et al. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol. Cancer Res. 2013;11:901–911. doi: 10.1158/1541-7786.MCR-12-0699. [DOI] [PubMed] [Google Scholar]
- 154.Ward S.J., Gratton H.E., Indrayudha P., Michavila C., Mukhopadhyay R., Maurer S.K., Caulton S.G., Emsley J., Dreveny I. The structure of the deubiquitinase USP15 reveals a misaligned catalytic triad and an open ubiquitin-binding channel. J. Biol. Chem. 2018;293:17362–17374. doi: 10.1074/jbc.RA118.003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee B.H., Lee M.J., Park S., Oh D.C., Elsasser S., Chen P.C., Gartner C., Dimova N., Hanna J., Gygi S.P., et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Boselli M., Lee B.H., Robert J., Prado M.A., Min S.W., Cheng C., Silva M.C., Seong C., Elsasser S., Hatle K.M., et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017;292:19209–19225. doi: 10.1074/jbc.M117.815126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang Y., Jiang Y., Ding S., Li J., Song N., Ren Y., Hong D., Wu C., Li B., Wang F., et al. Small molecule inhibitors reveal allosteric regulation of USP14 via steric blockade. Cell Res. 2018;28:1186–1194. doi: 10.1038/s41422-018-0091-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen X., Yang Q., Xiao L., Tang D., Dou Q.P., Liu J. Metal-based proteasomal deubiquitinase inhibitors as potential anticancer agents. Cancer Metastasis Rev. 2017;36:655–668. doi: 10.1007/s10555-017-9701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu N., Li X., Huang H., Zhao C., Liao S., Yang C., Liu S., Song W., Lu X., Lan X., et al. Clinically used antirheumatic agent auranofin is a proteasomal deubiquitinase inhibitor and inhibits tumor growth. Oncotarget. 2014;5:5453–5471. doi: 10.18632/oncotarget.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu N., Liu C., Li X., Liao S., Song W., Yang C., Zhao C., Huang H., Guan L., Zhang P., et al. A novel proteasome inhibitor suppresses tumor growth via targeting both 19S proteasome deubiquitinases and 20S proteolytic peptidases. Sci. Rep. 2014;4:5240. doi: 10.1038/srep05240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhao C., Chen X., Yang C., Zang D., Lan X., Liao S., Zhang P., Wu J., Li X., Liu N., et al. Repurposing an antidandruff agent to treating cancer: Zinc pyrithione inhibits tumor growth via targeting proteasome-associated deubiquitinases. Oncotarget. 2017;8:13942–13956. doi: 10.18632/oncotarget.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhao C., Chen X., Zang D., Lan X., Liao S., Yang C., Zhang P., Wu J., Li X., Liu N., et al. Platinum-containing compound platinum pyrithione is stronger and safer than cisplatin in cancer therapy. Biochem. Pharmacol. 2016;116:22–38. doi: 10.1016/j.bcp.2016.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhao C., Chen X., Zang D., Lan X., Liao S., Yang C., Zhang P., Wu J., Li X., Liu N., et al. A novel nickel complex works as a proteasomal deubiquitinase inhibitor for cancer therapy. Oncogene. 2016;35:5916–5927. doi: 10.1038/onc.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wrigley J.D., Gavory G., Simpson I., Preston M., Plant H., Bradley J., Goeppert A.U., Rozycka E., Davies G., Walsh J., et al. Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem. Biol. 2017;12:3113–3125. doi: 10.1021/acschembio.7b00334. [DOI] [PubMed] [Google Scholar]
- 165.Kluge A.F., Lagu B.R., Maiti P., Jaleel M., Webb M., Malhotra J., Mallat A., Srinivas P.A., Thompson J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018;28:2655–2659. doi: 10.1016/j.bmcl.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 166.Yue W., Chen Z., Liu H., Yan C., Chen M., Feng D., Yan C., Wu H., Du L., Wang Y., et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014;24:482–496. doi: 10.1038/cr.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable. No new data were created or analyzed in this study. Data sharing is not applicable to this article.